

Abstract
Cardiac metabolism is highly adaptive to changes in fuel availability and the energy demand of the heart. This metabolic flexibility is key for the heart to maintain its output during the development and in response to stress. Alterations in substrate preference have been observed in multiple disease states; a clear understanding of their impact on cardiac function in the long term is critical for the development of metabolic therapies. In addition, the contribution of cellular metabolism to growth, survival, and other signalling pathways through the generation of metabolic intermediates has been increasingly noted, adding another layer of complexity to the impact of metabolism on cardiac function. In a quest to understand the complexity of the cardiac metabolic network, genetic tools have been engaged to manipulate cardiac metabolism in a variety of mouse models. The ability to engineer cardiac metabolism in vivo has provided tremendous insights and brought about conceptual innovations. In this review, we will provide an overview of the cardiac metabolic network and highlight alterations observed during cardiac development and pathological hypertrophy. We will focus on consequences of altered substrate preference on cardiac response to chronic stresses through energy providing and non-energy providing pathways.
Keywords: Cardiac metabolism, Pathological hypertrophy, Metabolic signalling, Metabolic flexibility, Energy metabolism
1. Introduction
Cardiac function is highly dependent on adequate energy supply, as the heart must contract incessantly to deliver blood and to guarantee adequate oxygen supply to all organs of the body. The human heart produces and consumes on average 6 kg of ATP on a daily basis, which is over 15 times its own weight.1 In order to meet the high energetic demand, the heart possesses high flexibility to use any kind of substrates for ATP production.1–3 This becomes especially important under conditions of starvation, increased workload and acute and chronic stress, when the heart needs to adjust its substrate preference in order to meet the energetic demand. Shift of substrate preference can be beneficial in the short term to maintain energy supply, however, it can become detrimental and results in adverse cardiac function in the long-term. In addition, the role of cardiac substrate metabolism extends beyond energy provision as metabolites of multiple metabolic pathways can act as signalling molecules, affecting cardiomyocyte performance and cardiac function.
In this review article, we will focus on the importance of metabolic flexibility for maintaining cardiac function in the healthy and diseased state. We will discuss the contribution of different substrate classes to normal heart function and their role in the development and progression of pathological hypertrophy. We will further emphasize on the consequences of deranged substrate utilization on non-energy providing pathways.
2. Cardiac energy metabolism in health and disease
Although the heart is capable of using all classes of substrates, including carbohydrates, lipids, amino acids, and ketone bodies for energy provision, its substrate preference changes throughout the life cycle as well as under physiological and pathological stresses. The metabolic flexibility allows the heart to adapt to environmental changes but shifts in fuel selectivity can become detrimental in the diseased heart.4 Below, we will discuss major changes in substrate utilization that occur during cardiac development and in the diseased heart (Figure 1).
Figure 1.
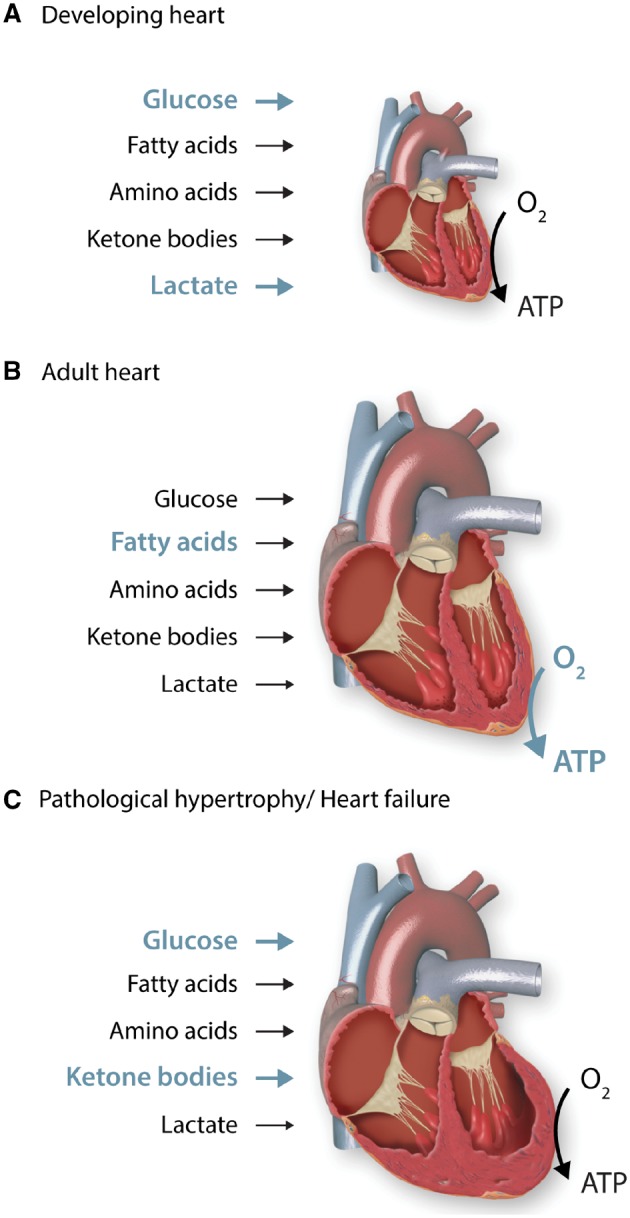
Contribution of different substrates to ATP production during cardiac development and disease progression. The heart is capable of using all classes of substrates, including carbohydrates, lipids, amino acids and ketone bodies to meet its energetic demands. Its substrate preference changes throughout the life cycle as well as under physiological and pathological conditions, which allows the heart to adapt to environmental changes. (A) The developing heart is highly dependent on aerobic glycolysis and lactate oxidation, while oxidation of fatty acids plays only a minor role. This metabolic phenotype is well suited for the biosynthesis of cellular building blocks, which are necessary for cardiomyocyte proliferation and cellular growth. (B) The adult heart is exposed to an increased haemodynamic load and oxygen tension. It mainly uses oxidative metabolism for ATP production, which is reflected by a substantial increase in mitochondrial volume mass. Fatty acids become the predominant fuel for the adult hearts. (C) Hearts with pathological hypertrophy revert to a foetal metabolic profile, demonstrating increased reliance on glucose and reduced oxidative capacity. In advanced heart failure, increased use of ketone bodies as fuel has been suggested.
2.1 Foetal development
The heart starts beating around day 23 in human embryogenesis and pumps blood in the 4th week.5,6 Circulating levels of glucose in foetuses are similar to those in newborns and adults, but circulating levels of fatty acids are very low.7 Lactate levels are also high in the foetal blood.8 Thus, the foetal heart mainly uses glucose and lactate, while oxidation of fatty acids plays only a minor role (Figure 1A).9–11
In contrast to the adult, the foetus resides in a low oxygen environment.12 Hypoxia-inducible factor 1 (Hif-1α) is a transcription factor activated at low oxygen levels such as hypoxic conditions. Besides controlling angiogenesis, Hif-1α regulates expression of genes involved in glucose metabolism, especially those in glycolysis.13 Consequently, various enzymes downstream of Hif-1α which are involved in glycolysis are highly expressed in the foetal heart (Glucose receptor 1 (GLUT1), hexokinase 1 (HKI), phosphofructokinase 1, lactate dehydrogenase A). In contrast, PGC-1α (peroxisome proliferator activated receptor γ (PPARγ) coactivator 1α) and PPARα, key regulators of fatty acid metabolism and mitochondrial biogenesis, are expressed at low levels in the fetal heart.14 Thus, enzymes of fatty acid oxidation (FAO) are expressed at low levels (carnitine palmiotyltransferase 1b (CPT-1b), malonyl-CoA decarboxylase).15 The hypoxic, highly glycolytic metabolic profile is well suited for the biosynthesis of cellular lipids, amino acids, nucleotides, and other macromolecules, which are needed to maintain cell growth during the phase of foetal development. The importance of metabolic reprogramming in cell growth is also supported by a recent study demonstrating that subjecting adult mice to hypoxia activates cell cycle entry in post-mitotic cardiomyocytes.16
2.2 Neonatal heart
During the transition from foetal to neonatal life, the heart is exposed to an increased haemodynamic load and oxygen tension, which, in turn, drives its metabolic transformation. Early after birth, cardiomyocytes stop proliferating but instead begin to hypertrophy in order to increase cardiac size and mass.10 In parallel, an explosive increase of mitochondrial biogenesis substantially increases the oxidative capacity of the heart.17 Within the first week after birth, cardiac reliance on glucose drops significantly.18 Circulating lactate levels drop as well, while levels of free fatty acids and triacylglycerol (TAG) increase significantly.7,19,20 Consequently, during postnatal development, FAO becomes the major source of ATP generation, while the contribution from lactate oxidation and glycolysis decreases significantly.10 These metabolic changes are not only mediated by substrate availability but also driven by increasing oxygen levels and enhanced cardiac workload. Furthermore, they are accompanied by changes in expression levels of various metabolic enzymes: Expression of GLUT1 and acetyl-CoA carboxylase 2 (ACC2) drop as a consequence of reduced Hif-1α expression, while expression of insulin-sensitive glucose transporter GLUT4 and PGC-1α/PPARα downstream genes increases.10
2.3 Adult heart
Metabolic changes that progressed in the neonatal period are completed in the adult heart in parallel to cardiomyocyte maturation. In the adult heart, mitochondria make up about 1/3 of cardiomyocyte volume, reflecting a substantial increase in the oxidative capacity for energy provision.2,10 Under normoxic conditions, more than 95% of ATP generated in the heart is derived from oxidative phosphorylation in the mitochondria whereas the remaining 5% come from glycolysis (Figure 1B).10 Lactate oxidation is markedly reduced in the adult heart, mainly because of low circulating lactate levels. Similarly, the contribution of ketone bodies (acetoacetate, β-hydroxybutyrate (βOHB), and acetone) and amino acids to overall cardiac oxidative metabolism is considered to be minor because of low availability of these substrates under normal physiological conditions in the adult heart.
