

Abstract
Aims
Chronic kidney disease (CKD) is a powerful independent risk factor for cardiovascular events, including vein graft failure. Because CKD impairs the clearance of small proteins, we tested the hypothesis that CKD exacerbates vein graft disease by elevating serum levels of critical cytokines that promote vein graft neointimal hyperplasia.
Methods and results
We modelled CKD in C57BL/6 mice with 5/6ths nephrectomy, which reduced glomerular filtration rate by 60%, and we modelled vein grafting with inferior-vena-cava-to-carotid interposition grafting. CKD increased vein graft neointimal hyperplasia four-fold, decreased vein graft re-endothelialization two-fold, and increased serum levels of interleukin-9 (IL-9) five-fold. By quantitative immunofluorescence and histochemical staining, vein grafts from CKD mice demonstrated a ∼two-fold higher prevalence of mast cells, and a six-fold higher prevalence of activated mast cells. Concordantly, vein grafts from CKD mice showed higher levels of TNF and NFκB activation, as judged by phosphorylation of NFκB p65 on Ser536 and by expression of VCAM-1. Arteriovenous fistula veins from humans with CKD also showed up-regulation of mast cells and IL-9. Treating CKD mice with IL-9-neutralizing IgG reduced vein graft neointimal area four-fold, increased vein graft re-endothelialization ∼two-fold, and reduced vein graft total and activated mast cell levels two- and four-fold, respectively. Treating CKD mice with the mast cell stabilizer cromolyn reduced neointimal hyperplasia and increased re-endothelialization in vein grafts. In vitro, IL-9 promoted endothelial cell apoptosis but had no effect on smooth muscle cell proliferation.
Conclusion
CKD aggravates vein graft disease through mechanisms involving IL-9 and mast cell activation.
Keywords: Neointimal hyperplasia, Cytokines, Mast cells, Endothelial cells, Smooth muscle cells
1. Introduction
Defined as a glomerular filtration rate (GFR) of <60 mL/min per 1.73 m2 of body surface area for 3 months or more, chronic kidney disease (CKD) is an important independent predictor of cardiovascular morbidity and mortality.1 CKD affects ∼20 million adults in the United States, and the prevalence of CKD is estimated to be 8 to 16% worldwide.2 Cardiovascular event rates increase 10% for every 10-unit fall in GFR below even 81 mL/min/1.73 sq m,3 and CKD patients are ∼40 times more likely to suffer a cardiovascular event and ∼15 times more likely to die than they are to suffer progression to renal replacement therapy.4 Furthermore, CKD substantially increases failure rates of coronary stenting5 and aortocoronary vein grafts: among patients with vein grafts that are patent 1–11 years after implantation, CKD increases the subsequent vein graft failure rate by 62%.6 This problem of vein graft failure in CKD patients is magnified by the observation that, in CKD patients, coronary artery bypass grafting appears to confer a lower long-term risk of myocardial infarction and revascularization than coronary stenting.7
Vein graft failure initiates with neointimal hyperplasia, a process involving proliferation and migration of medial smooth muscle cells (SMCs) into the sub-endothelial intima as well as an accumulation of extracellular matrix in the neointima.8 Vein graft neointimal hyperplasia has been shown to be exacerbated by inflammatory cytokines,9,10 and to predispose vein grafts to accelerated atherosclerosis.11 Because elevated serum levels of pro-inflammatory cytokines are found in humans with CKD,12 we hypothesized that inflammatory cytokines could mediate CKD-dependent acceleration of vein graft disease. To test whether identifying CKD-associated cytokines could facilitate therapies for CKD-associated vein graft disease, we used the 5/6ths nephrectomy model of CKD in mice and carotid interposition vein grafting.
2. Methods
2.1 Mice
C57BL/6 J mice were used in all experiments. All animal experiments were performed according to protocols approved by the Duke Institutional Animal Care and Use Committee; these protocols complied with the Guide for the Care and Use of Laboratory Animals (National Academies Press, 2011).
2.2 Nephrectomy model of CKD
5/6ths nephrectomy was performed in two sequential operations, modified from published protocols.13 Anesthesia with pentobarbital (50 mg/kg i.p.) was used for all surgeries; see Supplementary material online. After the second stage of the nephrectomy surgeries, mice recovered for 2 week before undergoing either GFR measurement or carotid interposition vein grafting.
GFR Measurement was performed under 2% isoflurane anesthesia, as we described.14
2.3 Carotid interposition vein grafting
Interposition vein graft surgery was performed as we described.15 Inferior venae cavae from WT donor mice were anastomosed end-to-side to the right common carotid artery of 5/6ths or 1/3rd-nephrectomized recipient mice.
2.4 Histology
All measurements and calculations on histologic specimens were made by observers blinded to specimen identity. Vein graft morphometry was performed with NIH ImageJ as we have described.9,15 Immunofluorescence microscopy and data analysis with ImageJ were performed as we described.9 The extent of vein graft re-endothelialization was determined as the percentage of luminal perimeter surfaced by von Willebrand factor-positive cells.16
Toluidine blue staining for mast cells was performed as described.17 Mast cells were identified by their metachromatic (purple) granules, which are distinct from the orthochromatic (blue) staining of all other cells. Mast cells were dichotomized as (a) quiescent, with densely packed granules in the cytoplasm, or (b) activated, or degranulating, with disgorged and loosely packed granules.17 A minimum of 100 mast cells per specimen were counted.
2.5 Serum cytokine assays
Serum was harvested by ventricular puncture from pentobarbital-anesthetized mice at the indicated time points. Mouse sera were analysed with the Bio-Plex Pro™ Mouse Cytokine Assay and with an ELISA for serum amyloid A18 (see Supplementary material online).
2.6 Systemic neutralization of IL-9
At the time of vein graft surgery, CKD mice were injected with one of two Armenian Hamster IgG2/κ molecules: (a) D9302C12, which neutralizes IL-9,19 or (b) control IgG, which binds no known mouse protein (BioLegend). Subsequently, mice were injected with the same IgG 3 times per week (300 μg i.p.),19 until 48–72 h before vein graft harvest at 4 week after implantation.
2.7 Systemic treatment with cromolyn
To prevent mast cell degranulation in CKD mice, we treated mice with the mast cell stabilizer cromolyn.17 One day prior to vein grafting, CKD mice were injected i.p. with either PBS or cromolyn (50 mg/kg). After vein grafting, mice were injected with cromolyn or PBS twice per week for 4 weeks, until vein graft harvest.
2.8 Endothelial and smooth muscle cell experiments
Mouse aortic endothelial cells (ECs) and smooth muscle cells (SMCs) were isolated by enzymatic digestion, then cultivated as we described.20 ECs for proliferation experiments were grown in medium containing 20% (vol/vol) serum from 5/6ths-nephrectomized or 1/3rd-nephrectomized mice; SMCs used 10% mouse serum. At each proliferation time point, cells were quantitated with a dye-binding assay, as we described.20 SMC proliferation and EC apoptosis assays were conducted ±IL-9 as described in Supplementary material online.
2.9 Human vein samples from arteriovenous fistulae (AVFs)
All procedures with human volunteers were approved by the Duke Institutional Review Board. Vein specimens were obtained by a single surgeon (J. H. L.) from five humans with end-stage renal disease when they underwent two-stage basilic vein transposition AVF surgery or radiocephalic AVF surgery.21 Samples of ‘native vein’ were obtained during the initial construction of the AVF, and ‘AVF vein’ samples were obtained during the surgery to transpose the arterialized basilic vein.
