

Abstract
The Wnt signaling pathway is critically involved in both the development and homeostasis of tissues via regulation of their endogenous stem cells. Aberrant Wnt signaling has been described as a key player in the initiation of and/or maintenance and development of many cancers, via affecting the behavior of Cancer Stem Cells (CSCs). CSCs are considered by most to be responsible for establishment of the tumor and also for disease relapse, as they possess inherent drug-resistance properties. The development of new therapeutic compounds targeting the Wnt signaling pathway promises new hope to eliminate CSCs and achieve cancer eradication. However, a major challenge resides in developing a strategy efficient enough to target the dysregulated Wnt pathway in CSCs, while being safe enough to not damage the normal somatic stem cell population required for tissue homeostasis and repair. Here we review recent therapeutic approaches to target the Wnt pathway and their clinical applications.
Keywords: Wnt-signaling, Cancer Stem Cells, Microenvironment
1) Introduction
Despite significant progress in cancer treatment and remission rates, numerous hurdles in the management of cancer persist. Resistance to treatment associated with disease relapse and metastasis still represent major critical problems that need to be addressed. A subset of cancer cells: the cancer stem cell (CSC) or cancer-initiating cell (CIC)1 populations are the key players associated with these problems. CSCs, by definition, share the same properties of self-renewal and pluripotency as normal somatic stem cells (SSCs). Self-renewal constitutes the ability to produce at least one daughter cell identical to the mother cell, thereby retaining its stem cell properties, while the pluripotency of stem cells allows them to differentiate into multiple divergent committed and specialized cell types. CSC may emerge from normal somatic stem cells in the affected tissue or organ system after genetic alterations acquired during DNA replication, via various insults and/or from microenvironmental factors and are believed to be responsible in cancer initiation1. Over the past few years, a major effort in cancer research has been to better characterize the CSC population and most importantly to efficiently and safely target these cells. One way to achieve this goal is the identification of major pathways involved in the stemness of CSCs and how to target them without affecting the normal somatic stem cells. As in SSC, CSC properties are governed by the evolutionarily conserved signaling pathways Notch2, Hedgehog3 and Wnt/β-catenin4,5. Here, we will review recent findings on Wnt signaling and its role in cancer initiation, maintenance and drug resistance and the promise of new compounds targeting this pathway.
2) Cancer Stem Cells and Their Role in Tumorigenesis
Over the past decades, the concept of cancer stem cells has re-emerged and led to impressive efforts in this field of research. Recent advances in the field have decreased doubt about CSC existence6-9 and their function in many cancers, however, the question of their origin remains highly controversial with two alternative theories. On the one hand, CSCs could result from a genetic alteration in a cancer cell from the tumor bulk, leading to the activation of one or more of the major signaling pathways previously cited, thereby acquiring self-renewal properties10,11. On the other hand, CSC could originate from acquired mutations of normal somatic stem cells transforming them into CSCs1,12. Both of these hypotheses may potentially apply according to the type of cancer and tissue affected. CSCs, represent a rare population of cells amongst the tumor bulk that are able to maintain the tumor via proliferation and self-renewal13 capabilities and telomerase expression13. They are also known to be more resistant to conventional treatment (chemotherapy and radiotherapy) and responsible for cancer relapse and metastasis14,15. Their quiescent state and specific interactions with their microenvironment play a significant role in their drug resistance properties16. All these characteristics aid in the explanation of why CSC are thought to be responsible for cancer establishment, progression, drug resistance and relapse17,18 and are strongly correlated to poor outcome in clinical reports19,20. Evidence of CSC existence and tumor initiating properties was first provided by Dick and colleagues in leukemia showing that only a very small proportion of primary acute myeloid leukemia (AML) cells, defined by the CD34+ 38- markers, was able to initiate disease in immunodeficient mice21. These Leukemic Stem Cells (LSC) possessed the self-renewal property of CSC and the capability of pluripotency leading to leukemia22 but could also give rise to non-LSC populations. Subsequently, over the past decade, a large number of studies have identified CSCs in multiple tumor types, including brain tumors23, melanoma24, breast25, liver26, pancreatic27 and colon cancer 28.
3) Wnt signaling in Embryonic Development and Homeostasis
Wnt signaling is involved in numerous fundamental processes essential for embryonic development and normal adult homeostasis. The first member of the Wnt family, initially discovered as the proto-oncogene “Int-1” in mice29, was found five years later to be the homolog of the “wingless” gene, one of the main regulators of Drosophila melanogaster segment polarity30. The term “wnt” was created by the fusion of these two gene names. The Wnt family is a highly evolutionarily conserved family of proteins as shown by ectopic expression of Wnt1 from Drosophila in other organisms, causing serious developmental issues31,32. The human Wnt family is composed of nineteen different cysteine-rich glycoproteins acting as ligands for more than 15 receptors or co-receptors33. This signaling pathway has already been shown to be involved in many cellular functions essential for normal organ development including cell proliferation, survival, self-renewal/differentiation etc34,35. Very rapidly after discovery of the pathway, multiple dysfunctions and mutation of these pathways were shown to be related to several diseases, including metabolic (e.g. type II diabetes36, degenerative (e.g. Parkinson’s37, Alzheimer’s38) and particularly cancers (hepatocarcinoma39,40, colon cancer41, leukemias42.
The Wnt signaling pathway has been extensively studied and reviewed4,43-44. The pathway is generally dissected into three sub-pathways: canonical, non-canonical planar cell polarity (PCP) pathway and non-canonical Wnt/calcium pathway. The canonical pathway requires Wnt ligand binding to Frizzled receptors as well as LRP5/6 co-receptors (low density lipoprotein receptor-related protein 5/6) to initiate intracellular signaling via β-catenin nuclear translocation. β-catenin is a highly unstable protein with a tightly controlled cytoplasmic presence. In the absence of Wnt ligands, cytoplasmic β-catenin is targeted by a so-termed degradation complex. This complex is composed of the tumor suppressor Adenomatous Polyposis Coli (APC), the scaffolding protein AXIN and two kinases CK1α (casein kinase 1α) and GSK-3β (glycogen synthase kinase 3 β)45 (Figure 1A). These last two components are able to phosphorylate β-catenin on several serine and threonine residues in its N-terminus. Phosphorylated β-catenin is then recognized by β-Transducin, which is part of an ubiquitin ligase complex, leading to poly-ubiquitination and proteasomal degradation of β-catenin46. Wnt ligand binding to Frizzled receptors in association with LRP5/6 induces Dishevelled (DVL) phosphorylation, which subsequently recruits Axin thereby deconstructing the degradation complex and achieving β-catenin stabilization and subsequent nuclear translocation. In the nucleus, β-catenin can bind members of the TCF/LEF (T-cell Factor/Lymphoid Enhancer Factor) family of transcription factors and recruit the transcriptional Kat3 co-activators p300 and/or CBP (CREB-binding protein) to transcribe Wnt target genes and engender chromatin modifications 47-50 (Figure 1B).
Figure 1.
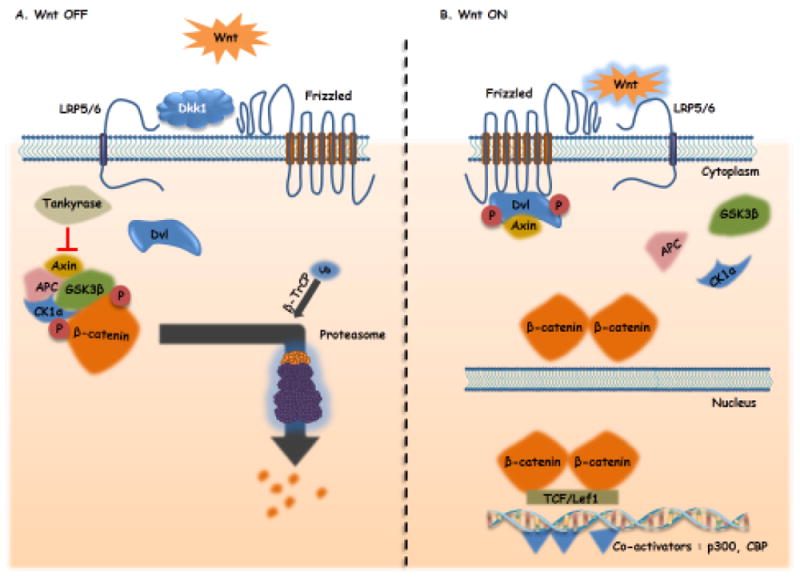
A: “Wnt Off”. In the absence of Wnt ligands, a destruction complex composed of Axin-1 and its tumor suppressor partners Adenomtous Polyposis Coli (APC), Glycogen synthase kinase 3 beta (GSK3B) and Casein kinase 1 (CK1α) is formed. The destruction complex phosphorylates ß-catenin and targets it for proteasomal degradation regulating the cytoplasmic level of ß-catenin.