2.4 Pathological Hypertrophy/Heart Failure
Pathological cardiac hypertrophy is observed in numerous forms of cardiovascular diseases and is considered a maladaptive response to chronic pathological stress, eventually resulting in cardiac failure. The hypertrophic process is complex, involving a vast array of structural and functional events within the growing cell.21 A striking change in pathological hypertrophy is the shift in fuel metabolism from FAO to increased reliance on glucose and an overall reduced oxidative metabolism and energy reserve (Figure 1C).22,23 Increased glucose utilization in the hypertrophied heart is characterized by an upregulation of glucose uptake and glycolysis with either no change or a decrease in glucose oxidation, resulting in an uncoupling of glucose uptake and its oxidation.23,24 These changes, combined with a decrease in FAO, likely represent a reduced capacity for mitochondrial oxidative metabolism, ultimately reducing cardiac energy provision. Concurrently, gene expression in the hypertrophied heart reverts to a foetal profile and thus changes in hypertrophied hearts represent a reappearance of the foetal metabolic profile.25,26
In recent times, evidence of increased ketone body oxidation has been reported in experimental animal models as well as in human heart failure.27,28 In addition, amino acid metabolism and especially, branched-chain amino acid (BCAA) catabolism is deranged in pathological hypertrophy, leading to an accumulation of BCAAs.29,30 The metabolic consequences and functional relevance of these changes in the pathogenesis of cardiac hypertrophy and heart failure remain to be understood. As discussed earlier, the contribution of these substrates to ATP production in the heart is very limited under normal conditions. However, the biological role of these metabolic pathways in cardiac remodelling certainly warrants further investigation (also see sections below).
It is known that cardiac hypertrophy also occurs under physiological conditions such as during pregnancy or exercise training. Molecular and metabolic changes associated with physiological and pathological hypertrophy are distinct. The role of cardiac metabolism in physiological hypertrophy has been summarized in a recent review.31 We will focus on pathological hypertrophy in this article.
3. Understanding consequences of altered metabolism
The contribution of metabolic remodelling to the development of cardiac dysfunction has been the topic of intensive studies in the past decade. It is now appreciated that changes in cardiac metabolism and mitochondrial function precede cardiac dysfunction, indicating that metabolic remodelling is an early event in disease progression.32,33 Consequently, current research focuses on the identification of critical metabolic pathways that might be able to prevent or attenuate disease progression. Numerous studies have used bioengineered mice to dissect the role of a variety of metabolic pathways in cardiac stress response. In this section, we will summarize a number of key findings in these mouse models (Table 1). We will also summarize the impact of metabolic remodelling that extends beyond energy supply.
Table 1.
Models of altered substrate metabolism and implications for cardiac function
| Pathway | Gene | Knock-out (KO) or Transgenic (TG) | Metabolism | Function (baseline) | Pathological hypertrophy | Reference |
|---|---|---|---|---|---|---|
| Glucose uptake | Glucose transporter 1 | Cardiac specific TG | ↑ glucose uptake ↑ glycolysis | Normal | Normal function | 37, 38 |
| Similar hypertrophy as control | ||||||
| Glucose uptake | Glucose transporter 1 | Cardiac specific KO | ↓ glucose uptake ↓ glycolysis | Normal | Similar dysfunction/hypertrophy as control | 40 |
| ↑ FAO | Maintained FAO/glycolysis | |||||
| Glucose uptake | Glucose transporter 4 | Cardiac specific KO | ↑ glucose uptake ↑ glycolysis | Compensated hypertrophy, normal function | – | 41 |
| Glucose utilization | Hexokinase II | Cardiac specific TG | ↑ glycolysis | Normal | Normal function | 42, 43 |
| ↑ glycogen production | prevention of hypertrophy | |||||
| Glucose oxidation | Pyruvate dehydrogenase kinase 4 | Cardiac specific TG | ↓ glucose oxidation | Normal | - | 44 |
| ↑ FAO | ||||||
| Lipid metabolism | Peroxisome proliferator-activated receptor alpha | Whole body KO | ↓ FAO | Impaired (Cardiomyopathy) | - | 62, 63 |
| ↑ glucose oxidation | ||||||
| Impaired energy generation | ||||||
| Lipid metabolism | Peroxisome proliferator-activated receptor alpha | Cardiac specific TG | ↓ glucose oxidation | Impaired (Cardiac hypertrophy) | - | 61 |
| ↑ FAO | ||||||
| Lipid metabolism | Peroxisome proliferator-activated receptor β/γ | Cardiac specific KO | ↓ FAO | Impaired (Cardiomyopathy) | - | 64 |
| ↑ glucose oxidation | ||||||
| Lipid accumulation | ||||||
| Lipid metabolism | Peroxisome proliferator-activated receptor β/γ | Inducible cardiac specific TG | ↑ oxidative metabolism (glucose + FA) | Normal | – | 65 |
| FA uptake | FAT/CD36 | Inducible cardiac specific KO | Increased energetics | Normal | Worsened function and hypertrophy | 66-68 |
| ↓ FAO | ||||||
| ↑ glucose oxidation | ||||||
| FAO | Carnitine palmitoyltransferase 1b | Heterozygous whole body KO | ↓ FAO | Normal | Worsened function and hypertrophy | 69 |
| ↑ glucose oxidation | Lipid accumulation | |||||
| Cardiomyocyte death | ||||||
| FAO | Acetyl-CoA carboxylase 2 | Cardiac specific KO | ↑ FAO | Normal | Normal function | 75, 76 |
| ↓ glucose oxidation | prevention of hypertrophy | |||||
| Keton body metabolism | Succinyl-CoA:3-oxoacid-CoA transferase | Cardiac specific KO | Impaired ketone body oxidation | Normal | Worsened function and hypertrophy | 91 |
| normal glucose/FA use | ||||||
| BCAA metabolism | Mitochondrial-targeted 2C-type protein phosphatase | Whole body KO | BCAA+BCKA accumulation | Normal | Accelerated heart failure | 96-98 |
| BCAA metabolism | mitochondrial branched-chain aminotransferase | Whole body KO | BCAA accumulation | Hypertrophy | – | 93 |
3.1 Glucose metabolism
Glucose that is used by the heart either comes from uptake of exogenous glucose or is derived from glycogen stores. The glycogen pool in the heart is relatively small, but subjected to rapid turnover, thereby actively contributing to energy provision.2 Exogenous glucose enters the cell through either insulin-independent glucose transporters (GLUT1) or insulin-sensitive glucose transporters (GLUT4). GLUT4 is the predominant glucose receptor in the adult heart and accounts for the majority of glucose transport into the cardiomyocyte.34 Glucose uptake follows the intracellular glucose gradient, since it gets rapidly phosphorylated to glucose-6-phosphate (G6P) by hexokinase II (HKII). This reaction operates at near equilibrium, due to the high affinity of HKII for glucose. G6P then serves as a precursor for glycolysis as well as the pentose phosphate pathway (PPP), glycogenesis (glycogen formation) and the hexosamine biosynthetic pathway (HBP) (Figure 2A).
Figure 2.

Consequences of altered cardiac metabolism on non-energy providing functions. The primary role of cardiac metabolism is to provide energy for cardiac contraction. However, altered substrate utilization and metabolic remodelling in the hypertrophied heart can affect various cellular functions beyond energy supply. (A) Glucose metabolism and cardiac growth: After entering the cell, glucose is phosphorylated to glucose-6-phosphate (G6P) before feeding into glycolysis, the hexosamine biosynthesis pathway (HBP) or the pentose phosphate pathway (PPP). In cardiac hypertrophy, increased glucose reliance results in increased flux through all three pathways. Accumulation of G6P has been linked to the activation of the mechanistic target of rapamycin complex 1 (mTORC1) and cell growth. Enhanced flux through the HBP pathway results in increased protein O-GlcNAcylation and modulation of enzyme activity. Intermediates of the PPP have been recognized to act as signalling molecules (Xylulose-5-phosphate) or modulate redox homeostasis (NADPH). Recently, identification of cardiac lactate receptor (GPR81) expression proposes a role of lactate as a signalling molecule. (B) Fatty acid metabolism and cardiotoxicity: Cytosolic free fatty acids are activated by acyl CoA synthetase (ACS) to form fatty acyl-CoAs. Besides entering mitochondria for oxidation, acyl-CoAs can also form ceramides, diacylglycerol (DAG) and triacylglycerol (TAG). Formation of DAG can also occur through phospholipid breakdown which will result in protein kinase C (PKC) activation. DAG as well as ceramide species are increased in the hypertrophied heart and have been associated with lipotoxicity, while redirecting these toxic lipid intermediates into the TAG pool prevents lipotoxicity. TAG can be hydrolyzed by adipose tissue triglyceride lipase (ATGL) and can either enter the fatty acid oxidation pathway or serve as ligand for nuclear receptor activation. Acyl-CoAs have recently been identified to contribute to epigenetic regulation by acting as endogenous histone deacetylase inhibitors. (C) Ketone body metabolism and cellular signalling: Ketone bodies are oxidized in the heart for ATP generation. After entering mitochondria, they rapidly from acetyl-CoA via βOHB dehydrogenase (BDH1), succinyl-CoA:3-oxoacid-CoA transferase (SCOT) and mitochondrial acetyl-CoA acetyltransferase 1 (ACAT1). βOHB can further act as endogenous histone deacetylase inhibitor or as signalling molecule through its G protein coupled receptor (GPR109A/GPR41). (D) Branched-chain amino acids catabolism in cardiomyocyte growth and survival: Catabolism of the branched-chain amino acids (BCAA) feeds into the TCA cycle although their contribution to energy provision is rather minor under normal conditions. The first steps in BCAA catabolism yields branched-chain keto acids (BCKA) through mitochondrial branched-chain aminotransferase (BCATm) reaction. BCKA is processed by a dehydrogenase complex (BCKDH) to form CoA compounds, before further breakdown for TCA cycle. BCKDH is the rate-limiting enzyme in BCAA catabolism, it is activated by dephosphorylation through a mitochondrial-localized 2C-type protein phosphatase (PP2Cm). Accumulation of BCAA results in mitochondrial dysfunction, characterized by loss of mitochondrial membrane potential (ΔΨ) and mitochondrial permeability transition pore (mPTP) opening. BCKA accumulation stimulates ROS formation. Additionally, leucine can directly activate mTORC1 and simulate cell growth.