2.10 Statistical analyses
All values were plotted in figures as means ± SE and cited in the text as means ± SD. GraphPad Prism® software was used to perform t tests (for comparing two groups), or 2-way ANOVA with Sidak’s post-hoc test for multiple comparisons. A P value of less than 0.05 was considered significant.
To analyse data from serum cytokine assays, the distributions of each cytokine were examined. We made two comparisons of ‘treatment’ vs. ‘control’: (1) CKD mice vs. control-GFR mice, and (2) anti-IL-9-IgG-injected CKD mice vs. control IgG-injected CKD mice. We compared the medians between the treatment and control groups using the non-parametric Wilcoxon Test with exact P value calculation, under the null hypothesis that there was no difference between the treatment and control groups. A conservative multiple comparisons correction for 23 independent cytokines was used to indicate a significant test: P < 0.05/23, or P < 0.00217.
3. Results
3.1 CKD increases vein graft neointimal hyperplasia and inflammation
To model CKD in mice, we performed 5/6ths nephrectomy as described,13 and assessed GFR 2 week later by FITC-inulin clearance. Compared with sham-operated control mouse GFR, CKD mouse GFR was 60 ± 10% less (Figure 1A)—a GFR reduction that models human chronic kidney disease stage 3.1
Figure 1.
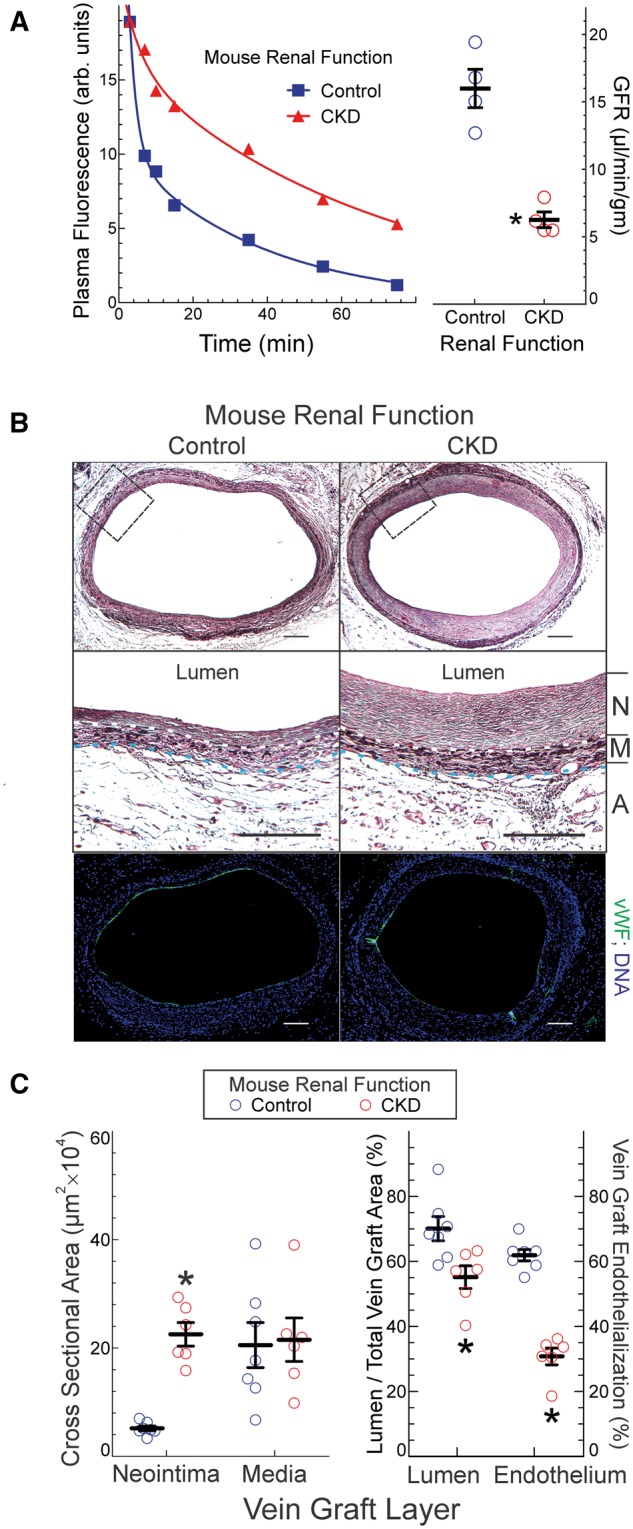
CKD exacerbates vein graft neointimal hyperplasia. (A), Mice were subjected to 5/6ths nephrectomy (CKD) or sham surgery (control); 2 week later, they were injected with FITC-inulin and subjected to sequential blood collections at the indicated times after injection; plasma fluorescence at each time point is plotted for individual CKD and control mice, at left. GFR was calculated from these decay curves and plotted (right panel) as means ±SE from 4 mice in each group. Compared with control: *P < 0.05 (t test). (B), CKD and 1/3rd-nephrectomized (control) mice were subjected to carotid interposition vein grafting, and vein grafts were harvested 4 week later after perfusion fixation. Serial cross sections were stained with a modified Masson trichrome (top) or with IgG targeting von Willebrand factor (vWF, green) and Hoechst 33 342 (DNA, blue). The area delineated by the dashed boxes (top) are enlarged and presented in the middle panels, in which dotted white and blue lines delineate the boundaries between neointima (‘N’) and media (‘M’), and between media and adventitia (‘A’), respectively. Scale bars = 50 μm. (C), Cross sectional areas for neointima, media, lumen and ‘total vein’ (excluding adventitia) were calculated by planimetry, and plotted as means ± SE from six mice in each group. ‘Vein graft endothelialization’ was calculated as the fraction of vein graft total luminal perimeter covered by vWF-positive (endothelial) cells, and plotted as the means ± SE from 6 mice in each group. Compared with control mice: *P < 0.05 (2-way ANOVA with post-hoc Sidak test).
To determine the effect of CKD on vein graft disease, we performed carotid interposition vein grafting15 on CKD and control-GFR mice, 2 week after 5/6ths or 1/3rd nephrectomy, respectively. Vein grafts were harvested 4 week post-operatively, and cross sections were evaluated by planimetry.15 Although vein graft tunica media areas were equivalent in control-GFR and CKD mice, the neointimal area was 4 ± 0.5-fold larger in CKD mice (Figure 1B and C). Despite their larger neointimal areas, vein grafts from CKD mice demonstrated a structural composition comparable to that found in vein grafts from control-GFR mice, as judged by the prevalence of SMCs and the density of collagen I20 (see Supplementary material online, Figure S1). However, the prevalence of proliferating SMCs was two-fold greater in vein grafts from CKD mice than in vein grafts from control-GFR mice (see Supplementary material online, Figure S1). Vein grafts from CKD mice also demonstrated (Figure 1B and C) a ∼50% reduction in the degree of vein graft re-endothelialization, which inversely correlates with vein graft neointimal hyperplasia.16,22
Because CKD is associated with increased plasma levels of inflammatory cytokines in humans,12,23 we tested whether vein grafts from CKD mice evinced greater levels of inflammatory signalling in the neointimal and medial layers. We adduced as evidence of inflammatory signalling the phosphorylation on Ser536 of the NFκB subunit p65; this phosphorylation is effected by IκB kinase-β, and augments NFκB transcriptional activity.24 Whereas vein grafts from control-GFR and CKD mice showed equivalent expression of total p65 NFκB, vein grafts from CKD mice showed 75 ± 9% higher levels of phospho-p65(Ser536) (Figure 2), and 60 ± 10% higher levels of p65 nuclear translocation (see Supplementary material online, Figure S2). Thus, vein grafts in CKD mice appeared to have more extensive activation of NFκB. Concordantly, vein grafts from CKD mice expressed ∼two-fold higher levels of the NFκB-dependent,24 pro-inflammatory gene product VCAM-1 (Figure 2), and showed ∼30% greater prevalence of adventitial macrophages—even though CKD and control vein grafts had equivalent levels of adventitial T lymphocytes (see Supplementary material online, Figure S1). Furthermore, vein grafts from CKD mice also had 60 ± 10% higher levels of TNF and 60 ± 10% greater expression of TNF receptor-1 (see Supplementary material online, Figure S1), which we previously found mediates most of the NFκB activation in vein graft SMCs.9 Taken together, these data suggest that CKD engenders greater vein graft inflammation.
