

Abstract
Aims
Fibroblast growth factor 1 (FGF1), a heparin/heparan sulfate-binding growth factor, is a potent cardioprotective agent against myocardial infarction (MI). The impact of heparin, the standard of care for MI patients entering the emergency room, on cardioprotective effects of FGF1 is unknown, however.
Methods and results
To address this, a rat model of MI was employed to compare cardioprotective potentials (lower infarct size and improve post-ischemic function) of native FGF1 and an engineered FGF1 (FGF1ΔHBS) with reduced heparin-binding affinity when given at the onset of reperfusion in the absence or presence of heparin. FGF1 and FGF1ΔHBS did not alter heparin‘s anticoagulant properties. Treatment with heparin alone or native FGF1 significantly reduced infarct size compared to saline (P < 0.05). Surprisingly, treatment with FGF1ΔHBS markedly lowered infarct size compared to FGF1 (P < 0.05). Both native and modified FGF1 restored contractile and relaxation function (P < 0.05 versus saline or heparin). Furthermore, FGF1ΔHBS had greater improvement in cardiac function compared to FGF1 (P < 0.05). Heparin negatively impacted the cardioprotective effects (infarct size, post-ischemic recovery of function) of FGF1 (P < 0.05) but not of FGF1ΔHBS. Heparin also reduced the biodistribution of FGF1, but not FGF1ΔHBS, to the left ventricle. FGF1 and FGF1ΔHBS bound and triggered FGFR1-induced downstream activation of ERK1/2 (P < 0.05); yet, heparin co-treatment decreased FGF1-produced ERK1/2 activation, but not that activated by FGF1ΔHBS.
Conclusion
These findings demonstrate that modification of the heparin-binding region of FGF1 significantly improves the cardioprotective efficacy, even in the presence of heparin, identifying a novel FGF ligand available for therapeutic use in ischemic heart disease.
Keywords: Cardioprotection, Structural biology, Growth factor, Heparin, Protein engineering
1. Introduction
Myocardial infarction (MI), resulting from coronary artery disease, is a devastating event that is still one of the leading causes of morbidity and mortality.1 Although there have been advances in clinical therapies for coronary artery disease, more than eight million individuals suffer a myocardial infarction every year.1 Myocardial ischemia/reperfusion (I/R) injury can result in reversible functional myocardial deterioration (i.e., stunning) and in irreversible tissue damage (i.e., infarction and fibrosis). Research on potential postconditioning agents and mechanisms for the clinical application to protect the myocardium from cellular damage after cardiac I/R injury could lead to novel therapies to reduce acute myocardial infarction-associated mortality and morbidity. Therefore, in vivo studies to evaluate the cardioprotective efficacy of molecules, such as fibroblast growth factor (FGF) in reperfusion injury are warranted before it can gain therapeutic utility in clinical scenarios of coronary artery syndromes and acute myocardial infarction.
Fibroblast growth factors (FGFs) are a family of 18 ligands (FGF1-10, FGF16–23) with the mouse FGF15 being ortholog of human FGF19.2–6 All FGF members are structurally and functionally related and are either involved in embryonic development and/or postnatal metabolism and disease.2–4,6 For example, a number of pharmacological and in vitro studies have suggested that FGF1 maintains the integrity/function of the myocardium by acting directly on cardiomyocytes or indirectly via its angiogenic properties.7–15 However, even though a wealth of data from pre-clinical studies demonstrate that FGF1 is a promising therapeutic strategy to improve myocardial survival and cardiac function, there exist several issues that complicate the clinical application of FGFs for acute myocardial infarction. One of the issues may be its interaction with heparin; exogenous heparin treatment, either with unfractionated or low molecular weight (enoxaparin), is standard medical practice for patients with acute MI.16–22
Heparin is a type of heparan sulfate (HS) made exclusively by mast cells that has the highest amount of iduronic acid and of N- and O-sulfate residues. Generally, it is acknowledged that FGF1 executes its pleiotropic actions by promoting FGFR dimerization and activation in a HS-dependent fashion.2,23–26 FGF1 binding to exogenous heparin or endogenous heparan sulfate proteoglycan (HSPG) protects it from proteases,24 alters its bioavailability and biodistribution,27 and aids in FGFR signaling.23,26,28 Previous reports showed that simultaneous intramyocardial injection of enoxaparin (low molecular weight heparin) combined with FGF1 promoted capillary growth and regional myocardial blood flow at one week after infarction;29 however, Hondermarck and colleagues30 demonstrated that increasing doses of heparin co-administered with FGF1 or FGF2 weakened their binding to blood vessels in a heparin dose-dependent manner. Additionally, Newman and group showed that, in rat AT2 cells, low to moderate concentrations of heparin enhanced FGF1-mediated signals compared to FGF1 treatment alone, while a high concentration of heparin inhibited FGF1 activity.31 In agreement with Newman and colleagues, Fannon and investigators found that, in Balb/c3T3 fibroblasts, a low concentration of heparin enhanced FGF2 receptor binding, while a high concentration of heparin inhibited binding.32 Up to now, however, there are no in vivo studies looking into the role of heparin in the cardioprotective effect of FGFs, in particular FGF1, in MI. Although FGF1 has been previously investigated in I/R injury as a preconditioning or postconditioning agent in small and large animal models,7–15,33 this research project is novel and innovative because, for the first time, the cardioprotective nature of this heparin-binding growth factor will be evaluated in the presence of heparin, a standard of care for acute coronary syndromes.16–18
In the present study, we initially set to modify interaction of FGF1 with heparin by mutating the FGF1 heparin-binding sites (FGF1ΔHBS) and then evaluated the cardioprotective efficacy properties of native and mutant FGF1 (FGF1ΔHBS) in the presence of heparin. This study reveals that heparin reduced the cardioprotective effect of native FGF1 against I/R injury; the impaired effect of FGF1 is partially due to its target redistribution by heparin away from the heart. Interestingly, reducing the heparin binding of FGF1 results in a marked decrease in infarct size and improvement in cardiac function, even in the presence of heparin co-therapy.
2. Methods
2.1 Pharmacological agents
Heparin sodium (1000 U/mL) was purchased from Sigma-Aldrich (MO, USA). Recombinant human hexa-histidine-tagged FGF1 (His-FGF1) were obtained from Dr. Moosa Mohammadi‘s laboratory. Non-tagged FGF1 was obtained from R&D system (USA). The FGF1ΔHBS mutant, which carries the K127D/K128Q/K133V triple mutation in its HS-binding region (Figure 1A) was expressed in E.coli and purified to homogeneity using anion exchange chromatography followed by size exclusion chromatography (Figure 1B) in Dr. M. Mohammadi‘s lab. Heparin and FGFs were diluted in phosphate-buffered saline immediately before use.
Figure 1.

Structure-guided design of the FGF1ΔHBS construct. (A) Expanded view of the hydrogen bonding interactions (dashed lines) between the HS-binding site of FGF2 and heparin hexasaccharide as observed in the crystal structure. FGF2 is shown as an orange ribbon diagram with HS-interacting residues rendered in sticks. The three FGF2 residues namely, K128, R129 and K134 that make important hydrogen bonding with HS are boxed. The corresponding three residues in FGF1 are K127, K128 and K133 which were mutated to aspartic acid, glutamine and valine, respectively, to engineer the FGF1ΔHBS construct. (B) FGF1ΔHBS elutes as a single symmetric peak at its predicted molecular weight from a Superdex 200 sizing column. Retention times of protein standards are given above the chromatogram. (C and D) Analysis of the interactions of wildtype FGF1 and FGF1ΔHBS with SOS. Indicated solutions of SOS were injected into solutions of wildtype FGF1 or FGF1ΔHBS in the cell. Wildtype FGF1 (panel C) binds SOS with a Kd of 4 μM; whereas, FGF1ΔHBS (panel D) fails to bind SOS.
2.2 Animals
Male Sprague Dawley rats weighing 240–300 g supplied by Harlan Laboratories were housed and handled according to the standards and guidelines set by the Care and Use of Laboratory Animals published by the US NIH (NIH Publication No. 85–23, revised 2011). All animal experimental protocols were approved by the University of Cincinnati Institutional Animal Care and Use Committee. All animals were acclimatized for at least 48 h before experimental use.
2.3 Experimental protocol and exclusion criteria
Rats were randomly divided into four sets of studies. Two sets were to assess the pharmacodynamics effects cardioprotective efficacy of native FGF1 and FGF1ΔHBS and FGF1 signaling and the other two sets were to evaluate the pharmacokinetic properties of native FGF1 and modified FGF1 (FGF1ΔHBS). The first set of experiments was for the ischemia/reperfusion study in which animals were divided into six groups as follows: (1) saline group: saline treatment given intravenously for 10 min immediately upon reperfusion, (2) heparin group: heparin, 60 U/kg intravenous (i.v.) bolus, given immediately at reperfusion + 12 U/kg/h i.v. infusion for 120 min of reperfusion, (3) FGF1 group: FGF1, 10 μg/kg i.v. infusion, given for 10 min immediately at reperfusion, (4) FGF1 + heparin: heparin, 60 U/kg bolus, given immediately upon reperfusion followed by FGF1, 10 μg/kg i.v. infusion, given for 10 min and 12 U/kg/h i.v. infusion given for 110 min of reperfusion,5 modified FGF1 (FGF1ΔHBS) group: FGF1ΔHBS, 10 μg/kg i.v. infusion, given for 10 min, or (6) FGF1ΔHBS + heparin: heparin, 60 U/kg bolus, given immediately at reperfusion followed by FGF1ΔHBS, 10 μg/kg i.v. infusion, given for 10 min and 12 U/kg/h i.v. infusion for 110 min of reperfusion. All rats were subjected to 30 min of regional ischemia and 120 min of reperfusion. Drugs were administered at the onset of reperfusion via the jugular vein (Figure 2A). The final dose (10 μg/kg, i.v.) of native and modified FGF1 was determined by performing preliminary dose response studies (1 μg/kg, 10 μg/kg, and 100 μg/kg; data not shown); the dose range was derived from published experiments (2.6 μg/kg, i.v. and 10 μg/kg, i.v.) that have previously been shown to be effective in protecting the heart against ischemia-reperfusion injury in rodent models.7,10–12,33,34 A total of ninety-six rats was completed for the I/R study. Sample size for each group ranged from 9 to 16 depending on the power analysis. A total of thirty-five rats were excluded from the study based on the lack of cyanosis of the heart or typical elevation of the ST segment in the electrocardiogram during ischemia, and death from malignant arrhythmia.
Figure 2.

Experimental design and protocols. (A) In vivo ischemia/reperfusion protocol for assessment of heparin on the cardioprotective effect of FGF1 and the novel FGF1 ligand with reduced heparin binding (FGF1ΔHBS). Sprague Dawley rats were subjected to 30 min of ischemia and 120 min of reperfusion. Hemodynamic parameters were measured at baseline, ischemia, and reperfusion (black arrows). (B) Schematic of FGF1 administration protocol for analysis of the tissue distribution or signaling of FGF1 or FGF1ΔHBS in the absence or presence of heparin. Solid organs and blood were collected at the point which mimics the full length of time of reperfusion for ELISA assay. The left ventricle was collected immediately post-treatment or the point which mimics the full length of time of reperfusion for FGF1 signaling.
For the biodistribution or FGF1 signaling study, rats were randomly divided into six groups as described above (Figure 2B). Following drug treatment (immediately following 10 min of treatment or 110 min post-treatment at the point which mimics the length of time of reperfusion), solid organs and blood (see sections “Tissue collection for exogenous FGF1 biodistribution” and “Cardiac preparation to detect the activation of FGF1 downstream molecules”) were collected. A total of 33 rats were used for biodistribution and FGF1 signaling studies, and 3 rats were excluded due to anesthetic overdose. Also, a total of 8 plasma samples were excluded because of hemolysis.
For elimination half-life of native FGF1 and FGF1ΔHBS, 12 rats were randomly divided into two groups and treated intraperitoneally with either FGF1 form as described in the section “Pharmacokinetic evaluation of exogenous FGF1 or FGF1ΔHBS”.
2.4 Assessment of blood clotting time
In sets of experiment, blood-clotting time was detected at baseline and 15 min after drug treatment. In brief, a nick 1–2-mm depth was made at 5 cm and 3 cm of the proximal tail for baseline and post-drug blood clotting time, respectively; the tail nick was blotted with 4 x 4 gauze every 15 s until bleeding totally stopped, and blood clotting time was recorded from the onset of bleeding until it stopped.
2.5 Invivo I/R model
The in vivo I/R model was established by ligating and loosening the left anterior descending artery (LAD) previously described by our laboratory.35–37 In brief, rats were anesthetized with thiobutabarbital sodium salt hydrate, Inactin® hydrate (100 mg/kg, i.p., Sigma-Aldrich), and intubated and artificially ventilated via a rodent respirator (respiratory rate: 75 strokes/min; tidal volume: 3 mL; Model 683, Harvard, USA). The heart was exposed through a left thoracotomy and pericardiotomy, and a 6–0 silk suture was placed around the left anterior descending (LAD) coronary artery. After equilibration for 15 min, hearts were subjected to 30 min of ischemia by ligating the LAD via a snare occluder and reperfusion occurred for 120 min via loosening the snare occluder. The appearance of cyanosis of the heart and significant ST-segment elevation in the electrocardiogram were used to verify successful ligation.
