

Abstract
Hyperekplexia, an inherited neuronal disorder characterized by exaggerated startle responses with unexpected sensory stimuli, is caused by dysfunction of glycinergic inhibitory transmission. From analysis of newly identified human hyperekplexia mutations in the glycine receptor (GlyR) α1 subunit, we found that an alanine-to-proline missense mutation (A384P) resulted in substantially higher desensitization level and lower agonist sensitivity of homomeric α1 GlyRs when expressed in HEK cells. The incorporation of the β subunit fully reversed the reduction in agonist sensitivity and partially reversed the desensitization of α1A384P. The heteromeric α1A384Pβ GlyRs showed enhanced desensitization but unchanged agonist-induced maximum responses, surface expression, main channel conductance, and voltage dependence compared with that of the wild-type α1β (α1WTβ) GlyRs. Coexpression of the R392H and A384P mutant α1 subunits, which mimic the expression of the compound heterozygous mutation in a hyperekplexia patient, resulted in channel properties similar to those with α1A384P subunit expression alone. In comparison, another human hyperekplexia mutation α1P250T, which was previously reported to enhance desensitization, caused a strong reduction in maximum currents in addition to the altered desensitization. These results were further confirmed by overexpression of α1P250T or α1A384P subunits in cultured neurons isolated from SD rats of either sex. Moreover, the IPSC-like responses of cells expressing α1A384Pβ induced by repeated glycine pulses showed a stronger frequency-dependent reduction than those expressing α1WTβ. Together, our findings demonstrate that A384 is associated with the desensitization site of the α1 subunit and its proline mutation produced enhanced desensitization of GlyRs, which contributes to the pathogenesis of human hyperekplexia.
SIGNIFICANCE STATEMENT Human startle disease is caused by impaired synaptic inhibition in the brainstem and spinal cord, which is due to either direct loss of GlyR channel function or reduced number of synaptic GlyRs. Considering that fast decay kinetics of GlyR-mediated inhibitory synaptic responses, the question was raised whether altered desensitization of GlyRs will cause dysfunction of glycine transmission and disease phenotypes. Here, we found that the α1 subunit mutation A384P, identified from startle disease patients, results in enhanced desensitization and leads to rapidly decreasing responses in the mutant GlyRs when they are activated repeatedly by the synaptic-like simulation. These observations suggest that the enhanced desensitization of postsynaptic GlyRs could be the primary pathogenic mechanism of human startle disease.
Keywords: cys-loop receptors, desensitization, glycine receptor, hyperekplexia
Introduction
Mutation in the glycine receptor (GlyR) α1 subunit is one of the major causes of human hyperekplexia, an inherited neurological disease characterized by exaggerated startle responses triggered by unexpected stimuli (Bode and Lynch, 2014). Understanding the functional roles of these mutations will not only uncover physiological roles of GlyRs at the behavioral and synaptic levels, but also bridge a connection between the molecular structure and functional output of GlyRs. Being the primary genetic target for hyperekplexia, new GlyR α1 mutations from patients have been reported (Bode and Lynch, 2014). However, functional analysis of these mutant GlyRs is still lagging behind, and the association between the mechanistic aspects of channel function and the pathogenesis of hyperekplexia remains largely unexplored.
Since the first identified mutation, R271L/Q, from hereditary hyperekplexia patients, researchers have revealed critical sites in the GlyR α1 subunit for channel gating, desensitization, allosteric modulation, and membrane expression (Shiang et al., 1993; Langosch et al., 1994; Rajendra et al., 1995; Villmann et al., 2009; Zhou et al., 2013; Schaefer et al., 2015). Functional analysis of missense hyperekplexia mutations and systematic screening of the flanking segments have helped to uncover the amino acid residues crucial to GlyR function. For example, the α1 subunit R271 residue, adjacent transmembrane segment 2 (TM2) and extracellular TM2-TM3 linker are involved in channel gating (Langosch et al., 1994; Rajendra et al., 1995). The intracellular TM1-TM2 linker containing the P250 residue is involved in both channel gating and desensitization (Saul et al., 1999). Loop D and β-strands 2–3 in the extracellular domain, including residues W68, D70, and R72, have been shown to determine forward trafficking of GlyRs to the neuronal surface (Schaefer et al., 2015). From these and other human mutations, it has been established that deficits in agonist affinity, channel gating, and membrane expression could directly impair the amplitude of GlyR-mediated currents at synapses, and were thereby causally linked to the pathogenesis of hyperekplexia. Channel desensitization is also an important electrophysiological property that is involved in shaping the time course of fast synaptic transmission and controlling current flow (Jones and Westbrook, 1996; Keramidas and Lynch, 2013; Zhang et al., 2013). However, GlyR mutations known to affect desensitization, including P250T and P230S, are also associated with deficits in other channel properties, such as agonist sensitivity or maximum responses (Saul et al., 1999; Bode et al., 2013). Therefore, whether desensitization could be the primary causative link to hyperekplexia is still unclear.
Interestingly, unlike glutamate receptors, the intracellular loops are considered one of the key entities for desensitization in several types of Cys-loop receptors, including GABAA receptors (GABAARs), 5-HT3A receptors and GlyRs (Hu et al., 2006; McKinnon et al., 2012; Gielen et al., 2015). In GlyRs, the TM1-TM2 linker is important for desensitization because several missense mutations in this segment affect desensitization (Lynch et al., 1997; Saul et al., 1999; Breitinger et al., 2001). Moreover, a group of studies has suggested involvement of the large TM3-TM4 intracellular loop (Nikolic et al., 1998; Papke and Grosman, 2014; Langlhofer et al., 2015), and a recent systematic study has further narrowed down the desensitization gate to the proximal region of the cytoplasmic end of TM3 (Gielen et al., 2015). Interestingly, TM4 does not appear to have a role because incorporating TM4 of GlyR α1 into the nondesensitized ρ1 GABAAR did not produce faster desensitization. However, as TM4 and its neighboring intracellular segment participate in the control of channel conductance and desensitization in other Cys-loop receptors (e.g., the 5-HT3A receptor) (Kelley et al., 2003; Hu et al., 2006), it is worth considering that the TM4 adjacent regions also contribute to GlyR desensitization.
In the present study, we performed functional analysis of a newly identified hyperekplexia mutation α1A384P located in the TM3-TM4 linker (Mine et al., 2015). Interestingly, we found that α1A384P strongly enhanced GlyR desensitization, indicating a new desensitization site at the cytoplasmic end of TM4 and a link between GlyR desensitization and the cause of human hyperekplexia.
Materials and Methods
cDNA constructs and transfection.
Wild-type (WT) human GlyR α1 (glra1) and GlyR β (glrb) cDNAs were subcloned into the pBK-CMV NB-200 expression vector (Liu et al., 2010; Zhou et al., 2013). Mutant α1 subunit plasmids were constructed by site-directed mutagenesis. The sequences of all plasmids were confirmed by automated DNA sequencing. HEK293T cells (6 × 105) were transfected with purified plasmids encoding the WT or mutant GlyR α1 subunit alone or with β subunits (1:5; total plasmid amount 1.2–2.5 μg) and were delivered into HEK293T cells either by using X-tremeGENE9 DNA transfection Reagent (Roche Diagnostics) or electroporation (NEPA21, NEPA GENE). A small amount (0.2 μg) of pcDNA3-GFP was cotransfected along with GlyR subunits to act as a transfection marker and facilitate the visualization of transfected cells during electrophysiological experiments. After transfection, cells were replated on poly-D-lysine-coated glass coverslips and were grown in DMEM in 24-well plates for 16–48 h before patch-clamp recordings. After all experiments, except single-channel recording and pulse application experiments, the α1 and α1β subunit compositions of GlyRs were confirmed by their shifted sensitivity to picrotoxin.
Neuronal culture and overexpression.
Cultured DRG neurons were prepared from DRGs of adult SD rats of either sex while deeply anesthetized with urethane. The spinal column was excised, and the DRGs were dissected out carefully. The meninges were removed, and DRGs were cut into small even pieces and incubated with 1% collagenase opti-MEM solution for 1 h and combined with 0.1% typsin for another 30 min. All isolated DRG neurons were centrifuged at 3000 rpm for 5 min and resuspended in 150 μl opti-MEM for electroporation with GlyR WT or A384P mutant α1 subunits with or without β subunits. Electroporation was performed by using NEPA21 electroporator with the following parameters: 2 pulses with 50 ms intervals (pulse voltage: 250 V, pulse length 1 ms) for poring pulse; 5 pulses with 50 ms interval (pulse voltage: 20 V, pulse length 50 ms) for transfer pulse. After electroporation, DRG neurons were seeded on coverslips precoated with poly-D-lysine. Electrophysiological recordings were performed after seeding 48 h.
Cultured cortical neurons were prepared from neocortex of day 18 fetal SD rats (from embryos of either sex) as described previously (Liu et al., 2010). Rat cerebral cortex was digested with a 0.25% trypsin solution (w/v) (Invitrogen) for 20 min at 37°C and then mechanically dissociated using a fire-polished Pasteur pipette. The cell suspension was then centrifuged at 2000 × g for 10 min, and the cell pellets were resuspended in DMEM containing 10% FBS (v/v). Cells were seeded onto poly-D-lysine-coated 24-well coverslips at a density of 6 × 104 cells per well. Cultures were maintained in a humidified incubator with 5% CO2 at 37°C. After 2 h, plating medium was changed to Neurobasal medium supplemented with B-27 and l-glutamine. The media was changed twice weekly thereafter. Three days after plating, neurons were transiently transfected with WT or mutant α1 subunits, β subunits (1:5), and GFP (total plasmid amount 1–1.5 μg) with Lipofectamine 2000 according to the manufacturer's protocols. Electrophysiological recordings were performed 24–48 h after transfection.
Whole-cell patch-clamp recordings.