3.1.1 Animal models of altered glucose metabolism
The hypertrophied heart shows increased reliance on glucose for energy provision. This is considered a compensatory mechanism in the early stage since it improves oxygen efficiency for ATP synthesis where oxygen supply might become limited.2,4 However, enhanced glucose utilization, especially increased glycolysis, has also been proposed to contribute to disease progression and becoming maladaptive in the long-term.35,36 A variety of transgenic (TG) and knock-out (KO) mice targeting glucose utilization pathways (uptake, glycolysis and oxidation) have been generated to answer the question if altered glucose reliance itself is a driving force for cardiac dysfunction.
Mice with cardiac-specific overexpression of GLUT1 have a substantial increase in basal glucose uptake and glycolysis, but demonstrate normal cardiac function and lifespan, suggesting that increased glucose use per se does not harm the adult heart in the long term.37–39 When subjected to pressure overload by ascending aortic constriction (AAC), GLUT1 TG mice displayed improved myocardial energetics and maintained cardiac function, but demonstrated a similar degree of cardiomyocyte hypertrophy as control mice after AAC. On the other hand, cardiomyocyte-specific GLUT1 KO mice have reduced glycolysis and glucose oxidation and a reciprocal increase in FAO.40 GLUT1 KO mice demonstrated maintained glycolysis and FAO rates after transverse aortic constriction (TAC) but exhibited equivalent degree of cardiac hypertrophy and cardiac dysfunction compared to controls. The importance of glucose metabolism to maintain cardiac function under stress conditions is further underscored by studies of mice with a cardiac-specific deletion of GLUT4.41 GLUT4 KO mice have a compensatory upregulation of GLUT1 and an increase in basal glucose uptake, which results in cardiac hypertrophy with preserved cardiac contractile function. However, since GLUT1 TG mice demonstrate no cardiac phenotype at baseline, the GLUT4 KO phenotype is most likely explained by the loss of GLUT4 expression and the failure to increase insulin-stimulated glucose uptake under physiological conditions. This also indicates that GLUT4-mediated glucose uptake is necessary to maintain normal cardiac function.
To manipulate glucose utilization pathways, cardiac-specific HKII TG mice have been generated.42,43 HKII expression is increased in cardiac hypertrophy and it has been proposed that this accounts for increased glycolysis in the hypertrophied heart. Although HKII TG mice have increased glycolysis and glycogen content, they maintain normal cardiac function, indicating that increased HKII expression is not a direct cause for cardiac dysfunction. Furthermore, chronic isoproterenol infusion exerted no adverse effect on cardiac function in HKII TG mice and HKII TG mice rather demonstrated reduced cardiomyocytes hypertrophy.43 To specifically target glucose oxidation, pyruvate dehydrogenase kinase 4 (PDK 4) TG mice have been generated.44 PDK4 expression is increased in cardiac hypertrophy, inhibiting the pyruvate dehydrogenase complex and subsequently, glucose oxidation. PDK4 TG have reduced glucose oxidation and reciprocally increased FAO rates, which renders them insensitive to insulin, but demonstrate normal cardiac function, even after ischemic insult.44
In summary, these studies show that increased glucose reliance per se is not detrimental as long as the energetic demand is met in the normal heart. However, in the long term, the metabolic remodelling associated with increased reliance on glucose could impair the flexibility of the heart to use other substrates and contribute to disease progression. For example, increasing glucose reliance in GLUT1 TG hearts renders the heart more susceptible to cardiomyopathy in diet-induced obesity.45 Consistent with this notion, it has been observed that the human failing heart lacks metabolic flexibility and consequently, responds poorly to stress.46
3.1.2 Linking glucose metabolism to cardiac growth
Hearts with pathological hypertrophy display a mismatch between glycolysis and glucose oxidation, which results in the accumulation of glycolytic intermediates. Accumulation of G6P has been proposed to promote cell growth by activating mechanistic target of rapamycin complex 1 (mTORC1).35,36 The specific site that G6P interacts with mTORC1, however, remains to be defined. mTORC1 activation has been well established in pathological hypertrophy and inhibition of mTORC1 by rapamycin has been shown to attenuate pathological hypertrophy and maintain cardiac function.47 Although this suggests that mTORC1 activation in the heart is maladaptive, mTOR KO mice demonstrate accelerated HF progression, indicating that a certain level of mTORC1 activity is necessary to maintain cardiac function.48
Glycolytic intermediates such as G6P or fructose-6-phosphate (F6P) feed into accessory pathways that have important biological functions (Figure 2A). The hexosamine biosynthetic pathway (HBP) utilizes F6P to generate the metabolite uridine diphosphate N-acetylglucosamine (UDP–GlcNAc). This and other charged nucleotide sugars serve as the basis for biosynthesis of glycoproteins and other glycoconjugates. Additionally, posttranslational protein modification by O-GlcNacylation regulates the activity of a vast variety of enzymes. Several studies have demonstrated increased flux through the HBP and increased protein O-GlcNacylation in hearts with pathological hypertrophy. As protein O-GlcNAcylation is clearly maladaptive after ischemic insults, increasing flux through HBP blunted hypertrophic response in cultured cardiomyocytes, indicating distinct roles of protein O-GlcNAcylation during ischemic and non-ischemic stresses.49
While only a small fraction of G6P enters the pentose phosphate pathway (PPP) in the normal heart, activation of the PPP has been reported in pathological hypertrophy.50 The PPP generates NADPH, which is important in combating reactive oxygen species.51 Excessive NADPH produced through this pathway has been suggested to fuel superoxide generation and contributing to cardiac dysfunction.51 However, deficiency of glucose 6-phosphate dehydrogenase (G6PD), the first enzyme of PPP, aggravates cardiac dysfunction after TAC, arguing for an adaptive role of increased PPP activity to maintain cardiac redox homeostasis.52
3.2 Fatty acid metabolism
Fatty acid supply for the heart comes from plasma free fatty acids and the hydrolysis of triglyceride-rich lipoproteins via the actions of lipoprotein lipase (LPL) as well as endogenous TAG.53–55 Uptake of free fatty acids is facilitated by plasma membrane transporters, fatty acid translocase/cluster of differentiation (FAT/CD36), fatty acid transport protein (FATP) and to a smaller extent by diffusion across the membrane. In the cytosol, fatty acids are rapidly activated to form acyl-coenzyme A (CoA) via acyl CoA synthetase (ACS). At this point, cytosolic acyl-CoAs have several fates: Entry of acyl-CoAs into the mitochondria through CPT-1 ultimately leads to their oxidation.56 Cytosolic acyl-CoAs can enter the de novo synthesis pathway for ceramide synthesis (sphingolipids) or form diacylglycerol (DAG) through a series of reactions in the endoplasmic reticulum (ER). DAG is an important precursor for phospholipid (PL) synthesis and can serve as a lipid signalling molecule.57 DAG can further combine with another free acyl-CoA to form TAG through the action of diacylglycerol acyltransferase (DGAT) (Figure 2B). The cardiac TAG pool is highly dynamic. Fatty acids derived from cardiac TAG is not only a source of endogenous fuel but also an important mechanism regulating gene transcription.58,59
3.2.1 Animal models of altered fatty acid metabolism
Fatty acids are the main fuel for the adult heart and decreased FAO reduces the capacity for ATP production. However, increased FAO has been associated with increased oxidative stress while impaired FAO leads to the accumulation of toxic lipid intermediates especially in a state of fatty acid overload such as obesity.10,60 Studies using mouse models of altered fatty acid metabolism also present a complex picture but nevertheless suggest that maintaining a balance of fatty acid supply and oxidation is critical for normal heart function.
A key observation made in rodent models of cardiac pathological hypertrophy and failure is the downregulation of PPARα.61 PPARα is an important transcriptional regulator of cellular lipid metabolism and is abundantly expressed in the heart. Activation of PPARα promotes uptake, utilization, and catabolism of fatty acids by upregulation of genes involved in fatty acid metabolism (e.g. FAT/CD36, malonyl-CoA decarboxylase, CPT-1b, medium and long chain acyl-CoA dehydrogenases). Consequently, PPARα KO mice show severely impaired FAO and a significant increase in cardiac glucose oxidation. PPARα KO mice develop cardiomyopathy at old age and are incapable of maintaining cardiac energy provision at high workload.62,63 On the other hand, PPARα TG mice demonstrate increased fatty acid uptake and oxidation, and decreased glucose utilization, which results in cardiac hypertrophy and impaired cardiac function.61 PPARβ/γ is another PPAR family member and a ubiquitously expressed nuclear receptor. As PPARα, PPARβ/γ is an important regulator of fatty acid metabolism and is also involved in the transcriptional control of many of the same enzymes in the FAO pathways. Cardiac-specific deletion of PPARβ/γ causes severe cardiomyopathy and an increase in cardiac lipid accumulation.64 In contrast, PPARβ/γ TG mice demonstrate increased expression of genes involved in glucose and fatty acid metabolism with no evidence of lipid accumulation or cardiac dysfunction. These mice showed an increase in general oxidative metabolism, and were protected against cardiac dysfunction after TAC, while still demonstrating the same degree of cardiomyocyte hypertrophy.65 These studies demonstrate that perturbation of cardiac lipid metabolism has profound effects on cardiac function and stress response, but they do not distinguish the role of fatty acid uptake, oxidation or other pathways of fatty acid metabolism.
