

Abstract
Neuronal information processing requires multiple forms of synaptic plasticity mediated by NMDARs and AMPA-type glutamate receptors (AMPARs). These plasticity mechanisms include long-term potentiation (LTP) and long-term depression (LTD), which are Hebbian, homosynaptic mechanisms locally regulating synaptic strength of specific inputs, and homeostatic synaptic scaling, which is a heterosynaptic mechanism globally regulating synaptic strength across all inputs. In many cases, LTP and homeostatic scaling regulate AMPAR subunit composition to increase synaptic strength via incorporation of Ca2+-permeable receptors (CP-AMPAR) containing GluA1, but lacking GluA2, subunits. Previous work by our group and others demonstrated that anchoring of the kinase PKA and the phosphatase calcineurin (CaN) to A-kinase anchoring protein (AKAP) 150 play opposing roles in regulation of GluA1 Ser845 phosphorylation and CP-AMPAR synaptic incorporation during hippocampal LTP and LTD. Here, using both male and female knock-in mice that are deficient in PKA or CaN anchoring, we show that AKAP150-anchored PKA and CaN also play novel roles in controlling CP-AMPAR synaptic incorporation during homeostatic plasticity in hippocampal neurons. We found that genetic disruption of AKAP-PKA anchoring prevented increases in Ser845 phosphorylation and CP-AMPAR synaptic recruitment during rapid homeostatic synaptic scaling-up induced by combined blockade of action potential firing and NMDAR activity. In contrast, genetic disruption of AKAP-CaN anchoring resulted in basal increases in Ser845 phosphorylation and CP-AMPAR synaptic activity that blocked subsequent scaling-up by preventing additional CP-AMPAR recruitment. Thus, the balanced, opposing phospho-regulation provided by AKAP-anchored PKA and CaN is essential for control of both Hebbian and homeostatic plasticity mechanisms that require CP-AMPARs.
SIGNIFICANCE STATEMENT Neuronal circuit function is shaped by multiple forms of activity-dependent plasticity that control excitatory synaptic strength, including LTP/LTD that adjusts strength of individual synapses and homeostatic plasticity that adjusts overall strength of all synapses. Mechanisms controlling LTP/LTD and homeostatic plasticity were originally thought to be distinct; however, recent studies suggest that CP-AMPAR phosphorylation regulation is important during both LTP/LTD and homeostatic plasticity. Here we show that CP-AMPAR regulation by the kinase PKA and phosphatase CaN coanchored to the scaffold protein AKAP150, a mechanism previously implicated in LTP/LTD, is also crucial for controlling synaptic strength during homeostatic plasticity. These novel findings significantly expand our understanding of homeostatic plasticity mechanisms and further emphasize how intertwined they are with LTP and LTD.
Keywords: AKAP150, Ca2+-permeable AMPA receptor, calcineurin, homeostatic plasticity, LTP, PKA
Introduction
Hebbian plasticity in the hippocampus and cortex is input-specific, bidirectional [long-term potentiation/long-term depression (LTP/LTD)], and can be induced rapidly (seconds-minutes) but expressed persistently (hours-days; Collingridge et al., 2010; Huganir and Nicoll, 2013). However, excitatory neurons in these two regions can also persistently scale-up or -down synaptic strength across all inputs in response to chronic (hours-days) decreases or increases, respectively, in overall input and firing. This global compensatory adjustment of synaptic strength is termed homeostatic synaptic plasticity and is thought to stabilize neuronal circuits by constraining firing and synaptic strength within ranges that maintain connectivity, while preventing hyperexcitability and preserving Hebbian plasticity (Turrigiano, 2012; Chen et al., 2014; K. F. Lee et al., 2014). Not surprisingly, alterations in both LTP/LTD and homeostatic plasticity are implicated in nervous system diseases (Wondolowski and Dickman, 2013). Thus, it is of great interest to understand how Hebbian and homeostatic plasticity interact (Keck et al., 2017).
Homeostatic plasticity, like Hebbian, can be induced by changes in Ca2+ signaling downstream of NMDARs and/or L-type voltage-gated Ca2+ channels and are both expressed through changes in AMPAR localization and activity (O'Brien et al., 1998; Turrigiano et al., 1998; Thiagarajan et al., 2005; Sutton et al., 2006; Aoto et al., 2008; Ibata et al., 2008; K. Y. Lee and Chung, 2014). Nonetheless, it was originally thought that downstream mechanisms mediating Hebbian and homeostatic AMPAR regulation would be largely nonoverlapping. However, accumulating evidence indicates that these processes use many of the same signaling pathways. Indeed, several studies have implicated ser/thr protein kinases and phosphatases in homeostatic plasticity that also have well characterized roles in LTP/LTD, including Ca2+-CaM-dependent protein kinase II (CaMKII; Thiagarajan et al., 2002, 2005; Groth et al., 2011), the cAMP-dependent protein kinase PKA (Goel et al., 2011; Diering et al., 2014), and the Ca2+-CaM-dependent protein phosphatase 2B/CaN (Kim and Ziff, 2014). These postsynaptic kinase/phosphatase signaling pathways may regulate synaptic strength during both LTP/LTD and homeostatic plasticity at least in part through regulating the synaptic incorporation of high-conductance GluA1 CP-AMPARs (Thiagarajan et al., 2005; Plant et al., 2006; Lu et al., 2007; Yang et al., 2010; Goel et al., 2011; Soares et al., 2013; Kim and Ziff, 2014; Sanderson et al., 2016), which are largely excluded from hippocampal synapses under basal conditions (Lu et al., 2009; Rozov et al., 2012). In addition, homeostatic synaptic plasticity, like LTP/LTD, can be induced and expressed at individual synapses in dendrites through local regulation of protein synthesis and GluA1 CP-AMPAR delivery, further suggesting common underlying mechanisms (Sutton et al., 2006; Hou et al., 2008; Maghsoodi et al., 2008; Soares et al., 2013).
Our laboratory and others have shown that PKA and CaN anchored to the scaffold AKAP79/150 (79 human/150 rodent; Akap5 gene) play key opposing roles regulating GluA1 Ser845 phosphorylation to control CP-AMPAR recruitment/removal at hippocampal synapses during LTP/LTD (Lu et al., 2007; Sanderson et al., 2012, 2016; Zhang et al., 2013). GluA1 Ser845 phosphorylation has also been linked to CP-AMPAR synaptic incorporation during homeostatic scaling-up in vitro in cultured cortical neurons in response to neuronal silencing and in vivo in visual cortex in response to visual deprivation, with GluA1 S845A mutant knock-in mice showing disrupted homeostatic potentiation in both cases (Goel et al., 2011; Diering et al., 2014; Kim and Ziff, 2014). Recently, increases in Ser845 phosphorylation and dendritic GluA1 expression that occur during scaling-up were also linked to PKA signaling that requires AKAP150 expression (Diering et al., 2014). In addition, decreased CaN signaling, which occurs during neuronal silencing, is sufficient to induce increased Ser845 phosphorylation and scaling-up in cortical neurons through CP-AMPARs (Kim and Ziff, 2014). However, the specific roles of AKAP150-anchored PKA and CaN during homeostatic plasticity have not been investigated. Thus, here we used hippocampal neurons cultured from AKAP150 knock-in mice that are specifically deficient in either PKA or CaN anchoring to show that the AKAP150-PKA/CaN complex regulates homeostatic synaptic plasticity through bidirectional control of CP-AMPAR synaptic incorporation.
Materials and Methods
Animal use and care.
All animal procedures were conducted in accordance with National Institutes of Health (NIH), United States Public Health Service guidelines and with the approval of the University of Colorado, Denver, Institutional Animal Care and Use Committee. Generation and characterization of AKAP150 knock-out (KO) mice (RRID: MGI_5448696) were previously described by Tunquist et al. (2008). Generation and characterization of AKAP150 CaN-anchoring deficient ΔPIX (RRID: MGI_5448682) and PKA-anchoring deficient ΔPKA (RRID: MGI_5635498) knock-in mice were previously described by Sanderson et al. (2012, 2016) and Murphy et al. (2014).
Primary culture of mouse hippocampal neurons.
Mouse hippocampal neurons were prepared from both male and female mice as described previously with slight modifications (Sanderson et al., 2012). Briefly, hippocampi were dissected from WT, AKAP150 KO, ΔPIX, or ΔPKA P0–P2 postnatal mice. Neurons were plated at medium density (140,000–150,000 cells/ml) on poly-d-lysine/laminin-coated 12 mm glass coverslips for electrophysiology and immunocytochemistry or at high density (240,000–250,000 cells/ml) on 6-well coated dishes for biochemistry. For the first 24 h, cultures were grown in MEM supplemented media. The above solution was replaced with neurobasal A medium supplemented with B-27 (Invitrogen-Life Technologies), mitotic inhibitors (fluorodeoxyuridine), 0.5 mm Glutamax (Invitrogen-Life Technologies), and 12.5 μm l-glutamate (Sigma-Aldrich). After 3–4 d in vitro (DIV), l-glutamate was removed and replaced with neurobasal supplemented with B27, Glutamax, and mitotic inhibitors. Neurons were fed every 3 or 4 d thereafter until 14–16 DIV.
Electrophysiology.
All chemicals were purchased from Sigma-Aldrich unless otherwise noted. Whole-cell voltage-clamp recordings of miniature EPSCs (mEPSCs) from DIV 14–16 mouse hippocampal neurons were performed at 31°C under infrared differential interference contrast microscopy on BX51 upright microscope (Olympus) using an Axopatch 200B amplifier (Molecular Devices). Microelectrodes had resistances of 3–6 MΩ. AMPAR mEPSCs were recorded from morphologically identified hippocampal pyramidal neurons at a holding potential of −65 mV using a CsCl-based internal solution internal solution containing the following (in mm): 140 CsCl, 2 MgCl2, 1 CaCl2, 10 HEPES, 10 EGTA, 2 Na2-ATP, 0.3 NaGTP, pH 7.3, in artificial CSF (ACSF) containing the following (in mm): 126 NaCl, 3 KCl, 2 CaCl2, 1.25 NaH2PO4, 1 MgSO4, 26 NaHCO3, 10 glucose (osmolarity, 290 mOsm) plus 1 μm TTX, 50 μm picrotoxin. Access resistances were between 15 and 35 MΩ; if the access resistance changed >20%, the recording was discarded. mEPSC data were analyzed using Mini Analysis Program (Synaptosoft). Values for mean mEPSC amplitude and frequency and binned (2 pA bins) cumulative distributions of mEPSCs were exported for each cell and plotted, and analyzed in Prism (GraphPad) as described under Experimental design and statistics below. However, for experiments in Figure 3, where recordings were made from the same cell before and then after the addition of IEM1460 to block CP-AMPARs, the Kolmogorov–Smirnoff (K–S) test was applied in Mini Analysis Program to compare the un-binned cumulative amplitude distributions before and after IEM1460 for each single cell recorded from.
Figure 3.

Rapid homeostatic synaptic potentiation in hippocampal neurons requires CP-AMPAR synaptic recruitment that is mediated by AKAP150-anchored PKA and opposed by AKAP150-anchored CaN. A, Representative mEPSC recordings, (B) bar graph of mean mEPSC amplitude, (C, D) cumulative plots of mEPSC amplitudes, and (E) bar graph of mean mEPSC frequency showing that the CP-AMPAR antagonist IEM1460 (70 μm) acutely inhibits expression of rapid homeostatic scaling-up in WT mouse neurons. The same neurons were recorded for a 4–5 min baseline period followed by 5–10 min of additional recording in IEM1460. IEM significantly depresses mEPSC amplitude and frequency after induction of homeostatic scaling up with 4 h TTX/MK (*p < 0.05, **p < 0.01 to +IEM by unpaired t test) but not under Control conditions. F, Representative mEPSC recordings, (G) bar graph of mean mEPSC amplitude,(H, I) cumulative plots of mEPSC amplitudes, and (J) bar graph of mean mEPSC frequency showing that neurons from AKAP150KO mice express IEM1460-sensitive synaptic CP-AMPARs that are responsible for basally increased mEPSC activity (*p < 0.05, **p < 0.01 to +IEM by paired t test) and occlude homeostatic scaling-up in response to 4 h TTX/MK (*p < 0.05, **p < 0.01 to +IEM by paired t test) and 24 h TTX/MK treatments (*p < 0.05, **p < 0.01 to +IEM by paired t test). K, Representative mEPSC recordings, (L) bar graph of mean mEPSC amplitude, (M, N) cumulative plots of mEPSC amplitudes, and (O) bar graph of mean mEPSC frequency showing that impaired homeostatic scaling-up in neurons from AKAP150ΔPKA mice is associated with a failure to recruit CP-AMPARs to synapses following TTX/MK treatments. P, Representative mEPSC recordings, (Q) bar graph of mean mEPSC amplitude, (R, S) cumulative plots of mEPSC amplitudes, and (T) bar graph of mean mEPSC frequency showing that neurons from AKAP150ΔPIX mice express IEM1460-sensitive synaptic CP-AMPARs under control conditions (**p < 0.01 to +IEM by paired t test) that prevent homeostatic scaling-up in response to subsequent TTX/MK treatments, despite not exhibiting any basal increases in mEPSC amplitude (*p < 0.05, **p < 0.01 to +IEM by paired t test). Error bars indicate SEM.
