

Abstract
sp3 C-H bond functionalization streamlines chemical synthesis by allowing the use of simple molecules and providing novel synthetic disconnection. Due to recent intense efforts devoted to develop new reactions based on the approach of C-H functionalization, its benefits in molecule derivatization and complex molecule synthesis have been recognized. This article discusses the strength and weakness of three main approaches, transition-metal catalyzed C-H activation, 1, n hydrogen atom transfer and transition-metal catalyzed carbene/nitrene transfer, for directed functionalization of unactivated sp3 C-H bonds. For each strategy, the scope, the reactivity of different C-H bonds, the position of the reacting C-H bonds relative to the directing group, and stereochemical outcomes are illustrated with examples in literature. The aim of this review is to provide guidance for users of C-H functionalization reactions and inspire future research in this area.
Keywords: C-H functionalization, transition metal, hydrogen atom transfer, carbene, nitrene
Graphical Abstract
1. Introduction
Nearly all organic molecules are extensively adorned with C-H bonds, whose lack of reactivity excludes them from being labeled a “functional group”. Methods to achieve direct C-H functionalization to derivatize organic molecules constitute a powerful tool.1 In contrast to conventional organic reactions, C-H functionalization obviates the need for a pre-existing functional handle, and represents an opportunity to modify a site of a molecule that was previously unattainable by traditional methodology. Maturation of this technology1 would lead to novel means to disconnect a molecule in retrosynthetic analyses and thus potentially provides more efficient synthetic routes from simple starting materials.
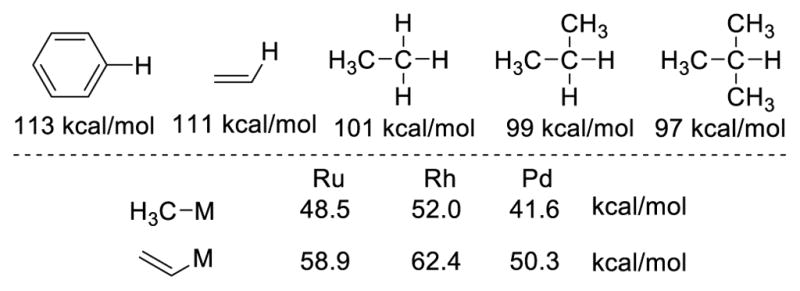
A cursory examination of typical C-H bond strengths underscores one of the main challenges associated with C-H functionalization: high enthalpic stability. The trend also suggests that aliphatic sp3 C-H bonds should be easiest to functionalize. While this is true in reactions that proceed via homolytic bond cleavage, it is not true for metal-catalyzed C-H bond activation as the latter ignores the relative strengths of M-C bonds generated. Noteworthy is that M-C(sp2) bonds tend to be much stronger than the corresponding metal alkyls (Fig. 1). 2,3 While sp2 C-H functionalization has received tremendous attention and has witnessed significant advances in recent decades, the functionalization of unactivated sp3 C-H bonds has been perceived as a more challenging topic. Furthermore, the ubiquity of sp3 C-H bonds in organic molecules poses a selectivity problem. While it seemed impossible to develop a synthetically useful transformation based on the functionalization of such bonds decades ago, some successes have emerged in this area in recent years, as exemplified by the number of publications and the applications of sp3 C-H functionalization reactions in natural product synthesis.
Figure 1.
Challenges in C-H Functionalization
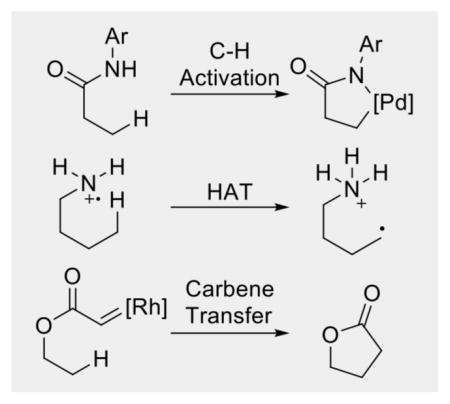
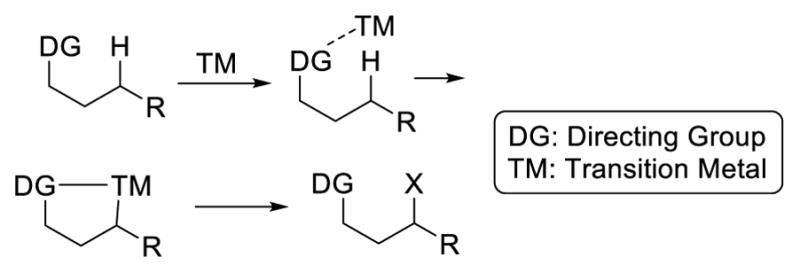
In most cases, the key to breaking inert sp3 C-H bonds and to address selectivity is the employment of a directing group (Fig. 2). The presence of the directing group lowers the energy barrier for cleaving the C-H bonds and directs a specific C-H bond for reaction through a cyclic transition state. This review aims to summarize the three main strategies (transition-metal catalyzed C-H activation, 1, n hydrogen atom transfer, and metal-catalyzed carbene/nitrene transfer) for directed functionalization of unactivated sp3 C-H bonds. The focus will be to compare and contrast the three strategies and discuss their complementarity in hopes that this review article will serve as a guide for users of directed C-H functionalization reactions, as well as for synthetic chemists to develop new transformations. In this review, unactivated sp3 C-H bonds are those primary, secondary, and tertiary C-H bonds not activated by a heteroatom (e.g. oxygen and nitrogen) or a π system (e.g. alkenes or aromatic systems). The functionalization of cyclopropyl and cyclobutyl C-H bonds is also not included in this review. In addition, while there is significant advance in undirected sp3 C-H functionalization in recent years, it is beyond the scope of this review. Readers who are interested in undirected sp3 C-H functionalization are encouraged to read the relevant reviews.4
Figure 2.
Use of a Directing Group in C-H Functionalization
2. Transition-metal Catalyzed C-H Activation
Transition-metal catalysis is currently the predominant focus for the functionalization of C-H bonds. The transition metal catalyst coordinates to a Lewis basic functional group on the molecule, which brings the catalyst into proximity of a specific C-H bond, lowering the energy barrier of the cleavage of this C-H bond. The resulting alkyl metal intermediate can be trapped with a reactive partner to form a C-X bond (Scheme 1). Since sp2 C-H bonds are in general stronger than their sp3 counterparts, it might appear that the activation of sp3 C-H bonds is less challenging. The literature suggests the opposite trend. In addition to the ubiquity of sp3 C-H bonds, this observation could be explained by the relative bond strength of the metal-carbon bonds. In general, a metal-sp3 bond is weaker than a metal-sp2 bond. This relative bond strength is illustrated with the iridium complexes in figure 3. For iridium(III) complexes 1 and 2, which are ligated with phosphines and cyclopentadienyl groups, the Ir-Ph bond is 30 kcal/mol stronger than the Ir-Cy bond.5 The rhodium complexes ligated by Tp also show that the Rh-Ph bond in 3 has a bond energy 16 kcal/mol higher than the Rh-Me bond in 4 (Figure 3). 6 This difference in bond energies affords a plausible explanation for the fact that it is more difficult to activate a C(sp3)-H bond than a C(sp2)-H bond in transition-metal catalysis. The following is a brief review on seminal reports on sp3 C-H activation catalyzed by transition metal complexes through the formation of a metal-carbon bond and does not represent an exhaustive list of all research in this area.
Scheme 1.
Transition-Metal Catalyzed C-H Activation
Figure 3.
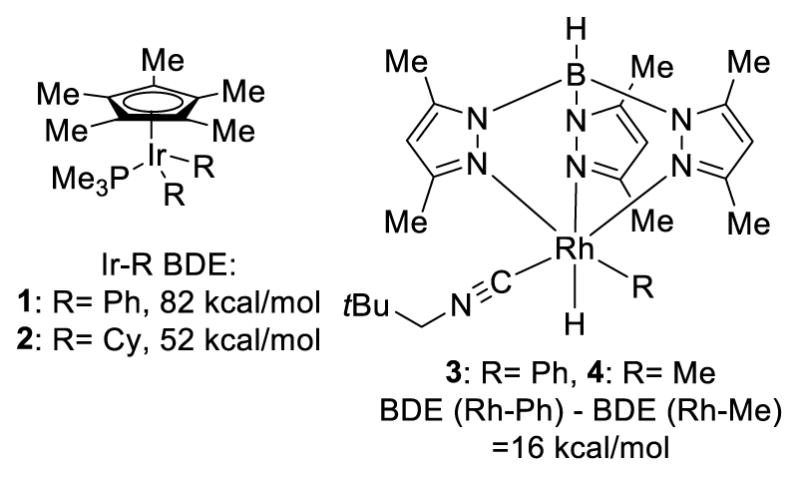
Weaker Carbon-Metal Bonds with sp3 Carbons
2.1. Palladium Catalysis7
2.1.1 Early Work
A seminal report from Dyker described Pd(II)-catalyzed synthesis of benzocyclobutene 7 from aryl iodide 5 and aryl bromide 6 featuring the activation of methyl C-H bonds (Scheme 2).8 In the proposed catalytic cycle, Pd(0) oxidatively adds to the C-I bond to form aryl Pd(II) intermediate A1. This oxidative addition brings the palladium catalyst close to the methyl group, facilitating the activation of the methyl C-H bond through cyclopalladation to give A2. Another oxidative addition into the aryl bromide leads to Pd(IV) intermediate A3, driving reductive elimination to form a C(sp2)-C(sp2) bond. Activation of the C(sp2)-H bond in A4 at the ortho position generates another palladacycle A5. The final C(sp2)-C(sp3) bond is forged by the reductive elimination of this Pd(II) species. Overall, the reaction represents the activation of C(sp3)-H bonds with an aryl iodide as a traceless directing group. This later inspired various groups to use vinyl halides and triflates as a traceless directing group in future development of C-H activation reactions.
Scheme 2.
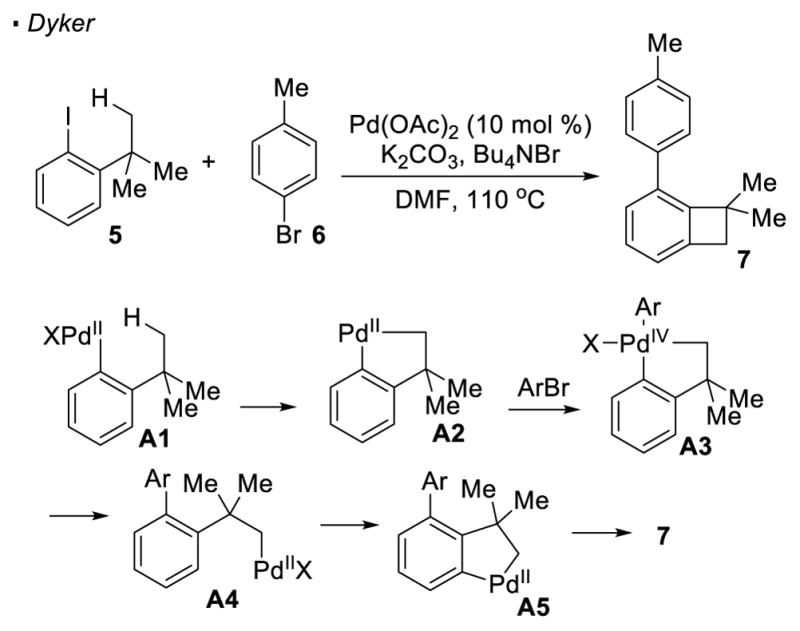
Aryl Halides as Directing Groups
Three other seminal reports on palladium catalyzed sp3 C-H activation, using nitrogen as directing groups, were contributed by Sanford, Yu and Daugulis. Sanford reported a palladium-catalyzed nitrogen-directed sp3 C-H oxygenation. This work was inspired by related stoichiometric studies by others9 and her previous work on the activation of sp2 C-H bonds (Scheme 3).10 Nitrogen, in the forms of imines and pyridines, was previously established as a competent directing group for acetoxylation of sp2 and benzylic sp3 C-H bonds with Pd(OAc)2 as the pre-catalyst and PhI(OAc)2 as the stoichiometric oxidant. For instance, imine 8 undergoes acetoxylation to form 9. Based on this established protocol, Sanford developed an oxime-directed acetoxylation of unactivated primary C-H bonds.11 It is believed that after the coordination of the oxime nitrogen to the palladium(II) catalyst, a concerted metalation-deprotonation event between the substrate and the Pd(II) catalyst leads to the formation of alkyl Pd(II) intermediate B1. Oxidation of Pd(II) to Pd(IV) by PhI(OAc)2 drives reductive elimination to furnish the C-O bond. In addition, Sanford found that pyridine can also be applied as a directing group for acetoxylation of primary C-H bonds, as illustrated by the conversion of pyridine 12 to 13. Yu12 reported palladium(II)-catalyzed iodination of unactivated primary sp3 C-H bonds of oxazolines, substrates that had previously been shown to undergo cyclopalladation with a stoichiometric amount of palladium13 (Scheme 4). Iodine is included as an additional oxidant, accounting for the observed iodination of oxazoline 14 to 15, instead of acetoxylation. In addition, Daugulis discovered that picolinamides can serve as a bidentate ligand for palladium-catalyzed C-H activation.14 In this report, unactivated sp3 C-H bonds are arylated with aryl iodides. For instance, picolinamide 16 can be arylated at the methyl C-H bond to yield 18. It is noteworthy that this work represents the first example of an intermolecular C-C bond forming reaction. Since these initial reports, various transformations based on Pd catalysis have been developed by different groups.
Scheme 3.

Nitrogen as Directing Group
Scheme 4.
Dehydrogenation and Cyclobutane Synthesis with Aryl Halides
2.1.2 Functionalization with Different Directing Groups
(I) Aryl/Vinyl Halides
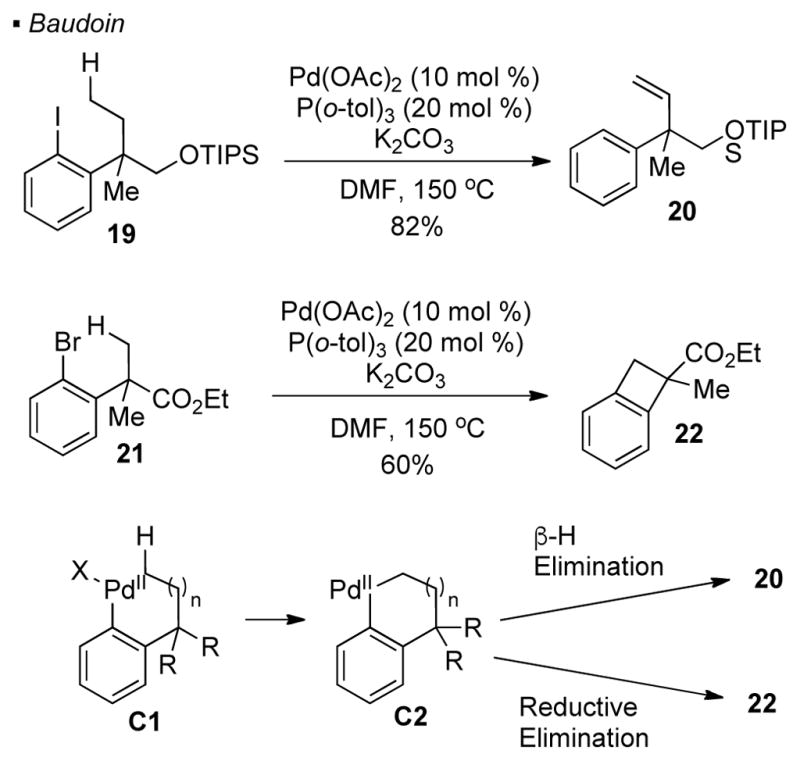
Various groups were inspired by reports from Dyker to use aryl halides or triflates as a traceless directing group. Baudoin discovered that dehydrogenation of the ethyl group in aryl iodide 19 takes place when it is heated under basic conditions with a catalytic system of Pd(OAc)2/P(o-tol)3 (Scheme 4).15 Compared to Dyker’s system, the terminal C-H bond that undergoes activation is one-bond further away from the directing vinyl halide. In the proposed mechanism, after oxidation addition into the C-I bond and C-H activation of the methyl group to form C2, β-hydride elimination results in the formation of an alkene and a palladium hydride. The observed product 20 is formed through a final reductive elimination, which furnishes a C-H bond. For aryl bromide 21 bearing one less carbon between the linker and the methyl C-H bond, the presence of a benzylic quaternary carbon renders β-H elimination impossible. In this case, benzocyclobutene 22 is the observed product because reductive elimination of intermediate C2 takes place. In addition, Buchwald showed that these putative palladium-alkyl intermediates can be intercepted by aryl boronic acids 16 or anilines, 17 resulting in arylation or amination of the methyl C-H bonds (Scheme 5). Thus, the arylated product 25 can be obtained from aryl bromide 23 and boronic acid 24, while the aminated product 28 arises from aryl bromide 26 and aniline 27 respectively. However, these reactions are only applicable to substrates bearing substitutions at 2, 4 and 6 positions relative to the halogen. The key to the success of these reactions are the sterics provided by these substituents to disfavor Suzuki cross-coupling and Buchwald-Hartwig amination.
Scheme 5.
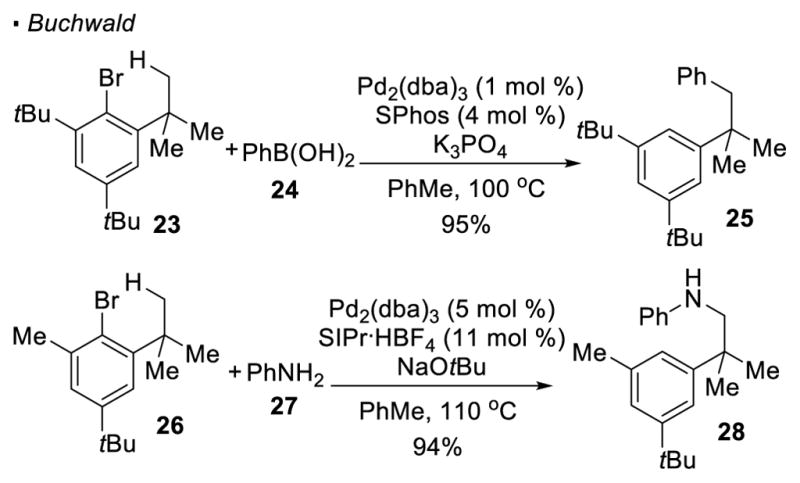
Trapping of Putative Palladium-Alkyl Intermediates
As demonstrated by reports from Fagnou, in contrast to Baudoin’s cyclobutene synthesis, 5-membered rings can be formed when the methyl group is placed one-bond further away from the aryl halides (Scheme 6).18 This reaction allows access to benzofuran 30 from aryl bromide 29. Extension of this strategy to the synthesis of indoline 32 from 31 is reported by Fuji and Ohno.19 In this reaction, the presence of a quaternary carbon to disfavor β-H elimination is not required to achieve the desired reactivity. Less reactive aryl chlorides are also competent substrates, as shown in the subsequent report from Rousseaux, Clot, Fagnou and Baudoin for the synthesis of cyclopentanes 34a and 34b from aryl chloride 33.20
Scheme 6.
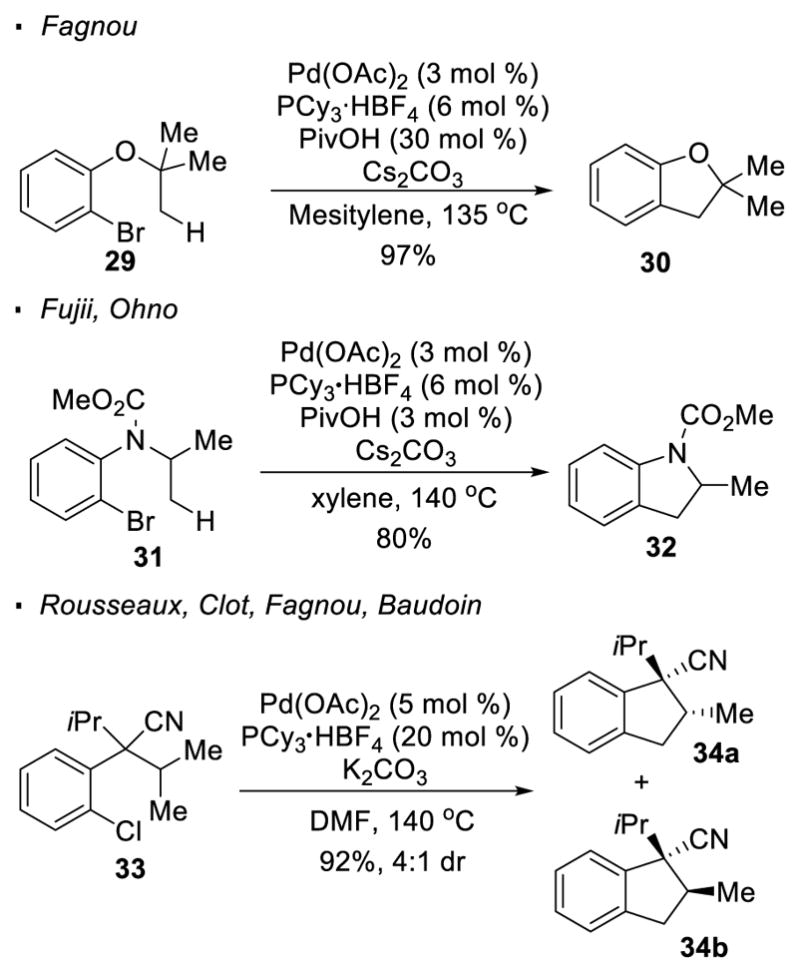
Synthesis of 5-Membered Rings with Aryl Halides
Baudoin successfully extended the scope of these reactions to embrace the use of vinyl halides as a traceless directing group. Thus, the methyl C-H bond in vinyl bromide 35 can be activated for the synthesis of bicyclic pyrrolidine 36 (Scheme 7). 21 In a subsequent report, Baudoin further demonstrated that α-bromo acrylamide 37 is also a competent substrate in this chemistry and showed that monocyclic pyrrolidine derivatives can be obtained from acyclic precusors.22 The mechanism of these transformations is believed to be the same as in the case of aryl halides.
Scheme 7.
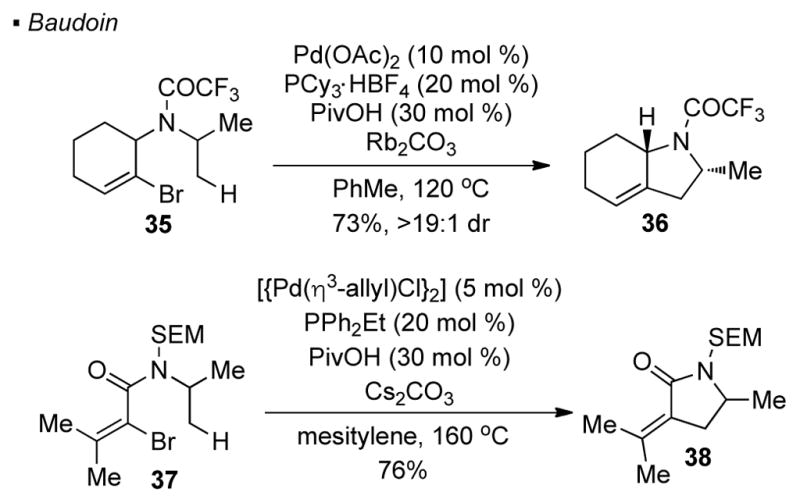
Vinyl Halides as Traceless Directing Group
In all previously discussed examples, the putative aryl palladium(II) intermediates prior to the activation of sp3 C-H bonds are generated from aryl or vinyl halides. On the contrary, Fagnou showed that sp2 C-H activation of pivaloylpyrrole 39 can generate the aryl palladium species D1, which can undergo sp3 C-H activation to form 6-membered palladacycle D2 (Scheme 8).23 Similar to previous examples, reductive elimination results in the formation of a 5-membered ring. In addition, as demonstrated by Minami and Hiyama, silylethynyl aryl ether 41 undergoes palladium-catalyzed intramolecular hydroalkylation featuring the activation of one of the methyl C-H bonds.24 In the proposed mechanism, the vinyl palladium intermediate E2, prior to C-H activation of the methyl C-H bond, is formed from the addition of PivOH across the alkyne in Pd(0) complex E1.25 The 7-membered palladacycle E3 generated through C-H activation undergoes reductive elimination to give the observed product 42.
Scheme 8.

