

Abstract
Aims
Plasmamembrane small conductance Ca2+-activated K+ (SK) channels were implicated in ventricular arrhythmias in infarcted and failing hearts. Recently, SK channels were detected in the inner mitochondria membrane (IMM) (mSK), and their activation protected from acute ischaemia-reperfusion injury by reducing intracellular levels of reactive oxygen species (ROS). We hypothesized that mSK play an important role in regulating mitochondrial function in chronic cardiac diseases. We investigated the role of mSK channels in Ca2+-dependent ventricular arrhythmia using rat model of cardiac hypertrophy induced by banding of the ascending aorta thoracic aortic banding (TAB).
Methods and results
Dual Ca2+ and membrane potential optical mapping of whole hearts derived from TAB rats revealed that membrane-permeable SK enhancer NS309 (2 μM) improved aberrant Ca2+ homeostasis and abolished VT/VF induced by β-adrenergic stimulation. Using whole cell patch-clamp and confocal Ca2+ imaging of cardiomyocytes derived from TAB hearts (TCMs) we found that membrane-permeable SK enhancers NS309 and CyPPA (10 μM) attenuated frequency of spontaneous Ca2+ waves and delayed afterdepolarizations. Furthermore, mSK inhibition enhanced (UCL-1684, 1 μM); while activation reduced mitochondrial ROS production in TCMs measured with MitoSOX. Protein oxidation assays demonstrated that increased oxidation of ryanodine receptors (RyRs) in TCMs was reversed by SK enhancers. Experiments in permeabilized TCMs showed that SK enhancers restored SR Ca2+ content, suggestive of substantial improvement in RyR function.
Conclusion
These data suggest that enhancement of mSK channels in hypertrophic rat hearts protects from Ca2+-dependent arrhythmia and suggest that the protection is mediated via decreased mitochondrial ROS and subsequent decreased oxidation of reactive cysteines in RyR, which ultimately leads to stabilization of RyR-mediated Ca2+ release.
Keywords: Ventricular arrhythmia, Small conductance Ca2+-activated K+ channels, Ryanodine receptor, Reactive oxygen species, Cardiac hypertrophy
1. Introduction
Sudden cardiac death due to ventricular tachyarrhythmias remains the major cause of mortality in the world.1 There is a well-established link of aberrant intracellular Ca2+ homeostasis with enhanced propensity of the heart to arrhythmias in hypertrophy, heart failure, myocardial infarct, and aging.2–4 Pro-arrhythmic spontaneous Ca2+ release from the sarcoplasmic reticulum (SR) in these conditions, accompanied by excessive production of reactive oxygen species (ROS), is largely ascribed to abnormally high activity of oxidized ryanodine receptors (RyRs), the SR Ca2+ release channels.4–8 During recent years mitochondria, located in close proximity to RyR clusters in cardiac cells, gained recognition as a major source of ROS in diseased, aged, or stressed hearts.4,9–12 Furthermore, targeted scavenging of mitochondrial ROS was proven to attenuate spontaneous Ca2+ release most probably via restoration of RyR redox state.4,9,13
Small conductance Ca2+-activated potassium channels (SK) mediate the feedback between intracellular Ca2+ dynamics and membrane repolarization in excitable cells.14–16 The SK channel family consists of three members: SK1 (KCa2.1, KCNN1), SK2 (KCa2.2, KCNN2), and SK3 (KCa2.3, KCNN3) that exhibit high degree of homology and conductance of 5–20 pS. The tetrameric SK channels are voltage independent and their gating is predominantly controlled by intracellular [Ca2+] in the submicromolar range conferred by constitutively bound calmodulin. In ventricular myocytes from healthy hearts plasmalemmal SK (pSK) channels are functionally dormant. However, enhanced SK expression and increased sensitivity to Ca2+ (from 500 to 200 nmol/L) was shown in myocytes from failing hearts and from myocardial infarct border zones.17–22 We recently demonstrated that RyR-mediated SR Ca2+ release is both necessary and sufficient for activation of pSK channels.23 Furthermore we showed that enhanced expression of SK channels plays an anti-arrhythmic role by countering the depolarizing force of the Na+/Ca2+ exchanger (NCX) during spontaneous Ca2+ release. Despite attenuating arrhythmia trigger in the form of Ca2+-dependent early or delayed afterdepolarizations (EADs and DADs, respectively),23,24 disease-associated SK upregulation was also shown to promote a substrate for arrhythmia because of action potential shortening and non-uniform tissue expression of the channels.18–21 It should be noted that previous studies extensively used SK blockers or transgenic mouse models to assess the role of SK channels in arrhythmogenesis.14,15 However, anti-arrhythmic potential of pharmacological enhancement of endogenous SK channels remains unexplored.
Importantly, in addition to plasmalemmal localization, the presence of functional SKs was demonstrated in the inner mitochondria membrane (mSK). Activation of mSK was found to prevent glutamate-induced oxytosis and mitochondrial dysfunction in a neuronal cell line.25 Subsequently, mSK channels were discovered in ventricular cardiomyocytes (CMs), where their pharmacological activation protects against ischaemic cardiac injury.26 In both cases, activation of mSK channels preserves mitochondrial function and reduces production of ROS in cells under stress. Previous studies of cardiac mitochondrial K+ channels such as mKATP and mBKCa have been mostly focused on their ‘pre-conditioning’ protection over ischaemic injury,27–29 whereas the physiological activation and function of these channels are less clear, particularly in light of their steep dependence on [ATP] or mitochondrial membrane potential (ΔΨm), respectively. Considering their submicromolar Ca2+ sensitivity, it is conceivable that mSK channels can respond to intracellular [Ca2+] and regulate mitochondrial function in healthy or diseased CMs. In this study, we demonstrate that SK channels modulators regulate mitochondrial ΔΨm in myocytes from rat hearts with hypertrophy induced by thoracic aortic banding (TAB)30,31 and affect mitochondria ROS production. Furthermore, we show that pharmacological enhancement of mSK protects from Ca2+-dependent arrhythmia by decreasing mitochondria ROS production leading to normalization of redox state of RyRs and stabilization of SR Ca2+ release.
2. Methods
The tissue, cellular, subcellular, and molecular effects of pharmacological manipulation with SK channels were studied at the organ level and in isolated adult rat ventricular myocytes from sham hearts and hearts with hypertrophy induced by TAB four weeks after procedure. In this well-established commercially available model (Charles River Labs), ligation of the ascending aorta results in pressure overload inducing development of cardiac hypertrophy and enhanced arrhythmogenesis in 4 weeks.30,31 We studied whole hearts using dual voltage-Ca2+ optical mapping. At the cellular level, cytosolic Ca2+ changes were monitored using confocal microscopy, and membrane potential was recorded with the whole cell patch-clamp technique using myocytes isolated from TAB hearts. Changes in levels of RNA, protein expression, and protein oxidation were studied using standard approaches.