2.6 Hemodynamic analysis
Hemodynamic measurement was assessed via a Millar MIKRO-TIP transducer (SPR-320, Houston, TX, USA) placed into the left ventricle via the right carotid artery. Left ventricular systolic (LVSP), end-diastolic pressure (LVEDP), heart rate (HR), rate of contraction (+dP/dt) as well as rate of relaxation (-dP/dt) were collected (Digi-Med data analysis) during specific timepoints of baseline, ischemia and reperfusion (Figure 2A).
2.7 Infarct size measurement
At the end of the ischemia-reperfusion injury study, the LAD was re-occluded with the suture used previously for the establishment of ischemia. 5% Evans blue dye solution was given via the jugular vein to define the area-at-risk (AAR). Then, the heart was arrested in diastole with 1 mL of 15% potassium chloride, and the animal was euthanized with an anesthetic overdose followed by a bilateral pneumothorax for removal of the heart. The heart was excised, washed with saline, frozen in −80 °C for 5–6 min, sliced transversely into 2–3 mm thick sections, incubated in 1% 2, 3, 5-triphenyl tetrazolium chloride (TTC, pH 7.4) for 30 min at 37 °C in the dark followed by fixing in 4% paraformaldehyde and digitally photographed. Infarct size (IS), depicted as the percentage of the area-at-risk, was determined by Image J software (NIH, 1.61 version).
2.8 Pharmacokinetic evaluation of exogenous FGF1 or FGF1ΔHBS
The in vivo half-life of FGF1 or FGF1ΔHBS (given as a single, intraperitoneal dose of 0.5 mg/kg body weight) was assessed, using the human FGF1 immunoassay ELISA kit (R&D System, MN), from blood (i.e., serum) collected at varying times as described previously.38 The pharmacokinetic parameters of FGF1 or FGF1ΔHBS were analyzed using the Drug and Statistics Software (DAS, v 2.0; Mathematical Pharmacology Professional Committee of China).
2.9 Tissue collection for exogenous FGF1 biodistribution
The rat was anesthetized with Inactin® hydrate (100 mg/kg, i.p.), and the external jugular vein was isolated for saline, FGF1, FGF1ΔHBS (mutant FGF1) and/or heparin administration. At the end of the experiment (based on half-life of FGF139,40), the animal was euthanized with an anesthetic overdose followed by excision and collection of the heart (including atria, left ventricle, and right ventricle), liver, lungs, kidneys, spleen, skeletal muscle, and brain (negative control as i.v. administered FGFs do not cross the blood-brain barrier41). Blood from the jugular vein was collected into the tubes containing 0.5 M EDTA and complete mini protease inhibitor cocktail (Roche), centrifuged at 2, 000 g for 15 min at 4°C, and the separated plasma was saved. Both tissue and plasma were snap-frozen in liquid nitrogen and stored at −80 °C for evaluation of exogenous FGF1 biodistribution.
2.10 Enzyme-linked immunosorbent assays (ELISA) of exogenous FGF1
Snap-frozen tissue samples were powdered and homogenized in protein extraction buffer (20 mM Tris, 2 mM EDTA, 2 M NaCl, 1% NP40 with Roche complete mini EDTA-free protease inhibitor cocktail (1 tablet/10 mL) and 1 mM PMSF added right before use) and protein was extracted as previously described.42 Exogenous levels of tissue and plasma FGF1 and FGF1ΔHBS were determined by ELISA per the manufacturer‘s instructions (FGF1 immunoassay kit, R&D Systems). In brief, 80 μg protein was incubated with assay diluent for 2 h at room temperature, shaking in a 96-well plate coated with a monoclonal antibody against FGF1. After 4 washes, conjugate buffer was added followed by incubation for 2 h at ambient temperature. All samples were incubated for 30 min in the dark with substrate solution, followed by stop solution. The optical density (O.D) was measured at 450 nm with correction wavelength of 570 nm in GENios Microplate Reader (Tecan, USA). The FGF1 standard in the kit was diluted with assay diluent to produce a dilution series of 2000, 1000, 500, 250, 125, 62.5, and 31.2 pg/mL. The assay diluent alone served as the zero standard (0 pg/mL). Standard and unknown samples were assayed in duplicate. Standard curve was created by using Excel via plotting the log of the average FGF1 concentrations versus the log of the O.D. The concentration of exogenous FGF1 (native or mutant) in tissue and plasma samples was calculated from the standard curve.
2.11 Cardiac preparation to detect the activation of FGF1 downstream molecules
Hearts from rats treated with saline, native FGF1 or FGF1ΔHBS in the presence or absence of heparin were collected immediately after administration and at 120-min post-treatment for evaluation of downstream signaling. One-half of each snap-frozen heart was powdered and homogenized in protein extraction buffer as previously described by our laboratory.43–45 For FGFR1, 150 μg of total protein were loaded onto a 8% SDS-PAGE gel, for ERK1/2 activation, 20 μg of whole cell protein homogenate was loaded onto a 15% SDS-PAGE gel, and for PKCα and PKCδ, 100 μg of whole cell protein homogenate was loaded onto a 10% SDS-PAGE gel and then transferred to nitrocellulose membrane. 0.1% Ponceau S in 5% acetic acid was used to examine the transfer efficiency and loading equality as described previously.43 Membranes were blocked with 5% dry milk and then incubated with primary antibody against phospho-FGFR1 (Y-654, 1: 500, Cell Signaling), phospho-ERK1/2 (1: 1000, Cell Signaling) or phospho-PKCα (1: 500, Santa Cruz) or phospho-PKCδ (1: 500, Santa Cruz) followed by incubation with HRP-conjugated secondary goat anti-rabbit antibody (1: 3000, Santa Cruz Biotechnology). Membranes were stripped and reprobed with antibodies against total FGFR1 (1: 500, Cell Signaling), total ERK (1: 1000, BD Transduction Labs) or total PKCα (1: 500, Santa Cruz) or PKCδ (1: 500, Santa Cruz). Bound antibodies were visualized by ECL system. Activation is indicated as a ratio of phosphorylated protein/total protein. β-actin (1: 1000, Cell Signaling) was also used as a loading control.
To detect levels of phospho-Akt, Akt, phospho-STAT3, STAT3, and GAPDH, an automated capillary Western blot was performed according to the WES user guide by ProteinSimple and all Wes reagents were purchased with the Wes Size Separation Master Kit (ProteinSimple, San Jose, CA). The cardiac samples (collected immediately post-treatment or 110-min post-treatment) were mixed together with 5 × Master Mix (DTT, fluorescence labeled maker, SDS) in a ratio of 5: 1 and then incubated at 70 °C for 5 min. The cardiac samples (0.4 mg/mL) and the biotin-labeled protein ladder (12 kDa, 40 kDa, 66 kDa, 116 kDa, 180 kDa and 230 kDa) were loaded into individual wells of the sample plate. Primary antibody against phospho-Akt (1: 50, Cell Signaling), Akt (1: 50, Cell Signaling), phospho-STAT3 (1: 50, Santa Cruz), STAT3 (1: 50, Millipore), and GAPDH (1: 50, Cell Signaling) were diluted with antibody diluent buffer. Loading conditions were determined by GAPDH. The experiment was then performed in Wes instrument as described by Wang and colleagues.46
2.12 Statistical analysis
Data were expressed as mean ± standard error of the mean (SEM). Differences between groups were assessed by one-way or two-way ANOVA with Student-Newman-Keul post-hoc test or Kruskal-Wallace non-parametric test with Tukey‘s post-hoc test, depending on the endpoint evaluated. P < 0.05 was considered statistically significant.
3. Results
3.1 FGF1 given immediately at reperfusion protects the heart against ischemia-reperfusion injury
Previous studies reported that pre-treatment with FGF1 prior to an ischemic insult improved cardiac function and enhanced cardiomyocyte survival.7,9,14,15 As these studies indicate that this growth factor intervention protects the heart only when given prior to an ischemic event, its utility for use in the clinical setting of acute MI is limited. Therefore, elucidation of (non-angiogenic) cardioprotective activity of FGF1 when administered following an acute ischemic (MI) event is a necessary step to develop FGF1 as a potential therapeutic agent. Consistent with Cuevas and investigators,8,10–12,47 our results demonstrate that human recombinant FGF1 given immediately at the onset of reperfusion rescued cardiomyocyte survival and cardiac function as represented by a reduction in infarct size and an improvement in post-ischemic contractility and relaxation (±dP/dt), respectively (P < 0.05, Figures 3 and 4, Table 1).
Figure 3.
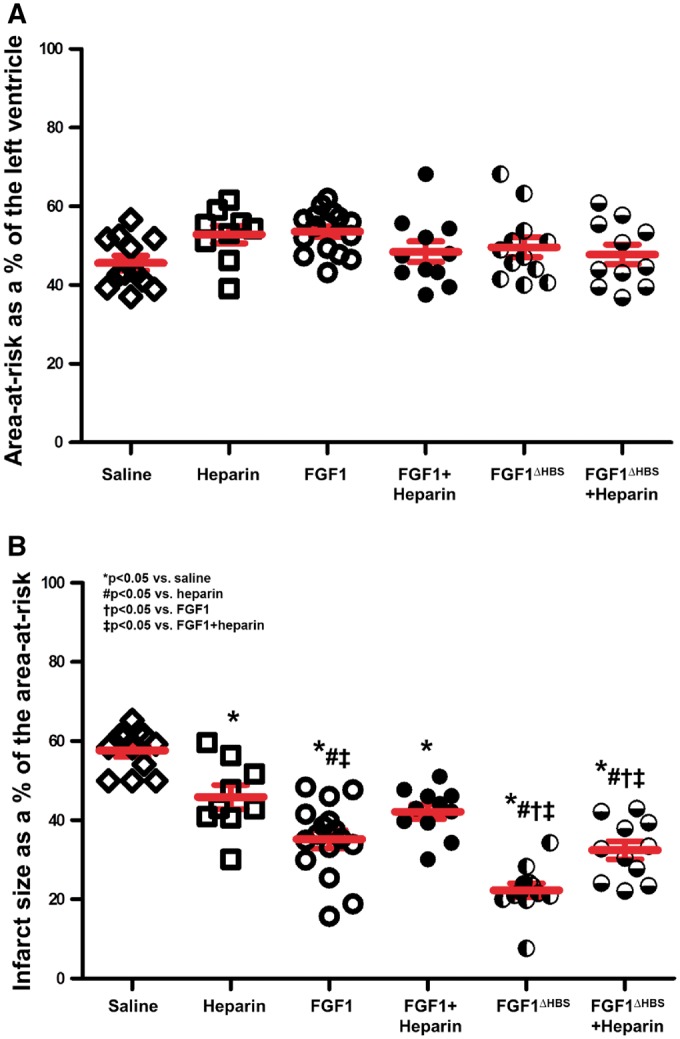
The role of heparin in the cardioprotective effect of FGF1 on preservation of cardiomyocytes after MI. (A) Percent (%) of area-at-risk normalized to the area of the left ventricle. (B) Percent (%) of infarct size normalized to the area-at-risk. n = 9–16. *P < 0.05 vs. saline cohort, #P < 0.05 vs. heparin, †P < 0.05 vs. FGF1, ‡P < 0.05 vs. FGF1 + heparin. The statistical test performed was 1-way ANOVA with Student-Newman-Keul post-hoc test.
Figure 4.

Post-ischemic recovery of contractile and relaxation function as measured by +dP/dt and –dP/dt, respectively. Contractile (A, C) and relaxation (B, D) function of FGF1 and modified FGF1 with reduced heparin binding (FGF1ΔHBS) in the absence or presence of heparin. Heparin reduced the post-ischemic improvement of cardiac function induced by FGF1 (panels A and B). FGF1ΔHBS improved post-ischemic cardiac function, even in the presence of heparin (panels C and D). Percent (%) recovery of contractility (panel E) or relaxation (panel F) is +dP/dt or -dP/dt, respectively, at 120-min post-reperfusion normalized to baseline. n = 5 (for heparin). n = 6 (for saline, FGF1, FGF1 + heparin, FGF1ΔHBS, and FGF1ΔHBS+ heparin). *P < 0.05 vs. saline cohort, #P < 0.05 vs. heparin, †P < 0.05 vs. FGF1, ‡P < 0.05 vs. FGF1 + heparin. For panels A-D, the statistical test performed was a repeated measures 2-way ANOVA with Student-Newman-Keul post-hoc test. For panel E-F, the statistical test performed was 1-way ANOVA with Student-Newman-Keul post-hoc test.
Table 1.
