

Abstract
Aims
Left-atrial (LA) fibrosis is an important feature of many atrial fibrillation (AF) substrates. The JAK-STAT system contributes to cardiac remodelling, but its role in AF is unknown. Here we investigated JAK-STAT changes in an AF-model and their potential contributions to LA-fibrosis.
Methods and results
LA-remodelling was studied in dogs with heart failure (HF) induced by ventricular tachypacing (VTP, 240 bpm), and in mice with left-ventricular (LV) dysfunction due to myocardial infarction (MI). The selective STAT-3 inhibitor S3I-201 was administered to fibroblasts in vitro or mice in vivo (10 mg/kg/d, osmotic mini-pump). HF-dogs developed LA-selective fibrosis and AF-susceptibility at 1-week VTP. The mRNA-expression of platelet-derived growth factor (PDGF, a JAK-STAT activator) isoforms A, C and D, as well as JAK2, increased in LA fibroblasts from 1-week VTP. HF upregulated protein-expression of PDGF-receptor-β and phosphorylated (activated) signal transducer and activator of transcription 3 (STAT3) in LA. PDGF-AB stimulation of LA fibroblasts increased PDGFR-α, STAT3 and phosphorylated-STAT3 expression, as well as collagen-1 and fibronectin-1 protein secretion (by 1.6- to 20-fold), with smaller changes in LV fibroblasts. Phosphorylated-STAT3 and collagen upregulation were suppressed by the JAK2 inhibitor AG-490, PDGF receptor inhibitor AG1296 and STAT3-inhibitor SI3-201. In vivo S3I-201 treatment of MI-mice attenuated LA-fibrosis, LA-dilation and P-wave duration changes versus vehicle-control.
Conclusions
HF activates the LA JAK-STAT system and enhances PDGF-signalling. JAK-STAT inhibition reduces the profibrotic effects of PDGF stimulation on canine fibroblasts in vitro while attenuating in vivo LA-fibrosis and remodelling in post-MI mice, suggesting that the JAK/STAT pathway contributes to LA-fibrogenesis and might be a potential target for LA-fibrosis prevention.
Keywords: Heart failure, PDGF, Atrial remodelling, Atrial fibrosis
1. Introduction
Atrial fibrillation (AF)-promoting atrial remodelling is induced by multiple stressors, including mechanical stretch and cytokine stimulation.1,2 Atrial fibrosis, a major component of atrial structural remodelling, can be induced by AF per se.3 In the fibrotic response, activated fibroblasts produce extracellular matrix (ECM) remodelling that negatively influences cardiac electrical, structural and contractile properties.3,4
A range of profibrotic signalling pathways has been implicated in myocardial fibrotic remodelling. Angiotensin II (Ang-II), transforming growth factor beta (TGF-β), and connective tissue growth factor (CTGF) are among the best-known.3,5–7 Mitogen-activated protein kinases (MAPKs) are important downstream effectors.3,8 Platelet-derived growth factor (PDGF) stimulates fibroblast proliferation and differentiation to myofibroblasts.9 PDGF blockade reduces interstitial fibrosis of infarcted hearts in rats10 and a PDGF inhibitor suppresses atrial-selective canine fibroblast activation, eliminating the characteristic atrial-ventricular fibroblast activation differences.11 Limited information is available about the role of PDGF and associated signalling pathways in cardiac fibrosis and congestive heart failure (CHF). PDGF activates RAS-ERK1/2 or PI3K-Akt/mTOR pathways, promoting proliferation and protein synthesis in vascular smooth muscle cells.12,13 Signal transducer and activator of transcription 3 (STAT3), a latent transcription factor, is an important determinant of cell survival and proliferation.14,15 PDGF directly activates the Janus kinase (JAK)-STAT pathway and induces mitogens in fibroblasts.16–18 JAK-STAT is activated in a pig model of electrically maintained AF.19 However, the contribution of the JAK-STAT system to atrial fibrosis and clinically relevant AF-substrates is unknown. In this study, we assessed the potential role of the PDGF-JAK-STAT system in regulating atrial fibroblast behaviour and atrial-selective fibrosis in CHF.
2. Methods
2.1 Animals
Animal-care procedures followed the guidelines of the Canadian Council on Animal Care and were approved by the Animals Research Ethics Committee of the Montreal Heart Institute. CHF was induced by ventricular tachypacing (VTP). VTP-dogs were anaesthetized under diazepam (0.25 mg/kg IV)/ketamine (5.0 mg/kg IV)/halothane (1–2% PI) anaesthesia, intubated, and ventilated. Bipolar pacing leads were fixed to the right-ventricular apex via left jugular-vein access, and connected to pacemakers (St Jude Medical, Minneapolis, MN) implanted subcutaneously in the neck. Following twenty-four hours for postoperative recovery, VTP was initiated at 240 bpm. Forty-seven adult mongrel dogs weighing 18.4–35.0 kg were divided into five groups: non-paced controls (n= 17), 12-h VTP (n= 5), 24-h VTP (n= 5), 1-week VTP (n= 5) and 2-week VTP (n= 15). In a first series of dogs, we examined tissue protein expression, with five control dogs and five dogs euthanized after 2-week VTP. In a second series, we analysed gene expression in fibroblasts from 12 control dogs, and 5 each at 12 h, 24 h, and 1 week VTP, and 10 dogs at 2 weeks of VTP. AF-duration measurement and tissue procurement were obtained under morphine (2 mg/kg im)/alpha-chloralose (120 mg/kg iv) anaesthesia, with the heart exposed through a left thoracotomy. Further details of dog preparation and handling for the VTP-model are provided in the Supplementary material online.
Twenty five male C57BL/6 mice (8–10 weeks) weighing 20–25 g (Charles River, Saint-Constant, Quebec) were divided into three groups: sham controls (n= 8), myocardial infarction (MI) + vehicle (n= 9), MI + S3I-201 (n= 8). MI was induced by left anterior descending coronary artery ligation (LAD), which was maintained for 2 weeks (methods detailed in the Supplementary material online). Vehicle (Sham and MI groups) or S3I-201 (10 mg/kg/d, MI + S3I group) were delivered via osmotic mini-pumps (Alzet 2001; Cupertino, CA) implanted subcutaneously one day before surgery.
2.2 Echocardiography
Transthoracic echocardiographic studies under 2%-isoflurane were obtained before and 14 days after sham or MI surgery with a phased-array 10S probe (4.5–11.5 MHz) in a Vivid 7 Dimension System (GE Healthcare Ultrasound, Horten, Norway). Echocardiographic methods and measures are detailed in the Supplementary material online.