Figure 2.
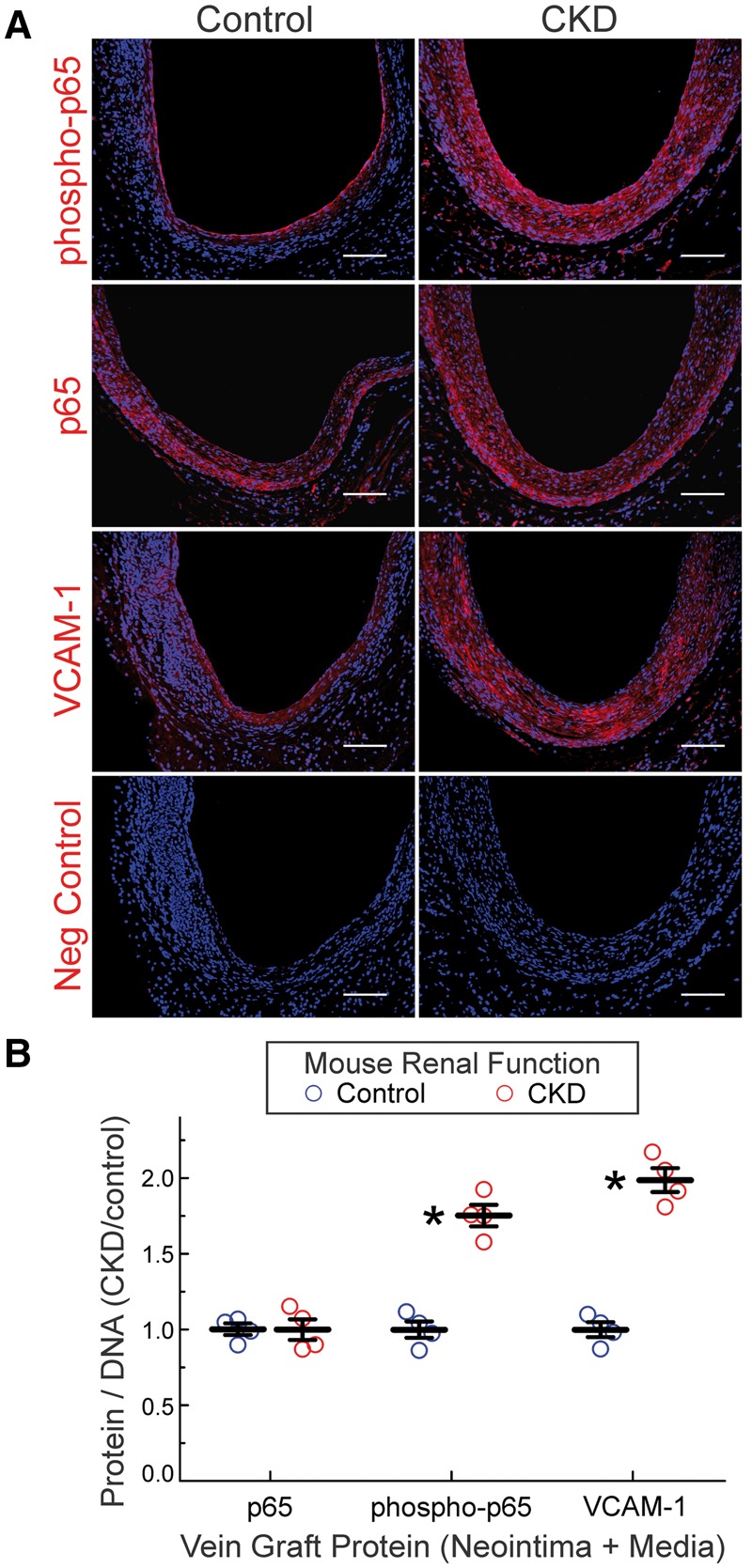
CKD augments NFκB activation in vein grafts. (A), Serial sections of vein grafts from Figure 1 were immunostained with rabbit IgG targeting the following proteins: NFκB p65 subunit phosphorylated on Ser536 (phospho-p65); NFκB p65 subunit, phosphorylated or not (p65); VCAM-1; or no specific protein (Neg Control). Alexa 546-conjugated anti-rabbit IgG and Hoechst 33 342 were used on all specimens. Specimens from control and CKD mice were stained concurrently. (B), The ratios of red (protein) to blue (DNA) pixels in the neointima plus media were quantitated by Image J, and normalized to the cognate ratios obtained for control specimens in each staining cohort, to obtain ‘CKD/control’, plotted individually and as the means ± SE for each group. Compared with control: *P < 0.01 (2-way ANOVA with post-hoc Sidak test).
3.2 CKD augments serum IL-9 levels
Does increased vein graft inflammation in CKD mice result from higher levels of plasma pro-inflammatory cytokines? To address this question, we analysed individual sera from 9 control-GFR and 12 CKD mice, matched for age and sex. Sera were harvested 2 week after 5/6ths nephrectomy, at a time when serum amyloid A levels were equivalent in both CKD and control mice (1.13 ± 0.02 vs. 1.19 ± 0.03 µg/mL), and thus when CKD mice had recovered from post-op inflammation.25 Of 23 cytokines tested, only five had serum levels that were significantly higher in CKD than in control mice (Figure 3). Of these, the cytokine with by far the greatest CKD-associated increment was interleukin-9 (IL-9) (Figure 3); consequently, we focused on IL-9 in subsequent studies.
Figure 3.
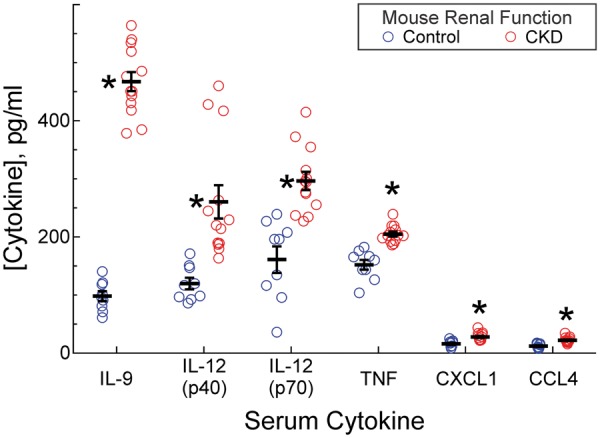
CKD increases serum cytokine levels. Mice subjected to 5/6ths nephrectomy (CKD) or sham surgery (control-GFR) were exsanguinated 2 weeks after the final surgery. Individual serum samples were analysed in triplicate for 23 cytokines. Serum cytokine levels were plotted as means ± SE from 9 (control-GFR) or 13 (CKD) individual mice in each group. Compared with control-GFR mouse sera: *P < 0.002 (Wilcoxon test). IL-12, interleukin-12 (p40 and p70 subunits). Cytokines not plotted (see Supplementary material online) were measured at equivalent levels in CKD and control mice.