B: “Wnt On”. Wnt ligands bind to the Frizzled/ Lrp5/6 (Low density lipoprotein receptor-related proteins 5 or 6) receptors leading to the phosphorylation of a negative regulator of the destruction complex, Dishevelled (Dvl). Dvl recruits Axin, inhibiting its interaction with other components of the destruction complex. ß-catenin is then free to accumulate in the cytoplasm and translocates to the nucleus, where it activates the trancscription of Wnt target genes after association with transcription factors of the TCF/Lef family and co-activators such as CBP (cyclic AMP response element-binding protein) and p300. Arrows indicate activation/induction, blunt ended lines indicate inhibition/blockade.
Two different Wnt pathways, qualified as “β-catenin-independent pathways” also co-exist with the canonical Wnt pathway and are more generally associated with differentiation, cell polarity and migration. In the non-canonical Planar Cell Polarity pathway (PCP), Wnt ligands can bind Frizzled receptors and activate small GTPases such as RhoA (Ras homolog gene family member A), RAC (Ras-related C3 botulinum toxin substrate) and Cdc42 (cell division control protein 42), via recruitment and activation of Dishvelled51 (Figure 2A). The PCP pathway affects the cytoskeleton and triggers the transcriptional activation of target genes responsible for cell adhesion and migration52. In the Calcium-dependent pathway, Wnt ligands utilize Frizzled receptors and RYK or ROR (alternative receptors) enhancing cell migration and Wnt canonical pathway inhibition through the management of intracellular calcium flux and activation of calmodulin kinase II (CaMK2), Jun kinase (JNK) and PKC53 (Figure 2B).
Figure 2.
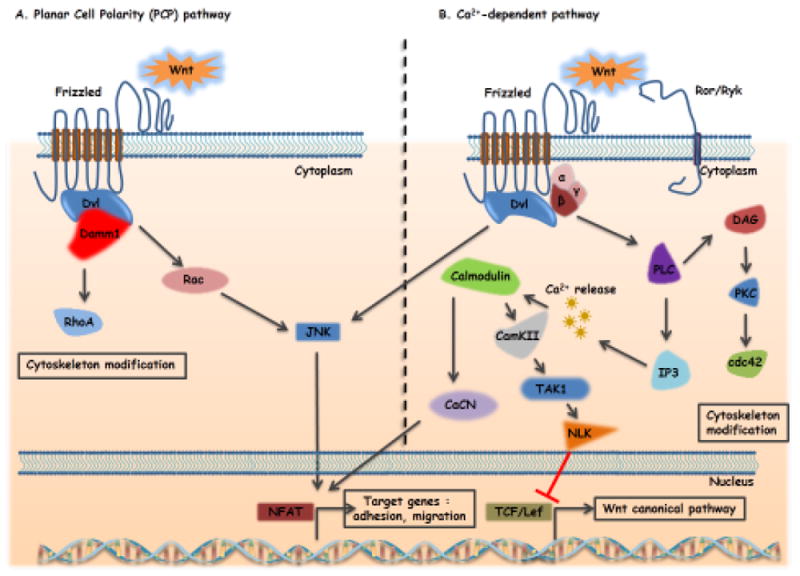
A: Non-Canonical Wnt-signaling: Non-canonical Wnt/PCP (planar cell polarity) pathway. Wnt ligand binding to frizzled receptors leads to activation of Dishevelled (Dvl) which recruits DAAM1 (Dishevelled associated activator of morphogenesis 1) enhancing the stimulation of GTPases Rac (Ras-related C3 botulinum toxin substrate) and RHOA (Ras homolog gene family member A) leading to actin cytoskeleton rearrangement. In addition, Dvl activates Rac and finally JNK (c-Jun-N-terminal-kinase) thereby modulating cell migration.
B: Non-canonical Wnt/calcium pathway. Wnt ligands bind to frizzled receptors and Ror/Ryk co-receptors, activating Dvl and trimeric G-proteins (Gα,β,γ). This leads to the generation of IP3 (inositol 1,4,5-triphosphate) and DAG2 (diacylglycerol) through PLC (Phospholipase C) activation. IP3 triggers the release of calcium ions (Ca2+) from the endoplasmic reticulum activating calmodulin and subsequently CAMKII (calcium/calmodulin- dependent kinase II), TAK-1 (TGF-β activated kinase 1) and NLK (Nemo-like kinase) thereby inhibiting the canonical Wnt pathway. Moreover, calmodulin activation stimulates calcineurin and NFAT (Nuclear Factor of Activated T-cells) involved in adhesion and migration processes. This pathway activates also PKC (Protein Kinase C) and Cdc42 (cell division control protein 42) rearranging the actin cytoskeleton. Arrows indicate activation, blunt ended lines indicate inhibition/blockade.
Although dissection into 3 different pathways facilitates our understanding of this highly complex signaling system, it has already been shown that in reality Wnt signaling involves the integration of these three pathways and they all need to be considered to derive a complete vision of the effects of Wnt signaling modulation54,55,56. These Wnt pathways are critical in major functions at the embryonic stage of development, including stem cell pool regulation, cell migration and specialization as well as at the adult stage in wound healing, and tissue homeostasis via SSC maintenance (including hair, skin57 and intestine58).
4) The Role of Wnt Signaling in Cancer Stem Cells
Dysfunctional Wnt signaling has been related to the evolution of and maintenance of leukemic stem cells as well as many other different cancers. This is not surprising given the importance of the Wnt pathway in stem cell homeostasis59. Examples of aberrant Wnt signaling in cancer stem cell development include the progression of chronic phase CML toward blastic crisis phase due to GSK3β mutations and β-catenin stabilization in GMP cells (granulocyte-macrophage progenitor cells)60. A recent study showed that despite the inhibitory effect of tyrosine kinase inhibitor (TKI) on Wnt signaling pathway in CML stem cells, relapses occur in patients at least in part by reactivation of the Wnt pathway61. TKI treatment induces a down-regulation of miR29 involved in CD70 promoter methylation. The overexpression of CD70 enhances the transcription of CD27 which is a known activator of the Wnt signaling pathway62. Wang et al. also showed also that constitutive activation of the canonical Wnt pathway, via expression of a stabilized β-catenin is necessary to generate AML leukemic stem cells from MLL-AF9-transduced progenitors cells11. This study suggests that aberrant Wnt pathway activation could give rise to leukemic stem cells (LSC) not only from hematopoietic stem cells (HSC) but additionally from more committed progenitors. Recently, Giambra and colleagues showed, using a Wnt reporter construct expressing GFP under the TCF promoter, that minor subpopulations of bulk T-cell acute lymphoblastic leukemia (T-ALL) had highly activated Wnt/β-catenin pathway signaling and that these cells were able to transplant the disease in a limiting dilution assay63. Leukemic stem cells were highly enriched in the GFP+ Wnt expressing population compared to the GFP- (ratio of over 200 fold) suggesting that Wnt signaling is also required for T-ALL stem cell self-renewal. In this model, the transcription of the β-catenin seems to be triggered by the transcription factor HIF1-alpha (Hypoxia-Induced Factor 1-alpha) and deletion of HIF1-alpha leads to LSC targeting63. Our group recently demonstrated the implication of the Wnt pathway in the self-renewal of B-cell acute lymphoblastic leukemia (B-ALL). The treatment of B-ALL cells with a small molecule that specifically binds to the N-terminal of CBP, ICG-001, inhibits the interaction between β-catenin and CBP leading to differentiation and loss of self-renewal64. iCRT14, a novel β-catenin-TCF interaction inhibitor, leads to a decrease in Wnt target gene expression, decreases viability of ALL cell lines in combination with chemotherapy and sensitizes chemoresistant patient samples-derived ALL cells responsible of relapse65.
In order to efficiently target CSCs, researchers initially focused on ways to identify them. Even if the normal SSCs and CSCs usually express the same cell surface markers66, some reports have successfully characterized cancer stem-like cells in breast cancer based upon specific marker sets (expression of CD44highCD24low)25. Both CD44 and CD24 are direct Wnt target genes67-70. CD44 acts like a positive regulator of the Wnt pathway by playing on LRP6 localization and activity 68,70,71. The Wnt signaling pathway also appears to play an important role in another hallmark of cancer stem cells and metastasis, i.e. the epithelial-to-mesenchymal transition (EMT)72,73,74. The down-regulation of E-Cadherin (usually tightly associated with β-catenin in normal epithelium) triggers the nuclear translocation of β-catenin and activation of canonical Wnt signaling75. The gene slug, a marker gene of EMT, also induces nuclear translocation of β-catenin76,76. Moreover, twist and slug, strong activators of EMT are both putative β-catenin targets77. Furthermore, a number of Wnt/β-catenin targets genes have been associated with invasion, migration and metastasis (including S100A4, fibronectin, L1CAM, CD44, MMP7, uPAR, etc.)78. Wnt signaling may also play an important role in the resistance of cancer stem cells to chemotherapy. The promoter sequence of the multidrug resistance gene ABCB1/MDR-1 contains several TCF binding elements triggering its transcription in colorectal cancer79. Inhibition of the β-catenin/CBP interaction using the small molecule ICG-001 decreases the expression of Survivin/BIRC5, which is an inhibitor of apoptosis and a target of CBP, leading to eradication of drug resistant ALL cells in vitro and prolonged survival of ALL engrafted mice64. Similar results have been obtained using ICG-001 with CML LSC (Zhao et al., 2015 Oncogene in the press). Wnt signaling has also been linked to hematopoietic CSC which seem to be dependent on this pathway11,80. In CML, Wnt pathway deregulation favors the progression of disease to more advanced phases81. The deregulation of Wnt signaling can also occur at the epigenetic level. For example, the promoters of several Wnt pathway inhibitors (i.e. SFRP, DKK and WIF-1) were found to be hypermethylated in ALL and AML, correlating negatively with the survival of these patients82,83.