Generation of Aryl/Vinyl Palladium through Other Means
(II) Oximes
After the seminal work by Sanford, more reactions with oximes as a directing group have appeared. Yu and Che reported palladium-catalyzed oxime-directed C-H amidation (Scheme 9).26 In this protocol, an amide or a sulfonamide is employed as a nitrogen source and potassium persulfate (K2S2O8) as an oxidant. Thus, the methyl C-H bond of 43 can be amidated with trifluoroacetamide 44 to afford 45. With the use of diphenyliodonium salt 47, Chen accomplished C-H arylation of oxime 46 to give 48 (Scheme 9).27
Scheme 9.
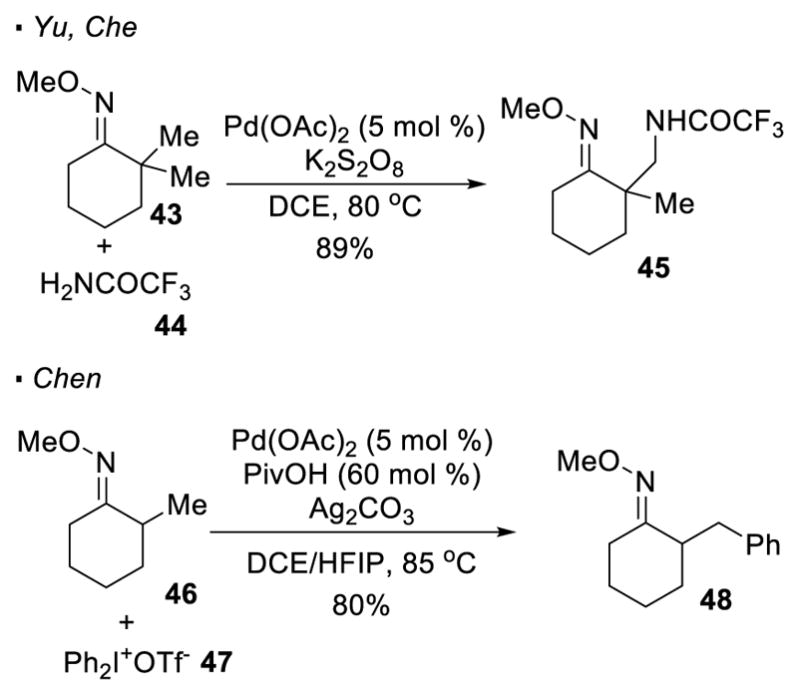
Oxime-Directed C-H Activation
(III) Pyridines
For the functionalization of pyridines, in addition to the aforementioned oxygenation by Sanford, formation of C-C bonds is also possible. Daugulis28 and Yu29 independently disclosed pyridine-directed arylation and alkylation of primary C-H bonds (Scheme 10). Sanford demonstrated that electrophilic alkenes can be installed at a primary C-H bond adjacent to a pyridine.30 For example, olefination of pyridine 12 with ethyl acrylate 54 gives pyridinium 55. In the proposed mechanism, after C-H activation, the alkyl palladium species adds across the electrophilic alkene. β-H elimination results in a net incorporation of the electrophilic alkene into the methyl C-H bond. The pyridine nitrogen can then cyclize onto the alkene through aza-Michael addition to yield the bicyclic pyridinium. Further manipulation yields valuable bicyclic amines.
Scheme 10.
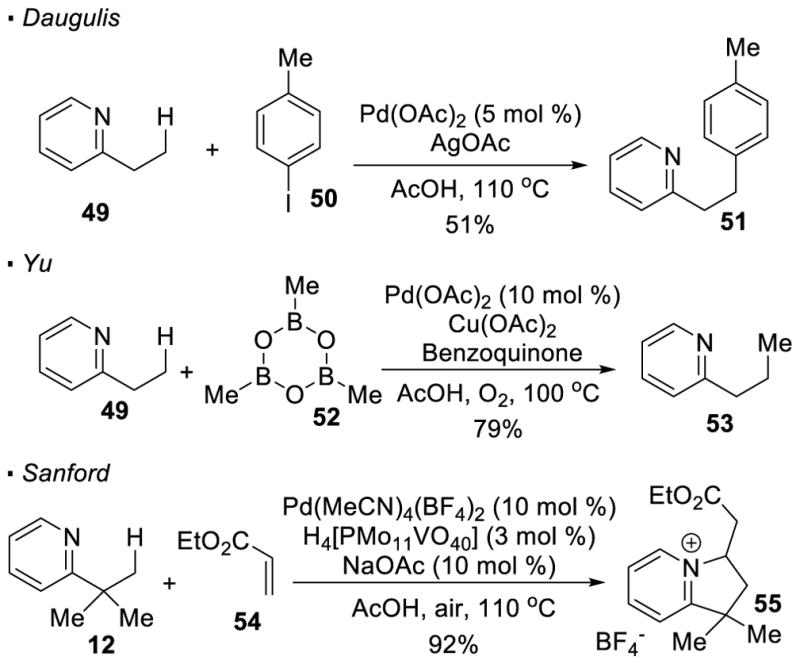
Derivatization of Pyridines through C-H Activation
(IVa) Amines (Amides)
In most cases for the functionalization of amines, an electron-withdrawing group is installed on the nitrogen to generate a directing group for C-H activation. Daugulis first demonstrated that bidentate picolinamides can serve as a directing group for palladium(II)-catalyzed C-H functionalization of amines (Scheme 3). Since then, the incorporation of different functionalities at unactivated primary C-H bonds with the same picolinamide directing group has been developed by various groups (Scheme 11). The key to success is the appropriate choice of a reaction partner that is used to trap the putative alkyl-palladium intermediates. For instance, by switching an aryl iodide to an alkyl iodide, Chen developed a protocol for the alkylation of the C-H bond in 56.31 Both Daugulis32 and Chen33 observed that when PhI(OAc)2 is applied as an oxidant, intramolecular C-H amination takes place, resulting in the synthesis of 4- or 5-membered nitrogen heterocycles 60 or 64. The ring size depends on the distance between the primary C-H bond and the nitrogen. Chen also realized that when an alcohol is used as a co-solvent, there is a switch in chemo-selectivity and intermolecular C-H oxygenation becomes the predominant pathway.34 Thus, methoxylation of amide 61 takes place to give 62 when methanol is applied as a solvent. In addition, Z.-J. Shi and B.-F. Shi reported the coupling of amides and diboron esters 35 or alkynes36 at unactivated C-H bonds respectively. These protocols allow for the borylation or alkenylation of the primary C-H bonds of picolinamides 65 and 68.
Scheme 11.

Use of Picolinamides for Amine Functionalization
To develop a protocol for the derivatization of medicinally relevant alicyclic amines, Sanford exploited palladium-catalyzed transannular C-H functionalization (Scheme 12). 37 The 4-position of piperidine 72 can be arylated after the nitrogen is alkylated and linked to a perfluorinated amide, a directing group first introduced by Yu (see Scheme 19), to afford its derivative 73.
Scheme 12.
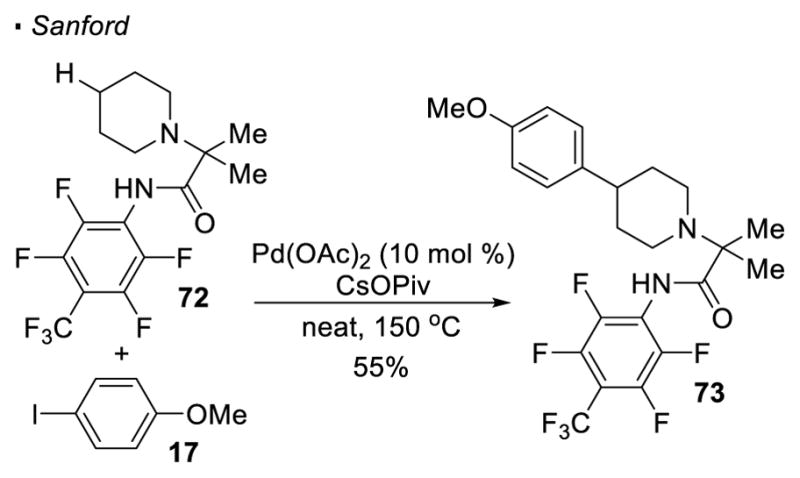
Transannular C-H Activation of Alicyclic Amines
Scheme 19.

Formation of C-C Bonds with Perfluorinated Amides
(IVb) Amines (Sulfonamides)
Yu reported that primary C-H bonds can be arylated with aryl boronic acids after installing a sulfonyl group on amines, as exemplified by the arylation of boronate ester 75 of trifluoromethanesulfonamide 74 (Scheme 13).38 Olefination of the C-H bond with an acrylate or styrene is another possibility.39 After the installation of the alkene at the C-H bond of amide 77, an intramolecular Heck reaction takes place to account for the formation of the cyclized product 79. It should be noted that in both cases, an exogenous ligand is the key to achieve the desired reactivity.
Scheme 13.
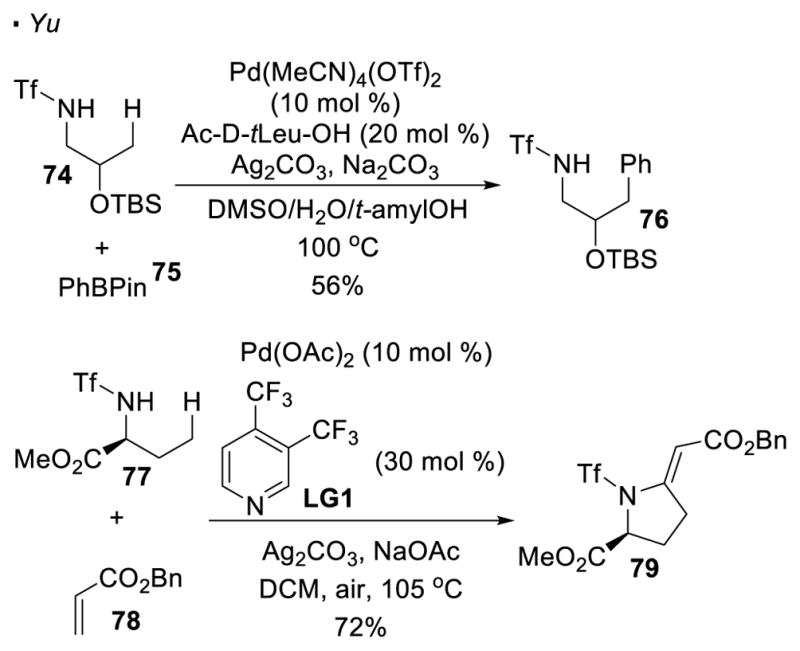
Sulfonamides as Directing Group for Functionalization of Amines
(IVc) Amines (Free Amines)
Sometimes pre-functionalization of amines is not required before the C-H activation step and a free amine can serve as a directing group. Gaunt reported the synthesis of strained aziridines (81) from bulky secondary amines (80) featuring the formation of a 4-membered palladacycle through nitrogen directed cleavage of a primary C-H bond (Scheme 14). 40 Oxidation of Pd(II) to Pd(IV) by PhI(OAc)2 drives reductive elimination, as in Sanford and Yu’s reaction, to form a C-N bond and afford aziridines. In the presence of CO, β-lactams (83) are the observed product. However, the formation of the β-lactams is mechanistically different from that of the aziridines. Carbonylation of the amine precedes the C-H activation step and generates a 5-membered palladacycle. Initially, only sterically hindered secondary amines were competent substrates since it is believed that the sterics of the amines was needed to disfavor the formation of an off-cycle bis(amine)Pd(II) complex. 41 Subsequent work showed with AdCO2H and benzoquinone as additives, even non-bulky secondary amines can be used as substrates to synthesize β-lactams. 42 The putative 4-membered palladacycle can also be intercepted by aryl boronic acids, resulting in net arylation of the C-H bonds.43 With amine 84, arylated product 85 can be accessed. When the methyl is an additional bond away from the nitrogen, C-H activation still occurs at the primary C-H bond. Thus, the methyl group in protected amino-alcohol 86 undergoes acetoxylation with Pd(OAc)2 catalyst and PhI(OAc)2 to afford 87.44
Scheme 14.
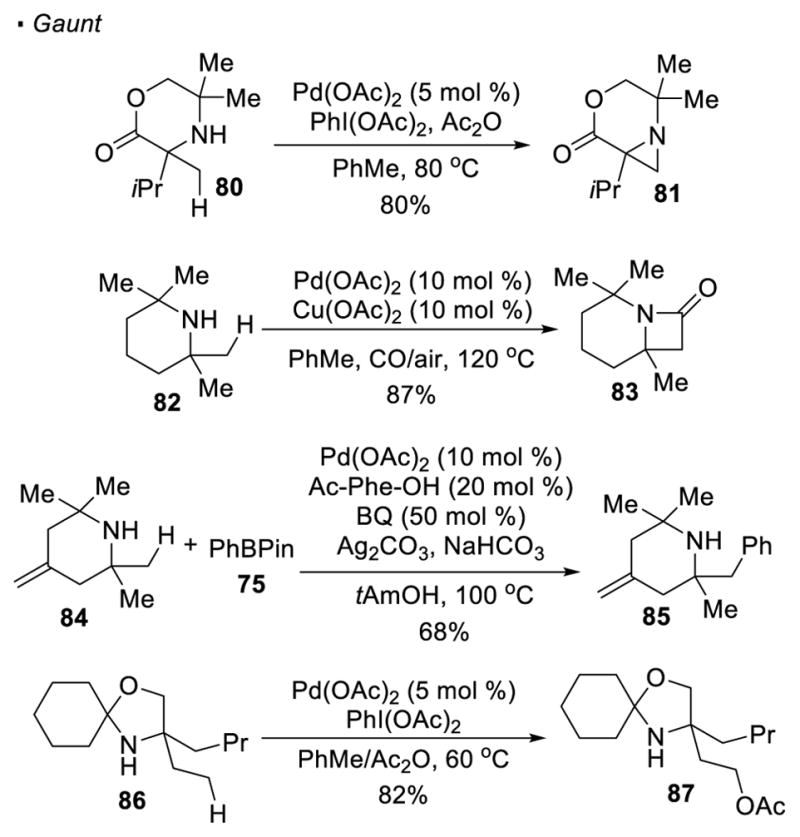
C-H Functionalization of Free Amines
Free primary amines have never been shown to direct C-H activation. Nevertheless, a directing group can be generated insitu to obviate the need to pre-functionalize the amine before the C-H activation step. Ge, 45 Dong 46 and Yu 47 independently showed that a stoichiometric or catalytic amount of an aldehyde can be used as an additive for palladium-catalyzed activation of unactivated sp3 C-H bonds of primary amines (Scheme 15). The condensation between the aldehyde and the primary amine generates a bidentate imine as a transient directing group. This strategy is successfully applied to the arylation of primary aliphatic amines or anilines such as 88 and 91 with aryl iodonium salts or aryl iodides. It should be noted that, in Yu’s report, even cyclic substrates, such as cyclohexyl amine 96, can be derivatized.
Scheme 15.
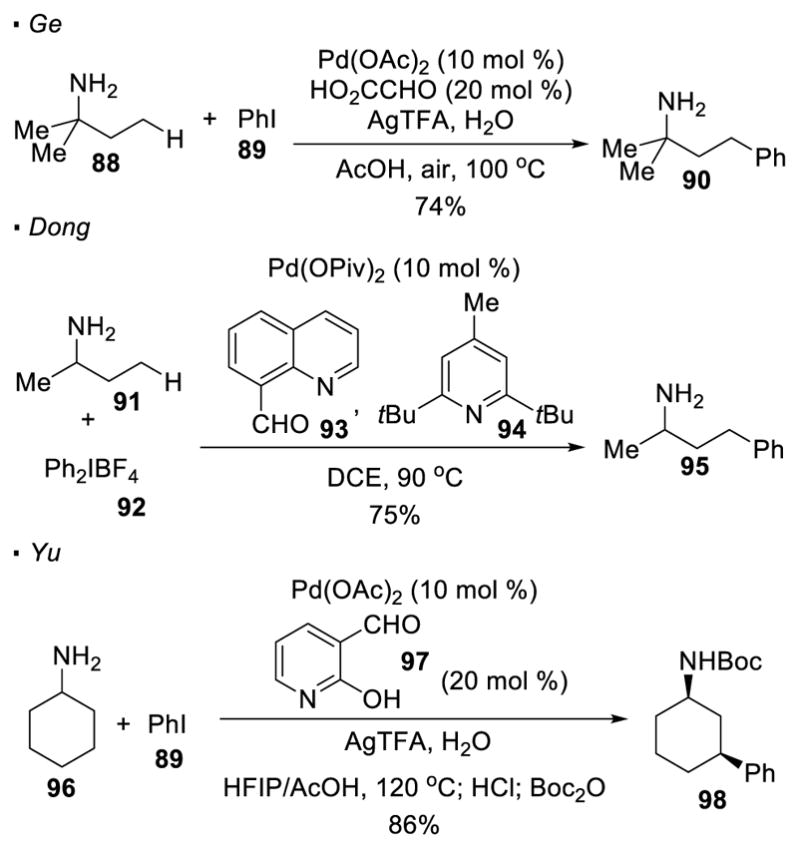
In-situ Generated Imines as Directing Groups
(IVd) Amines (Hydrazones)
Dong reported that hydrazones can be installed on sulfonamides and serve as a directing group for sp3 C-H activation (Scheme 16). 48 Thus, palladium-catalyzed acetoxylation of hydrazone 99 can be accomplished with PhI(OAc)2 as the oxidant. After the C-H activation, the N-N Bonds can be cleaved by Zn to afford sulfonamides.
Scheme 16.
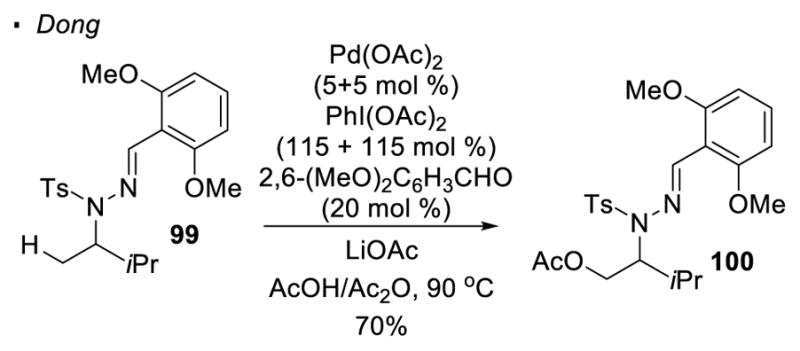
Hydrazone-Directed C-H Activation
(Va) Carbonyl Groups (8-aminoquinoline amides)
The functionalization of carbonyl compounds with palladium-catalyzed sp3 C-H activation is the most explored and well-developed. In most cases, it is the β C-H bonds of the carbonyl compounds that are functionalized. Daugulis first observed that 8-aminoquinoline amides can serve as a directing group for this purpose, where a β-methylene C-H bond of amide 101 is arylated to afford 102 (Scheme 17).14 The incorporation of various functional groups at these C-H bonds has been rendered possible due to the contributions of different research groups. For the formation of C-C bonds, alkylation of 103, alkynylation of 106, and alkenylation of 109 and 101 were developed by Daugulis, 49 Tobisu and Chatani, 50 Chen51 and Rao 52 respectively. The geometry of vinyl iodide 110 is preserved in Chen’s system for the alkenylation of amide 109.
Scheme 17.

Formation of C-C Bonds with 8-aminoquinolines
In addition to C-C bonds, reactions for the incorporation of various heteroatoms at the β-position have been developed (Scheme 18). Corey’s system for C-H acetoxylation of 114 relies on the use of oxone as an oxidant. 53 Besset applies N-thiotrifluoromethylphthalimide 117 as an oxidant to achieve C-H thiotrifluoromethylation of 116. 54 With sodium salts of arylsulfonates as a sulfur source and Ag2CO3 as an external oxidant, Shi developed a protocol for C-H sulfonylation. 55 Kuninobu and Kanai showed that C-H silylation of 121 can be achieved with disilane 122.56 As demonstrated by Xu, the use of NFSI as an electrophilic fluorine source results in C-F bond formation at the unactivated C-H bonds.57 To incoporate other halogens at the β-position, Rao modifies the directing group because 8-aminoquinoline is prone to electrophilic aromatic substitution. 58 Thus, 5-chloro-8-aminoquinoline 125, which is less reactive towards electrophilic aromatic substitution, is used to realize C-H chlorination and bromination with NCS or NBS respectively, to afford halogenated 126.
Scheme 18.

Formation of C-X Bonds with 8-aminoquinolines
(Vb) Carbonyl Groups (perfluorinated amides)
In addition to 8-aminoquinoline, perfluorinated amides are a versatile directing group for β functionalization of carbonyl compounds. With 2,3,5,6-tetrafluoro-4-trifluoromethylbenzamide as a directing group, Yu developed protocols that allow the incorporation of various functionalities at unactivated sp3 C-H bonds (Scheme 19). For C-C bond formation, arylation of amides 127 and 129 with aryl iodides,59 alkylation of amide 127 with ethyl iodide 57,60 and alkynylation of amide 132 with alkynyl bromide 10761 can be achieved. In these reactions, amides with or without α substitution are competent substrates, providing a method to functionalize both natural or unnatural amino-acids.
Using the same directing group, the formation of C-C bonds can be applied to the synthesis of nitrogen-heterocycles with an appropriate choice of reaction partners (Scheme 20). When electrophilic benzyl acrylate 78 is employed, olefination of the C-H bond in amide 134 is followed by aza-Michael addition, affording 5-membered pyrrolidinone 135 as the final product.62 In the presence of carbon monoxide, the putative palladium alkyl intermediate formed from 136 can be carbonylated. 63 The resulting 6-membered palladacycle then undergoes reductive elimination to afford succinimide 137.
Scheme 20.
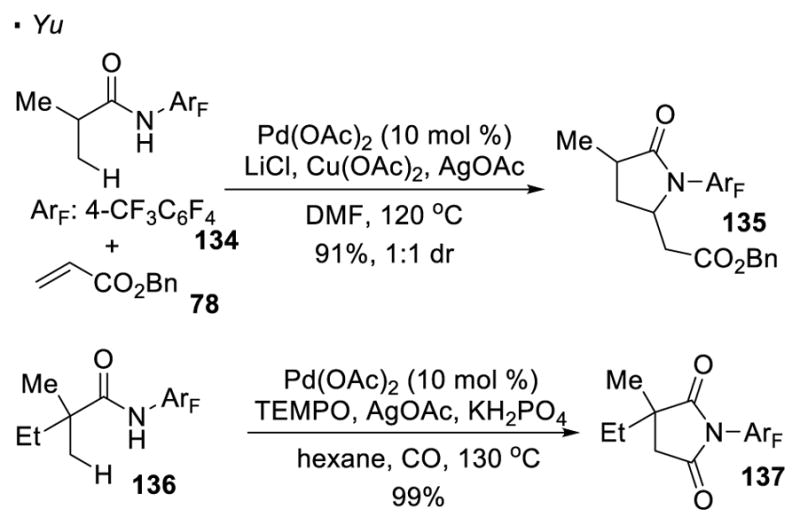
Synthesis of Nitrogen-Heterocycles from Perfluorinated Amides
Unsurprisingly, the formation of bonds other than C-C bonds can be achieved (Scheme 21). Using hydroxylamine 139 as an electrophilic nitrogen source, the intermolecular C-H amination of amide 138 yields aminated 140. 64 When the palladium alkyl intermediate is trapped with Selectfluor 141, a fluorine atom can be incorporated into the C-H bond of amide 127.65 Borylation can be realized with B2pin2 to afford amide 143.66 The resulting C-B bond can be converted to an array of functionalities. In all these examples with perfluorinated amines as the directing group, the exogenous ligands are essential to achieve the desired reactivity.
Scheme 21.