An expanded Methods section can be found in the Supplementary material online.
3. Results
3.1 Ex vivo dual voltage and Ca2+ optical mapping
To gain insight into the role of SK channels in arrhythmogenesis at the organ level we performed ex vivo whole heart studies using simultaneous membrane potential (Vm) and Ca2+ mapping with the voltage sensitive dye RH278 and the Ca2+ indicator Rhod-2.32 Exposure to the β-adrenergic agonist isoproterenol (ISO, 50 nmol/L) induced frequent premature ventricular complexes (PVCs, red arrows) and VT/VF in seven out of seven TAB hearts and one out of eight Sham hearts. Importantly, incubation of TAB hearts with the membrane permeable SK enhancer NS309 (2 μmol/L, 20 min)33 completely abolished PVCs and ventricular arrhythmia induced by ISO, as seen from ECG recordings in Figure1A. TAB rats showed electrical remodelling resulting in prolongation of APD (Figure1B) in agreement with earlier studies.30,31 Incubation with NS309 did not shorten APD in TAB rats, while subsequent application of the membrane impermeable SK peptide inhibitor apamin (10 nmol/L)15,17,18,20 in the presence of NS309 and ISO prolonged APD. These data suggest that SK channels are present in the plasma membrane of ventricular myocytes from TAB rats and the maximal contribution of SK channels to APD is achieved, such that NS309 does not produce any further effect on APD. Figure1C demonstrates representative activation maps recorded in Sham and TAB rat hearts exposed to ISO. The activation maps of VT/VF showed conduction blocks and reentry formation in addition to frequent PVCs. Membrane permeable SK enhancer NS309 alleviates wavebreaks and VT/VF without detectable effects on conduction velocity (Figure 1CandD). Subsequent application of apamin did not promote VF despite prolongation of APD (Figure1B) and presence of PVCs (Figure1A, red arrow in the bottom trace).
Figure 1.

Pharmacological enhancement of SK channels attenuates Ca2+-dependent arrhythmia in ex vivo optically mapped hypertrophic hearts. (A) Representative ECGs at baseline and in the presence of 50 nmol/L isoproterenol, membrane-permeable SK enhancer NS309 (2 μmol/L) and cell impermeable SK inhibitor apamin (10 nmol/L). (B) Representative traces of action potentials and corresponding pooled data, *#P < 0.05, vs Sham and TAB + NS309 + ISO, paired, and unpaired Student’s t-test where appropriate. (C) Representative activation maps in Sham and TAB hearts. (D) Pulled data for conduction velocities and conduction velocity heterogeneity, not significant at P < 0.05, unpaired Student’s t-test and One-way ANOVA where appropriate. (E) Representative Ca2+ traces in hearts paced at 150 ms cycle length before pause. Red arrows indicate spontaneous Ca2+ release. (F, G) Amplitude and rate of rise of spontaneous Ca2+ release during pause. *#P < 0.05, vs. Sham + ISO and TAB + NS309 + ISO, respectively, One-way ANOVA.
To study potential effects of SK modulators on spontaneous SR Ca2+ release we used burst stimulation followed by pause (Figure1E). Unlike Shams, TAB hearts subjected to this protocol under basal conditions demonstrated elevation in intracellular [Ca2+] during pause (red arrow on the third trace), indicating profound remodelling of Ca2+ homeostasis.34 Exposure to ISO promoted spontaneous elevation of Ca2+ in Sham hearts, while in TAB hearts ISO evoked much larger disturbances in Ca2+ handling with frequent VF induction. As a result, the stimulation protocol could not be finished in TAB group. Taken together, these results strongly implicate abnormal Ca2+ handling as an underlying cause for an increased arrhythmic potential in hypertrophied TAB rat hearts. Importantly, Ca2+ handling in ISO-treated TAB hearts was largely restored with NS309 (Figure 1E, F and G). Consequent application of apamin produced moderate increases in rate and amplitude of spontaneous Ca2+ release (Figure 1E, F and G) most likely because of an increased Ca2+ influx from the extracellular milieu through L-type Ca2+ channels due to APD prolongation. These data suggest that enhancement of SK activity in TAB rat hearts attenuates ventricular arrhythmias primarily due to its stabilizing effect on Ca2+ release.
3.2 Effects of SK channel modulators in hypertrophic rat ventricular CMs
To extend our studies to the cellular level we performed whole cell patch clamp experiments and confocal Ca2+ imaging (Figure2). We found that 10 min. exposure of the SK2- and SK3-specific enhancer, CyPPA (10 μmol/L),35 significantly decreased the ISO-induced incidence of EADs, DADs, and underlying spontaneous Ca2+ waves (SCWs) in TAB myocytes. Similar stabilizing effect on Ca2+ waves was achieved in a separate set of experiments by exposing TAB myocytes to 10 μmol/L NS309 for 10 min, while 10 μmol/L GW542473X (specific SK1 enhancer)36 showed no effect (Supplementary material online, Figure S2). Conversely, the SK inhibitor UCL-1684 (1 μmol/L)37 promoted SCWs and afterdepolarizations (Figure2). In addition, UCL-1684 significantly prolonged APD80 in TAB cells (Supplementary material online, Figure S3). It is conceivable that prolongation of APD upon SK block can contribute to increased spontaneous SR Ca2+ release, by promoting Ca2+ overload due to the increased Ca2+ influx through plasmalemmal L-type Ca2+ channels. However, given that SK enhancer CyPPA failed to shorten APD (Supplementary material online, Figure S3), the effects of SK modulators on Ca2+ waves cannot be fully attributed to changes in activity of plasmalemmal channels alone. This prompted us to focus on the potential roles of SKs recently found in IMM26 in regulation of Ca2+ homeostasis.
Figure 2.

Pharmacological enhancement of SK channels decreases while inhibition increases arrhythmic potential in isolated ventricular myocytes from hypertrophic (TAB) rat hearts. (A) Representative simultaneous recording of Ca2+ transients and Vm in hypertrophic myocyte exposed to 100 nmol/L ISO before and 10 min after application of SK ehancer CyPPA (10 μmol/L) or SK inhibitor UCL-1684 (1 μmol/L); (B, C) Number of ISO-treated hypertrophic cells field-stimulated at 0.5 Hz for 1 min exhibiting Ca2+ waves and wave frequency, respectively. Cell numbers are shown in the columns, *P < 0.05 vs. TAB, Exact Fisher test and hierarchical linear model where appropriate. Number of TAB animals used was 7.