5) Wnt signaling and the hematopoietic stem cell niche
Hematopoietic Stem Cell fate is tightly controlled by both internal and external signals, the latter coming mostly from the very specific HSC microenvironment, termed, the hematopoietic niche84-86. This niche is composed of various cell types (osteoblasts, osteoclasts, endothelial cells, mesenchymal stem cells, etc.) and communicates actively with HSC via direct contacts (integrins, N-Cadherin, etc.) and soluble factors including Wnt ligands87-89,90,91. Even after an extensive research, the role of the microenvironment and Wnt ligands on both HSC and LSC is highly controversial92,93. Under physiologic conditions, some studies show the critical role of the canonical Wnt signaling pathway for HSC quiescence and self-renewal maintenance: the expression of the Wnt pathway inhibitor Dickkopf1 (Dkk1), specifically in osteoblasts, leads to a decrease of Wnt signaling in HSC and loss of their stem cells properties, via uncontrolled proliferation and division94 while activation of Wnt signaling in the stroma induces Notch ligand secretion which activates self-renewal programs in HSC95,96. However, other researchers demonstrated that the canonical Wnt pathway was dispensable for the adult hematopoiesis in mouse models97-99. On the contrary, the overexpression of β-catenin in the hematopoietic system leads to failure in the maintenance of the HSC pool and to a leukemic-like differentiation block in both the myeloid and lymphoid compartments100. Under pathologic conditions, Wnt signaling seems to have different effects according to the leukemia studied. In the chronic myeloid leukemia (CML), Zhang et al showed that the microenvironment (mimicked by mesenchymal stem cells (MSC)) reduces apoptosis and improves the engraftment of CML LSC/progenitors treated with imatinib. This protective effect is mediated by direct interaction between leukemic cells and MSC through N-Cadherin and induces activation of the Wnt signaling pathway via stabilization of β-catenin101. To corroborate these findings, Heidel et al. showed that inhibition of β-catenin allows the targeting of CML LSC that are resistant to imatinib80. However, inhibition of extrinsic-Wnt signaling by Dkk1 does not impair homing and/or leukemogenesis of AML LSC or pre-LSC in vivo as the translocation t(9;11) induces sufficient cell-intrinsic Wnt signaling to promote leukemia development93. The microenvironment may be implicated in many ways as well, either in the initiation of the disease or by favoring LSC proliferation and drug resistance102,103, thus making it a potentially attractive new target for treatment. However, the role of the niche is still not completely understood. Kode et al showed the essential role of osteoblasts in acute myeloid leukemia initiation via Wnt and FoxO1 expression104. Recently, Bowers et al demonstrated the importance of the bone marrow microenvironment in HSC homeostasis and in leukemia development showing that osteoblast depletion impairs the quiescence and self-renewal of normal HSC and also leads to accelerated leukemia development in a mouse model of CML105. Taken together, these data suggest that the microenvironment may represent an attractive alternative to target LSC, notably via modulation of the Wnt signaling pathway. However, further studies are required to fully understand its fundamental role in leukemia establishment and maintenance.
6) Wnt Inhibiting Molecules: Biologics and clinics
After decades of research and discovery on the Wnt signaling pathway, few molecules are now considered to be relatively specific for targeting the Wnt pathway, and to date none has been approved by the US Food and Drug Administration (FDA). Some other FDA-approved molecules, like Non-Steroidal Anti-Inflammatory Drugs (NSAIDS, used for treatment of pain, fever) or vitamin derivatives, demonstrated interesting anti-cancer effects106,107 and particularly in Wnt-dependent cancers e.g colorectal cancer108,109. Cyclooxygenases (COX1 and 2) metabolize arachidonic acid into prostaglandins (PG) that via their G-protein Coupled Receptors can lead to β-catenin stabilization and activation of canonical Wnt signaling110,110-112. The inhibition of COX by NSAIDS (aspirin, sulindac or specific COX2 inhibitors like celecoxib) suppresses the synthesis of prostaglandins and thereby inhibits Wnt signaling. These compounds, especially celecoxib, also showed COX-independent anticancer effects notably in a xenograft model of COX-2-deficient tumors113-117. NSAIDS have the capacity to decrease the number of polyps in a mouse model of Familial Adenomatous Polyposis (FAP) mouse, where the APC gene is truncated and Wnt/β-catenin signaling constitutively actived118,119. FAP patients treated for 6 months with the NSAID sulindac showed a reduction in nuclear β-catenin in polyps and a reduction in polyp formation120-123. The aspirin derivative NO-ASA (NO-releasing aspirin) showed even better efficacy in reduction of polyp formation in vitro and in vivo possibly via disruption of the β-catenin/TCF complex without any observable toxicity to the normal intestine124-126.
Retinoids, produced from vitamin A metabolism, demonstrated anti-cancer effects at least in part via Wnt signaling pathway inhibition127. 1α,25-dihydroxy-vitamin D3, the active form of vitamin D, demonstrated tumor suppressor activity, notably by formation of a transcriptional complex able to bind β-catenin and thereby enhancing the expression of E-cadherin. These effects lead to retention of β-catenin in the cytoplasm, resulting in inhibition of the Wnt pathway in breast and colon cancers128. A novel humanized antibody (UC-961, cirmtuzumab) targeting the Receptor tyrosine kinase-like Orphan Receptor 1 (ROR1), expressed by the chronic lymphocytic leukemia cells (CLL) but not on normal tissues, showed anticancer effects in a CLL animal model129. This antibody recently entered a Phase I clinical trial to determine the safety and the effects of this antibody and is currently recruiting (NCT02222688).
Besides these FDA-approved non-specific Wnt inhibitors, several molecularly targeted agents have been developed and have entered pre-clinical or clinical trials. Dvl, being one of the key regulators of the Wnt canonical pathway, is a focus of numerous studies and has engendered the development of several inhibitors. The PDZ domain of Dvl plays an essential role in Dvl-Frizzled receptor interactions and the intracellular transduction of the Wnt signal. Some inhibitors of this PDZ domain (NSC 668036, FJ9, 3289-8625 – Figure 3), discovered by in silico screening, showed the ability to inhibit the Wnt pathway in vivo130-132. Other compounds, designed to inhibit key steps in the Wnt pathway have also been designed. LGK974 is a porcupine (PORCN) inhibitor, which entered into a phase I clinical trial in 2011 (Novartis, NCT01351103, recruitment phase). Porcupine is a member of the membrane-bound O-acetyltransferase (MBOAT) family and is responsible for lipid modification of Wnt and secretion133,134. The trial will investigate the effects of LGK974 on the Wnt signaling pathway in patients affected with Wnt-dependent cancers (pancreatic adenocarcinoma, BRAF mutant colorectal cancer) (clinicaltrials.gov). Recently, another PORCN inhibitor, ETC-1922159 (ETC-159), developed in a collaboration between the Agency for Science, Technology and Research (A*STAR) and Duke-National University of Singapore Graduate Medical School (Duke-NUS) entered into a phase I clinical trial in Singapore. The first patient was dosed on Jun 18, 2015. ETC-159 inhibits Wnt secretion and activity and is highly efficient preclinically in different cancers driven by Wnt signaling and notably in R-spondin translocation colorectal cancers135. The tankyrase inhibitors (XAV-939 and IWR-1) stabilize axin and induce the degradation of the β-catenin136 and may act as anti-tumor drugs also by participating in telomere shortening137.
Figure 3. Schematic of Wnt inhibitors currently in clinical trials.