2.3 Tissue and cell harvesting
On study-days, dogs were anaesthetized with morphine (2 mg/kg s.c.) and alpha-chloralose (120 mg/kg i.v.) and mechanically ventilated. Dogs were then euthanized by cardiac excision. Their hearts were removed via median thoracotomy and immediately immersed in oxygenated Tyrode’s solution. LA appendage, LA free wall and left ventricular (LV) free wall samples were fast-frozen in liquid-N2 for molecular biology and/or stored in formalin for histology. LA and LV were perfused for cell isolation and collagenase digestion (see detailed description in Supplementary material online). Freshly isolated fibroblasts and cardiomyocytes were snap-frozen in liquid-N2 and stored at −80 °C.
Mice were deeply anaesthetized with isoflurane, then euthanized by cervical dislocation. Hearts were then excised, briefly washed in cold 0.9% saline solution and weighed. Left atria were fixed in 10% formalin at room temperature.
2.4 Cell-culture and treatment
Paired LA and LV cell-samples from each control dog were plated in 6-well plastic plates (Corning Inc., Corning, NY) in parallel at equal density and cultured in DMEM containing 5%-FBS and 1%-penicillin/streptomycin. Fibroblasts were allowed to adhere for 2 days, rendered quiescent in serum-free medium for 16 hours, and then stimulated with PDGF-AB (Sigma-Aldrich, St Louis, MO) at different concentrations (1–50 ng/ml) for 24 h (mRNA quantification) or 48 h (protein quantification). Cultured fibroblasts with PDGF-stimulation were treated with the JAK2-selective inhibitor AG-490 (Tyrphostin B42, Selleckchem, Houston, TX, 20 μM), the PDGF-receptor inhibitor tyrphostin AG-1296 (Selleckchem, 10 μM), the STAT3-inhibitor S3I-201 (Selleckchem, 20, 50, or 100μM) or the non-selective JAK-inhibitor filgotinib (GLPG0634, Selleckchem, 10, 100, or 1000 nM). DMSO-treated fibroblasts served as vehicle controls. Fibroblast number was counted by hemocytometer before plating and after treatment. Supernatants of culture media were collected from the treated cell-cultures and fibroblasts were pelleted by centrifugation, followed by immediate freezing in liquid-N2 for RNA or protein extraction.
2.5 RNA extraction and quantitative real-time polymerase chain reaction analysis
RNA was isolated with mirVana kits (Ambion/Life Technology, Carlsbad, CA). Real-time quantitative polymerase chain reaction (qPCR) was performed with carboxy-fluorescein (FAM)-labelled fluorogenic TaqMan assay primers (Applied Biosystems, Foster City, CA) and TaqMan Universal Master Mix (Applied Biosystems). qPCR for PDGF isoforms A, B, C, and D, JAK1, JAK2, PDGF receptor A (PDGFRA) and B (PDGFRB), STAT3, and collagen isoforms 1-alpha-1 (COL1A1), and 3 (COL3A1) was performed with dog-specific TaqMan primers (Applied Biosystems) and relative quantities (2−ΔCt) calculated with the geometric mean of two reference genes (hypoxanthine phosphoribosyl transferase 1 [HPRT-1] and β2-microglobulin [B2M]) as internal standards.
2.6 Protein extraction and immunoblots
Protein extracts (30-μg) of snap-frozen LA and LV tissue from dogs or cultured fibroblasts were separated by electrophoresis on 12% SDS-PAGE (Bio-Rad) and transferred to PVDF membranes (EMD Millipore, Billerica, MA). For secreted collagen type I, the supernatants of the media (32 μl) from cultured fibroblasts were separated by electrophoresis on 7.5% SDS-PAGE and transferred to nitrocellulose membranes (Bio-Rad). Membranes were blocked and incubated overnight with the primary antibodies and then with secondary antibodies. All of the details regarding antibodies are provided in the Supplementary material online.
2.7 Histology
LA appendage and LV free wall tissues from dogs and entire LAs from mice were preserved in 10% buffered formalin for paraffin embedment. Transverse sections of tissue (12-μm thickness, 500-μm spacing) were stained with Masson Trichrome. Analysis of 400 × images (five to eight images from each animal) was performed with Image-Pro 7.0 software. Perivascular areas were avoided. Fibrous-tissue content was expressed as a percentage of field area and averaged across fields for each dog or mouse.
2.8 Statistical analysis
Data are mean ± S.E.M. For non-repeated measurements, one-way ANOVA was used for multiple-group comparisons, with post-hoc Bonferroni-corrected t-tests (between two selected groups) or Dunnett’s test (for comparisons between each intervention group and a single control group). For repeated measurements, two-way ANOVA with Bonferroni post-tests were used. When a significant interaction or main effect was found, pairwise comparisons were performed with Bonferroni-corrected t-tests. Statistical analyses were performed with GraphPad v5.0 (La Jolla, CA). A two-tailed P < 0.05 was considered statistically significant.
3. Results
3.1 PDGF/JAK-STAT pathway activation in experimental CHF
CHF dogs developed progressive AF susceptibility and atrial fibrosis. AF duration increased significantly from 1-week VTP and atrial-predominant fibrosis was noted (Supplementary material online, Figure S1), consistent with previous work.3,11 The baseline expression levels of PDGFC and PDGFD in atrial fibroblasts were greater than those in ventricular (Supplementary material online, Figure S2A). Fibroblast-selective expression (versus cardiomyocytes) was observed for PDGFB, PDGFC (in LA), and PDGFD. In particular, PDGFD expression was 10-fold higher in LA fibroblasts compared with LA cardiomyocytes (Supplementary material online, Figure S2A). mRNA expression of PDGFA, PDGFC, and PDGFD was upregulated in atrial fibroblasts from CHF dogs, whereas PDGFB did not change significantly (Figure 1A–D). No significant alterations were seen in ventricular fibroblasts, other than an isolated increase in PDGFC at 12-hour VTP (Figure 1A–D). In summary, PDGF isoforms showed fibroblast-selective expression at baseline and atrial-selective increases in CHF fibroblasts at time-points corresponding to in vivo AF-promotion and LA-selective fibrosis development. As PDGF is a secreted protein, we did not perform tissue Western blots.
Figure 1.

PDGF isoforms and PDGFRs were upregulated in CHF dogs. mRNA expression of PDGF A/B/C/D (A–D), PDGFRα and PDGFRβ (E, F) in LA and LV fibroblasts during as a function of VTP time. Results are mean ± S.E.M., n = 5–12/group, *P < 0.05, **P < 0.01, ***P < 0.001, vs. corresponding control (CTL). (G) Representative immunoblots for PDGFRα from atrial and ventricular tissue, and band-intensities for PDGFRα normalized to GAPDH. Mean ± S.E.M., n = 4/group, ***P < 0.001. (H) Representative immunoblots for PDGFRβ from atrial and ventricular tissue, and band-intensities for PDGFRβ normalized to GAPDH. Mean ± S.E.M., n = 5/group, **P < 0.01. One-way ANOVA with Dunnett’s tests (A–F) or Bonferroni-corrected t-tests (G and H) were used for statistical analysis. FB, fibroblast; LA, left atrial; LV, left ventricular; CTL, non-paced controls; CTL-A, control atrium; CTL-V, control ventricle; CHF-A, congestive heart failure atrium; CHF-V, congestive heart failure ventricle.