Cardiac parameters prior to and during I/R injury
| HR | LVSP | LVEDP | Tau | RT 1/2 | |
|---|---|---|---|---|---|
| (beats/min) | (mmHg) | (mmHg) | (msec) | (msec) | |
| Baseline | |||||
| Saline | 408 ± 20 | 148 ± 7 | 4 ± 1 | 17 ± 1 | 36 ± 3 |
| Heparin | 407 ± 17 | 139 ± 14 | 4 ± 1 | 17 ± 2 | 46 ± 9 |
| FGF1 | 381 ± 23 | 142 ± 14 | 5 ± 1 | 16 ± 2 | 39 ± 6 |
| FGF1+Heparin | 396 ± 47 | 144 ± 8 | 5 ± 1 | 19 ± 3 | 49 ± 11 |
| FGF1ΔHBS | 405 ± 14 | 148 ± 6 | 5 ± 1 | 18 ± 1 | 45 ± 5 |
| FGF1ΔHBS+Heparin | 423 ± 14 | 142 ± 7 | 4 ± 1 | 17 ± 2 | 38 ± 5 |
| 5' Ischemia | |||||
| Saline | 398 ± 20 | 63 ± 12 | 19 ± 3 | 33 ± 4 | 23 ± 3 |
| Heparin | 407 ± 38 | 54 ± 8 | 24 ± 2 | 32 ± 4 | 25 ± 4 |
| FGF1 | 372 ± 13 | 59 ± 10 | 23 ± 4 | 32 ± 6 | 24 ± 5 |
| FGF1+Heparin | 408 ± 30 | 55 ± 8 | 22 ± 4 | 31 ± 4 | 27 ± 2 |
| FGF1ΔHBS | 396 ± 18 | 59 ± 4 | 22 ± 3 | 32 ± 5 | 25 ± 4 |
| FGF1ΔHBS+Heparin | 409 ± 18 | 56 ± 5 | 21 ± 4 | 34 ± 4 | 28 ± 3 |
| 30' Ischemia | |||||
| Saline | 356 ± 32 | 55 ± 10 | 21 ± 3 | 31 ± 5 | 31 ± 2 |
| Heparin | 385 ± 9 | 52 ± 7 | 25 ± 3 | 34 ± 6 | 28 ± 2 |
| FGF1 | 365 ± 14 | 61 ± 11 | 23 ± 4 | 32 ± 4 | 31 ± 4 |
| FGF1+Heparin | 385 ± 38 | 57 ± 8 | 25 ± 3 | 32 ± 6 | 27 ± 2 |
| FGF1ΔHBS | 382 ± 11 | 51 ± 8 | 21 ± 4 | 20 ± 3 | 26 ± 3 |
| FGF1ΔHBS+Heparin | 392 ± 15 | 54 ± 7 | 22 ± 4 | 33 ± 4 | 27 ± 5 |
| 5' Reperfusion | |||||
| Saline | 382 ± 10 | 91 ± 8 | 16 ± 2 | 27 ± 2 | 31 ± 5 |
| Heparin | 395 ± 11 | 91 ± 6 | 16 ± 1 | 26 ± 2 | 31 ± 3 |
| FGF1 | 366 ± 13 | 111 ± 7* | 13 ± 3 | 23 ± 4 | 37 ± 4 |
| FGF1+Heparin | 373 ± 40 | 102 ± 9 | 17 ± 2 | 25 ± 5 | 38 ± 7 |
| FGF1ΔHBS | 380 ± 13 | 130 ± 5*#†‡ | 11 ± 1 | 21 ± 3 | 41 ± 9 |
| FGF1ΔHBS+Heparin | 395 ± 12 | 127 ± 6*#†‡ | 13 ± 3 | 22 ± 4 | 36 ± 6 |
| 30' Reperfusion | |||||
| Saline | 378 ± 11 | 106 ± 4 | 12 ± 2 | 25 ± 4 | 30 ± 4 |
| Heparin | 384 ± 10 | 108 ± 7 | 10 ± 1 | 23 ± 3 | 36 ± 7 |
| FGF1 | 332 ± 10 | 122 ± 4* | 8 ± 1 | 21 ± 4 | 40 ± 4 |
| FGF1+Heparin | 374 ± 27 | 115 ± 6 | 10 ± 2 | 21 ± 2 | 38 ± 9 |
| FGF1ΔHBS | 367 ± 16 | 140 ± 3*#‡ | 5 ± 1# | 17 ± 2 | 43 ± 8 |
| FGF1ΔHBS+Heparin | 377 ± 14 | 134 ± 6*#‡ | 7 ± 1 | 21 ± 2 | 34 ± 6 |
| 60' Reperfusion | |||||
| Saline | 360 ± 21 | 101 ± 3 | 10 ± 1 | 26 ± 3 | 29 ± 3 |
| Heparin | 373 ± 22 | 111 ± 2* | 10 ± 1 | 22 ± 2 | 33 ± 5 |
| FGF1 | 345 ± 24 | 126 ± 7* | 8 ± 1* | 21 ± 4 | 36 ± 6 |
| FGF1+Heparin | 354 ± 16 | 113 ± 7 | 9 ± 1 | 22 ± 2 | 35 ± 3 |
| FGF1ΔHBS | 370 ± 12 | 139 ± 4*#‡ | 5 ± 1*# | 18 ± 1 | 43 ± 6 |
| FGF1ΔHBS+Heparin | 363 ± 14 | 134 ± 3*#‡ | 7 ± 1* | 22 ± 3 | 35 ± 5 |
| 90' Reperfusion | |||||
| Saline | 353 ± 18 | 104 ± 3 | 12 ± 2 | 26 ± 4 | 28 ± 2 |
| Heparin | 366 ± 20 | 109 ± 7 | 11 ± 3 | 23 ± 3 | 34 ± 7 |
| FGF1 | 349 ± 18 | 128 ± 6* | 9 ± 1* | 20 ± 5 | 34 ± 3 |
| FGF1+Heparin | 367 ± 14 | 118 ± 9 | 11 ± 1 | 23 ± 5 | 37 ± 5 |
| FGF1ΔHBS | 361 ± 16 | 139 ± 5*# | 5 ± 2* | 18 ± 2 | 43 ± 6 |
| FGF1ΔHBS+Heparin | 373 ± 10 | 135 ± 4*# | 8 ± 2 | 20 ± 2 | 36 ± 3 |
| 120' Reperfusion | |||||
| Saline | 353 ± 14 | 104 ± 2 | 12 ± 5 | 26 ± 1 | 34 ± 6 |
| Heparin | 377 ± 11 | 108 ± 1 | 11 ± 2 | 24 ± 2 | 33 ± 4 |
| FGF1 | 332 ± 19 | 124 ± 8* | 9 ± 2 | 19 ± 4 | 38 ± 6 |
| FGF1+Heparin | 364 ± 20 | 113 ± 10 | 11 ± 2 | 22 ± 2 | 35 ± 3 |
| FGF1ΔHBS | 357 ± 21 | 141 ± 5*# | 6 ± 2# | 18 ± 2 | 46 ± 6 |
| FGF1ΔHBS+Heparin | 372 ± 13 | 132 ± 4*# | 7 ± 2 | 20 ± 2 | 39 ± 5 |
Data expressed as mean ± SEM. HR, heart rate. LVSP, left ventricular systolic pressure, LVEDP, left ventricular diastolic pressure, +dP/dt, rate of contraction, -dP/dt, rate of relaxation, RT1/2, half relaxation time. *P < 0.05 vs. saline.
P < 0.05 vs. heparin.
P < 0.05 vs. FGF1.
P < 0.05 vs. FGF1 + heparin. n = 5 (for heparin). n = 6 (for saline, FGF1, FGF1 + heparin, FGF1ΔHBS, and FGF1ΔHBS + heparin).
3.2 Heparin co-therapy reduces the cardioprotective effect of FGF1
Heparin treatment, either as unfractionated or low molecular weight (enoxaparin), is a standard medical practice for patients with acute MI.16–22 Since FGF1 is a heparin-binding protein,2,4 issues of heparin co-medication (per the 2010 NHLBI Workshop recommendations48) must be taken into consideration when developing FGF1 for acute MI therapy. To determine the effect of heparin on the cardioprotective activity of FGF1, infarct size and cardiac function was evaluated in the absence and presence of heparin co-administration (Figure 2A). Compared to saline treatment (58% infarct), heparin alone, at reperfusion, reduced infarct size (46% infarct), but not to the degree of FGF1 treatment (35% infarct, Figure 3B, P < 0.05); the heparin-induced reduction of myocardial infarct was similar to that first reported by Dr. Lucchesi’s group in the 1990s.49–52 Notably, heparin co-administration significantly inhibited FGF1-induced cardioprotection against MI (42% infarct, Figure 3B, P < 0.05). In addition, rats treated with FGF1 recovered to 69% of its baseline contractility compared to saline treatment (44%, Figure 4A and E, Table 1, P < 0.05). After 2 h of reperfusion, there was a significant difference in post-ischemic contractility between FGF1 treatment (69%) versus FGF1 + heparin (57%) co-therapy (Figure 4A and E, Table 1, P < 0.05), suggesting that heparin attenuates the FGF1-induced protection against post-ischemic cardiac dysfunction. There was no significant difference in post-ischemic contractility between the saline and heparin group (Figure 4A and E, Table 1, P < 0.05). Other LV measures are depicted in Table 1 (and Figure 4B and F) showing that rat hearts treated with FGF1 also have improved relaxation compared to saline treatment. Taken together, these data demonstrate that heparin reduces the cardioprotective effect of FGF1 against myocardial infarction and post-ischemic recovery of cardiac function.
3.3 Triple mutation in FGF1 heparin binding site (FGF1 mutant) is cardioprotective, leading to reduced infarct size and improved cardiac function even in the presence of heparin co-treatment compared to native FGF1
Based on the above data demonstrating that heparin inhibits the cardioprotective effect of FGF1 (Figures 3 and 4, Table 1), the question remains as to whether altering the HS-binding region of FGF1 to affect its binding affinity for heparin may change the cardioprotective efficacy of FGF1. From the crystallographic data, three basic residues at the HS binding site of FGF1, namely, K127, K128, and K133V, mediate the majority of hydrogen bonding with HS (Figure 1A). Accordingly, we reasoned that mutations of these three HS-binding residues should cause a major reduction in the HS-binding affinity of FGF1. The mutated FGF1 should retain some ability to promote HS-mediated FGFR dimerization and activation. A FGF1 mutant, termed FGF1ΔHBS, which carries the triple mutation in its HS-binding site, was designed. FGF1ΔHBS was expressed in E. coli and purified to homogeneity using anion exchange chromatography followed by size exclusion chromatography. FGF1ΔHBS elutes as a monodisperse peak at the predicted retention time, indicating that the mutations do not harm the tertiary folding of the ligand (Figure 1B). In fact, this was expected as K127, K128, and K133 are surface-exposed and do not play any role in ligand folding. To test the impact of the mutations on HS-binding ability of FGF1, isothermal titration calorimetry (ITC) was used to compare the interactions of native FGF1 and FGF1ΔHBS with Sucrose Octasulfate (SOS), a known surrogate for HS. As shown in Figure 1C and D, native FGF1 bound SOS with a Kd of 4 μM; whereas, binding of FGF1ΔHBS to SOS was negligible, demonstrating that the FGF1ΔHBS mutant sustains a substantial loss in HS binding.
Next, the cardioprotective potential of FGF1ΔHBS mutant when administered immediately upon reperfusion was evaluated in an in vivo rat model of myocardial infarction. FGF1ΔHBS was ∼2-fold more efficacious in reducing infarct size than native FGF1 (62% versus 39% reduction, respectively; Figure 3, P < 0.05). Importantly, heparin co-therapy had minor, and no statistically significant, inhibition on the cardioprotective effect of FGF1ΔHBS; whereas, the cardioprotective activity of native FGF1 was markedly attenuated in the presence of heparin (Figure 3). Of note, FGF1ΔHBS significantly reduced the infarct size to an extent that was lower than native FGF1 in the absence (22% vs 35%, respectively, P < 0.05, Figure 3) or presence of heparin (32% vs 42%, P < 0.05, Figure 4), which indicates that with the same dosage, FGF1ΔHBS was more efficacious than the native counterpart in protecting the heart from I/R injury. Both contractile and relaxation parameters were significantly improved +dP/dt and −dP/dt were markedly restored in FGF1ΔHBS and FGF1ΔHBS+heparin following ischemic injury (Figure 4C–F, Table 1, P < 0.05), demonstrating that the recovery of post-ischemic cardiac function is not compromised by the presence of heparin. Interestingly, the recovery of post-ischemic cardiac function of FGF1ΔHBS or FGF1ΔHBS+heparin was significantly improved compared to the native FGF1 cohorts (Figures 3 and 4, Table 1, P < 0.05). There were no significant differences in heart rate with the FGF1ΔHBS and FGF1ΔHBS+heparin groups compared to the saline group (Table 1).
3.4 FGF1 and FGF1ΔHBS do not affect the anticoagulant activity of heparin
When developing novel agents to treat ischemic heart disease, consideration needs to be made that any new therapy does not modify and/or interfere with heparin and its anticoagulant function, which is used as the standard of care for cardiac patients,16–22 and therefore, would make the new therapy not clinically applicable. Therefore, we evaluated the effect of native FGF1 and FGF1ΔHBS mutant on the coagulation time. There was no significant difference in blood clotting time during baseline (pre-treatment) or following administration of saline, FGF1 or FGF1ΔHBS (post-treatment). However, in the heparin, FGF1 + heparin and FGF1ΔHBS+heparin groups, the coagulation time was significantly longer post-treatment than at baseline or following administration of saline, FGF1 or FGF1ΔHBS (Table 2). These data demonstrate that neither native nor HS-binding mutant FGF1 co-administered with unfractionated heparin co-treatment affect the anticoagulant property of heparin therapy alone.
Table 2.
Blood clotting time of FGF1- and FGF1ΔHBS-treated rats in the presence or absence of heparin
| Group | Baseline (sec) | Post-Treatment (sec) |
|---|---|---|
| Saline | 178 ± 11 | 168 ± 10 |
| Heparin | 173 ± 8 | 415 ± 9*# |
| FGF1 | 163 ± 12 | 160 ± 12 |
| FGF1+Heparin | 154 ± 16 | 549 ± 53*# |
| FGF1ΔHBS | 153 ± 9 | 160 ± 9 |
| FGF1ΔHBS+Heparin | 181 ± 7 | 440 ± 10*# |
P < 0.05 vs. saline post-treatment.