Formation of C-X Bonds with Perfluorinated Amides
(Vc) Carbonyl Groups (O-Methyl Hydroxamic Acids and Carboxylic acids)
To provide an alternative method to functionalize carboxylic acid derivatives, Yu pioneered the use of O-methyl hydroxamic acids and free carboxylic acids as the directing groups (Scheme 22). The initial report from Yu allows arylation or alkylation of primary C-H bonds β to O-methyl hydroxamic acids67 and carboxylic acids68 bearing α quaternary centers. An improved protocol established from the same group tolerates the presence of α C-H bonds of O-methyl hydroxamic acids69 and carboxylic acids. 70 Thus, amino acids 145 and 147 can be derivatized using O-methyl hydroxamic acids and carboxylic acids as a directing group.
Scheme 22.
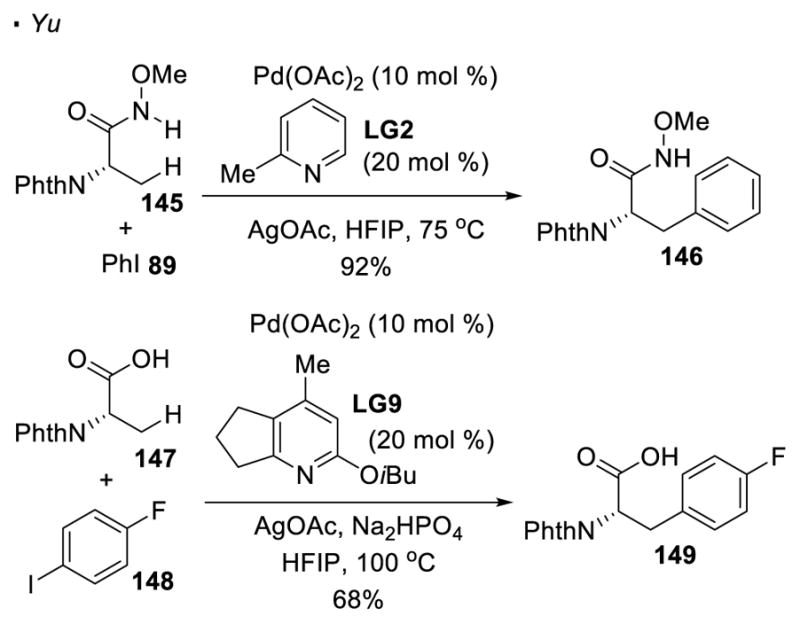
C-H Functionalization with O-Methyl Hydroxamic acids and Carboxylic Acids
(Vd) Carbonyl Groups (Peptides)
Yu demonstrated that peptides can be functionalized via palladium-catalyzed C-H activation at the N-Terminus.71 Such a strategy does not require the manipulation of directing groups since the native amino acid moiety can serve as a directing group (Scheme 23). For instance, dipeptide 150 and tripeptide 152 can be arylated at the β positions of the amino acids at the N-terminus.
Scheme 23.
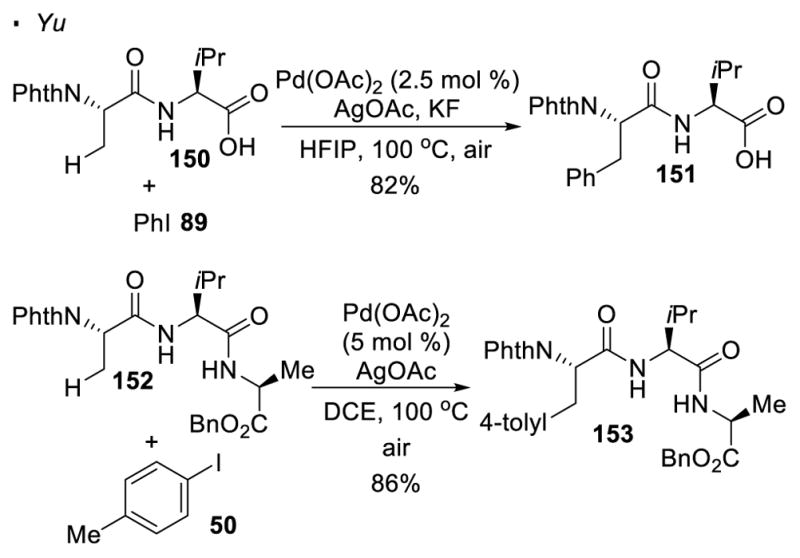
C-H Functionalization of Peptides
(Ve) Carbonyl Groups (ketones and aldehydes)
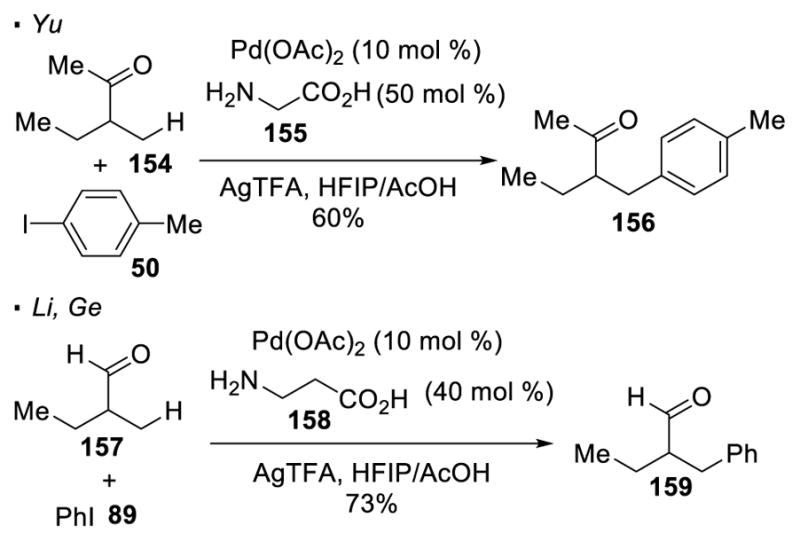
The carbonyl oxygen of aldehydes and ketones is weakly basic and does not have an appreciably acidic proton to generate an anion. These factors had inhibited the development of C-H activation with aldehydes and ketones until Yu, Li, and Ge’s recent contributions (Scheme 24). They addressed the low basicity of these carbonyl groups by converting them into a more basic functionality in-situ. By adding a sub-stoichiometric amount of amino-carboxylic acids such as 155 and 158, a transient bidentate imine directing group is formed under the reaction conditions. Yu,72 Li and Ge73 demonstrated that the β primary C-H bonds of ketone 154 and aldehyde 157 can be arylated with aryl iodides 50 and 89 with this strategy.
Scheme 24.
In-situ Generation of Directing Group from Carbonyl Groups
(Vf) Carbonyl Groups (γ Functionalization)
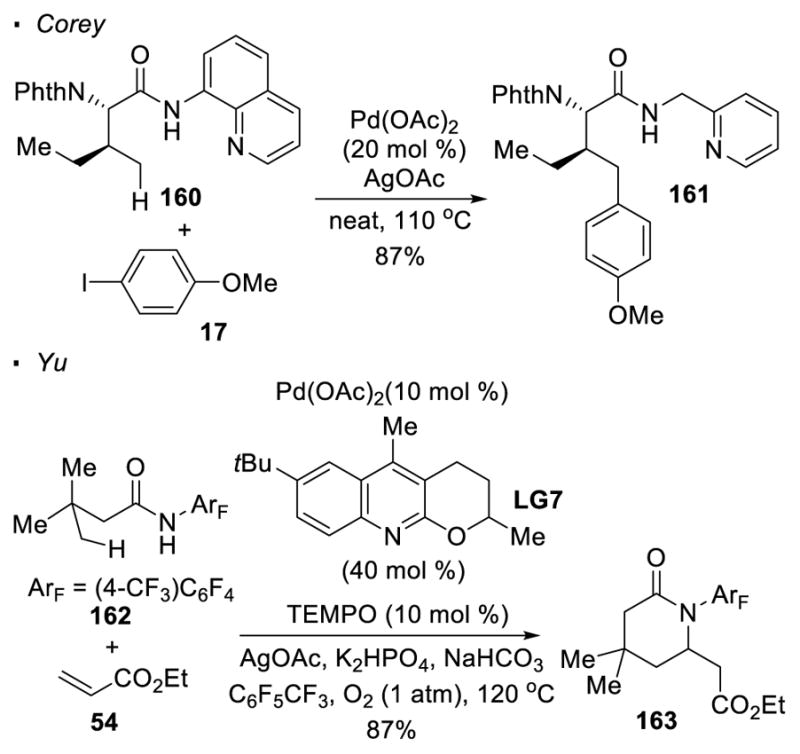
The above representative examples show that C-H bonds β to the carbonyl compounds are typically activated. However, the γ C-H bond of an amide can be occasionally functionalized (Scheme 25). Corey observed that carbonyl compound 160, which contains a sterically bulky substituent NPhth at the α position, experiences C-H activation preferentially at the γ primary C-H bond over a β tertiary C-H bond.53 It is proposed that the α substituent in this substrate leads to a conformational bias that favors the activation of the γ C-H bond. In this case, activation of the β C-H bonds is also disfavored because of the more challenging formation of a palladium alkyl species from a tertiary C-H bond than a primary C-H bond. Alternatively, to achieve the functionalization of a γ primary C-H bond, the β carbon is fully substituted such that β C-H activation is not a possibility. For instance, Yu showed that the γ C-H bonds of amide 162 can be olefinated74 or arylated.75
Scheme 25.
γ Functionalization of Carbonyl Compounds
(VI) Alcohol Derivatives
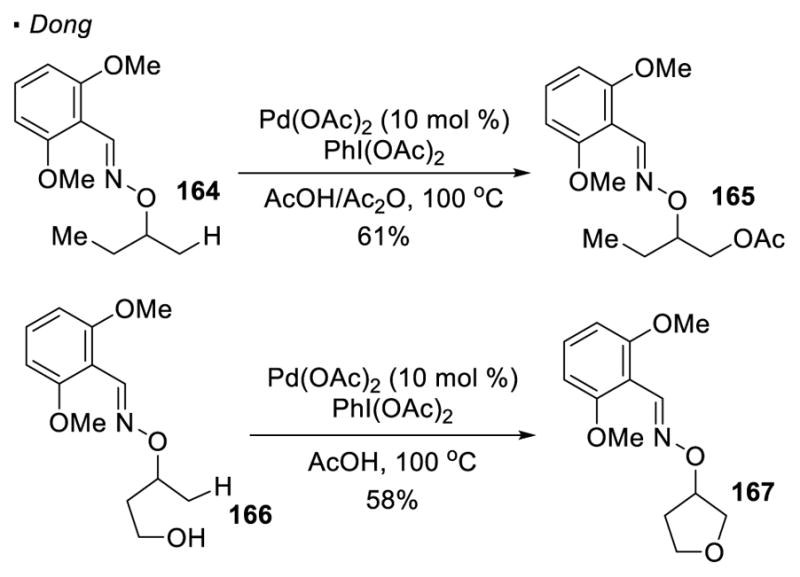
In order to functionalize alcohols (Scheme 26), Dong employs an oxime as a directing group. The system is analogous to that reported by Sanford, however the sp3 C-H bonds are located on the side of the oxygen, instead of the nitrogen. Oxygenation of the β primary C-H bonds can be accomplished with oxime 164 to afford 165.76 An intramolecular variant of the reaction with oxime 166 provides cyclic ether 167 as the product.77 The mechanism involving a Pd(II)/Pd(IV) is believed to be operative.
Scheme 26.
C-H Activation of Alcohol Derivatives
(VII) Pyrazoles
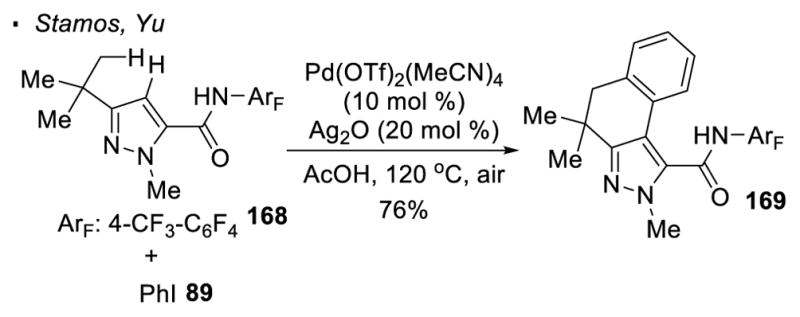
As demonstrated by Stamos and Yu, pyrazoles are also a competent directing group for sp3 C-H activation (Scheme 27).78 For the reaction of pyrazole 168, one of the methyl group is arylated with phenyl iodide. Subsequent activation of two sp2 C-H bonds resulting in the formation of tricyclic product 169.
Scheme 27.
Pyrazole-Directed C-H Activation
2.2. Catalysis with Other Noble Transition Metals
Other noble transition metals are also competent catalysts for the activation of unactivated sp3 C-H bonds, although they are less studied. Except in one case (Hartwig’s Ir(I)-catalyzed silylation and borylation, see below), all the reactions can be accomplished by palladium catalysis with different reaction conditions.
2.2.1 Ruthenium
When amide 170 is heated with a catalytic amount of Ru3(CO)12 under an atmosphere of carbon monoxide and ethylene, Chatani observed that succinimide 171 is formed (Scheme 28).79 In the proposed mechanism, the Ru catalyst forms Ru hydride from the amidyl N-H bond. A molecule of ethylene inserts into the Ru hydride bond to form Ru-ethyl intermediate F2. σ bond metathesis results in the activation of the methyl C-H bond to generate F3. Carbonylation takes place to give 6-membered metallacycle F4. A final reductive elimination yields the observed product 171.
Scheme 28.
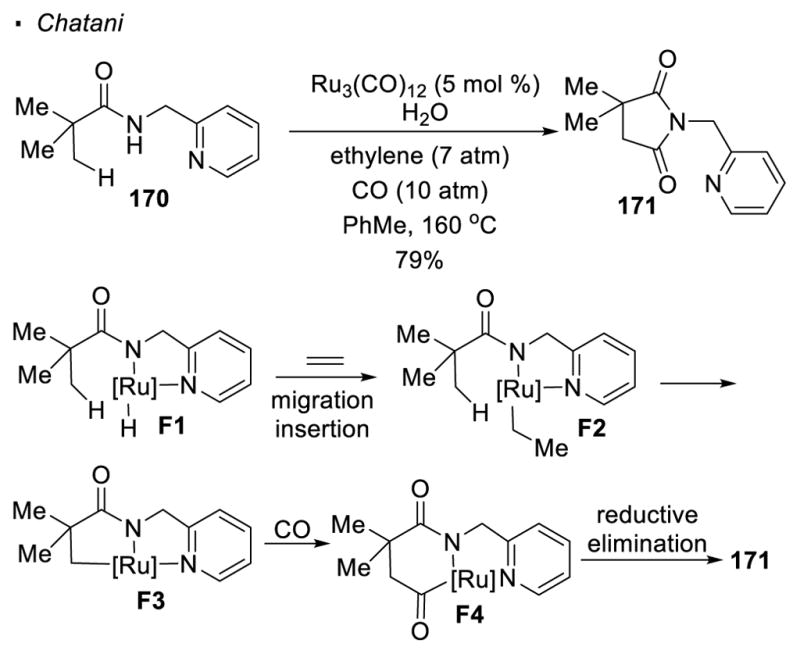
Ru-Catalyzed C-H Activation
2.2.2 Iridium
(I) Iridium(I)
A three-step protocol for Ir-catalyzed alcohol-directed silylation of primary C-H bonds was reported by Hartwig (Scheme 29).80 Similar to his previous work on sp2 C-H bond activation,81 an Ir(I)-catalyzed reaction is first applied to silylate the alcohol in order to install a hydrosilane as the directing group. Without the isolation of the hydrosilane intermediate, silylation of the C-H bonds is accomplished with a second Ir-catalyzed reaction. A C-O bond is formed by a Tamao-Fleming oxidation, affording a 1,3 diol as the final product. Therefore, 1,3 diol 174 can be synthesized from alcohol 172 with hydrosilane 173 as an intermediate. Hartwig later showed that Ir(III) catalyst 176 can be applied to silylate 82 or borylate 83 secondary C-H bonds, as exemplified by the borylation of the cyclohexyl C-H bond of hydrosilane 175 to afford boronate 177. The resulting C-B bond can be converted to a C-O, C-N or C-C bond with subsequent reactions.
Scheme 29.
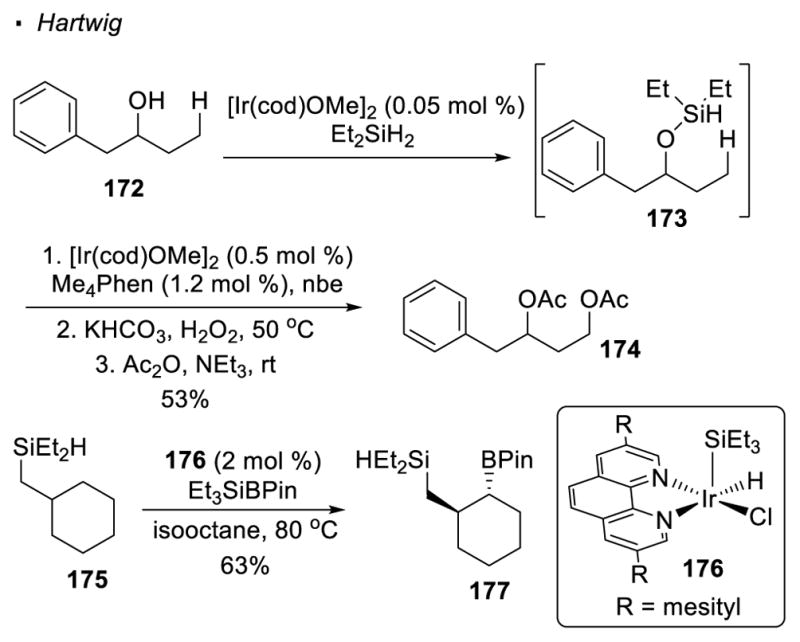
Ir(I)-Catalyzed Borylation
(II) Iridium(III)
Chang developed a protocol for Ir(III)-catalyzed oxime-directed amination of primary C-H bonds with tosyl azide as a nitrogen source (Scheme 30).84 Aminated oxime 179 can be obtained from oxime 178 with [Cp*IrCl2]2 as a pre-catalyst and AgBF4 as a co-catalyst. Zeng and Ke later showed that amide 180 is also a competent substrate for the same transformation with secondary C-H bonds under similar reaction conditions.85 As reported by Xia and Shi, arylation of primary C-H bonds can be accomplished with the same directing group and the same Ir pre-catalyst.86 Oxime 182 is arylated with diaryliodonium salt 47 to afford 183.
Scheme 30.
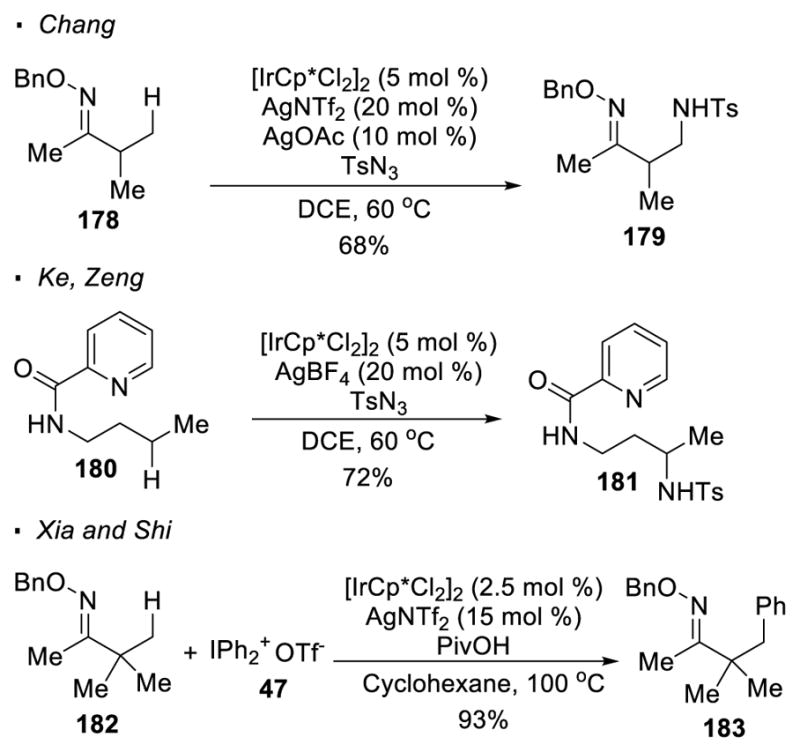
Ir(III)-catalyzed C-H Amination and Arylation
2.2.3 Rhodium
Rhodium(III)
You87 and Glorius88 accomplished pyridine directed C-H functionalization with Rh(III) precatalysts (Scheme 31). You achieves amination of pyridine 12 with nitrobenzenesulfonamide (NsNH2) as the nitrogen source and PhI(OAc)2 as an oxidant. Arylation of pyridine 185 relies on the use of boroxine 186 in Glorius’s protocol. Various groups subsequently reported rhodium(III)-catalyzed sp3 amination with other nitrogen sources or under different reaction conditions.89
Scheme 31.
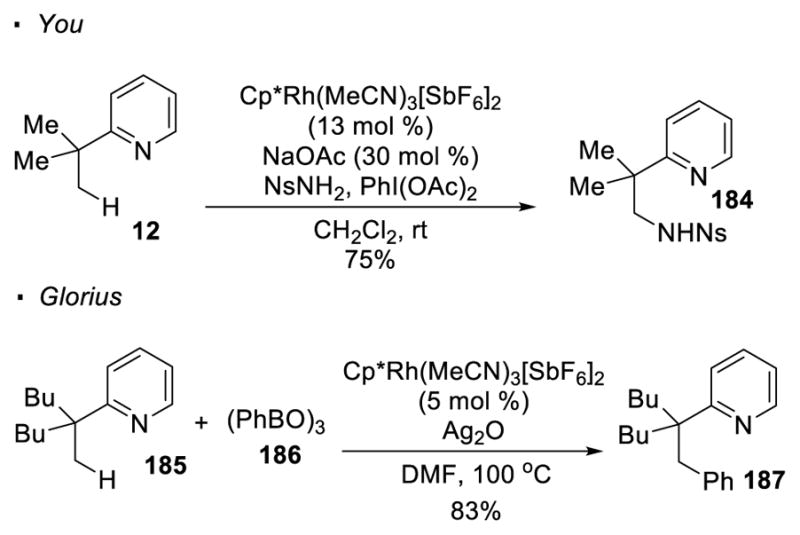
Rh(III)-Catalyzed C-H Amination of Pyridines
2.3. With Earth-Abundant Metal Catalysts
Recent efforts have been devoted to replace noble metals with less expensive first-row earth-abundant metals. However, from the following representative examples, it can be concluded that the scope of transformations that can be achieved with first-row transition metal catalysis is still very limited. No functional groups other than carbonyl compounds have been employed as competent directing groups. Therefore, at this time, only β functionalization of carbonyl groups can be accomplished. In addition, with the exception of Ni(0)-catalysis, only reactivity previously demonstrated with palladium catalysis has been reported.
2.3.1 Nickel
(I) Nickel(0)
Nickel, likely because of its position above palladium in the periodic table, was the initial focus. Nakao and Hiyama first employed low valent Ni(cod)2 as the precatalyst and AlMe3 as the co-catalyst for sp3 C-H activation (Scheme 32).90 Nickel(0)-catalyzed dehydrogenative [4+2] cycloaddition of formamide 188 and alkyne 189 can be accomplished to access piperidone 190. In the proposed mechanism, AlMe3 acts as a Lewis acid to activate the formamide. This allows the Ni catalyst to oxidatively add to the carbonyl C-H bond to form nickel hydride G2. Insertion of the alkyne into the metal-hydride bond results in the formation of vinyl nickel intermediate G3. C(sp3)-H activation takes place to give 5-membered nickelacycle G4. An alkyne can insert into one of the Ni-C bonds to expand the ring size of the nickelacycle. The observed piperidine product 190 is formed from reductive elimination of the final intermediate G5.
Scheme 32.
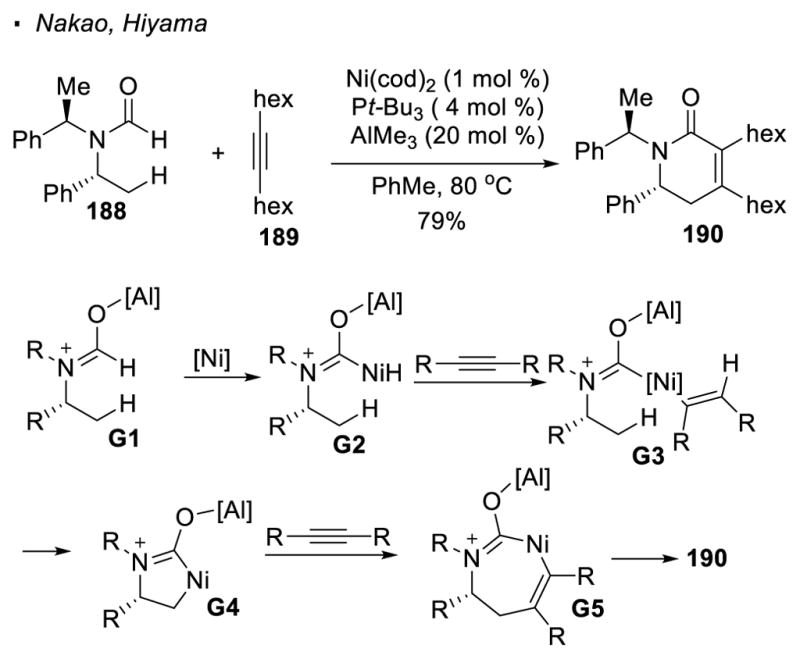
Nickel(0)-Catalyzed [4+2] Cycloaddition
(II) Nickel(II)
Compared to its low valent counterpart, nickel(II) is a more appealing catalyst because of its lower cost and ease of handling. In all these examples with nickel(II) catalysis, 8-aminoquinoline, first introduced by Daugulis for palladium catalysis, is used as a directing group (Scheme 33).14 Various functionalities can be incorporated into the molecule at these unactivated C-H bonds. This is exemplified by initial reports from Chatani and Ge. Using Ni(II) as a precatalyst in conjunction with carbonate bases, β primary C-H bonds of amide 191 can undergo arylation with aryl iodides 91 or alkylation with alkyl iodides.92 Shi showed Ni(II)-catalyzed alkenylation of amide 191 with vinyl iodides 93 to afford alkenylated products. As reported by Zhang, alkenylation can also accomplished with phenylacetylenes. 94 Various groups also simultaneously reported C-H thiolation using the same directing group.95
Scheme 33.
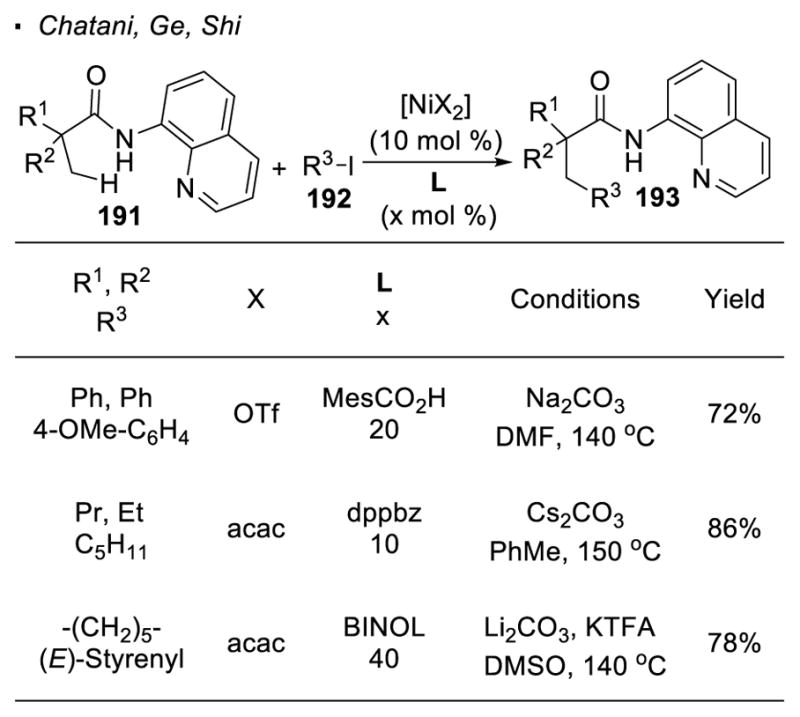
Nickel(II)-catalyzed C-H Functionalization
Synthesis of nitrogen-heterocycles from amides can also be catalyzed by nickel(II). As demonstrated by Ge, in the absence of an exogenous partner to trap the nickel-alkyl intermediate, reductive elimination can occur to give β-lactam 194 (Scheme 34).96 An alternative mechanism first involving C-H iodination and subsequent SN2 is proposed by Chatani.97 In addition, DMF can be used as a carbonylating agent. Amide 195 undergoes carbonylative cyclization to form succinimide 185 with NiBr2 catalyst and Cu(acac)2 cocatalyst.98
Scheme 34.
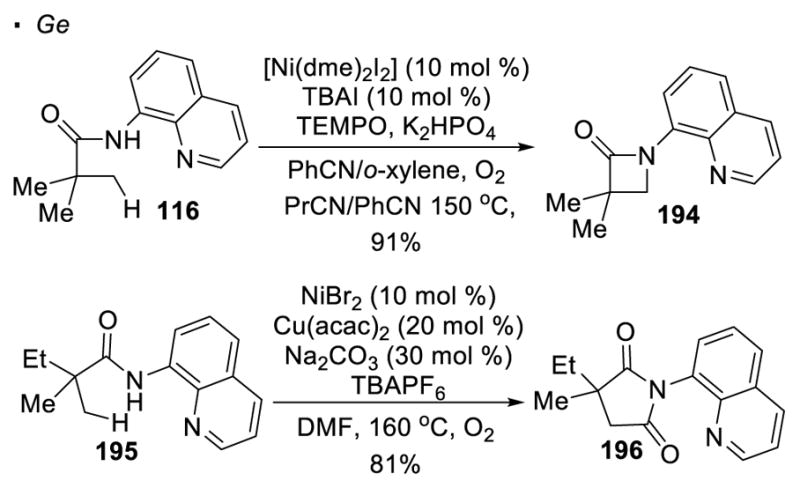
Synthesis of Nitrogen Heterocycles with Ni(II) Catalysis
2.3.2 Iron
The 8-aminoquinoline directing group also enabled Fe-catalyzed C-H activation. Nakamura and Ilies achieved iron-catalyzed β-arylation of primary C-H bonds of carbonyl compound 197 with ArMgBr and Ar2Zn (Scheme 35). 99 For instance, Subsequent reports from the same group demonstrate that methylation and vinylation of primary C-H bonds can be realized with AlMe3100 and vinyl boronates101 respectively. In all these reactions, chlorinated hydrocarbons 199 or 200 are added as an oxidant.
Scheme 35.
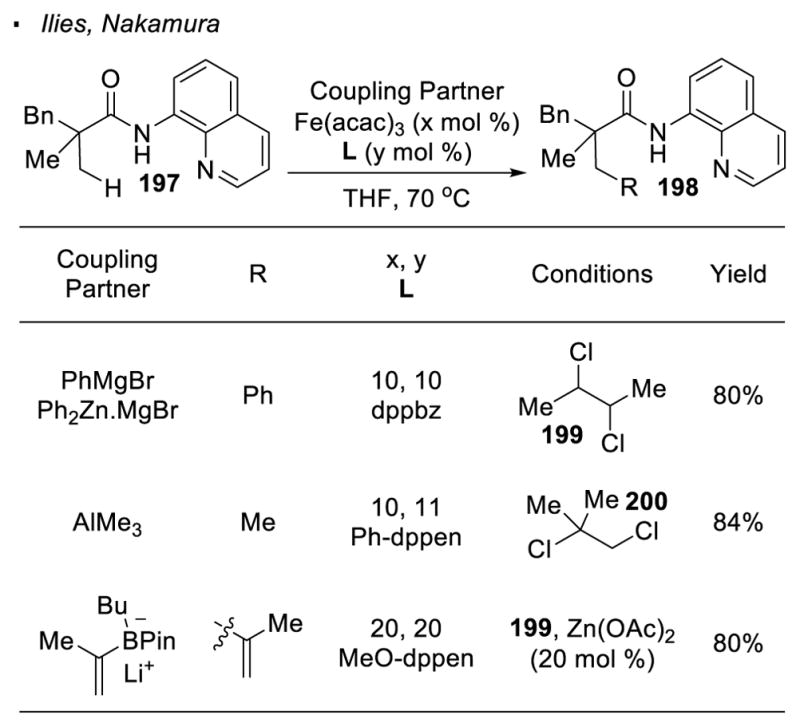
Fe-Catalyzed Formation of C-C Bonds
2.3.3 Cobalt
Cobalt has also been shown to be a competent catalyst for 8-aminoquinoline-directed C-H activation. As reported by Zhang, using Co(OAc)2 as the precatalyst, 5-membered pyrrolidinone 202 can be accessed from acyclic amide 116 and phenylacetylene 201 (Scheme 36).102 In this case, concomitant alkynylation and cyclization occurs. Li and Ge showed that amide 203 undergoes β-lactamization in the absence of an alkyne under similar reaction conditions to afford β-lactam 204. 103 Intermolecular amination can be achieved when a perfluorinated amide is added as a nitrogen source. Sundararaju104 and Gaunt105 independently found that Co(acac)2 catalyzes the carbonylative cyclization of amide 116 for access to succinimide 207 in the presence of carbon monoxide.
Scheme 36.