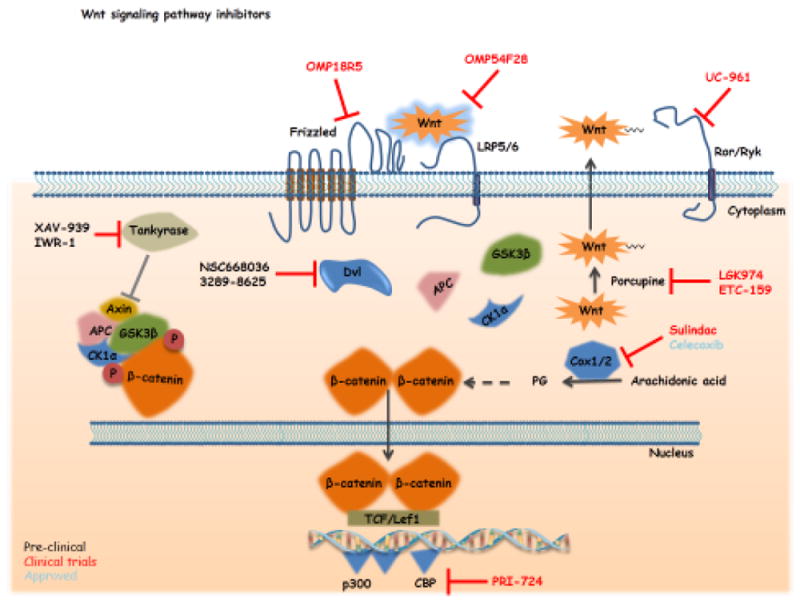
This figure summarizes the different Wnt pathway modulators with variable specificities and at different stages of development (fully described in the main text). The current agents in clinical trials are the porcupine inhibitor LGK974, which inhibits Wnt posttranslational palmitoylation and secretion. OMP18R5, is a fully humanized monoclonal antibody specifically binding to multiple Frizzled (Fzd) receptors and OMP-54F28 is a Fc fusion protein with Fzd8, which binds all Wnt ligands. Both inhibit the intracellular transduction of the Wnt signal. PRI-724 specifically targets the β-catenin transcriptional co-activator CBP thereby blocking their interaction. UC-961 (cirmtuzumab) is a humanized antibody targeting the targeting specifically the Wnt receptor ROR1. Arrows indicate activation/induction, blunt ended lines indicate inhibition/blockade.
Among the few agents already in clinical trials, two were developed by Oncomed Pharmaceuticals Inc. OMP-18R5 (Vantictumab) is a fully humanized antibody directed against minimally five different Frizzled receptors. In pre-clinical studies, OMP-18R5 demonstrated anti-proliferative effects in various human tumors model (lung, pancreas, breast and colon) and had synergistic effects with conventional chemotherapy138. The results of the first Phase Ia showed a decrease in Wnt pathway gene expression and increased expression of differentiation genes associated with some adverse events including fatigue, vomiting, diarrhea, constipation, nausea and abdominal pain (ASCO, 2013). This compound is now in Phase Ib trials in combination with standard chemotherapy for solid tumors (breast, lung and pancreas cancers). OMP-54F28, another agent developed by Oncomed Pharmaceuticals, is a recombinant fusion protein containing the extracellular ligand binding domain of human Frizzled 8 receptor fused to a human IgG1 Fc fragment139. OMP-54F28 can bind native Fzd8 receptor’s ligands and thereby inhibit Wnt signaling. Preclinical studies demonstrated the anti-tumor efficacy of OMP-54F28: reduction of tumor growth and decrease of CSC frequency as a single agent and in combination with other chemotherapeutic agents. A phase I trial (NCT01608867) is currently ongoing. It is a dose escalation study in patients with advanced solid tumors. Subjects will be assessed for safety, immunogenicity, pharmacokinetics, biomarkers, and efficacy (NCT01608867). It appears that the most common adverse events are fatigue, muscle spasms, alopecia, nausea, decreased appetite and dysgeusia (http://www.eurekalert.org/pub_releases/2014-05/uocd-rip053014.php). Additionally, patients are followed for bone density evolution, as bone fracture was observed in one patient at the highest tested dose (20mg/kg every three weeks after 6 cycles). Three Phase 1b studies have been started to check the dose escalation of OMP-54F28 in ovarian (NCT02092363), pancreatic (NCT02050178) and hepatocellular (NCT02069145) cancers in combination with respective conventional chemotherapy.
Wnt signaling can also be modulated very late in the pathway. Our group used a secondary structure-templated chemical library to identify ICG-001 which can efficiently modulate the Wnt pathway140. Despite the huge homology between the two Kat3 co-activator proteins CBP and p300, ICG-001 was shown to bind specifically to the cyclic AMP response element-binding protein (CBP) and not to the related transcriptional coactivator p300140,141. This molecule disrupts the ß-catenin/CBP complex and increases the proportion of ß-catenin/p300 leading to down-regulation of survivin/BIRC5 mRNA and specific apoptosis in colon cancer cells in vitro and in vivo. Recently, Prism Pharmaceuticals developed a second generation ß-catenin/CBP inhibitor PRI-724. In a Phase Ia safety study in colon cancer, this compound was able to decrease in a dose-dependent manner the expression of survivin/BIRC5 in circulating tumor cells, with an acceptable toxicity profile (ASCO, June 2013 and NCT01302405142). Three patients had stable disease for 8, 10 and 12 weeks. Three Phase I/II trials are ongoing in patients with AML/CML (NCT01606579, alone or in combination with AraC or dasatinib), with advanced or metastatic pancreatic adenorcarcinoma (NCT01764477, in combination with Gemcitabine) and in patients with newly diagnosed metastatic colorectal cancer (NCT02413853, in combination with bevacizumab, leucovorin calcium, oxaliplatin, and fluorouracil). A Phase I dose escalation trial in patients with HCV-induced cirrhosis is also on going (NCT02195440).
7) Concluding remarks
After more than 30 years of discovery and investigation, the complexity of the Wnt signaling pathway is clear. Many of its components have been revealed and its implications in a broad range of diseases have been described. However, to date no therapeutic agent is available on the market that specifically and efficiently targets this pathway. In the past 5 years, some Wnt signaling modulating agents, targeting different key steps in the pathway (Wnt secretion, signal transduction or ß-catenin transcriptional activity) entered the clinic to determine inhibitory efficacy and also critically safety. Indeed, Wnt signaling is a highly evolutionarily conserved pathway involved in multiple crucial homeostatic functions, suggesting that targeting this pathway may induce serious adverse events (e.g. OMP-54F28 trials, where all patients were monitored for bone mineral density modification (BMD) and turnover and received zoledronic acid when their BMD declines (NCT01608867)). Moreover, many of the potential targets like ß-catenin are also implicated in others critical functions (cell-cell adhesion, development, self-renewal…)143,144. Clearly, precise modulation of the Wnt pathway will be necessary to balance anti-tumor efficacy with adverse events and will be a challenge for ongoing and future clinical trials. Despite these concerns, new regulators of the Wnt signaling cascade offer the opportunity for us to increase our comprehension of this exceedingly complex pathway and potentially for the treatment of Wnt-related diseases including cancer.
Acknowledgments
YMK was supported by St. Baldrick’s Scholar Award, Nautica Foundation, Couples Against Leukemia and NIH R01CA172896 (YMK). MK is supported by USC Norris Comprehensive Cancer Center Support Grant P30 CA014089, NIH R01CA166161, R21NS074392, R21AI105057 and NIH R01 HL112638. We apologize for the omission of any of our colleagues work due to space limitations.
Biographies
Michael Kahn
USC Norris Comprehensive Cancer Center, USC Center for Molecular Pathways and Drug Discovery, University of Southern California, Los Angeles, California 90033, USA.
Michael Kahn is the Provost Professor of Medicine and Pharmacy at the University of Southern California (USC), USA. Prior to joining USC, he was at the University of Washington, Seattle, USA, as well as the scientific director at the Institute for Chemical Genomics. His laboratory utilizes forward chemical genomics strategies to dissect complex signalling pathways. His laboratory has been working extensively in the area of WNT signalling for the past 15 years.
Yong-Mi Kim
Yong-mi Kim is an Associate Professor of Pediatrics and Pathology at the University of Southern California, Children’s hospital Los Angeles. Her laboratory has been working in the area of drug resistance and the microenvironment of leukemia.
Yann Duchartre
Yann Duchartre graduated with a Ph.D. in Cell Biology and Physiology from University of Bordeaux (France) in December 2012. After a first postdoctoral position looking for new therapeutic approaches in CML at the Beckman research Institute of City of Hope, he joined the Children’s Hospital of Los Angeles to further study the pre B-ALL cells mechanisms of resistance to therapy.