P < 0.05 vs. baseline cohort.
n = 10-18 per group.
3.5 FGF1 and modified FGF1 (FGF1ΔHBS) have similar elimination half-life
One possibility to account for the enhanced cardioprotective activity of FGF1ΔHBS was that the elimination half-life was varied from native FGF1. Mean serum concentration-time profiles for FGF1 and FGF1ΔHBS are shown in Table 3. The non-compartmental in vivo pharmacokinetic properties of FGF1 and FGF1ΔHBS showed that the absorption phase half-life (t1/2α) and the elimination phase half-life (t1/2z) of FGF1ΔHBS were not significantly different from FGF1. The maximum serum concentration (Cmax) and area-under-curve (AUC) for FGF1ΔHBS were 4-fold and 2-fold greater than those for FGF1, respectively. However, the mean time to reach maximum serum concentration (Tmax) of FGF1ΔHBS was reached earlier than with FGF1. This result implies that the absorption rate of FGF1ΔHBS after intraperitoneal administration was relatively fast, which is to be expected as the weak HS binding affinity of FGF1ΔHBS should allow this ligand to avoid entrapment by HS in the pericellular/extracellular milieu.
Table 3.
Pharmacokinetics of native FGF1 and FGF1ΔHBS following intraperitoneally treatment
| Parameters | FGF1 (n = 6) | FGF1△HBS (n = 6) |
|---|---|---|
| AUC(0-9h) (μg·h/L) | 267.70 ± 37.40 | 559.70 ± 104.50 ** |
| AUC(0-∞) (μg·h/L) | 279.20 ± 37.80 | 565.60 ± 105.60** |
| R_AUC (t/∞) | 95.80 ± 1.00 | 99.00 ± 0.40 |
| t1/2α (h) | 0.13 ± 0.07 | 0.40 ± 0.18 |
| t1/2z (h) | 2.00 ± 0.20 | 1.99 ± 0.56 |
| Tmax (h) | 1.25 ± 0.27 | 0.50 ± 0.00 ** |
| Vz (L/kg) | 5.24 ± 0.84 | 2.60 ± 0.70** |
| CLz (L/h·kg) | 1.82 ± 0.27 | 0.92 ± 0.22** |
| Cmax (μg/L) | 93.90 ± 28.70 | 367.00 ± 82.40** |
AUC, area-under-the-curve. t1/2α, absorption phase half-life. t1/2z, elimination phase half-life. Tmax, mean time to reach maximum serum concentration. Vz, volume of distribution. CLz, clearance. Cmax, maximum serum concentration. **P < 0.01 vs. FGF1.
3.6 Heparin co-treatment modifies the biodistribution of exogenous FGF1 to the heart
It is reported that heparan sulfates on the cell surface or extracellular matrix can concentrate or sequester FGFs which accounts for some of the modulatory activities of heparin on FGFs.25,53–56 Therefore, the effect of heparin on exogenous FGF1 tissue localization was determined. Saline treatment is indicative of the endogenous level of FGF1 for each organ or in plasma. There was no difference in saline or heparin treatment with regard to FGF1 levels in plasma or organs evaluated. This suggests that heparin treatment does not lead to secretion of endogenous FGF1 from the organs. Compared to saline treatment, a significant amount of exogenous FGF1 was distributed to kidney, spleen, heart, liver, and plasma, while less went to lung and skeletal muscle (Figure 5A and C, P < 0.05). However, upon co-administration of FGF1 and heparin, exogenous FGF1 biodistribution was significantly decreased in kidney, spleen, heart, liver and plasma (Figure 5A and D, P < 0.05), suggesting heparin could change the biodistribution of exogenous FGF1 and influence the cardioprotective efficacy of FGF1. Most notably, when in the presence of heparin therapy, the greatest reduction in FGF1 biodistribution was at the heart (Figure 5A and D, P < 0.05). These findings are consistent with other observations that heparin modifies the distribution of FGF1 treatment.57,58
Figure 5.

The tissue distribution of FGF1 and FGF1ΔHBS in the absence or presence of heparin. (A) Total FGF1 levels (endogenous and exogenous) in saline treatment (endogenous FGF1, white bar), native FGF1 (includes endogenous + exogenous, gray bar), and native FGF1 + heparin (includes endogenous + exogenous, light brown bar). (B) Total FGF1 levels (endogenous and exogenous) in saline treatment (endogenous FGF1, white bar), modified FGF1, FGF1ΔHBS, (includes endogenous + exogenous, gray bar), and FGF1ΔHBS + heparin (includes endogenous + exogenous, light tan bar). (C) Exogenous native FGF1 (gray bar) and FGF1ΔHBS (orange bar) were both significantly targeted to the heart; whereas, exogenous native FGF1 was targeted also to the kidney, spleen, liver and plasma. (D) Targeting to heart of exogenous native FGF1 + heparin (white bar) was markedly reduced compared to FGF1 alone (panel C) and FGF1ΔHBS+ heparin (light tan bar). Tissue accumulation of exogenous native FGF1 or FGF1ΔHBS; exogenous FGF1 of each tissue is described as a subtraction of FGF1 concentration in native FGF1- or FGF1ΔHBS-treated rats from that of saline treatment. L-kidney, left kidney; R-kidney, right kidney; Skeletal-M, skeletal muscle; LV, left ventricle; RV, right ventricle. n = 5 (for saline, heparin), n = 7 (for FGF1, FGF1 + heparin, FGF1ΔHBS + heparin, FGF1ΔHBS). *P < 0.05 vs. saline. †P < 0.05 vs. FGF1, ‡P < 0.05 vs. FGF1 + heparin. The statistical test performed was 2-way ANOVA with Student-Newman-Keul post-hoc test.
The next question is whether heparin alters the biodistribution of FGF1ΔHBS. As shown in Figures 5B and C, in the absence of heparin, FGF1ΔHBS mainly went to the heart including atria, left ventricle, and right ventricle. In fact, there was a greater biodistribution of FGF1ΔHBS to the left ventricle compared to native FGF1. In the presence of heparin, the amount of FGF1ΔHBS to the heart was similar to that of FGF1ΔHBS alone (Figure 5B and D). These findings indicate that modifying the heparin-binding region of FGF1 retains and, even, enhances its biodistribution/targeting to the heart even in the presence of heparin therapy, and demonstrates that heparin can no longer sequester FGF1 from its target site, the heart.
3.7 Heparin co-therapy does not alter the activation of FGF1 downstream signaling triggered by modified FGF1 (FGF1ΔHBS)
To ensure that the mutations in the heparin-binding site did not impair FGF binding to its receptor, ITC was used to compare the binding interactions of native FGF1 and FGF1ΔHBS with the extracellular ligand-binding domain of FGFR3c, one of the cognate FGFRs of FGF1. Native FGF1 and FGF1ΔHBS affinities to FGFR3c ecodomain show that the HS-binding site mutations do not impact FGFR binding ability of FGF1 (Figures 6A and B). Importantly, the ITC data showing that the FGF1ΔHBS retains normal receptor binding affinity confirm that the HS mutations have no adverse effect on tertiary folding of the ligand. In support of this, FGFR1 activation (i.e., tyrosine phosphorylation) in vivo by FGF1 or FGF1ΔHBS was similar (Figure 6C).
Figure 6.

In vitro and in vivo characterization of the FGF1ΔHBS construct in FGF receptor binding and activation. (A and B) Analysis of the interactions of native FGF1 and FGF1ΔHBS with FGFR3c. Indicated solutions of native FGF1 or FGF1ΔHBS were injected into solutions of FGFR3c ectodomain in the cell. Native FGF1 (panel A) and FGF1ΔHBS (panel B) bind FGFR3c ectodomain with affinities of 704 nM and 432 nM, respectively. (C) In vivo activation (i.e., phosphorylation) of FGFR1 in the left ventricle is similar for native FGF1 and FGF1ΔHBS. n = 5–7. *P < 0.05 vs. saline. #P < 0.05 vs. heparin. The statistical test performed for panel C was Kruskal-Wallis non-parametric test with Tukey‘s post-hoc test.
It is reported that a low concentration of heparin restored the activity of FGF1 in HS-deficient cells in vitro, while a high concentration of heparin completely inhibited its function.31 To determine the influence of heparin on the activity of FGF1-FGFR1 signaling in the heart, ERK, PKC, Akt, and STAT3 activation, well-known pathways downstream of FGFR and RISK (reperfusion injury salvage kinase) and SAFE (survivor activating factor enhancement) mechanisms of cardioprotection, were assessed in left ventricle collected immediately after and 120-min post-drug administration. Immediately post-FGF1, FGF1 + heparin, FGF1ΔHBS, or FGF1ΔHBS+heparin treatment, phosphorylation of ERK1/2 was significantly increased compared to saline or heparin only treatment (Figure 7A, P < 0.05). However, heparin markedly reduced ERK1/2 activation stimulated by FGF1 (Figure 7A, P < 0.05). Surprisingly, there was a significantly higher level of phosphorylated ERK1/2 in the hearts collected from the FGF1ΔHBS-treated group compared to native FGF1 treatment whether in the absence or presence of heparin (Figure 7A, P < 0.05). By 2-h post-treatment, ERK activation returned to saline control levels (Figure 8A). Although heparin treatment had a minor cardioprotective effect (Figure 4), it was most likely not via ERK as its activation was not different than saline treatment (Figure 7A). Consistent with this finding, studies by Lucchesi’s laboratory demonstrated that heparin-mediated cardioprotection occurred via inhibition of complement activation of the immune system.49,52 Neither activation of PKCα or PKCδ nor activation of Akt of the RISK pathway and STAT3 of the SAFE pathway were different among the treatment groups or timepoints (Figure 7B–E and Figure 8B–E). These data suggest that both FGF1 and FGF1ΔHBS activated FGFR1 signaling (i.e., ERK1/2); yet, heparin co-therapy reduced FGF1 signaling, but not that of FGF1ΔHBS.
Figure 7.

FGFR signaling involved in cardioprotection. The activation (i.e., phosphorylation) of cardioprotective kinases in the RISK and SAFE pathways measured in the left ventricle collected immediately post-FGF1 or post-modified FGF1 with reduced heparin binding (FGF1ΔHBS) administration in the presence or absence of heparin. (A) ERK activation. (B) PKCα activation. (C) PKCδ activation. (D) Akt activation. (E) STAT3 activation. ERK activation (panel A) was markedly increased in native FGF1 and FGF1ΔHBS, with a significantly higher level of phosphorylated ERK1/2 in the hearts from the FGF1ΔHBS-treated group compared to native FGF1 treatment whether in the absence or presence of heparin. PKCalpha or delta (Panel B and C) or Akt (panel D) or STAT3 (panel E) activation was not different among treatment groups evaluated. Akt and STAT3 activation (panel D and panel E, respectively) were determined via an automated capillary Western blot (WES sytem) by ProteinSimple. For ERK activation, n = 4–6. For PKCα activation, n = 3–8. For PKCδ activation, n = 3–7. For Akt or STAT3 activation, n = 4. *P < 0.05. The statistical test performed for panels A-E was Kruskal-Wallis non-parametric test with Tukey‘s post-hoc test.
Figure 8.

FGFR signaling involved in cardioprotection. The activation (i.e., phosphorylation) of cardioprotective kinases in the RISK and SAFE pathways measured in the left ventricle collected 110-min post-FGF1 or post-modified FGF1 with reduced heparin binding (FGF1ΔHBS) administration in the presence or absence of heparin. (A) ERK activation. (B) PKCα activation. (C) PKCδ activation. (D) Akt activation. (E) STAT3 activation. ERK activation (panel A), PKCalpha or delta (Panel B and C) or Akt (panel D) or STAT3 (panel E) activation was not different among treatment groups evaluated. Akt and STAT3 activation (panel D and panel E, respectively) were determined via an automated capillary Western blot (WES sytem) by ProteinSimple. For ERK activation, n = 7–11. For PKCα activation, n = 4–8. For PKCδ activation, n = 3–6. For Akt or STAT3 activation, n = 5. The statistical test performed for panels A–E was Kruskal-Wallis non-parametric test with Tukey‘s post-hoc test.
4. Discussion
A number of key and clinically relevant findings were noted in the present study. First, heparin, the standard of care for MI patients entering the emergency room,16–22 abolishes the cardioprotective action (infarct size and cardiac function recovery) of FGF1 when both were co-administered at the onset of reperfusion. Second, a novel, rationally designed FGF1 ligand with reduced heparin binding (FGF1ΔHBS) has been demonstrated, for the first time, when given immediately upon reperfusion, to elicit a markedly greater cardioprotective efficacy than native FGF1 in the absence as well as in the presence of heparin. Third, heparin treatment changes the tissue distribution of exogenous FGF1 and reduces the availability of FGF1 to the heart; however, tissue distribution of the modified FGF1ΔHBS mutant to the heart still occurs even in the presence of heparin. Fourth, even in the presence of heparin, FGF1ΔHBS interaction with FGFR1 on the heart has enhanced ERK signaling, an important component of the Reperfusion Injury Salvage Kinase (RISK) pathway. Overall, these findings suggest that the attenuation of the cardioprotective effect of FGF1 by heparin co-therapy is most likely due to heparin‘s ability to alter the availability of FGF1 to the heart, and that the novel modified FGF1 ligand (with reduced heparin binding, FGF1ΔHBS) is of great promise as a co-medication strategy with heparin for patients exhibiting MI.