Increased serum levels of IL-9 in CKD mice correlated with 3.0 ± 0.6-fold higher vein graft levels of IL-9 in CKD mice (Figure 4). In addition, vein grafts from CKD mice demonstrated 1.7 ± 0.3-fold higher levels of the IL-9 receptor-α (IL-9Rα), which binds to and mediates signalling evoked by IL-9.26 Because IL-9 is a critical growth factor for mast cells,26 and because mast cells contribute to vascular inflammation in a variety of ways,17 we asked whether at least some of the excess IL-9Rα-expressing vein graft cells in CKD mice were mast cells.
Figure 4.

CKD augments vein graft mast cell prevalence and activation. (A), Serial sections of vein grafts from Figure 2 were stained with Hoechst 33 342 (DNA) and immunostained with rabbit IgG specific for IL-9, IL-9Rα, mouse mast cell proteases -6 and -7 (Tryptase), mouse mast cell protease-5 (mMCP5), or no particular protein (Neg Control). Scale bars = 50 μm. (B), Protein immunofluorescence throughout the vein graft wall was quantitated as in Figure 2, to yield ‘% of control,’ plotted individually and as means ± S.E. Compared with vein grafts from control-GFR mice: *P < 0.05 (2-way ANOVA with Sidak post-hoc test). (C), Serial sections were stained with toluidine blue, to identify metachromatic (purple) mast cell granules. Arrows indicate quiescent (upper panel) and activated (degranulating, lower panel) mast cells. Scale bars = 10 μm. (D), The prevalence of quiescent and activated mast cell in vein grafts were assessed by toluidine blue staining, and plotted as means ± S.E. of five vein grafts per group (≥100 adventitial mast cells were counted per specimen). Compared with vein grafts from control-GFR mice: *P < 0.05 (2-way ANOVA with Sidak post-hoc test).
To address this question, we used three complementary approaches to quantitate the prevalence of mast cells in the adventitia, where, like others,17 we found virtually all of the vein grafts’ mast cells: (a, b) immunostaining for the mast cell-specific proteases tryptase (mouse mast cell proteases 6 and 7)27 and mouse mast cell protease-5 (chymase),27 as well as (c) histochemically staining for mast cells with toluidine blue17 (Figure 4). By all three approaches, mast cells were 2–3 times more prevalent in vein grafts from CKD than from control-GFR mice (Figure 4). Furthermore, as assessed by toluidine blue staining, the prevalence of activated (degranulating) mast cells17 was six-fold higher in vein grafts from CKD mice than from control-GFR mice (Figure 4).
3.3 IL-9 promotes CKD-dependent vein graft disease through mast cell-dependent mechanisms
To discern whether IL-9 plays a causative role in CKD-induced vein graft disease, we injected CKD mice with IL-9-neutralizing or isotype control IgG for 4 week after vein grafting. Control IgG-injected CKD mice developed vein graft neointimal hyperplasia equivalent to that of untreated CKD mice (Figures 1 and 5). Remarkably, anti-IL-9 IgG treatment reduced this vein graft neointimal hyperplasia in CKD mice by four-fold, and reduced medial thickness by 1.6-fold (Figure 5). Consonant with its reduction in vein graft neointimal hyperplasia, anti-IL-9 IgG treatment also improved vein graft re-endothelialization, by 46 ± 5% (Figure 5), and reduced vein graft levels of NFκB phospho-p65(Ser536), IL-9Rα, and IL-9 by ∼40% (see Supplementary material online, Figure S3). Renal function was equivalent, however, in anti-IL-9-treated and control IgG-treated mice, as judged by creatinine values13 determined on post-mortem serum samples (∼25 μmol/L). Thus, IL-9 appears to contribute substantially to CKD-dependent vein graft inflammation and neointimal hyperplasia.
Figure 5.

IL-9 promotes CKD-induced vein graft disease and augments mast cell prevalence in CKD mouse vein grafts. Mice were subjected to 5/6ths nephrectomy and vein graft surgery as in Figure 1. At the time of vein graft surgery, mice were injected with hamster IgG2/κ—either anti-IL-9, or isotype control: 50 μg i.v. plus 250 μg i.p., followed by 300 μg i.p. 3×/week until vein graft harvest 4 week post-op. (A), Serial sections of perfusion-fixed vein grafts were stained with a modified trichrome or, immunofluorescently, for von Willebrand factor (vWF, green) and Hoechst 33 342 (blue, DNA). Scale bars = 200 μm. (B), The indicated cross sectional areas were calculated as in Figure 1 and plotted as means ± SE from seven mice in each group. ‘Vein graft endothelialization’ was calculated as in Figure 1, and plotted as means ± SE from five mice in each group. Compared with control mice: *P < 0.01. (C), The prevalence of quiescent and activated mast cells in vein grafts were assessed by toluidine blue staining, as in Figure 4. Shown are means ± S.E. from five vein grafts from each IgG-injected mouse group. Compared with control: *P < 0.05 (2-way ANOVA with Sidak post-hoc test).
Because anti-IL-9 IgG treatment so dramatically reduced CKD-dependent vein graft inflammation and neointimal hyperplasia, we tested whether anti-IL-9 IgG treatment also reduced the CKD-dependent increase in the prevalence and activation of vein graft mast cells. In vein grafts from control IgG-injected CKD mice, the prevalence of mast cells was comparable to that found in untreated CKD mice (Figures 5C and 4). However, in vein grafts from anti-IL-9 IgG-injected CKD mice, the prevalence of mast cells was 45 ± 8% lower than it was in vein grafts from isotype control IgG-injected CKD mice. Furthermore, in vein grafts from anti-IL-9 IgG-injected CKD mice the prevalence of activated mast cells was 75 ± 6% lower than it was in vein grafts from control IgG-injected CKD mice (Figure 5C).
Anti-IL-9 treatment of our CKD mice reduced the serum levels of five cytokines, among the 23 cytokines assayed: levels of IL-9, IL-1α, IL-6, IL-10, and IL-12 were 30-60% lower in anti-IL-9-treated than in control IgG-treated mice (see Supplementary material online, Figure S4). Each of these cytokines is secreted by mast cells.28 Thus, anti-IL-9 IgG treatment evoked changes in serum cytokine levels that are consistent with the changes it evoked in vein graft mast cell levels and mast cell activation.
To test more directly whether mast cell degranulation contributes to CKD-dependent vein graft disease, we treated vein-grafted CKD mice with cromolyn, an inhibitor of mast cell degranulation.17 As compared with vehicle-treated CKD mice, cromolyn-treated CKD mice developed ∼40% less vein graft neointimal hyperplasia and demonstrated ∼50% more vein graft re-endothelialization (see Supplementary material online, Figure S5). However, unlike anti-IL-9 IgG therapy, cromolyn therapy did not reduce the prevalence of total or quiescent mast cells, assessed by anti-tryptase immunostain or toluidine blue stain, respectively; rather, cromolyn therapy reduced the prevalence of activated (degranulating) mast cells, by 70 ± 5% (see Supplementary material online, Figure S5C). In contrast to anti-IL-9 IgG therapy, cromolyn therapy in CKD mice did not affect levels of 23 serum cytokines (see Supplementary material online, Figure S5D and data not shown). Thus, mast cell degranulation in CKD mice appears to augment vein graft neointimal hyperplasia and to inhibit vein graft re-endothelialization.