3.3 Expression and distribution of SK isoforms in rat ventricular myocytes
Despite a high degree of homology, three known SK channels isoforms (SK1-3) exhibit differential expression patterns in different tissues and in cardiac disease.14,15,17,18,38 Furthermore, it is still unclear which SK isoform out of three is expressed in the IMM of CMs.26 Using quantitative real time PCR (RT-qPCR) and Western blot analysis we assessed intracellular distribution and possible changes in expression of SK isoforms in myocytes from TAB hearts. Our RT-qPCR results confirm that all three isoforms are expressed in rat ventricular myocytes (Supplementary material online, Figure S1A). Hypertrophy induced by TAB did not change the mRNA levels of SK1 and SK2, while SK3 levels showed ∼two-fold increase (n = 4, P < 0.05, Student’s t-test). To assess specificity of anti-SK antibodies we constructed shRNAs to inhibit expression of SK1-3. Freshly isolated rat ventricular myocytes were infected with adenoviruses (MOI 100) carrying shRNA constructs and after 48 h in primary culture cell lysates were subjected to SDS PAGE (Figure3A). SK1 was identified as ∼66 kDa band (theoretical molecular weight 59.2 kDa), while SK2 and SK3 were running close to 100 kDa which is higher than their predicted molecular weights (62.2 and 81.4 kDa, respectively). Bioinformatic sequence analysis demonstrates that SK1 and SK2 but not SK3 have N-terminal mitochondria targeting sequences (Supplementary material online, Figure S4; also see MitoProt: http://ihg.gsf.de/ihg/mitoprot.html).39 In line with this prediction, our next experiments using differential centrifugation of cell lysates from isolated ventricular myocytes established SK3 as the major isoform localized to surface membrane, while SK1 was found predominantly in the mitochondrial fraction, and SK2 was present in both (Figure3B). Cytochrome C (Cyt C) and mitochondrial Ca2+ uniporter (MCU) were used as markers for mitochondria fraction. Pan-cadherin and α1c and α2 subunits of plasmalemmal Ca2+ channels were used as markers of membrane fraction. Analysis of protein expression levels demonstrated that SK3 and pSK2 are significantly reduced in TAB membranes when normalized to pan-cadherin (Figure 3BandC). SK1 and mitochondrial SK2 (mSK2) normalized to MCU showed no changes in TABs in comparison to Shams in mitochondrial fraction. Western blot analysis using total cell lysate samples from Sham and TAB ventricular myocytes showed no change in the protein levels of SK1, and SK3, while SK2 was ∼50% higher in TABs (Figure 3DandE).
Figure 3.

Expression and subcellular distribution of SK isoforms in ventricular myocytes from Sham and TAB rat hearts. (A) Identification of SK1, SK2, and SK3 using specific shRNAs expressed in cultured rat ventricular myocytes for 48 h. (B) SK channels distribution in membrane and mitochondrial fractions. Cyt C and MCU were used as the markers of mitochondria fraction. Pan-Cadherin and α1c and α2 subunits of L-type Ca2+ channels were used as the markers membrane fraction. (C) Pooled data for normalized optical density (OD, %) of SK1-3 Western Blot analysis in membrane and mitochondrial fractions from isolated myocytes from 5 to 7 Sham and TAB hearts, *P < 0.05, Student’s t-test. Surface membrane SKs (pSK2 and SK3) were normalized to Pan-Cadherin and mSKs SK1 and mSK2 were normalized to MCU. (D, E) Representative western blots and pooled data for OD of SK1-3 in unfractionated myocytes. GAPDH was used as loading control. *P < 0.05, n = 7–12, Student’s t-test.
3.4 Modulators of SK channels affect mitochondrial membrane potential in ventricular CMs
We next determined if fluctuations in intracellular [Ca2+] could modulate mSK activation to modify mitochondrial membrane potential (ΔΨm) using freshly isolated rat CMs with plasmamembrane permeabilized with saponin. Increasing cytosolic [Ca2+] from 50 nmol/L to 1 μmol/L and 50 μmol/L resulted in relatively slow Ca2+ uptake by mitochondria measured using mitochondria-entrapped Ca2+ indicator Rhod-2 (Supplementary material online, Figure S5A). To prevent myocyte contraction at high [Ca2+]cyt cells were pre-incubated with Cytochalasin D (10 μmol/L for 10 min).6 Parallel experiments with mitochondria stained with 1 nmol/L voltage sensitive dye TMRE showed small depolarization of ΔΨm even at 1 μmol/L [Ca2+]cyt, consistent with K+ influx due to activation of mitochondrial K+ channels with high affinity to Ca2+ and ability to open at very high ΔΨ (Supplementary material online, Figure S5B). Application of UCL-1684, a specific SK inhibitor (1 μmol/L), attenuated the drop in ΔΨm evoked by 1 μmol/L [Ca2+]cyt (Figure 4AandB). Importantly, neither UCL-1684 (1 μmol/L), nor SK activator CyPPA (10 μmol/L) had dramatic effects on mitochondrial Ca2+ uptake (Supplementary material online, Figure S6A and B). These data imply that Ca2+-mediated drop in ΔΨm at least in part is conferred by the activation of SK channels. Pharmacological activation of mito-K+ channels is expected to induce small ΔΨm depolarization.25,29 We were unable to detect statistically significant changes in ΔΨm upon application of SK activators in concentrations near reported EC50s (less or ∼10 μmol/L).33,35,36 However, application of 100 μmol/L CyPPA (selective to SK2 and SK3) resulted in ∼40% depolarization of ΔΨm as measured by TMRE fluorescence in permeabilized cells (Figure 4CandD). Depolarization of IMM evoked by 25 μmol/L of CyPPA or NS309 was fully reversible by consequent application of membrane permeable SK inhibitor UCL-1684 (1 μmol/L, Supplementary material online, Figure S7C and 1D) but not by application of membrane impermeable apamin (100 nmol/L, Supplementary material online, Figure S7A and B). Taken together, the effects of SK enhancers are most consistent with the activation of mSK channels, allowing K+ influx and depolarization of ΔΨm.27–29 Importantly, incubation of permeabilized myocytes with the SK1-specific SK activator GW542573X (100 μmol/L) failed to produce changes in ΔΨm. The latter suggests that the presence of SK1 in mitochondrial fraction (see Figure3B) does not result in proper retainment of the functional channel in the IMM, which leaves SK2 as the only functional SK subtype out of three targeted to mitochondria. For a mechanistic link between mSK channel activation and mitochondrial function in TAB rats, we measured the effect of the membrane-permeable SK inhibitor UCL-1684 (1 μmol/L) on ΔΨm in permeabilized Sham and TAB CMs. As shown in Figure 5A and B, UCL-1684 resulted in a small (∼10%) but significant hyperpolarization of ΔΨm in myocytes from TAB rats, while no effect was detected in Sham cells. This observation is consistent with mSK channels being activated at baseline in diseased hearts, causing a small depolarization of ΔΨm which is reversible by SK inhibition. Importantly, data presented in Figure 5CandD suggest that mSK channels were not maximally activated in TAB cells because both NS309 (selective to SK1, SK2, and SK3) and CyPPA (selective to SK2 and SK3) at high concentrations were able to evoke ΔΨm depolarization. Interestingly, the SK1-selective activator GW542573X failed to produce any changes in ΔΨm in TAB cells (Figure 5CandD) consistent with no effect on SCWs (see Supplementary material online, Figure S2).