Co-Catalyzed C-H Functionalization
2.3.4 Copper
Ge developed the synthesis of β-lactam 204 from amide 203 with copper(I) chloride as the catalyst (Scheme 37).106 In this reaction, duroquinone is believed to oxidize copper(II) to copper(III) to drive reductive elimination to form the C-N bond. Yang and You later showed that O2 can serve as an alternative oxidant with copper(I) iodide as the catalyst.107 The copper(II) acetate-catalyzed carbonylative cyclization of amide 195 to afford succinimide 196, reported by Li and Ge, relies on nitromethane 209 as a formal source of carbon monoxide.108 In the proposed mechanism, a nitronate ligand binds to the copper(III) alkyl H1 through ligand exchange to form intermediate H2. Reductive elimination forms a C-C bond to generate intermediate 210, which is further transformed to the observed cyclized product 189. Intermolecular C-H amination has also been established but a stoichiometric amount of a copper salt is required.109
Scheme 37.
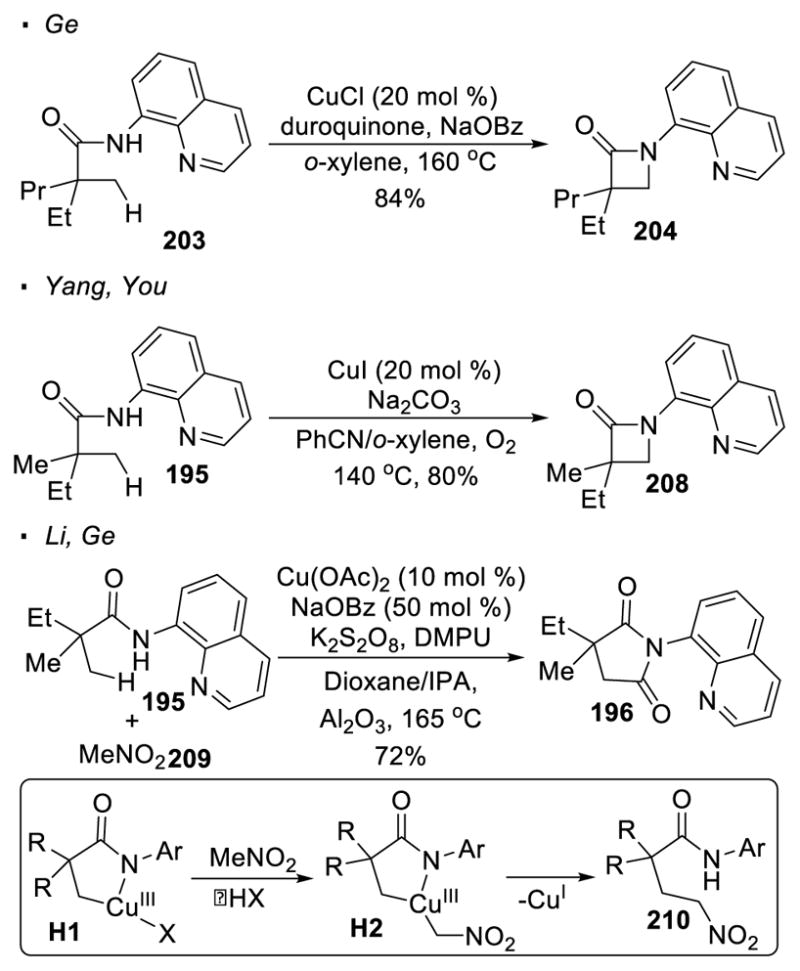
Cu-Catalyzed C-H Functionalization
3. Hydrogen Atom Transfer to Reactive Radical Species
A conceptually distinct approach to C(sp3)-H activation relies on established radical reactivity. 110 In this case, a hydrogen atom is transferred from the carbon center to a highly reactive radical species. The driving force of this hydrogen atom transfer is the formation of a X-H bond which is stronger than the breaking C-H bond. Radicals that are capable of abstracting a hydrogen atom from an unactivated sp3 C-H bond are typically oxygen radicals, nitrogen radicals, and aryl/vinyl radicals (Scheme 38). The resulting alkyl radical from the C-H bond can be trapped with a radical partner, resulting in the installation of a functional group at the original C-H bond.
Scheme 38.
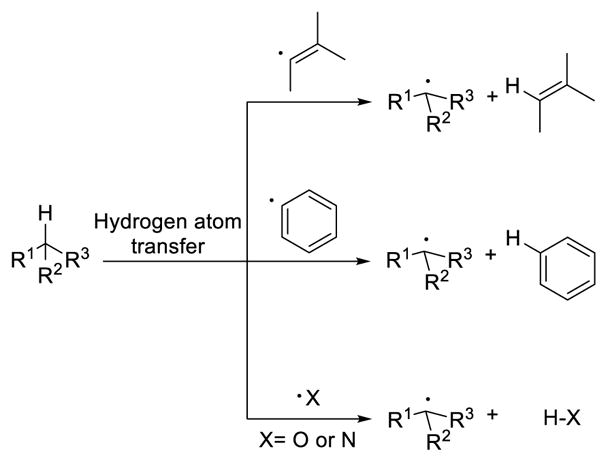
Hydrogen Atom Transfer
Intramolecular hydrogen atom transfer is often favored, compared to an intermolecular event, due to a lower entropic cost. In this case, the reactive radical species can be viewed as a directing group. In most cases, 1,5 hydrogen atom transfer (1,5 HAT) is the predominant pathway, in which the cleaved C-H bond is located five bonds away from the reactive radical (Scheme 39).111 The functionalities that can be incorporated into the C-H bonds is contingent on the identity of the reactive radical (oxygen/nitrogen/vinyl/aryl), as well as the method to generate the radical. Therefore, these aspects will be the primary focus of the following discussion.
Scheme 39.
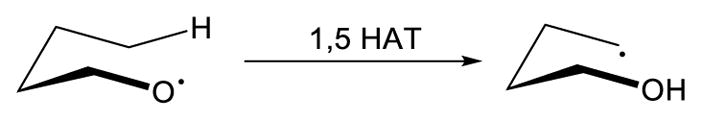
1,5 Hydrogen Atom Transfer
3.1. Nitrogen Radicals
The first class of radicals that are capable of abstracting a hydrogen atom from inert sp3 C-H bonds is nitrogen radicals.112 The N-H bond of a neutral amine has a comparable bond energy to that of a sp3 C-H bond, so there is a lack of appreciable driving force for hydrogen atom transfer to occur.113 To realize 1,5 HAT, the nitrogen radical is rendered more electrophilic through either protonation or with an electron-withdrawing group.
3.1.1 Generation and Transformations
(I) Homolysis of N-X Bonds
The most common method to generate a nitrogen radical is through the homolytic cleavage of a nitrogen-heteroatom bond (Scheme 40). In the Hofmann-Löffler-Freytag (HLF) reaction, a nitrogen-halogen bond is subjected to thermal or photochemical conditions in the presence of a strong acid.114 For example, the nitrogen-bromine bond in amine 211 breaks homolytically to give the nitrogen radical 212 and a bromine radical. The protonated nitrogen radical abstracts a hydrogen atom from the δ C-H bond to give alkyl radical 213. The combination of the alkyl radical and the halogen radical results in the formation of a carbon-halogen bond. Subsequent basic workup leads to a SN2 reaction and affords pyrrolidine 215 as the final product. In all these cases, the nitrogen radical is generated concurrently with a halogen radical. Therefore, the alkyl radical formed from 1,5 HAT is inevitably intercepted by the halogen radical. This invariably leads to the formation of a carbon-halogen bond.
Scheme 40.
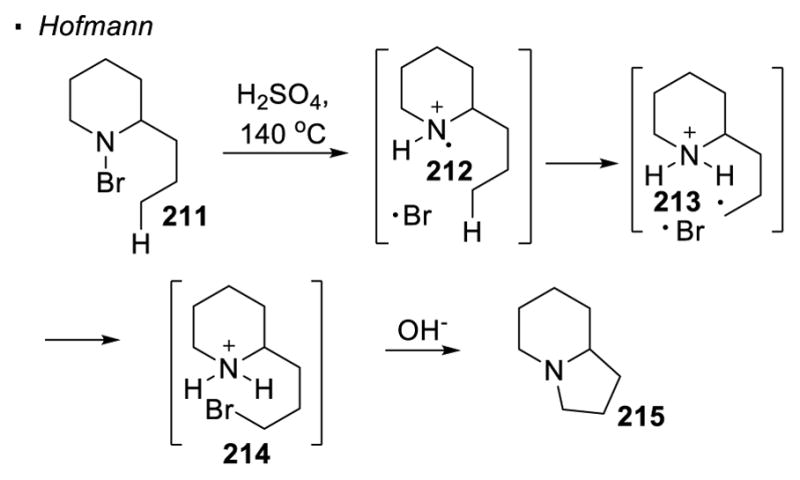
Generation of Nitrogen Radicals by Homolysis of N-X Bonds
More recent research has been devoted to the investigation of in-situ generation of a nitrogen-halogen bond from a N-H bond (Scheme 41). This obviates the handling of unstable nitrogen-halogen bonds. With the Suarez modification, elemental halogen, along with Pb(OAc)4 or PhI(OAc)2, is used to generate a nitrogen-halogen bond from a N-H bond.115 The presence of an electron-withdrawing group (nitro, cyano, phosphonyl and carbonyl groups) on nitrogen makes the nitrogen radical reactive enough to realize HAT. Thus, amine derivative 216, bearing various electron-withdrawing groups, is a competent substrate for the HLF reaction. Reports from Togo and Yokoyama,116 and Fan117 demonstrated that sulfonyl groups on nitrogen are also compatible with this chemistry. Muniz reported a catalytic variant of the Suarez modification. In his protocol, iodine is employed as a catalyst for the reaction of sulfonamide 220 to afford HLF product 221.118 For all these transformations described above, the formation of the nitrogen radical is also accompanied by a halogen radical, and as in the traditional HLF reaction, a carbon-halogen bond is therefore formed. A subsequent step would afford a pyrrolidine as the final product. In addition, Yu showed that when amide 222 is subjected to NIS and TMSN3, the δ C-H bond is aminated and the ε C-H bond is iodinated to give γ-lactam 223 (Scheme 42).119 It is proposed that an azide radical can abstract a hydrogen atom from the original HLF product 224 to give a nitrogen radical and an alkene. Cyclization of the nitrogen radical onto the alkene generates an alkyl radical which captures an iodine atom from NIS. As exemplified by the aforementioned reactions, only halogenation of C-H bonds has been accomplished with in-situ generation of a nitrogen-halogen bond as the nitrogen radical precursor.
Scheme 41.
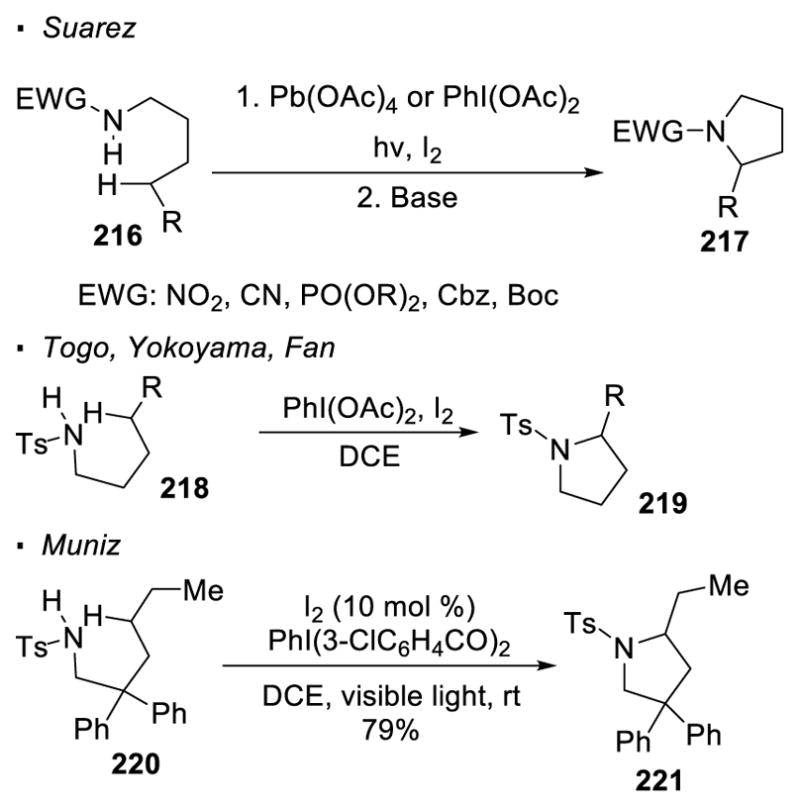
In-situ Generation of N-X Bonds
Scheme 42.
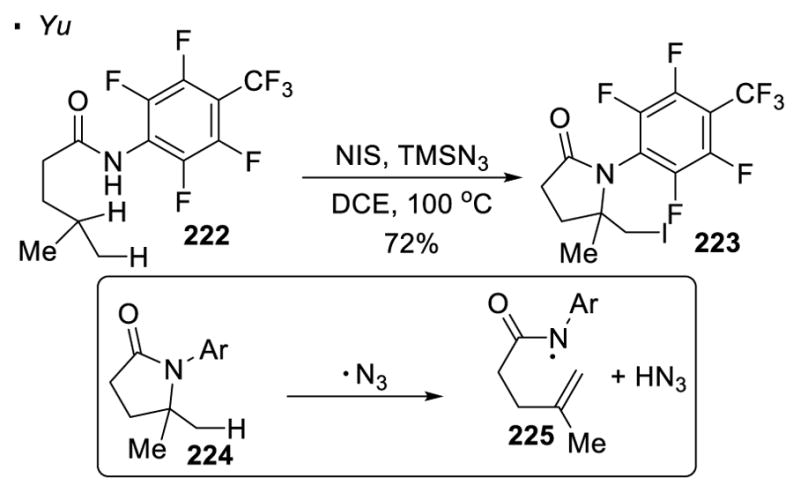
γ, ε Functionalization with Nitrogen Radicals
(II) Reduction of N-X Bonds
When the nitrogen is at a higher oxidation level, reduction provides a means to generate a nitrogen radical. Decomposition of organic azides with various reducing agents is not discussed here since the resulting neutral nitrogen radicals are not reactive enough to break unactivated sp3 C-H bonds (Scheme 43).120 Chiba reported a copper-catalyzed intramolecular amination of the tertiary C-H bond of amidoxime 228 (Scheme 44).121 In the proposed catalytic cycle, the copper(I) catalyst reduces the N-O bond to amidinyl radical 230. 1,5 HAT generates alkyl radical 231 which is oxidized by copper(II) to carbocation 232. Cyclization then occurs to give dihydroimidazole 229. An analogous route to generate a nitrogen radical is demonstrated by Yu with photoredox catalysis. 122 The N-Cl bond in sulfonamide 233 is reduced by the excited photocatalyst [Ir(ppy)2(dtbpy)]PF6. Through 1,5 HAT, an alkyl radical is generated. The oxidation of the alkyl radical to a carbocation turns over the photocatalyst and allows cyclization to afford the HLF product 234. Overall, only intramolecular amination to give the HLF products has been achieved when nitrogen at a higher oxidation level is employed as the nitrogen radical precursor.
Scheme 43.
Generation of Nitrogen Radicals by Reduction of Organic Azides
Scheme 44.
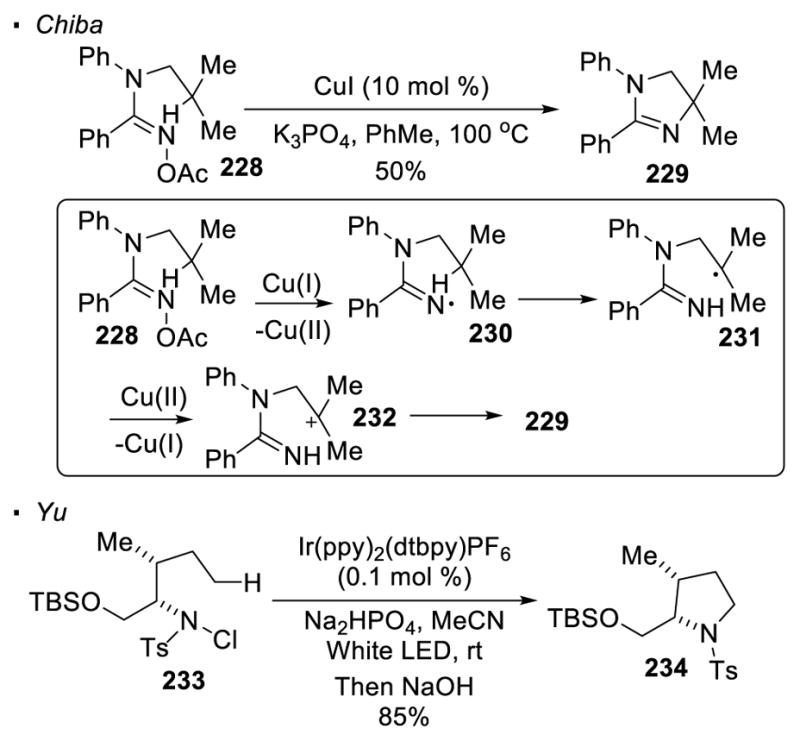
Catalytic Reduction of N-X Bonds
(III) Abstraction of Hydrogen Atoms
The abstraction of a hydrogen atom provides another means to form a nitrogen radical from a N-H bond. Chiba showed that the reaction between the N-H bond in hydrazone 235 with TEMPO can generate a nitrogen radical (Scheme 45).123 Although the nitrogen radical generated in this case is stabilized by the alpha nitrogen atom, the equilibrium between the nitrogen radical 237 and the alkyl radical 238 provides a small concentration of the alkyl radical. The trapping of the alkyl radical by TEMPO results in the formation of a C-O bond. Subsequent elimination of TEMPO-H in 239 gives aza-diene 240, which can cyclize to give dihydropyrazole 236. Yu also demonstrated that sulfonamide 241 undergoes C-H bromination with NBS and TMSN3 in the presence of a copper(II) catalyst (Scheme 45).124 In the proposed mechanism, an azide radical generated in-situ abstracts a hydrogen atom from the N-H bond to form nitrogen radical 243. The alkyl radical 244 formed by 1,5 HAT can react with in-situ generated copper(III) bromide to afford the observed brominated 242.
Scheme 45.