To induce homeostatic scaling, DIV 14–16 neurons were treated in cultured media for designated time intervals with tetrodotoxin (TTX; 2 μm; 4, 24, or 48 h), MK801 (10 μm; 3 h), or various combinations of TTX and MK801 as indicated in the Results and figure legends. Coverslips were removed and placed in a recording chamber. mEPSCs were recorded from individual neurons for 4–5 min. For experiments in Figure 1 examining the impacts of NASPM (Tocris Bioscience), NASPM (30 μm) was added either for the entire durations of the 4, 24, or 48 h TTX treatments or only during the 3 h MK801 portions of the combined TTX/MK treatments and found to prevent scaling-up equivalently in both cases. Thus, these NASPM data were pooled for presentation in Figure 1, C, D, and I–K. For experiments in Figure 1I–K, 70 μm IEM1460 (Tocris Bioscience) was added after the indicated scaling-up treatments but before recording mEPSC amplitudes. In Figure 3, mEPSCs were recorded for a baseline of 4–5 min and then for an additional 5–10 min in the presence of IEM1460 (70 μm) from the same neurons. Statistical differences between means values for different experimental conditions were determined using Prism by either Student's t test (for pairwise comparisons) or one-way ANOVA with Dunnett's post hoc (for multiple comparisons to a control condition). For graphical comparisons between aggregate mEPSC amplitude cumulative distributions, the binned (2 pA bins) cumulative distributions for each cell were averaged for each condition in Prism and then are plotted as mean ± SEM for each condition.
Figure 1.

Homeostatic synaptic potentiation requires synaptic recruitment of CP-AMPARs in hippocampal neurons. A, Representative mEPSC recordings and (B) cumulative plots of mEPSC amplitudes showing scaling-up induced by 4 or 24 h TTX (2 μm) treatment with MK801 (10 μm) also present for the last 3 h (4 h TTX/MK or 24 h TTX/MK) in 15–16 DIV WT mouse hippocampal neurons. C, Representative mEPSC recordings and (D) cumulative plots of mEPSC amplitudes showing that inclusion of NASPM (20 μm) prevents scaling up induced by 4 or 24 h TTX/MK and 48 h TTX treatments. E, Representative mEPSC recordings and (F) cumulative plots of mEPSC amplitudes showing that 48 h TTX but not 4 or 24 h TTX alone induces robust scaling-up. G, Representative mEPSC recordings and (H) cumulative plots of mEPSC amplitudes showing that 3 or 24 h MK801 alone does not induce robust scaling up. I, Cumulative plots of mEPSC amplitudes showing that 48 h TTX induced scaling-up is prevented by NASPM cotreatment and partially reversed by acute treatment with IEM1460 (70 μm). J, Bar graph summaries of data in A–I for regulation of mean mEPSC amplitude and (K) frequency. *p < 0.05, **p < 0.01 to Control by one-way ANOVA; #p < 0.05, ∧p < 0.05 by t test. Error bars indicate SEM.
Immunoblotting.
Mouse hippocampal neurons were collected and sonicated in buffer (1% SDS, 10 mm EDTA, 100 mm Tris, pH 8). This homogenate was heated to 95°C for 5 min and then frozen at −70°C until SDS-PAGE. For pS845 GluA1 immunoblotting analysis, 10 μg of extract was loaded on gels and blots were probed first overnight at 4°C with rabbit anti-GluA1-S845 (1:1000; Millipore, AB5849; RRID:AB_2532143) followed by stripping and re-probing of the same blots overnight at 4°C with rabbit anti-GluA1 (1:1000; Millipore, PC2461; RRID:AB_253142), as above, to detect total receptor levels. Detection was performed with HRP-coupled secondary antibodies (Bio-Rad; 1:1000) and enhanced chemiluminescence (West Pico or West Dura Chemiluminescent Substrate, Pierce). Chemiluminescence was imaged using a Fluorchem gel documentation system (Alpha Innotech), and band intensities were analyzed using ImageJ software (NIH). For GluA1 pS845 analysis, band intensities for pS845 were normalized to the total GluA1 intensity for the same band after stripping and re-probing the same blot to give a normalized pS845/GluA1 ratio. pS845/GluA1 ratio values were then normalized to the ratio value obtained using the same exposure conditions for WT control samples that were always run on the same blot, as previously described (Sanderson et al., 2012, 2016). Statistical differences between means values in pairwise comparisons of two different conditions were determined by Student's t test using Prism.
Immunocytochemistry.
To measure total GluA1 AMPAR colocalization with PSD-95, neurons were fixed for 10 min at room temp in 4% paraformaldehyde, rinsed with PBS, permeabilized with 0.2% Triton X-100 for 10 min, and then blocked for 48 h in 10% BSA in PBS. Coverslips were then incubated with rabbit anti-GluA1 (Millipore #PC2461; RRID:AB_253142; 1:500) and mouse anti-PSD-95 antibodies (NeuroMab, 75-028; RRID:AB_2292909; 1:1000) in 1% BSA followed by goat anti-rabbit Texas Red and goat anti-mouse AlexaFluor 647-conjugated fluorescent secondary antibodies (Invitrogen). For measurements of synaptic surface GluA1 AMPAR levels, neurons were live-labeled at 37°C for 15 min in culture media with a rabbit polyclonal antibody (Millipore, ABN241; RRID:AB_2721164; 1:250) that recognizes the extracellular GluA1 N-terminal domain followed by washing in culture media, washing in PBS, fixation in 4% paraformaldehyde/4% sucrose, and labeling with goat anti-rabbit Texas Red secondary antibody before permeabilization and subsequent labeling with mouse anti-PSD-95 primary and goat anti-mouse AlexaFluor 647 fluorescent secondary antibodies as above.
Fluorescence microscopy and image analysis.
Deconvolved images of immunostained coverslips were obtained on a Axiovert 200M microscope (Carl Zeiss) equipped with a 300W xenon lamp (Sutter Instrument), 63× Plan-Apo/1.4 NA objective, Cy3/Texas Red and Cy5/AlexaFluor 647 filter sets (Chroma), Coolsnap CCD camera (Photometrics), and SlideBook 5.5–6.0 software (Intelligent Imaging Innovations) as described previously (K. E. Smith et al., 2006; Horne and Dell'Acqua, 2007; Sanderson et al., 2012). Briefly, images were acquired with 2 × 2 binning. Three-dimensional z-stack images of x, y planes with 0.5 μm steps were collected and deconvolved to the nearest neighbor to generate confocal x, y sections. Two-dimensional maximum-intensity projection images were generated from these deconvolved image stacks for quantitative mask analysis in SlideBook 6.0 to measure GluA1/PSD-95 colocalization in manually drawn masked dendritic sections using Pearson's correlation (r): scale of 1, perfect correlation; 0, no correlation/random overlap; to −1, inverse correlation. To quantify changes in total or surface synaptic GluA1 localization following homeostatic scaling, total and surface GluA1/PSD-95 correlation values were then normalized to corresponding control values and are expressed as fold-change. Statistical differences between means values in pairwise comparisons of two different experimental conditions were determined by Student's t test using Prism. For statistical comparisons across three or more conditions, one-way ANOVA with Dunnett's post hoc analysis was used in Prism.
Experimental design and statistics.
The design of each experiment and statistical analysis methods used are described above under each type of experiment. However, we briefly summarize those methods here: all data in bar graphs are plotted as mean ± SEM. For statistical comparisons of mean values for two or more experimental conditions to a control condition one-way ANOVA with Dunnett's post hoc was used in Prism. Statistical differences between means values in pairwise comparisons of two different experimental conditions were determined by Student's t test using Prism. Unpaired t tests were used in all cases except for experiments in Figure 3, where paired t tests were used for mean values from recordings made from the same cell before and then after the addition of IEM1460 to block CP-AMPARs. In all cases a statistically significant difference between means is considered with values of *p < 0.05. Values of p > 0.05 are considered not significant (NS). Binned (2 pA bins) cumulative distributions of mEPSC amplitude were exported from Mini Analysis Program for each cell and then averaged in Prism to generate plots as mean ± SEM. For experiments in Figure 3 where recordings were made from the same cell before and then after the addition of IEM1460 to block CP-AMPARs, we also applied the K–S test in Mini Analysis Program to compare differences in the raw, un-binned cumulative amplitude distributions before and after IEM1460 for each single cell recorded from with statistical significance between distributions reported as ***p < 0.001. Thus, in the Results section for these experiments, we report how many cells out of the total number of cells recorded from for each condition showed significantly different K–S test p values (i.e., ***p < 0.001) for before vs after IEM addition.
Results
Homeostatic synaptic potentiation in WT hippocampal neurons requires CP-AMPAR activity
Previous studies implicating AKAP150, PKA, and/or CaN in regulating GluA1 S845 phosphorylation and CP-AMPAR synaptic recruitment during homeostatic plasticity used prolonged (48 h) inhibition of action potential (AP) firing with the Na+ channel TTX to induce scaling-up in cultured cortical neurons (Diering et al., 2014; Kim and Ziff, 2014). However, prior studies in hippocampal neurons showed that similar homeostatic scaling-up is induced and expressed more rapidly (3–4 h) in response to combined inhibition of AP firing with TTX and of NMDAR activity with the competitive antagonist AP-V (Sutton et al., 2006). Furthermore, this rapid scaling-up is also expressed by CP-AMPARs and can be induced even by local application of AP-V in dendrites to promote protein synthesis-dependent local delivery of GluA1 to the plasma membrane surface and synapses (Sutton et al., 2006). However, it is not known whether AKAP150, PKA, and/or CaN signaling and regulation of GluA1 Ser845 phosphorylation are also involved in CP-AMPAR synaptic incorporation during this rapid homeostatic scaling.
Thus, we first evaluated the impact of AP firing blockade with TTX for 4 h combined with inhibition of NMDAR Ca2+ influx with the open channel blocker MK801 for the last 3 h on AMPAR-mediated mEPSCs in hippocampal neurons cultured from WT mice (4 h TTX/MK; Fig. 1A). In agreement with prior studies using TTX/AP-V treatment (Sutton et al., 2006), this 4 h TTX/MK treatment caused a prominent rightward shift in the cumulative distribution of mEPSC amplitudes (Fig. 1B) and a significant increase in mean mEPSC amplitude compared with untreated controls (Fig. 1J; Control: 15.8 ± 0.6 pA, n = 45; 4 h TTX/MK: 20.1 ± 1.5 pA, n = 16; **p < 0.01 by one-way ANOVA). In agreement with prior findings that rapid homeostatic scaling requires CP-AMPAR activity (Sutton et al., 2006), inclusion of the CP-AMPAR open-channel blocker NASPM along with TTX and MK801 largely prevented the observed increases in AMPAR activity (Fig. 1C), as shown by only a small rightward shift in amplitude cumulative distribution (Fig. 1D) and no significant increase in mean mEPSC amplitude compared with controls (Fig. 1J; 4 h TTX/MK/NASPM: 17.6 ± 0.7, n = 15; NS p > 0.05 to Control by one-way ANOVA). Similarly robust scaling-up of mEPSC amplitudes was also observed when AP firing was blocked with TTX for 24 h and MK801 was added only for the last 3 h (Fig. 1A,B,I; 24 h TTX/MK: 21.2 ± 1.2 pA, n = 23; **p < 0.01 to Control by one-way ANOVA; NS p = 0.56 compared with 4 h TTX/MK by unpaired t test). Likewise, this homeostatic increase in AMPAR mEPSC amplitudes in response to 24 h TTX/MK treatment was also prevented by NASPM (Fig. 1C,D,J; 24 h TTX/MK/NASPM: 15.3 ± 0.7 pA, n = 14; NS p > 0.05 to Control by one-way ANOVA). As also seen in previous studies (Sutton et al., 2006), neither blockade of AP firing alone with TTX (Fig. 1E) or NMDARs alone with MK801 (Fig. 1G) for 4 or 24 h was able to induce substantial scaling-up of mEPSC amplitude distribution (Fig. 1F,H) or significantly increase mean mEPSC amplitude (Fig. 1J; 4 h TTX: 15.9 ± 0.9 pA, n = 13; 24 h TTX: 14.9 ± 0.7 pA, n = 13; 3 h MK: 16.4 ± 0.7 pA, n = 10; 24 h MK801: 16.1 ± 1.1 pA, n = 10; all NS p > 0.05 to Control by one-way ANOVA).