PDGF receptor α (PDGFRα) and PDGF receptor β (PDGFR β) transcripts were predominantly expressed in fibroblasts compared with cardiomyocytes in both LA and LV (Supplementary material online, Figure S2B). Small, statistically non-significant changes (with the exception of a weakly significant PDGFRα decrease in LA at 2-week VTP) in mRNA expression of PDGFRα and PDGFR β occurred with CHF (Figure 1E and F). Protein expression of PDGFRα in LA-tissue was higher than in LV. However, no significant differences were seen between control and fully developed CHF (2-week VTP; Figure 1G). PDGFRβ showed similar expression levels in control LA versus LV and was increased by CHF in LA (but not LV, Figure 1H). The PDGF and PDGFRβ expression data suggest the involvement of PDGF in atrial-selective fibrogenesis with CHF.
The expression levels of the downstream effectors of PDGF signalling, JAK1/2 and STAT3, in control and CHF fibroblasts are shown in Figure 2. The mRNA expression of JAK1 did not change significantly with CHF (Figure 2A). JAK2 expression, however, was upregulated with significant changes beginning at 1-week VTP, and similar changes in LA and LV fibroblasts (Figure 2B). We were unable to perform successful Western blots for JAK 1 and 2, because the commercially available antibodies (all raised against non-canine epitopes) proved to be inadequate. Protein-expression of phosphorylated and total STAT3 (t-STAT3) were increased by CHF in both LA and LV (Figures 2C–E). The ratio of phosphorylated-STAT3 to total-STAT3 was significantly upregulated, indicating STAT3 activation, in LA but not LV tissue (Figure 2F). A two-fold greater expression of JAK2 was seen in fibroblasts vs. cardiomyocytes (Supplementary material online, Figure S2C), but the baseline expression of STAT3 was similar in fibroblasts and cardiomyocytes. In all, our results show that PDGFA/C/D, PDGFRβ, JAK2, and phosphorylated-STAT3 are upregulated in CHF-dogs, and that the changes are atrial-selective.
Figure 2.

CHF upregulates JAK-STAT components in dogs. (A, B) mRNA expression of JAK1 and JAK2 in LA and LV fibroblasts as a function of VTP time. Mean ± S.E.M., n = 5–12/group, *P < 0.05, **P < 0.01, vs. CTL. (C) Representative immunoblots for phosphorylated STAT3 (p-STAT3) and t-STAT3 from atrial and ventricular tissue. (D–F) Band-intensities for p-STAT3, t-STAT3 (normalized to GAPDH), and phosphorylated-STAT3/t-STAT3 ratio. Mean ± S.E.M., n = 5/group, *P < 0.05, **P < 0.01, ***P < 0.001. One-way ANOVA with Dunnett’s tests (A and B) or Bonferroni-corrected t-tests (D–F) were used for statistical analysis. FB, fibroblast; LA, left atrial; LV, left ventricular; CTL, non-paced controls; CTL-A, control atrium; CTL-V, control ventricle; CHF-A, congestive heart failure atrium; CHF-V, congestive heart failure ventricle.
3.2 JAK-STAT pathway activation and ECM production changes in fibroblasts after PDGF-stimulation
Our results indicate LA-selective enhancement of the expression of PDGF and its β-receptor isoform in CHF. It is tempting to speculate that the PDGF changes are responsible for the JAK-STAT activation that we noted. In order to study the potential effects of the PDGF-system on cardiac fibroblast function and JAK-STAT signalling, freshly isolated fibroblasts from control dogs were cultured and exposed to recombinant PDGF-AB. PDGF-AB caused an augmentation of mRNA-expression of PDGFRα and PDGFRβ (Figure 3A and B). The protein-expression of PDGFRα increased with PDGF-AB exposure (Figure 3C and D), showing preferential responses in LA versus LV at low concentrations (note that effects are shown as fold-changes relative to vehicle-control).
Figure 3.

Exposure to PDGF upregulates PDGFRs in canine fibroblasts. (A,B) mRNA expression of PDGFRα and PDGFRβ in atrial and ventricular fibroblasts stimulated with PDGF-AB at different concentrations. (C) Representative immunoblots for PDGFRα in LA and LV fibroblasts stimulated with PDGF-AB. (D) Band-intensities for PDGFRα normalized to GAPDH and presented as fold-change versus corresponding vehicle. Mean ± S.E.M., n = 4–7/group for A, B, and D. All values were normalized to corresponding vehicle on the same blot for D. *P < 0.05, **P < 0.01, ***P < 0.001 vs. corresponding vehicles. One-way ANOVA with Dunnett’s tests were used for statistical analysis. LA, left atrial; LV, left ventricular; FB, fibroblast; Veh: vehicle; P, PDGF.
PDGF increased the mRNA-expression of JAK2 significantly, to a similar level in LA and LV fibroblasts (Figure 4A), consistent with the increases in JAK2-expression in both LA and LV fibroblasts with CHF (Figure 2B). PDGF increased mRNA expression of STAT3 significantly in LA fibroblasts, without significant changes in LV fibroblasts (Figure 4B). Phosphorylated-STAT3/total-STAT3 ratio increased significantly in LA fibroblasts by 4.2-fold at 5 ng/ml and 5-fold at 10 ng/ml PDGF vs. vehicle (Figure 4C and D). There were no significant changes in LV fibroblasts at either concentration. These results confirm the ability of PDGF to activate the JAK-STAT system in atrial fibroblasts. PDGF-stimulation increased the cell number by 2.2-fold in LA fibroblasts, and by 1.9-fold in LV fibroblasts (Supplementary material online, Figure S3). Fibroblast secretion of collagen type I (Collagen-1) into the culture supernatant was increased by PDGF-stimulation in LA fibroblasts; however, no statistically significant changes were seen in LV fibroblasts (Figure 4E and F). Fibronectin-1 secretion increased in a dose-dependent manner with PDGF-stimulation, and greater changes were seen in LA vs. LV fibroblasts (Figure 4G and H).
Figure 4.