Generation of Nitrogen Radicals by Abstraction of Hydrogen
(IV) Oxidation of N-H Bonds
A less common method to generate a nitrogen radical is through the oxidation of a N-H bond. Nikishin reported that in the presence of a stoichiometric amount of strongly oxidizing Na2S2O8, the HLF product can be obtained from sulfonamide 245 (Scheme 46). 125 It is believed that the oxidizing agent oxidizes the N-H bond to give nitrogen radical 247 that can realize 1,5 HAT to generate an alkyl radical δ to the nitrogen. Oxidation of the alkyl radical 248 to carbocation 249 promotes cyclization to pyrrolidine 246 as the final product. Although both the conversion and yield are moderate, this reaction suggests the feasibility of using a N-H bond as a nitrogen radical precursor. Another HLF reaction that possibly involves the oxidation of a N-H bond to generate a nitrogen radical is reported by Shi.126 With Ag catalysis and a hypervalent iodine reagent as the terminal oxidant, sulfonamide 250 undergoes a HLF reaction to form cyclized product 253. Although the authors propose a concerted metalation/deprotonation mechanism with silver to account for the cleavage of the C-H bond, a nitrogen radical intermediate is proposed in their subsequent report under essentially the same reaction conditions.127 In addition, Chiba reported the generation of nitrogen radicals from the N-H bond of amidine 252 with copper(II) catalysts and oxygen.128 Instead of being oxidized, the alkyl radical intermediate 254 in this case is trapped by O2 to form superoxo radical 255. A Fenton-type fragmentation is proposed to give copper(II) alkoxide intermediate 256. Dihydrooxazole 257 is formed after subsequent nucleophilic substitution. Except for Chiba’s system with amidines, in all the reactions relying on the oxidation of the N-H bond to generate the nitrogen radical, 5-membered-ring products through intramolecular C-H amination are obtained.
Scheme 46.

Generation of Nitrogen Radicals by Oxidation of N-H Bonds
Most recent research incorporates photoredox catalysis into the oxidation of N-H bonds. Rovis 129 and Knowles 130 simultaneously and independently disclosed remote alkylation of unactivated sp3 C-H bonds directed by an amide (Scheme 47). In the Rovis system, trifluoroacetamide 263 undergoes alkylation at the tertiary C-H bond with methyl methacrylate 264 to form amide 265. Similarly, alkylated product 271 can be obtained from amide 269 with methyl vinyl ketone 270 using Knowles’ protocol. In the proposed catalytic cycle of these two reactions, the excited photocatalyst oxidizes amide 258 to yield nitrogen radical 259 in the presence of a base. 1,5 HAT and trapping of the alkyl radical 260 with the electrophilic alkene generates a radical α to an electron-withdrawing group (261). Reduction of 261 turns over the photocatalyst and final protonation affords the alkylated product 262. In Rovis’ system, mechanistic work suggests a stepwise deprotonation/oxidation event for the generation of the nitrogen radical. Thus, the intermediacy of amidyl anion 267 is proposed. This accounts for the necessity of the strong electron-withdrawing trifluoroacetyl group on nitrogen to acidify the N-H bond and the use of a strong base, K3PO4. On the other hand, a concerted proton-coupled-electron-transfer event is believed to operate in Knowles’ system. Therefore, amides bearing less acidic N-H bonds are competent substrates in the presence of a more oxidizing photocatalyst. This set of conditions allows a broader substrate scope at the expense of selectivity. Thus, the two systems developed by Rovis and Knowles are complementary.
Scheme 47.

Formation of C-C Bonds with Nitrogen Radicals
3.2. Oxygen Radicals
Oxygen radicals are more electrophilic and reactive than nitrogen radicals due to the higher electronegativity of oxygen; thus, neutral oxygen radical is able to abstract hydrogen atoms from inert C-H bonds without protonation.131 This process has a favorable thermodynamic force, as indicated by the stronger bond energy of O-H bonds (105 kcal/mol) than sp3 C-H bonds. However, because of the strength of the C=O bond, β-scission occasionally out-competes intramolecular hydrogen atom transfer. In β-scission, alkyl radical 276 and ketone 277 are formed from the cleavage of the Cα-Cβ bond (Scheme 48).
Scheme 48.
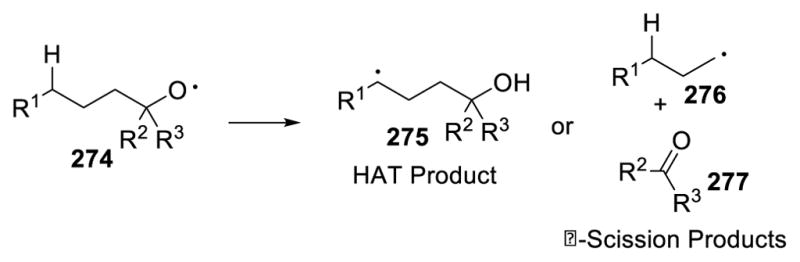
Reactivity of Oxygen Radicals
3.2.1 Generation and Transformations
(I) Homolysis of O-X Bonds
One of the most common oxygen radical precursors is an oxygen-heteroatom bond. In most cases, the identity of the heteroatom determines the coupling partner at the C-H bond. Similar to the HLF reaction with nitrogen radicals, the oxygen radical is formed from the homolytic cleavage of the oxygen radical precursor. The first reported C-H functionalization reaction with oxygen radicals is the Barton reaction (Scheme 49).132 Cleavage of the N-O bond of nitrite ester 278 under photochemical conditions generates oxygen radical 280 and a nitrosyl radical. The oxygen radical abstracts a hydrogen atom from the δ–C-H bond in 1,5 fashion. The resulting alkyl radical 281 combines with the nitrosyl radical, which after subsequent tautomerization and acetoxylation of 282 affords oxime 279 as the final product. Other examples with an oxygen-heteroatom bond as an oxygen radical precursor are illustrated in Scheme 50. Walling and Padwa demonstrated that with an oxygen-halogen bond, chlorination of the C-H bond can be accomplished.133 Similarly, Cekovic achieved the formation of a C-S bond using a O-S precursor.134 In these examples, the mechanisms are similar and a resulting C-X bond is formed at the C-H bond δ to the directing oxygen radical.
Scheme 49.
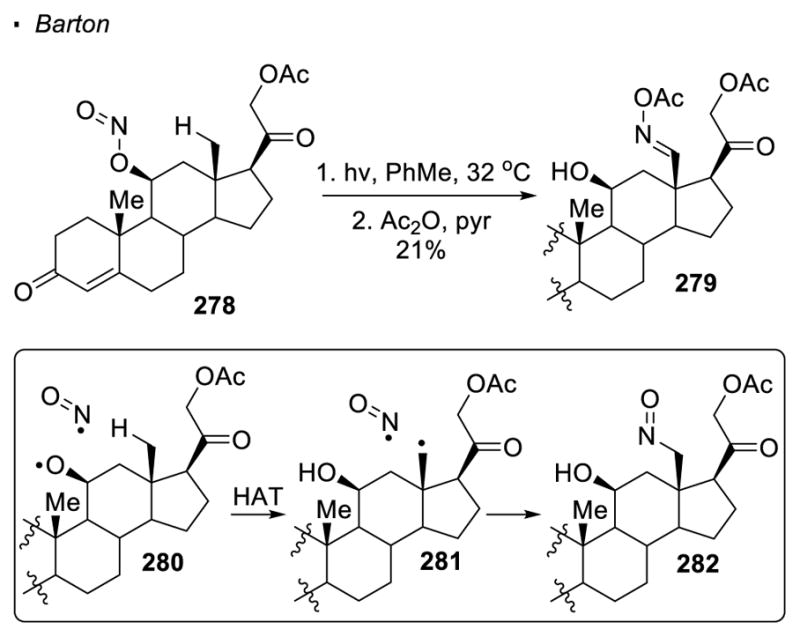
The Barton Reaction
Scheme 50.
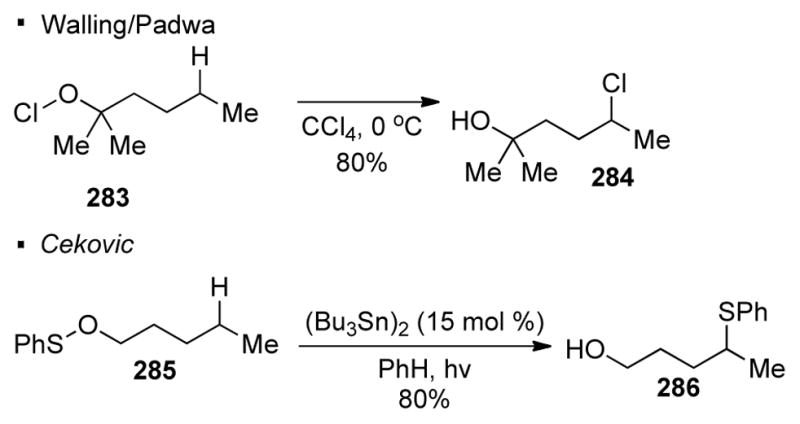
Use of O-X Bonds for C-X Bonds Formation
When the oxygen radical is formed through homolytic cleavage of an O-X bond, there is limited success in trapping the alkyl radicals with an external partner (Scheme 51). Cekovic observed that when a large excess of an electrophilic alkene is employed, alkylation of the alkyl radical can out-compete the trapping of the X radical formed during homolysis of the O-X bond. 135 When 80 equiv. of acrylonitrile 288 is used in the photochemical decomposition of nitrile ester 287, trapping of the alkyl radical δ to the oxygen by acrylonitrile can be accomplished. The resulting radical 290 combines with the nitrosyl radical to afford intermediate 291, which tautomerizes to form oxime 289. Similarly, the alkyl radical generated using O-S bond as the oxygen radical precursor can be trapped by ethyl acrylate 54. In these cases, the alkyl radical α to the carboxylate abstracts a hydrogen atom from Bu3SnH. The tributyltin radical formed is believed to scavenge any sulfur radical or abstract a sulfur atom from an O-S bond.
Scheme 51.
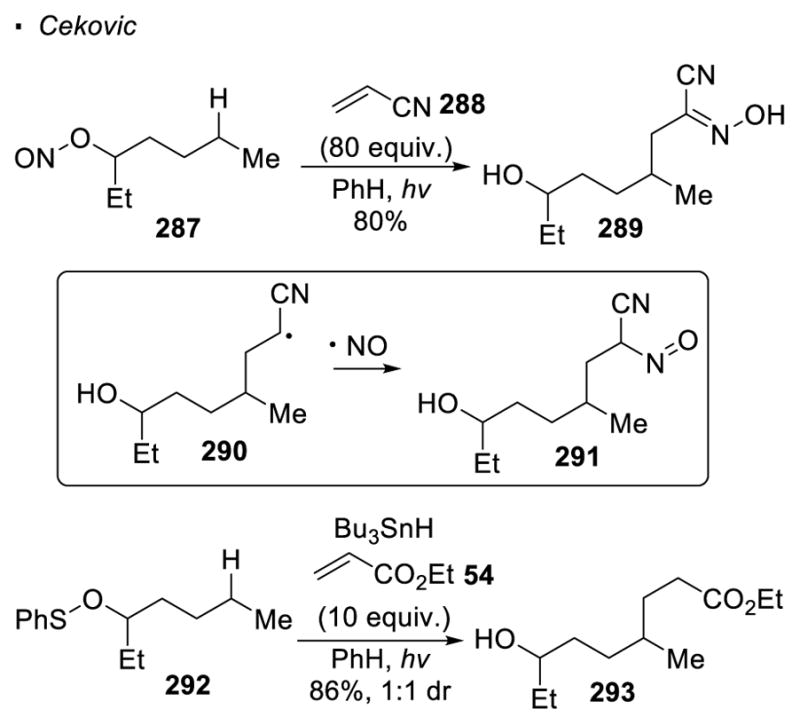
Use of Excess Alkenes for C-H Alkylation
(II) Reduction of O-X Bonds
A strategy to avoid simultaneous formation of an oxygen radical and another radical from an O-X bond is the use of a stoichiometric reductant. As reported by Cekovic, when alkyl hydroperoxide 294 is treated with a stoichiometric amount of FeSO4, the O-O bond is reduced to give the oxygen radical without the concomitant formation of a hydroxyl radical (Scheme 52).136 The alkyl radical formed is thus free to react with a copper(II) salt. With the appropriate choice of the copper(II) salt, thiocyanation, azidation and halogenation of the C-H bond can be accomplished.
Scheme 52.
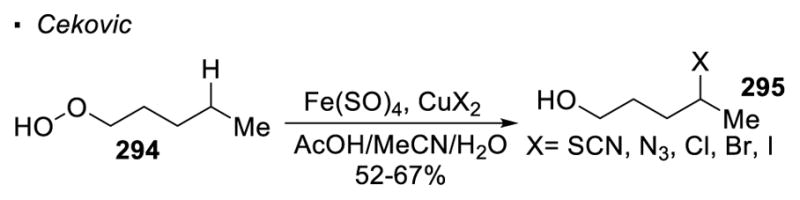
Generation of Oxygen Radicals by Reduction of O-X Bonds
The most recent research incorporates photoredox catalysis such that an oxygen-heteroatom bond is cleaved by reduction to generate the oxygen radical. This strategy wisely avoids the formation of another radical such that the alkyl radical from 1,5 HAT is free to react with a radical partner (Scheme 53). In Chen’s system, the Hantzsch ester 298 (HE) is oxidized by the excited photocatalyst Ir(ppy)3 to generate radical cation 300.137 The photocatalyst then reduces the phthalimide in 296 to form radical anion 301, which can undergo proton transfer with 300 and decompose to an oxygen radical. 1,5 HAT generates an alkyl radical which is then coupled with electrophilic alkene 297. Elimination of the sulfonyl radical affords alkylated product 299.
Scheme 53.
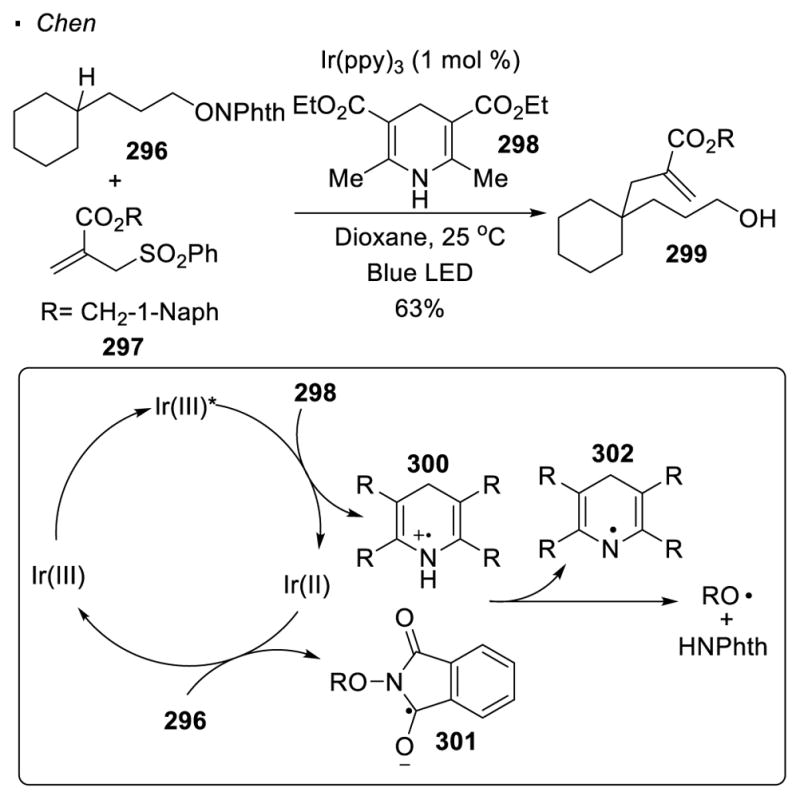
Generation of Oxygen Radicals with Photoredox Catalysis
It can be concluded from Cekovic’s and Chen’s work that when the oxygen radical is generated through single-electron reduction of an O-X bond, trapping of the alkyl radical formed from 1,5 HAT with an external partner is a possibility, providing a strategy to form various bonds at the C-H bond with the use of different reaction partners. However, some O-X bonds are unstable to handle and access to these are generally non-trivial.
(III) Free Alcohols as Precursors
An O-H bond is another common class of oxygen radical precursors. Pb(OAc)4 is shown to be a competent oxidant for the generation of an oxygen radical in this regard, allowing the formation of ether 304 from alcohol 303 (Scheme 54).138 Ligand exchange between one of the acetates and the alcohol provides lead(IV) alkoxide species 305. It is proposed that homolytic cleavage of the lead-oxygen bond gives oxygen radical 306 and lead(III) acetate. After 1,5 HAT, the alkyl radical 307 generated is intercepted by the lead(III) acetate to form Pb-alkyl species 308. Heterolytic cleavage of the Pb-C bond yields carbocation 309, which is prone to cyclization to afford tetrahydrofuran 304. Alternatively, Pd-alkyl intermediate 308 can undergo an intramolecular ligand transfer reaction to give 304 without the involvement of carbocation 309.
Scheme 54.
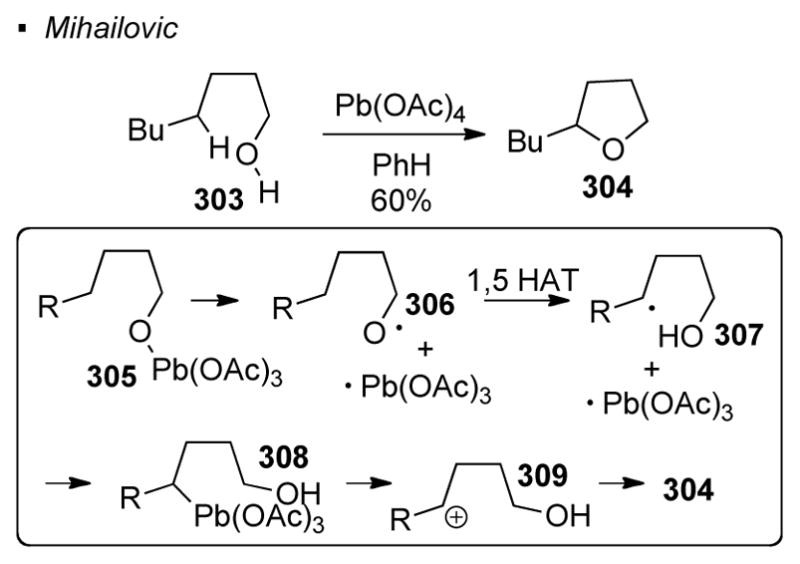
Generation of Oxygen Radicals by Oxidation of O-H Bonds
An alternative method to generate an oxygen radical from a O-H bond is to generate an oxygen-halogen bond in-situ followed by its homolytic cleavage. For instance, with the conditions reported by Suarez (I2 and PhI(OAc)2), tetrahydrofuran 310 can be formed from steroid 311 (Scheme 55). 139 The mechanism is analogous to the HLF reaction discussed previously.
Scheme 55.
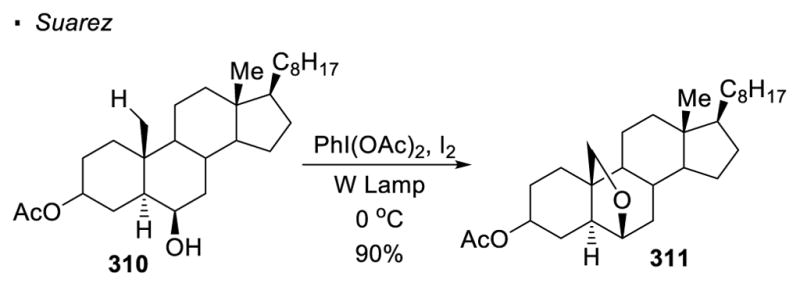
In-Situ Generation of O-I Bonds
The transfer of the hydrogen atom from the O-H bond is another means to generate an oxygen radical. In Chiba’s example where TEMPO is used to generate an oxygen radical through hydrogen atom transfer from the O-H bond of an oxime, intramolecular oxygenation of the C-H bond is observed (Scheme 56).123 In this system, TEMPO abstracts a hydrogen atom from oxime 312 to give oxygen radical 314. The alkyl radical 315 formed by 1,5 HAT is trapped by TEMPO to generate intermediate 316. Elimination of TEMPO-H gives intermediate 317, which cyclizes to afford dihydroisoxazole 313 through either an ionic or radical mechanism. In the radical pathway, TEMPO abstracts a hydrogen atom from oxime 317. The resulting oxygen radical 318 effects cyclization to give carbon-center radical 319, which abstract a hydrogen atom from TEMPO-H to afford 313.
Scheme 56.