However, consistent with many other previous studies of homeostatic plasticity induced by more prolonged neuronal silencing (Stellwagen and Malenka, 2006; Gainey et al., 2009, 2015; Diering et al., 2014; Kim and Ziff, 2014), blockade of AP firing with TTX alone for 48 h was able to induce scaling-up (Fig. 1E), as reflected by both a robust rightward shift in mEPSC amplitude distribution (Fig. 1F) and a significant increase in mean mEPSC amplitude (Fig. 1J; 48 h TTX: 23.7 ± 1.3 pA, n = 22, **p < 0.01 to Control by one-way ANOVA; NS p > 0.05 to both 4 and 24 h TTX/MK by one-way ANOVA). Importantly, as also seen for the rapid 4 and 24 h TTX/MK scaling-up conditions, cotreatment with NASPM (Fig. 1C) prevented the 48 h TTX treatment from shifting the distribution of mEPSC amplitudes to the right (Fig. 1D) and from increasing mean mEPSC amplitude (Fig. 1J; 48 h TTX/NASPM: 13.7 ± 0.7 pA, n = 7; NS p > 0.05 to Control by one-way ANOVA). Thus, homeostatic potentiation induced in WT mouse hippocampal neurons either rapidly by combined inhibition of AP firing and NMDARs or more slowly by inhibition AP firing alone requires postsynaptic CP-AMPAR activity.
Finally, the combined 4 and 24 h TTX/MK treatments that led to rapid scaling-up of mEPSC amplitudes also significantly increased mEPSC frequency in a NASPM-sensitive manner, whereas all other treatments, including the more prolonged 48 h TTX scaling-up treatment, did not significantly change mEPSC frequency (Fig. 1K; Control: 6.4 ± 0.7 Hz, n = 16; 4 h TTX/MK: 12.5 ± 2.5 Hz, n = 16; 24 h TTX/MK: 19.2 ± 3.0 Hz, n = 23; *p < 0.05, **p < 0.01 to Control by one-way ANOVA, NS p = 0.11, 4 h TTX/MK compared with 24 h TTX/MK; 4 h TTX/MK/NASPM: 4.7 ± 1.2 Hz, n = 15; 24 h TTX/MK/NASPM: 5.4 ± 0.9 Hz, n = 14; 4 h TTX: 5.3 ± 1.0 Hz, n = 13; 24 h TTX: 4.6 ± 0.9 Hz, n = 13; 3 h MK: 2.8 ± 0.5 Hz, n = 9; 24 h MK801: 5.6 ± 1.3 Hz, n = 11; 48 h TTX: 7.3 ± 1.2 Hz, n = 22; 48 h TTX/NASPM: 3.7 ± 0.8 Hz, n = 7; all others NS p > 0.05 to Control by one-way ANOVA). Based on the developmental age of our cultures (14–16 DIV) and prior work by others, this increase in mEPSC frequency accompanying rapid homeostatic scaling-up of mEPSC amplitudes induced by TTX/MK801 treatment is likely mediated by both some amount of postsynaptic unsilencing due to recruitment of high conductance CP-AMPARs to synapses that previously lacked detectable AMPAR responses as well as GluA1 and BDNF-mediated retrograde communication enhancing presynaptic glutamate release at all synapses where CP-AMPARs were inserted (Jakawich et al., 2010; Lindskog et al., 2010; Groth et al., 2011). Interestingly, chemical induction of LTP in cultured rodent neurons also increases both mEPSC amplitude and frequency through postsynaptic GluA1 CP-AMPAR recruitment (Lu et al., 2001; Fortin et al., 2010; Keith et al., 2012).
Knock-out of AKAP150 increases basal mEPSC amplitude and prevents rapid homeostatic scaling-up in hippocampal neurons
A previous study found that knock-down of AKAP150 expression using RNAi inhibited the increases in Ser845 phosphorylation and GluA1 surface expression that accompany homeostatic scaling-up in cortical neurons (Diering et al., 2014). However, the role of AKAP150 in directly regulating AMPAR synaptic strength (i.e., mEPSC amplitude) was not examined in this previous study nor were the specific roles of AKAP-anchored PKA versus CaN signaling, both of which would be inhibited by RNAi knockdown of AKAP150. Accordingly, to compare to previous work using RNAi knockdown, we first examined homeostatic scaling-up in hippocampal neurons cultured from AKAP150 KO mice (Tunquist et al., 2008). In contrast to the robust scaling-up of mEPSC amplitudes and increased mEPSC frequency seen in WT mouse neurons in response to either the 4 or 24 h TTX/MK treatments (Fig. 2A,B,I,J), AKAP150 KO neurons failed to show any increases in mEPSC amplitude (Fig. 2C,D,I; KO Control: 18.6 ± 0.9 pA, n = 28; KO 4 h TTX/MK: 19.7 ± 1.3 pA, n = 22; KO 24 h TTX/MK: 18.6 ± 1.0 pA, n = 28; all NS p > 0.05 to KO Control by one-way ANOVA) or frequency compared with the untreated controls (Fig. 2J; KO Control: 6.5 ± 0.9 Hz, n = 28; KO 4 h TTX/MK: 7.2 ± 1.35 Hz, n = 22; KO 24 h TTX/MK: 7.6 ± 1.4 Hz, n = 28; all NS p > 0.05 to KO Control by one-way ANOVA). However, under control conditions mEPSC frequency was similar (Fig. 2J), but, surprisingly, the cumulative distribution of mEPSC amplitudes was already right-shifted (Fig. 2D) and mEPSC mean amplitude was already significantly increased in AKAP150 KO compared with WT controls. This basal amplitude increase was then maintained after the 4 and 24 h TTX/MK treatments (Fig. 2D,I; all #p < 0.05 to WT Control by one-way ANOVA). Thus, the basal increase in mEPSC amplitude in AKAP150 KO neurons may be occluding homeostatic scaling-up.
Figure 2.

Rapid homeostatic synaptic potentiation in hippocampal neurons requires AKAP150-anchoring of both PKA and CaN: disruption by knock-out of AKAP150 (KO) and genetic deletion of PKA-anchoring (ΔPKA) or CaN-anchoring (ΔPIX). A, Representative mEPSC recordings and (B) cumulative plots of mEPSC amplitudes showing scaling-up induced by 4 or 24 h TTX (2 μm) with MK801 (10 μm) present for the last 3 h (4 h TTX/MK or 24 h TTX/MK) in 15–16 DIV WT mouse hippocampal neurons (data are reproduced from Fig. 1A and B). C, Representative mEPSC recordings and (D) cumulative plots of mEPSC amplitudes showing that scaling-up induced by 4 or 24 h TTX/MK treatment is absent in neurons from AKAP150 KO mice. Under basal conditions AKAP150 KO neurons already exhibit a rightward shift in mEPSC amplitude distribution toward larger amplitudes compared with WT controls (black line) and the 4 and 24 h TTX/MK treatments fail to produce any additional shifts. E, Representative mEPSC recordings and (F) cumulative plots of mEPSC amplitudes showing that scaling-up induced by 4 or 24 h TTX/MK is absent in neurons from PKA anchoring-deficient AKAP150ΔPKA mice. G, Representative mEPSC recordings and (H) cumulative plots of mEPSC amplitudes showing that scaling-up induced by 4 or 24 h TTX/MK is absent in neurons from CaN anchoring-deficient AKAP150ΔPIX mice. Bar graph summaries of data in A–H showing the 4 and 24 h TTX/MK conditions induce significant increases in mean mEPSC (I) amplitude and (J) frequency in hippocampal neurons from WT but not from AKAP150 KO, ΔPKA, or ΔPIX mice. *p < 0.05, **p < 0.01 to corresponding Controls by one-way ANOVA; #p < 0.05 to WT Control by one-way ANOVA. Error bars indicate SEM.
AKAP150-anchored PKA and CaN are both required for rapid homeostatic scaling-up
As alluded to above, AKAP150 KO/RNAi knockdown eliminates/suppresses all of the many functions of this multifunctional scaffold (Fig. 2A), including structural interactions with membrane lipids (Dell'Acqua et al., 1998; Keith et al., 2012), the actin cytoskeleton (Gomez et al., 2002), cell adhesion molecules (Gorski et al., 2005), and other postsynaptic scaffold proteins (Colledge et al., 2000; Bhattacharyya et al., 2009; Robertson et al., 2009), anchoring of adenylyl cyclase (Bauman et al., 2006; Efendiev et al., 2010; Willoughby et al., 2010) and PKC (Klauck et al., 1996; Hoshi et al., 2003; Tavalin, 2008; Hoshi et al., 2010), as well as the opposing signaling functions of anchored PKA and CaN (Carr et al., 1992; Glantz et al., 1992; Coghlan et al., 1995; Dell'Acqua et al., 2002). Thus, given that both increased PKA and decreased CaN signaling have been linked to regulation of GluA1 phosphorylation and synaptic function during scaling-up, as discussed above, AKAP150 KO/RNAi alone is not sufficient to determine the role of this signaling complex in homeostatic plasticity or other previously characterized neuronal functions (Hoshi et al., 2005; Oliveria et al., 2007; Jurado et al., 2010; Keith et al., 2012; Zhang and Shapiro, 2012; Dittmer et al., 2014; Murphy et al., 2014). To specifically investigate the roles of anchored PKA versus CaN in homeostatic synaptic potentiation, we used hippocampal neurons cultured from our previously generated AKAP150 knock-in mice that are selectively deficient in PKA anchoring (Fig. 2E; ΔPKA, in-frame deletion of residues 709–718 in the amphipathic α-helical PKA-RII anchoring domain; Murphy et al., 2014; Sanderson et al., 2016) or CaN anchoring (Fig. 2G; ΔPIX, in-frame deletion of residues 655–661 constituting the PxIxIT-like CaNA anchoring motif; Oliveria et al., 2007; Sanderson et al., 2012). AKAP150 ΔPKA and ΔPIX mice (as well as PKA-anchoring deficient AKAP150 D36 mice; Lu et al., 2007) have been instrumental in elucidating the specific roles of AKAP150-anchored PKA and CaN in regulating Hebbian plasticity where LTP/LTD (and in some cases behavioral) phenotypes in these knock-in mice were found to be not only distinct from one another but also from those observed in AKAP150 KO mice (Lu et al., 2007, 2008; Tunquist et al., 2008; Weisenhaus et al., 2010; Sanderson et al., 2012, 2016; Zhang et al., 2013).
Interestingly, genetic deletion of either PKA anchoring in ΔPKA mice (Fig. 2E,F) or CaN anchoring in ΔPIX mice (Fig. 2G,H) was sufficient to prevent homeostatic scaling-up of mEPSC amplitude in response to both the 4 and 24 h TTX/MK treatments (Fig. 2I; ΔPKA Control: 14.8 ± 1.0 pA, n = 26; ΔPKA 4 h TTX/MK: 16.4 ± 0.9 pA, n = 14; ΔPKA 24 h TTX/MK: 15.3 ± 0.9 pA, n = 22; ΔPIX Control: 15.7 ± 0.7 pA, n = 31; 4 h ΔPIX TTX/MK: 15.2 ± 1.2 pA, n = 20; ΔPIX 24 h TTX/MK: 15.0 ± 0.8 pA, n = 23; all NS p > 0.05 compared with respective controls by one-way ANOVA). In addition, neither the 4 nor 24 h TTX/MK treatment increased mEPSC frequency in neurons from ΔPKA or ΔPIX mice (Fig. 2J; ΔPKA Control: 8.9 ± 1.5 Hz, n = 24; ΔPKA 4 h TTX/MK: 8.8 ± 2.2 Hz, n = 14; ΔPKA 24 h TTX/MK: 8.8 ± 1.4 Hz, n = 22; ΔPIX Control: 9.9 ± 1.7 Hz, n = 31; ΔPIX 4 h TTX/MK: 6.2 ± 1.3 Hz, n = 20; ΔPIX 24 h TTX/MK: 11.7 ± 2.2 Hz, n = 23; all NS p > 0.05 compared with respective controls by one-way ANOVA). However, in contrast to AKAP150 KO neurons, basal mEPSC amplitudes in untreated controls were not increased in ΔPKA and ΔPIX neurons compared with WT neurons (Fig. 2F,H,I; both NS p > 0.05 compared with WT Control by one-way ANOVA). Thus, unlike with complete loss of all AKAP150 signaling functions in KO neurons, the impaired homeostatic plasticity observed with selective loss of PKA or CaN anchoring in ΔPKA and ΔPIX neurons cannot be attributed to increased basal AMPAR transmission.