Footnotes
Conflict of interest
None
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- 1.Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001;414:105–111. doi: 10.1038/35102167. [DOI] [PubMed] [Google Scholar]
- 2.Liu J, Sato C, Cerletti M, Wagers A. Notch signaling in the regulation of stem cell self-renewal and differentiation. Curr Top Dev Biol. 2010;92:367–409. doi: 10.1016/S0070-2153(10)92012-7. [DOI] [PubMed] [Google Scholar]
- 3.Merchant AA, Matsui W. Targeting Hedgehog--a cancer stem cell pathway. Clin Cancer Res. 2010;16:3130–3140. doi: 10.1158/1078-0432.CCR-09-2846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Miki T, Yasuda SY, Kahn M. Wnt/beta-catenin signaling in embryonic stem cell self-renewal and somatic cell reprogramming. Stem Cell Rev. 2011;7:836–846. doi: 10.1007/s12015-011-9275-1. [DOI] [PubMed] [Google Scholar]
- 5.Takahashi-Yanaga F, Kahn M. Targeting Wnt signaling: can we safely eradicate cancer stem cells? Clin Cancer Res. 2010;16:3153–3162. doi: 10.1158/1078-0432.CCR-09-2943. [DOI] [PubMed] [Google Scholar]
- 6.Driessens G, Beck B, Caauwe A, Simons BD, Blanpain C. Defining the mode of tumour growth by clonal analysis. Nature. 2012;488:527–530. doi: 10.1038/nature11344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chen J, et al. A restricted cell population propagates glioblastoma growth after chemotherapy. Nature. 2012;488:522–526. doi: 10.1038/nature11287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schepers AG, et al. Lineage tracing reveals Lgr5+ stem cell activity in mouse intestinal adenomas. Science. 2012;337:730–735. doi: 10.1126/science.1224676. [DOI] [PubMed] [Google Scholar]
- 9.McCarthy N. Cancer stem cells: Tracing clones. Nat Rev Cancer. 2012;12:579. doi: 10.1038/nrc3354. [DOI] [PubMed] [Google Scholar]
- 10.Espinoza I, Miele L. Deadly crosstalk: Notch signaling at the intersection of EMT and cancer stem cells. Cancer Lett. 2013;341:41–45. doi: 10.1016/j.canlet.2013.08.027. [DOI] [PubMed] [Google Scholar]
- 11.Wang Y, et al. The Wnt/beta-catenin pathway is required for the development of leukemia stem cells in AML. Science. 2010;327:1650–1653. doi: 10.1126/science.1186624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Krivtsov AV, et al. Transformation from committed progenitor to leukaemia stem cell initiated by MLL-AF9. Nature. 2006;442:818–822. doi: 10.1038/nature04980. [DOI] [PubMed] [Google Scholar]
- 13.O’Brien CA, Kreso A, Jamieson CH. Cancer stem cells and self-renewal. Clin Cancer Res. 2010;16:3113–3120. doi: 10.1158/1078-0432.CCR-09-2824. [DOI] [PubMed] [Google Scholar]
- 14.Li L, et al. SIRT1 activation by a c-MYC oncogenic network promotes the maintenance and drug resistance of human FLT3-ITD acute myeloid leukemia stem cells. Cell Stem Cell. 2014;15:431–446. doi: 10.1016/j.stem.2014.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sadarangani A, et al. GLI2 inhibition abrogates human leukemia stem cell dormancy. J Transl Med. 2015;13:98. doi: 10.1186/s12967-015-0453-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vermeulen L, et al. Wnt activity defines colon cancer stem cells and is regulated by the microenvironment. Nat Cell Biol. 2010;12:468–476. doi: 10.1038/ncb2048. [DOI] [PubMed] [Google Scholar]
- 17.Clevers H. The cancer stem cell: premises, promises and challenges. Nat Med. 2011;17:313–319. doi: 10.1038/nm.2304. [DOI] [PubMed] [Google Scholar]
- 18.Visvader JE, Lindeman GJ. Stem cells and cancer - the promise and puzzles. Mol Oncol. 2010;4:369–372. doi: 10.1016/j.molonc.2010.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wicha MS. Migratory gene expression signature predicts poor patient outcome: are cancer stem cells to blame? Breast Cancer Res. 2012;14:114. doi: 10.1186/bcr3338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hussenet T, Dembele D, Martinet N, Vignaud JM, du MS. An adult tissue-specific stem cell molecular phenotype is activated in epithelial cancer stem cells and correlated to patient outcome. Cell Cycle. 2010;9:321–327. doi: 10.4161/cc.9.2.10421. [DOI] [PubMed] [Google Scholar]
- 21.Lapidot T, et al. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature. 1994;367:645–648. doi: 10.1038/367645a0. [DOI] [PubMed] [Google Scholar]
- 22.Jamieson CH, Weissman IL, Passegue E. Chronic versus acute myelogenous leukemia: a question of self-renewal. Cancer Cell. 2004;6:531–533. doi: 10.1016/j.ccr.2004.12.005. [DOI] [PubMed] [Google Scholar]
- 23.Singh SK, et al. Identification of human brain tumour initiating cells. Nature. 2004;432:396–401. doi: 10.1038/nature03128. [DOI] [PubMed] [Google Scholar]
- 24.Fang D, et al. A tumorigenic subpopulation with stem cell properties in melanomas. Cancer Res. 2005;65:9328–9337. doi: 10.1158/0008-5472.CAN-05-1343. [DOI] [PubMed] [Google Scholar]
- 25.Al-Hajj M, Wicha MS, ito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A. 2003;100:3983–3988. doi: 10.1073/pnas.0530291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ma S, et al. Identification and characterization of tumorigenic liver cancer stem/progenitor cells. Gastroenterology. 2007;132:2542–2556. doi: 10.1053/j.gastro.2007.04.025. [DOI] [PubMed] [Google Scholar]
- 27.Li C, et al. Identification of pancreatic cancer stem cells. Cancer Res. 2007;67:1030–1037. doi: 10.1158/0008-5472.CAN-06-2030. [DOI] [PubMed] [Google Scholar]
- 28.O’Brien CA, Pollett A, Gallinger S, Dick JE. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature. 2007;445:106–110. doi: 10.1038/nature05372. [DOI] [PubMed] [Google Scholar]
- 29.Nusse R, Varmus HE. Many tumors induced by the mouse mammary tumor virus contain a provirus integrated in the same region of the host genome. Cell. 1982;31:99–109. doi: 10.1016/0092-8674(82)90409-3. [DOI] [PubMed] [Google Scholar]
- 30.Baker NE. Molecular cloning of sequences from wingless, a segment polarity gene in Drosophila: the spatial distribution of a transcript in embryos. EMBO J. 1987;6:1765–1773. doi: 10.1002/j.1460-2075.1987.tb02429.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McMahon AP, Moon RT. Ectopic expression of the proto-oncogene int-1 in Xenopus embryos leads to duplication of the embryonic axis. Cell. 1989;58:1075–1084. doi: 10.1016/0092-8674(89)90506-0. [DOI] [PubMed] [Google Scholar]
- 32.Mlodzik M. Planar cell polarization: do the same mechanisms regulate Drosophila tissue polarity and vertebrate gastrulation? Trends Genet. 2002;18:564–571. doi: 10.1016/s0168-9525(02)02770-1. [DOI] [PubMed] [Google Scholar]
- 33.Niehrs C. The complex world of WNT receptor signalling. Nat Rev Mol Cell Biol. 2012;13:767–779. doi: 10.1038/nrm3470. [DOI] [PubMed] [Google Scholar]
- 34.Willert K, Jones KA. Wnt signaling: is the party in the nucleus? Genes Dev. 2006;20:1394–1404. doi: 10.1101/gad.1424006. [DOI] [PubMed] [Google Scholar]
- 35.Schambony A, Wedlich D. Wnt-5A/Ror2 regulate expression of XPAPC through an alternative noncanonical signaling pathway. Dev Cell. 2007;12:779–792. doi: 10.1016/j.devcel.2007.02.016. [DOI] [PubMed] [Google Scholar]
- 36.Welters HJ, Kulkarni RN. Wnt signaling: relevance to beta-cell biology and diabetes. Trends Endocrinol Metab. 2008;19:349–355. doi: 10.1016/j.tem.2008.08.004. [DOI] [PubMed] [Google Scholar]
- 37.L’Episcopo F, et al. Plasticity of subventricular zone neuroprogenitors in MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine) mouse model of Parkinson’s disease involves cross talk between inflammatory and Wnt/beta-catenin signaling pathways: functional consequences for neuroprotection and repair. J Neurosci. 2012;32:2062–2085. doi: 10.1523/JNEUROSCI.5259-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu CC, et al. Deficiency in LRP6-mediated Wnt signaling contributes to synaptic abnormalities and amyloid pathology in Alzheimer’s disease. Neuron. 2014;84:63–77. doi: 10.1016/j.neuron.2014.08.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chua HH, et al. RBMY, a novel inhibitor of glycogen synthase kinase 3beta, increases tumor stemness and predicts poor prognosis of hepatocellular carcinoma. Hepatology. 2015 doi: 10.1002/hep.27996. [DOI] [PubMed] [Google Scholar]