PDGF upregulates JAK2, STAT3 and collagen-1/fibronectin-1 secretion in canine fibroblasts. (A,B) mRNA expression of JAK2 and STAT3 in LA and LV fibroblasts stimulated with PDGF-AB at 10 ng/ml. (C) Representative immunoblots of phosphorylated-STAT3 and total-STAT3 in LA and LV fibroblasts stimulated with PDGF-AB at concentrations indicated. (D) Band-intensities for p-STAT3/t-STAT3 ratio. (E,F) Representative immunoblots and band-intensities (normalized to cell number) of secreted collagen-1 from culture media of fibroblasts stimulated with PDGF-AB for 48 h. (G,H) Representative immunoblots and band-intensities (normalized to cell number) of secreted fibronectin-1 from culture media of fibroblasts stimulated with PDGF-AB for 48 h. Mean ± S.E.M., n = 4–8/group for A, B, D, and F; and n = 3–4/group for H. All values were normalized to corresponding vehicle on the same blot for D, F, and H. *P < 0.05, **P < 0.01 vs. corresponding vehicle. t-tests (A,B) and one-way ANOVA with Dunnett’s tests (D, F, and H) were used for statistical analysis. LA, left atrial; LV, left ventricular; FB, fibroblast; Veh: vehicle; P, PDGF; COL-1, collagen type-I; FN-1, fibronectin-1.
3.3 Effects of JAK-inhibitors on results of PDGF-stimulation in fibroblasts
The actions of PDGF-stimulation to increase the expression of phosphorylated-STAT3 and phosphorylated-STAT3/total-STAT3 ratio in atrial fibroblasts were abolished by the JAK2-selective inhibitor AG-490 or the PDGF-receptor inhibitor AG 1296 (Figure 5A–D). Similarly, PDGF-upregulation of fibroblast proliferation and collagen-1 secretion was reversed by treatment with AG-490 or AG 1296 (Figure 5E–G). These results show that JAK2 is involved in the downstream signalling between PDGF-induced activation via its receptor and atrial fibroblast proliferation/collagen-secretion.
Figure 5.

The JAK2 inhibitor AG490 and PDGF-receptor inhibitor AG1296 reverse PDGF-AB action in canine atrial fibroblasts. (A) Representative immunoblots of p-STAT3 and t-STAT3 in fibroblasts stimulated with PDGF-AB with or without AG490 (20 µM) or AG1296 (10 µM). (B–D) Quantitative protein-expression of p-STAT3, t-STAT3, and p-STAT3/t-STAT3 ratio. (E) Representative immunoblots of secreted collagen-1 from culture media from LA fibroblasts stimulated with PDGF-AB with or without AG490 (20 µM) or AG1296 (10 µM). (F) Fibroblast cell-count (cells/culture-dish) after 48-hour incubation with PDGF-AB with or without AG490 (20 µM) or AG1296 (10 µM). (G) Band-intensities for secreted collagen-1 (normalized to cell number). All values were normalized to controls (without vehicle) on the same blot. Mean ± S.E.M., *P < 0.05, **P < 0.01, ***P < 0.001. n = 8–14/group. One-way ANOVA with Bonferroni-corrected t-tests were used for statistical analysis. COL-1, collagen type I; Veh, vehicle; P, PDGF.
Filgotinib shows selectivity for JAK/STAT signalling through JAK1 at 10 nM.20 At higher concentrations, filgotinib can also inhibit JAK2 (IC50 = 28 nM), TYK2 (IC50 = 116 nM), and JAK3 (IC50 = 810 nM). At a JAK1-selective concentration (10 nM), filgotinib did not affect the changes induced by PDGF-stimulation (Supplementary material online, Figure S4). However, at 100 and 1000 nM, the drug substantially attenuated PDGF effects on STAT3 phosphorylation and fibroblast function. These results suggest that JAK1 is unlikely to contribute to JAK/STAT signalling downstream to PDGF.
3.4 Effects of the STAT3 inhibitor S3I-201 on PDGF-stimulation of fibroblasts
The mRNA expression-levels of STAT3, collagen-1α1, and collagen 3α1 in the presence of PDGF-stimulation were all reduced by S3I-201 treatment in a dose-dependent manner (Figure 6A–C). The effect of PDGF-stimulation on phosphorylated-STAT3 was reversed by S3I-201-treatment, while S3I-201 did not affect the expression level of total-STAT3 (Figure 6D), so that the PDGF-induced increase in the ratio of phosphorylated-STAT3 to total-STAT3 was suppressed by S3I-201 (Figure 6E). Furthermore, S3I-201 reversed the augmentation in collagen-1 secretion caused by PDGF-stimulation (Figure 6F and G). PDGF-exposure and S3I-201 had smaller and statistically non-significant effects on LV fibroblasts (Figure 6F and G), consistent with atrial-selective actions. Our results indicate that the in vitro fibroblast-activating effect of PDGF requires intact STAT3 signalling.
Figure 6.

The STAT3 inhibitor S3I-201 reverses PDGF-AB action in canine fibroblasts. (A–C) mRNA expression of STAT3, collagen 1α1, and collagen 3α1 in fibroblasts stimulated with PDGF-AB and incubated with S3I-201 at different concentrations. n = 3–8/group. Mean ± S.E.M., *P < 0.05, **P < 0.01, ***P < 0.001. (D) Representative immunoblots of p-STAT3 and t-STAT3 in fibroblasts stimulated with PDGF-AB (50 ng/ml) with or without S3I-201 (20 µM). (E) Quantitative protein expression of p-STAT3/t-STAT3 ratio (n = 4/group). (F) Representative immunoblots of secreted collagen-1 from culture media of LA and LV fibroblasts stimulated with PDGF-AB (50ng/ml) with or without S3I-201 (50 µM). (G) Band-intensities for secreted collagen-1 (normalized to cell number). n = 5–8/group. Mean ± SEM, **P < 0.01, vs. Veh, #P < 0.05, ##P < 0.01 vs. corresponding PDGF-treatment. One-way ANOVA with Dunnett’s tests (A–C) or Bonferroni-corrected t-tests (E and G) were used for statistical analysis. COL1A1, collagen 1α1; COL-1, collagen type I COL3A1, collagen 3α1; LA, left atrium; LV, left ventricle; Veh, vehicle; P, PDGF; S3I-20/50/100, S3I-201 at 20/50/100 μM.
3.5 Effects of S3I-201 in mice with post-MI LV dysfunction
Mice studied 2 weeks post-MI showed LV hypertrophy manifested as increased heart weight and increased LV mass/body-weight ratio (black bars in Supplementary material online, Figure S5A and B). Their LVs were dilated (Supplementary material online, Figure S5C and D) and showed significant functional impairment (Supplementary material online, Figure S5E–H). Considerable LA fibrosis was noted (Figure 7A and B) and electrocardiographic P-wave durations were prolonged (Figure 7C), indicating LA conduction abnormalities. The LA was dilated in LV systole and diastole (Figure 7D and E). These results indicate significant LA-disease due to LV dysfunction post-MI. The effects of the STAT3 blocker S3I-201 are illustrated in Figure 7 and Supplementary material online, Figure S5. S3I-201 did not significantly alter post-MI LV remodelling and dysfunction (Supplementary material online, Figure S5). However, adverse LA remodelling post-MI was significantly attenuated, with nearly complete reversal of changes in LA fibrous-tissue content and P-wave duration (Figure 7A–C). LA dilation was modestly but significantly attenuated when measured at LV end-systole (Figure 7D), but not significantly altered at end-diastole (Figure 7E). Overall, these results indicate an important role of the JAK-STAT system in LA structural remodelling due to LV dysfunction in a clinically relevant in vivo model.