AKAP150-anchored PKA and CaN control homeostatic synaptic plasticity through opposing regulation of CP-AMPAR synaptic recruitment
Inhibition of scaling-up by inclusion of NASPM during the 4 and 24 h TTX/MK and 48 h TTX treatments in WT neurons in Figure 1 is consistent with a requirement for CP-AMPARs in induction and/or expression of homeostatic plasticity. To independently confirm a requirement for CP-AMPAR in expression of rapid homeostatic scaling (as seen in previous work; Sutton et al., 2006), we also performed recordings from the same cells before and after application of the CP-AMPAR antagonist IEM1460 for WT neurons under control conditions or following induction of scaling with 4 or 24 h TTX/MK treatment (Fig. 3A). In WT control neurons, IEM1460 had no measurable impact on mEPSC amplitude (Fig. 3B; WT Control before vs after +IEM only 1 of 7 neurons: ***p < 0.001 by K–S test; Fig. 3C; WT Control: 14.3 ± 1.4 pA, +IEM 13.9 ± 1.0 pA, n = 7; NS p > 0.05 by paired t test) or frequency (Fig. 3E; WT Control: 7.3 ± 1.7 Hz, +IEM 7.1 ± 1.8 Hz, n = 7; NS p > 0.05 by paired t test) consistent with GluA2-containing AMPARs supporting the majority of basal transmission in hippocampal neurons (Lu et al., 2009; Rozov et al., 2012). In contrast, following induction of scaling-up with 4 or 24 h TTX/MK treatment, IEM application essentially completely reversed the observed increases in mEPSC amplitude [Fig. 3B; WT 24 h TTX/MK before vs after +IEM 6 of 6 neurons: ***p < 0.001 by K–S test; WT 4 h TTX/MK before vs after IEM 8 of 14 neurons: ***p < 0.001 by K–S test (amplitude distributions not shown); Fig. 3D; 4 h TTX/MK: 21.2 ± 1.8 pA, +IEM 15.8 ± 0.8 pA, n = 14, **p < 0.01 by paired t test; 24 h TTX/MK: 19.5 ± 1.7 pA, +IEM 14.3 ± 0.9 pA, n = 6, **p < 0.01 by paired t test] and frequency (Fig. 2E; 4 h TTX/MK: 15.8 ± 5.0 Hz, +IEM 7.9 ± 1.9 Hz; n = 14; *p < 0.05 by paired t test; 24 h TTX/MK: 18.4 ± 5.3 Hz, +IEM 8.5 ± 2.5 Hz; n = 6; *p < 0.05 by paired t test). Application of IEM1460 also substantially reduced the increase in mean mEPSC amplitude (Fig. 1J; 48 h TTX: 23.7 ± 1.3 pA, n = 22; 48 h TTX+IEM: 16.8 ± 1.1 pA, n = 6; #p = 0.015 by t test) and the rightward-shift in mEPSC amplitude distribution (Fig. 1I) observed above for scaling-up induced with 48 h TTX treatment; IEM had no impact on mEPSC frequency compared with 48 h TTX alone (Fig. 1K; 48 h TTX: 7.3 ± 1.2 Hz, n = 22; 48 h TTX+IEM: 7.4 ± 1.2, n = 6; NS by t test). However, unlike for 4 and 24 h TTX/MK scaling-up, IEM was not as effective as NASPM in completely reversing the right-shifted mEPSC amplitude distribution (Fig. 1I) or the increased mean mEPSC amplitude for 48 h TTX scaling-up (Fig. 1J; 48 h TTX/NASPM compared with 48 h TTX+IEM ∧p = 0.022 by t test). This incomplete effect of IEM in reversing scaling-up compared with the complete effect of NASPM in preventing scaling-up indicates that CP-AMPARs are important for both the induction and expression of homeostatic potentiation in response to 48 h TTX treatment but that GluA2-containing AMPARs are also likely making contributions to expression. This result is consistent with several previous studies showing that interfering with GluA2 expression and its regulators can also disrupt homeostatic synaptic plasticity, especially when induced with prolonged (48–72 h) TTX treatments (Gainey et al., 2009; 2015; Anggono et al., 2011; Ancona Esselmann et al., 2017; but see Altimimi and Stellwagen, 2013).
Based on our confirmation that synaptic incorporation of CP-AMPARs is crucial for not only induction but also expression of scaling-up in WT mouse hippocampal neurons, we hypothesized that the impaired homeostatic plasticity observed in AKAP150 KO, ΔPKA, and ΔPIX neurons could be due to alterations in CP-AMPAR regulation in these mutant mice. Consistent with this idea, previous work on Hebbian plasticity mechanisms found that AKAP150-anchored PKA and CaN oppose each other in regulating synaptic incorporation and removal of CP-AMPARs during LTP and LTD at CA1 synapses. In particular, PKA anchoring-deficient ΔPKA and D36 mice exhibited impaired CP-AMPAR synaptic recruitment during LTP and LTD, whereas CaN anchoring-deficient ΔPIX mice exhibited enhanced CP-AMPAR synaptic recruitment during LTP and impaired removal during LTD (Lu et al., 2007; Sanderson et al., 2012, 2016). Consistent with the idea that impaired scaling-up in ΔPKA neurons is due to a failure to recruit CP-AMPARs to synapses, IEM1460 had no impact on mEPSC transmission in ΔPKA neurons under basal conditions (Fig. 3L; ΔPKA Control: 13.2 ± 0.7 pA, +IEM 12.8 ± 0.6 pA, n = 14, NS p > 0.05 by paired t test; Fig. 3M; before vs after +IEM only 1 of 14 neurons: ***p < 0.001 by K–S test; Fig. 3O; ΔPKA Control: 5.5 ± 1.0 Hz, +IEM 4.3 ± 0.9 Hz, n = 14, NS p > 0.05 by paired t test) or after the 4 and 24 h TTX/MK treatments [Fig. 3L; 4 h TTX/MK: 15.2 ± 1.0 pA, +IEM 14.9 ± 0.7 pA, n = 9, NS p > 0.05 by paired t test; 24 h TTX/MK: 14.2 ± 1.0 pA, +IEM 13.8 ± 0.8 pA, n = 7, NS p > 0.05 by paired t test; Fig. 3N; 4 h TTX/MK before vs after +IEM only 1 of 9 neurons: ***p < 0.001 by K–S test (cumulative distributions not plotted); 24 h TTX/MK before vs after +IEM only 1 of 7 neurons: ***p < 0.001 by K–S test; Fig. 3O; 4 h TTX/MK: 6.8 ± 2.6 Hz, +IEM 5.4 ± 1.7 Hz, n = 9, NS p > 0.05 by paired t test; 24 h TTX/MK: 6.9 ± 1.5 Hz, +IEM 4.8 ± 0.9 Hz, n = 7; NS p > 0.05 by paired t test]. Thus, genetic disruption of AKAP150-PKA anchoring prevents CP-AMPAR synaptic incorporation, not only during LTP/LTD, but also during homeostatic potentiation.
We previously found that ΔPIX mice, unlike WT or ΔPKA, have substantially increased contributions of CP-AMPARs to otherwise normal basal AMPAR transmission at CA1 synapses (Sanderson et al., 2012, 2016). Accordingly, application of IEM1460 to ΔPIX cultured neurons (Fig. 3P) under basal control conditions caused significant decreases in mEPSC amplitude (Fig. 3Q; ΔPIX Control: 15.6 ± 0.8 pA, +IEM 12.5 ± 0.8 pA, n = 20; **p < 0.01 by paired t test; Fig. 3R; before vs after +IEM 15 of 20 neurons: ***p < 0.001 by K–S test) and frequency (Fig. 3T; ΔPIX Control: 9.8 ± 2.4 Hz, +IEM 5.9 ± 1.1 Hz, n = 20; **p < 0.01 by paired t test). Interestingly, the increased basal mEPSC transmission observed in AKAP150 KO neurons was also sensitive to inhibition by IEM1460 (Fig. 3G; KO Control: 19.5 ± 1.6 pA, +IEM 14.4 ± 0.7 pA, n = 10; **p < 0.01 by paired t test; Fig. 3H; before vs after +IEM 9 of 10 neurons: ***p < 0.001 by K–S test; Fig. 3J; KO Control: 7.8 ± 1.7 Hz, +IEM 4.1 ± 0.9 Hz, n = 10; *p < 0.05 by paired t test). Furthermore, impaired homeostatic scaling-up was correlated with maintained sensitivity to IEM1460 after the 4 and 24 h TTX/MK treatments, not only in ΔPIX neurons [Fig. 3R; 4 h TTX/MK: 15.3 ± 1.6 pA, +IEM 16.3 ± 1.3 pA, n = 10, *p < 0.05 by paired t test; 24 h TTX/MK: 14.1 ± 1.1 pA, +IEM 11.3 ± 0.6 pA, n = 8, **p < 0.01 by paired t test; Fig. 3S; 4 h TTX/MK before vs after +IEM 6 of 10 neurons: ***p < 0.001 by K–S test (cumulative distributions not plotted); 24 h TTX/MK before vs after +IEM 8 of 8 neurons: ***p < 0.001 by K–S test; Fig. 3T; 4 h TTX/MK: 5.9 ± 2.2 Hz, +IEM 3.4 ± 1.3 Hz, n = 10, *p < 0.05 by paired t test; 24 h TTX/MK: 14.5 ± 4.6 Hz, +IEM 5.4 ± 2.2 Hz, n = 8, *p < 0.05 by paired t test], but also in AKAP150 KO neurons [Fig. 3G; 4 h TTX/MK: 20.5 ± 2.1 pA, +IEM 16.3 ± 1.3 pA, n = 10, **p < 0.01 by paired t test; 24 h TTX/MK: 20.3 ± 1.8 pA, +IEM 17.4 ± 1.2 pA, n = 10, *p < 0.05 by paired t test; Fig. 3I; 4 h TTX/MK: 20.5 ± 2.1 pA before vs after +IEM 8 of 10 neurons: ***p < 0.001 by K–S test (cumulative distributions not plotted); 24 h TTX/MK before vs after +IEM 6 of 10 neurons: ***p < 0.001 by K–S test; Fig. 3J; 4 h TTX/MK: 5.4 ± 1.4 Hz, +IEM 3.2 ± 1.1 Hz, n = 10, *p < 0.05 by paired t test; 24 h TTX/MK: 9.0 ± 2.5 Hz, +IEM 4.0 ± 1.1 Hz, n = 10, *p < 0.05 by paired t test]. Thus, basally increased synaptic CP-AMPAR activity in both ΔPIX and KO neurons appears to be occluding subsequent homeostatic scaling-up, even in the absence of increased overall basal mEPSC transmission in the case of ΔPIX.