Figure 4.

Pharmacological enhancers and inhibitors of SK channels modulate ΔΨm in Sham myocytes. (A) Representative recording of changes in mitochondrial membrane potential in response to increase in intracellular Ca2+ from 50 nmol/L to 1 μmol/L followed by application of 1 μmol/L UCL-1684, a selective SK inhibitor. Myocytes from healthy hearts were permeabilized with saponin and immobilized with 10 μmol/L Cytochalasin D. TMRE signal was normalized to minimum fluorescence obtained by application of 50 μmol/L FCCP and presented as a % of baseline. (B) Pooled data for normalized TMRE signal, *#P < 0.05 vs. 50 nmol/L [Ca2+]cyt and 1 μmol/L [Ca2+]cyt, respectively, hierarchical linear modelling. Numbers of cells are presented in columns; number of Sham animals was 3. (C) Representative recordings of ΔΨm in permeabilized cells exposed to GW542573X (selective SK1 enhancer, black) and CyPPA (selective SK2 and SK3 enhancer, red); (D) Pooled data for C, *P < 0.05 vs. baseline, hierarchical linear modelling. Numbers of cells are presented in columns; number of Sham animals was 5.
Figure 5.

Pharmacological inhibition of SK channels increases while enhancement decreases ΔΨm in TAB myocytes. (A) Representative recording of changes in ΔΨm in response to SK channels block by 1 μmol/L UCL-1684 in permeabilized Sham (black) and TAB (grey) myocytes. (B) Pooled data for A, *P < 0.05, hierarchical linear model. Numbers of cells are presented in columns, number of Sham animals was 4, and number of TABs was 5. (C) Representative recordings of ΔΨm in permeabilized cells exposed to 100 μmol/L GW542573X (selective SK1 enhancer, black), CyPPA (selective SK2 and SK3 enhancer, red), and NS309 (enhancer of SK1, SK2, and SK3, blue). (D) Pooled data for C, *P < 0.05 vs. baseline, hierarchical linear model. Numbers of cells are presented in columns; number of TABs was 6.
3.5 Effects of SK modulators on mitochondrial ROS production and RyR hyperactivity in hypertrophic CMs
Excessive ROS production by mitochondria is a hallmark of hypertrophy, heart failure or aging; conditions that are often accompanied by pro-arrhythmic disturbances in Ca2+ handling. Our data demonstrate that increased ROS production is likely responsible for the increased frequency of RyR-mediated SCWs in CMs from TAB rats, as direct scavenging of mitochondria derived ROS with mito-TEMPO (mitochondria specific-scavenger, 10 μmol/L, 10 min.)4,13 also inhibits the proarrhythmic SCWs (Supplementary material online, Figure S8). The monobromobimane (mBB) fluorescence assay shows that incubation of hypertrophied CMs with CyPPA or NS309 reduces thiol-oxidation of a protein band with MW similar to RyRs (Figure 6AandB). Experiments using immunoprecipitated channels from isolated TCMs further confirmed that incubation with SK enhancers decrease oxidation of RyRs (Supplementary material online, Figure S10) consistent with our hypothesis that mSK enhancement eliminates SCWs by reducing ROS production. Furthermore, experiments using the mitochondria-specific ROS indicator MitoSOX demonstrated that inhibition of SK channels with UCL-1684 increased ROS production, while enhancers NS309 and CyPPA reduced it in freshly isolated TAB CMs (Figure6C).
Figure 6.

Reversal of RyR oxidation in TAB myocytes by SK-enhancers via attenuation of mitochondrial ROS. (A) CyPPA reduced thiol-oxidation of RyR in TAB ventricular myocytes. Cells were incubated with 10 μmol/L CyPPA for 10 min before lysis. ROS scavenger dithiothreitol (10 mmol/L) and oxidant 2,2′-dithiodipyridine (200 μmol/L) were used to obtain minimal and maximal oxidation for normalization. Top: Representative fluorescence images of oxidation-sensing dye mBB. Bottom: RyR western for normalization. (B) Pooled data for A. Additional group of cells was treated with 10 μmol/L NS309 for 10 min in the same way as CyPPA, *P < 0.05 vs. Sham and #P < 0.05 vs. TAB, n = 4 hearts for both Sham and TAB, one-way ANOVA. (C) SK modulators alter rate of ROS production measured with MitoSOX in CMs. Cell numbers are shown in the columns. *P < 0.05 vs. Sham and #P < 0.05 vs. TAB, hierarchical linear model. Number of Sham animals was 3; number of Tab animals was 4.
To assess the direct effects of SK activation on RyR-mediated SR Ca2+ leak, we performed measurements of Ca2+ sparks in myocytes permeabilized with saponin to eliminate any potential contribution of effects caused by plasma membrane SKs that can confound interpretation of the results. As shown in Figure7, Ca2+ spark frequency is increased, while SR Ca2+ content measured using rapid application of 20 mmol/L caffeine is decreased in myocytes from TAB rats vs. Shams. Ten-minute pre-incubation with 10 μmol/L CyPPA or NS309 resulted in higher SR Ca2+ load in TAB myocytes and in case of NS309, which is more potent SK enhancer35 significantly reduced Ca2+ spark frequency. It is well known that RyR activity is controlled by [Ca2+] not only at its cytosolic activation sites, but at luminal sites as well.40,41 Oxidation of RyRs was linked to the enhanced sensitivity of RyRs to intra-SR [Ca2+] resulting in substantial decrease in SR Ca2+ content.7 A decrease and even lack of increase in spark frequency at higher SR Ca2+ loads in SK-enhancer-treated TAB myocytes is consistent with at least partial stabilization of RyRs due to restoration of their luminal Ca2+ sensitivity. This recovery of RyR function might be sufficient to substantially reduce propagating SCWs in myocytes from hypertrophic hearts (see Figure2, Supplementary material online, Figure S2) and thereby propensity to Ca2+-dependent arrhythmia.
Figure 7.

SK enhancement stabilizes RyRs in TAB myocytes. Pre-incubation of TAB myocytes with 10 μmol/L CyPPA or 10 μmol/L NS309 for 10 min normalizes SR Ca2+ content in permeabilized TAB myocytes. (A, B) Representative line scan images and pooled data for spark frequency, respectively. (C, D) Representative traces of caffeine-induced Ca2+ transients (20 mmol/L) and pooled data for caffeine-induced Ca2+ transients amplitude, respectively. Ca2+ sparks and Ca2+ transients were measured in saponin-permeabilized myocytes using Fluo-4. Cell numbers are shown in the columns, *#P < 0.05 vs. Sham and TAB respectively, hierarchical linear model. Number of Sham animals was 4; number of TAB animals was 6.