To determine if reduced FAO is adaptive in pathological hypertrophy, cellular and mitochondrial fatty acid transporters have been targeted in mouse models. Cardiac-specific deletion of CD36, a plasma membrane fatty acid transporter, reduces cardiac FAO and consequently, results in a compensatory increase in glucose oxidation in mice.66 Cardiac function is maintained in CD36 KO mice and cardiac efficiency is improved.67 However, CD36 KO mice demonstrate increased cardiomyocyte hypertrophy and greater cardiac dysfunction after TAC.68 Deletion of muscle specific isoform of carnitine palmityltransferase 1 (CPT-1b), the rate-limiting enzyme for the long-chain fatty acids entry into the mitochondria, is lethal. Heterozygous CPT-1b KO mice demonstrate normal cardiac function but when subject to pressure overload, exhibit exacerbated cardiac dysfunction and hypertrophy, accompanied by cardiac lipid accumulation and cardiomyocyte death.69 Although these mice demonstrate no upregulation of the liver isoform of the CPT-1 (CPT-1a), downregulation of CPT-1b in pathological hypertrophy is often accompanied by upregulation of CPT-1a.70,71 Acute CPT-1a overexpression in rat hearts resulted in reduced FAO and enhanced ANP expression, suggesting that increased expression of CPT-1a is responsible for the reduced mitochondrial FA uptake and thus contributing to the maladaptive remodelling.72
Specifically increasing mitochondrial FAO was achieved by deleting ACC2.73 ACC2 carboxylates acetyl-CoA to form malonyl-CoA, a potent inhibitor of CPT-1b.74 Deletion of ACC2 enhances the entry of long-chain fatty acids into mitochondria for oxidation. Cardiac-specific ACC2 KO mice are resistant to metabolic remodelling after pressure overload and are protected against cardiac hypertrophy. They also demonstrate preserved systolic and diastolic function during chronic pressure overload or angiotensin II treatment.75–77
Together, these studies demonstrate that the outcome of manipulating fatty acid metabolism is pathway specific. Reducing fatty acid supply and/or oxidation appears to be tolerated in unstressed heart but compromises the ability of the heart to respond to stress. Increasing fatty acid uptake and oxidation by overexpression of PPARα causes cardiomyopathy while specifically increasing mitochondrial fatty acid oxidation protects against pathological hypertrophy and cardiac dysfunction.
3.2.2 Endogenous lipid metabolism and cardiotoxicity
Cardiac TAG levels are increased in hearts of animal models of obesity but decreased in hearts with pressure overload.78,79 Despite the fact that increased TAG levels correlate with insulin resistance, data from animal models suggest that TAG is likely only a marker for cellular lipid overload.60,80 Supporting this notion, overexpression of DGAT1 in hearts of lipotoxic models protects against cardiac dysfunction despite increased lipid accumulation, supporting a role for TAG as non-toxic lipid storage.81 TAG turnover has recently been shown as an important contributor to lipid signalling pathways. Using ATGL KO mice, Haemmerle et al. showed that lipolysis of TAG was a prerequisite to activate fatty acids for ligand binding, whereas endogenous free FA were incapable of activating PPARs. ATGL KO mice displayed reduced PPARα target gene expression, lower FAO and massive TAG accumulation, which could be rescued by synthetic PPARα agonists.82
Several lipid species have been implicated in cardiotoxicity. Both DAGs and ceramides have been suggested to mediate cardiac lipotoxicity.80,83 Increased DAG formation ultimately results in protein kinase C (PKC) activation.84 PKC downstream signalling has been implicated in apoptosis, insulin resistance, autophagy and the development of pathological cardiac hypertrophy.85 On the other hand, ceramide signalling has also been linked to apoptosis and cellular stress response.80 Ceramide accumulation correlates with cardiac dysfunction and inhibition of de-novo ceramide synthesis can rescue lipid-induced cardiac dysfunction, supporting a detrimental role of ceramide accumulation on cardiac function.86,87 However, inhibition of de-novo ceramide biosynthesis by deleting a subunit of serine palmitoyltransferase in mouse hearts also resulted in lipotoxic cardiomyopathy suggesting other lipid species could also be toxic.88
3.3 Ketone body metabolism
Ketone bodies are produced by hepatic ketogenesis and used in peripheral tissues, especially heart and brain, as an energy source under starving conditions or glucose deprivation.27,89 Once entering the cell and mitochondria, ketone bodies rapidly form acetyl-CoA via a series of reactions catalyzed by βOHB dehydrogenase (BDH1), succinyl-CoA:3-oxoacid-CoA transferase (SCOT), and mitochondrial acetyl-CoA acetyltransferase 1 (ACAT1) (Figure 2C). BDH1 and SCOT expression are increased in the hearts of animal models and human heart failure.27,28 Increased circulating ketone levels have been reported in heart failure patients, supporting the notion that the failing heart increases ketone body utilization.90
An important question raised by these observations is whether the increased reliance on ketone bodies is adaptive or maladaptive for the failing heart.89,91 The heart oxidizes ketone bodies in proportion to their availability, as SCOT operates at near equilibrium. SCOT KO mice demonstrate impaired ketone body utilization, but normal substrate utilization and cardiac function. However, after pressure overload, SCOT KO mice exhibited exacerbated cardiac dysfunction and worsened pathological remodelling.91 This suggests that the ability to oxidize ketone bodies is important for the diseased heart, indicating that an increased reliance on ketone bodies is an adaptive process for energy provision during stress.
3.4 Branched-chain amino acid metabolism
Cardiac metabolism of amino acids, especially BCAAs, has recently received increasing attention. BCAAs include leucine, isoleucine, and valine; they share structural features in side-chain and are metabolized through a common catabolic pathway. BCAAs enter the cell through specialized amino acid transporters.92 They are catabolized in the mitochondria and eventually oxidized in the TCA cycle (Figure 2D). The rate-limiting enzyme of BCAA catabolism is the branched-chain α-keto acids dehydrogenase complex (BCKDH), whose activity is inhibited by phosphorylation. Dephosphorylation of BCKDH by the mitochondrial-localized 2C-type protein phosphatase (PP2Cm) promotes the BCAA catabolism (Figure 2D).30 In hearts with pathological hypertrophy, BCAA catabolism is impaired and the expression of the catabolic enzymes reduced, resulting in accumulation of BCAAs and branched-chain a-keto-acids (BCKAs).29
To elucidate if impaired BCAA catabolism contributes to cardiac dysfunction, two mouse models have been utilized. First, PP2Cm KO mice demonstrate accumulation of BCAAs and BCKAs.30 Although these mice had normal cardiac function at baseline, they exhibited accelerated transition to heart failure after pressure overload. In the second model, deletion of mitochondrial branched-chain aminotransferase (BCATm KO) results in accumulation of BCAAs but not BCKAs.93 BCATm KO mice had increased mTORC1 activation and cardiac hypertrophy.93 Both studies support the notion that impaired BCAA catabolism is maladaptive for the heart, while the second animal model suggests that accumulation of BCAAs is responsible for cardiac hypertrophy. The mechanistic link between impaired BCAA catabolism and the adverse outcome in animal models of heart failure is poorly understood. As discussed above, leucine is a potent regulator of mTORC1 signalling. Leucine accumulation could therefore stimulate cardiomyocyte growth or alter autophagy.94,95 Additionally, it has been suggested that BCCA accumulation impairs mitochondrial function by triggering loss of mitochondrial membrane potential and opening of the mitochondrial permeability transition pore,30,96,97 whereas BCKAs have been shown to trigger reactive oxygen species formation (ROS).98,99
4. New roles of metabolites beyond energy provision
4.1 Metabolites as signalling molecules
It is increasingly recognized that the heart can secrete bioactive molecules, referred to as cardiokines, which can have autocrine, paracrine and/or endocrine functions. One of the well-known cardiokines is atrial natriuretic peptide (ANP), which is produced and secreted from the heart but can also affect non-cardiac cells.100 Interestingly, several metabolites have recently been identified to function like cardiokines, acting via G-protein coupled receptor (GPCR) signalling mechanisms (Table 2).101 Additionally, metabolites can have intracellular signalling roles, expanding the classical secretory pathway for cardiokines.
Table 2.
Signalling metabolites and their receptor
| Ligand | Receptor | Receptor expression | Cardiac function | Reference |
|---|---|---|---|---|
| Succinate | GPR91/Sucnr1 | Kidney, liver, intestine, central nervous system, heart | Increased expression in hypertrophy | 105-107 |
| α-Ketoglutarate | GPR99/Oxgr1 | Skeletal muscle, central nervous system, adrenal gland, testis, kidney, heart | Increased expression in hypertrophy | 109, 110-113 |
| β-Hydroxybutyrate | GPR109A/Niacr1 | Adipocytes, immunce cells, intestinal epithelium | N/A | 115, 116 |
| GPR41/FFAR3 | N/A | |||
| Lactate | GPR81/HCAR1 | adipocytes, brain, cancer cells, kidney, skeletal muscle, heart | N/A | 117, 119 |
Succinate is a TCA cycle intermediate that also plays important regulatory roles. For example, succinate stabilizes the transcription factor Hif-1α in tumour cells; it can post-translationally modify proteins by being covalently attached to a lysine residue.102,103 In the heart, protein succinylation has been reported to contribute to cardiomyocyte maturation, and increased protein succinylation also correlates with cardiac dysfunction.103,104 Most recently, succinate and its newly identified receptor GPCR 91 (GPR91/Sucnr1) have been shown to be increased in pathological hypertrophy.105–108 Oral administration of succinate was able to activate GPR91 signalling and subsequently induced cardiomyocyte hypertrophy via the PI3K/Akt cascade, supporting its role as a cardiokine.107 Although succinate levels are increased in pathological hypertrophy, it remains unclear if succinate is actively secreted by cardiomyocytes.108
Another TCA cycle intermediate, α-ketoglutarate (αKG), has also been identified to play an important role in mTORC1 regulation and cardiomyocyte growth.109–111 Recently, its receptor (GPR99/Oxgr1) has been demonstrated to be increased in cardiac pathological hypertrophy, while cellular αKG levels are reduced.108,112,113 αKG itself is cell impermeable, supporting the idea of receptor-mediated signalling. Yet, even though a recent report demonstrated that αKG administration activated GPR99 signalling in skeletal muscle hypertrophy, a direct role of αKG in cardiomyocyte hypertrophy remains to be determined.109 It also needs to be established if αKG gets actively secreted from cardiomyocytes.