Whole-cell recordings were performed under voltage-clamp mode using an Axopatch 200B (Molecular Devices) as described previously (Zhou et al., 2013). Whole-cell currents were recorded with a holding potential of −60 mV, and signals were low-pass filtered at 2 kHz and digitized at 10 kHz (Digidata 1440A). Recording pipettes (3–5 mΩ) were filled with intracellular solution that contained the following (in m): 140 CsCl, 10 HEPES, 4 Mg-ATP, and 0.5 BAPTA (pH 7.20, osmolarity, 290–295 mOsm). The coverslips were continuously superfused with the extracellular solution containing the following (in m): 140 NaCl, 5.4 KCl, 10 HEPES, 1.0 MgCl2, 1.3 CaCl2, and 20 glucose, pH 7.4 (305–315 mOsm). To evoke glycine currents, we used fast perfusion of glycine and other agonists with a computer-controlled multibarrel fast perfusion system (Warner Instruments). All experiments were performed at 23°C–25°C. Maximum currents (Imax) were evoked by the saturated concentration of the agonist as determined by the concentration-response curves.
Single-channel recording.
Single-channel currents from cell-attached patches were recorded in an external solution containing the following (in m): 140 NaCl, 5 KCl, 1 MgCl2, 2 CaCl2, 10 glucose, and 10 HEPES, pH 7.4. During recording, 1 m glycine was present in the pipette solution that consisted of the following (in m): 120 NaCl, 5 KCl, 10 MgCl2, 0.1 CaCl2, 10 glucose, and 10 HEPES, pH 7.4. Micropipette potential was held at 100 mV. Single-channel currents were amplified and low-pass filtered at 2 kHz using an Axopatch 200B amplifier, digitized at 20 kHz using Digidata 1550, and saved using pCLAMP 10.4 (Molecular Devices). Data were analyzed offline using TAC 4.2 and TACFit 4.2 (Bruxton) software. Single-channel open and closed events were analyzed using the 50% threshold detection method and visually inspected before accepting the events. Single-channel openings occurred as bursts of one or more openings or clusters of bursts. Duration and amplitude histograms were generated using TACFit 4.2 (Bruxton). Single-channel amplitudes (i) were calculated by fitting all-point histograms with single- or multi-Gaussian curves. The difference between the fitted “closed” and “open” peaks was taken as i. Duration histograms were fitted with exponential components in the form: ∑(ai/τi)exp(−t/τi), where a and τ represent the relative area and time constant of the ith component, respectively, and t is the time.
Surface biotinylation.
HEK293T cells were transfected with α1, β, and GFP at a ratio of 1:5:0.5, and surface biotinylation was performed 24 h after transfection. The cells were rinsed with cold PBS and incubated with PBS containing 0.5 mg/ml Sulfo-NHS-SS-biotin (Thermo Fisher Scientific) for 1 h at 4°C with gentle shaking. Nonreacted biotinylation reagent was quenched by 100 m glycine in PBS, and cells were lysed in RIPA buffer (150 m NaCl, 50 m Tris-HCl, 1 m EDTA, 1% Triton X-100, pH 7.4) supplied with protease mixture (Roche Diagnostics). After centrifugation, the supernatant containing total cell proteins was collected, and 20 μg of total protein was subsequently incubated with NeutrAvidin Agarose (Thermo Fisher Scientific) overnight at 4°C. The precipitated beads were collected by centrifuge and resuspended with loading buffer for detecting the surface proteins. The supernatant was collected as the cytosol fraction. The fractionated proteins, together with an aliquot of 20 μg of total proteins, were then subjected to SDS-PAGE, and Western blots were incubated with antibodies raised against GlyRα1 subunits (Enzo) and GFP (Gentex). The amount of GlyRα1 and GFP proteins was detected by ECL (Advansta) and analyzed by ImageJ software.
Experimental design and data analysis.
The WT or the A384P mutation-containing GlyRs were overexpressed in HEK cells or cultured neurons, and their electrophysiological properties were examined by patch-clamp recordings. Values were expressed as mean ± SEM. One-way ANOVA or a two-tailed Student's t test was used for statistical analysis, and p values <0.05 were considered to be statistically significant. Imax was determined as the amplitude of peak currents induced by saturated concentration or indicated concentration of agonists. Concentration-response curves were created by fitting data to the Hill equation: I = Imax/[1 + (EC50/[A])nH], where I is the current, [A] is a given concentration of agonist, and nH is the Hill coefficient (GraphPad, Prism 6). Weighted time constants for desensitization were calculated by fitting the desensitization currents with one or two exponential components by Clampfit 10 (Molecular Devices). The extent of desensitization was calculated with the equation: % desensitization = 1 − Ires/Ipeak, where Ires is the residual currents at the end of agonist application and Ipeak is the peak amplitude of the current. According to our previous study (Zhou et al., 2013), young cortical neurons mainly express α2 subunits, which exhibit slower activation kinetics (mean 10%–50% rise time >200 ms). Neurons overexpressing α1 subunits showed significantly faster activation kinetics (mean 10%–50% rise time <100 ms) and 10%–50% rise time was analyzed by Clampfit. Therefore, only neurons that had glycine currents with rise times <100 ms were included for further analysis. All 3D images were rendered by using the UCSF Chimera package.
Results
Receptor desensitization was strongly enhanced in homomeric α1A384P and heteromeric α1A384Pβ GlyRs
Functional GlyRs can be formed by either homomeric assembly of α1 subunits or heteromeric assembly of α1 and β subunits, the latter of which represents the endogenous form of GlyRs at postsynaptic sites (Lynch, 2004). We performed whole-cell recordings to assess the functions of α1A384P subunit-containing GlyRs overexpressed in HEK cells. Interestingly, homomeric α1A384P subunit-containing GlyR-mediated whole-cell currents revealed drastically enhanced receptor desensitization during a 3 s application of glycine at saturating concentration (10 m; Fig. 1A,B). The desensitization profile was evaluated by two properties: the weighted desensitization time constant (τW) and the extent of desensitization (% desensitization). We found that homomeric α1A384P GlyRs exhibited profound desensitization (Fig. 1A,B), with an 8.2-fold faster τW than that of α1WT GlyRs and a nearly complete extent (97%) of desensitization (Fig. 1C,D; Table 1). The heteromeric α1A384Pβ GlyRs showed a 1.6-fold faster τW and a significantly increased extent of desensitization compared with that of α1WTβ GlyRs (Fig. 1E–H). Picrotoxin sensitivity was used as a functional assay of β subunits incorporation because α1 homomers are more sensitive to picrotoxin blockade than α1β heteromers (Pribilla et al., 1992; Lynch, 2004). Our data showed that the inhibition by picrotoxin (100 μm) was significantly reduced with expression of both α1WT and α1A384P subunits when the β subunits were coexpressed (Fig. 1I–K), indicating successful incorporation of β subunits in heteromeric GlyRs. Therefore, the significantly enhanced desensitization, which supposedly restricts Cl− current flow, appeared to be one of the major functional alterations in both α1A384P and α1A384Pβ GlyRs.
Figure 1.
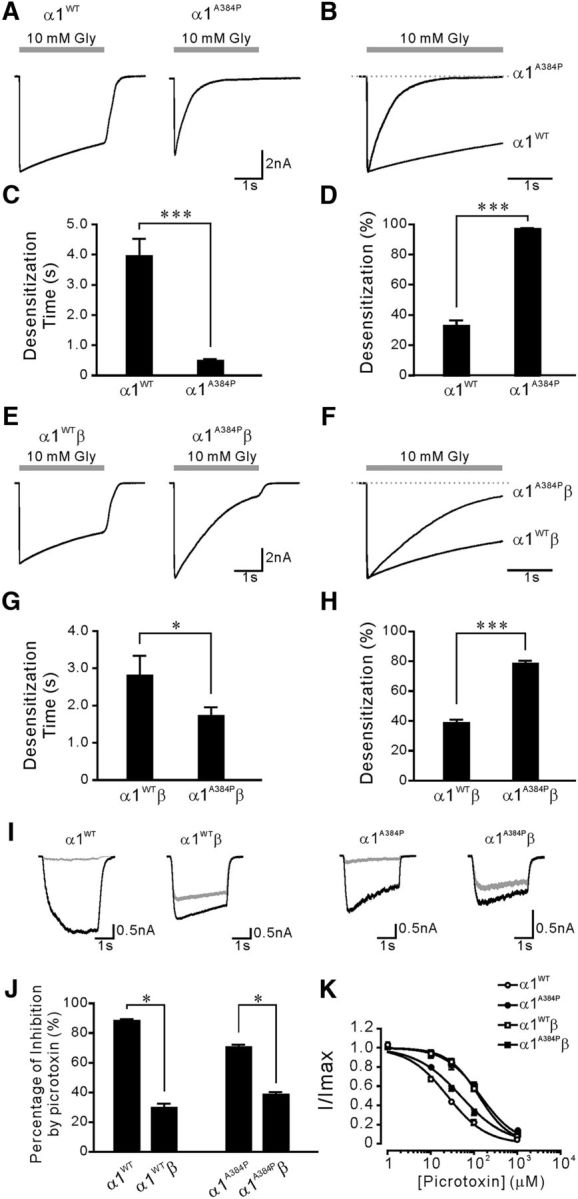
The α1 subunit A384P mutation enhanced desensitization of α1 GlyRs. A, Representative traces of α1WT (left) and α1A384P GlyR currents (right) induced by 10 m glycine. B, Peak-scaled currents from A showing the desensitization phase of α1A384P compared with α1WT receptor currents. The recovery phase is omitted. C, The weighted desensitization time constant of α1WT and α1A384P GlyR currents. D, The extent of desensitization of α1WT and α1A384P GlyR currents. E, Representative traces of α1WTβ (left) and α1A384Pβ GlyR currents (right) induced by 10 m glycine. F, Peak-scaled currents from E showing the desensitization phase of α1A384Pβ compared with of α1WTβ GlyR currents. G, The weighted desensitization time constant of α1WTβ and α1A384Pβ GlyR currents. H, The extent of desensitization of α1WTβ and α1A384Pβ GlyR currents. I, Representative traces showing GlyR currents before (black) and after (gray) application of 100 μm picrotoxin. Top traces, Picrotoxin sensitivity of GlyR currents from the α1WT homomers or α1WTβ heteromers. Bottom traces, Picrotoxin sensitivity of GlyR currents from the α1A384P GlyR homomers or α1A384Pβ GlyR heteromers. For all traces, an EC20 concentration of glycine of was used; 100 μm picrotoxin was applied both in the bath and together with glycine. J, Summarized data showing picrotoxin sensitivity of GlyR currents from α1WT (n = 16), α1WTβ (n = 16), α1A384P (n = 28), and α1A384Pβ GlyRs (n = 35). Incorporation of the β subunit significantly decreased the picrotoxin sensitivity of both WT and A384P mutant GlyRs. K, Concentration-inhibition curves for inhibition of α1WTβ and α1A384Pβ GlyR currents by picrotoxin. Glycine concentrations at EC10-EC20 for each receptor were used to induce glycine currents. *p < 0.05 (Student's t test). ***p < 0.001 (Student's t test).