CKD mice demonstrated impaired vein graft re-endothelialization, and this defect was substantially mitigated by treatment with anti-IL-9 IgG or the mast cell inhibitor cromolyn (Figures 1 and 5; see Supplementary material online, Figure S5). To address whether the serum of CKD mice could be directly toxic to ECs, we cultured mouse ECs in the presence or absence of serum derived from mice with either normal GFR or CKD, and quantitated EC number after 4 days. Serum from control-GFR mice promoted EC growth, but serum from CKD mice did not (Figure 6). The addition of IL-9-neutralizing IgG to control-GFR mouse serum had no effect on EC proliferation. However, the addition of IL-9-neutralizing IgG to CKD mouse serum augmented EC proliferation (Figure 6). Thus, it appears that CKD mouse serum may be directly toxic to ECs, and that part of this toxicity may be mediated by IL-9 itself. To test this possibility more directly, we cultured ECs in the absence or presence of the [IL-9] observed in CKD mouse serum (600 pg/mL). Under these conditions, IL-9 engendered a three-fold increase in EC apoptosis, judged by the prevalence of ECs positive for cleaved caspase-316 (Figure 6B and C). Despite the apparent toxicity to ECs of CKD serum and IL-9, neither one adversely affected SMC proliferation (Figure 6D and E).
Figure 6.

CKD mouse serum inhibits endothelial cell proliferation. Mice were subjected to 5/6ths (CKD) or 1/3rd nephrectomy (Control); 2 week after the final surgery, serum was harvested and pooled from 12 mice in each GFR group. (A), Mouse ECs were cultured in medium containing 20% (v/v) serum from either CKD or control mice, along with isotype control (Non-immune) or anti-IL-9 IgG, each at 2 μg/mL. After 4 days, EC number was quantitated (see Methods). Within each experiment, the number of ECs was divided by that obtained with control serum and control IgG, and multiplied by 100 to obtain ‘% of control’. Plotted are means ±SE from three independent experiments performed with distinct mouse EC lines. Compared with control serum: *P < 0.05; compared with isotype control IgG: #, P < 0.05 (2-way ANOVA with post-hoc Sidak test). (B and C), Mouse ECs were grown on culture slides in the absence (None) or presence of IL-9 (600 pg/mL) for 48 h, and then stained for von Willebrand factor (vWF) and the apoptosis-associated cleaved caspase-3 (scale bar = 25 μm). The prevalence of cleaved caspase-3-positive ECs was plotted (mean ± SE) from three independent experiments with two independent lines of mouse ECs. Compared with ‘none’: *P < 0.05 (t test). (D), Mouse SMCs were grown in medium containing 10% serum from CKD or control mice; SMC numbers were quantitated at the indicated time points and plotted as means ± SE from three independent experiments performed with distinct SMC lines. (E), Mouse SMCs used in panel D were grown in medium containing 2.5% fetal bovine serum lacking (None) or containing IL-9 at 600 pg/mL; SMC numbers were quantitated at the indicated time points and plotted as means ± SE from three independent experiments.
To determine whether IL-9-related mechanisms could be involved in CKD-related vein neointimal hyperplasia in humans, we enrolled human subjects with chronic kidney disease stage V who were undergoing basilic vein transposition surgery to create arteriovenous fistulae (AVFs) for haemodialysis access (see Supplementary material online, Table S1).21 This surgery is performed in two stages; consequently, we were able to collect native vein during the first stage of the surgery and arterialized vein during the second stage of the surgery.21 By using paired samples from each end-stage-CKD human subject, we were able to compare vein samples that were identical genetically but distinct by virtue of adaptation to haemodynamic conditions. With arterialization of the vein in these end-stage CKD subjects, there was a 45–65% increase in the expression of IL-9, IL-9Rα, and mast cell tryptase (Figure 7 and see Supplementary material online, Table S1). These data are consistent with the possibility that IL-9 and mast cells may play a role in human CKD-dependent vein pathology in AVFs, in which vein neointimal hyperplasia is a principal cause of AVF failure.21
Figure 7.
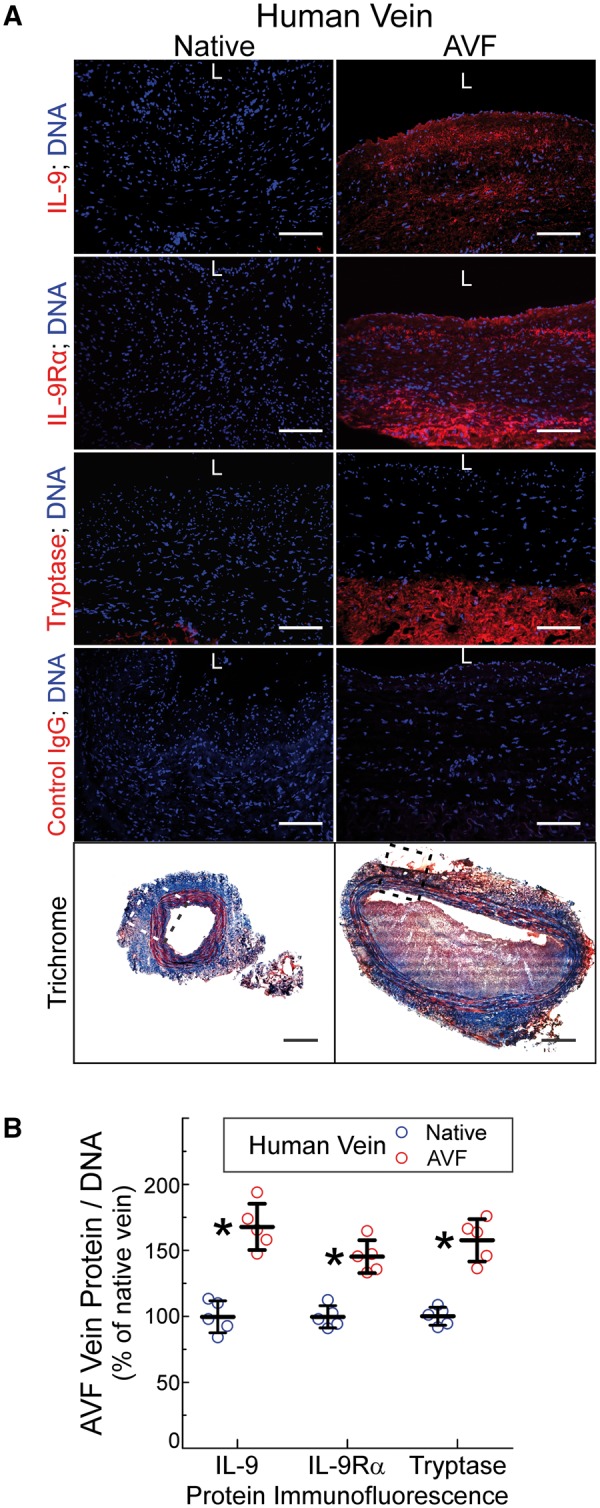
Human veins demonstrate increased IL-9, IL-9Rα, and mast cell prevalence during maturation of arteriovenous fistulas (AVFs) in patients with end-stage renal disease. Vein specimens were obtained from five humans with end-stage renal disease undergoing two-stage basilic vein transposition or radiocephalic AVF surgery (see Supplementary material online, Table S1). (A), Serial frozen sections were stained with trichrome (bottom) or, pairwise, with primary IgGs targeting IL-9, IL-9Rα, mast cell tryptase, or no particular protein (Control); sections were counterstained with Hoechst 33 342 (DNA, blue). ‘L’ designates the luminal surface. Scale bars = 100 μm for immunofluorescence panels and 500 μm for the trichrome panels. Shown are images from Subject 3 (see Supplementary material online, Table S1); the boxed areas indicate the regions imaged for immunofluorescence. (B), From five independent pairs of native and AVF vein specimens, protein immunofluorescence from three random 200× fields was quantitated as in Figure 2, normalized to values obtained for the native vein, and plotted as means ± S.E. Compared with values from the native vein: *P < 0.05.