4. Discussion
Recently SK channels gained recognition as an attractive new target for treatment of malignant cardiac arrhythmias.14–16 Previous reports were focused on delineating the role of aberrant expression/function of pSK channels and the potential cardioprotective role of mSK channels is only beginning to emerge.26 In this work, using rat model of pressure-overload-induced hypertrophy,30,31 we provide the first evidence that mSK channels modulators can influence SR Ca2+ release via regulation of mitochondrial ROS production, which determines oxidative state and activity of RyRs in diseased hearts.
4.1 SK modulation and ventricular arrhythmia
Previous studies using bee-venom peptide apamin, a specific SK inhibitor, demonstrated that inhibition of SK channels in rabbit hearts with heart failure induced by rapid pacing can attenuate arrhythmia via prolongation of APD and reducing heterogeneity, thus limiting reentry.16,18,19,20 However, experiments in single cells from human and canine failing hearts demonstrated that apamin application not only further prolongs already long APD caused by electrical remodelling, but also induces profound instabilities in repolarization suggesting an anti-arrhythmic role of SKs.17 Furthermore, we recently showed in rat myocytes with adenovirus-mediated overexpression of SK2 that pro-arrhythmic spontaneous Ca2+ release effectively induces repolarizing SK current which attenuate NCX-driven afterdepolarizations.23 Accordingly, in failing rabbit hearts, apamin exacerbated EADs associated with disturbances in Ca2+ cycling.24 Taken together, results of these studies imply that in conditions accompanied with aberrant Ca2+ handling, enhancement rather than inhibition of SK channels must play a protective role by limiting Ca2+-dependent triggering events in the form of EADs and DADs. Indeed, our experiments in whole hearts and single cells demonstrate beneficial anti-arrhythmic effects of SK enhancers and pro-arrhythmic effects of SK inhibitors (Figures 1and2 and Supplementary material online, Figure S2). SK inhibitors prolonged APD in TABs providing an evidence of the presence of functional SK channels in plasma membrane of TAB CMs (Figure1B, Supplementary material online, Figure S3). Remarkably, SK enhancers NS309 and CyPPA (which increase SK activity by sensitizing channels to Ca2+)35 failed to shorten APD both in whole hearts (Figure1B) and in isolated CMs (Supplementary material online, Figure S3A). This implies that pSK channels in TABs are already hypersensitive to Ca2+, which is similar to human18 and rabbit HF.19 SK-enhancer-mediated leftward shift to Kd to [Ca2+] as high as 200 nmol/L characteristic of cardiac disease is unlikely to cause any additional effects on APD, because SK channels will be already open during Ca2+ transient when [Ca2+]cyt reaches micromolar range. Lack of effects of membrane-permeable NS309 and CyPPA also implies that their anti-arrhythmic effects are largely mediated by mSKs through regulation of SR Ca2+ release. The latter is further confirmed by the inability of membrane-impermeable apamin to reverse protective stabilization of Ca2+ cycling by the membrane permeable NS309 in TAB rat hearts despite apamin-induced prolongation of APD (Figure1).
4.2 Expression and distribution of SK channels in ventricular myocytes
In addition to enhanced sensitivity of pSKs to Ca2+, increased expression levels of the channels were demonstrated in humans and several animal models of heart failure and myocardial infarct.16–22 Our results using RT-qPCR demonstrate that SK1 and SK2 mRNA levels are unchanged and SK3 mRNA levels are ∼two-fold higher in isolated CMs from TABs than Shams. However, Western blot analysis shows no change in SK3 levels in whole CMs lysates (Figure 3DandE). One possibility why SK3 protein levels remain unchanged in TAB CMs at increased mRNA levels is translation interference by microRNAs. To test this possibility we assessed the expression levels of muscle-specific microRNA 499 (miR-499), showed to target SK3.42 We found that the levels of miR-499 are significantly higher in TABs vs. Shams (Supplementary material online, Figure S1B). SK1 protein levels were unchanged in CMs which is consistent with RT-qPCR results, while SK2 was moderately ∼50% higher in TABs vs Shams despite no change in mRNA levels. Our results markedly differ from reported two-to four-fold upregulation of SK2 and SK3 protein levels in tissues from rat,43 human and canine failing hearts.15,17 This discrepancy can be attributed to the existence of SK channels in cell types other than myocytes that are present in cardiac tissue and also undergo disease-associated remodelling. For example, SK channels are likely present in fibroblasts44 and endothelial cells45 and can contribute to substantial increase in expression levels detected in ventricular tissues from HF patients. Moreover, our experiments with fractionation of CMs demonstrated significant reduction of pSK2 and SK3 (Figure 3BandD). Ren et al.46 demonstrated that phosphorylation of SK2 by protein kinase A at three C-terminal serines leads to the channel removal from the plasma membrane. It is possible that in conditions when blood levels of catecholamines are increased like in HF, plasmalemmal localization of SK2 and structurally similar SK3 channels is diminished rather than increased as previously thought.
Our further experiments with CMs fractionation confirmed presence of SK channels in mitochondria of ventricular myocytes as described by Stowe etal.26 However, this study did not provide definite answer which SK isoform resides in the IMM in CMs. We demonstrate that SK1 and SK2 subtypes are present in the mitochondrial fraction in rat CMs, while SK3 is restricted to the plasmalemma (Figure3B). These results are in agreement with bioinformatics prediction, which suggested that SK1 and SK2 but not SK3 have N-terminal mitochondria-targeting sequences (MitoProt39: http://ihg.gsf.de/ihg/mitoprot.html, 26 January 2017, date last accessed; Supplementary material online, Figure S4). SK1 and SK2 localized in mitochondria show no change in expression levels in TABs (Figure3C). Importantly, the bioinformatics programme also predicts that SK2 exhibits much higher probability of mitochondria targeting than SK1 (0.90 vs. 0.17, respectively). In line with this, our data show lack of effect of SK1-specific activator GW542573X on ΔΨm (Figures 4C, D, 5CandD) and SCWs (Supplementary material online, Figure S2). Although we cannot completely rule out potential role of SK1, our results establish SK2 as the major SK isoform residing in the inner mitochondrial membrane.