Table 1.
Weighted desensitization time constant for desensitization and extent of desensitization of WT and mutant GlyRsa
| Construct | τW (s) | % desensitization | n |
|---|---|---|---|
| Glycine-induced response | |||
| α1WT | 3.95 ± 0.57 | 33.0 ± 3.5 | 16 |
| α1WTβ | 2.80 ± 0.53 | 38.7 ± 2.3 | 16 |
| α1A384P | 0.48 ± 0.06*** | 96.9 ± 0.7*** | 28 |
| α1A384Pβ | 1.73 ± 0.23†† | 78.3 ± 2.0††† | 35 |
| α1P250T | 0.22 ± 0.03*** | 95.6 ± 1.6*** | 10 |
| α1P250Tβ | 0.23 ± 0.02††† | 94.8 ± 0.9††† | 9 |
| α1R392H | 2.05 ± 0.20** | 56.0 ± 2.2*** | 4 |
| α1WT/α1R392H | 5.00 ± 1.67 | 33.2 ± 4.6 | 11 |
| α1WT/α1R392Hβ | 3.11 ± 0.95 | 31.7 ± 3.9 | 8 |
| α1A384P/α1R392H | 0.40 ± 0.09 | 98.6 ± 0.4 | 7 |
| α1A384P/α1R392Hβ | 1.36 ± 0.27 | 76.0 ± 4.2 | 5 |
| α1WT/α1P250T | 0.92 ± 0.09*** | 55.4 ± 11.3 | 4 |
| β-alanine-induced response | |||
| α1WT | 4.22 ± 0.68 | 21.8 ± 3.3 | 8 |
| α1WTβ | 3.73 ± 0.92 | 25.2 ± 3.6 | 8 |
| α1A384P | 0.66 ± 0.16*** | 98.2 ± 0.4*** | 8 |
| α1A384Pβ | 1.13 ± 0.24† | 82.0 ± 3.7††† | 6 |
| α1P250T | 0.21 ± 0.02*** | 97.8 ± 0.2*** | 6 |
| α1P250Tβ | 0.54 ± 0.15† | 83.8 ± 3.4†† | 6 |
| Taurine-induced responses | |||
| α1WT | 4.26 ± 0.85 | 20.2 ± 2.0 | 8 |
| α1WTβ | 3.88 ± 1.15 | 28.8 ± 5.0 | 10 |
| α1A384P | 0.69 ± 0.11** | 91.2 ± 3.1*** | 8 |
| α1A384Pβ | 1.41 ± 0.11 | 71.3 ± 5.3††† | 6 |
| α1P250T | 0.71 ± 0.10** | 82.3 ± 2.8*** | 6 |
| α1P250Tβ | 1.76 ± 0.45 | 72.1 ± 8.95† | 4 |
aData from whole-cell recordings from WT or mutant α1 GlyRs expressed in HEK293T cells in response to glycine (10 mm), β-alanine (5 mm), or taurine (10 mm). Data are mean ± SEM. Two-tailed Student's t test was used to compare the data of α1WT with that of the mutants, including α1A384P, α1P250T, α1R392H, α1WT/α1R392H, and α1WT/α1P250T; to compare the data of α1WTβ with that of the mutants, including α1A384Pβ, α1P250Tβ, and α1WT/α1R392Hβ; to compare the data between α1A384P/α1R392H and α1A384P; and to compared the data between α1A384P/α1R392Hβ and α1A384Pβ.
**p < 0.01,
***p < 0.001, significant difference from α1WT.
†p < 0.05,
††p < 0.01,
†††p < 0.001, significant difference from α1WTβ.
Pharmacological properties were altered in homomeric α1A384P GlyRs but not in heteromeric α1A384Pβ GlyRs
We further tested the pharmacological profiles of GlyRs that contain the A384P mutation. In homomeric α1 GlyRs, the mutant α1A384P subunit caused significant changes in sensitivity to three endogenous agonists: glycine, β-alanine, and taurine (Fig. 2A; Table 2). The α1A384P subunit produced a significant right shift of the concentration-response curve for glycine (2.9-fold increase in EC50, p < 0.001), β-alanine (2.3-fold increase in EC50, p < 0.01), and taurine (2.6-fold increase in EC50, p < 0.001). Interestingly, however, in the postsynaptic form of heteromeric α1β GlyRs, α1A384Pβ and α1WTβ receptors exhibited indistinguishable concentration-response curves and EC50 to glycine (p = 0.62), β-alanine (p = 0.42), or taurine (p = 0.47) (Fig. 2B,C). Considering that fast inhibitory synaptic transmission is mainly mediated by synaptically released glycine and requires activation of postsynaptic GlyRs, the unchanged sensitivity of α1A384Pβ receptors to glycine indicates that the agonist binding property is unlikely a major functional deficit with the A384P mutant subunit.
Figure 2.

The α1 subunit A384P mutation in α1β GlyRs produced no significant alterations in basic channel properties. A, Concentration-response curves for homomeric α1WT and α1A384P GlyRs normalized to maximal currents induced by glycine, β-alanine, or taurine. B, Concentration-response curves for heteromeric α1WTβ and α1A384Pβ GlyRs normalized to maximal currents induced by glycine, β-alanine, or taurine. C, Representative traces showing currents from α1WTβ (top) and α1A384Pβ GlyRs (bottom). D, Scatter plot with mean ± SEM to show the maximal current responses (Imax) induced by glycine from α1WT and α1A384P GlyRs, and Imax induced by glycine, β-alanine, or taurine in α1WTβ and α1A384Pβ GlyRs. E, The biotinylated surface proteins and total cell lysate from HEK cells expressing α1WTβ or α1A384Pβ GlyRs were assayed using Western blot analysis. GFP was assayed as a control for nonsurface proteins. The quantified results at the bottom show the relative expression levels of α1 subunits from three independent experiments. F, Left, Representative traces of 1 m GlyR currents at different holding potentials from −60 to 60 mV. Right, Peak I–V relations (normalized to the current recorded at −60 mV) of α1WTβ and α1A384Pβ GlyR currents. G, Concentration-inhibition curves for inhibition of α1WTβ and α1A384Pβ GlyR currents by strychnine. Glycine concentrations at EC10-EC20 for each receptor were used to induce glycine currents. Statistical significance was determined by unpaired Student's t test and was defined as not significant (p > 0.05).
Table 2.
Sensitivities to agonists for WT and mutant GlyRsa
| Construct | EC50 (μm) | Hill coefficient | n | Imax (nA) | n |
|---|---|---|---|---|---|
| Glycine-induced responses | |||||
| α1WT | 86.1 ± 7.4 | 1.75 | 9 | 8.68 ± 0.97 | 21 |
| α1WTβ | 55.1 ± 3.8 | 1.69 | 14 | 10.37 ± 0.88 | 28 |
| α1A384P | 250.6 ± 17.6*** | 2.00 | 10 | 8.55 ± 0.97 | 9 |
| α1A384Pβ | 62.1 ± 5.2 | 1.44 | 15 | 7.75 ± 1.07 | 21 |
| α1P250T | 571.5 ± 15.8*** | 1.69 | 6 | 0.66 ± 0.17*** | 10 |
| α1P250Tβ | — | — | — | 0.41 ± 0.08††† | 11 |
| α1R392H | 159.5 ± 10.3 | 2.14 | 4 | 2.05 ± 0.45** | 4 |
| α1WT/α1R392H | 56.6 ± 3.2 | 2.55 | 11 | 7.57 ± 0.89 | 11 |
| α1WT/α1R392Hβ | 36.4 ± 5.5 | 1.35 | 8 | 7.34 ± 0.70 | 8 |
| α1A384P/α1R392H | 242.5 ± 15.4 | 1.82 | 10 | 9.88 ± 2.16 | 7 |
| α1A384P/α1R392Hβ | 47.2 ± 3.5 | 1.37 | 8 | 7.54 ± 1.33 | 5 |
| α1WT/α1P250T | 175.3 ± 30.5** | 1.59 | 4 | 2.74 ± 0.60*** | 4 |
| β-alanine-induced responses | |||||
| α1WT | 166.3 ± 11.4 | 2.08 | 8 | 5.91 ± 1.29 | 8 |
| α1WTβ | 141.3 ± 10.8 | 1.56 | 8 | 9.67 ± 0.97 | 8 |
| α1A384P | 384.2 ± 35.6** | 1.70 | 8 | 9.14 ± 1.82 | 8 |
| α1A384Pβ | 203.7 ± 14.7 | 1.62 | 6 | 9.21 ± 1.55 | 6 |
| α1P250T | 1174 ± 47*** | 1.79 | 6 | 1.43 ± 0.82** | 6 |
| α1P250Tβ | — | — | — | 0.28 ± 0.07††† | 6 |
| Taurine-induced responses | |||||
| α1WT | 611.9 ± 42.3 | 1.53 | 16 | 7.04 ± 0.64 | 8 |
| α1WTβ | 373.6 ± 23.3 | 1.63 | 9 | 8.01 ± 0.63 | 12 |
| α1A384P | 1580 ± 109*** | 1.53 | 16 | 6.38 ± 1.13 | 8 |
| α1A384Pβ | 428.2 ± 19.5 | 1.63 | 10 | 6.45 ± 0.73 | 12 |
| α1P250T | 2205 ± 145*** | 1.52 | 6 | 0.19 ± 0.03*** | 6 |
| α1P250Tβ | — | — | — | 0.07 ± 0.03††† | 6 |
aData from whole-cell recordings made from WT or mutant α1 GlyRs expressed in HEK293T cells. Maximum currents (Imax) were induced by glycine (10 mm), β-alanine (5 mm), or taurine (10 mm). The EC50 and Hill coefficient were not measured in α1P250Tβ due to the small current amplitude of this construct. Data are mean ± SEM. Two-tailed Student's t test was used to compare the data of α1WT with that of the mutants, including α1A384P, α1P250T, α1R392H, α1WT/α1R392H, and α1WT/α1P250T; to compare the data of α1WTβ with that of the mutants, including α1A384Pβ, α1P250Tβ, and α1WT/α1R392Hβ; to compare the data between α1A384P/α1R392H and α1A384P; and to compare the data between α1A384P/α1R392Hβ and α1A384Pβ.