4. Discussion
This study provides the first direct evidence that CKD promotes vein graft disease, through mechanisms requiring IL-9 and mast cell activation. We found that either anti-IL-9 or mast cell-inhibiting interventions mitigated CKD-dependent vein graft disease and promoted vein graft re-endothelialization in mice. Furthermore, the possibility that these findings pertain to human disease is suggested by our observation that IL-9 and mast cell prevalence both up-regulate in the arterializing vein of AVFs in humans with end-stage renal disease.
That CKD promotes vascular disease generally is not only suggested by abundant observational data in humans3–6 but also directly demonstrated in animal models. With the 5/6ths nephrectomy model in Apoe-/- mice, CKD exacerbates atherosclerosis through mechanisms involving augmented hypercholesterolemia29 and angiotensin II-promoted LDL oxidation.30 However, the pro-atherogenic role of CKD-augmented cytokine levels has not been dissected from the role of CKD-augmented hypercholesterolemia in the pathogenesis of accelerated atherosclerosis.13
Although they differ substantially from vein grafts haemodynamically because they carry ≥3 times more blood flow,31 AVFs have been used in mice to demonstrate the effects of CKD on vein arterialization. CKD promotes neointimal hyperplasia in the venous segment of jugulocarotid AVFs32–34 through mechanisms that—unlike those we found in vein grafts—may be independent of SMC proliferation.32 This vein neointimal hyperplasia in AVFs can be reduced by bone morphogenetic protein-7,32 calcitriol (or genetic deficiency of intermediate-early response gene X-1),35 glucagon-like peptide-1 receptor agonist,36 VEGF-A-targeting shRNA,37 heme oxygenase activity,31 and inhibition of endothelial cell Notch signalling.34 Our study extends these previous studies by demonstrating that CKD also potentiates neointimal hyperplasia and endothelial toxicity in vein grafts, and that it does so largely by augmenting IL-9 levels.
IL-9 is a 14.2 kDa polypeptide (∼32 kDa when fully glycosylated) secreted by CD4+ T cells (Th9, Th2) and, to a lesser degree, granulocytes and mast cells.26,38 By binding to its heterodimeric receptor comprising the IL-9Rα and the common γ-chain receptor, IL-9 affects mast cells, lymphocytes (Th2, Th9, Th17, Treg, B cells), airway epithelial cells and SMCs.26,38 IL-9 is a critical growth factor for mast cells; it also promotes proliferation of B lymphocytes and eosinophils.26 Whereas IL-9 is important for immunity to parasites, it also contributes to allergic inflammation38 and perhaps to other diseases: levels of IL-9 are elevated in the plasma of humans with atherosclerosis and in human atherosclerotic plaques39; the prevalence of (IL-9-secreting) Th9 cells among all CD4+ lymphocytes is higher among acute coronary syndrome patients than among control subjects40; and IL-9 promotes atherosclerosis in Apoe-/- mice.40 Although CKD elevates plasma IL-9 levels in mice (Figure 3) and may elevate plasma levels of IL-9 in diabetic humans,41 it remains to be determined how CKD elevates plasma IL-9 levels: by augmenting IL-9 secretion from particular cell types, by reducing glomerular filtration of IL-9, or through a combination of these processes.
The role of mast cells in vascular diseases has been explored with a variety of models. In dinitrofluorobenzene-sensitized Apoe-/- mice, systemic or focal activation of mast cells augments atherosclerosis through mechanisms that can be inhibited by cromolyn and that appear to involve mast cell-dependent increases in leucocyte adhesion and plaque microvessel permeability.17 Jugulocarotid AVFs develop markedly less vein neointimal hyperplasia in mast cell-deficient42 c-KitW/Wv mice than in cognate wild-type mice.43 Congruent with these findings—and with our own study of mast cells in human AVFs (Figure 7)—c-Kit+ cells (i.e. predominantly mast cells42) are ∼50% more numerous in arterialized veins of human brachiobasilic AVFs than in corresponding native veins harvested from the same subjects.43 In carotid interposition vein grafts, cromolyn has been used to reduce total wall thickness of vein grafts placed in Apoe-/- mice.44 However, this study’s vein graft model is of questionable relevance to vein grafts placed in humans: the vein grafts use end-to-end vein-to-artery attachments achieved by slipping the vein (telescopically) over the carotid artery’s cut ends, which have been everted over polyethylene tubing segments, and then tying the veins in place with circumferential ligatures (rather than sutured anastomoses); consequently, neointimal hyperplasia in this model is exceptionally aggressive (discussed in Ref. 15). In contrast, our vein grafts employ sutured, end-to-side vein-to-carotid anastomoses that model surgery performed on human subjects.15
CKD reduced vein graft re-endothelialization in our vein graft model, and re-endothelialization correlated inversely with the degree of vein graft neointimal hyperplasia. Similarly, CKD delays re-endothelialization of jugular veins used in AVFs of mice undergoing 5/6ths nephrectomy,33 and reduces endothelial barrier function.34 In our CKD mice, anti-IL-9 IgG and cromolyn therapies both augmented vein graft re-endothelialization and diminished neointimal hyperplasia. These findings accord with our previous work: by genetically manipulating TNF receptors9,16 or by administering endothelial cells intravenously,22 we found that accelerating vein graft re-endothelialization diminishes neointimal hyperplasia.
CKD-associated endothelial deficits may help to explain why our CKD mouse vein grafts demonstrate IL-9 immunostaining throughout the neointima, media, and adventitia (Figure 4, see Supplementary material online, Figure S3), even though the primary IL-9-responsive mast cells and primary IL-9-producing T lymphocytes reside almost exclusively in the adventitia (see Supplementary material online, Figure S1). Plasma IL-9—at levels five-fold higher in CKD than in normal mice—permeates the damaged vein graft endothelium and diffuses throughout all layers of the vein graft. The resultant high level of vein graft IL-9 does not appear to promote SMC proliferation (Figure 6), but does promote adventitial mast cell proliferation and degranulation (Figure 5), a process that itself could subsequently promote SMC proliferation by increasing vein graft levels of TNF9 (see Supplementary material online, Figure S1), among other cytokines that can promote neointimal hyperplasia.27 In addition, mast cell protease-5 (‘chymase’) activates transforming growth factor β1 and angiotensin II,45 both of which contribute to neointimal hyperplasia in a variety of models.46–48 Indeed, inhibiting mast cell chymase reduces canine arteriovenous fistula neointimal hyperplasia.49 Whether targeting IL-9 therapeutically will prove practical in reducing vein graft disease or arteriovenous fistula vein disease in humans with CKD remains to be determined.
Supplementary material
Supplementary material is available at Cardiovascular Research online.
Conflict of interest: none declared.
Funding
This work was supported by the National Institutes of Health [P30DK096493 to SBG, LZ, ERH, and NJF; DK103082 to JHL, and HL121689 to NJF], the American Heart Association [15GRNT25710194 to LZ], and the Edna and Fred L. Mandel, Jr. Foundation.