Homeostatic synaptic plasticity is associated with opposing regulation of GluA1 S845 phosphorylation by AKAP150-anchored PKA and CaN
As mentioned, previous studies found that phosphorylation of GluA1 Ser845 plays a key role in regulating CP-AMPAR trafficking to control synaptic insertion/removal dynamics during LTP/LTD (H. K. Lee et al., 2003, 2010; Lu et al., 2007; He et al., 2009; Qian et al., 2012; Sanderson et al., 2012, 2016; Zhang et al., 2013). In addition, several studies indicate that increased Ser845 phosphorylation is also required for homeostatic scaling-up induced by 48 h TTX treatment and that the phospho-deficient GluA1 S845A mutation prevents this scaling-up (Diering et al., 2014; Kim and Ziff, 2014; Kim et al., 2015a,b). To examine whether Ser845 phosphorylation is also regulated during the rapid homeostatic scaling-up examined here, we analyzed whole-cell extracts from WT neurons under control conditions and following 4 h TTX/MK treatment by immunoblotting with antibodies recognizing phospho-Ser845 followed by stripping and re-probing to detect total GluA1 levels (Fig. 4A). Importantly, measurement of phospho-Ser845/total GluA1 normalized to the WT control condition revealed that induction of homeostatic scaling-up with 4 h TTX/MK resulted in an ∼2-fold increase in Ser845 phosphorylation in WT neurons (Fig. 4B; WT Control: 1.0 ± 0.1, n = 7; 4 h TTX/MK: 2.2 ± 0.5, n = 8; *p < 0.019 by unpaired t test). In contrast, 4 h TTX/MK treatment failed to significantly increase Ser845 phosphorylation in neurons cultured from AKAP150 KO (Fig. 4A,B; KO Control: 1.3 ± 0.2, n = 9; KO 4 h TTX/MK: 1.9 ± 0.4, n = 9; NS p = 0.10 by unpaired t test) and ΔPKA mice (Fig. 4A,B; ΔPKA Control: 1.1 ± 0.4, n = 6; ΔPKA 4 h TTX/MK: 1.4 ± 0.5, n = 6; NS p = 0.31 by unpaired t test). Neurons from AKAP150 ΔPIX mice already exhibited significantly increased (∼2-fold) basal S845 phosphorylation (#p = 0.049 to WT Control by unpaired t test) and did not show any further increase after 4 h TTX/MK treatment (Fig. 4A,B; ΔPIX Control: 2.3 ± 0.7, n = 7; ΔPIX 4 h TTX/MK: 2.2 ± 0.5, n = 7; NS p = 0.46 to Control; #p = 0.028 to WT Control by unpaired t test). Thus, ΔPIX neurons exhibited constitutively elevated levels of both CP-AMPAR activity and GluA1 phosphorylation that were similar to those observed in WT neurons only after induction of scaling-up, consistent with some sort of occlusion mechanism. In contrast, despite no basal decrease in Ser845 phosphorylation, ΔPKA neurons appeared unable to effectively engage PKA signaling to increase Ser845 phosphorylation in response to induction of homeostatic scaling. Interestingly, the elevated basal CP-AMPAR activity in AKAP150 KO neurons was not accompanied by significantly enhanced basal GluA1 phosphorylation, and 4 h TTX/MK treatment produced a variable but nonsignificant trend (p = 0.10) toward increased phosphorylation, suggesting that simultaneous loss of anchored PKA and CaN may somewhat offset each other but with the net result still being less effective control of GluA1 phosphorylation and CP-AMPAR activity both basally and during homeostatic plasticity.
Figure 4.

Induction of rapid homeostatic synaptic potentiation in hippocampal neurons increases phosphorylation of GluA1–S845 by AKAP150-anchored PKA in opposition to AKAP150-anchored CaN. A, Representative immunoblots of whole-cell extracts of hippocampal neurons cultured from WT, AKAP150 KO, ΔPKA, or ΔPIX mice to detect phospho-GluA1-S845 (pS845) levels followed by stripping and re-probing to detect total GluA1 levels. Note: Left, Extracts from WT and KO were analyzed on the same blot using the same exposures, but then the lanes were rearranged for presentation. B, Quantification of the ratio of pS845/total GluA1 normalized to WT control (always present on the same blot) shows that induction of rapid homeostatic scaling-up with 4 h TTX/MK significantly increases S845 phosphorylation in neurons from WT (*p < 0.05 by unpaired t test) but not AKAP150 KO or ΔPKA mice. Neurons from AKAP150ΔPIX mice already exhibit increased basal S845 phosphorylation and do not show any further increase after 4 h TTX/MK treatment (#p < 0.05 to WT Control by unpaired t test). Error bars indicate SEM.
AKAP150-anchored PKA and CaN are both required for regulation of GluA1 postsynaptic localization during homeostatic synaptic plasticity
Finally, we investigated whether the alterations in regulation of GluA1 phosphorylation and synaptic CP-AMPAR activity observed above in AKAP150 mutant mice during homeostatic potentiation were also reflected in corresponding changes in GluA1 synaptic localization. We used immunofluorescence microscopy to image the localization of total GluA1 (both intracellular and cell surface pools) in hippocampal dendrites relative to its major postsynaptic density (PSD) scaffold protein PSD-95 under control conditions and after 4 h TTX/MK treatment (Fig. 5A). Quantification of total GluA1/PSD-95 colocalization from images of dendrites using Pearson's correlation (r) revealed that induction of rapid homeostatic scaling in WT neurons with 4 h TTX/MK treatment increased total GluA1 localization ∼40% at postsynaptic sites defined by PSD-95 [Fig. 5B; WT 4 h TTX/MK 1.39 ± 0.06 fold-change normalized to Control, n = 24 (n = 26 Control); ***p < 0.0001 by unpaired t test]. However, 4 h TTX/MK failed to increase total GluA1/PSD-95 colocalization in neurons cultured from AKAP150 KO [Fig. 5B; KO 4 h TTX/MK: 0.80 ± 0.10 fold-change normalized to Control, n = 7 (n = 14 Control), NS p = 0.25 by unpaired t test] or ΔPKA mice [Fig. 5B; ΔPKA 4 h TTX/MK: 1.05 ± 0.06 fold-change normalized to Control, n = 28 (n = 24 Control), NS p = 0.59 by unpaired t test]. Total GluA1/PSD-95 colocalization actually decreased slightly in ΔPIX mouse neurons after 4 h TTX/MK [Fig. 5B; ΔPIX 4 h TTX/MK: 0.83 ± 0.06, n = 28 (n = 45 Control); *p = 0.048 to Control by unpaired t test]. Similar results were obtained when this postsynaptic colocalization analysis was repeated using immunostaining to detect only the plasma membrane cell surface pool of GluA1 (Fig. 5C,D). Accordingly, we found that 4 h TTX/MK treatment robustly increased (∼50%) synaptic surface GluA1 localization in WT neurons compared with untreated controls [Fig. 5D; WT: 1.53 ± 0.04-fold normalized to Control, n = 23 (n = 31 Control), ***p < 0.0001 by t test]. However, synaptic surface GluA1 localization was unchanged in ΔPIX neurons and only modestly increased (∼20%) in AKAP150 KO and ΔPKA neurons after 4 h TTX/MK treatment [Fig. 5D; KO: 1.24 ± 0.06-fold normalized to Control, n = 9 (n = 11 Control), *p = 0.017 by t test; ΔPKA: 1.19 ± 0.07-fold normalized to Control, n = 13 (n = 24 Control), *p = 0.019 by t test; ΔPIX: 0.92 ± 0.06-fold normalized to Control, n = 13 (n = 28 Control), NS p = 0.21 by t test]. Thus, the failure of 4 h TTX/MK treatment to robustly increase total and surface postsynaptic GluA1/PSD-95 colocalization in all three AKAP150 mutant mouse lines compared with WT mice also corresponds with the inability of these mutants to effectively regulate Ser845 phosphorylation (Fig. 4) and induce homeostatic increases in AMPAR mEPSC activity (Figs. 2, 3) in response to this same treatment above.
Figure 5.
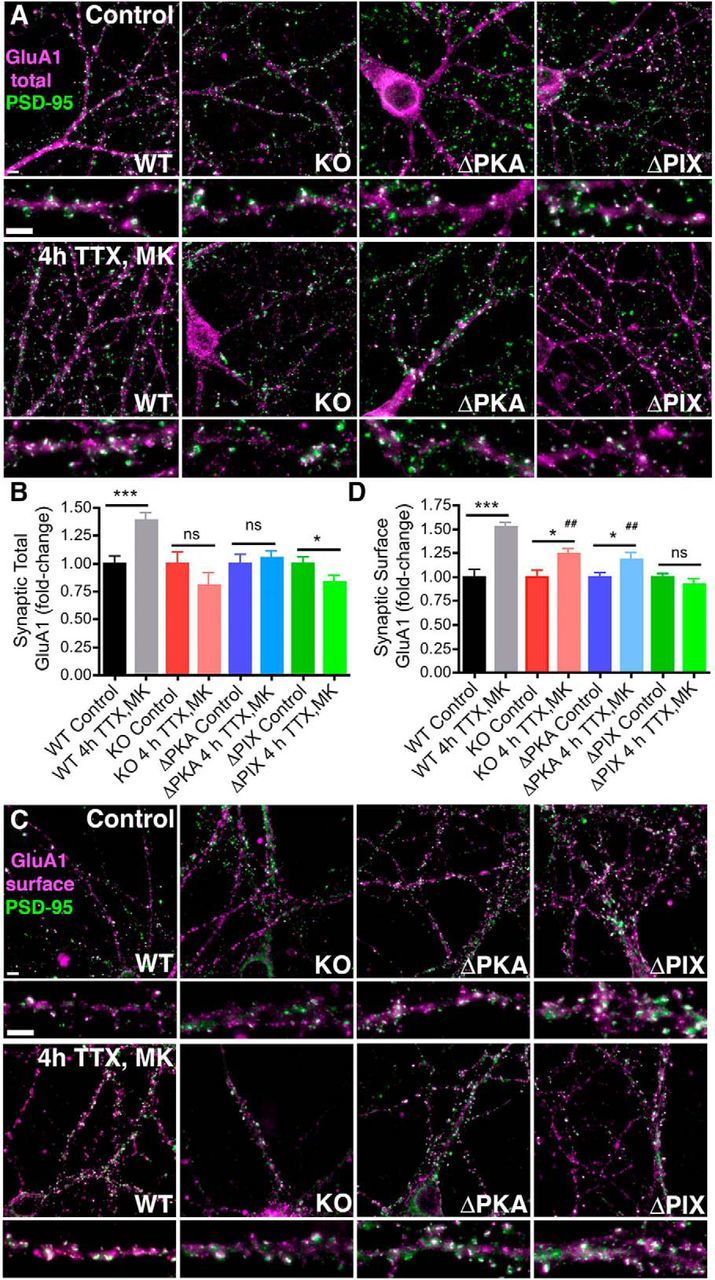
Induction of rapid homeostatic synaptic potentiation increases synaptic GluA1 localization in hippocampal neurons cultured from WT but not AKAP150 KO, ΔPKA, or ΔPIX mice. A, Representative images showing total GluA1 (magenta) and PSD-95 (green) colocalization (white) in 16 DIV hippocampal neurons cultured from WT, AKAP150 KO, ΔPKA, or ΔPIX mice under control conditions or after induction of rapid homeostatic scaling-up with 4 h TTX/MK treatment. Bottom, Magnifications of dendrites. Scale bars, 5 μm. B, Quantification of fold-change in total synaptic GluA1 colocalization with PSD-95 from images as in A using Pearson's correlation (r) shows that induction of homeostatic scaling-up with 4 h TTX/MK significantly increases GluA1/PSD-95 colocalization in WT (***p < 0.001 to Control by unpaired t test) but not KO or ΔPKA mouse neurons (both NS p > 0.05 to Controls by unpaired t test). Total GluA1/PSD-95 colocalization actually decreases slightly in ΔPIX mouse neurons after 4 h TTX/MK (*p < 0.05 to Control by unpaired t test). Error bars indicate SEM. C, Representative images showing surface GluA1 (magenta) and PSD-95 (green) colocalization (white) in 16 DIV hippocampal neurons cultured from WT, AKAP150 KO, ΔPKA, or ΔPIX mice under control conditions or after induction of rapid homeostatic scaling-up with 4 h TTX/MK treatment. Bottom, Magnifications of dendrites. Scale bars, 5 μm. D, Quantification of fold-change in synaptic surface GluA1 colocalization with PSD-95 from images as in C using Pearson's correlation (r) shows that induction of homeostatic scaling-up with 4 h TTX/MK treatment increases surface GluA1/PSD-95 colocalization in WT mouse neuron (***p < 0.0001 to Control by unpaired t test) significantly more than in KO or ΔPKA mouse neurons (both *p < 0.05 to corresponding Controls by unpaired t test and ##p < 0.01 to WT 4 h TTX/MK by one-way ANOVA). Surface GluA1/PSD-95 colocalization is unchanged in ΔPIX mouse neurons after 4 h TTX/MK (NS p > 0.05 to Control by unpaired t test). Error bars indicate SEM.