**p < 0.01,
***p < 0.001, significant difference from α1WT.
††† p < 0.001, significant difference from α1WTβ.
We next examined whether the maximum current (Imax) was reduced in α1A384P subunit-containing GlyRs. The A384P mutant homomers exhibited no significant reduction in the Imax induced by glycine, β-alanine, or taurine (Fig. 2D; Table 2). Consistent with the Imax results, total protein expression and surface expression of α1A384Pβ GlyRs were not significantly different from those of α1WTβ subunits (Fig. 2E). Because A384P is located near the TM3–4 intracellular loop, which is involved in the rectification of other Cys-loop receptors, such as 5-HT3A receptors (McKinnon et al., 2011; Baptista-Hon et al., 2013), we next examined the I–V relationships to illustrate whether the A384P mutation produces different rectification properties that might affect the chloride conductance upon depolarization. Our results showed no obvious alterations in the I–V relationships or reversal potentials in α1A384Pβ GlyRs compared with those in α1WTβ GlyRs (Fig. 2F). These data suggest that the altered channel function of α1A384Pβ GlyRs is not due to impaired chloride conductance or rectification properties.
Strychnine and picrotoxin are both GlyR antagonists, but they target the receptor differently at the extracellular domain and the transmembrane domains, respectively. Homomeric α1A384P GlyRs had reduced picrotoxin sensitivity (2.0-fold increase in IC50, p < 0.01) and increased strychnine sensitivity (2.4-fold decrease in IC50, p < 0.01) compared with WT GlyRs, whereas α1A384Pβ heteromeric GlyRs exhibited no significant change in either strychnine (p = 0.99) or picrotoxin sensitivity (p = 0.50) compared with WT αβ GlyRs. By comparing homomeric α1WT or α1A384P GlyRs with heteromeric α1WTβ or α1A384Pβ GlyRs, we found that incorporation of the β subunit significantly reduced picrotoxin sensitivity for both α1WT (5.3-fold increase in IC50, p < 0.01) and α1A384P GlyRs (2.8-fold increase in IC50, p < 0.01) (Figs. 1K, 2G). These data are consistent with previous reports that the β subunit is a major determinant for picrotoxin resistance, and further indicates that the A384P mutation does not affect the binding of α1 to β subunits. Collectively, these data reveal that the α1A384Pβ GlyR, the postsynaptic form of mutant GlyR, showed no significant deficit in agonist or antagonist sensitivity, as well as Imax, surface expression, or rectification properties.
Mutant α1A384Pβ GlyRs had lower open probability, but unchanged main conductance levels, compared with WT α1β GlyRs
To determine whether glycine receptors carrying the A384P mutation resulted in changes in receptor gating, we measured glycine-evoked single-channel currents of WT α1β and mutant α1A384P β GlyRs. Single channels from WT α1β receptors opened to clusters of openings with a main conductance level of ∼38 pS (Fig. 3A,C; Table 3). Conversely, single channels from α1A384Pβ receptors opened to two distinct conductance levels, ∼35 pS, and ∼15 pS (Fig. 3B). The open probability and the mean open time are, respectively, 0.195 ± 0.08, 47.0 ± 10.7 ms for high conductance openings, and 0.20 ± 0.07, 38.3 ± 7.51 ms for low conductance openings of α1A384Pβ. While clusters of α1A384Pβ receptor openings displaying the main conductance level had no significant differences to those observed from wild-type α1β receptors, low conductance openings displayed a conductance level significantly different from the main conductance level (15.1 ± 1.67, p < 0.0001, one-way ANOVA). Because wild-type α1β receptors lack two conductance levels, to simplify the analysis the measurements shown for α1A384Pβ receptors were the result of the combined data of main and low conductance openings (Table 3). Both α1A384Pβ and wild-type α1β receptors opened to at least three different open states (O1, O2, and O3). The open time distributions were fitted best by three weighted (ao1, ao2, and ao3) exponential functions with three open time constants (τo1, τo2, and τo3) (Fig. 3D). Mutant α1A384Pβ receptors did not alter any of the open time constants (p = 0.911) but favored an increase in the relative occurrence of O2 openings while decreasing the relative occurrence of O3 openings (p = 0.0003). The difference between the relative occurrence of O2 and O3 openings led to a concomitant decrease in mean open time and open probability of α1A384Pβ receptors (Fig. 3C; Table 3).
Figure 3.
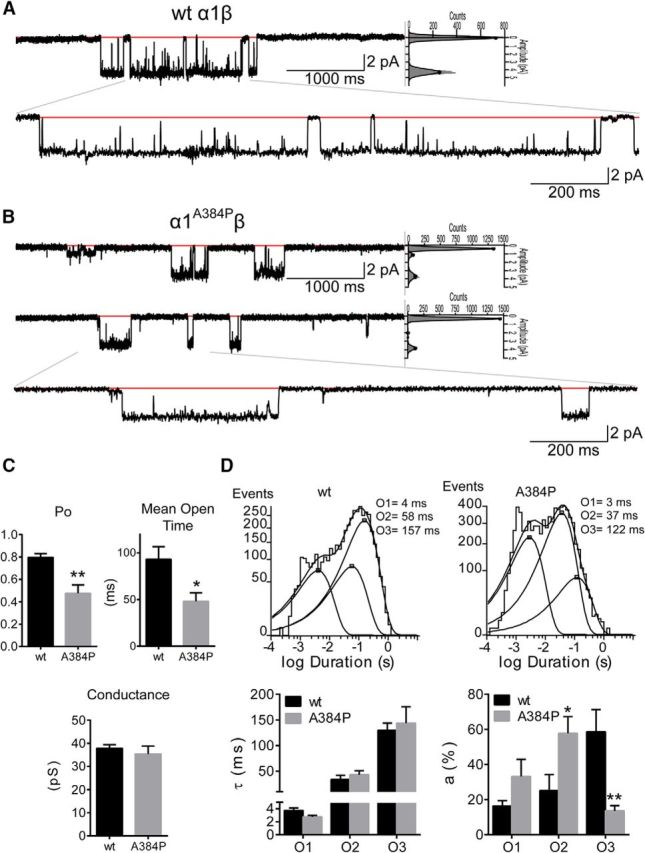
Single-channel properties of α1WTβ and α1A384Pβ GlyRs. Representative single-channel current traces from cell-attached patches of α1WTβ (A) and α1A384Pβ (B) GlyRs. Openings are downward, and each representative nonenlarged trace was a continuous 5000 ms recording. Red lines indicate the closed state of the receptor. Sweep histograms of closed (at 0 pA) and open (≠0 pA) events and the relative contribution (gray area under the fitting lines) of each level were displayed at the right side of the 5000 ms traces. Bottom traces, Enlarged 1600 ms section as indicated by the gray lines. C, Bar graphs summarize the open probability (Po), mean open time, and conductance of α1WTβ and α1A384Pβ GlyR single-channel currents. Data are mean ± SEM (n = 6). **p < 0.01 (unpaired Student's t test relative to wt). *p < 0.05 (unpaired Student's t test relative to wt). D, Average open time histograms of α1WTβ (4515 events) and α1A384Pβ (7729 events) GlyRs are shown with open time constants for O1, O2, and O3 openings. Summary bar graphs of time constants (τo1, τo2, and τo3) and relative areas (ao1, ao2, and ao3) of open time histograms for α1WTβ (wt, black bars) and α1A384Pβ (A384P, gray bars) GlyRs. Data are mean ± SEM (n = 6). **p < 0.01 (two-way ANOVA with Sidak's multiple comparisons test relative to wt). *p < 0.05 (two-way ANOVA with Sidak's multiple comparisons test relative to wt).
Table 3.
Single-channel properties of α1A384Pβ receptorsa
| α1β | α1A384Pβ | pb | |
|---|---|---|---|
| Open probability | 0.80 ± 0.03 | 0.24 ± 0.07 | 0.0027 |
| Mean open time, ms | 93.2 ± 13.4 | 48.2 ± 9.11 | 0.0197 |
| Main conductance, pS | 37.9 ± 1.55 | 35.4 ± 3.40 | 0.5069 |
| pc | |||
| Open time constants | |||
| τO1, ms | 3.74 ± 0.38 | 2.75 ± 0.24 | 0.9110 |
| τO2, ms | 33.8 ± 8.14 | 43.2 ± 7.84 | |
| τO3, ms | 130 ± 14 | 144 ± 32 | |
| aO1, % | 16 ± 3 | 33 ± 10 | 0.0003 |
| aO2, % | 25 ± 9 | 58 ± 9 | |
| aO3, % | 59 ± 13 | 14 ± 3 | |
aData are mean ± SEM (n = 6).
bUnpaired two-tailed Student's t test relative to WT α1β receptors.
cTwo-way ANOVA with Sidak's multiple comparisons test relative to WT α1β receptors.
Heterogeneous expression of α1A384P and α1R392H subunits exhibited receptors with similar channel properties to homomeric α1A384P receptors
The A384P mutation was first identified in a patient who was a compound heterozygote (A384P/R392H) (Mine et al., 2015). The surface expression of homomeric α1R392H GlyRs has been reported to be highly reduced (Villmann et al., 2009). However, α1R392H/α1WT GlyRs produced indistinguishable EC50 and Imax compared with α1WT GlyRs (Rea et al., 2002). Consistent with previous studies, homomeric α1R392H GlyRs had a mild reduction in glycine sensitivity (1.9-fold increase in EC50, p = 0.07) and a substantial reduction in Imax (4.2-fold decrease, p < 0.01; Table 2; see Fig. 5A). Coexpression of α1R392H and α1WT subunits at a 1:1 ratio (α1WT/α1R392H) resulted in fully functional homomeric GlyRs, showing indistinguishable EC50 (p = 0.42) and Imax (p = 0.50) between homomeric α1WT and α1R392H/α1WT GlyRs (Fig. 4A,B; Table 2). In GlyR heteromers, coexpression of α1R392H and β subunits caused no change in EC50 (p = 0.89) and a modest decrease of Imax (by ∼30%, p = 0.05) compared with α1WTβ GlyRs (Fig. 4B). These results could be explained by at least two possibilities which include the following: (1) α1R392H was incorporated with α1WT to form α1R392H/α1WT (or α1R392H/α1WTβ) GlyRs, whose functions are indistinguishable from the WT GlyRs; and (2) α1R392H could not be incorporated with α1WT and thereby resulted in the expression of α1R392H and αWT separately (or α1R392Hβ and α1WTβ when the β subunit was cotransfected) in the same cell. In this case, the α1R392H-containing GlyRs might be largely absent from the cell surface and the WT GlyRs are still the only functional receptors. Despite this, our data indicate that coexpression of α1R392H and α1WT subunits had only a minor effect on GlyR function at the whole-cell level, which is in line with the fact that the heterozygous R392H carrier did not show hyperekplexia symptoms.