Figure 7.
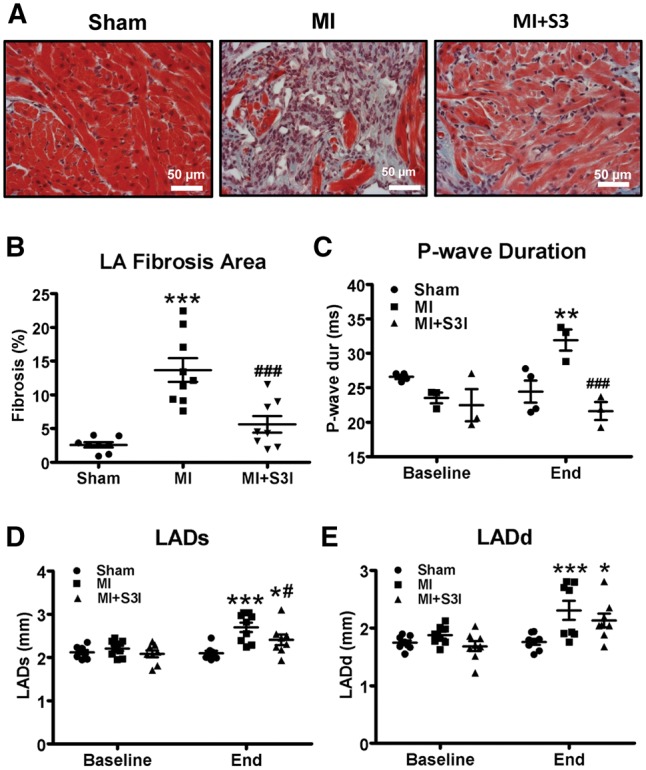
In vivo S3I-201 administration reverses atrial remodelling in myocardial-infarction mice. (A) Representative histological tissue sections (Masson Trichrome staining) of left atria. (B) Quantification of LA fibrous tissue content as %-cross-sectional area. (C) P-wave duration at end-study was decreased by S3I-201 vs. vehicle-control (MI). (D–E) Systolic (LADs) and diastolic (LADd) left-atrial dimension. n = 8–9/group for B, D, and E; n = 3–4/group for C. Mean ± S.E.M., *P < 0.05, **P < 0.01, ***P < 0.001 vs. sham; #P < 0.05, ##P < 0.01, ###P < 0.001 vs. MI. Sham, sham surgery without ligation; MI, myocardial infarction; MI + S3I, myocardial infarction with S3I-201 treatment. Two-way ANOVA with Bonferroni post-tests was used for statistical analysis.
4. Discussion
This study provides new insights into the role of PDGF/JAK-STAT signalling in the fibrotic process leading to the AF-substrate in CHF. Our findings are summarized schematically in Figure 8. We found upregulation of key components of the PDGF/JAK-STAT pathway in CHF-related atrial fibrotic remodelling (Figure 8, red arrows). We noted that PDGF-stimulation of atrial fibroblasts up-regulated JAK-STAT expression and activity, and enhanced ECM-protein production (Figure 8 blue arrows). The profibrotic cellular effects of PDGF were attenuated by the PDGF-receptor inhibitor AG 1296 (Figure 8, orange symbols), the JAK2-selective inhibitor AG-490, the JAK-inhibitor filgotinib at concentrations that inhibit JAK2 (Figure 8, grey symbols), and the STAT3 inhibitor S3I-201 (Figure 8, green symbols). Finally, we showed that in vivo administration of S3I-201 reduces atrial structural, electrical, and fibrotic remodelling in mice with LV-dysfunction post-MI. Our results suggest that the JAK-STAT pathway plays a potentially important role in the atrial fibrosis associated with AF-promoting LV-dysfunction.
Figure 8.

Schematic illustration of our findings. CHF (VTP-dogs or MI-mice) increased PDGF-JAK-STAT components and atrial fibrosis (red upward arrows); PDGF-stimulation activated JAK-STAT system and altered fibroblast behaviour in vitro (blue upward arrows). AG 1296, AG-490, and filgotinib reduced STAT3 activation and collagen secretion in atrial fibroblasts. S3I-201 reduced STAT3 expression/activation and collagen deposition, and attenuated atrial fibrosis (Orange, grey, or green ‘╩’ indicate inhibitory effects).
4.1 JAK-STAT in cardiac remodelling
Mechanical stretch, pressure overload,21 MI,22 and Ang-II treatment23 activate cardiac JAK-STAT signalling. JAK-STAT activation enhances cardiomyocyte survival and reduces apoptosis by regulating the expression of multiple cardioprotective, anti-inflammatory, or growth-related genes.14 The activation of STAT3 during early heart-failure might be a protective response,14 and cardiomyocyte-specific STAT3 knockout mice show enhanced susceptibility to cardiac injury.24 IL-10 and IL-11 attenuate cardiac dysfunction, preventing apoptotic cell death and reducing inflammation, by activating STAT3.22,25 On the other hand, JAK-STAT signalling activated by IL-1β and Ang-II leads to cardiomyocyte hypertrophy in vitro and in the mouse heart with pressure-overload.26,27 S3I-201 protects against Ang-II induced oxidative stress, endothelial dysfunction and hypertension in rats.28 In the present study, S3I-210 attenuated atrial enlargement in mice with LV-dysfunction 2 weeks post-MI, but did not significantly affect ventricular remodelling.
Most studies analysing the effects of JAK-STAT signalling on cardiac cells have been performed in cardiomyocytes, with much less known about JAK-STAT actions in cardiac fibroblasts. JAK-STAT activation was found to correlate with early cell trans-differentiation, proliferation, and differentiation of fibroblasts in human hepatic and cutaneous scar tissue.29,30 Bowman et al.18 pointed out the importance of PDGF-Src-Stat3–Myc signalling in PDGF-induced mitogenesis of NIH 3T3 fibroblasts. Tsai et al.31 found that Ang-II activates STAT3 via Rac1 in atrial cardiomyocytes and fibroblasts. In this study, we noted PDGF/JAK-STAT activation in CHF-induced atrial remodelling and demonstrated that the fibroblast-activating effects of PDGF to enhance proliferation and ECM-protein production are mediated through JAK-STAT activation. JAK-STAT activation was atrial-selective, consistent with the greater degree of fibrosis in LA versus LV (Supplementary material online, Figure S1), and was also selective for fibroblasts over cardiomyocytes. Our findings indicate that the JAK-STAT system is an important regulator of atrial fibroblast behaviour.