4.3 SK channels and mitochondrial ROS
Cardioprotection mediated by mitochondrial KATP and BK Ca2+-activated channels is well established, while the cardioprotective potential of mSK channels is only beginning to emerge. BK and KATP channel openers are known to reduce damaging ROS production by mitochondria (under stress conditions), decrease arrhythmic potential, and improve overall cardiac function after ischaemia-reperfusion or hydroxyl radical-induced stunning in failing hearts.14,27–29,47 Recently Stowe etal.26 demonstrated that pharmacological pre-conditioning using SK enhancers resulted in dramatically improved function of guinea pig hearts subjected to ischaemia-reperfusion. This improvement was linked to normalized mitochondria bioenergetics accompanied by reduced levels of superoxide production. Our pharmacological analysis confirms presence of functional mSKs in ventricular myocytes responsive to changes in [Ca2+] within the physiological range, determined in experiments assessing ΔΨm (Figure4). Furthermore, increased activity of mSK (Figure 5AandB) may be an adaptive response to preserve mitochondria function as inhibition of mSK channels in hypertrophic myocytes results in increased mito-ROS production measured with MitoSOX (Figure6C). Importantly, pharmacologic enhancement of mSK channels in TAB CMs reduced mito-ROS rendering them similar to control CMs (Figure6C). It should be noted that activation of mito-K+ channels during pre-conditioning was well established to increase rather than decrease mito-ROS emission by accelerating electron flux along the respiratory chain. In line with this, our experiments revealed that treatment of rat myocytes from healthy hearts with 10 μmol/L CyPPA increased rate of ROS production (not shown). Interestingly, our experiments showed that neither SK enhancer nor SK inhibitor can dramatically affect mitochondrial Ca2+ uptake in TAB CMs (Supplementary material online, Figure S6), thus SK-mediated changes in mito-ROS production are likely not dependent on changes in mito-Ca2+ homeostasis. Apparently, the exact mechanisms of mSK- mediated protection in cells from diseased hearts, where ROS levels are already high, are yet to be determined and their delineation will require extensive efforts in the future.
4.4 SK channels and RyR-mediated Ca2+ release
Overactive RyRs play a central role in arrhythmias associated with hypertrophy, heart failure, MI, and aging.4–7 Excessive activity of RyRs in these conditions is routinely attributed to posttranslational modifications including phosphorylation at RyR’s CaMKII and PKA sites and oxidation of reactive cysteines.8,40,41 Reducing agents were shown to produce stabilizing effects on RyRs resulting in attenuation of SR Ca2+ leak, pro-arrhythmic SCWs and Ca2+ alternans in many experimental settings.5–8,13 We recently demonstrated that incubation of ventricular myocytes derived from old rabbit hearts with mitochondria-specific ROS scavenger mito-TEMPO normalizes RyR redox state and thereby SR Ca2+ release,4 which corroborates the theory of a tight control of RyR function by mitochondria mediated by ROS.4,10,11,13 Our data demonstrate that pharmacological enhancement of SKs reverses oxidation of RyRs in myocytes from hypertrophic hearts via normalization of ROS production by mitochondria (Figure6, Supplementary material online, Figure S10), which results in stabilization of RyR-mediated Ca2+ release (Figure7), attenuation of pro-arrhythmic SCWs at the single cell level (Figure2, Supplementary material online, Figure S2) and rescue of Ca2+ homeostasis in the whole heart experiments (Figure1). Importantly, while blockade of pSK channels produces anti-arrhythmic effects in certain conditions by reducing arrhythmic substrate, our data imply that usage of membrane-permeable SK inhibitors may affect mitochondrial function thus promoting Ca2+-dependent triggers for arrhythmia.
5. Limitations
Usage of pharmacological modulators of ion channels and small peptides can raise concerns of their specificity and potential off-target effects. To address these concerns we performed experiments in rat myocytes infected with adenoviral constructs carrying rSK2 and maintained in primary culture for 48 h. SK2 overexpression significantly reduced the number of SCWs (Supplementary material online, Figure S9) and mito-ROS measured with MitoSOX (SK: 842 ± 99 a.u., n = 4; Control: 1338 ± 80 a.u., n = 5, P < 0.05, Student’s t-test), similar to the effects of pharmacological activators of SK2 in freshly isolated TAB cells. The other limitation is that we cannot completely rule out a possible role of pSK channels in the regulation of mitochondrial function and thereby Ca2+ release. Finally, presented data does not provide the exact mechanism by which mSKs can regulate mitochondrial ROS production, which is the subject of future research.
6. Conclusions
Our results suggest that functional upregulation of SK channels in hypertrophic hearts serve protective, albeit incomplete, roles against Ca2+-dependent arrhythmia via two independent mechanisms (Figure8): (i) Plasmalemmal SKs protect from Ca2+ overload by shortening APD limiting Ca2+ influx and attenuate NCX-mediated EADs and DADs during SCWs; (ii) mSKs stabilize RyR-mediated Ca2+ release by reducing mitochondrial production of ROS. We conclude that facilitation of mSK channels in diseased hearts may serve as a new strategy to treat Ca2+-dependent arrhythmia in hypertrophy and HF.
Figure 8.
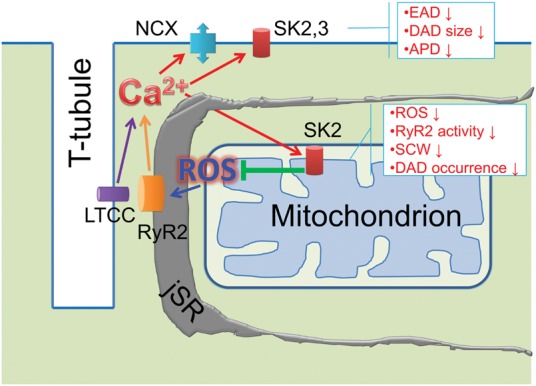
Schematic presentation of proposed cellular mechanisms of SK-mediated protection from Ca2+-dependent ventricular arrhythmia. (i) Functional upregulation of SK2 and 3 in the plasma membrane limits disease-related prolongation of APD and reduces Ca2+-dependent EADs and DADs countering INCX; (ii) Functional upregulation of mitochondria-targeted SK2 attenuates disease-related increase in mito-ROS. Pharmacological enhancement of SK2 reduces mito-ROS resulting in normalization of the oxidative state of RyR, stabilization of RyR-mediated Ca2+ release and abolishment of DAD-driving SCWs.
Supplementary material
Supplementary material is available at Cardiovascular Research online.
Supplementary Material
Acknowledgements
The authors are thankful to Dr. Jin O-Uchi for his help in establishing the protocol for separation of membrane and mitochondrial fractions from isolated ventricular myocytes.
Conflict of interest: none declared.
Funding
This work was supported by American Heart Association Grant in Aid 15GRNT25650002 and NIH R01 HL121796-01A1 (D.T.); T32HL094300-05 and R01HL110791-04 (G.K.), R01 HL1024025 and R01 HL106592 (S.C.D.).