Discussion
Our findings significantly expand the understanding of homeostatic synaptic plasticity mechanisms and provide new appreciation for how interconnected they are with those controlling LTP/LTD. Specifically, we confirm and extend previous work showing that PKA/CaN regulation of GluA1 phosphorylation and CP-AMPAR synaptic incorporation are associated not only with LTP/LTD (Kameyama et al., 1998; H. K. Lee et al., 1998; 2000, 2003; 2010; Lu et al., 2007; Qian et al., 2012; Sanderson et al., 2012, 2016; Zhang et al., 2013) but also with homeostatic synaptic potentiation (Goel et al., 2011; Diering et al., 2014; Kim and Ziff, 2014). Most importantly, we show that homeostatic plasticity, like LTP/LTD, critically depends on PKA and CaN targeting to CP-AMPARs by the AKAP150 scaffold. Remarkably, by using a series of AKAP150 mutant mice, we found that just delocalizing PKA or CaN from this AKAP signaling complex profoundly alters regulation of GluA1 phosphorylation and CP-AMPAR synaptic function to impact both forms of plasticity.
As shown here, selective disruption of AKAP150-PKA anchoring prevented increased GluA1 Ser845 phosphorylation and CP-AMPAR incorporation to mediate homeostatic synaptic potentiation in hippocampal neurons, as previously shown for CA1 LTP ex vivo in acute slices from adult mice (Lu et al., 2007). However, LTP can still be induced in ΔPKA mice (and related D36 mice; Lu et al., 2007; Sanderson et al., 2016) under conditions where GluA1 subunits and CP-AMPAR synaptic incorporation are not strictly required, such as in juveniles (Jensen et al., 2003; Kolleker et al., 2003; H. K. Lee et al., 2003; Plant et al., 2006; Adesnik and Nicoll, 2007; Granger et al., 2013). In the cases of complete AKAP150 KO and selective CaN anchoring disruption, the situation becomes more complicated. Neurons from AKAP150 KO and ΔPIX mice both exhibited increased basal CP-AMPAR activity that apparently occluded additional CP-AMPAR recruitment during homeostatic scaling. Yet, increased basal CP-AMPAR activity was only associated with increased synaptic strength in AKAP150 KO neurons and was only associated with increased Ser845 phosphorylation in ΔPIX neurons. Thus, just a basal shift in synaptic subunit composition toward CP-AMPARs may be sufficient to prevent subsequent homeostatic CP-AMPAR recruitment, which could in part explain why knockdown or knock-out of GluA2 that basally increases CP-AMPARs can impair scaling-up, although additional impacts on proteins that bind the GluA2 C-terminal domain are also likely involved (Gainey et al., 2009; Ancona Esselmann et al., 2017; but see Altimimi and Stellwagen, 2013).
The failures in homeostatic potentiation observed in AKAP150 KO and ΔPIX neurons are also related to, but distinct from, the impacts of these mutants on LTP and GluA1 phosphorylation in vivo/ex vivo. The ΔPIX mutation significantly enhanced basal Ser845 phosphorylation both when analyzed here in vitro in cultured neurons, as well as when analyzed previously in vivo in hippocampal tissue and ex vivo in acute slices (Sanderson et al., 2012). In the case of AKAP150 KO mice, basal Ser845 phosphorylation was either unchanged (in vitro here and ex vivo; Lu et al., 2007; Zhang et al., 2013) or decreased (in vivo; Tunquist et al., 2008). Nonetheless, AKAP150 KO mice express normal NMDAR-dependent CA1 LTP in both juvenile and adult mice (Weisenhaus et al., 2010), and juvenile ΔPIX mice express enhanced CA1 LTP through CP-AMPAR recruitment (Sanderson et al., 2012). Thus, although it was not determined whether AKAP150 KO mice have altered basal CA1 synaptic AMPAR subunit composition, it is clear in the case of ΔPIX mice that, unlike with scaling-up, basal CP-AMPAR incorporation does not occlude LTP mediated by additional CP-AMPAR recruitment. Interestingly, both AKAP150 KO and PKA anchoring-deficient D36 mice do show deficits in another L-type Ca2+ channel-dependent form of LTP that requires GluA1 Ser845 phosphorylation and CP-AMPAR recruitment to synapses (Qian et al., 2012, 2017; Zhang et al., 2013; Patriarchi et al., 2016). Thus, overall, the roles of AKAP150-anchored PKA and CaN in regulating CP-AMPARs during homeostatic potentiation and LTP are overlapping but clearly not identical.
Nonetheless, this convergence of PKA/CaN regulation of LTP and homeostatic plasticity is still surprising, given that LTP is induced by brief, but strong, postsynaptic depolarization and increased NMDAR Ca2+ influx, while here we induced scaling-up with prolonged blockade of neuronal firing and NMDAR Ca2+ influx. Yet, in both cases what might be crucial is the relative balance of local PKA and CaN signaling near GluA1, first basally and then how that balance changes with alterations in neuronal activity. Because of four S845 phosphorylation sites in a GluA1 homomer (compared with 2 in a GluA1/2 heteromer), the importance of this PKA/CaN signaling may be amplified in the case of CP-AMPAR regulation. Previous work by others found that pharmacologic inhibition of CaN phosphatase activity was sufficient to increase Ser845 phosphorylation and CP-AMPAR synaptic incorporation and mimic scaling-up in cortical neurons (Kim and Ziff, 2014). This prior study is in part consistent with our observations for ΔPIX mice, where loss of local CaN signaling leads to a dramatic shift toward phosphorylation of Ser845 and basal CP-AMPAR synaptic incorporation. However, this basal CP-AMPAR recruitment in ΔPIX did not increase basal synaptic strength and fully mimic scaling-up, indicating that GluA1 CP-AMPARs are replacing existing GluA2-containing AMPARs in ΔPIX neurons, rather than adding to them as with CaN inhibition. Nonetheless, subsequent induction of homeostatic potentiation was impaired in ΔPIX likely due to this already imbalanced PKA/CaN signaling in favor of PKA. AKAP150 KO mice in part phenocopy loss of CaN anchoring and/or inhibition of CaN activity, in that basal CP-AMPAR activity and synaptic strength are increased. However, Ser845 phosphorylation analysis in AKAP150 KO mice suggests more similarity to loss of PKA anchoring in ΔPKA mice in that, despite normal basal phosphorylation, the ability to increase S845 phosphorylation and efficiently deliver GluA1 to synapses is impaired.
Basal AKAP150-anchored PKA activity toward GluA1 is likely at least in part cAMP-independent, as shown for other AKAP-linked substrates (F. D. Smith et al., 2013), but can be further stimulated by cAMP downstream of either Gs or Ca2+-CaM activation of adenylyl cyclases 5 and 8, respectively, which are directly bound to the AKAP (Bauman et al., 2006; Efendiev et al., 2010; Willoughby et al., 2010). However, recent studies show that AKAP-anchored type-II PKA holoenzymes do not substantially release the catalytic C subunits from RII regulatory subunits even after cAMP activation, resulting in PKA phosphorylation being restricted to a ∼25 nm nanodomain surrounding the AKAP (F. D. Smith et al., 2013, 2017). Thus, both basal and cAMP-stimulated activities of AKAP150-anchored PKA are extremely localized in nature. Indeed, Diering et al. (2014) observed increased PKA substrate phosphorylation during homeostatic scaling-up that was localized to synaptic fractions. Accordingly, the ΔPKA deletion should strongly shift the local PKA/CaN balance in favor of CaN-dephosphorylation such that, even with a reduction in Ca2+-CaN signaling upon neuronal silencing, local PKA signaling (whether cAMP-dependent or independent) would not be effectively engaged to increase S845 phosphorylation or recruit CP-AMPARs. However, in the case of LTP induction in ΔPKA mice, the accompanying strong increase in NMDAR Ca2+ influx could (depending on the strength of the induction stimulus and developmental age) bypass any requirements for PKA phosphorylation of Ser845 and recruit GluA2-containing AMPARs through other pathways, consistent with the CP-AMPAR/GluA1 phosphorylation dependence of CA1 LTP being flexible and influenced by both age and induction stimulus (H. K. Lee et al., 2003; 2010; Plant et al., 2006; Adesnik and Nicoll, 2007; Gray et al., 2007; Lu et al., 2007; Guire et al., 2008; Yang et al., 2010; Qian et al., 2012; Sanderson et al., 2012, 2016; Whitehead et al., 2013; Park et al., 2016).
Overall, our findings paint a more complex picture than previously appreciated of the interplay between signaling mechanisms underlying Hebbian and homeostatic plasticity. Importantly, this complex interplay was only revealed by studying the specific loss of AKAP150 anchored PKA versus CaN in knock-in mouse models, as opposed to loss of all AKAP150 functions in KO mice, further underscoring the limitation of relying on just KO or RNAi to understand the functions of multivalent scaffold proteins. Our past and present results now indicate that AKAP-anchored PKA and CaN oppose each other in regulation of GluA1 phosphorylation and CP-AMPAR synaptic incorporation during both LTP/LTD and homeostatic scaling-up. However, it remains to be seen whether AKAP-anchored PKA and CaN also regulate other mechanisms implicated in homeostatic plasticity, such as retinoic acid signaling (Aoto et al., 2008; Wang et al., 2011; Arendt et al., 2015), dendritic protein synthesis (Sutton et al., 2006; Maghsoodi et al., 2008), and gene transcription (Thiagarajan et al., 2002; Ibata et al., 2008; Groth et al., 2011; Joseph and Turrigiano, 2017). As mentioned in the Introduction, L-type voltage-gated Ca2+ channels are implicated in homeostatic plasticity, and interestingly, AKAP150 also targets PKA and CaN to L-channels to control their activity and downstream signaling to the nucleus to regulate transcription (Oliveria et al., 2007; Dittmer et al., 2014; Murphy et al., 2014). Thus, perhaps we have only just begun to unravel the role of this multifunctional AKAP150 signaling complex in homeostatic plasticity.
Footnotes
This work was supported by NIH Grant R01NS040701 (M.L.D.). Contents are the authors' sole responsibility and do not necessarily represent official NIH views. We thank Dr. Jason Aoto for critical reading of the paper.
The authors declare no competing financial interests.