Figure 5.

Comparison of activation and desensitization profiles of α1A384P and α1P250T GlyR currents. A, Schematic representation of a GlyR α1 subunit topology showing the location of hyperekplexia mutations P250T and A384P. B, Bottom view of the crystal structure of three adjacent subunits (in gray or bronze) in the human α3 GlyR showing the location of the homologous residues of A384 and P250 of the α1 subunit. The illustrated structure was displayed from the TM domains of GlyR as indicated by the regions between the yellow lines in the inset. Inset, Side view of the α3 GlyR indicating the viewing angle. C, Top, Representative traces of α1A384P and α1P250T GlyR currents evoked by glycine. Bottom, Peak-scaled currents from the top traces showing the difference in the desensitization phase of α1A384P and α1P250T currents. D, The Imax, weighted desensitization time constant, and extent of desensitization of α1A384P (black bars) and α1P250T (gray bars) GlyR currents. E, Representative traces of homomeric α1A384P and heteromeric α1A384Pβ GlyR currents. F, Representative traces of homomeric or heteromeric α1P250T and α1P250Tβ or α1P250T and α1P250Tβ GlyR currents. G, Comparison of Imax, weighted desensitization time constant, and extent of desensitization, and picrotoxin sensitivity between homomeric α1A384P (solid bars) and heteromeric α1A384Pβ (striped bars) (left pair) or homomeric α1P250T (solid bars) and heteromeric α1P250Tβ (striped bars) (right pair). C, E, F, Gray bars represent the time course of application of 1 m glycine. *p < 0.05 (unpaired Student's t test). **p < 0.01 (unpaired Student's t test). ***p < 0.001 (unpaired Student's t test).
Figure 4.

Heterozygous expression of α1R392H with α1A384P subunits did not alter the properties of the α1A384P GlyRs. A, Representative currents evoked by glycine from HEK cells expressing α1R392H, α1WT/α1R392H, or α1WT GlyRs. Gray bars represent the time course of application of 1 m glycine. Calibration: 2 nA, 1 s. B, Imax of glycine evoked currents recorded from cells expressing either homomeric or heteromeric α1WT or α1WT/α1R392H (1:1 coexpression) GlyRs. C, D, Representative currents induced by glycine from HEK cells expressing either homomeric (C) or heteromeric (D) α1A384P or α1384P/α1R392H (1:1 coexpression) GlyRs. Gray bars represent the time course of application of 1 m glycine. Calibration: 2 nA, 1 s. E–G, Quantitative results showing the difference in GlyR Imax (E), weighted desensitization time constant (F), and desensitization extent (G) between homomeric or heteromeric α1A384P and α1384P/α1R392H GlyR. H, Incorporation of the β subunit reduced the inhibition by picrotoxin of both α1A384P and α1A384P/α1R392H GlyR currents. *p < 0.05. **p < 0.01. ***p < 0.001.
In a similar way, coexpression of α1A384P and α1R392H subunits (at a ratio of 1:1) resulted in functional channels that bear a resemblance to homomeric α1A384P GlyRs alone (Fig. 4C,E), without any effect on glycine sensitivities, Imax (Table 2) or desensitization profiles (Fig. 4F,G; Table 1). When coexpressed with the β subunit, which was confirmed by the reduced inhibition by picrotoxin (Fig. 4H), the α1A384P/α1R392Hβ heteromer exhibited Imax and desensitization parameters that were indistinguishable from those of α1A384Pβ GlyRs (Fig. 4D–G). These data reveal that the electrophysiological properties of homomeric α1A384P and heteromeric α1A384Pβ GlyRs were unlikely affected by coexpression of the α1R392H. Similarly, this could be attributed to either the formation of functional α1A384P/α1R392H receptors or lack of incorporation of α1R392H into α1A384P that leads to only functional α1A384P on the cell surface.
Comparison of the effects of hyperekplexia mutant α1A384P and α1P250T subunits on GlyR desensitization
In addition to A384P, two other α1 subunit missense mutations associated with human hyperekplexia have been reported to alter GlyR desensitization, including P250T and P230S (Saul et al., 1999; Bode et al., 2013). We compared electrophysiological properties of homomeric α1A384P with α1P250T GlyRs because these two mutations are both located in the intracellular loop and might share more similarities than P230S, which is located in the TM1 (Fig. 5A,B). P250T is located in the TM1-TM2 linker and is one amino acid residue before the TM2 segment. In contrast, A384P is located in the TM3-TM4 linker and three residues before the TM4 segment. Homomeric α1P250T GlyRs had drastically reduced Imax to glycine (13.2-fold, p < 0.001), β-alanine (4.1-fold, p < 0.01), and taurine (37.1-fold, p < 0.001) compared with α1WT GlyRs, and increased EC50 to glycine (6.6-fold, p < 0.001), β-alanine (7.1-fold, p < 0.001), and taurine (3.6-fold, p < 0.01) (Table 2). Our results were consistent with previous reports that agonist sensitivity and channel conductance were impaired by P250T (Saul et al., 1999). The Imax of homomeric α1P250T GlyRs was also significantly smaller than α1A384P GlyRs upon activation with all three types of agonists (Fig. 5D). Furthermore, alignments of peak-scaled currents showed that homomeric α1P250T GlyRs exhibited a faster desensitization rate than α1A384P GlyRs in response to glycine (Fig. 5C) or β-alanine activation; however, the faster τ was not observed in taurine-induced currents (Fig. 5D; Table 1). Despite the difference in desensitization rate, the desensitization extent of α1P250T and α1A384P receptor currents reached similar levels in response to all three agonists (Fig. 5D; Table 1).
The incorporation of β subunits had different regulatory effects on the activation and desensitization properties of α1A384Pβ and α1P250Tβ GlyRs. When coexpressed with β subunits, both α1P250Tβ and α1A384Pβ receptors had significantly reduced sensitivity to picrotoxin inhibition (Fig. 5G). The incorporation of β subunits did not significantly change the Imax in either α1P250Tβ or α1A384Pβ receptors. Interestingly, coexpression of the β subunits significantly rescued desensitization observed in homomeric α1A384P (Fig. 5E) but not in α1P250T (Fig. 5F) receptors, causing a 3.5-fold slower τ and lower extent of desensitization than the homomeric α1A384P receptors (Fig. 5G). Despite the partial rescue by the β subunits, α1A384Pβ receptors still exhibited enhanced desensitization compared with α1WTβ receptors (Fig. 1; Table 1). Together, these results indicated that the P250T mutation is involved in both channel activation and desensitization pathways, whereas the A384P mutation mainly causes enhanced desensitization, even in the presence of the β subunits at postsynaptic sites. Therefore, the strongly reduced Imax, the reduced agonist sensitivity, and the enhanced desensitization could all contribute to the causes of hyperekplexia in the patients with the P250T mutation.
Transfection of α1A384P or α1P250T subunits accelerated desensitization of GlyR currents in cultured DRG neurons
Because the functional alterations of GlyRs expressed in HEK cells might not represent changes in neurons, we tested electrophysiological effects of transfection of α1WT, α1A384P, or α1P250T subunits in cultured DRG neurons, which are known to lack endogenous GlyRs (Kung et al., 2001). DRG neurons expressing α1A384P subunits had concentration-response curves (p = 0.11) and Imax (p = 0.07) insignificant from those expressing α1WT subunits (Fig. 6C,E). In line with the results from HEK cells, α1A384P GlyRs still exhibited enhanced desensitization in neurons (Fig. 6A,F,G). DRG neurons expressing α1P250T subunits had substantially reduced Imax (p < 0.01), right-shifted glycine concentration-response curves (p < 0.05), and enhanced desensitization (Fig. 6C,E–G). When the β subunit was coexpressed with WT or mutant α1 subunits, there was still no significant difference in the concentration-response relationship (p = 0.93) or Imax (p = 0.79) between α1WTβ and α1A384Pβ receptors (Fig. 6B,D,E). Although the incorporation of β subunits partially restored the desensitization time and the extent of desensitization, heteromeric α1A384Pβ receptors still exhibited stronger desensitization compared with α1WTβ receptors (Fig. 6F,G). The α1P250Tβ receptors had an even faster desensitization time and higher percentage of desensitization than α1A384Pβ receptors; however, its concentration-response curve was significantly right-shifted (p < 0.01) and the Imax (p < 0.05) was strongly reduced (Fig. 6D,E). Cotransfection of α1A384P or α1WT subunits with β subunits significantly reduced picrotoxin sensitivity relative to the homomeric receptors. α1P250Tβ also exhibited reduced picrotoxin sensitivity than α1P250T; however, the difference did not reach a significant level (p = 0.08), probably because the small current amplitude of P250T mutants influenced the precise measurement of picrotoxin inhibition or the protein expression profiles were different between HEK cells and DRG neurons. Moreover, we also expressed α1WT or α1A384P subunits in cultured cortical neurons. Under this condition, α1A384P receptors still exhibited 4.2-fold faster τW (p < 0.01) and higher extent of desensitization (p < 0.05) without significant alterations in Imax or EC50 (Table 4). Together, our results suggest that the A384P mutation resulted in enhanced desensitization without apparent deficits in agonist affinity or channel conductance in α1A384P and α1A384Pβ receptors in neurons.
Figure 6.