References
- 1. Zheng ZJ, Croft JB, Giles WH, Mensah GA.. Sudden cardiac death in the United States, 1989 to 1998. Circulation 2001;104:2158–2163. [DOI] [PubMed] [Google Scholar]
- 2. Ter Keurs HE, Boyden PA.. Calcium and arrhythmogenesis. Physiol Rev 2007;87:457–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Laurita KR, Rosenbaum DS.. Mechanisms and potential therapeutic targets for ventricular arrhythmias associated with impaired cardiac calcium cycling. J Mol Cell Cardiol 2008;44:31–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cooper LL, Li W, Lu Y, Centracchio J, Terentyeva R, Koren G, Terentyev D.. Redox modification of ryanodine receptors by mitochondria-derived reactive oxygen species contributes to aberrant Ca2+ handling in ageing rabbit hearts. J Physiol 2013;591:5895–5911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Belevych AE, Terentyev D, Terentyeva R, Nishijima Y, Sridhar A, Hamlin RL, Carnes CA, Gyorke S.. The relationship between arrhythmogenesis and impaired contractility in heart failure: role of altered ryanodine receptor function. Cardiovasc Res 2011;90: 493–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Belevych AE, Terentyev D, Viatchenko-Karpinski S, Terentyeva R, Sridhar A, Nishijima Y, Wilson LD, Cardounel AJ, Laurita KR, Carnes CA, Billman GE, Gyorke S.. Redox modification of ryanodine receptors underlies calcium alternans in a canine model of sudden cardiac death. Cardiovasc Res 2009;84:387–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Terentyev D, Gyorke I, Belevych AE, Terentyeva R, Sridhar A, Nishijima Y, de Blanco EC, Khanna S, Sen CK, Cardounel AJ, Carnes CA, Gyorke S.. Redox modification of ryanodine receptors contributes to sarcoplasmic reticulum Ca2+ leak in chronic heart failure. Circ Res 2008;103:1466–1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Niggli E, Ullrich ND, Gutierrez D, Kyrychenko S, Poláková E, Shirokova N.. Posttranslational modifications of cardiac ryanodine receptors: Ca(2+) signaling and EC-coupling. Biochim Biophys Acta 2013;1833:866–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Schwarz K, Siddiqi N, Singh S, Neil CJ, Dawson DK, Frenneaux MP.. The breathing heart - Mitochondrial respiratory chain dysfunction in cardiac disease. Int J Cardiol 2014;171:134–143 [DOI] [PubMed] [Google Scholar]
- 10. Akar FG, Mitochondrial targets for arrhythmia suppression: is there a role for pharmacological intervention? J Interv Card Electrophysiol 2013;37:249–258. [DOI] [PubMed] [Google Scholar]
- 11. Zhou L, Aon MA, Liu T, O'Rourke B.. Dynamic modulation of Ca2+ sparks by mitochondrial oscillations in isolated guinea pig cardiomyocytes under oxidative stress. J Mol Cell Cardiol 2012;51:632–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zorov DB, Juhaszova M, Sollott SJ.. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiol Rev 2014;94:909–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bovo E, Lipsius SL, Zima AV.. Reactive oxygen species contribute to the development of arrhythmogenic Ca2+ waves during β-adrenergic receptor stimulation in rabbit cardiomyocytes. J Physiol 2012;590:3291–3304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Clements RT, Terentyev D, Sellke FW.. Ca(2+)-activated K(+) channels as therapeutic targets for myocardial and vascular protection. Circ J 2015;79:455–462. [DOI] [PubMed] [Google Scholar]
- 15. Mahida S. Expanding role of SK channels in cardiac electrophysiology. Heart Rhythm 2014;11:1233–1238. [DOI] [PubMed] [Google Scholar]
- 16. Chang PC, Chen PS.. SK channels and ventricular arrhythmias in heart failure. Trends Cardiovasc Med 2015;25:508–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bonilla IM, Long VP 3rd, Vargas-Pinto P, Wright P, Belevych A, Lou Q, Mowrey K, Yoo J, Binkley PF, Fedorov VV, Györke S, Janssen PM, Kilic A, Mohler PJ, Carnes CA.. Calcium-activated potassium current modulates ventricular repolarization in chronic heart failure. PLoS One 2014:9:e108824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chang PC, Turker I, Lopshire JC, Masroor S, Nguyen BL, Tao W, Rubart M, Chen PS, Chen Z, Ai T.. Heterogeneous upregulation of apamin-sensitive potassium currents in failing human ventricles. J Am Heart Assoc 2013;2: e004713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chua SK, Chang PC, Maruyama M, Turker I, Shinohara T, Shen MJ, Chen Z, Shen C, Rubart-von der Lohe M, Lopshire JC, Ogawa M, Weiss JN, Lin SF, Ai T, Chen PS.. Small-conductance calcium-activated potassium channel and recurrent ventricular fibrillation in failing rabbit ventricles. Circ Res 2011;108:971–979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hsieh YC, Chang PC, Hsueh CH, Lee YS, Shen C, Weiss JN, Chen Z, Ai T, Lin SF, Chen PS.. Apamin-sensitive potassium current modulates action potential duration restitution and arrhythmogenesis of failing rabbit ventricles. Circ Arrhythm Electrophysiol 2013;6:410–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gui L, Bao Z, Jia Y, Qin X, Cheng ZJ, Zhu J, Chen QH.. Ventricular tachyarrhythmias in rats with acute myocardial infarction involves activation of small-conductance Ca2+-activated K+ channels. Am J Physiol Heart Circ Physiol 2013;304:H118–H130. [DOI] [PubMed] [Google Scholar]
- 22. Lee YS, Chang PC, Hsueh CH, Maruyama M, Park HW, Rhee KS, Hsieh YC, Shen C, Weiss JN, Chen Z, Lin SF, Chen PS.. Apamin-sensitive calcium-activated potassium currents in rabbit ventricles with chronic myocardial infarction. J Cardiovasc Electrophysiol 2013;24:1144–1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Terentyev D, Rochira JA, Terentyeva R, Roder K, Koren G, Li W.. Sarcoplasmic reticulum Ca(2)(+) release is both necessary and sufficient for SK channel activation in ventricular myocytes. Am J Physiol Heart Circ Physiol 2014;306:H738–H746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chang PC, Hsieh YC, Hsueh CH, Weiss JN, Lin SF, Chen PS.. Apamin induces early afterdepolarizations and torsades de pointes ventricular arrhythmia from failing rabbit ventricles exhibiting secondary rises in intracellular calcium. Heart Rhythm 2013;10:1516–1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Dolga AM, Netter MF, Perocchi F, Doti N, Meissner L, Tobaben S, Grohm J, Zischka H, Plesnila N, Decher N, Culmsee C.. Mitochondrial small conductance SK2 channels prevent glutamate-induced oxytosis and mitochondrial dysfunction. J Biol Chem 2013;288:10792–10804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Stowe DF, Gadicherla AK., Zhou Y, Aldakkak M, Cheng Q, Kwok WM, Jiang MT, Heisner JS, Yang M, Camara AK.. Protection against cardiac injury by small Ca(2+)-sensitive K(+) channels identified in guinea pig cardiac inner mitochondrial membrane. Biochim Biophys Acta 2013;1828:427–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Malinska D, Mirandola SR, Kunz WS.. Mitochondrial potassium channels and reactive oxygen species. FEBS Lett 2010;584:2043–2048. [DOI] [PubMed] [Google Scholar]