Supplementary Material
References
- 1. Levey AS, de Jong PE, Coresh J, El Nahas M, Astor BC, Matsushita K, Gansevoort RT, Kasiske BL, Eckardt KU.. The definition, classification, and prognosis of chronic kidney disease: a KDIGO Controversies Conference report. Kidney Int 2011;80:17–28. [DOI] [PubMed] [Google Scholar]
- 2. Jha V, Garcia-Garcia G, Iseki K, Li Z, Naicker S, Plattner B, Saran R, Wang AY, Yang CW.. Chronic kidney disease: global dimension and perspectives. Lancet 2013;382: 260–272. [DOI] [PubMed] [Google Scholar]
- 3. Anavekar NS, McMurray JJ, Velazquez EJ, Solomon SD, Kober L, Rouleau JL, White HD, Nordlander R, Maggioni A, Dickstein K, Zelenkofske S, Leimberger JD, Califf RM, Pfeffer MA.. Relation between renal dysfunction and cardiovascular outcomes after myocardial infarction. N Engl J Med 2004;351:1285–1295. [DOI] [PubMed] [Google Scholar]
- 4. Go AS, Chertow GM, Fan D, McCulloch CE, Hsu CY.. Chronic kidney disease and the risks of death, cardiovascular events, and hospitalization. N Engl J Med 2004;351:1296–1305. [DOI] [PubMed] [Google Scholar]
- 5. Appleby CE, Ivanov J, Lavi S, Mackie K, Horlick EM, Ing D, Overgaard CB, Seidelin PH, von Harsdorf R, Dzavik V.. The adverse long-term impact of renal impairment in patients undergoing percutaneous coronary intervention in the drug-eluting stent era. Circ Cardiovasc Interv 2009;2:309–316. [DOI] [PubMed] [Google Scholar]
- 6. Wellenius GA, Mukamal KJ, Winkelmayer WC, Mittleman MA.. Renal dysfunction increases the risk of saphenous vein graft occlusion: results from the Post-CABG trial. Atherosclerosis 2007;193:414–420. [DOI] [PubMed] [Google Scholar]
- 7. Bangalore S, Guo Y, Samadashvili Z, Blecker S, Xu J, Hannan EL.. Revascularization in patients with multivessel coronary artery disease and chronic kidney disease: everolimus-eluting stents versus coronary artery bypass graft surgery. J Am Coll Cardiol 2015;66:1209–1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cai X, Freedman NJ.. New therapeutic possibilities for vein graft disease in the post-edifoligide era. Future Cardiol 2006;2:493–501. [DOI] [PubMed] [Google Scholar]
- 9. Zhang L, Peppel K, Brian L, Chien L, Freedman NJ.. Vein graft neointimal hyperplasia is exacerbated by tumor necrosis factor receptor-1 signaling in graft-intrinsic cells. Arterioscler Thromb Vasc Biol 2004;24:2277–2283. [DOI] [PubMed] [Google Scholar]
- 10. Zhang L, Brian L, Freedman NJ.. Vein graft neointimal hyperplasia is exacerbated by CXCR4 signaling in vein graft-extrinsic cells. J Vasc Surg 2012;56:1390–1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mann MJ, Gibbons GH, Kernoff RS, Diet FP, Tsao PS, Cooke JP, Kaneda Y, Dzau VJ.. Genetic engineering of vein grafts resistant to atherosclerosis. Proc Natl Acad Sci USA 1995;92:4502–4506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gupta J, Mitra N, Kanetsky PA, Devaney J, Wing MR, Reilly M, Shah VO, Balakrishnan VS, Guzman NJ, Girndt M, Periera BG, Feldman HI, Kusek JW, Joffe MM, Raj DS.. Association between albuminuria, kidney function, and inflammatory biomarker profile in CKD in CRIC. Clin J Am Soc Nephrol 2012;7:1938–1946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Pedersen TX, Madsen M, Junker N, Christoffersen C, Vikesa J, Bro S, Hultgardh-Nilsson A, Nielsen LB.. Osteopontin deficiency dampens the pro-atherogenic effect of uraemia. Cardiovasc Res 2013;98:352–359. [DOI] [PubMed] [Google Scholar]
- 14. Gurley SB, Mach CL, Stegbauer J, Yang J, Snow KP, Hu A, Meyer TW, Coffman TM.. Influence of genetic background on albuminuria and kidney injury in Ins2(+/C96Y) (Akita) mice. Am J Physiol Renal Physiol 2010;298:F788–F795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zhang L, Hagen PO, Kisslo J, Peppel K, Freedman NJ.. Neointimal hyperplasia rapidly reaches steady state in a novel murine vein graft model. J Vasc Surg 2002;36:824–832. [PubMed] [Google Scholar]
- 16. Zhang L, Sivashanmugam P, Wu JH, Brian L, Exum ST, Freedman NJ, Peppel K.. Tumor necrosis factor receptor-2 signaling attenuates vein graft neointima formation by promoting endothelial recovery. Arterioscler Thromb Vasc Biol 2007;28:284–289. [DOI] [PubMed] [Google Scholar]
- 17. Bot I, de Jager SC, Zernecke A, Lindstedt KA, van Berkel TJ, Weber C, Biessen EA.. Perivascular mast cells promote atherogenesis and induce plaque destabilization in apolipoprotein E-deficient mice. Circulation 2007;115:2516–2525. [DOI] [PubMed] [Google Scholar]
- 18. Zhang L, Peppel K, Sivashanmugam P, Orman ES, Brian L, Exum ST, Freedman NJ.. Expression of tumor necrosis factor receptor-1 in arterial wall cells promotes atherosclerosis. Arterioscler Thromb Vasc Biol 2007;27:1087–1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Li H, Nourbakhsh B, Ciric B, Zhang GX, Rostami A.. Neutralization of IL-9 ameliorates experimental autoimmune encephalomyelitis by decreasing the effector T cell population. J Immunol 2010;185:4095–4100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wu JH, Zhang L, Fanaroff AC, Cai X, Sharma KC, Brian L, Exum ST, Shenoy SK, Peppel K, Freedman NJ.. G protein-coupled receptor kinase-5 attenuates atherosclerosis by regulating receptor tyrosine kinases and 7-transmembrane receptors. Arterioscler Thromb Vasc Biol 2012;32:308–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Conte MS, Nugent HM, Gaccione P, Roy-Chaudhury P, Lawson JH.. Influence of diabetes and perivascular allogeneic endothelial cell implants on arteriovenous fistula remodeling. J Vasc Surg 2011;54:1383–1389. [DOI] [PubMed] [Google Scholar]
- 22. Brown MA, Zhang L, Levering VW, Wu JH, Satterwhite LL, Brian L, Freedman NJ, Truskey GA.. Human umbilical cord blood-derived endothelial cells reendothelialize vein grafts and prevent thrombosis. Arterioscler Thromb Vasc Biol 2010;30:2150–2155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Pruijm M, Ponte B, Vollenweider P, Mooser V, Paccaud F, Waeber G, Marques-Vidal P, Burnier M, Bochud M.. Not all inflammatory markers are linked to kidney function: results from a population-based study. Am J Nephrol 2012;35:288–294. [DOI] [PubMed] [Google Scholar]