- 40.Monga SP. beta-Catenin Signaling and Roles in Liver Homeostasis, Injury, and Tumorigenesis. Gastroenterology. 2015;148:1294–1310. doi: 10.1053/j.gastro.2015.02.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gregorieff A, Clevers H. Wnt signaling in the intestinal epithelium: from endoderm to cancer. Genes Dev. 2005;19:877–890. doi: 10.1101/gad.1295405. [DOI] [PubMed] [Google Scholar]
- 42.Jamieson CH, et al. Granulocyte-macrophage progenitors as candidate leukemic stem cells in blast-crisis CML. N Engl J Med. 2004;351:657–667. doi: 10.1056/NEJMoa040258. [DOI] [PubMed] [Google Scholar]
- 43.Voronkov A, Krauss S. Wnt/beta-catenin signaling and small molecule inhibitors. Curr Pharm Des. 2013;19:634–664. doi: 10.2174/138161213804581837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kahn M. Can we safely target the WNT pathway? Nat Rev Drug Discov. 2014;13:513–532. doi: 10.1038/nrd4233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nakamura T, et al. Axin, an inhibitor of the Wnt signalling pathway, interacts with beta-catenin, GSK-3beta and APC and reduces the beta-catenin level. Genes Cells. 1998;3:395–403. doi: 10.1046/j.1365-2443.1998.00198.x. [DOI] [PubMed] [Google Scholar]
- 46.Kimelman D, Xu W. beta-catenin destruction complex: insights and questions from a structural perspective. Oncogene. 2006;25:7482–7491. doi: 10.1038/sj.onc.1210055. [DOI] [PubMed] [Google Scholar]
- 47.Veeman MT, Axelrod JD, Moon RT. A second canon. Functions and mechanisms of beta-catenin-independent Wnt signaling. Dev Cell. 2003;5:367–377. doi: 10.1016/s1534-5807(03)00266-1. [DOI] [PubMed] [Google Scholar]
- 48.Mosimann C, Hausmann G, Basler K. Beta-catenin hits chromatin: regulation of Wnt target gene activation. Nat Rev Mol Cell Biol. 2009;10:276–286. doi: 10.1038/nrm2654. [DOI] [PubMed] [Google Scholar]
- 49.Moon RT. Wnt/beta-catenin pathway. Sci STKE. 2005;2005:cm1. doi: 10.1126/stke.2712005cm1. [DOI] [PubMed] [Google Scholar]
- 50.Teo JL, Kahn M. The Wnt signaling pathway in cellular proliferation and differentiation: A tale of two coactivators. Adv Drug Deliv Rev. 2010;62:1149–1155. doi: 10.1016/j.addr.2010.09.012. [DOI] [PubMed] [Google Scholar]
- 51.Lai SL, Chien AJ, Moon RT. Wnt/Fz signaling and the cytoskeleton: potential roles in tumorigenesis. Cell Res. 2009;19:532–545. doi: 10.1038/cr.2009.41. [DOI] [PubMed] [Google Scholar]
- 52.Yamamoto S, et al. Cthrc1 selectively activates the planar cell polarity pathway of Wnt signaling by stabilizing the Wnt-receptor complex. Dev Cell. 2008;15:23–36. doi: 10.1016/j.devcel.2008.05.007. [DOI] [PubMed] [Google Scholar]
- 53.van AR, Nusse R. Towards an integrated view of Wnt signaling in development. Development. 2009;136:3205–3214. doi: 10.1242/dev.033910. [DOI] [PubMed] [Google Scholar]
- 54.Moon RT, Kohn AD, De Ferrari GV, Kaykas A. WNT and beta-catenin signalling: diseases and therapies. Nat Rev Genet. 2004;5:691–701. doi: 10.1038/nrg1427. [DOI] [PubMed] [Google Scholar]
- 55.Thrasivoulou C, Millar M, Ahmed A. Activation of intracellular calcium by multiple Wnt ligands and translocation of beta-catenin into the nucleus: a convergent model of Wnt/Ca2+ and Wnt/beta-catenin pathways. J Biol Chem. 2013;288:35651–35659. doi: 10.1074/jbc.M112.437913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Florian MC, et al. A canonical to non-canonical Wnt signalling switch in haematopoietic stem-cell ageing. Nature. 2013;503:392–396. doi: 10.1038/nature12631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Alonso L, Fuchs E. Stem cells in the skin: waste not, Wnt not. Genes Dev. 2003;17:1189–1200. doi: 10.1101/gad.1086903. [DOI] [PubMed] [Google Scholar]
- 58.Pinto D, Clevers H. Wnt control of stem cells and differentiation in the intestinal epithelium. Exp Cell Res. 2005;306:357–363. doi: 10.1016/j.yexcr.2005.02.022. [DOI] [PubMed] [Google Scholar]
- 59.Klaus A, Birchmeier W. Wnt signalling and its impact on development and cancer. Nat Rev Cancer. 2008;8:387–398. doi: 10.1038/nrc2389. [DOI] [PubMed] [Google Scholar]
- 60.Abrahamsson AE, et al. Glycogen synthase kinase 3beta missplicing contributes to leukemia stem cell generation. Proc Natl Acad Sci U S A. 2009;106:3925–3929. doi: 10.1073/pnas.0900189106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Riether C, et al. Tyrosine kinase inhibitor-induced CD70 expression mediates drug resistance in leukemia stem cells by activating Wnt signaling. Sci Transl Med. 2015;7:298ra119. doi: 10.1126/scitranslmed.aab1740. [DOI] [PubMed] [Google Scholar]
- 62.Schurch C, Riether C, Matter MS, Tzankov A, Ochsenbein AF. CD27 signaling on chronic myelogenous leukemia stem cells activates Wnt target genes and promotes disease progression. J Clin Invest. 2012;122:624–638. doi: 10.1172/JCI45977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Giambra V, et al. Leukemia stem cells in T-ALL require active Hif1alpha and Wnt signaling. Blood. 2015;125:3917–3927. doi: 10.1182/blood-2014-10-609370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gang EJ, et al. Small-molecule inhibition of CBP/catenin interactions eliminates drug-resistant clones in acute lymphoblastic leukemia. Oncogene. 2014;33:2169–2178. doi: 10.1038/onc.2013.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Dandekar S, et al. Wnt inhibition leads to improved chemosensitivity in paediatric acute lymphoblastic leukaemia. Br J Haematol. 2014;167:87–99. doi: 10.1111/bjh.13011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Klonisch T, et al. Cancer stem cell markers in common cancers - therapeutic implications. Trends Mol Med. 2008;14:450–460. doi: 10.1016/j.molmed.2008.08.003. [DOI] [PubMed] [Google Scholar]
- 67.Han J, et al. Small interfering RNA-mediated downregulation of beta-catenin inhibits invasion and migration of colon cancer cells in vitro. Med Sci Monit. 2012;18:BR273–BR280. doi: 10.12659/MSM.883205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wielenga VJ, et al. Expression of CD44 in Apc and Tcf mutant mice implies regulation by the WNT pathway. Am J Pathol. 1999;154:515–523. doi: 10.1016/S0002-9440(10)65297-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ahmed MA, et al. CD24 is upregulated in inflammatory bowel disease and stimulates cell motility and colony formation. Inflamm Bowel Dis. 2010;16:795–803. doi: 10.1002/ibd.21134. [DOI] [PubMed] [Google Scholar]
- 70.Shulewitz M, et al. Repressor roles for TCF-4 and Sfrp1 in Wnt signaling in breast cancer. Oncogene. 2006;25:4361–4369. doi: 10.1038/sj.onc.1209470. [DOI] [PubMed] [Google Scholar]
- 71.Schmitt M, Metzger M, Gradl D, Davidson G, Orian-Rousseau V. CD44 functions in Wnt signaling by regulating LRP6 localization and activation. Cell Death Differ. 2015;22:677–689. doi: 10.1038/cdd.2014.156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ksiazkiewicz M, Markiewicz A, Zaczek AJ. Epithelial-mesenchymal transition: a hallmark in metastasis formation linking circulating tumor cells and cancer stem cells. Pathobiology. 2012;79:195–208. doi: 10.1159/000337106. [DOI] [PubMed] [Google Scholar]
- 73.DiMeo TA, et al. A novel lung metastasis signature links Wnt signaling with cancer cell self-renewal and epithelial-mesenchymal transition in basal-like breast cancer. Cancer Res. 2009;69:5364–5373. doi: 10.1158/0008-5472.CAN-08-4135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Moreno-Bueno G, Portillo F, Cano A. Transcriptional regulation of cell polarity in EMT and cancer. Oncogene. 2008;27:6958–6969. doi: 10.1038/onc.2008.346. [DOI] [PubMed] [Google Scholar]