References
- Adesnik H, Nicoll RA (2007) Conservation of glutamate receptor 2-containing AMPA receptors during long-term potentiation. J Neurosci 27:4598–4602. 10.1523/JNEUROSCI.0325-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altimimi HF, Stellwagen D (2013) Persistent synaptic scaling independent of AMPA receptor subunit composition. J Neurosci 33:11763–11767. 10.1523/JNEUROSCI.1102-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ancona Esselmann SG, Díaz-Alonso J, Levy JM, Bemben MA, Nicoll RA (2017) Synaptic homeostasis requires the membrane-proximal carboxy tail of GluA2. Proc Natl Acad Sci U S A 114:13266–13271. 10.1073/pnas.1716022114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anggono V, Clem RL, Huganir RL (2011) PICK1 loss of function occludes homeostatic synaptic scaling. J Neurosci 31:2188–2196. 10.1523/JNEUROSCI.5633-10.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoto J, Nam CI, Poon MM, Ting P, Chen L (2008) Synaptic signaling by all-trans retinoic acid in homeostatic synaptic plasticity. Neuron 60:308–320. 10.1016/j.neuron.2008.08.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arendt KL, Zhang Z, Ganesan S, Hintze M, Shin MM, Tang Y, Cho A, Graef IA, Chen L (2015) Calcineurin mediates homeostatic synaptic plasticity by regulating retinoic acid synthesis. Proc Natl Acad Sci U S A 112:E5744–E5752. 10.1073/pnas.1510239112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauman AL, Soughayer J, Nguyen BT, Willoughby D, Carnegie GK, Wong W, Hoshi N, Langeberg LK, Cooper DM, Dessauer CW, Scott JD (2006) Dynamic regulation of cAMP synthesis through anchored PKA-adenylyl cyclase V/VI complexes. Mol Cell 23:925–931. 10.1016/j.molcel.2006.07.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharyya S, Biou V, Xu W, Schlüter O, Malenka RC (2009) A critical role for PSD-95/AKAP interactions in endocytosis of synaptic AMPA receptors. Nat Neurosci 12:172–181. 10.1038/nn.2249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carr DW, Stofko-Hahn RE, Fraser ID, Cone RD, Scott JD (1992) Localization of the cAMP-dependent protein kinase to the postsynaptic densities by A-kinase anchoring proteins: characterization of AKAP 79. J Biol Chem 267:16816–16823. [PubMed] [Google Scholar]
- Chen L, Lau AG, Sarti F (2014) Synaptic retinoic acid signaling and homeostatic synaptic plasticity. Neuropharmacology 78:3–12. 10.1016/j.neuropharm.2012.12.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coghlan VM, Perrino BA, Howard M, Langeberg LK, Hicks JB, Gallatin WM, Scott JD (1995) Association of protein kinase A and protein phosphatase 2B with a common anchoring protein. Science 267:108–111. 10.1126/science.7528941 [DOI] [PubMed] [Google Scholar]
- Colledge M, Dean RA, Scott GK, Langeberg LK, Huganir RL, Scott JD (2000) Targeting of PKA to glutamate receptors through a MAGUK-AKAP complex. Neuron 27:107–119. 10.1016/S0896-6273(00)00013-1 [DOI] [PubMed] [Google Scholar]
- Collingridge GL, Peineau S, Howland JG, Wang YT (2010) Long-term depression in the CNS. Nat Rev Neurosci 11:459–473. 10.1038/nrn2867 [DOI] [PubMed] [Google Scholar]
- Dell'Acqua ML, Faux MC, Thorburn J, Thorburn A, Scott JD (1998) Membrane-targeting sequences on AKAP79 bind phosphatidylinositol-4, 5-bisphosphate. EMBO J 17:2246–2260. 10.1093/emboj/17.8.2246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dell'Acqua ML, Dodge KL, Tavalin SJ, Scott JD (2002) Mapping the protein phosphatase-2B anchoring site on AKAP79: binding and inhibition of phosphatase activity are mediated by residues 315–360. J Biol Chem 277:48796–48802. 10.1074/jbc.M207833200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diering GH, Gustina AS, Huganir RL (2014) PKA-GluA1 coupling via AKAP5 controls AMPA receptor phosphorylation and cell-surface targeting during bidirectional homeostatic plasticity. Neuron 84:790–805. 10.1016/j.neuron.2014.09.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dittmer PJ, Dell'Acqua ML, Sather WA (2014) Ca2+/calcineurin-dependent inactivation of neuronal L-type Ca2+ channels requires priming by AKAP-anchored protein kinase A. Cell Rep 7:1410–1416. 10.1016/j.celrep.2014.04.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efendiev R, Samelson BK, Nguyen BT, Phatarpekar PV, Baameur F, Scott JD, Dessauer CW (2010) AKAP79 interacts with multiple adenylyl cyclase (AC) isoforms and scaffolds AC5 and -6 to alpha-amino-3-hydroxyl-5-methyl-4-isoxazole-propionate (AMPA) receptors. J Biol Chem 285:14450–14458. 10.1074/jbc.M110.109769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortin DA, Davare MA, Srivastava T, Brady JD, Nygaard S, Derkach VA, Soderling TR (2010) Long-term potentiation-dependent spine enlargement requires synaptic Ca2+-permeable AMPA receptors recruited by CaM-kinase I. J Neurosci 30:11565–11575. 10.1523/JNEUROSCI.1746-10.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gainey MA, Hurvitz-Wolff JR, Lambo ME, Turrigiano GG (2009) Synaptic scaling requires the GluR2 subunit of the AMPA receptor. J Neurosci 29:6479–6489. 10.1523/JNEUROSCI.3753-08.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gainey MA, Tatavarty V, Nahmani M, Lin H, Turrigiano GG (2015) Activity-dependent synaptic GRIP1 accumulation drives synaptic scaling up in response to action potential blockade. Proc Natl Acad Sci U S A 112:E3590–E3599. 10.1073/pnas.1510754112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glantz SB, Amat JA, Rubin CS (1992) cAMP signaling in neurons: patterns of neuronal expression and intracellular localization for a novel protein, AKAP 150, that anchors the regulatory subunit of cAMP-dependent protein kinase II beta. Mol Biol Cell 3:1215–1228. 10.1091/mbc.3.11.1215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goel A, Xu LW, Snyder KP, Song L, Goenaga-Vazquez Y, Megill A, Takamiya K, Huganir RL, Lee HK (2011) Phosphorylation of AMPA receptors is required for sensory deprivation-induced homeostatic synaptic plasticity. PLoS One 6:e18264. 10.1371/journal.pone.0018264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez LL, Alam S, Smith KE, Horne E, Dell'Acqua ML (2002) Regulation of A-kinase anchoring protein 79/150-cAMP-dependent protein kinase postsynaptic targeting by NMDA receptor activation of calcineurin and remodeling of dendritic actin. J Neurosci 22:7027–7044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorski JA, Gomez LL, Scott JD, Dell'Acqua ML (2005) Association of an A-kinase-anchoring protein signaling scaffold with cadherin adhesion molecules in neurons and epithelial cells. Mol Biol Cell 16:3574–3590. 10.1091/mbc.E05-02-0134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granger AJ, Shi Y, Lu W, Cerpas M, Nicoll RA (2013) LTP requires a reserve pool of glutamate receptors independent of subunit type. Nature 493:495–500. 10.1038/nature11775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray EE, Fink AE, Sariñana J, Vissel B, O'Dell TJ (2007) Long-term potentiation in the hippocampal CA1 region does not require insertion and activation of GluR2-lacking AMPA receptors. J Neurophysiol 98:2488–2492. 10.1152/jn.00473.2007 [DOI] [PubMed] [Google Scholar]
- Groth RD, Lindskog M, Thiagarajan TC, Li L, Tsien RW (2011) Beta Ca2+/CaM-dependent kinase type II triggers upregulation of GluA1 to coordinate adaptation to synaptic inactivity in hippocampal neurons. Proc Natl Acad Sci U S A 108:828–833. 10.1073/pnas.1018022108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guire ES, Oh MC, Soderling TR, Derkach VA (2008) Recruitment of calcium-permeable AMPA receptors during synaptic potentiation is regulated by CaM-kinase I. J Neurosci 28:6000–6009. 10.1523/JNEUROSCI.0384-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He K, Song L, Cummings LW, Goldman J, Huganir RL, Lee HK (2009) Stabilization of Ca2+-permeable AMPA receptors at perisynaptic sites by GluR1–S845 phosphorylation. Proc Natl Acad Sci U S A 106:20033–20038. 10.1073/pnas.0910338106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horne EA, Dell'Acqua ML (2007) Phospholipase C is required for changes in postsynaptic structure and function associated with NMDA receptor-dependent long-term depression. J Neurosci 27:3523–3534. 10.1523/JNEUROSCI.4340-06.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshi N, Zhang JS, Omaki M, Takeuchi T, Yokoyama S, Wanaverbecq N, Langeberg LK, Yoneda Y, Scott JD, Brown DA, Higashida H (2003) AKAP150 signaling complex promotes suppression of the M-current by muscarinic agonists. Nat Neurosci 6:564–571. 10.1038/nn1062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshi N, Langeberg LK, Scott JD (2005) Distinct enzyme combinations in AKAP signalling complexes permit functional diversity. Nat Cell Biol 7:1066–1073. 10.1038/ncb1315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshi N, Langeberg LK, Gould CM, Newton AC, Scott JD (2010) Interaction with AKAP79 modifies the cellular pharmacology of PKC. Mol Cell 37:541–550. 10.1016/j.molcel.2010.01.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou Q, Zhang D, Jarzylo L, Huganir RL, Man HY (2008) Homeostatic regulation of AMPA receptor expression at single hippocampal synapses. Proc Natl Acad Sci U S A 105:775–780. 10.1073/pnas.0706447105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huganir RL, Nicoll RA (2013) AMPARs and synaptic plasticity: the last 25 years. Neuron 80:704–717. 10.1016/j.neuron.2013.10.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibata K, Sun Q, Turrigiano GG (2008) Rapid synaptic scaling induced by changes in postsynaptic firing. Neuron 57:819–826. 10.1016/j.neuron.2008.02.031 [DOI] [PubMed] [Google Scholar]
- Jakawich SK, Nasser HB, Strong MJ, McCartney AJ, Perez AS, Rakesh N, Carruthers CJ, Sutton MA (2010) Local presynaptic activity gates homeostatic changes in presynaptic function driven by dendritic BDNF synthesis. Neuron 68:1143–1158. 10.1016/j.neuron.2010.11.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen V, Kaiser KM, Borchardt T, Adelmann G, Rozov A, Burnashev N, Brix C, Frotscher M, Andersen P, Hvalby Ø, Sakmann B, Seeburg PH, Sprengel R (2003) A juvenile form of postsynaptic hippocampal long-term potentiation in mice deficient for the AMPA receptor subunit GluR-A. J Physiol 553:843–856. 10.1113/jphysiol.2003.053637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joseph A, Turrigiano GG (2017) All for one but not one for all: excitatory synaptic scaling and intrinsic excitability are coregulated by CaMKIV, whereas inhibitory synaptic scaling is under independent control. J Neurosci 37:6778–6785. 10.1523/JNEUROSCI.0618-17.2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurado S, Biou V, Malenka RC (2010) A calcineurin/AKAP complex is required for NMDA receptor-dependent long-term depression. Nat Neurosci 13:1053–1055. 10.1038/nn.2613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kameyama K, Lee HK, Bear MF, Huganir RL (1998) Involvement of a postsynaptic protein kinase A substrate in the expression of homosynaptic long-term depression. Neuron 21:1163–1175. 10.1016/S0896-6273(00)80633-9 [DOI] [PubMed] [Google Scholar]
- Keck T, Toyoizumi T, Chen L, Doiron B, Feldman DE, Fox K, Gerstner W, Haydon PG, Hübener M, Lee HK, Lisman JE, Rose T, Sengpiel F, Stellwagen D, Stryker MP, Turrigiano GG, van Rossum MC (2017) Integrating Hebbian and homeostatic plasticity: the current state of the field and future research directions. Philos Trans R Soc Lond B Biol Sci 372:20160158. 10.1098/rstb.2016.0158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keith DJ, Sanderson JL, Gibson ES, Woolfrey KM, Robertson HR, Olszewski K, Kang R, El-Husseini A, Dell'acqua ML (2012) Palmitoylation of A-kinase anchoring protein 79/150 regulates dendritic endosomal targeting and synaptic plasticity mechanisms. J Neurosci 32:7119–7136. 10.1523/JNEUROSCI.0784-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Ziff EB (2014) Calcineurin mediates synaptic scaling via synaptic trafficking of Ca2+-permeable AMPA receptors. PLoS Biol 12:e1001900. 10.1371/journal.pbio.1001900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Violette CJ, Ziff EB (2015a) Reduction of increased calcineurin activity rescues impaired homeostatic synaptic plasticity in presenilin 1 M146V mutant. Neurobiol Aging 36:3239–3246. 10.1016/j.neurobiolaging.2015.09.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Titcombe RF, Zhang H, Khatri L, Girma HK, Hofmann F, Arancio O, Ziff EB (2015b) Network compensation of cyclic GMP-dependent protein kinase II knockout in the hippocampus by Ca2+-permeable AMPA receptors. Proc Natl Acad Sci U S A 112:3122–3127. 10.1073/pnas.1417498112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klauck TM, Faux MC, Labudda K, Langeberg LK, Jaken S, Scott JD (1996) Coordination of three signaling enzymes by AKAP79, a mammalian scaffold protein. Science 271:1589–1592. 10.1126/science.271.5255.1589 [DOI] [PubMed] [Google Scholar]