Formation of Isoxazolines
In almost all reactions in which an O-H bond is used to generate an oxygen radical, intramolecular C-O oxygenation occurs to give a 5-membered oxygen heterocycle. The only exception is reported by Ryu and Sonoda, where a high pressure of CO is applied under the reaction conditions such that alkyl radical 322 undergoes carbonylation before it is oxidized. The oxidation yields acylium ion 324 and affords γ-lactone 321 after cyclization (Scheme 57).140
Scheme 57.
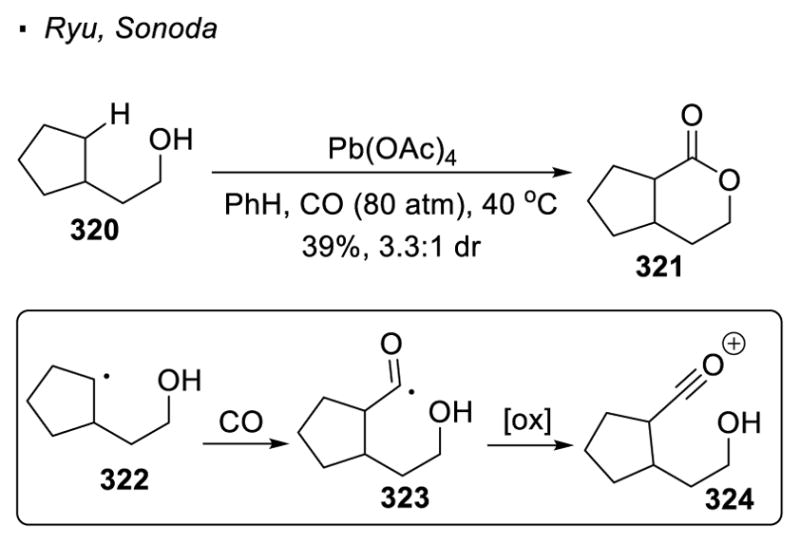
Formation of γ-lactones through Carbonylation
(IV) Cleavage of Epoxides
Epoxides are also sometimes used to generate oxygen radicals. Rawal 141 and Kim 142 independently disclosed the functionalization of sp3 C-H bonds with oxygen radicals formed from epoxides (Scheme 58). In both reports, a radical that is α to the epoxide is first generated. Subsequent homolytic cleavage of the C-O bond is driven by the ring strain of the epoxide and gives the oxygen radical. 143 In Rawal’s work, Barton-McCombie deoxygenation is applied to epoxide 325 to generate the alkyl radical α to the epoxide (327). For Kim, such an intermediate is generated by the addition of the tributyltin radical to the alkene in 330. The alkyl radicals generated from 1,5 HAT in these two cases are trapped by the tethered alkenes that are formed during the reaction to afford cyclized products 326 and 331 respectively.
Scheme 58.
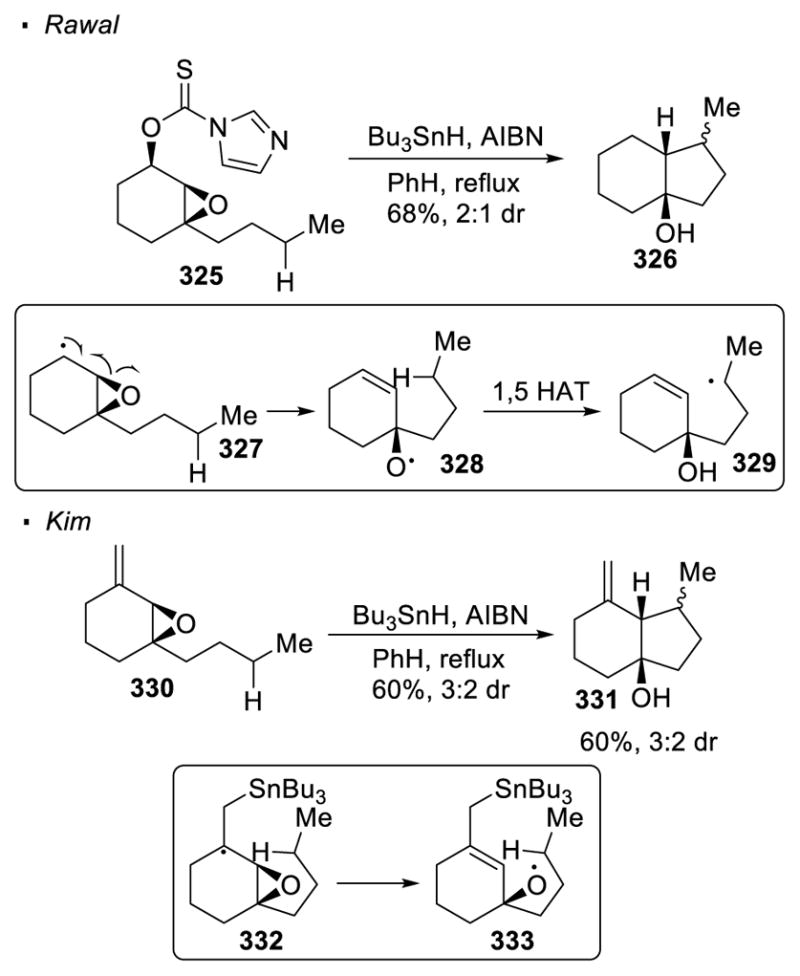
Epoxides as Oxygen Radical Precursors
(V) Electronic Excitation of Carbonyl Groups
The final class of oxygen radical precursors are carbonyl groups. When a carbonyl group is illuminated with a UV light, an electron is excited from the n orbital of the oxygen atom to the π* of the carbonyl group (Scheme 59). This results in the formation of a diradical species, with both the carbonyl carbon and oxygen possessing radical character. The carbonyl oxygen radical is able to abstract a hydrogen atom through 1,5 HAT. The resulting alkyl radical re-combines with the carbonyl carbon radical, leading to the formation of cyclobutanol. This is typically known as Norrish type II reactivity or Norrish-Yang cyclization.144 The formation of cyclobutanol 337 from ketone 334 is an example illustrating this reactivity. To date, no successful attempts to trap the alkyl radical with a species other than the carbonyl carbon radical have been reported. Therefore, a cyclobutanol is always the product obtained when a carbonyl group is used to form an oxygen radical for intramolecular sp3 C-H functionalization.
Scheme 59.
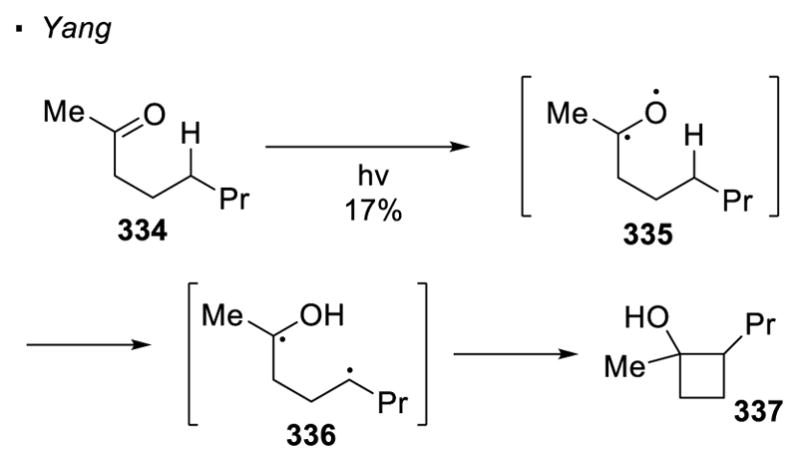
Norrish-Yang Cyclization
3.3 Vinyl and Aryl Radicals
Vinyl and Aryl radicals are the final class of radicals that can cleave unactivated sp3 C-H bonds through hydrogen atom transfer. The higher bond strength of a C(sp2)-H bond (113 kcal/mol) compared to a C(sp3)-H bond (95–105 kcal/mol) provides a thermodynamic driving force for the process.
3.3.1 Generation and Transformations
(I) Reduction of Aryl or Vinyl Halides
Generally, a vinyl or aryl radical is generated through the reduction of the corresponding halide with Bu3SnH under photochemical or thermal conditions. The reactive tributyltin radical cleaves the carbon-halogen through abstraction of the halogen atom. Sometimes AIBN is added to facilitate the formation of the tributyltin radical. The vinyl or aryl halide transfers the halogen to the resulting tributyltin radical to form a vinyl or halogen radical. Parsons first demonstrated that vinyl radicals generated from vinyl halides and tributyltin hydride can be used to functionalize sp3 C-H bonds (Scheme 60).145 In this case, an allylic C-H bond in 340 is cleaved in a 1,5 fashion to give intermediate 341. The allylic radical cyclizes onto the pendent alkene to afford the alkyl radical 342, which is reduced by Bu3SnH to give the final product 339.
Scheme 60.
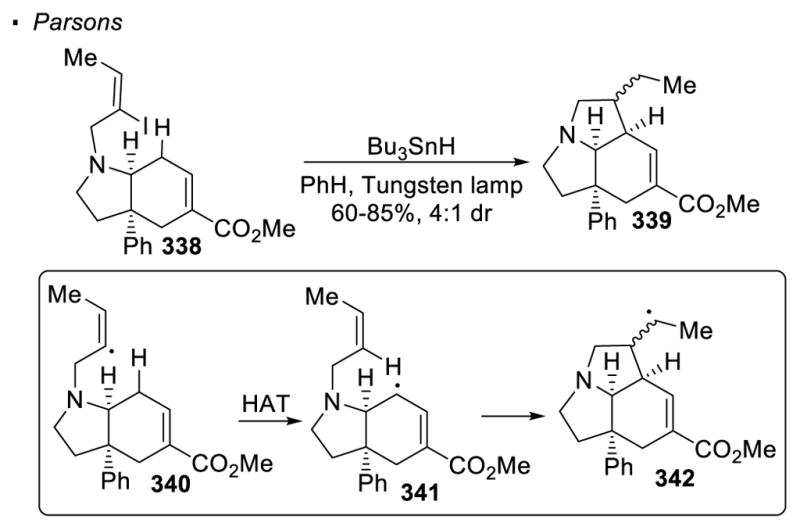
Initial report on 1,5 HAT with Vinyl Halides
Later, Curran improved this system by using a catalytic amount of Bu3SnCl and a stoichiometric amount of NaBH3(CN) in tBuOH (Scheme 61). 146 This system provides a catalytic amount of Bu3SnH under the reaction conditions and is particularly beneficial for the functionalization of unactivated sp3 C-H bonds. Since hydrogen atom transfer from unactivated sp3 C-H bonds to a vinyl radical is relatively slow, the reduction of the vinyl radical by Bu3SnH becomes a competitive process. Therefore, a low concentration of Bu3SnH is sometimes required to avoid the net reduction of the vinyl halide.
Scheme 61.
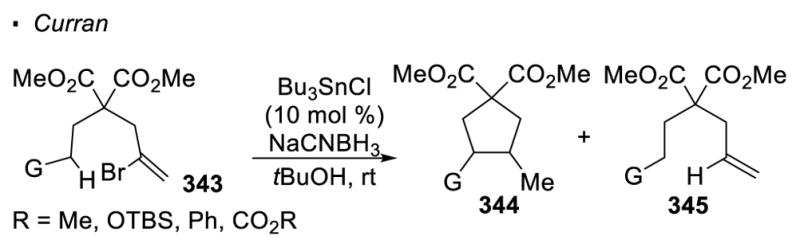
Catalytic System for 1,5 HAT with sp3 C-H Bonds
An alcohol directed sp3 C-H functionalization using aryl radicals is reported by Curran (Scheme 62).147 In this case, an aryl bromide is installed onto the alcohol through the formation of an aryl ether. In the presence of Bu3SnH and AIBN under thermal conditions, aryl bromide 346 decomposes to give an aryl radical that can abstract a hydrogen atom from a C-H bond β to the oxygen atom. The resulting alkyl radical is trapped by a tethered alkene. Hydrogen atom transfer from Bu3SnH to the final alkyl radical intermediate affords cyclized product 347. This transformation serves as a strategy to derivatize an alcohol with sp3 C-H functionalization.
Scheme 62.
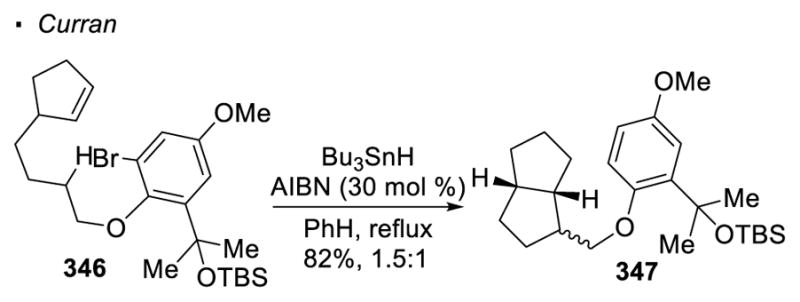
Derivatization of Alcohols with Vinyl Radicals for 1,5 HAT
(II) Radical Addition to Alkynes
An alternative method to generate a vinyl radical is through the radical addition to an alkyne. For the conversion of alkyne 348 to cyclopentane 349, Malacria showed that the addition of an α-silyl radical to an alkyne generates vinyl radical 350 (Scheme 63).148 This vinyl radical can undergo 1,5 HAT to give primary alkyl radical 351. As in Curran’s reaction, radical cyclization onto the alkene takes place to afford the cyclized product 349. Malacria also demonstrated that tetracyclic framework 354 can be accessed from acyclic precursor 353and acrylonitrile 288.149 The alkyl radical in intermediate 355 adds to the alkyne to give vinyl radical 356. The alkyl radical generated from 1,5 HAT with this resulting vinyl radical is located at the position β to the sulfonyl group. Elimination then takes place to give the alkene in the observed tetracyclic product 354.
Scheme 63.

Generation of Vinyl Radicals through Radical Addition to Alkynes
(III) Reduction of Aryl Triazenes
Reduction of aryl triazenes under acidic conditions is another means to generate aryl radicals. Baran demonstrated that alkene 359 can be obtained from aryl triazene 358 in the presence of TFA and TEMPO (Scheme 64).150 In the proposed mechanism, elimination of HNEt2 from 358 yields diazonium 360, which is reduced by TEMPO to give aryl radical 361. 1,7 HAT gives tertiary radical 362. Subsequent oxidation and deprotonation accounts for the formation of alkene Y.
Scheme 64.
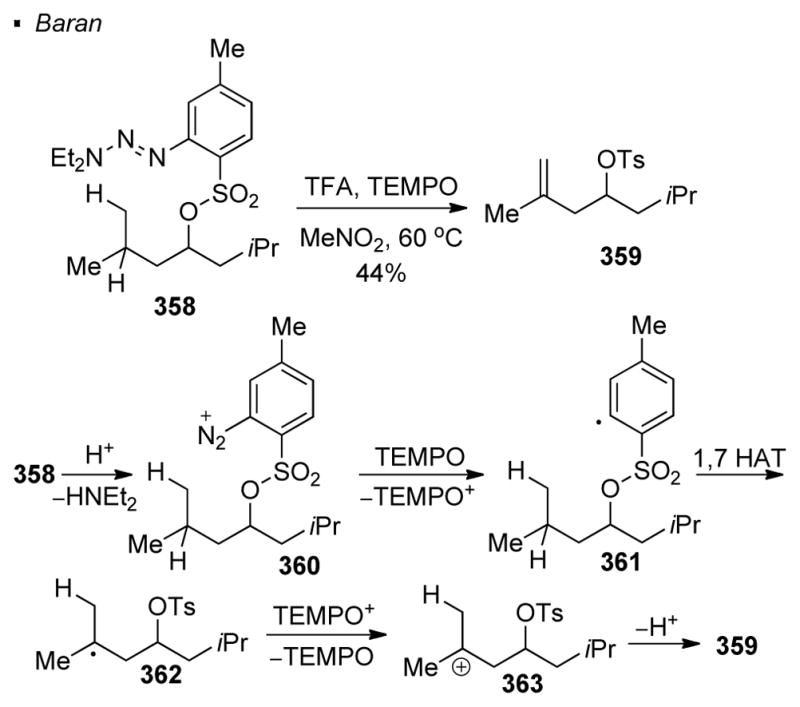
Generation of Aryl Radicals from Aryl Triazenes
From the above representative examples, it can be observed that the alkyl radicals generated from 1,5 HAT with aryl or vinyl radicals thus far only undergo intramolecular events, such as cyclizing onto tethered alkenes or elimination of an α-leaving group, or oxidation to form carbocations.
4. Metal-Catalyzed Carbene/Nitrene Transfer
Carbenes and nitrenes are carbon and nitrogen atoms with only 6 electrons in the outermost electron shell. The lack of an octet configuration renders them highly unstable and able to break unactivated sp3 C-H bonds despite their bond strength. Modern methods rely on transition-metal catalysis such that such reactive species are generated under relatively mild conditions. Along with the interaction between these species and the transition metal catalysts to form metal carbenoids/nitrenoids, control of selectivity can be achieved (Scheme 65). 151 The following discussion only includes intramolecular carbene/nitrene transfer because the functional groups serving as the precursors of these reactive species (usually diazo compounds or sulfonamides/carbamates respectively) can be considered as a directing group. Various reviews on this topic have appeared. 152,153 Therefore, with dirhodium(II) catalysis as an illustration, the following discussion aims to provide readers with a fundamental understanding of intramolecular C-H insertion with carbene transfer and C-H amination with nitrene transfer. Readers who seek a deeper and more thorough insight in this area are encouraged to access these recent reviews.
Scheme 65.
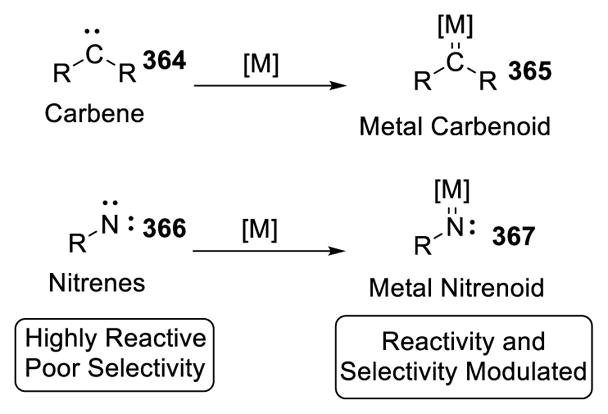
Metal Carbenoids and Nitrenoids
4.1. Carbene Transfer
A diazo functionality is the typical precursor of a transition metal carbenoid. Generally, an electron-withdrawing group, often a carbonyl, is needed to facilitate the installation of the diazo group, and increase the stability and ease of handling of the resulting diazo-containing molecules.154 A dirhodium(II) salt can be used to catalyze the decomposition of the diazo group. The resulting rhodium carbenoid intermediate can insert into an alkyl C-H bond, resulting in the formation of a C-C bond.
The potential of dirhodium catalysts for C-H insertion was first reported by Teyssie (Scheme 66).155 He showed that ethyl diazoacetate 368 is decomposed in the presence of a catalytic amount of dirhodium(II) trifluoroacetate. The resulting rhodium carbenoid undergoes C-H insertion with the solvent, cyclopentane 339. Wenkert first applied this reactivity in the context of an intramolecular reaction.156 In this case, C-C bond formation occurs at the allylic C-H bond of α-diazo ketone 371 with dirhodium acetate as a catalyst to give cyclopentanone 372. Taber found that unactivated sp3 C-H bonds also undergo the reaction, as exemplified by the formation of cyclopentanone 374 from α-diazo-β-keto ester 373 through the transfer of carbene.157 The mechanism of the C-H insertion step is believed to be concerted, yet asynchronous, leaving a partial positive charge at the carbon of the C-H bond.
Scheme 66.
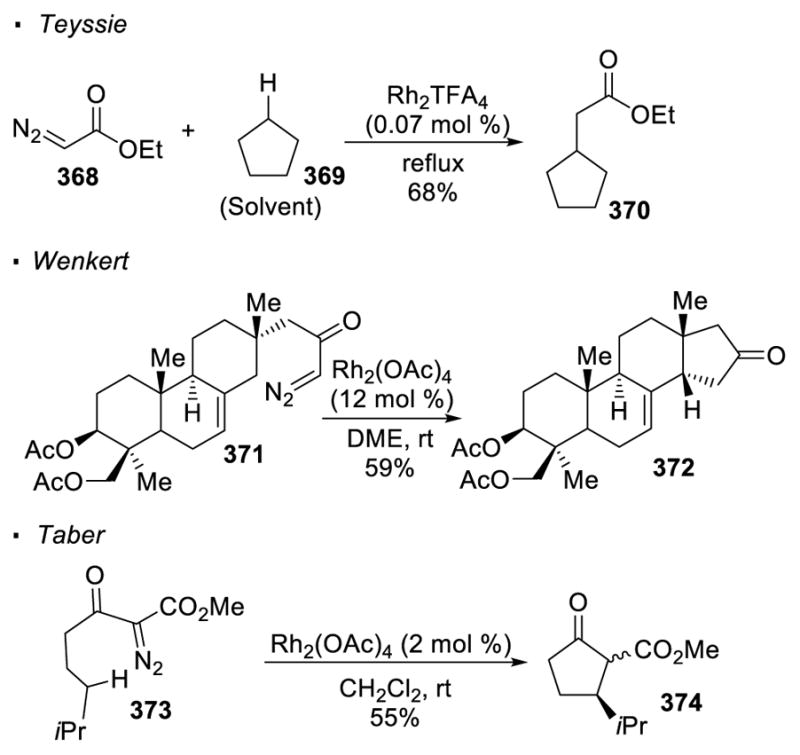
Rh(II)-Catalyzed Intramolecular C-H Insertion
4.2. Nitrene Transfer
Sulfonamides and carbamates are the common precursors to metal nitrenoids. Although intermolecular nitrene transfer was first accomplished with porphyrin-ligated manganese catalysts,158 Du Bois discovered that Rh(II) dimers show obvious advantages in the chemistry of C-H amination (Scheme 67).159 Both carbamate 375 and sulfamate 377 are competent substrates although the two substrate classes offer different regioselectivity. The formation of a five-membered ring is favoured in the former case while a six-membered ring is the predominant product in the latter case. In these reactions, the carbamate or the sulfamate reacts with PhI(OAc)2 in the presence of the base MgO to form iminobenzene 380, which reacts with the rhodium(II) catalyst to form rhodium nitrenoid 381. A plausible explanation in the case of sulfamates is offered by Du Bois, who suggests that the formation of the 5-membered ring product is disfavored due to strain that compresses the N-S-O bond angle. It is proposed that the insertion of the rhodium nitrenoid into the C-H bonds also takes place through a concerted asynchronous mechanism.
Scheme 67.
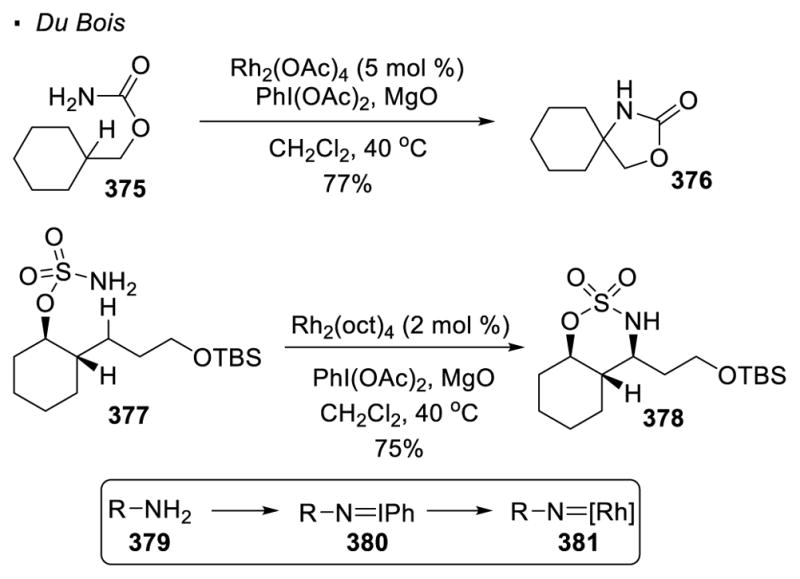
Rh(II)-Catalyzed Intramolecular C-H Amination
4.3. Synthetic Applications
(I) Carbene Transfer
Carbene transfer has been used to facilitate natural product synthesis in the context of 5- and 6-membered rings (Scheme 68). For instance, Taber used Rh(II)-catalyzed C-H insertion as a key reaction to access pentalenolactone E 386.160 With a catalytic amount of dirhodium acetate, α-diazo-β-keto ester 382 undergoes intramolecular C-H insertion to form cyclopentanone 383, affording the tricyclic framework of the natural product. Taber also illustrated the power of carbene transfer in the synthesis of alkaloid 251F 387.161 Cyclopentane 385, which can be further elaborated to the natural product, is generated from an intramolecular C-H insertion reaction of α-diazo ester 384 with a catalytic amount of dirhodium(II) octanoate. It should be noted that Cane accessed the same core of pentalenolactone E 387 with Rh(II)-catalyzed C-H insertion with a different disconnection strategy.162 In this case, the sixmembered lactone 389 is obtained from precursor α-diazoketone 388.
Scheme 68.
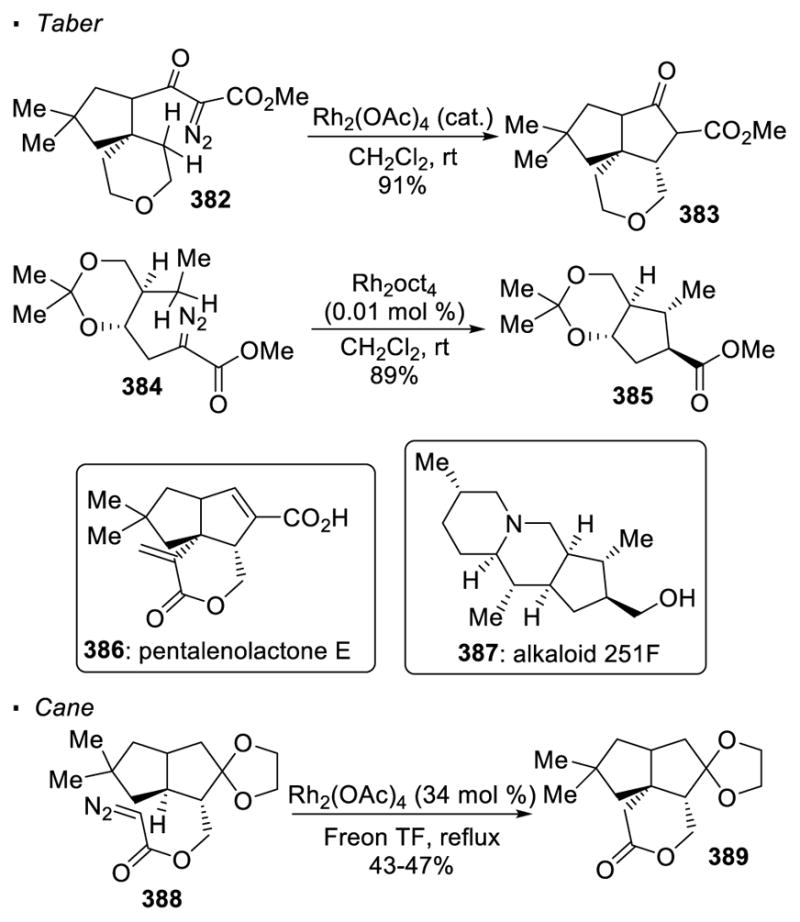
Application of Intramolecular C-H insertion
(II) Nitrene Transfer
Nitrogen is one of the most common heteroatoms present in natural products. Du Bois demonstrated the power of nitrenoid transfer for installing amine functionalities in total synthesis (Scheme 69). In the synthesis of manzacidin A 392, an intramolecular Rh(II)-catalyzed nitrene transfer of sulfamate 390 installs the required amine functionality at a nearby tertiary C-H bond.163 The resulting cyclic sulfamate in 391 can be cleaved to accomplish the synthesis of the natural product. Not only can this C-H amination strategy be applied to simple precursors, molecules of high complexity are also competent substrates. For instance, in the synthesis of (−)-tetrodotoxin 395, intramolecular nitrene transfer of carbamate 393 affords 394 with Rh(II) catalysis. 164 It is noteworthy that other functional groups, including acetals, a primary alkyl chloride and a lactone, are tolerated, and the C-H bond that undergoes amination is very sterically-encumbered.
Scheme 69.