4.2 JAK-STAT in atrial fibrosis associated with AF
Tsai et al.19 showed that STAT1 and STAT3 were both activated in pigs with pacing-inducing sustained AF. Xue et al.32 showed that STAT3 protein expression was increased in atrial tissue from permanent AF patients with rheumatic heart disease compared with those in sinus rhythm, paralleling changes in atrial fibrous tissue content. Neither study addressed the role of STAT3-activation by blocking the system in vivo. Our finding that STAT3 inhibition reduces atrial fibrosis associated with LV-dysfunction in post-MI mice indicates the functional importance of STAT3-change in AF. This result is consistent with observations that STAT3 inhibition has anti-fibrotic effects in other systems. For example, Pang et al.33 showed that inhibition of STAT3 attenuates renal interstitial fibroblast-activation and interstitial fibrosis in obstructive nephropathy. Mir et al.34 noted that inhibition of STAT3 attenuates collagen synthesis and hypertrophy in a rat cardiac hypertrophy model.
4.3 Novelty and potential significance
To the best of our knowledge, this is the first study to elucidate the role of the JAK-STAT system in AF-promoting atrial fibrotic remodelling. Although the JAK-STAT system is indicated in schemas of potential profibrotic pathways in AF,2 the pathway has not previously been directly addressed in studies of atrial profibrillatory fibrotic remodelling. JAK-STAT signalling can be activated downstream to either Ang-II or PDGF receptors; the present study points to PDGF as an important component of the atrial profibrotic JAK-STAT axis.
This is also the first study to demonstrate atrial-selective activation of the PDGF/JAK-STAT system in cardiac remodelling. In addition to PDGF, various other profibrotic pathways, such as those mediated via Ang-II, IL-6, and CTGF, can involve the participation of JAK-STAT.29,31 Ang-II activates JAK-STAT through Rac1 in rat fibroblasts,31 and CTGF induces JAK-STAT activation in human hypertrophic scar fibroblasts.29 JAK2 activation also leads to the phosphorylation of other proteins such as MAPKs.35 In addition, JAK-STAT signalling represents one limb of an autocrine loop for Ang-II generation, which serves to amplify the action of Ang-II in cardiac remodelling.36 Therefore, the JAK-STAT system is well positioned to serve as a central mediator in fibrotic responses and contribute to atrial remodelling.
Although PDGF is secreted by both fibroblasts and cardiomyocytes, PDGF receptors are predominantly expressed in fibroblasts (Supplementary material online, Figure S2B). Thus, the PDGF/JAK-STAT system is a potential target for fibroblast-selective treatment. PDGF-receptor inhibitors attenuate fibrosis in multiple studies10,37,38 and suppress atrial-selective cardiac fibroblast activation.11 However, most PDGFR inhibitors are non-selective tyrosine kinase inhibitors, limiting their clinical use. Therefore, the identification of JAK-STAT as an alternative target, and the development of JAK-STAT inhibitors like S3I-201, may point to novel therapeutic approaches for AF-prevention.
4.4 Potential limitations
It has been shown that during myocardial fibrosis in salt-sensitive hypertensive rats, PDGFRα acts at early stages, but PDGFRβ function is enhanced throughout the remodelling process.9 This might explain why we observed upregulation of PDGFRβ protein in CHF but no changes in PDGFRα (Figure 1G and H), as we analysed tissue samples from dogs with 2-week VTP only because of a lack of protein-sample availability from earlier time-points. In addition, whereas PDGFRβ protein increased significantly in dog atrium (Figure 1H), PDGFRB mRNA did not change significantly (Figure 1F). This discrepancy between mRNA and protein results may be due to post-transcriptional regulation (e.g. by microRNA).
In this study, we focused on changes in fibroblasts since we were interested in the role of the JAK-STAT system in profibrillatory fibrotic remodelling. We did study the evolution of JAK2 and STAT3 mRNA expression over VTP time in cardiomyocytes from CHF dogs. As shown in Supplementary material online, Figure S6, the only change we saw was an isolated increase in STAT3 expression at 12-h VTP. However, a detailed exploration of possible JAK-STAT changes in CHF cardiomyocytes and its importance is beyond the scope of this study. Similarly, it is possible that JAK-STAT signalling involves downstream pathways additional to those that we examined here. Atrial fibrosis is known to play an important role in AF pathophysiology, particularly in the model we studied here,3,5 but JAK-STAT signalling could also be acting via the regulation of cardiomyocyte ion channels or Ca2+ handling.
The mouse studies were conducted as a proof of principle for the involvement of STAT3-signalling in atrial fibrosis associated with myocardial dysfunction. Because of the small size of mouse atria, all of the LA tissue samples from mice were used for Masson Trichrome staining (Figure 7A). We did have limited right-atrial tissue samples on which we performed Western blots for PDGFRα, PDGFRβ, STAT3 and phosphorylated-STAT3. The PDGFR changes were in qualitative agreement with the dog data but no statistically significant differences were seen (Supplementary material online, Figure S7), and the STAT3 data showed no consistent trends (Supplementary material online, Figure S8). The discrepancies from our dog data may be due to the fact that the mouse work was performed in right atrium, which may be much less affected by MI than LA, and/or to species differences in relative contributions of fibroblasts versus cardiomyocytes to atrial-tissue expression of PDGFRs and STAT3. Since fibroblasts comprise only a small part of the myocardial mass, contamination by other cell types could account for the lack of consistent changes. We did not have fibroblast samples from the intervention study in the mice, and even if we repeated the full experimental series the quantity of freshly isolated fibroblasts from the very small mouse atrium would not have been enough to perform Western blots.
The role of atrial JAK-STAT signalling pathways in CHF patients remains to be investigated. We were unable to perform such studies because of a lack of human atrial samples from patients with CHF, but we hope to investigate this issue in future work.
STAT3 regulates a broad range of genes controlling cell survival, cell cycle progression, inflammation, ECM homeostasis and cell-to-cell communication. Thus, STAT3-inhibition may have significant extra-cardiac effects on other cell types and organs; thus, systemic STAT3 inhibition might not be a viable anti-AF strategy because of adverse effect potential.39 No toxic effects of STAT3 inhibition were seen in MI mice of our study, or reported in other animal models of cardiac hypertrophy or tissue fibrosis.40,41 However, the safety profile of STAT3 inhibition in other animal models and with longer term administration remains to be assessed. Heart-specific or cardiac fibroblast-targeting STAT3-inhibition, e.g. by cardiac-directed biological therapy, might be a possible approach to achieve efficacy without unwanted effects. Although we demonstrated that STAT3 activation causes fibroblast proliferation and increases ECM production, we did not study the downstream mechanisms of STAT3 fibroblast regulation. Further work is thus needed to identify the range of STAT-responsive genes and signalling pathways responsible for the contribution of STAT3 to atrial fibrogenesis.