- 24. Jean-Charles PY, Zhang L, Wu JH, Han SO, Brian L, Freedman NJ, Shenoy SK.. Ubiquitin-specific protease 20 regulates the reciprocal functions of β-arrestin2 in Toll-like receptor 4-promoted NFκB Activation. J Biol Chem 2016;291:7450–7464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Fattori E, Cappelletti M, Costa P, Sellitto C, Cantoni L, Carelli M, Faggioni R, Fantuzzi G, Ghezzi P, Poli V.. Defective inflammatory response in interleukin 6-deficient mice. J Exp Med 1994;180:1243–1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Steenwinckel V, Louahed J, Orabona C, Huaux F, Warnier G, McKenzie A, Lison D, Levitt R, Renauld JC.. IL-13 mediates in vivo IL-9 activities on lung epithelial cells but not on hematopoietic cells. J Immunol 2007;178:3244–3251. [DOI] [PubMed] [Google Scholar]
- 27. Wernersson S, Pejler G.. Mast cell secretory granules: armed for battle. Nat Rev Immunol 2014;14:478–494. [DOI] [PubMed] [Google Scholar]
- 28. Galli SJ, Nakae S, Tsai M.. Mast cells in the development of adaptive immune responses. Nat Immunol 2005;6:135–142. [DOI] [PubMed] [Google Scholar]
- 29. Bro S, Bentzon JF, Falk E, Andersen CB, Olgaard K, Nielsen LB.. Chronic renal failure accelerates atherogenesis in apolipoprotein E-deficient mice. J Am Soc Nephrol 2003;14:2466–2474. [DOI] [PubMed] [Google Scholar]
- 30. Bro S, Binder CJ, Witztum JL, Olgaard K, Nielsen LB.. Inhibition of the renin-angiotensin system abolishes the proatherogenic effect of uremia in apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol 2007;27:1080–1086. [DOI] [PubMed] [Google Scholar]
- 31. Kang L, Grande JP, Hillestad ML, Croatt AJ, Barry MA, Katusic ZS, Nath KA.. A new model of an arteriovenous fistula in chronic kidney disease in the mouse: beneficial effects of upregulated heme oxygenase-1. Am J Physiol Renal Physiol 2016;310:F466–F476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kokubo T, Ishikawa N, Uchida H, Chasnoff SE, Xie X, Mathew S, Hruska KA, Choi ET.. CKD accelerates development of neointimal hyperplasia in arteriovenous fistulas. J Am Soc Nephrol 2009;20:1236–1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Liang A, Wang Y, Han G, Truong L, Cheng J.. Chronic kidney disease accelerates endothelial barrier dysfunction in a mouse model of an arteriovenous fistula. Am J Physiol Renal Physiol 2013;304:F1413–F1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wang Y, Liang A, Luo J, Liang M, Han G, Mitch WE, Cheng J.. Blocking Notch in endothelial cells prevents arteriovenous fistula failure despite CKD. J Am Soc Nephrol 2014;25:773–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Brahmbhatt A, NievesTorres E, Yang B, Edwards WD, Roy Chaudhury P, Lee MK, Kong H, Mukhopadhyay D, Kumar R, Misra S, Sen U.. The role of Iex-1 in the pathogenesis of venous neointimal hyperplasia associated with hemodialysis arteriovenous fistula. PLoS One 2014;9:e102542.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Chien CT, Fan SC, Lin SC, Kuo CC, Yang CH, Yu TY, Lee SP, Cheng DY, Li PC.. Glucagon-like peptide-1 receptor agonist activation ameliorates venous thrombosis-induced arteriovenous fistula failure in chronic kidney disease. Thromb Haemost 2014;112:1051–1064. [DOI] [PubMed] [Google Scholar]
- 37. Yang B, Janardhanan R, Vohra P, Greene EL, Bhattacharya S, Withers S, Roy B, Nieves Torres EC, Mandrekar J, Leof EB, Mukhopadhyay D, Misra S.. Adventitial transduction of lentivirus-shRNA-VEGF-A in arteriovenous fistula reduces venous stenosis formation. Kidney Int 2014;85:289–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Goswami R, Kaplan MH.. A brief history of IL-9. J Immunol 2011;186:3283–3288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Gregersen I, Skjelland M, Holm S, Holven KB, Krogh-Sørensen K, Russell D, Askevold ET, Dahl CP, Ørn S, Gullestad L, Mollnes TE, Ueland T, Aukrust P, Halvorsen B, Khoury JE.. Increased systemic and local interleukin 9 levels in patients with carotid and coronary atherosclerosis. PLoS One 2013;8:e72769.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zhang W, Tang T, Nie D, Wen S, Jia C, Zhu Z, Xia N, Nie S, Zhou S, Jiao J, Dong W, Lv B, Xu T, Sun B, Lu Y, Li Y, Cheng L, Liao Y, Cheng X.. IL-9 aggravates the development of atherosclerosis in Apoe-/- mice. Cardiovasc Res 2015;106:453–464. [DOI] [PubMed] [Google Scholar]
- 41. Vasanthakumar R, Mohan V, Anand G, Deepa M, Babu S, Aravindhan V.. Serum IL-9, IL-17, and TGF-beta levels in subjects with diabetic kidney disease (CURES-134). Cytokine 2015;72:109–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Dahlin JS, Hallgren J.. Mast cell progenitors: origin, development and migration to tissues. Mol Immunol 2015;63:9–17. [DOI] [PubMed] [Google Scholar]
- 43. Skartsis N, Martinez L, Duque JC, Tabbara M, Velazquez OC, Asif A, Andreopoulos F, Salman LH, Vazquez-Padron RI.. c-Kit signaling determines neointimal hyperplasia in arteriovenous fistulae. Am J Physiol Renal Physiol 2014;307:F1095–F1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. de Vries MR, Wezel A, Schepers A, van Santbrink PJ, Woodruff TM, Niessen HW, Hamming JF, Kuiper J, Bot I, Quax PH.. Complement factor C5a as mast cell activator mediates vascular remodelling in vein graft disease. Cardiovasc Res 2013;97:311–320. [DOI] [PubMed] [Google Scholar]
- 45. Doggrell SA, Wanstall JC.. Vascular chymase: pathophysiological role and therapeutic potential of inhibition. Cardiovasc Res 2004;61:653–662. [DOI] [PubMed] [Google Scholar]
- 46. Wolff RA, Ryomoto M, Stark VE, Malinowski R, Tomas JJ, Stinauer MA, Hullett DA, Hoch JR.. Antisense to transforming growth factor-beta1 messenger RNA reduces vein graft intimal hyperplasia and monocyte chemotactic protein 1. J Vasc Surg 2005;41:498–508. [DOI] [PubMed] [Google Scholar]
- 47. Otsuka G, Agah R, Frutkin AD, Wight TN, Dichek DA.. Transforming growth factor beta 1 induces neointima formation through plasminogen activator inhibitor-1-dependent pathways. Arterioscler Thromb Vasc Biol 2006;26:737–743. [DOI] [PubMed] [Google Scholar]
- 48. Yamada T, Kondo T, Numaguchi Y, Tsuzuki M, Matsubara T, Manabe I, Sata M, Nagai R, Murohara T.. Angiotensin II receptor blocker inhibits neointimal hyperplasia through regulation of smooth muscle-like progenitor cells. Arterioscler Thromb Vasc Biol 2007;27:2363–2369. [DOI] [PubMed] [Google Scholar]
- 49. Jin D, Ueda H, Takai S, Okamoto Y, Muramatsu M, Sakaguchi M, Shibahara N, Katsuoka Y, Miyazaki M.. Effect of chymase inhibition on the arteriovenous fistula stenosis in dogs. J Am Soc Nephrol 2005;16:1024–1034. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.