Current activation and desensitization of homomeric α1WT, α1A384P, and α1P250T and heteromeric α1WTβ, α1A384Pβ, and α1P250Tβ GlyRs in cultured DRG neurons. A, Representative traces showing 10 m glycine-induced current responses in untransfected DRG neurons, or DRG neurons expressing homomeric α1WT, α1A384P, or α1P250T GlyRs. B, Representative traces showing 10 m glycine-induced current responses in DRG neurons expressing heteromeric α1WTβ, α1A384Pβ, or α1P250Tβ GlyRs. Gray bars represent the time course of application of 10 m glycine. C, D, Glycine concentration-response curves in DRG neurons expressing (C) homomeric α1WT (n = 11), α1A384P (n = 14), and α1P250T (n = 5) or (D) heteromeric α1WTβ (n = 6), α1A384Pβ (n = 5), and α1P250Tβ GlyRs (n = 4) normalized to maximal currents. E, Glycine-induced Imax in DRG neurons expressing homomeric α1WT, α1A384P, and α1P250T or heteromeric α1WTβ, α1A384Pβ, and α1P250Tβ GlyRs. F, G, The weighted desensitization time constant (F) and desensitization extent (G) of currents from homomeric α1WT, α1A384P, and α1P250T or heteromeric α1WTβ, α1A384Pβ, and α1P250Tβ GlyRs recorded from transfected DRG neurons. H, Picrotoxin sensitivity of α1WT, α1A384P, and α1P250T GlyRs expressed in DRG neurons. *p < 0.05 (unpaired Student's t test). **p < 0.01 (unpaired Student's t test). ***p < 0.001 (unpaired Student's t test).
Table 4.
Activation and desensitization profiles of WT and the A384P mutant GlyRs overexpressed in cultured cortical neuronsa
| Construct | 10%–50% rise time (ms) | EC50 (μm) | Hill coefficient | Imax (nA) | τW (s) | % desensitization | n |
|---|---|---|---|---|---|---|---|
| Nontransfected | 248.2 ± 29.8 | 64.9 ± 3.2 | 1.487 | 2.67 ± 0.41 | 1.92 ± 0.14 | 49.8 ± 4.0 | 18 |
| α1WT | 65.3 ± 10.2 | 80.4 ± 7.4 | 1.931 | 5.74 ± 1.15 | 1.88 ± 0.39 | 62.0 ± 4.4 | 9 |
| α1A384P | 61.5 ± 7.0 | 122.5 ± 4.6 | 2.035 | 7.79 ± 0.64 | 0.45 ± 0.04 | 77.5 ± 3.1 | 9 |
aData from whole-cell recordings made from transfected or nontransfected neurons. Successful transfection of α1 construct was first identified by the GFP fluorescence and then confirmed by the 10%–50% rise time, which was used to distinguish from the endogenous α2 GlyRs with slower activation kinetics (mean 10–50 rise time >200 ms) expressed by cortical neurons. Data are mean ± SEM.
A384P accelerated frequency-dependent desensitization
Considering that the glycine receptor-mediated IPSCs have a fast decay constant from 4.9 to 11.2 ms (Takahashi et al., 1992; Muller et al., 2006; Graham et al., 2011), we next addressed the question of whether the α1 subunit A384P mutation could affect desensitization of synaptic glycine currents. Inhibitory interneurons in the spinal cord central pattern generator network are able to generate rhythmic activity during locomotion (Butt and Kiehn, 2003; Zhong et al., 2010), which could lead to pulsatile release of glycine. We applied trains of brief glycine pulses with different frequencies to mimic IPSC-like responses in HEK cells expressing α1WTβ or α1A384Pβ GlyRs. The results showed that the repeated glycine pulses produced a greater decreasing trend in the peak currents in α1A384Pβ than α1WTβ receptors. After an 8 s train of glycine application at 1 Hz (Fig. 7A,D), the reduction of peak current amplitude was 3.8% in α1WTβ but was 13.5% in α1A384Pβ (Fig. 7G). The difference in current reduction between α1A384Pβ and α1WTβ was even more obvious at higher frequencies. After an 8 s train stimulation at 5 Hz, the reduction was 12.2% in α1WTβ versus 67.2% in α1A384Pβ (Fig. 7B,E,H); and at 10 Hz, the reduction was 19.5% in α1WTβ versus 83.4% in α1A384Pβ (Fig. 7C,F,I). The stronger reduction of peak current amplitude in α1A384Pβ was not due to irreversible suppression of glycine responses because, in the same cell, the amplitude of the first response in each train was unchanged. When normalized to the first peak of the 1 Hz train, the first peak of the 5 Hz and 10 Hz train was 93.0 ± 8.3% and 92.4 ± 8.9% in WT, and 96.6 ± 4.4% and 95.3 ± 3.6% in A384P, respectively (p > 0.05 in all groups by paired t test), suggesting that the first peak response in each train had fully recovered from the reduction caused by the previous stimulation. Together, these data indicate that α1A384Pβ strongly reduced the IPSC-like responses induced by repetitive pulses of glycine, most likely through a mechanism of enhanced desensitization.
Figure 7.

Current responses of α1WTβ and α1A384Pβ GlyRs to repetitive application of brief pulses of glycine at different frequencies. A–C, Example current traces from two cells transfected with α1WTβ (left traces) or α1A384Pβ (right traces) evoked by application of high-frequency trains of brief glycine (1 mm) pulses at 1 Hz (A), 5 Hz (B), and 10 Hz (C). D–F, Peak current values show a more rapidly decreasing trend in α1A384Pβ (n = 7, gray) compared with α1WTβ (n = 7, black) GlyRs upon repetitive stimulation with glycine (1 m) pulses at 1 Hz (D), 5 Hz (E), and 10 Hz (F). Peak current values were normalized with respect to the first peak in each series and averaged. G–I, Comparison of the normalized peak current values between α1WTβ (n = 7, black) and α1A384Pβ (n = 7, gray) GlyRs at the 2nd, 4th, 6th, and 8th s of the repetitive stimulation with glycine at 1 Hz (G), 5 Hz (H), and 10 Hz (I). *p < 0.05 (unpaired Student's t test). **p < 0.01 (unpaired Student's t test). ***p < 0.001 (unpaired Student's t test).
Discussion
In the present study, we found that the GlyR α1 subunit mutation A384P associated with hyperekplexia caused enhanced desensitization in both rate and extent. The α1A384P homomers showed reduced agonist sensitivity. However, the α1A384Pβ GlyR heteromers showed no significant difference in agonist sensitivity, maximal currents, main single-channel conductance, and membrane expression compared with the α1WTβ heteromers in both HEK cells and cultured neurons. The R392H mutation, which was identified in a compound heterozygote patient with A384P, did not alter the functional properties of a1A384P subunit-containing GlyRs. Moreover, in the postsynaptic α1β form, the α1A384P subunit-containing GlyRs showed frequency-dependent reduction of peak amplitude induced by repetitive pulse of glycine stimulation. Our results demonstrate that the α1 subunit mutation, A384P, enhanced desensitization of GlyR, which is highly associated with the pathogenesis of human hyperekplexia.
A384 and the adjacent region in a1 subunits are involved in the desensitization site of homomeric a1 and heteromeric a1β GlyRs
The desensitization state can be modulated by various structures of GlyRs ranging from the extracellular domain, the TM domains to the intracellular domains. The pre-TM1 domain mutation R218Q displayed reduced desensitization of GlyRs (Castaldo et al., 2004), whereas mutations in TM1 (P230S) or in the TM1-TM2 linker (P250T, W243A, I247A) showed enhanced desensitization (Lynch et al., 1997; Bode et al., 2013). A naturally occurring splicing variant α3K, which lacks 15 amino acids at the beginning of the TM3-TM4 loop, enhanced desensitization compared with the α3L GlyR variant (Nikolic et al., 1998). A deletion or replacement of the TM3-TM4 intracellular loop with short peptide sequences also enhanced homomeric α1 GlyR desensitization (Papke and Grosman, 2014; Langlhofer et al., 2015), indicating that the TM3-TM4 linker might determine the α1 subunit GlyR desensitization state. More detailed studies have shown that the intracellular end of TM2 and TM3 determined the properties of α1 subunit GlyR desensitization. The A384P mutation is located just three amino acids before the beginning of TM4, indicating that the cytoplasmic end of TM4 also contributes to the modulation of desensitization of GlyRs under certain circumstances.
The molecular mechanism of A384P effects on GlyR α1 desensitization remains unclear. Interestingly, the A384 residue is located adjacent to or within a membrane-associated helix structure, known as the “MA-stretch,” which has been observed from structures of ACh receptors and 5-HT3A receptors and is presumably considered homologous in many Cys-loop receptors (Unwin, 2005; Hassaine et al., 2014). The MA-stretch contains multiple charged residues and is located in the TM3-TM4 linker closely adjacent to the cytoplasmic end of TM4 (Peters et al., 2005). In 5-HT3A receptors, mutations of the three arginine residues (R432, R436, and R440) in the MA-stretch increased the channel conductance and hydrophobic residue substitution of R440 slowed desensitization (Kelley et al., 2003; Hu et al., 2006). In GlyRs, however, mutation of residues R377, K378, K385, and K386 in the MA-stretch or the adjacent region decreased channel conductance but did not seem to affect desensitization (Carland et al., 2009). The sequence variability of the TM3-TM4 segments among different Cys-loop receptors may lead to the functional differences between 5-HT3A and glycine receptors. Interestingly, A384 is embedded in the MA-stretch and A384P likely contributes to the modulation of GlyR desensitization. Considering the strongly restricted torsional angle between proline and the neighboring amino acid residues, our findings indicate that the desensitization of A384P might be caused by the abnormal turning angles of MA-stretch before M4 in the mutant GlyRs.