One of the ketone bodies, βOHB, can bind to at least two cell surface receptors, GPR109A/Niacr1 and free fatty acid receptor 3 (FFAR3/GPR41).114,115 At the present, the βOHB-induced GPR109A signalling has mainly been characterized in adipocytes116; expression of either receptor in the heart has not been confirmed, neither has been the secretion of βOHB from cardiomyocytes. Thus, a direct role of βOHB in receptor-mediated signalling in the heart needs to be determined. However, in light of increased ketone body availability in late stage heart failure, the signalling role of βOHB in the heart is worth attention.
It has been shown that circulating lactate can be taken up by distal tissues and organs or stimulate GPCR signalling through its receptor, GPR81/HCAR1.115,117 Lactate has also been referred to as ‘lactormone’, executing classical hormonal functions.118 Although GPR81 is mainly expressed on adipocytes, is can be detected at lower levels in the heart, suggesting a potential autocrine role of locally produced lactate on cardiac function.119
4.2 Metabolites as cofactors for the epigenetic machinery
It is increasingly recognized that products of intermediary metabolism play important biological roles other than energy provision, for example, in regulating epigenetic modifications through histone acetylation and DNA methylation. By serving as cofactors for chromatin- or DNA-modifying enzymes, important intermediates of cell metabolism allow the integration of metabolic information and transcriptional control. Fluctuating metabolite concentrations might therefore provide signalling cues for continual adjustment of gene expression by modulating the epigenome or influencing chromatin dynamics.120,121 Here, we will briefly outline a few studies on metabolites that regulate the epigenetic machinery.
One of the key metabolites generated from substrate metabolism, acetyl-CoA, serves as substrate for histone acetyltransferases (HATs) or more general, lysine acetyltransferase (KAT) in addition to feed into the TCA cycle, thereby directly linking energy metabolism with gene expression and protein activity.120 A direct relationship between changes in intracellular acetyl-CoA concentrations and histone acetylation has been demonstrated in yeast, yet this has not been confirmed in mammals or in the heart.122 However, increased mitochondrial export of αKG and cytosolic acetyl-CoA formation from citrate by ATP citrate lyase have recently been linked to epigenetic remodelling and impaired cardiac function.123 αKG itself is a co-substrate for chromatin demethylases and might regulate chromatin methylation.120 Increased fat intake has also been link to widespread chromatin remodelling and a recent study has suggested that multiple acyl-CoAs can act as endogenous KAT inhibitors.124 Similarly, βOHB was recently found to be an endogenous inhibitor of histone deacetylases (HDACs), however there has been no evidence so far for this regulatory pathway in the heart.125
5. Conclusion
It has long been recognized that substrate metabolism is altered in the heart under stress conditions. The shift of fuel preference from FAO towards glucose in pathological hypertrophy is well documented. Studies in the past decades have focused on the functional significance of the shift using bioengineered mouse models. As discussed above, the results of these studies do not provide a simple answer as to whether the metabolic remodelling is adaptive or maladaptive. Instead, they show that increased utilization of one particular fuel is not harmful as long as the uptake and utilization is balanced and the energetic demand of the heart met. However, the loss of the metabolic flexibility associated with the shift of substrate preference can impair the stress response, either during a change of substrate availability or a change in energy supply. Furthermore, recent studies have revealed novel and interesting role of substrate metabolism beyond energy provision. These include amino acid and ketone body metabolism, as well as multiple compounds of intermediary metabolism that play intriguing roles in cell growth and survival. Thus, every substrate has a role in the development of cardiomyopathy; a shift in substrate preference impacts the flexibility of the metabolic network for energy generation and other regulatory functions.
Conflict of interest: none declared.
Funding
This work was supported by grants from the National Institutes of Health (HL088634, HL110349, HL118989, HL122199 and HL129510 to R.T.) and by a grant from the Deutsche Forschungsgemeinschaft (2764/1-1 to J.R.).
References
- 1. Ingwall JS. ATP and the Heart. Boston: Kluwer Academic Publishers; 2002. [Google Scholar]
- 2. Doenst T, Nguyen TD, Abel ED.. Cardiac metabolism in heart failure: implications beyond ATP production. Circul Research 2013;113:709–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kolwicz SC Jr., Purohit S, Tian R.. Cardiac metabolism and its interactions with contraction, growth, and survival of cardiomyocytes. Circul Res 2013;113:603–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Neubauer S. The failing heart – an engine out of fuel. New Eng J Med 2007;356:1140–1151. [DOI] [PubMed] [Google Scholar]
- 5. Abdulla R, Blew GA, Holterman MJ.. Cardiovascular embryology. Pediatr Cardiol 2004;25:191–200. [DOI] [PubMed] [Google Scholar]
- 6. Harvey R, Rosenthal N.. Heart Development. London, UK: Academic Press; 1999. [Google Scholar]
- 7. Girard J, Ferre P, Pegorier JP, Duee PH.. Adaptations of glucose and fatty acid metabolism during perinatal period and suckling-weaning transition. Physiol Rev 1992;72:507–562. [DOI] [PubMed] [Google Scholar]
- 8. Fisher DJ, Heymann MA, Rudolph AM.. Myocardial consumption of oxygen and carbohydrates in newborn sheep. Pediatr Res 1981;15:843–846. [DOI] [PubMed] [Google Scholar]
- 9. Lopaschuk GD, Spafford MA, Marsh DR.. Glycolysis is predominant source of myocardial ATP production immediately after birth. Am J Physiol 1991;261:H1698–1705. [DOI] [PubMed] [Google Scholar]
- 10. Lopaschuk GD, Jaswal JS.. Energy metabolic phenotype of the cardiomyocyte during development, differentiation, and postnatal maturation. J Cardiovasc Pharmacol 2010;56:130–140. [DOI] [PubMed] [Google Scholar]
- 11. Itoi T, Lopaschuk GD.. The contribution of glycolysis, glucose oxidation, lactate oxidation, and fatty acid oxidation to ATP production in isolated biventricular working hearts from 2-week-old rabbits. Pediatr Res 1993;34:735–741. [DOI] [PubMed] [Google Scholar]
- 12. Patterson AJ, Zhang L.. Hypoxia and fetal heart development. Curr Mol Med 2010;10:653–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Semenza GL. Hypoxia-inducible factor 1 and cardiovascular disease. Annu Rev Physiol 2014;76:39–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Liang H, Ward WF.. PGC-1alpha: a key regulator of energy metabolism. Adv Physiol Educ 2006;30:145–151. [DOI] [PubMed] [Google Scholar]
- 15. Lopaschuk GD, Collins-Nakai RL, Itoi T.. Developmental changes in energy substrate use by the heart. Cardiovasc Res 1992;26:1172–1180. [DOI] [PubMed] [Google Scholar]
- 16. Nakada Y, Canseco DC, Thet S, Abdisalaam S, Asaithamby A, Santos CX, Shah A, Zhang H, Faber JE, Kinter MT, Szweda LI, Xing C, Deberardinis R, Oz O, Lu Z, Zhang CC, Kimura W, Sadek HA.. Hypoxia induces heart regeneration in adult mice. Nature 2017;541:222–227. [DOI] [PubMed] [Google Scholar]
- 17. Dorn GW 2nd, Vega RB, Kelly DP.. Mitochondrial biogenesis and dynamics in the developing and diseased heart. Genes Develop 2015;29:1981–1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lopaschuk GD, Spafford MA.. Energy substrate utilization by isolated working hearts from newborn rabbits. Am J Physiol 1990;258:H1274–1280. [DOI] [PubMed] [Google Scholar]
- 19. Portman OW, Behrman RE, Soltys P.. Transfer of free fatty acids across the primate placenta. Am J Physiol 1969;216:143–147. [DOI] [PubMed] [Google Scholar]
- 20. Hamosh M, Simon MR, Canter H Jr., Hamosh P.. Lipoprotein lipase activity and blood triglyceride levels in fetal and newborn rats. Pediatr Res 1978;12:1132–1136. [DOI] [PubMed] [Google Scholar]
- 21. Glennon PE, Sugden PH, Poole-Wilson PA.. Cellular mechanisms of cardiac hypertrophy. Br Heart J 1995;73:496–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Akki A, Smith K, Seymour AM.. Compensated cardiac hypertrophy is characterised by a decline in palmitate oxidation. Mol Cell Biochem 2008;311:215–224. [DOI] [PubMed] [Google Scholar]
- 23. Allard MF, Schonekess BO, Henning SL, English DR, Lopaschuk GD.. Contribution of oxidative metabolism and glycolysis to ATP production in hypertrophied hearts. Am J Physiol 1994;267:H742–H750. [DOI] [PubMed] [Google Scholar]
- 24. Kagaya Y, Kanno Y, Takeyama D, Ishide N, Maruyama Y, Takahashi T, Ido T, Takishima T.. Effects of long-term pressure overload on regional myocardial glucose and free fatty acid uptake in rats. A quantitative autoradiographic study. Circulation 1990;81:1353–1361. [DOI] [PubMed] [Google Scholar]