Application of Intramolecular C-H Amination
As exemplified by the above examples, transition-metal catalyzed transfer of carbenes and nitrenes represents a powerful tool in synthesis. An outstanding feature of this strategy is that very sterically-shielded C-H bonds can be functionalized. Tertiary C-H bonds, including those in very hindered positions, can be targeted for reactions to afford quaternary carbons, which are traditionally considered challenging to access. The tolerance of a wide array of functional groups further enhances the utility of this strategy. Carbene or nitrene transfer continues to be a useful method for the construction of difficult targets.
5. Comparison and Complementarity
5.1. Reactivity of Different C-H Bonds
(I) Transition-metal Catalyzed C-H Activation
The thermodynamic stability of the corresponding alkyl metal species is an indicator of the reactivity of different kinds of sp3 C-H bonds to transition-metal catalyzed C-H activation. Primary alkyl metal species are more stable than their secondary counterparts, which are in turn more stable than the tertiary counterparts.165 This trend is illustrated in Scheme 70. The equilibrium positions lie far left on the side of the primary alkyl Pt species 396 and 398, suggesting their higher thermodynamic stability than their secondary or tertiary counterparts.
Scheme 70.
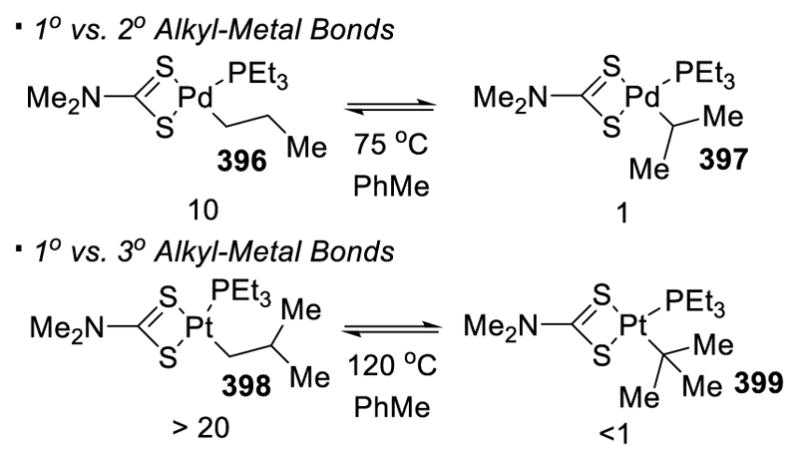
Relative Stability of Pd-Alkyl Species
In parallel to the thermodynamics of the corresponding transition metal alkyl species, activation of secondary C-H bonds is more difficult than primary C-H bonds. This trend is illustrated with a report by Hartwig. In his Ir-catalyzed silylation of C-H bonds of 400, primary C-H bonds are approximately 40 times more reactive than secondary C-H bonds, as reflected by the product distribution of 401 and 402 (Scheme 71).82 However, the activation of secondary C-H bonds with transition-metal catalysis can be achieved, as exemplified by many recent reports.
Scheme 71.
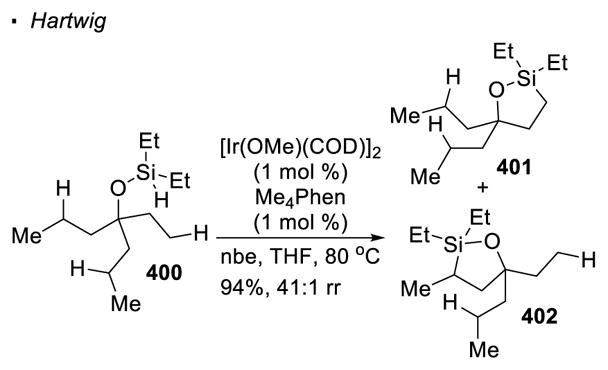
The Relative Reactivity of Primary and Secondary C-H Bonds
In contrast, transition-metal catalyzed activation of tertiary C-H bonds is rare. Dong recently demonstrated Pd-catalyzed acetoxylation of tertiary C-H bonds is possible at the bridgehead of [2.2.1] bicycloheptane (Scheme 72)76, 166. In addition, it is noteworthy that the activation of tertiary C-H bonds of cyclopropanes167 and cyclobutanes168 have been achieved.
Scheme 72.
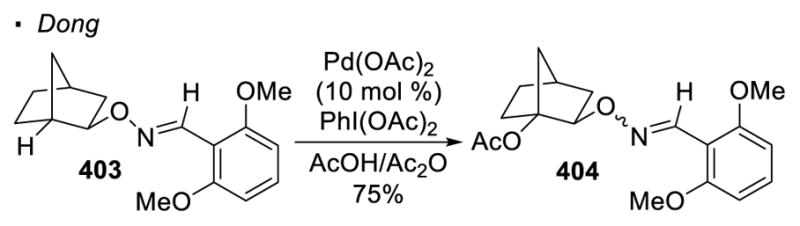
Pd-catalyzed C-H Activation of Tertiary C-H Bonds
(II) Hydrogen Atom Transfer
Hydrogen atom transfer represents a complementary approach to transition metal catalysis. Tertiary C-H bonds are the most reactive among all types of unactivated sp3 C-H bonds partly because of their relatively low bond energy. In addition, alkyl radicals are nucleophilic radicals and react well with electron-deficient radical partners.169 The higher electron density of tertiary alkyl radicals accelerates their trapping with electron-deficient radical partners, therefore suppressing undesired side reactions. Secondary and primary C-H bonds and are less reactive than their tertiary counterparts but also can give reasonable yields. Unfortunately, the functionalization of primary C-H bonds with hydrogen atom transfer was not demonstrated in the most recent research with photoredox catalysis.129,130,137
(IIIa) Transfer of Carbenes
For dirhodium(II)-catalyzed carbene transfer, it is generally observed that more electron-rich C-H bonds are more reactive. This difference in reactivity is attributed to a concerted, yet asynchronous, mechanism in which the transition state has a developing positive charge at the carbon atom of the C-H bond. Therefore, tertiary C-H bonds are more reactive than the secondary or primary counterparts. Taber observed this selectivity with dirhodium(II) catalysis (Scheme 73). When α-diazo-β-keto ester 405 is treated with Rh2(OAc)4 catalyst, cyclopentanones 406 and 407 are formed in a regioisomeric ratio of 2.3:1, indicating that the tertiary C-H bonds are approximately 4.6 times more reactive than the secondary C-H bonds.170
Scheme 73.
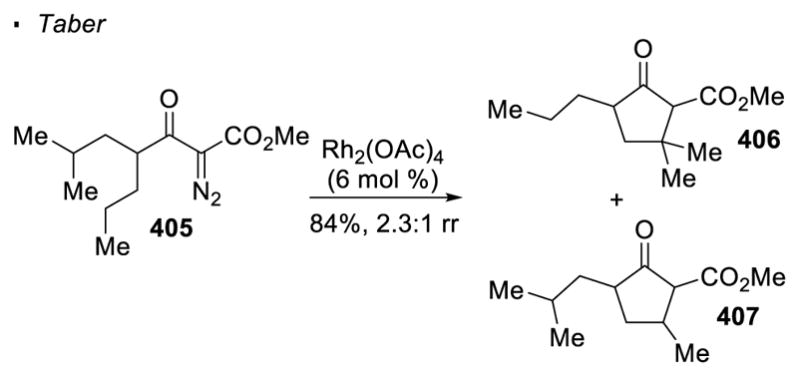
Reactivity of 2° vs 3° C-H Bonds
The electronic differentiation of different C-H bonds sometimes can be enhanced by choice of a different dirhodium(II) catalyst. Scheme 74 shows that when α-diazo-β-keto ester 408 undergoes intramolecular C-H insertion with the Rh2(OAc)4 catalyst, lactone 409 is formed preferentially to lactone 410, reflecting the higher reactivity of more electron-rich tertiary C-H bonds than primary C-H bonds.171 This ratio of 9:1 is improved to >99:1 with dirhodium(II) tetraacetamide, Rh2(acam)4 411, since dirhodium carboxamides are believed to give rise to a tighter transition state.
Scheme 74.
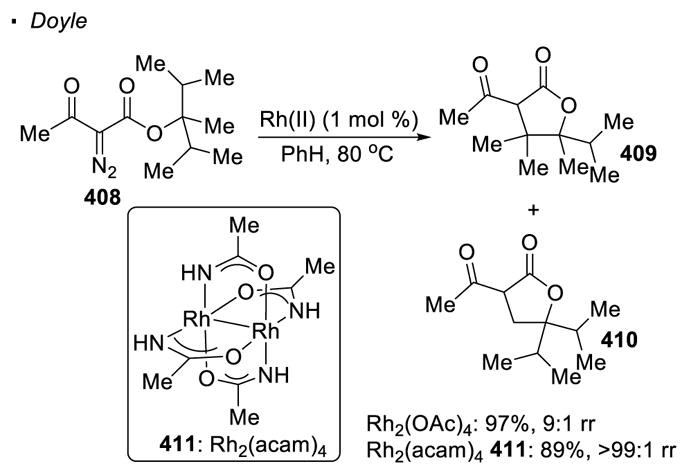
Electronic Effects of Ligands on Regioselectivity
However, sterics sometimes override electronics and becomes the governing factor. With Rh2(OAc)4 as the catalyst, the C-H insertion of α-diazo-β-keto ester 412 gives cyclopentanones 413 and 414 in a ratio of 1:7 (Scheme 75).172 This product distribution follows the trend predicted by the electronics of the C-H bonds since the benzyl group is more capable of stabilising the developing positive charge in the transition state. On the other hand, when sterically bulky dirhodium(II) tetra(triphenylacetate), Rh2(TPA)4 415, is employed as the catalyst, a reversal of regioselectivity is observed and cyclopentanone 413 is formed exclusively. The sterically less hindered primary C-H bonds show a higher reactivity with the use of bulky dirhodium(II) catalyst in this case.
Scheme 75.
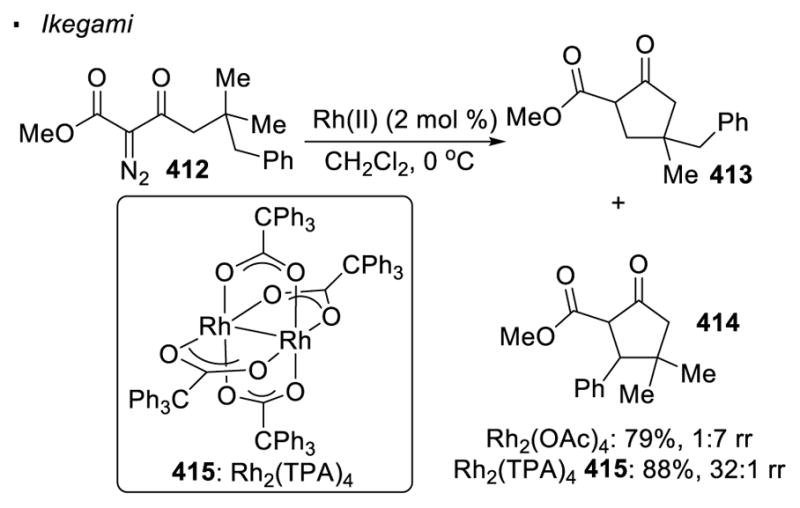
Sterics Effects of Ligands on Regioselectivity
(IIIb) Transfer of Nitrenes
Similarly, a higher reactivity of more electron-rich C-H bonds for metal-catalyzed nitrene transfer is observed. Du Bois found that in the intramolecular C-H bond amination of sulfamate ester 416 with Rh2(OAc)4 as a catalyst, the tertiary C-H bond is 40 times more reactive than the secondary C-H bonds, as reflected by the 20:1 ratio of products 417 and 418 (Scheme 76).173 The same explanation for the higher reactivity of more electron-rich C-H bonds in dirhodium(II) catalyzed C-H amination as in C-H insertion is proposed. The product distribution can be adjusted with the choice of catalysts. With significantly more sterically-demanding Rh2(TPA)4 415, a diminished 4.5:1 ratio of 418 to 418 is obtained.
Scheme 76.
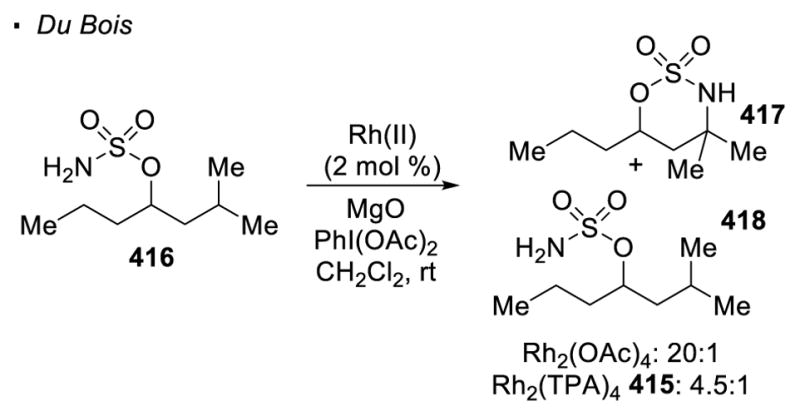
Regioselectivity of C-H Amination
5.2. Position of C-H Bonds Relative to Directing Groups
(I) Transition-Metal Catalyzed C-H Activation
The stability of the ring size of the metallocycles has a significant impact on the site-selectivity of the reactions. Formation of 4-, 5-, and 6-membered ring is possible with 5-membered ring being the most common. The logical extension of the feasibility to form these ring sizes is that C-H bonds β, γ, δ to the directing groups can potentially be activated (Scheme 77).
Scheme 77.
Transition-metal Catalyzed Activation of β, γ, δ C-H Bonds
Sometimes the regioselectivity of transition-metal catalyzed C-H activation is governed by the relative reactivity of different C-H bonds. As discussed earlier, for the reaction of aryl halide 21 with palladium catalysis, the methyl C-H bond is activated through the formation of a 5-membered palladacycle (Scheme 4).15 However, for aryl halide 19, which bears one additional carbon atom, cyclopalladation takes place at the more reactive methyl C-H bond, instead of the methylene C-H bond. In this case, a 6-membered palladacycle is formed preferentially to a 5-membered ring. In addition, for the functionalization of carbonyl groups, β C-H bonds are usually activated.14 However, a γ methyl group can be activated over the less reactive β tertiary C-H bond (Scheme 25).53
(II) Hydrogen Atom Transfer
1,5 HAT is almost always the predominant pathway due to a favourable 6-membered transition state. 1,4 HAT has never been reported to be synthetically useful for the functionalization of unactivated sp3 C-H bonds. Products from 1,6 HAT are sometimes observed in small amounts in conjunction with the 1,5 HAT products. For instance, Mihailovic observed that for the formation of cyclic ethers with oxygen radicals, the ratio of 1,5 HAT to 1,6 HAT is around 12:1 (Scheme 78).174 Long-range HAT is accompanied by a high entropy cost and is uncommon. A successful example is reported by Baran for the desaturation of aliphatic compounds (Scheme 64).150 Another example is reported by Nishio (Scheme 79) for the synthesis of amide 423 from ketone 422.175 Upon irradiation with UV light, diradical 424 is formed. The oxygen radical abstracts a hydrogen atom from the methyl C-H bond through 1,8 HAT, leading to the formation of alkyl 425, which cyclizes to give 426. Amide 423 is formed via either intermediate 427 or 428. The key to the success is the lack of hydrogen atoms for 1,5, 1,6 and 1,7 HAT and the geometry constraint imposed by the sp2 hybridized atoms which significantly reduces the entropy cost for 1,8 HAT. Breslow also investigated long-range HAT for the C-H functionalization of long hydrocarbon chains 176 and complex steroid molecules 177 (Scheme 80). In these reactions, the geometry constraint of the molecules (429 and 432) directs the oxygen radical to specific C-H bonds, giving rise to the observed selectivity. Although this work represents a novel concept, it has not been extended to functionalize organic molecules in a general way. Overall, 1,5 HAT is the predominant pathway in the functionalization of unactivated sp3 C-H bonds.
Scheme 78.
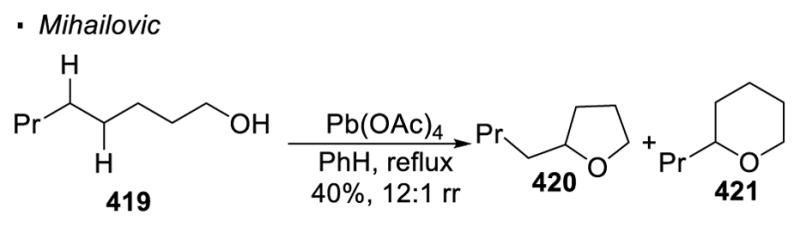
Predominance of 1,5 HAT
Scheme 79.
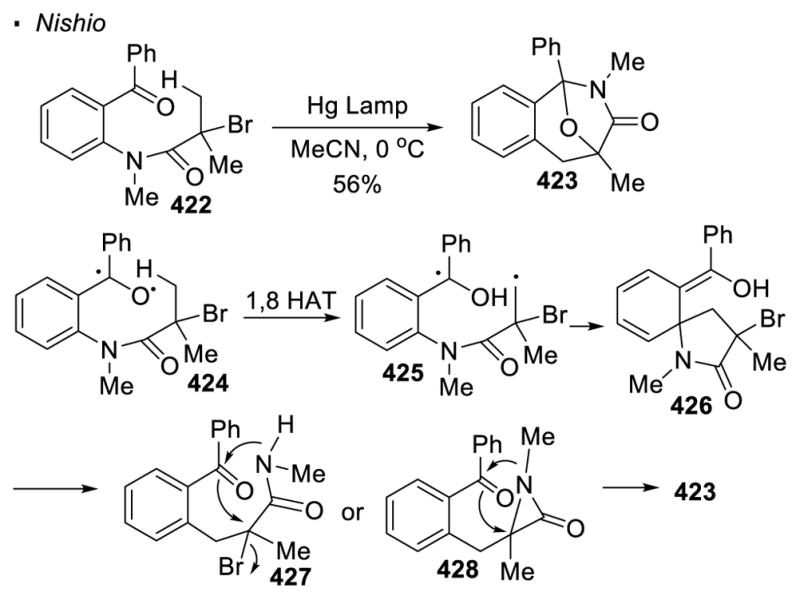
1,8 Hydrogen Atom Transfer
Scheme 80.
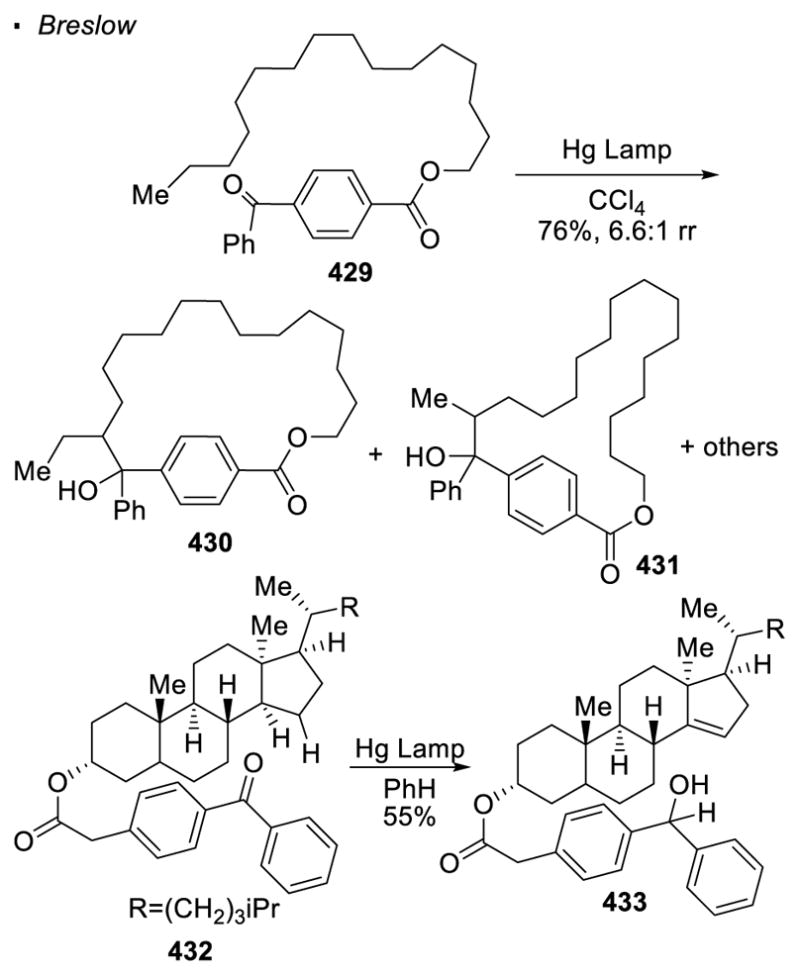
Long-Range HAT
The ratio of 1,5 HAT to 1,6 HAT can be adjusted by the geometry of the substrates although this strategy lacks generality. Wille observed that the vinyl radical generated by nitrate radical addition to cyclooctyne 434 could undergo competitive 1,5 and 1,6 HAT (Scheme 81).178 The resulting alkyl radicals 436 and 438 can add to the alkene to give either [5.3.0]-bicyclodecane 437 or [4.4.0]-bicyclodecane 439. The ratio of the two products reflects a 3:1 ratio of 1,5 HAT and 1,6 HAT. Moreover, Baran reported a HLF reaction with carbamates for the synthesis of 1,3-diols featuring 1,6 HAT (Scheme 82).179 Possibly due to the presence of three sp2 hybridized atoms, the relative energy of 1,5 HAT and 1,6 HAT changes. As reflected by the 1.1 to 1 product ratio of 441 and 442, 1,5 and 1,6 HAT are almost equally favorable. By disfavouring 1,5 HAT with stronger primary or secondary C-H bonds, 1,6 HAT becomes the dominant pathway and provides access to 1,3-diol 444 from 443. Synthetically useful transformations featuring 1,6 HAT are also possible when C-H bonds are not present for 1,5 HAT. For instance, in Penenory’s reaction of aryl iodide 447, the aryl radical 450 can abstract a hydrogen atom through 1,6 HAT to generate primary radical 451 (Scheme 83).180 The alkyl radical adds to the aromatic ring, resulting in arylation of the ε C-H bond. Summarizing from these representative examples, synthetically useful transformations based on 1,6 HAT are possible if geometric or electronic bias is in place to disfavor 1,5 HAT.
Scheme 81.
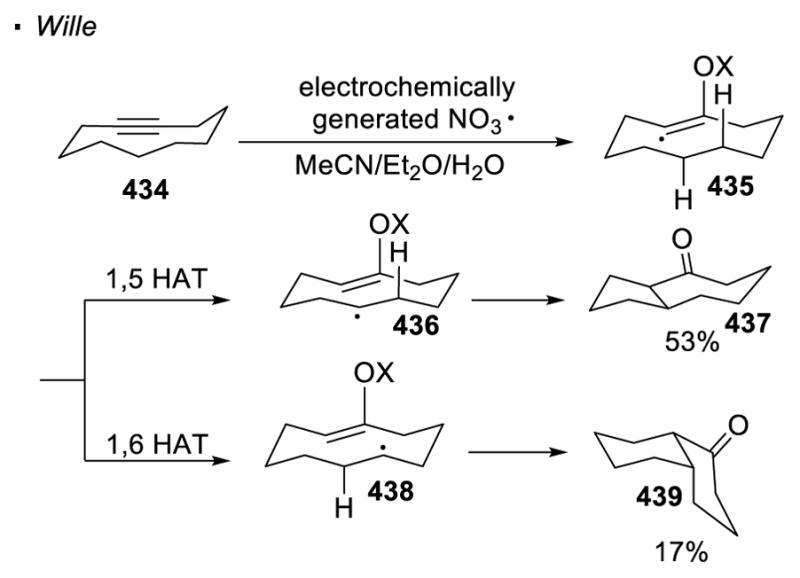
Effects of Geometry on 1,5 and 1,6 HAT
Scheme 82.
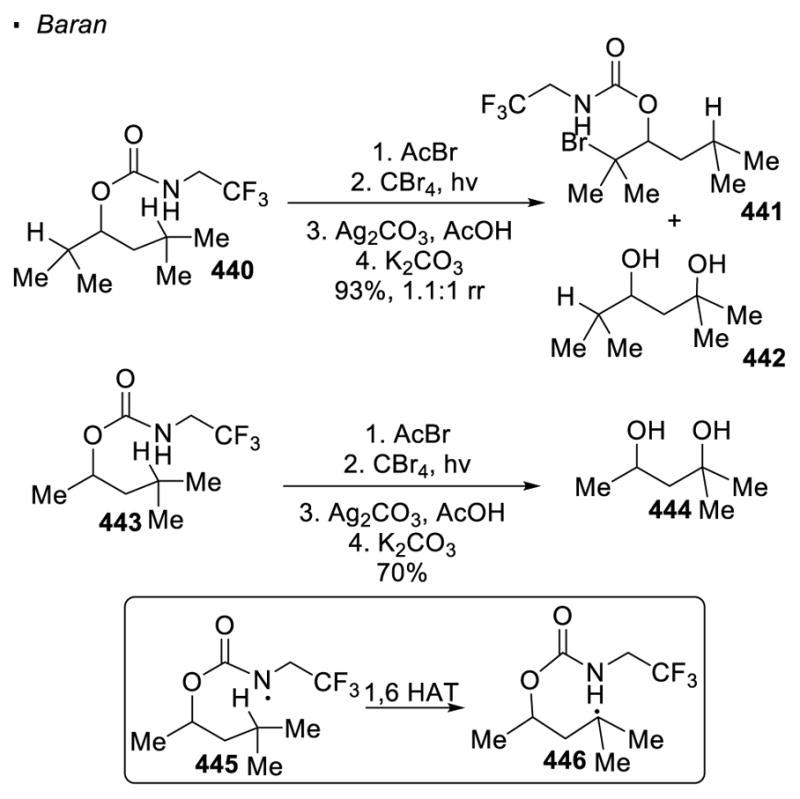
1,6 HAT Favored by Geometry and Bond Strengths
Scheme 83.
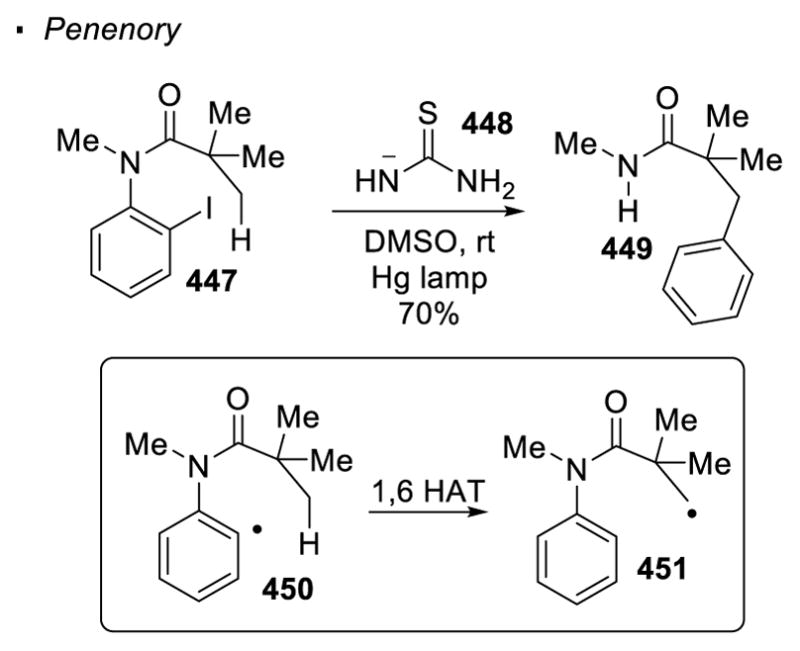
1,6 HAT with no C-H Bonds for 1,5 HAT
Although the predominance of 1,5 HAT and 1,6 might appear as a limitation, it should provide a means for rational design to target a particular C-H bond from the directing group. For instance, in Chiba’s synthesis of oxazolines (Scheme 46), the amine is first converted to an amidine such that the iminyl NH responsible for 1,5 HAT is located at the β-position relative to the original nitrogen.128 Therefore, in contrast to the HLF reaction in which the δ position is preferentially functionalized, the β C-H bond relative to the amine is cleaved and oxygenated.
(IIIa) Transfer of Carbenes
For intramolecular C-H insertion with dirhodium(II) catalysis, cyclopentanes are preferentially formed over cyclobutanes and cyclohexanes while the formation of other ring sizes is uncommon. That is, the C-H bond that is four bonds away from the diazo functionality is usually functionalized. However, the selectivity is complicated by other factors including the electronics/sterics of the competing C-H bonds and the choice of the dirhodium catalyst. This complication can be illustrated by the following example from Ikegami (Scheme 84). α-diazo-β-keto ester 452 undergoes intramolecular C-H insertion with a catalytic amount of Rh2(OAc)4 to give cyclopentanone 453 and cyclobutanone 454 in a ratio of 1:1.7.172 The preference to form a 5-membered ring is diminished because of the higher reactivity of the more electron-rich tertiary C-H bond. With Rh2(acam)4 411 as the catalyst, the electronic effect is reinforced, increasing the selectivity for cyclobutanone 453. On the other hand, with bulky Rh2(TPA)4 catalyst 415, the reaction at the more sterically hindered tertiary C-H bond is completely shut down, resulting in exclusive formation of cyclopentanone 490.
Scheme 84.
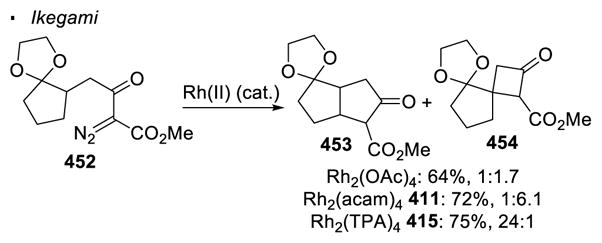
Regioselectivity of C-H Insertion
(IIIb) Transfer of Nitrenes
As discussed earlier, for intramolecular rhodium-catalyzed nitrene transfer, formation of 5-membered and 6-membered rings is the most common (Scheme 67). For carbamates, 5-membered oxazolidinones are the predominant product. Sulfamates and phosphoramidates show complementary selectivity and favour the formation of 6-membered oxathiazines and oxazaphosphinanes.159 The formation of other ring sizes, which involves the functionalization of C-H bonds at other positions, remains rare.
5.3. Stereochemical Control
(I) Transition-Metal Catalyzed C-H Activation
The prospect of controlling absolute stereochemistry in transition-metal catalyzed C-H activation has been demonstrated by various groups. Initial reports focused on the use of aryl halides or triflates as a traceless directing group for the enantioselective formation of 5-membered rings.
Kündig first demonstrated that chiral NHC ligand LG10 on palladium can induce asymmetry for the synthesis of tricyclic 456 from vinyl bromide 455 through the activation of a secondary C-H bond (Scheme 85).181 Cramer later showed that the same transformation can be achieved with chiral phosphine ligand LG11.182 Thus, tricyclic 459 can be obtained with vinyl triflate 457. For the activation of primary C-H bonds, Kagan showed that the use of chiral phosphine Me-DUPHOS L12 allows for the desymmetrization of vinyl bromide 460 to access indoline 461 (Scheme 86). 183 For the synthesis of enantiomerically-enriched dihydroindene 463 from vinyl bromide 462, Clot and Baudoin utilized axially chiral phosphine L13 (Scheme 87).184
Scheme 85.
Asymmetric Activation of Secondary C-H Bonds
Scheme 86.
Asymmetric Activation of Primary C-H Bonds
Scheme 87.
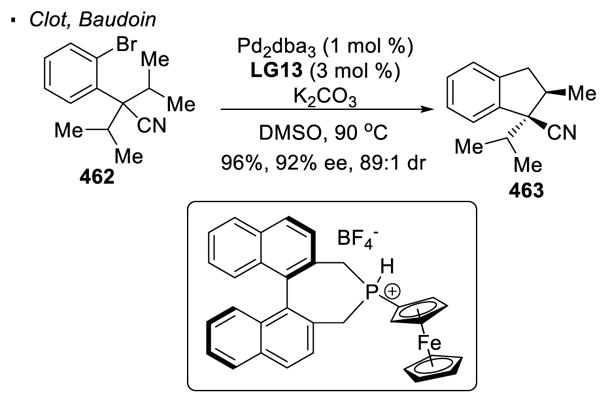
Asymmetric Synthesis of Cyclopentanes
Yu demonstrated enantioselective palladium-catalyzed carbonyl-directed sp3 C-H activation. With a modified amino acid as a chiral ligand on palladium, differentiation of the two pro-stereogenic methyl groups can be accomplished, albeit with moderate stereoselectivity in most cases (Scheme 88).185 For instance, in the γ-arylation of amide 464, the ee obtained was 29% with chiral ligand L14. This initial report confirms that the chiral ligands on transition metals can exert stereochemical control in carbonyl-directed C-H activation. In subsequent work, Yu showed that in the event of arylation of β secondary C-H bonds of carbonyl compounds, a high level of stereochemical control can be obtained by using chiral bidentate acetyl-protected aminoethyl quinoline ligand L15 on palladium.186 Thus, amide 129 can be arylated to access enantiomerically-enriched product 467. In addition, desymmetrization of amide 134 can be achieved with L16 such that the new stereogenic center is established at the α position of the carbonyl group. 187 Enantiomerically-enriched 468 can undergo C-H arylation, alkynylation, alkylation, oxygenation, amination or bromination at the methyl group with another C-H activation reaction, allowing access to products with various functionalities.
Scheme 88.