Compared with A384P, another hyperekplexia missense mutation, P250T, showed significant changes not only in desensitization but also in other channel properties (Saul et al., 1999). The basic channel properties, particularly Imax and agonist sensitivities, were largely impaired by P250T but not by A384P. As proposed by previous studies that the desensitization gate is different from the channel activation gate in Cys-loop receptors (Purohit and Grosman, 2006; Hernandez et al., 2017), P250T likely affects both gating and desensitization pathway, whereas A384P is only involved in the desensitization. This is likely due to their different conformational relationships to the channel pore (Hernandez et al., 2017). Based on the crystal structure of α3 GlyRs (Huang et al., 2015), the side chain of P250 extends toward the channel lumen, whereas that of A384 is toward the M3/M4 interface (Fig. 5B). In addition, P250 is not only a conserved residue among many Cys-loop receptors, such as α1–3 subunits of GlyRs, the α subunit of glutamate-activated chloride channel (GluCl), and α1–6, γ1–3, δ, ε, and ρ subunits of GABAARs, but also is the common constriction gate of channel lumen as indicated by the crystal structures of GABAAR β3, GluCl, GlyR α1, and α3 (Hibbs and Gouaux, 2011; Althoff et al., 2014; Miller and Aricescu, 2014; Du et al., 2015; Huang et al., 2015). These observations suggest that the homologous site of P250 is generally important for channel gating not only in GlyR α1 but also in other Cys-loop receptors.
Both P250T and A384P mutations enhanced GlyR α1 desensitization. GlyRs with the P250T mutation had a faster desensitization rate than GlyRs with the A384P mutation. This is consistent with the conformational analysis of the two residues. In contrast to P250, which directly controls the constriction of channel lumen at the cytoplasmic end, A384 possibly needs to interact with TM3 or the TM1-TM2 linker to indirectly influence TM2 movement through allosteric transduction process. The deficits caused by some α1 subunit mutations can be partially rescued by incorporation of the β subunit (Xiong et al., 2014). In our study, incorporation of β subunits also partially reversed the accelerated desensitization rate and increased extent caused by the A384P, but not the P250T, mutation.
Possible pathogenesis of hyperekplexia in patients carrying the a1 subunit A384P mutation
The A384P mutation was identified in a compound heterozygote (A384P/R392H) (Mine et al., 2015) hyperekplexia patient. Accordingly, mutation analysis of the index patient's family pedigree showed that individuals homozygous, but not heterozygous, for R392H were affected by hyperekplexia (Vergouwe et al., 1999; Hmami et al., 2014). Expression of R392H alone in HEK293T cells substantially reduced the membrane expression of GlyRs and thus consequently reduced the glycine-induced Imax (Rea et al., 2002; Villmann et al., 2009), whereas coexpression of α1R392H and α1WT subunits exhibited channel properties similar to those found with expression of α1WT subunits alone (Rea et al., 2002). Consistent with previous studies, we found that coexpression of α1R392H and α1WT subunits did not affect glycine sensitivity or glycine-induced Imax. This indicates that, in humans, loss of function produced by the R392H mutation could also be completely compensated for by the WT α1 subunit from the WT allele (Vergouwe et al., 1999). To study the functional consequence of heterozygous expression of both α1A384P and α1R392H subunits that occurs in the hyperekplexia patient, we coexpressed α1A384P and α1R392H subunits in HEK cells. We found that coexpression of the two mutant subunits resulted in channel properties similar to those of the α1A384P subunit alone, showing enhancement in desensitization without major impairments in agonist sensitivity or Imax. These findings suggested that α1 subunit mutation, A384P, alone can lead to hyperekplexia in humans.
Different from receptors with α1P250T subunits that cause both strong desensitization and reduced Imax, enhanced desensitization is the major deficit of receptors with α1A384P subunits and also a major cause of hyperekplexia. However, glycinergic IPSCs usually have a fast decay time constant (4.9–11.2 ms) (Takahashi et al., 1992; Muller et al., 2006; Graham et al., 2011), while the desensitization time constant of GlyRs containing α1A384P subunits is slower than 1 s, especially when the β subunit is incorporated. With repeated pulses of glycine at different frequencies to mimic synaptic responses, the peak response amplitude was reduced in a frequency-dependent manner, which was more significant in α1A384Pβ than in α1WTβ GlyRs. A similar phenomenon has been reported for GABAA receptors (Bianchi and Macdonald, 2002). Interestingly, even at 1 Hz, the pulse responses were still reduced by 13.5% after an 8 s stimulation in α1A384Pβ receptors, indicating that the desensitization states cannot be fully recovered within a 1 s interval. Nevertheless, the first peak in each train had similar amplitudes in the same patch, which was due to the longer intertrain intervals (>10 s) that allowed sufficient recovery from desensitization. It also indicates that the decreasing trend induced by train stimulation is not a long-term rundown of GlyR responses caused by other irreversible mechanisms, such as intracellular accumulation of ROS-induced sustained inhibition of ACh receptors (Campanucci et al., 2008). Together, these data indicate that desensitization caused by the A384P mutation could influence synaptic responses in the nervous system.
Presynaptic homomeric GlyRs might be one of the primary causes of hyperekplexia (Xiong et al., 2014). In the present study, we were not able to evaluate whether the deficit in presynaptic or postsynaptic α1A384P subunit-containing GlyRs contributed more to the pathogenesis. However, both presynaptic and postsynaptic desensitization may be involved in pathogenesis. We infer that the enhanced desensitization is the major channel deficit produced by the A384P mutation, and that it is likely to be one of the primary causes for hyperekplexia in the patient.
Footnotes
This work was supported by Taiwan National Science Council Grant NSC 102-2320-B-039-038-MY3, Ministry of Science and Technology Grant MOST 104-2320-B-039-045-MY3, MOST 106-2632-B-039-001, and MOST 104-2320-B-039-048-MY3, and China Medical University Hospital Grant DMR-106-112 to N.Z. and D.C.W. This work was supported in part by Taiwan Ministry of Health and Welfare Clinical Trial and Research Center of Excellence Grant MOHW106-TDU-B-212-113004, and National Institutes of Health Grant R01 NS33300 to R.L.M. and C.C.H.
The authors declare no competing financial interests.
References
- Althoff T, Hibbs RE, Banerjee S, Gouaux E (2014) X-ray structures of GluCl in apo states reveal a gating mechanism of cys-loop receptors. Nature 512:333–337. 10.1038/nature13669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baptista-Hon DT, Deeb TZ, Lambert JJ, Peters JA, Hales TG (2013) The minimum M3–M4 loop length of neurotransmitter-activated pentameric receptors is critical for the structural integrity of cytoplasmic portals. J Biol Chem 288:21558–21568. 10.1074/jbc.M113.481689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianchi MT, Macdonald RL (2002) Slow phases of GABA(A) receptor desensitization: structural determinants and possible relevance for synaptic function. J Physiol 544:3–18. 10.1113/jphysiol.2002.020255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bode A, Wood SE, Mullins JG, Keramidas A, Cushion TD, Thomas RH, Pickrell WO, Drew CJ, Masri A, Jones EA, Vassallo G, Born AP, Alehan F, Aharoni S, Bannasch G, Bartsch M, Kara B, Krause A, Karam EG, Matta S, et al. (2013) New hyperekplexia mutations provide insight into glycine receptor assembly, trafficking, and activation mechanisms. J Biol Chem 288:33745–33759. 10.1074/jbc.M113.509240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bode A, Lynch JW (2014) The impact of human hyperekplexia mutations on glycine receptor structure and function. Mol Brain 7:2. 10.1186/1756-6606-7-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breitinger HG, Villmann C, Becker K, Becker CM (2001) Opposing effects of molecular volume and charge at the hyperekplexia site alpha 1(P250) govern glycine receptor activation and desensitization. J Biol Chem 276:29657–29663. 10.1074/jbc.M100446200 [DOI] [PubMed] [Google Scholar]
- Butt SJ, Kiehn O (2003) Functional identification of interneurons responsible for left-right coordination of hindlimbs in mammals. Neuron 38:953–963. 10.1016/S0896-6273(03)00353-2 [DOI] [PubMed] [Google Scholar]
- Campanucci VA, Krishnaswamy A, Cooper E (2008) Mitochondrial reactive oxygen species inactivate neuronal nicotinic acetylcholine receptors and induce long-term depression of fast nicotinic synaptic transmission. J Neurosci 28:1733–1744. 10.1523/JNEUROSCI.5130-07.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carland JE, Cooper MA, Sugiharto S, Jeong HJ, Lewis TM, Barry PH, Peters JA, Lambert JJ, Moorhouse AJ (2009) Characterization of the effects of charged residues in the intracellular loop on ion permeation in alpha1 glycine receptor channels. J Biol Chem 284:2023–2030. 10.1074/jbc.M806618200 [DOI] [PubMed] [Google Scholar]
- Castaldo P, Stefanoni P, Miceli F, Coppola G, Del Giudice EM, Bellini G, Pascotto A, Trudell JR, Harrison NL, Annunziato L, Taglialatela M (2004) A novel hyperekplexia-causing mutation in the pre-transmembrane segment 1 of the human glycine receptor alpha1 subunit reduces membrane expression and impairs gating by agonists. J Biol Chem 279:25598–25604. 10.1074/jbc.M311021200 [DOI] [PubMed] [Google Scholar]