A key factor to facilitate the translation of novel cardioprotective therapies into the clinical setting is the timing of administration of the cardioprotective agent. Given the clear benefit of establishing prompt reperfusion on myocardial salvage and clinical outcomes,59–67 it is important to identify and develop cardioprotective agents that will be given at the time of reperfusion without delaying reperfusion, and that is the rationale for administering either FGF1 or FGF1ΔHBS immediately upon reperfusion. A clinical study (J-WIND trial) demonstrated that the atrial natriuretic peptide analogue, carperitide, administered at the time of primary percutaneous coronary intervention (PPCI) lowered creatinine kinase, a clinical measure of myocardial infarct size.68 Similar to the J-WIND trial68 and supportive and consistent with a handful of previously published in vivo studies from the late 1990 s by Cuevas and colleagues,8,10–12,47 which identified FGF1 as a postconditioning agent, our current study demonstrates that FGF1 or FGF1ΔHBS administered immediately upon reperfusion protected the heart from MI (Figure 3, P < 0.05) and LV dysfunction (Figure 4, P < 0.05). On the other hand, there are proof-of-concept clinical studies demonstrating that drug treatment (such as cyclosporine, metropolol, exenatide, adenosine) during late ischemia to early reperfusion, and prior to PPCI or thrombolytic therapy, reduces infarct size and improves cardiac function.69–73 There also is evidence that the acute beneficial effects of FGF1 against the steps involved (i.e., oxidative stress, apoptosis, neutrophil infiltration) in myocardial infarction (and cardiac dysfunction) have been observed pre-clinically when FGF1 was given prior to an ischemic event.7,9,14,15 Yet, it is unknown whether FGF1 and FGF1ΔHBS can (or cannot) elicit cardioprotection when given during ischemia, thereby adding to it efficacy as a clinical therapeutic for ischemic disease, and this is a limitation of the current study.
The FGF-heparin/heparan sulfate interactions modulate the multiple biological outcomes of FGFs, including FGF1. Heparin/heparan sulfate (HS) is implicated to prevent FGF1 from degradation and facilitate the active formation of FGF1-FGFR complex.2,6,23–26,28 There is also evidence that heparin alters the bioavailability,27 inhibits FGF-mediated signaling31,32 as well as weakens FGF binding to tissue.30 The current study demonstrates that, when given immediately upon reperfusion, heparin or FGF1 alone protected the heart against myocardial infarction as evidenced by reduced infarct size and improved cardiac contractile and diastolic function at 2-h post-ischemia (Figures 3 and 4 and Table 1), which supports previous work that heparin49–52 or FGF17–15 protects from ischemia-reperfusion injury. However, with heparin and FGF1 co-therapy, the cardioprotective effect of FGF1 was completely abolished (Figures 3 and 4). Further evidence that anticoagulant or antithrombotic agents used in coronary artery disease modulate the cardioprotective effect of other pharmacological agents is provided by Gross and colleagues.74 These investigators showed that aspirin co-treatment, a prophylactic for ischemic heart disease, abrogates morphine‘s cardioprotective effect.
Heparin (unfractionated or low molecular weight form) is the standard of care for patients with MI per the guidelines of the American Heart Association and American College of Cardiology.16–22 Therefore, new cardioprotective therapeutics need to demonstrate efficacy in the presence of heparin without altering heparin‘s anticoagulant activity. Although heparin inhibited FGF1-induced cardioprotection against myocardial infarction and dysfunction, FGF1 did not alter anticoagulant activity of heparin (Table 2).
Our observation that FGF1-induced cardioprotection is abrogated by heparin co-therapy may be, in part, attributed to exogenous heparin affecting the bioavailability of exogenous FGF1 to the heart (see Figures 3–5). This finding from our data is supported by a number of studies. Ligand bioavailability at the target site of action is a significant limitation for an intravenous FGF therapeutic in acute MI because of FGF’s heparin-binding properties. Free heparin/HS can trap or sequester FGFs in the blood and other extracellular spaces and inhibit FGF activity.75–77 Most intravenously administered FGF is expected to be cleared by heparan sulfate proteoglycans (HSPG) ubiquitously expressed in all tissues/organs, thus limiting FGF bioavailability at cardiac tissue sites.27 For example, the biodistribution of i.v. administered rhFGF2 in rat to liver, kidneys, and spleen and to a lesser extent, heart and lungs is a reflection of FGF-HS proteoglycan interactions.78 Xia and investigators evaluated pharmacokinetic parameters of modified FGF1 ligands with altered protein stability or heparin-binding and demonstrated that the distribution and redistribution profiles were determined by HSPG affinity and that heparin competes with HS for binding to FGF1.79 In fact, Hondermarck and colleagues30 demonstrated that increasing doses of heparin co-administered with FGF weakened the binding of FGF to blood vessels in a heparin dose-dependent manner. Furthermore, Xue and colleagues reported that exogenous heparin treatment resulted in a redistribution of the heparin–FGF1 complex from the cell surface to the medium, thus leading to reduced effectiveness of FGF1.77 Similarly, in vivo evaluation of 99mTc-labeled FGF1 tissue biodistribution showed that heparin prevented its binding to liver, but not kidney as well as increased FGF1 excretion.57 Our results support some of these effects of heparin on FGF1 tissue biodistribution such that left ventricular or plasma levels of FGF1 were less in the presence of heparin, suggesting a redistribution of exogenous FGF1 from target organs and/or an increase in FGF1 excretion (although not measured in the current study), respectively, with heparin co-therapy. However, there is evidence that FGF-heparan sulfate/heparin interactions are also of benefit. HSPGs aid with the proper presentation of FGFs to FGFRs and formation of stable FGF/FGFR complexes.25,75,80 HSPGs of the ECM can act as reservoirs for FGFs, prevent proteolytic degradation and increase local gradients of FGF during stimulation of endothelial cells.25,75,80 For example, heparin regulated the in vitro activity of FGF1 on neurite outgrowth by altering its proteolytic degradation, thereby increasing its biological half-life from 7 to 39 h.40 Since FGF1 biodistribution is not simply restricted to the organs studied, other tissue including eye, adrenal glands, and bone marrow are also potential “target organs.”78
Site-directed mutagenesis is an important technique for altering cytokine function and affecting its efficacy and/or potency. Recently, FGFs have been modified to eliminate undesirable properties, but still keep or potentiate the beneficial actions. For example, native FGF1 was manipulated to increase its thermostability and half-life.79,81–85 In addition, a truncated form of FGF1 has been created to remove its mitogenic activity and protect cardiomyocytes in vitro and in vivo without tumorigenesis.7 Furthermore, protease resistant variants of FGF1 have been developed to prolong its biological activity.79,81–85 Encouraged by these achievements, we employed a novel, modified FGF1 with reduced heparin-binding affinity (FGF1ΔHBS) (Figures 1C and D), which is still compatible with FGFR binding (Figure 6) and signaling (Figures 7 and 8), in the treatment of MI. In the current study, heparin had little inhibitory effect on FGF1ΔHBS compared to native FGF1 as measured by the preservation of cardiac survival and muscle function (Figures 3 and 4, respectively). Furthermore, similar to native FGF1, FGF1ΔHBS targeted largely to the heart (2282 ± 97 pg/mL vs. 3073 ± 101 pg/mL, respectively) compared to other tissue types (Figure 5C); however, unlike native FGF1 which in the presence of heparin co-treatment led to a re-distribution away from the heart, FGF1ΔHBS even with heparin administration still directed mostly to the heart as FGF1ΔHBS alone (4336 ± 775 pg/mL vs. 3073 ± 101 pg/mL, respectively) compared to other organs evaluated (see Figure 5C and D). Although FGF1ΔHBS has reduced heparin/heparan sulfate-binding, it still accumulates significantly to the heart like its native form; this may most likely be due to the rat heart being highly sulfated, composed of 60% heparan sulfate.86 This increased FGF1ΔHBS sequestration to the heart most likely resulted in the enhanced ERK signaling observed (Figure 7A). These findings demonstrate that FGF1 ligand with reduced heparin binding (FGF1ΔHBS) may be a promising strategy for acute treatment of MI; yet, future studies are still needed to address its long-term effects post-MI. This is in light of previously published observations that FGF1 plus enoxaparin has been reported to promote capillary growth and increased regional myocardial blood flow one week after infarction; the reasons may be due to that heparin protects FGF1 from degradation, increases the expression of FGF2 and enhance angiogenic potential of FGF1 in the left ventricle.29,87–89 Overall, these studies suggest that heparin plays biphasic roles in ischemia; it may reduce the acute protective effect of FGF1 in MI, but long-term, the combination of heparin + FGF1 may be of great promise for MI treatment by increasing angiogenesis activity.
The heparin-binding site of FGF is distinct from the FGF receptor (FGFR) binding site.90,91 The ITC data (Figure 6A and B) show that the HS-binding site mutations do not impact FGFR binding ability of FGF1ΔHBS (i.e., retains normal receptor binding affinity), and confirm that the HS mutations have no adverse effect on tertiary folding of the ligand. Moreover, the in vivo data (Figures 6C and 7A) demonstrate that the mutant FGF1 (FGF1ΔHBS) activates (as measured by phosphorylation status) downstream FGF signaling (e.g., ERK), providing further evidence that normal receptor binding activity is preserved in vivo with FGF1ΔHBS. Although it is well-documented that binding to heparan sulfate aids with the proper presentation of FGFs to FGFRs and formation of stable FGF/FGFR complexes and signaling,24–26,75,80,91–94 this appears to not be the case for the cardioprotective activity of FGF1ΔHBS. Even in the absence of heparin binding, FGF1ΔHBS activated FGFR1 and ERK signaling in the heart, possibly due to the elevated concentration of FGF1ΔHBS to the heart. Supporting our finding, several studies have reported that FGF1 can interact with FGFR and trigger downstream signaling pathways even in the absence of heparan sulfate binding.81,95–99 This enhanced affinity to the receptor, and higher ERK activation of FGF1ΔHBS suggests that in conjunction with the re-distribution of FGF1ΔHBS to the heart, elevated ERK signaling may be the mechanism by which FGF1ΔHBS elicits a greater protection against ischemia-reperfusion injury. Surprisingly, there was no difference in activation of the other cardioprotective kinases studied (Figures 7B–E and 8B–E), including PKC, Akt or STAT3 of which the latter two kinases are involved in RISK and SAFE pathways of cardioprotection and post-conditioning.100–103 Normally, FGF1 activates FGFR which is coupled to intracellular signaling pathways including the RAS-MAPK, PI3K-Akt, PLCγ-PKC, and STAT pathways.104–107 The stimulated receptor then recruits and activates several docking proteins containing src homology (SH-2) domains, e.g., Phospholipase C (PLC) γ and Shb, or phosphotyrosine binding domains, e.g., SHC and FRS2 (FGFR substrate 2).108–111 The phosphorylation of the phosphotyrosine site, Y-654, is elevated in the presence of FGF1 or FGF1ΔHBS (Figure 6C), which leads a binding complex of SHC-FRS2-GRB2-SOS-RAS-Raf-1 and activation of MAPK signaling.112–116 In our study, FGF1ΔHBS triggers increased ERK activation, but not PKC, Akt or STAT3 signaling. It is currently unknown whether this selective activation of ERK is indicative of biased agonism117–123 or biases in the formation of heterodimer versus homodimer which can occur receptor tyrosine kinases124,125 and significant further study would need to occur to demonstrate this.
Taken together, the findings in this study, for the first time, demonstrate that: 1) intravenous administration of FGF1 at the onset of reperfusion protects the heart against cardiac ischemia injury, while heparin reduces the protective effect of FGF1; 2) heparin reduces the availability of FGF1 to the heart, which may be potential mechanism(s) of why heparin lessens the cardioprotective effect of FGF1, 3) novel FGF1 ligand with reduced heparin binding (FGF1ΔHBS) lowers infarct size and improves cardiac function in the presence of heparin, although it has a similar elimination half-life profile of native FGF1, and 4) FGF1ΔHBS enhances the cardioprotective signaling (e.g., ERK activation) even in the presence of heparin to a greater extent than native FGF1, suggesting another potential mechanism of how this novel FGF1 ligand may be a promising therapeutic strategy for the treatment of myocardial infarction.
Acknowledgements
We thank Y. Zhang for technical assistance with the biodistribution studies and the laboratory of Dr. W. Keith Jones and Xiaoping Ren for the use of their camera to image the rat heart infarcts.
Conflict of interest: J. Schultz and M. Mohammadi have an University of Cincinnati invention disclosure filed on “Novel FGF therapeutics for use in ischemic diseases” – 113-084 (May 2013).
Funding
National Heart, Lung and Blood Institute at the National Institutes of Health Grant [grant number R01 HL-075633 to J. J. Schultz and grant number R01 HL-74314 to G. Gross], National Institute of Dental and Craniofacial Research at the National Institutes of Health Grant [grant number R01 DE-13686 to M. Mohammadi], Natural Science Foundation of China [grant numbers 81102486 and 81273509 to Z. Huang], Science Technology Department of Zhejiang Province [grant number 2016C03G2090419 to Z. Huang], and American Heart Association Great Rivers Affiliate, Grant-In-Aid [grant number 14GRNT20450150 to J. J. Schultz]. We also thank X. Cheng and his department (Department of Cardiology, Second affiliated hospital, Nanchang University) for sponsorship of C. Huang. This study was supported by scholarship (C. Huang) under the State Scholarship Fund of China.