Although S3I-201 is a widely used JAK-STAT inhibitor, like all pharmacological agents there are concerns about specificity. It would be of interest to use mice with genetically inactivated JAK-STAT signalling to test our hypothesis. Systemic genetic knockdown of STAT3 might produce indirect effects mediated by JAK-STAT ventricular cardiomyocyte signalling. In vivo inducible genetic inhibition of STAT3 in atrial fibroblasts would be of great interest but is technically challenging and out of the scope of this study.
Supplementary material
Supplementary material is available at Cardiovascular Research online.
Supplementary Material
Acknowledgements
The authors wish to thank Nathalie L’Heureux and Chantal St-Cyr for expert technical assistance.
Conflict of interest: none declared.
Funding
This work was supported by the Canadian Institutes of Health Research (MGP6957), the Quebec Heart and Stroke Foundation, and the Foundation Leducq (European-North American Atrial Fibrillation Research Alliance, ENAFRA).
References
- 1. Corradi D, Callegari S, Maestri R, Benussi S, Alfieri O.. Structural remodelling in atrial fibrillation. Nat Clin Pract Cardiovas Med. 2008;5:782–796. [DOI] [PubMed] [Google Scholar]
- 2. Burstein B, Nattel S.. Atrial structural remodelling as an antiarrhythmic target. J Cardiovasc Pharmacol 2008;52:4–10. [DOI] [PubMed] [Google Scholar]
- 3. Burstein B, Nattel S.. Atrial fibrosis: mechanisms and clinical relevance in atrial fibrillation. J Am Coll Cardiol 2008;51:802–809. [DOI] [PubMed] [Google Scholar]
- 4. Pellman J, Lyon RC, Sheikh F.. Extracellular matrix remodelling in atrial fibrosis: mechanisms and implications in atrial fibrillation. J Mol Cell Cardiol 2010;48:461–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Nattel S, Burstein B, Dobrev D.. Atrial remodelling and atrial fibrillation: mechanisms and implications. Circ Arrhythm Electrophysiol 2008;1:62–73. [DOI] [PubMed] [Google Scholar]
- 6. Creemers EE, Pinto YM.. Molecular mechanisms that control interstitial fibrosis in the pressure-overloaded heart. Cardiovasc Res 2011;89:265–272. [DOI] [PubMed] [Google Scholar]
- 7. Lin CS, Pan CH.. Regulatory mechanisms of atrial fibrotic remodelling in atrial fibrillation. Cell Mol Life Sci. 2008;65:1489–1508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Booz GW, Baker KM.. Molecular signaling mechanisms controlling growth and function of cardiac fibroblasts. Cardiovasc Res 1995;30:537–543. [PubMed] [Google Scholar]
- 9. Fan B, Ma L, Li Q, Wang L, Zhou J, Wu J.. Role of PDGFs/PDGFRs signaling pathway in myocardial fibrosis of DOCA/salt hypertensive rats. Int J Clin Exp Pathol 2014;7:16–27. [PMC free article] [PubMed] [Google Scholar]
- 10. Liu C, Zhao W, Meng W, Zhao T, Chen Y, Ahokas RA, Liu H, Sun Y.. Platelet-derived growth factor blockade on cardiac remodelling following infarction. Mol Cell Biochem 2014;397:295–304. [DOI] [PubMed] [Google Scholar]
- 11. Burstein B, Libby E, Calderone A, Nattel S.. Differential behaviors of atrial versus ventricular fibroblasts: a potential role for platelet-derived growth factor in atrial-ventricular remodelling differences. Circulation 2008;117:1630–1641. [DOI] [PubMed] [Google Scholar]
- 12. Mulvany MJ. Small artery remodelling and significance in the development of hypertension. News Physiol Sci 2002;17:105–109. [DOI] [PubMed] [Google Scholar]
- 13. Ogawa A, Firth AL, Smith KA, Maliakal MV, Yuan JX.. PDGF enhances store-operated Ca2+ entry by upregulating STIM1/Orai1 via activation of Akt/mTOR in human pulmonary arterial smooth muscle cells. Am J Physiol Cell Physiol 2012;302:C405–C411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kishore R, Verma SK.. Roles of STATs signaling in cardiovascular diseases. JAKSTAT 2012;1:118–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mohri T, Iwakura T, Nakayama H, Fujio Y.. JAK-STAT signaling in cardiomyogenesis of cardiac stem cells. JAKSTAT 2012;1:125–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Patel BK, Wang LM, Lee CC, Taylor WG, Pierce JH, LaRochelle WJ.. Stat6 and Jak1 are common elements in platelet-derived growth factor and interleukin-4 signal transduction pathways in NIH 3T3 fibroblasts. J Biol Chem 1996;271:22175–22182. [DOI] [PubMed] [Google Scholar]
- 17. Vignais ML, Sadowski HB, Watling D, Rogers NC, Gilman M.. Platelet-derived growth factor induces phosphorylation of multiple JAK family kinases and STAT proteins. Mol Cell Biol 1996;16:1759–1769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bowman T, Broome MA, Sinibaldi D, Wharton W, Pledger WJ, Sedivy JM, Irby R, Yeatman T, Courtneidge SA, Jove R.. Stat3-mediated Myc expression is required for Src transformation and PDGF-induced mitogenesis. Proc Natl Acad Sci U S A 2001;98:7319–7324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tsai CT, Lin JL, Lai LP, Lin CS, Huang SK.. Membrane translocation of small GTPase Rac1 and activation of STAT1 and STAT3 in pacing-induced sustained atrial fibrillation. Heart Rhythm 2008;5:1285–1293. [DOI] [PubMed] [Google Scholar]
- 20. Van Rompaey L, Galien R, van der Aar EM, Clement-Lacroix P, Nelles L, Smets B, Lepescheux L, Christophe T, Conrath K, Vandeghinste N, Vayssiere B, De Vos S, Fletcher S, Brys R, van 't Klooster G, Feyen JH, Menet C.. Preclinical characterization of GLPG0634, a selective inhibitor of JAK1, for the treatment of inflammatory diseases. J Immunol 2013;191:3568–3577. [DOI] [PubMed] [Google Scholar]
- 21. Pan J, Fukuda K, Saito M, Matsuzaki J, Kodama H, Sano M, Takahashi T, Kato T, Ogawa S.. Mechanical stretch activates the JAK/STAT pathway in rat cardiomyocytes. Circ Res 1999;84:1127–1136. [DOI] [PubMed] [Google Scholar]