- 25. Barger PM, Kelly DP.. Fatty acid utilization in the hypertrophied and failing heart: molecular regulatory mechanisms. Am J Med Sci 1999;318:36–42. [DOI] [PubMed] [Google Scholar]
- 26. Razeghi P, Young ME, Alcorn JL, Moravec CS, Frazier OH, Taegtmeyer H.. Metabolic gene expression in fetal and failing human heart. Circulation 2001;104:2923–2931. [DOI] [PubMed] [Google Scholar]
- 27. Aubert G, Martin OJ, Horton JL, Lai L, Vega RB, Leone TC, Koves T, Gardell SJ, Kruger M, Hoppel CL, Lewandowski ED, Crawford PA, Muoio DM, Kelly DP.. The failing heart relies on ketone bodies as a fuel. Circulation 2016;133:698–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bedi KC Jr, Snyder NW, Brandimarto J, Aziz M, Mesaros C, Worth AJ, Wang LL, Javaheri A, Blair IA, Margulies KB, Rame JE.. Evidence for intramyocardial disruption of lipid metabolism and increased myocardial ketone utilization in advanced human heart failure. Circulation 2016;133:706–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Huang Y, Zhou M, Sun H, Wang Y.. Branched-chain amino acid metabolism in heart disease: an epiphenomenon or a real culprit? Cardiovasc Res 2011;90:220–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sun H, Lu G, Ren S, Chen J, Wang Y.. Catabolism of branched-chain amino acids in heart failure: insights from genetic models. Pediatr Cardiol 2011;32:305–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Abel ED, Doenst T.. Mitochondrial adaptations to physiological vs. pathological cardiac hypertrophy. Cardiovasc Res 2011;90:234–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Buchanan J, Mazumder PK, Hu P, Chakrabarti G, Roberts MW, Yun UJ, Cooksey RC, Litwin SE, Abel ED.. Reduced cardiac efficiency and altered substrate metabolism precedes the onset of hyperglycemia and contractile dysfunction in two mouse models of insulin resistance and obesity. Endocrinology 2005;146:5341–5349. [DOI] [PubMed] [Google Scholar]
- 33. Taegtmeyer H, Golfman L, Sharma S, Razeghi P, van Arsdall M.. Linking gene expression to function: metabolic flexibility in the normal and diseased heart. Ann N Y Acad Sci 2004;1015:202–213. [DOI] [PubMed] [Google Scholar]
- 34. Shao D, Tian R.. Glucose transporters in cardiac metabolism and hypertrophy. Compr Physiol 2015;6:331–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sen S, Kundu BK, Wu HC, Hashmi SS, Guthrie P, Locke LW, Roy RJ, Matherne GP, Berr SS, Terwelp M, Scott B, Carranza S, Frazier OH, Glover DK, Dillmann WH, Gambello MJ, Entman ML, Taegtmeyer H.. Glucose regulation of load-induced mTOR signaling and ER stress in mammalian heart. J Am Heart Assoc 2013;2:e004796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sharma S, Guthrie PH, Chan SS, Haq S, Taegtmeyer H.. Glucose phosphorylation is required for insulin-dependent mTOR signalling in the heart. Cardiovasc Res 2007;76:71–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Liao R, Jain M, Cui L, D'Agostino J, Aiello F, Luptak I, Ngoy S, Mortensen RM, Tian R.. Cardiac-specific overexpression of GLUT1 prevents the development of heart failure attributable to pressure overload in mice. Circulation 2002;106:2125–2131. [DOI] [PubMed] [Google Scholar]
- 38. Pereira RO, Wende AR, Olsen C, Soto J, Rawlings T, Zhu Y, Anderson SM, Abel ED.. Inducible overexpression of GLUT1 prevents mitochondrial dysfunction and attenuates structural remodeling in pressure overload but does not prevent left ventricular dysfunction. J Am Heart Assoc 2013;2:e000301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Luptak I, Yan J, Cui L, Jain M, Liao R, Tian R.. Long-term effects of increased glucose entry on mouse hearts during normal aging and ischemic stress. Circulation 2007;116:901–909. [DOI] [PubMed] [Google Scholar]
- 40. Pereira RO, Wende AR, Olsen C, Soto J, Rawlings T, Zhu Y, Riehle C, Abel ED.. GLUT1 deficiency in cardiomyocytes does not accelerate the transition from compensated hypertrophy to heart failure. J Mol Cell Cardiol 2014;72:95–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Abel ED, Kaulbach HC, Tian R, Hopkins JC, Duffy J, Doetschman T, Minnemann T, Boers ME, Hadro E, Oberste-Berghaus C, Quist W, Lowell BB, Ingwall JS, Kahn BB.. Cardiac hypertrophy with preserved contractile function after selective deletion of GLUT4 from the heart. J Clin Invest 1999;104:1703–1714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Liang Q, Donthi RV, Kralik PM, Epstein PN.. Elevated hexokinase increases cardiac glycolysis in transgenic mice. Cardiovasc Res 2002;53:423–430. [DOI] [PubMed] [Google Scholar]
- 43. McCommis KS, Douglas DL, Krenz M, Baines CP.. Cardiac-specific hexokinase 2 overexpression attenuates hypertrophy by increasing pentose phosphate pathway flux. J Am Heart Assoc 2013;2:e000355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Chambers KT, Leone TC, Sambandam N, Kovacs A, Wagg CS, Lopaschuk GD, Finck BN, Kelly DP.. Chronic inhibition of pyruvate dehydrogenase in heart triggers an adaptive metabolic response. J Biol Chem 2011;286:11155–11162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Yan J, Young ME, Cui L, Lopaschuk GD, Liao R, Tian R.. Increased glucose uptake and oxidation in mouse hearts prevent high fatty acid oxidation but cause cardiac dysfunction in diet-induced obesity. Circulation 2009;119:2818–2828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Neglia D, De Caterina A, Marraccini P, Natali A, Ciardetti M, Vecoli C, Gastaldelli A, Ciociaro D, Pellegrini P, Testa R, Menichetti L, L'Abbate A, Stanley WC, Recchia FA.. Impaired myocardial metabolic reserve and substrate selection flexibility during stress in patients with idiopathic dilated cardiomyopathy. Am J Physiol Heart Circul Physiol 2007;293:H3270–H3278. [DOI] [PubMed] [Google Scholar]
- 47. Shioi T, McMullen JR, Tarnavski O, Converso K, Sherwood MC, Manning WJ, Izumo S.. Rapamycin attenuates load-induced cardiac hypertrophy in mice. Circulation 2003;107:1664–1670. [DOI] [PubMed] [Google Scholar]
- 48. Zhang D, Contu R, Latronico MV, Zhang J, Rizzi R, Catalucci D, Miyamoto S, Huang K, Ceci M, Gu Y, Dalton ND, Peterson KL, Guan KL, Brown JH, Chen J, Sonenberg N, Condorelli G.. MTORC1 regulates cardiac function and myocyte survival through 4E-BP1 inhibition in mice. J Clin Invest 2010;120:2805–2816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Marsh SA, Dell'Italia LJ, Chatham JC.. Activation of the hexosamine biosynthesis pathway and protein O-GlcNAcylation modulate hypertrophic and cell signaling pathways in cardiomyocytes from diabetic mice. Amino Acids 2011;40:819–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ussher JR, Jaswal JS, Lopaschuk GD.. Pyridine nucleotide regulation of cardiac intermediary metabolism. Circul Res 2012;111:628–641. [DOI] [PubMed] [Google Scholar]
- 51. Yang HC, Wu YH, Liu HY, Stern A, Chiu DT.. What has passed is prologue: new cellular and physiological roles of G6PD. Free Radic Res 2016:1–58. [DOI] [PubMed] [Google Scholar]
- 52. Hecker PA, Lionetti V, Ribeiro RF Jr., Rastogi S, Brown BH, O'Connell KA, Cox JW, Shekar KC, Gamble DM, Sabbah HN, Leopold JA, Gupte SA, Recchia FA, Stanley WC.. Glucose 6-phosphate dehydrogenase deficiency increases redox stress and moderately accelerates the development of heart failure. Circul Heart Failure 2013;6:118–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. van der Vusse GJ, van Bilsen M, Glatz JF.. Cardiac fatty acid uptake and transport in health and disease. Cardiovasc Res 2000;45:279–293. [DOI] [PubMed] [Google Scholar]
- 54. Lopaschuk GD, Ussher JR, Folmes CD, Jaswal JS, Stanley WC.. Myocardial fatty acid metabolism in health and disease. Physiol Rev 2010;90:207–258. [DOI] [PubMed] [Google Scholar]
- 55. Schulze PC, Drosatos K, Goldberg IJ.. Lipid use and misuse by the heart. Circulation research 2016;118:1736–1751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Lopaschuk GD, Belke DD, Gamble J, Itoi T, Schonekess BO.. Regulation of fatty acid oxidation in the mammalian heart in health and disease. Biochim Biophys Acta 1994;1213:263–276. [DOI] [PubMed] [Google Scholar]
- 57. Sarri E, Sicart A, Lazaro-Dieguez F, Egea G.. Phospholipid synthesis participates in the regulation of diacylglycerol required for membrane trafficking at the Golgi complex. J Bioll Chem 2011;286:28632–28643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Banke NH, Wende AR, Leone TC, O'Donnell JM, Abel ED, Kelly DP, Lewandowski ED.. Preferential oxidation of triacylglyceride-derived fatty acids in heart is augmented by the nuclear receptor PPARalpha. Circul Res 2010;107:233–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Kienesberger PC, Pulinilkunnil T, Sung MM, Nagendran J, Haemmerle G, Kershaw EE, Young ME, Light PE, Oudit GY, Zechner R, Dyck JR.. Myocardial ATGL overexpression decreases the reliance on fatty acid oxidation and protects against pressure overload-induced cardiac dysfunction. Mol Cell Biol 2012;32:740–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Pillutla P, Hwang YC, Augustus A, Yokoyama M, Yagyu H, Johnston TP, Kaneko M, Ramasamy R, Goldberg IJ.. Perfusion of hearts with triglyceride-rich particles reproduces the metabolic abnormalities in lipotoxic cardiomyopathy. Am J Physiol Endocrinol Metabol 2005;288:E1229–E1235. [DOI] [PubMed] [Google Scholar]