- 75.Huels DJ, et al. E-cadherin can limit the transforming properties of activating beta-catenin mutations. EMBO J. 2015;34:2321–2333. doi: 10.15252/embj.201591739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Conacci-Sorrell M, et al. Autoregulation of E-cadherin expression by cadherin-cadherin interactions: the roles of beta-catenin signaling, Slug, and MAPK. J Cell Biol. 2003;163:847–857. doi: 10.1083/jcb.200308162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Heuberger J, Birchmeier W. Interplay of cadherin-mediated cell adhesion and canonical Wnt signaling. Cold Spring Harb Perspect Biol. 2010;2:a002915. doi: 10.1101/cshperspect.a002915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Brabletz T, et al. Invasion and metastasis in colorectal cancer: epithelial-mesenchymal transition, mesenchymal-epithelial transition, stem cells and beta-catenin. Cells Tissues Organs. 2005;179:56–65. doi: 10.1159/000084509. [DOI] [PubMed] [Google Scholar]
- 79.Yamada T, et al. Transactivation of the multidrug resistance 1 gene by T-cell factor 4/beta-catenin complex in early colorectal carcinogenesis. Cancer Res. 2000;60:4761–4766. [PubMed] [Google Scholar]
- 80.Heidel FH, et al. Genetic and pharmacologic inhibition of beta-catenin targets imatinib-resistant leukemia stem cells in CML. Cell Stem Cell. 2012;10:412–424. doi: 10.1016/j.stem.2012.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Radich JP, et al. Gene expression changes associated with progression and response in chronic myeloid leukemia. Proc Natl Acad Sci U S A. 2006;103:2794–2799. doi: 10.1073/pnas.0510423103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Valencia A, et al. Wnt signaling pathway is epigenetically regulated by methylation of Wnt antagonists in acute myeloid leukemia. Leukemia. 2009;23:1658–1666. doi: 10.1038/leu.2009.86. [DOI] [PubMed] [Google Scholar]
- 83.Roman-Gomez J, et al. Epigenetic regulation of Wnt-signaling pathway in acute lymphoblastic leukemia. Blood. 2007;109:3462–3469. doi: 10.1182/blood-2006-09-047043. [DOI] [PubMed] [Google Scholar]
- 84.Morrison SJ, Scadden DT. The bone marrow niche for haematopoietic stem cells. Nature. 2014;505:327–334. doi: 10.1038/nature12984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wang LD, Wagers AJ. Dynamic niches in the origination and differentiation of haematopoietic stem cells. Nat Rev Mol Cell Biol. 2011;12:643–655. doi: 10.1038/nrm3184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Boulais PE, Frenette PS. Making sense of hematopoietic stem cell niches. Blood. 2015;125:2621–2629. doi: 10.1182/blood-2014-09-570192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.De TF, et al. A crosstalk between the Wnt and the adhesion-dependent signaling pathways governs the chemosensitivity of acute myeloid leukemia. Oncogene. 2006;25:3113–3122. doi: 10.1038/sj.onc.1209346. [DOI] [PubMed] [Google Scholar]
- 88.Van Den Berg DJ, Sharma AK, Bruno E, Hoffman R. Role of members of the Wnt gene family in human hematopoiesis. Blood. 1998;92:3189–3202. [PubMed] [Google Scholar]
- 89.Austin TW, Solar GP, Ziegler FC, Liem L, Matthews W. A role for the Wnt gene family in hematopoiesis: expansion of multilineage progenitor cells. Blood. 1997;89:3624–3635. [PubMed] [Google Scholar]
- 90.Malhotra S, Kincade PW. Wnt-related molecules and signaling pathway equilibrium in hematopoiesis. Cell Stem Cell. 2009;4:27–36. doi: 10.1016/j.stem.2008.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Malhotra S, Kincade PW. Canonical Wnt pathway signaling suppresses VCAM-1 expression by marrow stromal and hematopoietic cells. Exp Hematol. 2009;37:19–30. doi: 10.1016/j.exphem.2008.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Luis TC, Ichii M, Brugman MH, Kincade P, Staal FJ. Wnt signaling strength regulates normal hematopoiesis and its deregulation is involved in leukemia development. Leukemia. 2012;26:414–421. doi: 10.1038/leu.2011.387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Lane SW, et al. Differential niche and Wnt requirements during acute myeloid leukemia progression. Blood. 2011;118:2849–2856. doi: 10.1182/blood-2011-03-345165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Fleming HE, et al. Wnt signaling in the niche enforces hematopoietic stem cell quiescence and is necessary to preserve self-renewal in vivo. Cell Stem Cell. 2008;2:274–283. doi: 10.1016/j.stem.2008.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kim JA, et al. Identification of a stroma-mediated Wnt/beta-catenin signal promoting self-renewal of hematopoietic stem cells in the stem cell niche. Stem Cells. 2009;27:1318–1329. doi: 10.1002/stem.52. [DOI] [PubMed] [Google Scholar]
- 96.Ahn JY, Park G, Shim JS, Lee JW, Oh IH. Intramarrow injection of beta-catenin-activated, but not naive mesenchymal stromal cells stimulates self-renewal of hematopoietic stem cells in bone marrow. Exp Mol Med. 2010;42:122–131. doi: 10.3858/emm.2010.42.2.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Cobas M, et al. Beta-catenin is dispensable for hematopoiesis and lymphopoiesis. J Exp Med. 2004;199:221–229. doi: 10.1084/jem.20031615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Jeannet G, et al. Long-term, multilineage hematopoiesis occurs in the combined absence of beta-catenin and gamma-catenin. Blood. 2008;111:142–149. doi: 10.1182/blood-2007-07-102558. [DOI] [PubMed] [Google Scholar]
- 99.Kabiri Z, et al. Wnts are dispensable for differentiation and self-renewal of adult murine hematopoietic stem cells. Blood. 2015;126:1086–1094. doi: 10.1182/blood-2014-09-598540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kirstetter P, Anderson K, Porse BT, Jacobsen SE, Nerlov C. Activation of the canonical Wnt pathway leads to loss of hematopoietic stem cell repopulation and multilineage differentiation block. Nat Immunol. 2006;7:1048–1056. doi: 10.1038/ni1381. [DOI] [PubMed] [Google Scholar]
- 101.Zhang B, et al. Microenvironmental protection of CML stem and progenitor cells from tyrosine kinase inhibitors through N-cadherin and Wnt-beta-catenin signaling. Blood. 2013;121:1824–1838. doi: 10.1182/blood-2012-02-412890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Manabe A, Coustan-Smith E, Behm FG, Raimondi SC, Campana D. Bone marrow-derived stromal cells prevent apoptotic cell death in B-lineage acute lymphoblastic leukemia. Blood. 1992;79:2370–2377. [PubMed] [Google Scholar]
- 103.Meads MB, Gatenby RA, Dalton WS. Environment-mediated drug resistance: a major contributor to minimal residual disease. Nat Rev Cancer. 2009;9:665–674. doi: 10.1038/nrc2714. [DOI] [PubMed] [Google Scholar]
- 104.Kode A, et al. FoxO1-dependent induction of acute myeloid leukemia by osteoblasts in mice. Leukemia. 2015 doi: 10.1038/leu.2015.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Bowers M, et al. Osteoblast ablation reduces normal long-term hematopoietic stem cell self-renewal but accelerates leukemia development. Blood. 2015;125:2678–2688. doi: 10.1182/blood-2014-06-582924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.DuBois RN, Giardiello FM, Smalley WE. Nonsteroidal anti-inflammatory drugs, eicosanoids, and colorectal cancer prevention. Gastroenterol Clin North Am. 1996;25:773–791. doi: 10.1016/s0889-8553(05)70274-0. [DOI] [PubMed] [Google Scholar]
- 107.De MC, et al. Dual Cyclooxygenase and Carbonic Anhydrase Inhibition by Nonsteroidal Anti-Inflammatory Drugs for the Treatment of Cancer. Curr Med Chem. 2015;22:2812–2818. doi: 10.2174/0929867322666150716113501. [DOI] [PubMed] [Google Scholar]
- 108.Smalley WE, DuBois RN. Colorectal cancer and nonsteroidal anti-inflammatory drugs. Adv Pharmacol. 1997;39:1–20. doi: 10.1016/s1054-3589(08)60067-8. [DOI] [PubMed] [Google Scholar]
- 109.Thun MJ, Henley SJ, Patrono C. Nonsteroidal anti-inflammatory drugs as anticancer agents: mechanistic, pharmacologic, and clinical issues. J Natl Cancer Inst. 2002;94:252–266. doi: 10.1093/jnci/94.4.252. [DOI] [PubMed] [Google Scholar]
- 110.Castellone MD, Teramoto H, Williams BO, Druey KM, Gutkind JS. Prostaglandin E2 promotes colon cancer cell growth through a Gs-axin-beta-catenin signaling axis. Science. 2005;310:1504–1510. doi: 10.1126/science.1116221. [DOI] [PubMed] [Google Scholar]