References
- 1. Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M, Das SR, de Ferranti S, Despres JP, Fullerton HJ, Howard VJ, Huffman MD, Isasi CR, Jimenez MC, Judd SE, Kissela BM, Lichtman JH, Lisabeth LD, Liu S, Mackey RH, Magid DJ, McGuire DK, Mohler ER 3rd, Moy CS, Muntner P, Mussolino ME, Nasir K, Neumar RW, Nichol G, Palaniappan L, Pandey DK, Reeves MJ, Rodriguez CJ, Rosamond W, Sorlie PD, Stein J, Towfighi A, Turan TN, Virani SS, Woo D, Yeh RW, Turner MB.. Heart disease and stroke statistics-2016 update: A report from the american heart association. Circulation 2016;133:e38–e360. [DOI] [PubMed] [Google Scholar]
- 2. Beenken A, Mohammadi M.. The fgf family: Biology, pathophysiology and therapy. Nat Rev Drug Discov 2009;8:235–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ornitz DM, Itoh N.. Fibroblast growth factors. Genome Biol 2001;2:3005.1–3005.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bikfalvi A, Klein S, Pintucci G, Rifkin DB.. Biological roles of fibroblast growth factor-2. Endocr Rev 1997;18:26–45. [DOI] [PubMed] [Google Scholar]
- 5. Yamashita T, Yoshioka M, Itoh N.. Identification of a novel fibroblast growth factor, fgf-23, preferentially expressed in the ventrolateral thalamic nucleus of the brain. Biochem Biophys Res Commun 2000;277:494–498. [DOI] [PubMed] [Google Scholar]
- 6. Powers CJ, McLeskey SW, Wellstein A.. Fibroblast growth factors, their receptors and signaling. Endocr Relat Cancer 2000;7:165–197. [DOI] [PubMed] [Google Scholar]
- 7. Li XK, Lin ZF, Li Y, Hu S, Tan Y, Huang Z, Meng J, Liang LM, Xiao J, Qu J, Cai L.. Cardiovascular protection of nonmitogenic human acidic fibroblast growth factor from oxidative damage in vitro and in vivo. Cardiovasc Pathol 2007;16:85–91. [DOI] [PubMed] [Google Scholar]
- 8. Cuevas P, Carceller F, Martinez-Coso V, Cuevas B, Fernandez-Ayerdi A, Reimers D, Asin-Cardiel E, Gimenez-Gallego G.. Cardioprotection from ischemia by fibroblast growth factor: Role of inducible nitric oxide synthase. Eur J Med Res 1999;4:517–524. [PubMed] [Google Scholar]
- 9. Cuevas P, Carceller F, Martinez-Coso V, Asin-Cardiel E, Gimenez-Gallego G.. Fibroblast growth factor cardioprotection against ischemia-reperfusion injury may involve k+ atp channels. Eur J Med Res 2000;5:145–149. [PubMed] [Google Scholar]
- 10. Cuevas P, Carceller F, Lozano RM, Crespo A, Zazo M, Gimenez-Gallego G.. Protection of rat myocardium by mitogenic and non-mitogenic fibroblast growth factor during post-ischemic reperfusion. Growth Factors 1997;15:29–40. [DOI] [PubMed] [Google Scholar]
- 11. Cuevas P, Carceller F, Hernandez-Madrid A, Cuevas B, Martinez-Coso V, Gimenez-Gallego G.. Protective effects of acidic fibroblast growth factor against cardiac arrhythmias induced by ischemia and reperfusion in rats. Eur J Med Res 1997;2:33–36. [PubMed] [Google Scholar]
- 12. Cuevas P, Carceller F, Cuevas B, Gimenez-Gallego G, Martinez-Coso V.. A non-mitogenic form of acidic fibroblast growth factor reduces neutrophil infiltration in rat ischemic reperfused heart. Eur J Med Res 1997;2:139–143. [PubMed] [Google Scholar]
- 13. Cuevas P, Barrios V, Gimenez-Gallego G, Martinez-Coso V, Cuevas B, Benavides J, Garcia-Segovia J, Asin-Cardiel E.. Serum levels of basic fibroblast growth factor in acute myocardial infarction. Eur J Med Res 1997;2:282–284. [PubMed] [Google Scholar]
- 14. Buehler A, Martire A, Strohm C, Wolfram S, Fernandez B, Palmen M, Wehrens XH, Doevendans PA, Franz WM, Schaper W, Zimmermann R.. Angiogenesis-independent cardioprotection in fgf-1 transgenic mice. Cardiovasc Res 2002;55:768–777. [DOI] [PubMed] [Google Scholar]
- 15. Palmen M, Daemen MJ, De Windt LJ, Willems J, Dassen WR, Heeneman S, Zimmermann R, Van Bilsen M, Doevendans PA.. Fibroblast growth factor-1 improves cardiac functional recovery and enhances cell survival after ischemia and reperfusion: A fibroblast growth factor receptor, protein kinase c, and tyrosine kinase-dependent mechanism. J Am Coll Cardiol 2004;44:1113–1123. [DOI] [PubMed] [Google Scholar]
- 16. Wright RS, Anderson JL, Adams CD, Bridges CR, Casey DE Jr., Ettinger SM, Fesmire FM, Ganiats TG, Jneid H, Lincoff AM, Peterson ED, Philippides GJ, Theroux P, Wenger NK, Zidar JP, Jacobs AK.. 2011 accf/aha focused update of the guidelines for the management of patients with unstable angina/non-st-elevation myocardial infarction (updating the 2007 guideline): A report of the american college of cardiology foundation/american heart association task force on practice guidelines. Circulation 2011;123:2022–2060. [DOI] [PubMed] [Google Scholar]
- 17. Lee LV. Anticoagulants in coronary artery disease. Cardiol Clin 2008;26:615–628. [DOI] [PubMed] [Google Scholar]
- 18. Anderson JL, Adams CD, Antman EM, Bridges CR, Califf RM, Casey DE Jr., Chavey WE 2nd, Fesmire FM, Hochman JS, Levin TN, Lincoff AM, Peterson ED, Theroux P, Wenger NK, Wright RS, Smith SC. Jr.,. 2011 accf/aha focused update incorporated into the acc/aha 2007 guidelines for the management of patients with unstable angina/non-st-elevation myocardial infarction: A report of the american college of cardiology foundation/american heart association task force on practice guidelines. Circulation 2011;123:e426–e579. [DOI] [PubMed] [Google Scholar]
- 19. Gray HH, Henderson RA, de Belder MA, Underwood SR, Camm AJ.. Early management of unstable angina and non-st-segment elevation myocardial infarction: Summary of nice guidance. Heart 2010;96:1662–1668. [DOI] [PubMed] [Google Scholar]
- 20. Pranckeviciene G, Kadusevicius E, Putniene A.. Influence of coadministration of antithrombotic medicines, warfarin, and nsaids on heparin safety: Data from a prospective observational study. Jmcp 2013;19:478–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Montalescot G, Zeymer U, Silvain J, Boulanger B, Cohen M, Goldstein P, Ecollan P, Combes X, Huber K, Pollack C Jr., Benezet JF, Stibbe O, Filippi E, Teiger E, Cayla G, Elhadad S, Adnet F, Chouihed T, Gallula S, Greffet A, Aout M, Collet JP, Vicaut E.. Intravenous enoxaparin or unfractionated heparin in primary percutaneous coronary intervention for st-elevation myocardial infarction: The international randomised open-label atoll trial. Lancet 2011;378:693–703. [DOI] [PubMed] [Google Scholar]
- 22. Jneid H, Anderson JL, Wright RS, Adams CD, Bridges CR, Casey DE, Ettinger SM, Fesmire FM, Ganiats TG, Lincoff AM, Peterson ED, Philippides GJ, Theroux P, Wenger NK, Zidar JP, Anderson JL.. 2012 accf/aha focused update of the guideline for the management of patients with unstable angina/non-st-elevation myocardial infarction (updating the 2007 guideline and replacing the 2011 focused update): A report of the american college of cardiology foundation/american heart association task force on practice guidelines. Circulation 2012;126:875–910. [DOI] [PubMed] [Google Scholar]
- 23. Mohammadi M, Olsen SK, Goetz R.. A protein canyon in the fgf-fgf receptor dimer selects from an a la carte menu of heparan sulfate motifs. Curr Opin Struct Biol 2005;15:506–516. [DOI] [PubMed] [Google Scholar]
- 24. Spivak-Kroizman T, Lemmon MA, Dikic I, Ladbury JE, Pinchasi D, Huang J, Jaye M, Crumley G, Schlessinger J, Lax I.. Heparin-induced oligomerization of fgf molecules is responsible for fgf receptor dimerization, activation, and cell proliferation. Cell 1994;79:1015–1024. [DOI] [PubMed] [Google Scholar]
- 25. Belov AA, Mohammadi M.. Molecular mechanisms of fibroblast growth factor signaling in physiology and pathology. Cold Spring Harb Perspect Biol 2013;5:1–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mohammadi M, Olsen SK, Ibrahimi OA.. Structural basis for fibroblast growth factor receptor activation. Cytokine Growth Factor Rev 2005;16:107–137. [DOI] [PubMed] [Google Scholar]
- 27. Logan A, Hill DJ.. Bioavailability: Is this a key event in regulating the actions of peptide growth factors?. J Endocrinol 1992;134:157–161. [DOI] [PubMed] [Google Scholar]
- 28. Yayon A, Klagsbrun M, Esko JD, Leder P, Ornitz DM.. Cell surface, heparin-like molecules are required for binding of basic fibroblast growth factor to its high affinity receptor. Cell 1991;64:841–848. [DOI] [PubMed] [Google Scholar]
- 29. Geist A, Marx J, Muller S, Uzan A, von Specht BU, Haberstroh J.. Combination of enoxaparin and fibroblast growth factor-1 increases myocardial blood flow and capillary density after myocardial infarction in rabbits. Eur Surg Res 2005;37:191–198. [DOI] [PubMed] [Google Scholar]
- 30. Hondermarck H, Courty J, Boilly B, Thomas D.. Distribution of intravenously administered acidic and basic fibroblast growth factors in the mouse. Experientia 1990;46:973–974. [DOI] [PubMed] [Google Scholar]
- 31. Newman DR, Li CM, Simmons R, Khosla J, Sannes PL.. Heparin affects signaling pathways stimulated by fibroblast growth factor-1 and -2 in type ii cells. Am J Physiol Lung Cell Mol Physiol 2004;287:L191–L200. [DOI] [PubMed] [Google Scholar]
- 32. Fannon M, Forsten KE, Nugent MA.. Potentiation and inhibition of bfgf binding by heparin: A model for regulation of cellular response. Biochemistry 2000;39:1434–1445. [DOI] [PubMed] [Google Scholar]
- 33. Zhao YZ, Lu CT, Li XK, Tang QQ, Tian XQ, Zhao YP, Zhang Y, Tian JL, Yang W, Ge S, Nair CK, Shen X.. Improving the cardio protective effect of afgf in ischemic myocardium with ultrasound-mediated cavitation of heparin modified microbubbles: Preliminary experiment. J Drug Target 2012;20:623–631. [DOI] [PubMed] [Google Scholar]
- 34. Cuevas P, Reimers D, Carceller F, Martinez-Coso V, Redondo-Horcajo M, Saenz de Tejada I, Gimenez-Gallego G.. Fibroblast growth factor-1 prevents myocardial apoptosis triggered by ischemia reperfusion injury. Eur J Med Res 1997;2:465–468. [PubMed] [Google Scholar]
- 35. Schultz JE, Rose E, Yao Z, Gross GJ.. Evidence for involvement of opioid receptors in ischemic preconditioning in rat hearts. Am J Physiol Heart Circ Physiol 1995;268:H2157–H2161. [DOI] [PubMed] [Google Scholar]
- 36. Schultz JE, Qian YZ, Gross GJ, Kukreja RC.. The ischemia-selective katp channel antagonist, 5-hydroxydecanoate, blocks ischemic preconditioning in the rat heart. J Mol Cell Cardiol 1997;29:1055–1060. [DOI] [PubMed] [Google Scholar]
- 37. Schultz JE, Hsu AK, Gross GJ.. Morphine mimics the cardioprotective effect of ischemic preconditioning via a glibenclamide-sensitive mechanism in the rat heart. Circ Res 1996;78:1100–1104. [DOI] [PubMed] [Google Scholar]
- 38. Cai Y, Zhang Z, Fan K, Zhang J, Shen W, Li M, Si D, Luo H, Zeng Y, Fu P, Liu C.. Pharmacokinetics, tissue distribution, excretion, and antiviral activity of pegylated recombinant human consensus interferon-alpha variant in monkeys, rats and guinea pigs. Regul Pept 2012;173:74–81. [DOI] [PubMed] [Google Scholar]
- 39. Mueller SN, Thomas KA, Di Salvo J, Levine EM.. Stabilization by heparin of acidic fibroblast growth factor mitogenicity for human endothelial cells in vitro. J Cell Physiol 1989;140:439–448. [DOI] [PubMed] [Google Scholar]
- 40. Damon DH, Lobb RR, D'amore PA, Wagner JA.. Heparin potentiates the action of acidic fibroblast growth factor by prolonging its biological half-life. J Cell Physiol 1989;138:221–226. [DOI] [PubMed] [Google Scholar]
- 41. Wu D, Song BW, Vinters HV, Pardridge WM.. Pharmacokinetics and brain uptake of biotinylated basic fibroblast growth factor conjugated to a blood-brain barrier drug delivery system. J Drug Target 2002;10:239–245. [DOI] [PubMed] [Google Scholar]