- Du J, Lü W, Wu S, Cheng Y, Gouaux E (2015) Glycine receptor mechanism elucidated by electron cryo-microscopy. Nature 526:224–229. 10.1038/nature14853 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gielen M, Thomas P, Smart TG (2015) The desensitization gate of inhibitory cys-loop receptors. Nat Commun 6:6829. 10.1038/ncomms7829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham BA, Tadros MA, Schofield PR, Callister RJ (2011) Probing glycine receptor stoichiometry in superficial dorsal horn neurones using the spasmodic mouse. J Physiol 589:2459–2474. 10.1113/jphysiol.2011.206326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassaine G, Deluz C, Grasso L, Wyss R, Tol MB, Hovius R, Graff A, Stahlberg H, Tomizaki T, Desmyter A, Moreau C, Li XD, Poitevin F, Vogel H, Nury H (2014) X-ray structure of the mouse serotonin 5-HT3 receptor. Nature 512:276–281. 10.1038/nature13552 [DOI] [PubMed] [Google Scholar]
- Hernandez CC, Kong W, Hu N, Zhang Y, Shen W, Jackson L, Liu X, Jiang Y, Macdonald RL (2017) Altered channel conductance states and gating of GABAA receptors by a pore mutation linked to Dravet syndrome. eNeuro February 4:ENEURO.0251-16.2017. 10.1523/ENEURO.0251-16.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hibbs RE, Gouaux E (2011) Principles of activation and permeation in an anion-selective cys-loop receptor. Nature 474:54–60. 10.1038/nature10139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hmami F, Wood SE, Chaouki S, Oulmaati A, Hida M, Rees MI, Chung SK, Bouharrou A (2014) Neonatal hyperekplexia with homozygous p.R392H mutation in GLRA1. Epileptic Disord 16:354–357. 10.1684/epd.2014.0663 [DOI] [PubMed] [Google Scholar]
- Huang X, Chen H, Michelsen K, Schneider S, Shaffer PL (2015) Crystal structure of human glycine receptor-alpha3 bound to antagonist strychnine. Nature 526:277–280. 10.1038/nature14972 [DOI] [PubMed] [Google Scholar]
- Hu XQ, Sun H, Peoples RW, Hong R, Zhang L (2006) An interaction involving an arginine residue in the cytoplasmic domain of the 5-HT3A receptor contributes to receptor desensitization mechanism. J Biol Chem 281:21781–21788. 10.1074/jbc.M600676200 [DOI] [PubMed] [Google Scholar]
- Jones MV, Westbrook GL (1996) The impact of receptor desensitization on fast synaptic transmission. Trends Neurosci 19:96–101. 10.1016/S0166-2236(96)80037-3 [DOI] [PubMed] [Google Scholar]
- Kelley SP, Dunlop JI, Kirkness EF, Lambert JJ, Peters JA (2003) A cytoplasmic region determines single-channel conductance in 5-HT3 receptors. Nature 424:321–324. 10.1038/nature01788 [DOI] [PubMed] [Google Scholar]
- Keramidas A, Lynch JW (2013) An outline of desensitization in pentameric ligand-gated ion channel receptors. Cell Mol Life Sci 70:1241–1253. 10.1007/s00018-012-1133-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kung AY, Rick C, O'Shea S, Harrison NL, McGehee DS (2001) Expression of glycine receptors in rat sensory neurons vs. HEK293 cells yields different functional properties. Neurosci Lett 309:202–206. 10.1016/S0304-3940(01)02066-3 [DOI] [PubMed] [Google Scholar]
- Langlhofer G, Janzen D, Meiselbach H, Villmann C (2015) Length of the TM3–4 loop of the glycine receptor modulates receptor desensitization. Neurosci Lett 600:176–181. 10.1016/j.neulet.2015.06.017 [DOI] [PubMed] [Google Scholar]
- Langosch D, Laube B, Rundström N, Schmieden V, Bormann J, Betz H (1994) Decreased agonist affinity and chloride conductance of mutant glycine receptors associated with human hereditary hyperekplexia. EMBO J 13:4223–4228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Wu DC, Wang YT (2010) Allosteric potentiation of glycine receptor chloride currents by glutamate. Nat Neurosci 13:1225–1232. 10.1038/nn.2633 [DOI] [PubMed] [Google Scholar]
- Lynch JW. (2004) Molecular structure and function of the glycine receptor chloride channel. Physiol Rev 84:1051–1095. 10.1152/physrev.00042.2003 [DOI] [PubMed] [Google Scholar]
- Lynch JW, Rajendra S, Pierce KD, Handford CA, Barry PH, Schofield PR (1997) Identification of intracellular and extracellular domains mediating signal transduction in the inhibitory glycine receptor chloride channel. EMBO J 16:110–120. 10.1093/emboj/16.1.110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinnon NK, Reeves DC, Akabas MH (2011) 5-HT3 receptor ion size selectivity is a property of the transmembrane channel, not the cytoplasmic vestibule portals. J Gen Physiol 138:453–466. 10.1085/jgp.201110686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinnon NK, Bali M, Akabas MH (2012) Length and amino acid sequence of peptides substituted for the 5-HT3A receptor M3M4 loop may affect channel expression and desensitization. PLoS One 7:e35563. 10.1371/journal.pone.0035563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller PS, Aricescu AR (2014) Crystal structure of a human GABAA receptor. Nature 512:270–275. 10.1038/nature13293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mine J, Taketani T, Yoshida K, Yokochi F, Kobayashi J, Maruyama K, Nanishi E, Ono M, Yokoyama A, Arai H, Tamaura S, Suzuki Y, Otsubo S, Hayashi T, Kimura M, Kishi K, Yamaguchi S (2015) Clinical and genetic investigation of 17 Japanese patients with hyperekplexia. Dev Med Child Neurol 57:372–377. 10.1111/dmcn.12617 [DOI] [PubMed] [Google Scholar]
- Muller E, Le Corronc H, Triller A, Legendre P (2006) Developmental dissociation of presynaptic inhibitory neurotransmitter and postsynaptic receptor clustering in the hypoglossal nucleus. Mol Cell Neurosci 32:254–273. 10.1016/j.mcn.2006.04.007 [DOI] [PubMed] [Google Scholar]
- Nikolic Z, Laube B, Weber RG, Lichter P, Kioschis P, Poustka A, Mülhardt C, Becker CM (1998) The human glycine receptor subunit alpha3. Glra3 gene structure, chromosomal localization, and functional characterization of alternative transcripts. J Biol Chem 273:19708–19714. 10.1074/jbc.273.31.19708 [DOI] [PubMed] [Google Scholar]
- Papke D, Grosman C (2014) The role of intracellular linkers in gating and desensitization of human pentameric ligand-gated ion channels. J Neurosci 34:7238–7252. 10.1523/JNEUROSCI.5105-13.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters JA, Hales TG, Lambert JJ (2005) Molecular determinants of single-channel conductance and ion selectivity in the cys-loop family: insights from the 5-HT3 receptor. Trends Pharmacol Sci 26:587–594. 10.1016/j.tips.2005.09.011 [DOI] [PubMed] [Google Scholar]
- Pribilla I, Takagi T, Langosch D, Bormann J, Betz H (1992) The atypical M2 segment of the beta subunit confers picrotoxinin resistance to inhibitory glycine receptor channels. EMBO J 11:4305–4311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purohit Y, Grosman C (2006) Block of muscle nicotinic receptors by choline suggests that the activation and desensitization gates act as distinct molecular entities. J Gen Physiol 127:703–717. 10.1085/jgp.200509437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajendra S, Lynch JW, Pierce KD, French CR, Barry PH, Schofield PR (1995) Mutation of an arginine residue in the human glycine receptor transforms beta-alanine and taurine from agonists into competitive antagonists. Neuron 14:169–175. 10.1016/0896-6273(95)90251-1 [DOI] [PubMed] [Google Scholar]
- Rea R, Tijssen MA, Herd C, Frants RR, Kullmann DM (2002) Functional characterization of compound heterozygosity for GlyRalpha1 mutations in the startle disease hyperekplexia. Eur J Neurosci 16:186–196. 10.1046/j.1460-9568.2002.02054.x [DOI] [PubMed] [Google Scholar]
- Saul B, Kuner T, Sobetzko D, Brune W, Hanefeld F, Meinck HM, Becker CM (1999) Novel GLRA1 missense mutation (P250T) in dominant hyperekplexia defines an intracellular determinant of glycine receptor channel gating. J Neurosci 19:869–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaefer N, Kluck CJ, Price KL, Meiselbach H, Vornberger N, Schwarzinger S, Hartmann S, Langlhofer G, Schulz S, Schlegel N, Brockmann K, Lynch B, Becker CM, Lummis SC, Villmann C (2015) Disturbed neuronal ER-golgi sorting of unassembled glycine receptors suggests altered subcellular processing is a cause of human hyperekplexia. J Neurosci 35:422–437. 10.1523/JNEUROSCI.1509-14.2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiang R, Ryan SG, Zhu YZ, Hahn AF, O'Connell P, Wasmuth JJ (1993) Mutations in the alpha 1 subunit of the inhibitory glycine receptor cause the dominant neurologic disorder, hyperekplexia. Nat Genet 5:351–358. 10.1038/ng1293-351 [DOI] [PubMed] [Google Scholar]
- Takahashi T, Momiyama A, Hirai K, Hishinuma F, Akagi H (1992) Functional correlation of fetal and adult forms of glycine receptors with developmental changes in inhibitory synaptic receptor channels. Neuron 9:1155–1161. 10.1016/0896-6273(92)90073-M [DOI] [PubMed] [Google Scholar]
- Unwin N. (2005) Refined structure of the nicotinic acetylcholine receptor at 4A resolution. J Mol Biol 346:967–989. 10.1016/j.jmb.2004.12.031 [DOI] [PubMed] [Google Scholar]
- Vergouwe MN, Tijssen MA, Peters AC, Wielaard R, Frants RR (1999) Hyperekplexia phenotype due to compound heterozygosity for GLRA1 gene mutations. Ann Neurol 46:634–638. 10.1002/1531-8249(199910)46:4%3C634::AID-ANA12%3E3.0.CO;2-9 [DOI] [PubMed] [Google Scholar]
- Villmann C, Oertel J, Melzer N, Becker CM (2009) Recessive hyperekplexia mutations of the glycine receptor alpha1 subunit affect cell surface integration and stability. J Neurochem 111:837–847. 10.1111/j.1471-4159.2009.06372.x [DOI] [PubMed] [Google Scholar]
- Xiong W, Chen SR, He L, Cheng K, Zhao YL, Chen H, Li DP, Homanics GE, Peever J, Rice KC, Wu LG, Pan HL, Zhang L (2014) Presynaptic glycine receptors as a potential therapeutic target for hyperekplexia disease. Nat Neurosci 17:232–239. 10.1038/nn.3615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Xue F, Liu Y, Yang H, Wang X (2013) The structural mechanism of the cys-loop receptor desensitization. Mol Neurobiol 48:97–108. 10.1007/s12035-013-8420-z [DOI] [PubMed] [Google Scholar]
- Zhong G, Droho S, Crone SA, Dietz S, Kwan AC, Webb WW, Sharma K, Harris-Warrick RM (2010) Electrophysiological characterization of V2a interneurons and their locomotor-related activity in the neonatal mouse spinal cord. J Neurosci 30:170–182. 10.1523/JNEUROSCI.4849-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou N, Wang CH, Zhang S, Wu DC (2013) The GLRA1 missense mutation W170S associates lack of Zn2+ potentiation with human hyperekplexia. J Neurosci 33:17675–17681. 10.1523/JNEUROSCI.3240-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]