- 111.Brudvik KW, Paulsen JE, Aandahl EM, Roald B, Tasken K. Protein kinase A antagonist inhibits beta-catenin nuclear translocation, c-Myc and COX-2 expression and tumor promotion in Apc(Min/+) mice. Mol Cancer. 2011;10:149. doi: 10.1186/1476-4598-10-149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Jansen SR, et al. Prostaglandin E2 promotes MYCN non-amplified neuroblastoma cell survival via beta-catenin stabilization. J Cell Mol Med. 2015;19:210–226. doi: 10.1111/jcmm.12418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Grosch S, Tegeder I, Niederberger E, Brautigam L, Geisslinger G. COX-2 independent induction of cell cycle arrest and apoptosis in colon cancer cells by the selective COX-2 inhibitor celecoxib. FASEB J. 2001;15:2742–2744. doi: 10.1096/fj.01-0299fje. [DOI] [PubMed] [Google Scholar]
- 114.Tegeder I, et al. Inhibition of NF-kappaB and AP-1 activation by R- and S-flurbiprofen. FASEB J. 2001;15:2–4. doi: 10.1096/fj.00-0130fje. [DOI] [PubMed] [Google Scholar]
- 115.Yamazaki R, Kusunoki N, Matsuzaki T, Hashimoto S, Kawai S. Selective cyclooxygenase-2 inhibitors show a differential ability to inhibit proliferation and induce apoptosis of colon adenocarcinoma cells. FEBS Lett. 2002;531:278–284. doi: 10.1016/s0014-5793(02)03535-4. [DOI] [PubMed] [Google Scholar]
- 116.Zhang Z, Lai GH, Sirica AE. Celecoxib-induced apoptosis in rat cholangiocarcinoma cells mediated by Akt inactivation and Bax translocation. Hepatology. 2004;39:1028–1037. doi: 10.1002/hep.20143. [DOI] [PubMed] [Google Scholar]
- 117.Maier TJ, Janssen A, Schmidt R, Geisslinger G, Grosch S. Targeting the beta-catenin/APC pathway: a novel mechanism to explain the cyclooxygenase-2-independent anticarcinogenic effects of celecoxib in human colon carcinoma cells. FASEB J. 2005;19:1353–1355. doi: 10.1096/fj.04-3274fje. [DOI] [PubMed] [Google Scholar]
- 118.Steinbach G, et al. The effect of celecoxib, a cyclooxygenase-2 inhibitor, in familial adenomatous polyposis. N Engl J Med. 2000;342:1946–1952. doi: 10.1056/NEJM200006293422603. [DOI] [PubMed] [Google Scholar]
- 119.Yang K, et al. Regional response leading to tumorigenesis after sulindac in small and large intestine of mice with Apc mutations. Carcinogenesis. 2003;24:605–611. doi: 10.1093/carcin/24.3.605. [DOI] [PubMed] [Google Scholar]
- 120.Boon EM, et al. Sulindac targets nuclear beta-catenin accumulation and Wnt signalling in adenomas of patients with familial adenomatous polyposis and in human colorectal cancer cell lines. Br J Cancer. 2004;90:224–229. doi: 10.1038/sj.bjc.6601505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Baron JA, et al. A randomized trial of aspirin to prevent colorectal adenomas. N Engl J Med. 2003;348:891–899. doi: 10.1056/NEJMoa021735. [DOI] [PubMed] [Google Scholar]
- 122.Sandler RS, et al. A randomized trial of aspirin to prevent colorectal adenomas in patients with previous colorectal cancer. N Engl J Med. 2003;348:883–890. doi: 10.1056/NEJMoa021633. [DOI] [PubMed] [Google Scholar]
- 123.Phillips RK, et al. A randomised, double blind, placebo controlled study of celecoxib, a selective cyclooxygenase 2 inhibitor, on duodenal polyposis in familial adenomatous polyposis. Gut. 2002;50:857–860. doi: 10.1136/gut.50.6.857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Williams JL, et al. Nitric oxide-releasing nonsteroidal anti-inflammatory drugs (NSAIDs) alter the kinetics of human colon cancer cell lines more effectively than traditional NSAIDs: implications for colon cancer chemoprevention. Cancer Res. 2001;61:3285–3289. [PubMed] [Google Scholar]
- 125.Williams JL, et al. NO-donating aspirin inhibits intestinal carcinogenesis in Min (APC(Min/+)) mice. Biochem Biophys Res Commun. 2004;313:784–788. doi: 10.1016/j.bbrc.2003.12.015. [DOI] [PubMed] [Google Scholar]
- 126.Nath N, Kashfi K, Chen J, Rigas B. Nitric oxide-donating aspirin inhibits beta-catenin/T cell factor (TCF) signaling in SW480 colon cancer cells by disrupting the nuclear beta-catenin-TCF association. Proc Natl Acad Sci U S A. 2003;100:12584–12589. doi: 10.1073/pnas.2134840100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Xiao JH, et al. Adenomatous polyposis coli (APC)-independent regulation of beta-catenin degradation via a retinoid X receptor-mediated pathway. J Biol Chem. 2003;278:29954–29962. doi: 10.1074/jbc.M304761200. [DOI] [PubMed] [Google Scholar]
- 128.Palmer HG, et al. Vitamin D(3) promotes the differentiation of colon carcinoma cells by the induction of E-cadherin and the inhibition of beta-catenin signaling. J Cell Biol. 2001;154:369–387. doi: 10.1083/jcb.200102028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Choi MY, et al. Pre-clinical Specificity and Safety of UC-961, a First-In-Class Monoclonal Antibody Targeting ROR1. Clin Lymphoma Myeloma Leuk. 2015;15(Suppl):S167–S169. doi: 10.1016/j.clml.2015.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Fujii N, et al. An antagonist of dishevelled protein-protein interaction suppresses beta-catenin-dependent tumor cell growth. Cancer Res. 2007;67:573–579. doi: 10.1158/0008-5472.CAN-06-2726. [DOI] [PubMed] [Google Scholar]
- 131.Shan J, Shi DL, Wang J, Zheng J. Identification of a specific inhibitor of the dishevelled PDZ domain. Biochemistry. 2005;44:15495–15503. doi: 10.1021/bi0512602. [DOI] [PubMed] [Google Scholar]
- 132.Grandy D, et al. Discovery and characterization of a small molecule inhibitor of the PDZ domain of dishevelled. J Biol Chem. 2009;284:16256–16263. doi: 10.1074/jbc.M109.009647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Kadowaki T, Wilder E, Klingensmith J, Zachary K, Perrimon N. The segment polarity gene porcupine encodes a putative multitransmembrane protein involved in Wingless processing. Genes Dev. 1996;10:3116–3128. doi: 10.1101/gad.10.24.3116. [DOI] [PubMed] [Google Scholar]
- 134.Rios-Esteves J, Resh MD. Stearoyl CoA desaturase is required to produce active, lipid-modified Wnt proteins. Cell Rep. 2013;4:1072–1081. doi: 10.1016/j.celrep.2013.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Madan B, et al. Wnt addiction of genetically defined cancers reversed by PORCN inhibition. Oncogene. 2015 doi: 10.1038/onc.2015.280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Huang SM, et al. Tankyrase inhibition stabilizes axin and antagonizes Wnt signalling. Nature. 2009;461:614–620. doi: 10.1038/nature08356. [DOI] [PubMed] [Google Scholar]
- 137.Kulak O, et al. Disruption of Wnt/beta-Catenin Signaling and Telomeric Shortening Are Inextricable Consequences of Tankyrase Inhibition in Human Cells. Mol Cell Biol. 2015;35:2425–2435. doi: 10.1128/MCB.00392-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Gurney A, et al. Wnt pathway inhibition via the targeting of Frizzled receptors results in decreased growth and tumorigenicity of human tumors. Proc Natl Acad Sci U S A. 2012;109:11717–11722. doi: 10.1073/pnas.1120068109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Le PN, McDermott JD, Jimeno A. Targeting the Wnt pathway in human cancers: therapeutic targeting with a focus on OMP-54F28. Pharmacol Ther. 2015;146:1–11. doi: 10.1016/j.pharmthera.2014.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Emami KH, et al. A small molecule inhibitor of beta-catenin/CREB-binding protein transcription [corrected] Proc Natl Acad Sci U S A. 2004;101:12682–12687. doi: 10.1073/pnas.0404875101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.McMillan M, Kahn M. Investigating Wnt signaling: a chemogenomic safari. Drug Discov Today. 2005;10:1467–1474. doi: 10.1016/S1359-6446(05)03613-5. [DOI] [PubMed] [Google Scholar]
- 142.El-Khoueiry AB, N Y, Y D, C S, K M, Z M, B J, F M, I T, K H, L H. A phase I first-in-human study of PRI-724 in patients (pts) with advanced solid tumors. J Clin Oncol. 2013;31(suppl; abstract 2501) [Google Scholar]
- 143.Brembeck FH, Rosario M, Birchmeier W. Balancing cell adhesion and Wnt signaling, the key role of beta-catenin. Curr Opin Genet Dev. 2006;16:51–59. doi: 10.1016/j.gde.2005.12.007. [DOI] [PubMed] [Google Scholar]
- 144.Sawa H. Control of cell polarity and asymmetric division in C. elegans. Curr Top Dev Biol. 2012;101:55–76. doi: 10.1016/B978-0-12-394592-1.00003-X. [DOI] [PubMed] [Google Scholar]