- 22. Krishnamurthy P, Rajasingh J, Lambers E, Qin G, Losordo DW, Kishore R.. IL-10 inhibits inflammation and attenuates left ventricular remodelling after myocardial infarction via activation of STAT3 and suppression of HuR. Circ Res 2009;104:e9–e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Marrero MB, Schieffer B, Paxton WG, Heerdt L, Berk BC, Delafontaine P, Bernstein KE.. Direct stimulation of Jak/STAT pathway by the angiotensin II AT1 receptor. Nature 1995;375:247–250. [DOI] [PubMed] [Google Scholar]
- 24. Jacoby JJ, Kalinowski A, Liu MG, Zhang SS, Gao Q, Chai GX, Ji L, Iwamoto Y, Li E, Schneider M, Russell KS, Fu XY.. Cardiomyocyte-restricted knockout of STAT3 results in higher sensitivity to inflammation, cardiac fibrosis, and heart failure with advanced age. Proc Natl Acad Sci U S A 2003;100:12929–12934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Obana M, Maeda M, Takeda K, Hayama A, Mohri T, Yamashita T, Nakaoka Y, Komuro I, Takeda K, Matsumiya G, Azuma J, Fujio Y.. Therapeutic activation of signal transducer and activator of transcription 3 by interleukin-11 ameliorates cardiac fibrosis after myocardial infarction. Circulation 2010;121:684–691. [DOI] [PubMed] [Google Scholar]
- 26. Honsho S, Nishikawa S, Amano K, Zen K, Adachi Y, Kishita E, Matsui A, Katsume A, Yamaguchi S, Nishikawa K, Isoda K, Riches DW, Matoba S, Okigaki M, Matsubara H.. Pressure-mediated hypertrophy and mechanical stretch induces IL-1 release and subsequent IGF-1 generation to maintain compensative hypertrophy by affecting Akt and JNK pathways. Circ Res 2009;105:1149–1158. [DOI] [PubMed] [Google Scholar]
- 27. Chang L, Yang R, Wang M, Liu J, Wang Y, Zhang H, Li Y.. Angiotensin II type-1 receptor-JAK/STAT pathway mediates the induction of visfatin in angiotensin II-induced cardiomyocyte hypertrophy. Am J Med Sci 2012;343:220–226. [DOI] [PubMed] [Google Scholar]
- 28. Ma Y, Li H, Yue Z, Guo J, Xu S, Xu J, Jia Y, Yu N, Zhang B, Liu S, Liu M, Shao W, Chen S, Liu P.. Cryptotanshinone attenuates cardiac fibrosis via downregulation of COX-2, NOX-2, and NOX-4. J Cardiovasc Pharmacol 2014;64:28–37. [DOI] [PubMed] [Google Scholar]
- 29. Lakner AM, Moore CC, Gulledge AA, Schrum LW.. Daily genetic profiling indicates JAK/STAT signaling promotes early hepatic stellate cell transdifferentiation. World J Gastroenterol 2010;16:5047–5056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tao L, Liu J, Li Z, Dai X, Li S.. Role of the JAK-STAT pathway in proliferation and differentiation of human hypertrophic scar fibroblasts induced by connective tissue growth factor. Mol Med Rep 2010;3:941–945. [DOI] [PubMed] [Google Scholar]
- 31. Tsai CT, Lai LP, Kuo KT, Hwang JJ, Hsieh CS, Hsu KL, Tseng CD, Tseng YZ, Chiang FT, Lin JL.. Angiotensin II activates signal transducer and activators of transcription 3 via Rac1 in atrial myocytes and fibroblasts: implication for the therapeutic effect of statin in atrial structural remodelling. Circulation 2008;117:344–355. [DOI] [PubMed] [Google Scholar]
- 32. Xue XD, Huang JH, Wang HS.. Angiotensin II activates signal transducers and activators of transcription 3 via Rac1 in the atrial tissue in permanent atrial fibrillation patients with rheumatic heart disease. Cell biochem Biophys 2015;71:205–213. [DOI] [PubMed] [Google Scholar]
- 33. Pang M, Ma L, Gong R, Tolbert E, Mao H, Ponnusamy M, Chin YE, Yan H, Dworkin LD, Zhuang S.. A novel STAT3 inhibitor, S3I-201, attenuates renal interstitial fibroblast activation and interstitial fibrosis in obstructive nephropathy. Kidney Int 2010;78:257–268. [DOI] [PubMed] [Google Scholar]
- 34. Mir SA, Chatterjee A, Mitra A, Pathak K, Mahata SK, Sarkar S.. Inhibition of signal transducer and activator of transcription 3 (STAT3) attenuates interleukin-6 (IL-6)-induced collagen synthesis and resultant hypertrophy in rat heart. Biol Chem 2012;287:2666–2677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Rawlings JS, Rosler KM, Harrison DA.. The JAK/STAT signaling pathway. J Cell Sci 2004;117:1281–1283. [DOI] [PubMed] [Google Scholar]
- 36. Booz GW, Day JN, Baker KM.. Interplay between the cardiac renin angiotensin system and JAK-STAT signaling: role in cardiac hypertrophy, ischemia/reperfusion dysfunction, and heart failure. J Mol Cell Cardiol 2002;34:1443–1453. [DOI] [PubMed] [Google Scholar]
- 37. Leipner C, Grun K, Muller A, Buchdunger E, Borsi L, Kosmehl H, Berndt A, Janik T, Uecker A, Kiehntopf M, Bohmer FD.. Imatinib mesylate attenuates fibrosis in coxsackievirus b3-induced chronic myocarditis. Cardiovasc Res 2008;79:118–126. [DOI] [PubMed] [Google Scholar]
- 38. Jang SW, Ihm SH, Choo EH, Kim OR, Chang K, Park CS, Kim HY, Seung KB.. Imatinib mesylate attenuates myocardial remodelling through inhibition of platelet-derived growth factor and transforming growth factor activation in a rat model of hypertension. Hypertension 2014;63:1228–1234. [DOI] [PubMed] [Google Scholar]
- 39. Haghikia A1, Ricke-Hoch M, Stapel B, Gorst I, Hilfiker-Kleiner D.. STAT3, a key regulator of cell-to-cell communication in the heart. Cardiovasc Res 2014;102:281–289. [DOI] [PubMed] [Google Scholar]
- 40. Zhang L, Xu X, Yang R, Chen J, Wang S, Yang J, Xiang X, He Z, Zhao Y, Dong Z, Zhang D.. Paclitaxel attenuates renal interstitial fibroblast activation and interstitial fibrosis by inhibiting STAT3 signaling. Drug Des Devel Ther 2015;9:2139–2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Mir SA, Chatterjee A, Mitra A, Pathak K, Mahata SK, Sarkar S.. Inhibition of signal transducer and activator of transcription 3 (STAT3) attenuates interleukin-6 (IL-6)-induced collagen synthesis and resultant hypertrophy in rat heart. J Biol Chem 2012;287:2666–2677. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.