- 28. Szabo I, Leanza L, Gulbins E, Zoratti M.. Physiology of potassium channels in the inner membrane of mitochondria. Pflugers Arch 2012;463:231–246. [DOI] [PubMed] [Google Scholar]
- 29. Aon M, Cortossa S, Wei A, Grunnet M, O’Rourke B.. Energetic performance is improved by specific activation of K+ fluxes through K Ca channels in heart mitochodria. Biochim Biophys Acta 2011;1797:71–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Del Monte F, Butler K, Boecker W, Gwathmey JK, Hajjar RJ.. Novel technique of aortic banding followed by gene transfer during hypertrophy and heart failure. Physiol Genomics 2002;9:49–56. [DOI] [PubMed] [Google Scholar]
- 31. Jin H, Chemaly ER, Lee A, Kho C, Hadri L, Hajjar RJ, Akar FG.. Mechanoelectrical remodelling and arrhythmias during progression of hypertrophy. Faseb J 2010;24:451–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lau E, Kossidas K, Kim TY, Kunitomo Y, Ziv O, Song Z, Taylor C, Schofield L, Yammine J, Liu G, Peng X, Qu Z, Koren G, Choi BR.. Spatially discordant alternans and arrhythmias in tachypacing-induced cardiac myopathy in transgenic LQT1 rabbits: the importance of IKs and Ca2+ cycling. PLoS One 2015;10:e0122754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Strobaek D, Teuber L, Jorgensen TD, Ahring PK, Kjaer K, Hansen RS, Olesen SP, Christophersen P, Skaaning-Jensen B.. Activation of human IK and SK Ca2+ -activated K+ channels by NS309 (6,7-dichloro-1H-indole-2,3-dione 3-oxime). Biochim Biophys Acta 2004;1665:1–5. [DOI] [PubMed] [Google Scholar]
- 34. Erickson JR, Pereira L, Wang L, Han G, Ferguson A, Dao K, Copeland RJ, Despa F, Hart GW, Ripplinger CM, Bers DM.. Diabetic hyperglycaemia activates CaMKII and arrhythmias by O-linked glycosylation. Nature 2013;502:372–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hougaard C, Eriksen BL, Jorgensen S, Johansen TH, Dyhring T, Madsen LS, Strobaek D, Christophersen P.. Selective positive modulation of the SK3 and SK2 subtypes of small conductance Ca2+-activated K+ channels. Br J Pharmacol 2007;151:655–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hougaard C, Jensen ML, Dale TJ, Miller DD, Davies DJ, Eriksen BL, Strøbaek D, Trezise DJ, Christophersen P.. Selective activation of the SK1 subtype of human small-conductance Ca2+-activated K+ channels by 4-(2-methoxyphenylcarbamoyloxymethyl)-piperidine-1-carboxylic acid tert-butyl ester (GW542573X) is dependent on serine 293 in the S5 segment. Mol Pharmacol 2009;76:569–578. [DOI] [PubMed] [Google Scholar]
- 37. Rosa JC, Galanakis D, Ganellin CR, Dunn PM, Jenkinson DH.. Bis-quinolinium cyclophanes: 6,10-diaza-3(1,3),8(1,4)-dibenzena-1,5(1,4)- diquinolinacyclodecaphane (UCL 1684), the first nanomolar, non-peptidic blocker of the apamin-sensitive Ca(2+)-activated K+ channel. J Med Chem 1998;41:2–5. [DOI] [PubMed] [Google Scholar]
- 38. Tuteja D, Xu D, Timofeyev V, Lu L, Sharma D, Zhang Z, Xu Y, Nie L, Vazquez AE, Young JN, Glatter KA, Chiamvimonvat N.. Differential expression of small-conductance Ca2+-activated K+ channels SK1, SK2, and SK3 in mouse atrial and ventricular myocytes. Am J Physiol Heart Circ Physiol 2005;289:H2714–H2723. [DOI] [PubMed] [Google Scholar]
- 39. Claros MG, Vincens P.. Computational method to predict mitochondrial proteins and their targeting sequences. Eur J Biochem 1996;241:779–786. [DOI] [PubMed] [Google Scholar]
- 40. Györke S, Carnes C.. Dysregulated sarcoplasmic reticulum calcium release: potential pharmacological target in cardiac disease. Pharmacol Ther 2008;119:340–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zima AV, Bovo E, Mazurek SR, Rochira JA, Li W, Terentyev D.. Ca handling during excitation-contraction coupling in heart failure. Pflugers Arch 2014;466:1129–1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ling TY, Wang XL, Chai Q, Lau TW, Koestler CM, Park SJ, Daly RC, Greason KL, Jen J, Wu LQ, Shen WF, Shen WK, Cha YM, Lee HC.. Regulation of the SK3 channel by microRNA-499–potential role in atrial fibrillation. Heart Rhythm 2013;10:1001–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Yang D, Wang T, Ni Y, Song B, Ning F, Hu P, Luo L, Wang Y, Ma A.. Apamin-sensitive K+ current upregulation in volume-overload heart failure is associated with the decreased interaction of CK2 with SK2. J Membr Biol 2015;248:1181–1189. [DOI] [PubMed] [Google Scholar]
- 44. Choi S, Lee W, Yun J, Seo J, Lim I.. Expression of Ca-activated K channels and their role in proliferation of rat cardiac fibroblasts. Korean J Physiol Pharmacol 2008;12:51–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Liu Y, Xie A, Singh AK, Ehsan A, Choudhary G, Dudley S, Sellke FW, Feng J.. Inactivation of endothelial small/intermediate conductance of calcium-activated potassium channels contributes to coronary arteriolar dysfunction in diabetic patients. J Am Heart Assoc 2015;4:e002062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ren Y, Barnwell LF, Alexander JC, Lubin FD, Adelman JP, Pfaffinger PJ, Schrader LA, Anderson AE.. Regulation of surface localization of the small conductance Ca2+-activated potassium channel, Sk2, through direct phosphorylation by cAMP-dependent protein kinase. J Biol Chem 2006;281:11769–11779. [DOI] [PubMed] [Google Scholar]
- 47. Maack C, Dabew ER, Hohl M, Schäfers HJ, Böhm M.. Endogenous activation of mitochondrial KATP channels protects human failing myocardium from hydroxyl radical-induced stunning. Circ Res 2009;105:811–817. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.