- Kolleker A, Zhu JJ, Schupp BJ, Qin Y, Mack V, Borchardt T, Köhr G, Malinow R, Seeburg PH, Osten P (2003) Glutamatergic plasticity by synaptic delivery of GluR-B(long)-containing AMPA receptors. Neuron 40:1199–1212. 10.1016/S0896-6273(03)00722-0 [DOI] [PubMed] [Google Scholar]
- Lee HK, Kameyama K, Huganir RL, Bear MF (1998) NMDA induces long-term synaptic depression and dephosphorylation of the GluR1 subunit of AMPA receptors in hippocampus. Neuron 21:1151–1162. 10.1016/S0896-6273(00)80632-7 [DOI] [PubMed] [Google Scholar]
- Lee HK, Barbarosie M, Kameyama K, Bear MF, Huganir RL (2000) Regulation of distinct AMPA receptor phosphorylation sites during bidirectional synaptic plasticity. Nature 405:955–959. 10.1038/35016089 [DOI] [PubMed] [Google Scholar]
- Lee HK, Takamiya K, Han JS, Man H, Kim CH, Rumbaugh G, Yu S, Ding L, He C, Petralia RS, Wenthold RJ, Gallagher M, Huganir RL (2003) Phosphorylation of the AMPA receptor GluR1 subunit is required for synaptic plasticity and retention of spatial memory. Cell 112:631–643. 10.1016/S0092-8674(03)00122-3 [DOI] [PubMed] [Google Scholar]
- Lee HK, Takamiya K, He K, Song L, Huganir RL (2010) Specific roles of AMPA receptor subunit GluR1 (GluA1) phosphorylation sites in regulating synaptic plasticity in the CA1 region of hippocampus. J Neurophysiol 103:479–489. 10.1152/jn.00835.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KF, Soares C, Béïque JC (2014) Tuning into diversity of homeostatic synaptic plasticity. Neuropharmacology 78:31–37. 10.1016/j.neuropharm.2013.03.016 [DOI] [PubMed] [Google Scholar]
- Lee KY, Chung HJ (2014) NMDA receptors and L-type voltage-gated Ca2+ channels mediate the expression of bidirectional homeostatic intrinsic plasticity in cultured hippocampal neurons. Neuroscience 277:610–623. 10.1016/j.neuroscience.2014.07.038 [DOI] [PubMed] [Google Scholar]
- Lindskog M, Li L, Groth RD, Poburko D, Thiagarajan TC, Han X, Tsien RW (2010) Postsynaptic GluA1 enables acute retrograde enhancement of presynaptic function to coordinate adaptation to synaptic inactivity. Proc Natl Acad Sci U S A 107:21806–21811. 10.1073/pnas.1016399107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu W, Man H, Ju W, Trimble WS, MacDonald JF, Wang YT (2001) Activation of synaptic NMDA receptors induces membrane insertion of new AMPA receptors and LTP in cultured hippocampal neurons. Neuron 29:243–254. 10.1016/S0896-6273(01)00194-5 [DOI] [PubMed] [Google Scholar]
- Lu W, Shi Y, Jackson AC, Bjorgan K, During MJ, Sprengel R, Seeburg PH, Nicoll RA (2009) Subunit composition of synaptic AMPA receptors revealed by a single-cell genetic approach. Neuron 62:254–268. 10.1016/j.neuron.2009.02.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y, Allen M, Halt AR, Weisenhaus M, Dallapiazza RF, Hall DD, Usachev YM, McKnight GS, Hell JW (2007) Age-dependent requirement of AKAP150-anchored PKA and GluR2-lacking AMPA receptors in LTP. EMBO J 26:4879–4890. 10.1038/sj.emboj.7601884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y, Zhang M, Lim IA, Hall DD, Allen M, Medvedeva Y, McKnight GS, Usachev YM, Hell JW (2008) AKAP150-anchored PKA activity is important for LTD during its induction phase. J Physiol 586:4155–4164. 10.1113/jphysiol.2008.151662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maghsoodi B, Poon MM, Nam CI, Aoto J, Ting P, Chen L (2008) Retinoic acid regulates RARalpha-mediated control of translation in dendritic RNA granules during homeostatic synaptic plasticity. Proc Natl Acad Sci U S A 105:16015–16020. 10.1073/pnas.0804801105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy JG, Sanderson JL, Gorski JA, Scott JD, Catterall WA, Sather WA, Dell'Acqua ML (2014) AKAP-anchored PKA maintains neuronal L-type calcium channel activity and NFAT transcriptional signaling. Cell Rep 7:1577–1588. 10.1016/j.celrep.2014.04.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Brien RJ, Kamboj S, Ehlers MD, Rosen KR, Fischbach GD, Huganir RL (1998) Activity-dependent modulation of synaptic AMPA receptor accumulation. Neuron 21:1067–1078. 10.1016/S0896-6273(00)80624-8 [DOI] [PubMed] [Google Scholar]
- Oliveria SF, Dell'Acqua ML, Sather WA (2007) AKAP79/150 anchoring of calcineurin controls neuronal L-type Ca2+ channel activity and nuclear signaling. Neuron 55:261–275. 10.1016/j.neuron.2007.06.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park P, Sanderson TM, Amici M, Choi SL, Bortolotto ZA, Zhuo M, Kaang BK, Collingridge GL (2016) Calcium-permeable AMPA receptors mediate the induction of the protein kinase A-dependent component of long-term potentiation in the hippocampus. J Neurosci 36:622–631. 10.1523/JNEUROSCI.3625-15.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patriarchi T, Qian H, Di Biase V, Malik ZA, Chowdhury D, Price JL, Hammes EA, Buonarati OR, Westenbroek RE, Catterall WA, Hofmann F, Xiang YK, Murphy GG, Chen CY, Navedo MF, Hell JW (2016) Phosphorylation of Cav1.2 on S1928 uncouples the L-type Ca2+ channel from the beta2 adrenergic receptor. EMBO J 35:1330–1345. 10.15252/embj.201593409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plant K, Pelkey KA, Bortolotto ZA, Morita D, Terashima A, McBain CJ, Collingridge GL, Isaac JT (2006) Transient incorporation of native GluR2-lacking AMPA receptors during hippocampal long-term potentiation. Nat Neurosci 9:602–604. 10.1038/nn1678 [DOI] [PubMed] [Google Scholar]
- Qian H, Matt L, Zhang M, Nguyen M, Patriarchi T, Koval OM, Anderson ME, He K, Lee HK, Hell JW (2012) β2-Adrenergic receptor supports prolonged theta tetanus-induced LTP. J Neurophysiol 107:2703–2712. 10.1152/jn.00374.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian H, Patriarchi T, Price JL, Matt L, Lee B, Nieves-Cintrón M, Buonarati OR, Chowdhury D, Nanou E, Nystoriak MA, Catterall WA, Poomvanicha M, Hofmann F, Navedo MF, Hell JW (2017) Phosphorylation of Ser1928 mediates the enhanced activity of the L-type Ca2+ channel Cav1.2 by the β2-adrenergic receptor in neurons. Sci Signal 10:eaaf9659. 10.1126/scisignal.aaf9659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson HR, Gibson ES, Benke TA, Dell'Acqua ML (2009) Regulation of postsynaptic structure and function by an A-kinase anchoring protein-membrane-associated guanylate kinase scaffolding complex. J Neurosci 29:7929–7943. 10.1523/JNEUROSCI.6093-08.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozov A, Sprengel R, Seeburg PH (2012) GluA2-lacking AMPA receptors in hippocampal CA1 cell synapses: evidence from gene-targeted mice. Front Mol Neurosci 5:22. 10.3389/fnmol.2012.00022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanderson JL, Gorski JA, Gibson ES, Lam P, Freund RK, Chick WS, Dell'Acqua ML (2012) AKAP150-anchored calcineurin regulates synaptic plasticity by limiting synaptic incorporation of Ca2+-permeable AMPA receptors. J Neurosci 32:15036–15052. 10.1523/JNEUROSCI.3326-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanderson JL, Gorski JA, Dell'Acqua ML (2016) NMDA receptor-dependent LTD requires transient synaptic incorporation of Ca2+-permeable AMPARs mediated by AKAP150-anchored PKA and calcineurin. Neuron 89:1000–1015. 10.1016/j.neuron.2016.01.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith FD, Reichow SL, Esseltine JL, Shi D, Langeberg LK, Scott JD, Gonen T (2013) Intrinsic disorder within an AKAP-protein kinase A complex guides local substrate phosphorylation. eLife 2:e01319. 10.7554/eLife.01319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith FD, Esseltine JL, Nygren PJ, Veesler D, Byrne DP, Vonderach M, Strashnov I, Eyers CE, Eyers PA, Langeberg LK, Scott JD (2017) Local protein kinase A action proceeds through intact holoenzymes. Science 356:1288–1293. 10.1126/science.aaj1669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith KE, Gibson ES, Dell'Acqua ML (2006) cAMP-dependent protein kinase postsynaptic localization regulated by NMDA receptor activation through translocation of an A-kinase anchoring protein scaffold protein. J Neurosci 26:2391–2402. 10.1523/JNEUROSCI.3092-05.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soares C, Lee KF, Nassrallah W, Béïque JC (2013) Differential subcellular targeting of glutamate receptor subtypes during homeostatic synaptic plasticity. J Neurosci 33:13547–13559. 10.1523/JNEUROSCI.1873-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stellwagen D, Malenka RC (2006) Synaptic scaling mediated by glial TNF-alpha. Nature 440:1054–1059. 10.1038/nature04671 [DOI] [PubMed] [Google Scholar]
- Sutton MA, Ito HT, Cressy P, Kempf C, Woo JC, Schuman EM (2006) Miniature neurotransmission stabilizes synaptic function via tonic suppression of local dendritic protein synthesis. Cell 125:785–799. 10.1016/j.cell.2006.03.040 [DOI] [PubMed] [Google Scholar]
- Tavalin SJ. (2008) AKAP79 selectively enhances protein kinase C regulation of GluR1 at a Ca2+-calmodulin-dependent protein kinase II/protein kinase C site. J Biol Chem 283:11445–11452. 10.1074/jbc.M709253200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiagarajan TC, Piedras-Renteria ES, Tsien RW (2002) α- and βCaMKII. inverse regulation by neuronal activity and opposing effects on synaptic strength. Neuron 36:1103–1114. 10.1016/S0896-6273(02)01049-8 [DOI] [PubMed] [Google Scholar]
- Thiagarajan TC, Lindskog M, Tsien RW (2005) Adaptation to synaptic inactivity in hippocampal neurons. Neuron 47:725–737. 10.1016/j.neuron.2005.06.037 [DOI] [PubMed] [Google Scholar]
- Tunquist BJ, Hoshi N, Guire ES, Zhang F, Mullendorff K, Langeberg LK, Raber J, Scott JD (2008) Loss of AKAP150 perturbs distinct neuronal processes in mice. Proc Natl Acad Sci U S A 105:12557–12562. 10.1073/pnas.0805922105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrigiano G. (2012) Homeostatic synaptic plasticity: local and global mechanisms for stabilizing neuronal function. Cold Spring Harb Perspect Biol 4:a005736. 10.1101/cshperspect.a005736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrigiano GG, Leslie KR, Desai NS, Rutherford LC, Nelson SB (1998) Activity-dependent scaling of quantal amplitude in neocortical neurons. Nature 391:892–896. 10.1038/36103 [DOI] [PubMed] [Google Scholar]
- Wang HL, Zhang Z, Hintze M, Chen L (2011) Decrease in calcium concentration triggers neuronal retinoic acid synthesis during homeostatic synaptic plasticity. J Neurosci 31:17764–17771. 10.1523/JNEUROSCI.3964-11.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisenhaus M, Allen ML, Yang L, Lu Y, Nichols CB, Su T, Hell JW, McKnight GS (2010) Mutations in AKAP5 disrupt dendritic signaling complexes and lead to electrophysiological and behavioral phenotypes in mice. PLoS One 5:e10325. 10.1371/journal.pone.0010325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitehead G, Jo J, Hogg EL, Piers T, Kim DH, Seaton G, Seok H, Bru-Mercier G, Son GH, Regan P, Hildebrandt L, Waite E, Kim BC, Kerrigan TL, Kim K, Whitcomb DJ, Collingridge GL, Lightman SL, Cho K (2013) Acute stress causes rapid synaptic insertion of Ca2+-permeable AMPA receptors to facilitate long-term potentiation in the hippocampus. Brain 136:3753–3765. 10.1093/brain/awt293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willoughby D, Masada N, Wachten S, Pagano M, Halls ML, Everett KL, Ciruela A, Cooper DM (2010) AKAP79/150 interacts with AC8 and regulates Ca2+-dependent cAMP synthesis in pancreatic and neuronal systems. J Biol Chem 285:20328–20342. 10.1074/jbc.M110.120725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wondolowski J, Dickman D (2013) Emerging links between homeostatic synaptic plasticity and neurological disease. Front Cell Neurosci 7:223. 10.3389/fncel.2013.00223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Wang XB, Zhou Q (2010) Perisynaptic GluR2-lacking AMPA receptors control the reversibility of synaptic and spines modifications. Proc Natl Acad Sci U S A 107:11999–12004. 10.1073/pnas.0913004107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Shapiro MS (2012) Activity-dependent transcriptional regulation of M-type (Kv7) K+ channels by AKAP79/150-mediated NFAT actions. Neuron 76:1133–1146. 10.1016/j.neuron.2012.10.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M, Patriarchi T, Stein IS, Qian H, Matt L, Nguyen M, Xiang YK, Hell JW (2013) Adenylyl cyclase anchoring by a kinase anchor protein AKAP5 (AKAP79/150) is important for postsynaptic beta-adrenergic signaling. J Biol Chem 288:17918–17931. 10.1074/jbc.M112.449462 [DOI] [PMC free article] [PubMed] [Google Scholar]