- 42. House SL, Bolte C, Zhou M, Doetschman T, Klevitsky R, Newman G, Schultz Jel J.. Cardiac-specific overexpression of fibroblast growth factor-2 protects against myocardial dysfunction and infarction in a murine model of low-flow ischemia. Circulation 2003;108:3140–3148. [DOI] [PubMed] [Google Scholar]
- 43. Manning JR, Perkins SO, Sinclair EA, Gao X, Zhang Y, Newman G, Pyle WG, Schultz JEJ.. Low molecular weight fibroblast growth factor-2 signals via protein kinase c and myofibrillar proteins to protect against postischemic cardiac dysfunction. Am J Physiol Heart Circ Physiol 2013;304:H1382–H1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Liao S, Porter D, Scott A, Newman G, Doetschman T, Schultz JEJ.. The cardioprotective effect of the low molecular weight isoform of fibroblast growth factor-2: The role of jnk signaling. J Mol Cell Cardiol 2007;42:106–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Liao S, Bodmer JR, Azhar M, Newman G, Coffin JD, Doetschman T, Schultz JJ.. The influence of fgf2 high molecular weight (hmw) isoforms in the development of cardiac ischemia-reperfusion injury. J Mol Cell Cardiol 2010;48:1245–1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wang J, Valdez A, Chen Y.. Evaluation of automated wes system as an analytical and characterization tool to support monoclonal antibody drug product development. J Pharm Biomed Anal 2016;139:263–268. [DOI] [PubMed] [Google Scholar]
- 47. Cuevas P, Martinez-Coso V, Fu X, Orte L, Reimers D, Gimenez-Gallego G, Forssmann WG, Saenz D, Tejada I.. Fibroblast growth factor protects the kidney against ischemia- reperfusion injury. Eur J Med Res 1999;4:403–410. [PubMed] [Google Scholar]
- 48. Schwartz Longacre L, Kloner RA, Arai AE, Baines CP, Bolli R, Braunwald E, Downey J, Gibbons RJ, Gottlieb RA, Heusch G, Jennings RB, Lefer DJ, Mentzer RM, Murphy E, Ovize M, Ping P, Przyklenk K, Sack MN, Vander Heide RS, Vinten-Johansen J, Yellon DM.. New horizons in cardioprotection: Recommendations from the 2010 national heart, lung, and blood institute workshop. Circulation 2011;124:1172–1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Park JL, Kilgore KS, Naylor KB, Booth EA, Murphy KL, Lucchesi BR.. N-acetylheparin pretreatment reduces infarct size in the rabbit. Pharmacology 1999;58:120–131. [DOI] [PubMed] [Google Scholar]
- 50. Kilgore KS, Tanhehco EJ, Naylor KB, Lucchesi BR.. Ex vivo reversal of heparin-mediated cardioprotection by heparinase after ischemia and reperfusion. J Pharmacol Exp Ther 1999;290:1041–1047. [PubMed] [Google Scholar]
- 51. Gralinski MR, Driscoll EM, Friedrichs GS, DeNardis MR, Lucchesi BR.. Reduction of myocardial necrosis after glycosaminoglycan administration: Effects of a single intravenous administration of heparin or n-acetylheparin 2 hours before regional ischemia and reperfusion. J Cardiovasc Pharmacol Ther 1996;1:219–228. [DOI] [PubMed] [Google Scholar]
- 52. Black SC, Gralinski MR, Friedrichs GS, Kilgore KS, Driscoll EM, Lucchesi BR.. Cardioprotective effects of heparin or n-acetylheparin in an in vivo model of myocardial ischaemic and reperfusion injury. Cardiovasc Res 1995;29:629–636. [PubMed] [Google Scholar]
- 53. Shah MM, Sakurai H, Gallegos TF, Sweeney DE, Bush KT, Esko JD, Nigam SK.. Growth factor-dependent branching of the ureteric bud is modulated by selective 6-o sulfation of heparan sulfate. Dev Biol 2011;356:19–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Guerrini M, Hricovini M, Torri G.. Interaction of heparins with fibroblast growth factors: Conformational aspects. Curr Pharm Design 2007;13:2045–2056. [DOI] [PubMed] [Google Scholar]
- 55. Gallagher JT, Turnbull JE.. Heparan sulphate in the binding and activation of basic fibroblast growth factor. Glycobiology 1992;2:523–528. [DOI] [PubMed] [Google Scholar]
- 56. Turnbull JE, Gallagher JT.. Heparan sulphate: Functional role as a modulator of fibroblast growth factor activity. Biochem Soc Trans 1993;21:477–482. [DOI] [PubMed] [Google Scholar]
- 57. Zinn KR, Kelpke S, Chaudhuri TR, Sugg T, Mountz JM, Thompson JA.. Imaging tc-99m-labeled fgf-1 targeting in rats. Nucl Med Biol 2000;27:407–414. [DOI] [PubMed] [Google Scholar]
- 58. Rosengart TK, Kuperschmid JP, Maciag T, Clark RE.. Pharmacokinetics and distribution of heparin-binding growth factor i (endothelial cell growth factor) in the rat. Circ Res 1989;64:227–234. [DOI] [PubMed] [Google Scholar]
- 59. Lønborg J, Schoos MM, Kelbæk H, Holmvang L, Steinmetz J, Vejlstrup N, Jørgensen E, Helqvist S, Saunamäki K, Bøtker HE, Kim WY, Terkelsen CJ, Clemmensen P, Engstrøm T.. Impact of system delay on infarct size, myocardial salvage index, and left ventricular function in patients with st-segment elevation myocardial infarction. Am Heart J 2012;164:538–546. [DOI] [PubMed] [Google Scholar]
- 60. Varcoe RW, Clayton TC, Gray HH, de Belder MA, Ludman PF, Henderson RA.. Impact of call-to-balloon time on 30-day mortality in contemporary practice. Heart 2017;103:117–124. [DOI] [PubMed] [Google Scholar]
- 61. Topol EJ, Van de Werf FJ, Acute myocardial infarction: Early diagnosis and management Textbook of cardiovascular medicine, 2nd edition Philadelphia: Lippincott, Williams, and Wilkins; 2002: 385-419. [Google Scholar]
- 62. Brodie BR, Webb J, Cox DA, Qureshi M, Kalynych A, Turco M, Schultheiss HP, Dulas D, Rutherford B, Antoniucci D, Stuckey T, Krucoff M, Gibbons R, Lansky A, Na Y, Mehran R, Stone GW.. Impact of time to treatment on myocardial reperfusion and infarct size with primary percutaneous coronary intervention for acute myocardial infarction (from the emerald trial). Am J Cardiol 2007;99:1680–1686. [DOI] [PubMed] [Google Scholar]
- 63. Guerchicoff A, Brener SJ, Maehara A, Witzenbichler B, Fahy M, Xu K, Gersh BJ, Mehran R, Gibson CM, Stone GW.. Impact of delay to reperfusion on reperfusion success, infarct size, and clinical outcomes in patients with st-segment elevation myocardial infarction: The infuse-ami trial (infuse-anterior myocardial infarction). JACC. Cardiovasc Interventions 2014;7:733–740. [DOI] [PubMed] [Google Scholar]
- 64. Bainey KR, Ferguson C, Ibrahim QI, Tyrrell B, Welsh RC.. Impact of reperfusion strategy on aborted myocardial infarction: Insights from a large canadian st-elevation myocardial infarction clinical registry. Can J Cardiol 2014;30:1570–1575. [DOI] [PubMed] [Google Scholar]
- 65. Puymirat E, Caudron J, Steg PG, Lemesle G, Cottin Y, Coste P, Schiele F, de Labriolle A, Bataille V, Ferrieres J, Simon T, Danchin N.. Prognostic impact of non-compliance with guidelines-recommended times to reperfusion therapy in st-elevation myocardial infarction. The fast-mi 2010 registry. Eur Heart J Acute Cardiovasc Care 2017;6:26–33. [DOI] [PubMed] [Google Scholar]
- 66. Magalhaes P, Mateus P, Carvalho S, Leao S, Cordeiro F, Moreira JI.. Relationship between treatment delay and type of reperfusion therapy and mechanical complications of acute myocardial infarction. Eur Heart J Acute Cardiovasc Care 2016;5:468–474. [DOI] [PubMed] [Google Scholar]
- 67. Kloner RA, Forman MB, Gibbons RJ, Ross AM, Alexander RW, Stone GW.. Impact of time to therapy and reperfusion modality on the efficacy of adenosine in acute myocardial infarction: The amistad-2 trial. Eur Heart J 2006;27:2400–2405. [DOI] [PubMed] [Google Scholar]
- 68. Kitakaze M, Asakura M, Kim J, Shintani Y, Asanuma H, Hamasaki T, Seguchi O, Myoishi M, Minamino T, Ohara T, Nagai Y, Nanto S, Watanabe K, Fukuzawa S, Hirayama A, Nakamura N, Kimura K, Fujii K, Ishihara M, Saito Y, Tomoike H, Kitamura S.. Human atrial natriuretic peptide and nicorandil as adjuncts to reperfusion treatment for acute myocardial infarction (j-wind): Two randomised trials. Lancet 2007;370:1483–1493. [DOI] [PubMed] [Google Scholar]
- 69. Piot C, Croisille P, Staat P, Thibault H, Rioufol G, Mewton N, Elbelghiti R, Cung TT, Bonnefoy E, Angoulvant D, Macia C, Raczka F, Sportouch C, Gahide G, Finet G, Andre-Fouet X, Revel D, Kirkorian G, Monassier JP, Derumeaux G, Ovize M.. Effect of cyclosporine on reperfusion injury in acute myocardial infarction. N Engl J Med 2008;359:473–481. [DOI] [PubMed] [Google Scholar]
- 70. Ibanez B, Macaya C, Sanchez-Brunete V, Pizarro G, Fernandez-Friera L, Mateos A, Fernandez-Ortiz A, Garcia-Ruiz JM, Garcia-Alvarez A, Iniguez A, Jimenez-Borreguero J, Lopez-Romero P, Fernandez-Jimenez R, Goicolea J, Ruiz-Mateos B, Bastante T, Arias M, Iglesias-Vazquez JA, Rodriguez MD, Escalera N, Acebal C, Cabrera JA, Valenciano J, Perez de Prado A, Fernandez-Campos MJ, Casado I, Garcia-Rubira JC, Garcia-Prieto J, Sanz-Rosa D, Cuellas C, Hernandez-Antolin R, Albarran A, Fernandez-Vazquez F, de la Torre-Hernandez JM, Pocock S, Sanz G, Fuster V.. Effect of early metoprolol on infarct size in st-segment-elevation myocardial infarction patients undergoing primary percutaneous coronary intervention: The effect of metoprolol in cardioprotection during an acute myocardial infarction (metocard-cnic) trial. Circulation 2013;128:1495–1503. [DOI] [PubMed] [Google Scholar]
- 71. Woo JS, Kim W, Ha SJ, Kim JB, Kim SJ, Kim WS, Seon HJ, Kim KS.. Cardioprotective effects of exenatide in patients with st-segment-elevation myocardial infarction undergoing primary percutaneous coronary intervention: Results of exenatide myocardial protection in revascularization study. Arteriosclero, Thromb Vasc Biol 2013;33:2252–2260. [DOI] [PubMed] [Google Scholar]
- 72. Mahaffey KW, Puma JA, Barbagelata NA, DiCarli MF, Leesar MA, Browne KF, Eisenberg PR, Bolli R, Casas AC, Molina-Viamonte V, Orlandi C, Blevins R, Gibbons RJ, Califf RM, Granger CB.. Adenosine as an adjunct to thrombolytic therapy for acute myocardial infarction: Results of a multicenter, randomized, placebo-controlled trial: The acute myocardial infarction study of adenosine (amistad) trial. J Am Coll Cardiol 1999;34:1711–1720. [DOI] [PubMed] [Google Scholar]
- 73. Ross AM, Gibbons RJ, Stone GW, Kloner RA, Alexander RW.. A randomized, double-blinded, placebo-controlled multicenter trial of adenosine as an adjunct to reperfusion in the treatment of acute myocardial infarction (amistad-ii). J Am Coll Cardiol 2005;45:1775–1780. [DOI] [PubMed] [Google Scholar]
- 74. Gross ER, Hsu AK, Gross GJ.. Acute aspirin treatment abolishes, whereas acute ibuprofen treatment enhances morphine-induced cardioprotection: Role of 12-lipoxygenase. J Pharmacol Exp Ther 2004;310:185–191. [DOI] [PubMed] [Google Scholar]
- 75. Presta M, Dell’era P, Mitola S, Moroni E, Ronca R, Rusnati M.. Fibroblast growth factor/fibroblast growth factor receptor system in angiogenesis. Cytokine Growth Factor Rev 2005;16:159–178. [DOI] [PubMed] [Google Scholar]
- 76. Rusnati M, Presta M.. Fibroblast growth factors/fibroblast growth factor receptors as targets for the development of anti-angiogenesis strategies. Curr Pharm Design 2007;13:2025–2044. [DOI] [PubMed] [Google Scholar]
- 77. Xue L, Shireman PK, Hampton B, Burgess WH, Greisler HP.. The cysteine-free fibroblast growth factor 1 mutant induces heparin-independent proliferation of endothelial cells and smooth muscle cells. J Surgi Res 2000;92:255–260. [DOI] [PubMed] [Google Scholar]
- 78. Colin S, Jeanny JC, Mascarelli F, Vienet R, Al-Mahmood S, Courtois Y, Labarre J.. In vivo involvement of heparan sulfate proteoglycan in the bioavailability, internalization, and catabolism of exogenous basic fibroblast growth factor. Mol Pharmacol 1999;55:74–82. [DOI] [PubMed] [Google Scholar]