- 61. Finck BN, Lehman JJ, Leone TC, Welch MJ, Bennett MJ, Kovacs A, Han X, Gross RW, Kozak R, Lopaschuk GD, Kelly DP.. The cardiac phenotype induced by PPARalpha overexpression mimics that caused by diabetes mellitus. J Clin Invest 2002;109:121–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Loichot C, Jesel L, Tesse A, Tabernero A, Schoonjans K, Roul G, Carpusca I, Auwerx J, Andriantsitohaina R.. Deletion of peroxisome proliferator-activated receptor-alpha induces an alteration of cardiac functions. American journal of physiology Heart and circulatory physiology 2006;291:H161–166. [DOI] [PubMed] [Google Scholar]
- 63. Luptak I, Balschi JA, Xing Y, Leone TC, Kelly DP, Tian R.. Decreased contractile and metabolic reserve in peroxisome proliferator-activated receptor-alpha-null hearts can be rescued by increasing glucose transport and utilization. Circulation 2005;112:2339–2346. [DOI] [PubMed] [Google Scholar]
- 64. Liu J, Wang P, He L, Li Y, Luo J, Cheng L, Qin Q, Brako LA, Lo WK, Lewis W, Yang Q.. Cardiomyocyte-Restricted Deletion of PPARbeta/delta in PPARalpha-Null Mice Causes Impaired Mitochondrial Biogenesis and Defense, but No Further Depression of Myocardial Fatty Acid Oxidation. PPAR Res 2011;2011:372854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Liu J, Wang P, Luo J, Huang Y, He L, Yang H, Li Q, Wu S, Zhelyabovska O, Yang Q.. Peroxisome proliferator-activated receptor beta/delta activation in adult hearts facilitates mitochondrial function and cardiac performance under pressure-overload condition. Hypertension 2011;57:223–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Nagendran J, Pulinilkunnil T, Kienesberger PC, Sung MM, Fung D, Febbraio M, Dyck JR.. Cardiomyocyte-specific ablation of CD36 improves post-ischemic functional recovery. Journal of molecular and cellular cardiology 2013;63:180–188. [DOI] [PubMed] [Google Scholar]
- 67. Nakatani K, Watabe T, Masuda D, Imaizumi M, Shimosegawa E, Kobayashi T, Sairyo M, Zhu Y, Okada T, Kawase R, Nakaoka H, Naito A, Ohama T, Koseki M, Oka T, Akazawa H, Nishida M, Komuro I, Sakata Y, Hatazawa J, Yamashita S.. Myocardial energy provision is preserved by increased utilization of glucose and ketone bodies in CD36 knockout mice. Metabolism 2015;64:1165–1174. [DOI] [PubMed] [Google Scholar]
- 68. Nakatani K, Masuda D, Oka T, Okada T, Kawase R, Nakaoka H, Ohama T, Nishida M, Komuro I, Yamashita S.. Abstract 12900: Pressure Overload Induces Hypertrophy and Impaired Cardiac Function in Long-chain Fatty Acid Transporter CD36 Knockout Mice. Circulation 2013;128:A12900–A12900. [Google Scholar]
- 69. He L, Kim T, Long Q, Liu J, Wang P, Zhou Y, Ding Y, Prasain J, Wood PA, Yang Q.. Carnitine palmitoyltransferase-1b deficiency aggravates pressure overload-induced cardiac hypertrophy caused by lipotoxicity. Circulation 2012;126:1705–1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Sorokina N, O'Donnell JM, McKinney RD, Pound KM, Woldegiorgis G, LaNoue KF, Ballal K, Taegtmeyer H, Buttrick PM, Lewandowski ED.. Recruitment of compensatory pathways to sustain oxidative flux with reduced carnitine palmitoyltransferase I activity characterizes inefficiency in energy metabolism in hypertrophied hearts. Circulation 2007;115:2033–2041. [DOI] [PubMed] [Google Scholar]
- 71. Lahey R, Wang X, Carley AN, Lewandowski ED.. Dietary fat supply to failing hearts determines dynamic lipid signaling for nuclear receptor activation and oxidation of stored triglyceride. Circulation 2014;130:1790–1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Lewandowski ED, Fischer SK, Fasano M, Banke NH, Walker LA, Huqi A, Wang X, Lopaschuk GD, O'Donnell JM.. Acute liver carnitine palmitoyltransferase I overexpression recapitulates reduced palmitate oxidation of cardiac hypertrophy. Circulation research 2013;112:57–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Abu-Elheiga L, Matzuk MM, Abo-Hashema KA, Wakil SJ.. Continuous fatty acid oxidation and reduced fat storage in mice lacking acetyl-CoA carboxylase 2. Science 2001;291:2613–2616. [DOI] [PubMed] [Google Scholar]
- 74. Reszko AE, Kasumov T, David F, Thomas KR, Jobbins KA, Cheng JF, Lopaschuk GD, Dyck JR, Diaz M, Des Rosiers C, Stanley WC, Brunengraber H.. Regulation of malonyl-CoA concentration and turnover in the normal heart. The Journal of biological chemistry 2004;279:34298–34301. [DOI] [PubMed] [Google Scholar]
- 75. Essop MF, Camp HS, Choi CS, Sharma S, Fryer RM, Reinhart GA, Guthrie PH, Bentebibel A, Gu Z, Shulman GI, Taegtmeyer H, Wakil SJ, Abu-Elheiga L.. Reduced heart size and increased myocardial fuel substrate oxidation in ACC2 mutant mice. Am J Physiol Heart Circul Physiol 2008;295:H256–H265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Kolwicz SC Jr., Olson DP, Marney LC, Garcia-Menendez L, Synovec RE, Tian R.. Cardiac-specific deletion of acetyl CoA carboxylase 2 prevents metabolic remodeling during pressure-overload hypertrophy. Circul Res 2012;111:728–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Choi YS, de Mattos AB, Shao D, Li T, Nabben M, Kim M, Wang W, Tian R, Kolwicz SC Jr.. Preservation of myocardial fatty acid oxidation prevents diastolic dysfunction in mice subjected to angiotensin II infusion. J Mol Cell Cardiol 2016;100:64–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. O'Donnell JM, Fields AD, Sorokina N, Lewandowski ED.. The absence of endogenous lipid oxidation in early stage heart failure exposes limits in lipid storage and turnover. J Mol Cell Cardiol 2008;44:315–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. O'Donnell JM, Zampino M, Alpert NM, Fasano MJ, Geenen DL, Lewandowski ED.. Accelerated triacylglycerol turnover kinetics in hearts of diabetic rats include evidence for compartmented lipid storage. Am J Physiol Endocrinol Metabol 2006;290:E448–E455. [DOI] [PubMed] [Google Scholar]
- 80. Zhang L, Ussher JR, Oka T, Cadete VJ, Wagg C, Lopaschuk GD.. Cardiac diacylglycerol accumulation in high fat-fed mice is associated with impaired insulin-stimulated glucose oxidation. Cardiovasc Res 2011;89:148–156. [DOI] [PubMed] [Google Scholar]
- 81. Listenberger LL, Han X, Lewis SE, Cases S, Farese RV Jr., Ory DS, Schaffer JE.. Triglyceride accumulation protects against fatty acid-induced lipotoxicity. Proc Natl Acad Sci USA 2003;100:3077–3082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Haemmerle G, Moustafa T, Woelkart G, Buttner S, Schmidt A, van de Weijer T, Hesselink M, Jaeger D, Kienesberger PC, Zierler K, Schreiber R, Eichmann T, Kolb D, Kotzbeck P, Schweiger M, Kumari M, Eder S, Schoiswohl G, Wongsiriroj N, Pollak NM, Radner FP, Preiss-Landl K, Kolbe T, Rulicke T, Pieske B, Trauner M, Lass A, Zimmermann R, Hoefler G, Cinti S, Kershaw EE, Schrauwen P, Madeo F, Mayer B, Zechner R.. ATGL-mediated fat catabolism regulates cardiac mitochondrial function via PPAR-alpha and PGC-1. Nat Med 2011;17:1076–1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Park TS, Goldberg IJ.. Sphingolipids, lipotoxic cardiomyopathy, and cardiac failure. Heart Fail Clin 2012;8:633–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Drosatos K, Bharadwaj KG, Lymperopoulos A, Ikeda S, Khan R, Hu Y, Agarwal R, Yu S, Jiang H, Steinberg SF, Blaner WS, Koch WJ, Goldberg IJ.. Cardiomyocyte lipids impair beta-adrenergic receptor function via PKC activation. Am J Physiol Endocrinol Metabol 2011;300:E489–E499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Bowman JC, Steinberg SF, Jiang T, Geenen DL, Fishman GI, Buttrick PM.. Expression of protein kinase C beta in the heart causes hypertrophy in adult mice and sudden death in neonates. J Clin Invest 1997;100:2189–2195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Park TS, Hu Y, Noh HL, Drosatos K, Okajima K, Buchanan J, Tuinei J, Homma S, Jiang XC, Abel ED, Goldberg IJ.. Ceramide is a cardiotoxin in lipotoxic cardiomyopathy. J Lipid Res 2008;49:2101–2112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Russo SB, Baicu CF, Van Laer A, Geng T, Kasiganesan H, Zile MR, Cowart LA.. Ceramide synthase 5 mediates lipid-induced autophagy and hypertrophy in cardiomyocytes. J Clin Invest 2012;122:3919–3930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Lee SY, Kim JR, Hu Y, Khan R, Kim SJ, Bharadwaj KG, Davidson MM, Choi CS, Shin KO, Lee YM, Park WJ, Park IS, Jiang XC, Goldberg IJ, Park TS.. Cardiomyocyte specific deficiency of serine palmitoyltransferase subunit 2 reduces ceramide but leads to cardiac dysfunction. J Biol Chem 2012;287:18429–18439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Kolwicz SC Jr., Airhart S, Tian R.. Ketones step to the plate: a game changer for metabolic remodeling in heart failure? Circulation 2016;133:689–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Lommi J, Kupari M, Koskinen P, Naveri H, Leinonen H, Pulkki K, Harkonen M.. Blood ketone bodies in congestive heart failure. J Am College Cardiol 1996;28:665–672. [DOI] [PubMed] [Google Scholar]