- 79. Xia X, Babcock JP, Blaber SI, Harper KM, Blaber M, Houtman JCD.. Pharmacokinetic properties of 2nd-generation fibroblast growth factor-1 mutants for therapeutic application. PLoS One 2012;7:e48210.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Bishop JR, Schuksz M, Esko JD.. Heparan sulphate proteoglycans fine-tune mammalian physiology. Nature 2007;446:1030–1037. [DOI] [PubMed] [Google Scholar]
- 81. Zakrzewska M, Wiedlocha A, Szlachcic A, Krowarsch D, Otlewski J, Olsnes S.. Increased protein stability of fgf1 can compensate for its reduced affinity for heparin. J Biol Chem 2009;284:25388–25403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Zakrzewska M, Krowarsch D, Wiedlocha A, Olsnes S, Otlewski J.. Highly stable mutants of human fibroblast growth factor-1 exhibit prolonged biological action. J Mol Biol 2005;352:860–875. [DOI] [PubMed] [Google Scholar]
- 83. Culajay JF, Blaber SI, Khurana A, Blaber M.. Thermodynamic characterization of mutants of human fibroblast growth factor 1 with an increased physiological half-life. Biochemistry 2000;39:7153–7158. [DOI] [PubMed] [Google Scholar]
- 84. Chen G, Gulbranson DR, Yu P, Hou Z, Thomson JA.. Thermal stability of fibroblast growth factor protein is a determinant factor in regulating self-renewal, differentiation, and reprogramming in human pluripotent stem cells. Stem Cells 2012;30:623–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Kobielak A, Zakrzewska M, Kostas M, Jakimowicz P, Otlewski J, Krowarsch D.. Protease resistant variants of fgf1 with prolonged biological activity. Ppl 2014;21:434–443. [DOI] [PubMed] [Google Scholar]
- 86. Dietrich CP, Sampaio LO, Toledo OM.. Characteristic distribution of sulfated mucopolysaccharides in different tissues and in their respective mitochondria. Biochem Biophys Res Commun 1976;71:1–10. [DOI] [PubMed] [Google Scholar]
- 87. Flanagan MF, Aoyagi T, Arnold LW, Maute C, Fujii AM, Currier J, Bergau D, Warren HB, Rakusan K.. Effects of chronic heparin administration on coronary vascular adaptation to hypertension and ventricular hypertrophy in sheep. Circulatio 1999;100:981–987. [DOI] [PubMed] [Google Scholar]
- 88. Zhao T, Zhao W, Chen Y, Ahokas RA, Sun Y.. Acidic and basic fibroblast growth factors involved in cardiac angiogenesis following infarction. International J Cardiol 2011;152:307–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Bombardini T, Picano E.. The coronary angiogenetic effect of heparin: Experimental basis and clinical evidence. Angiology 1997;48:969–976. [DOI] [PubMed] [Google Scholar]
- 90. Saxena K, Schieborr U, Anderka O, Duchardt-Ferner E, Elshorst B, Gande SL, Janzon J, Kudlinzki D, Sreeramulu S, Dreyer MK, Wendt KU, Herbert C, Duchaussoy P, Bianciotto M, Driguez PA, Lassalle G, Savi P, Mohammadi M, Bono F, Schwalbe H.. Influence of heparin mimetics on assembly of the fgf.Fgfr4 signaling complex. J Biol Chem 2010;285:26628–26640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Ornitz DM, Xu J, Colvin JS, McEwen DG, MacArthur CA, Coulier F, Gao G, Goldfarb M.. Receptor specificity of the fibroblast growth factor family. J Biol Chem 1996;271:15292–15297. [DOI] [PubMed] [Google Scholar]
- 92. Wu ZL, Zhang L, Yabe T, Kuberan B, Beeler DL, Love A, Rosenberg RD.. The involvement of heparan sulfate (hs) in fgf1/hs/fgfr1 signaling complex. J Biol Chem 2003;278:17121–17129. [DOI] [PubMed] [Google Scholar]
- 93. Waksman G, Herr AB.. New insights into heparin-induced fgf oligomerization. Nat Struct Biol 1998;5:527–530. [DOI] [PubMed] [Google Scholar]
- 94. Ornitz DM, Herr AB, Nilsson M, Westman J, Svahn CM, Waksman G.. Fgf binding and fgf receptor activation by synthetic heparan-derived di- and trisaccharides. Science 1995;268:432–436. [DOI] [PubMed] [Google Scholar]
- 95. Nugent MA, Edelman ER.. Kinetics of basic fibroblast growth factor binding to its receptor and heparan sulfate proteoglycan: A mechanism for cooperactivity. Biochemistry 1992;31:8876–8883. [DOI] [PubMed] [Google Scholar]
- 96. Klingenberg O, Wiedlocha A, Rapak A, Muñoz R, Falnes PØ, Olsnes S.. Inability of the acidic fibroblast growth factor mutant k132e to stimulate DNA synthesis after translocation into cells. J Biol Chem 1998;273:11164–11172. [DOI] [PubMed] [Google Scholar]
- 97. Klingenberg O, Wiedlocha A, Olsnes S.. Effects of mutations of a phosphorylation site in an exposed loop in acidic fibroblast growth factor. J Biol Chem 1999;274:18081–18086. [DOI] [PubMed] [Google Scholar]
- 98. Fannon M, Nugent MA.. Basic fibroblast growth factor binds its receptors, is internalized, and stimulates DNA synthesis in balb/c3t3 cells in the absence of heparan sulfate. J Biol Chem 1996;271:17949–17956. [DOI] [PubMed] [Google Scholar]
- 99. Roghani M, Mansukhani A, Dell'era P, Bellosta P, Basilico C, Rifkin DB, Moscatelli D.. Heparin increases the affinity of basic fibroblast growth factor for its receptor but is not required for binding. J Biol Chem 1994;269:3976–3984. [PubMed] [Google Scholar]
- 100. Hausenloy DJ, Yellon DM.. Survival kinases in ischemic preconditioning and postconditioning. Cardiovasc Res 2006;70:240–253. [DOI] [PubMed] [Google Scholar]
- 101. Hausenloy DJ, Tsang A, Yellon DM.. The reperfusion injury salvage kinase pathway: A common target for both ischemic preconditioning and postconditioning. Trends Cardiovasc Med 2005;15:69–75. [DOI] [PubMed] [Google Scholar]
- 102. Lecour S. Multiple protective pathways against reperfusion injury: A safe path without aktion?. J Mol Cell Cardiol 2009;46:607–609. [DOI] [PubMed] [Google Scholar]
- 103. Lecour S. Activation of the protective survivor activating factor enhancement (safe) pathway against reperfusion injury: Does it go beyond the risk pathway?. J Mol Cell Cardiol 2009;47:32–40. [DOI] [PubMed] [Google Scholar]
- 104. Mohammadi M, Honegger AM, Rotin D, Fischer R, Bellot F, Li W, Dionne CA, Jaye M, Rubinstein M, Schlessinger J.. A tyrosine-phosphorylated carboxy-terminal peptide of the fibroblast growth factor receptor (flg) is a binding site for the sh2 domain of phospholipase c-gamma 1. Mol Cell Biol 1991;11:5068–5078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Ong SH, Hadari YR, Gotoh N, Guy GR, Schlessinger J, Lax I.. Stimulation of phosphatidylinositol 3-kinase by fibroblast growth factor receptors is mediated by coordinated recruitment of multiple docking proteins. Proc Natl Acad Sci USA 2001;98:6074–6079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Hart KC, Robertson SC, Kanemitsu MY, Meyer AN, Tynan JA, Donoghue DJ.. Transformation and stat activation by derivatives of fgfr1, fgfr3, and fgfr4. Oncogene 2000;19:3309–3320. [DOI] [PubMed] [Google Scholar]
- 107. Kouhara H, Hadari YR, Spivak-Kroizman T, Schilling J, Bar-Sagi D, Lax I, Schlessinger J.. A lipid-anchored grb2-binding protein that links fgf-receptor activation to the ras/mapk signaling pathway. Cell 1997;89:693–702. [DOI] [PubMed] [Google Scholar]
- 108. Dhalluin C, Yan KS, Plotnikova O, Lee KW, Zeng L, Kuti M, Mujtaba S, Goldfarb MP, Zhou MM.. Structural basis of snt ptb domain interactions with distinct neurotrophic receptors. Mol Cell 2000;6:921–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Larsson H, Klint P, Landgren E, Claesson-Welsh L.. Fibroblast growth factor receptor-1-mediated endothelial cell proliferation is dependent on the src homology (sh) 2/sh3 domain-containing adaptor protein crk. J Biol Chem 1999;274:25726–25734. [DOI] [PubMed] [Google Scholar]
- 110. Landgren E, Klint P, Yokote K, Claesson-Welsh L.. Fibroblast growth factor receptor-1 mediates chemotaxis independently of direct sh2-domain protein binding. Oncogene 1998;17:283–291. [DOI] [PubMed] [Google Scholar]
- 111. Karlsson T, Songyang Z, Landgren E, Lavergne C, Di Fiore PP, Anafi M, Pawson T, Cantley LC, Claesson-Welsh L, Welsh M.. Molecular interactions of the src homology 2 domain protein shb with phosphotyrosine residues, tyrosine kinase receptors and src homology 3 domain proteins. Oncogene 1995;10:1475–1483. [PubMed] [Google Scholar]
- 112. Williams NG, Paradis H, Agarwal S, Charest DL, Pelech SL, Roberts TM.. Raf-1 and p21v-ras cooperate in the activation of mitogen-activated protein kinase. Proc Natl Acad Sci USA 1993;90:5772–5776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Ryan PJ, Paterno GD, Gillespie LL.. Identification of phosphorylated proteins associated with the fibroblast growth factor receptor type i during early xenopus development. Biochem Biophys Res Commun 1998;244:763–767. [DOI] [PubMed] [Google Scholar]
- 114. Mohammadi M, Dikic I, Sorokin A, Burgess WH, Jaye M, Schlessinger J.. Identification of six novel autophosphorylation sites on fibroblast growth factor receptor 1 and elucidation of their importance in receptor activation and signal transduction. Mol Cell Biol 1996;16:977–989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Klint P, Kanda S, Claesson-Welsh L.. Shc and a novel 89-kda component couple to the grb2-sos complex in fibroblast growth factor-2-stimulated cells. J Biol Chem 1995;270:23337–23344. [DOI] [PubMed] [Google Scholar]
- 116. Curto M, Frankel P, Carrero A, Foster DA.. Novel recruitment of shc, grb2, and sos by fibroblast growth factor receptor-1 in v-src-transformed cells. Biochem Biophys Res Commun 1998;243:555–560. [DOI] [PubMed] [Google Scholar]
- 117. Luttrell LM, Maudsley S, Bohn LM.. Fulfilling the promise of “biased” g protein-coupled receptor agonism. Mol Pharmacol 2015;88:579–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Holloway AC, Qian H, Pipolo L, Ziogas J, Miura S, Karnik S, Southwell BR, Lew MJ, Thomas WG.. Side-chain substitutions within angiotensin ii reveal different requirements for signaling, internalization, and phosphorylation of type 1a angiotensin receptors. Mol Pharm 2002;61:768–777. [DOI] [PubMed] [Google Scholar]
- 119. Gray JA, Roth BL.. Paradoxical trafficking and regulation of 5-ht(2a) receptors by agonists and antagonists. Brain Res Bull 2001;56:441–451. [DOI] [PubMed] [Google Scholar]
- 120. Berg KA, Maayani S, Goldfarb J, Scaramellini C, Leff P, Clarke WP.. Effector pathway-dependent relative efficacy at serotonin type 2a and 2c receptors: Evidence for agonist-directed trafficking of receptor stimulus. Mol Pharm 1998;54:94–104. [PubMed] [Google Scholar]
- 121. Takasu H, Gardella TJ, Luck MD, Potts JT Jr, Bringhurst FR.. Amino-terminal modifications of human parathyroid hormone (pth) selectively alter phospholipase c signaling via the type 1 pth receptor: Implications for design of signal-specific pth ligands. Biochemistry 1999;38:13453–13460. [DOI] [PubMed] [Google Scholar]
- 122. Sneddon WB, Magyar CE, Willick GE, Syme CA, Galbiati F, Bisello A, Friedman PA.. Ligand-selective dissociation of activation and internalization of the parathyroid hormone (pth) receptor: Conditional efficacy of pth peptide fragments. Endocrinology 2004;145:2815–2823. [DOI] [PubMed] [Google Scholar]
- 123. Sagan S, Chassaing G, Pradier L, Lavielle S.. Tachykinin peptides affect differently the second messenger pathways after binding to cho-expressed human nk-1 receptors. J Pharm Exp Ther 1996;276:1039–1048. [PubMed] [Google Scholar]
- 124. Macdonald-Obermann JL, Pike LJ.. Different epidermal growth factor (egf) receptor ligands show distinct kinetics and biased or partial agonism for homodimer and heterodimer formation. J Biol Chem 2014;289:26178–26188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. De Smet F, Christopoulos A, Carmeliet P.. Allosteric targeting of receptor tyrosine kinases. Nat Biotechnol 2014;32:1113–1120. [DOI] [PubMed] [Google Scholar]