- 91. Schugar RC, Moll AR, Andre d'Avignon D, Weinheimer CJ, Kovacs A, Crawford PA.. Cardiomyocyte-specific deficiency of ketone body metabolism promotes accelerated pathological remodeling. Mol Metab 2014;3:754–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Hyde R, Taylor PM, Hundal HS.. Amino acid transporters: roles in amino acid sensing and signalling in animal cells. Biochem J 2003;373:1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Neishabouri SH, Hutson SM, Davoodi J.. Chronic activation of mTOR complex 1 by branched chain amino acids and organ hypertrophy. Amino Acids 2015;47:1167–1182. [DOI] [PubMed] [Google Scholar]
- 94. Shimobayashi M, Hall MN.. Multiple amino acid sensing inputs to mTORC1. Cell Res 2016;26:7–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Meijer AJ, Lorin S, Blommaart EF, Codogno P.. Regulation of autophagy by amino acids and MTOR-dependent signal transduction. Amino Acids 2015;47:2037–2063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Sun H, Olson KC, Gao C, Prosdocimo DA, Zhou M, Wang Z, Jeyaraj D, Youn JY, Ren S, Liu Y, Rau CD, Shah S, Ilkayeva O, Gui WJ, William NS, Wynn RM, Newgard CB, Cai H, Xiao X, Chuang DT, Schulze PC, Lynch C, Jain MK, Wang Y.. Catabolic defect of branched-chain amino acids promotes heart failure. Circulation 2016;133:2038–2049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Lu G, Ren S, Korge P, Choi J, Dong Y, Weiss J, Koehler C, Chen JN, Wang Y.. A novel mitochondrial matrix serine/threonine protein phosphatase regulates the mitochondria permeability transition pore and is essential for cellular survival and development. Genes Dev 2007;21:784–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Lu G, Sun H, She P, Youn JY, Warburton S, Ping P, Vondriska TM, Cai H, Lynch CJ, Wang Y.. Protein phosphatase 2Cm is a critical regulator of branched-chain amino acid catabolism in mice and cultured cells. J Clin Inves 2009;119:1678–1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Funchal C, Latini A, Jacques-Silva MC, Dos Santos AQ, Buzin L, Gottfried C, Wajner M, Pessoa-Pureur R.. Morphological alterations and induction of oxidative stress in glial cells caused by the branched-chain alpha-keto acids accumulating in maple syrup urine disease. Neurochem Int 2006;49:640–650. [DOI] [PubMed] [Google Scholar]
- 100. Doroudgar S, Glembotski CC.. The cardiokine story unfolds: ischemic stress-induced protein secretion in the heart. Trends Mol Med 2011;17:207–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Shimano M, Ouchi N, Walsh K.. Cardiokines: recent progress in elucidating the cardiac secretome. Circulation 2012;126:e327–e332. [DOI] [PubMed] [Google Scholar]
- 102. Selak MA, Armour SM, MacKenzie ED, Boulahbel H, Watson DG, Mansfield KD, Pan Y, Simon MC, Thompson CB, Gottlieb E.. Succinate links TCA cycle dysfunction to oncogenesis by inhibiting HIF-alpha prolyl hydroxylase. Cancer Cell 2005;7:77–85. [DOI] [PubMed] [Google Scholar]
- 103. Hirschey MD, Zhao Y.. Metabolic regulation by lysine malonylation, succinylation, and glutarylation. Mol Cell Proteomics 2015;14:2308–2315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Sadhukhan S, Liu X, Ryu D, Nelson OD, Stupinski JA, Li Z, Chen W, Zhang S, Weiss RS, Locasale JW, Auwerx J, Lin H.. Metabolomics-assisted proteomics identifies succinylation and SIRT5 as important regulators of cardiac function. Proc Natl Acad Sci USA 2016;113:4320–4325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Yang L, Yu D, Fan HH, Feng Y, Hu L, Zhang WY, Zhou K, Mo XM.. Triggering the succinate receptor GPR91 enhances pressure overload-induced right ventricular hypertrophy. Int J Clin Exp Pathol 2014;7:5415–5428. [PMC free article] [PubMed] [Google Scholar]
- 106. Yang L, Yu D, Mo R, Zhang J, Hua H, Hu L, Feng Y, Wang S, Zhang WY, Yin N, Mo XM.. The succinate receptor GPR91 is involved in pressure overload-induced ventricular hypertrophy. PLoS one 2016;11:e0147597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Aguiar CJ, Rocha-Franco JA, Sousa PA, Santos AK, Ladeira M, Rocha-Resende C, Ladeira LO, Resende RR, Botoni FA, Barrouin Melo M, Lima CX, Carballido JM, Cunha TM, Menezes GB, Guatimosim S, Leite MF.. Succinate causes pathological cardiomyocyte hypertrophy through GPR91 activation. Cell Commun Signal 2014;12:78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Lai L, Leone TC, Keller MP, Martin OJ, Broman AT, Nigro J, Kapoor K, Koves TR, Stevens R, Ilkayeva OR, Vega RB, Attie AD, Muoio DM, Kelly DP.. Energy metabolic reprogramming in the hypertrophied and early stage failing heart: a multisystems approach. Circul Heart Failure 2014;7:1022–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Cai X, Zhu C, Xu Y, Jing Y, Yuan Y, Wang L, Wang S, Zhu X, Gao P, Zhang Y, Jiang Q, Shu G.. Alpha-ketoglutarate promotes skeletal muscle hypertrophy and protein synthesis through Akt/mTOR signaling pathways. Sci Rep 2016;6:26802. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 110. Yao K, Yin Y, Li X, Xi P, Wang J, Lei J, Hou Y, Wu G.. Alpha-ketoglutarate inhibits glutamine degradation and enhances protein synthesis in intestinal porcine epithelial cells. Amino Acids 2012;42:2491–2500. [DOI] [PubMed] [Google Scholar]
- 111. Chin RM, Fu X, Pai MY, Vergnes L, Hwang H, Deng G, Diep S, Lomenick B, Meli VS, Monsalve GC, Hu E, Whelan SA, Wang JX, Jung G, Solis GM, Fazlollahi F, Kaweeteerawat C, Quach A, Nili M, Krall AS, Godwin HA, Chang HR, Faull KF, Guo F, Jiang M, Trauger SA, Saghatelian A, Braas D, Christofk HR, Clarke CF, Teitell MA, Petrascheck M, Reue K, Jung ME, Frand AR, Huang J.. The metabolite alpha-ketoglutarate extends lifespan by inhibiting ATP synthase and TOR. Nature 2014;510:397–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Omede A,, Zi M,, Prehar S,, Maqsood A,, Cartwright E,, mamas m, Oceandy D.. Abstract 12399: The G-Protein Coupled Receptor 99 (gpr99), a Receptor for Heart Failure Metabolite Alpha-Ketoglutarate, Regulates Pressure Overload Induced Hypertrophy in Mice. Circulation 2014;130:A12399–A12399. [Google Scholar]
- 113. Diehl J, Gries B, Pfeil U, Goldenberg A, Mermer P, Kummer W, Paddenberg R.. Expression and localization of GPR91 and GPR99 in murine organs. Cell Tissue Res 2016;364:245–262. [DOI] [PubMed] [Google Scholar]
- 114. Newman JC, Verdin E.. Ketone bodies as signaling metabolites. Trends Endocrinol Metab 2014;25:42–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Ahmed K, Tunaru S, Offermanns S.. GPR109A, GPR109B and GPR81, a family of hydroxy-carboxylic acid receptors. Trends Pharmacol Sci 2009;30:557–562. [DOI] [PubMed] [Google Scholar]
- 116. Taggart AK, Kero J, Gan X, Cai TQ, Cheng K, Ippolito M, Ren N, Kaplan R, Wu K, Wu TJ, Jin L, Liaw C, Chen R, Richman J, Connolly D, Offermanns S, Wright SD, Waters MG.. (D)-beta-Hydroxybutyrate inhibits adipocyte lipolysis via the nicotinic acid receptor PUMA-G. J Biol Chem 2005;280:26649–26652. [DOI] [PubMed] [Google Scholar]
- 117. Morland C, Lauritzen KH, Puchades M, Holm-Hansen S, Andersson K, Gjedde A, Attramadal H, Storm-Mathisen J, Bergersen LH.. The lactate receptor, G-protein-coupled receptor 81/hydroxycarboxylic acid receptor 1: Expression and action in brain. J Neurosci Res 2015;93:1045–1055. [DOI] [PubMed] [Google Scholar]
- 118. Brooks GA. Cell-cell and intracellular lactate shuttles. J Physiol 2009;587:5591–5600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Liu C, Wu J, Zhu J, Kuei C, Yu J, Shelton J, Sutton SW, Li X, Yun SJ, Mirzadegan T, Mazur C, Kamme F, Lovenberg TW.. Lactate inhibits lipolysis in fat cells through activation of an orphan G-protein-coupled receptor, GPR81. J Biol Chem 2009;284:2811–2822. [DOI] [PubMed] [Google Scholar]
- 120. Keating ST, El-Osta A.. Epigenetics and metabolism. Circul Res 2015;116:715–736. [DOI] [PubMed] [Google Scholar]
- 121. Janke R, Dodson AE, Rine J.. Metabolism and epigenetics. Annu Rev Cell Dev Biol 2015;31:473–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Cai L, Tu BP.. Acetyl-CoA drives the transcriptional growth program in yeast. Cell Cycle 2011;10:3045–3046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Karlstaedt A, Zhang X, Vitrac H, Harmancey R, Vasquez H, Wang JH, Goodell MA, Taegtmeyer H.. Oncometabolite d-2-hydroxyglutarate impairs alpha-ketoglutarate dehydrogenase and contractile function in rodent heart. Proc Natl Acad Sci USA 2016;113:10436–10441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124. Montgomery DC, Sorum AW, Guasch L, Nicklaus MC, Meier JL.. Metabolic regulation of histone acetyltransferases by endogenous Acyl-CoA cofactors. ChemBiol 2015;22:1030–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Shimazu T, Hirschey MD, Newman J, He W, Shirakawa K, Le Moan N, Grueter CA, Lim H, Saunders LR, Stevens RD, Newgard CB, Farese RV Jr., de Cabo R, Ulrich S, Akassoglou K, Verdin E.. Suppression of oxidative stress by beta-hydroxybutyrate, an endogenous histone deacetylase inhibitor. Science 2013;339:211–214. [DOI] [PMC free article] [PubMed] [Google Scholar]