Asymmetric C-H Functionalization of Carbonyl Compounds
A recent report from Gaunt indicates that desymmetrization of sterically hindered secondary amines can be achieved to form enantiomerically-enriched aziridines with chiral phosphoric acid TRIP as a chiral ligand on the palladium catalyst (Scheme 89).188 For instance, C-H activation of a methyl C-H bond of secondary amine 469 can be accomplished to access aziridine 470.
Scheme 89.
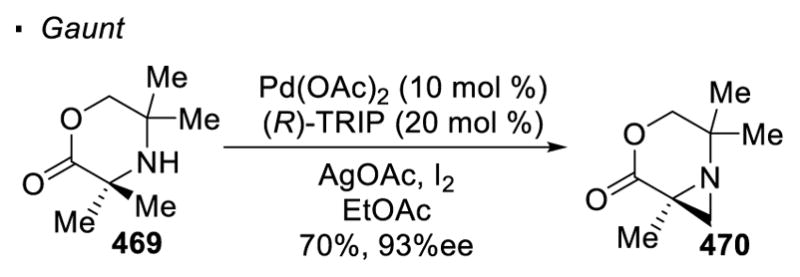
Enantioselective Aziridation
Despite these advances, the stereochemical outcome for the activation of a C-H bond of a tertiary stereogenic center has not been studied due to the challenging formation of tertiary alkyl metal species. However, some alkyl transition metal intermediates are configurationally stable and give enantiomerically enriched products, suggesting the possibility of preserving the stereochemistry of a tertiary stereogenic center in transition-metal catalyzed C-H activation.189
The effect of a pre-existing stereogenic center on the stereochemical outcome of metal-catalyzed sp3 C-H activation has been studied. A high level of diastereomeric control has been observed in some reactions of palladium-catalyzed C-H activation. In the C-H activation of carbonyl groups, the α stereogenic centers are found to exert a large effect on the forming stereogenic center (Scheme 90). For instance, the palladium-catalyzed β-fluorination of amino-acid 471 proceeds with excellent diastereoselectivity to afford 472.65 In addition, a high level of stereochemical control is observed in the γ arylation of amino acids, as exemplified by the functionalization of amide 473 to afford arylated 474.75 To account for the observed diastereomer in this reaction, the cyclic 6-membered alkyl palladium intermediate I1, which places both the NPhth and the methyl groups at equatorial positions, is proposed. The key to success in achieving high diastereoselectivity in transition-metal catalyzed C-H activation is a cyclic intermediate that enforces a conformational bias to favor one of the diastereomers.
Scheme 90.
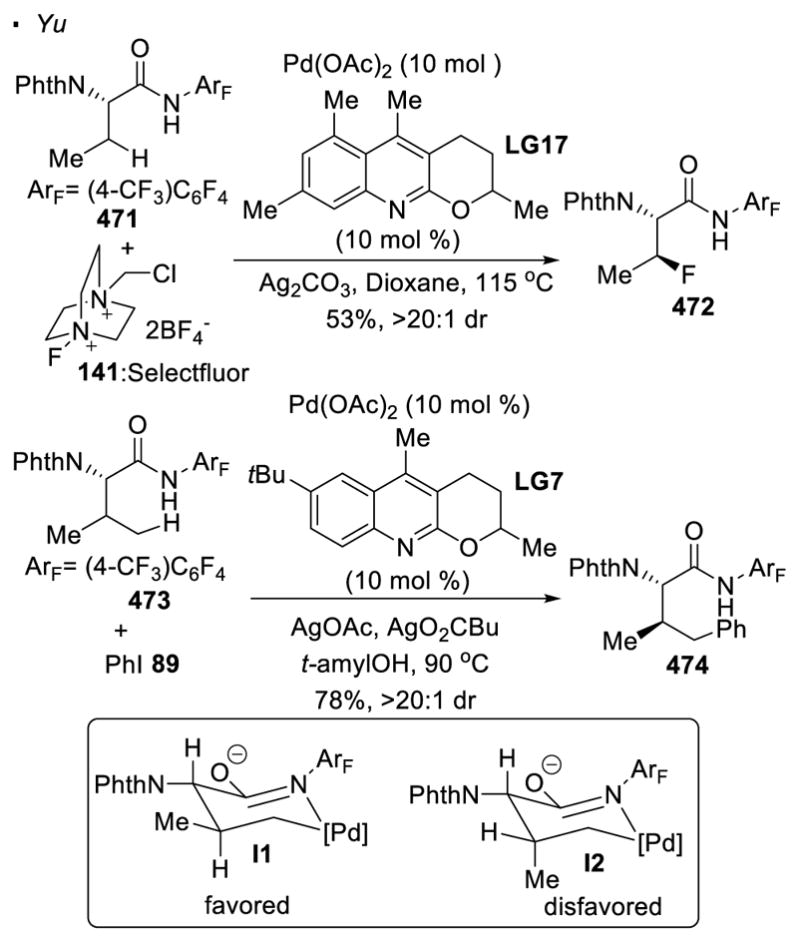
Diastereoselective C-H Activation
(II) Hydrogen Atom Transfer
Asymmetric variants of hydrogen atom transfer transformations have not yet been developed. However, previously-established asymmetric reactions of alkyl radicals generated by other means suggest the possibility of such a control.
One potential strategy to render a reaction with hydrogen atom transfer asymmetric is to employ a chiral auxiliary. The successful application of a chiral auxiliary in the chemistry of intermolecular addition of alkyl radicals was disclosed by Porter, Giese and Lindner (Scheme 91).190 They found that a tert-butyl radical can add to chiral electrophilic alkene 475 with excellent diastereoselectivity. Hydrolysis of the two amides could provide an enantiomerically-enriched compound.
Scheme 91.
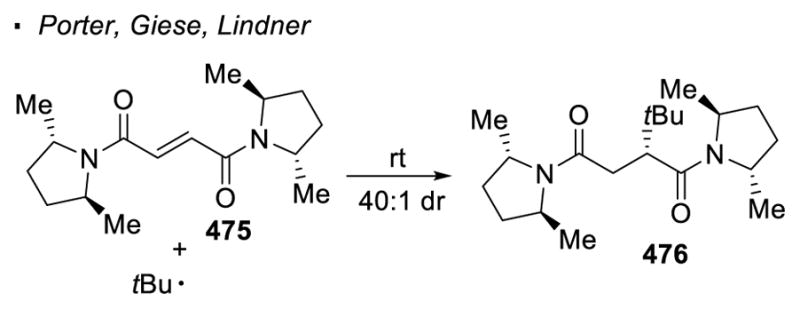
Use of Chiral Auxiliary in Chemistry of Alkyl Radicals
The use of a chiral Lewis acid could potentially serve as an alternative strategy for enantioselective C-H functionalization with hydrogen atom transfer. Since alkyl radicals are nucleophilic, the increase in electrophilicity of the radical partner leads to a higher rate of radical addition.169 This opens the possibility of using a chiral Lewis acid to activate a radical partner. Sibi reported that the addition of isopropyl radical to electrophilic alkene 477 proceeds with an excellent level of stereochemical control in the presence of a chiral bisoxazoline-ligated magnesium catalyst (Scheme 92).191 In addition, tert-butyl radical adds to vinylogous ester 480 with excellent enantio- and diastereoselectivity with a sub-stoichiometric amount of chiral aluminium(III) complex 482.192 The use of a chiral Lewis acid for functionalization of unactivated sp3 C-H bonds has not been explored although Meggers recently applied this concept to functionalize sp3 C-H bonds activated by oxygen (Scheme 93).193 In this case, an oxygen radical is generated from an N-O bond and is used to abstract a hydrogen atom of the C-H bond, as in Chen’s system.137 The addition of the radical to electrophilic alkene 485 is mediated by chiral Rh(III) catalyst 487. It is likely that this strategy can be extended to systems involving unactivated sp3 C-H bonds.
Scheme 92.

Lewis Acid-Catalyzed Asymmetric Addition of Alkyl Radicals
Scheme 93.
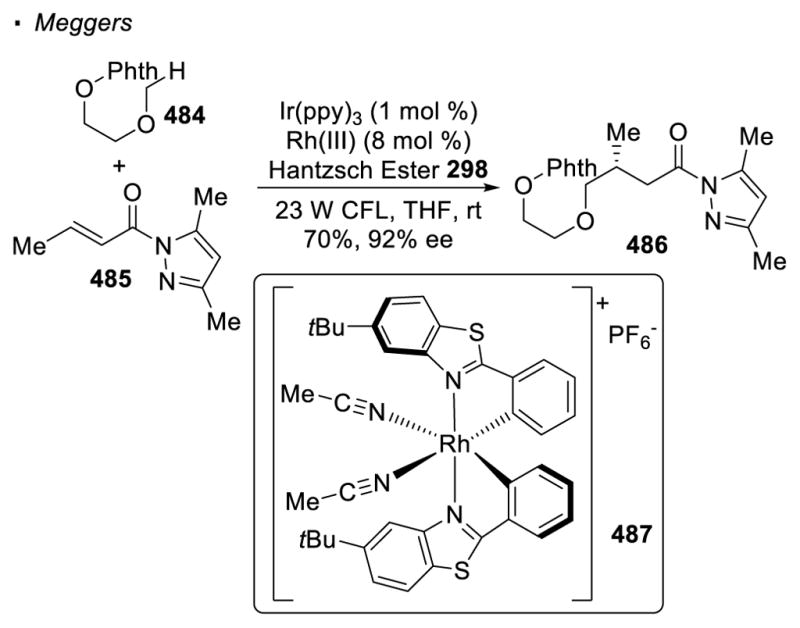
Use of Chiral Lewis Acid in HAT
It should be noted that the newly formed stereogenic centers thus far in the chemistry of alkyl radicals are at the radical trap but not at the carbon atom of the alkyl radical. Transformations that successfully differentiate two prochiral hydrogens or groups in the substrate with alkyl radical remain to be developed.
Consistent with other radical reactions, when a hydrogen atom from a stereogenic center is abstracted, the resulting alkyl radical is not configurationally stable. Consequently, the integrity of the stereochemistry is lost. For instance, when Rovis examined the use of enantiomerically-enriched substrate 488 for nitrogen-directed C-C bond formation at unactivated sp3 C-H bonds, the alkylated product 489 is obtained as a racemic mixture (Scheme 94).129
Scheme 94.
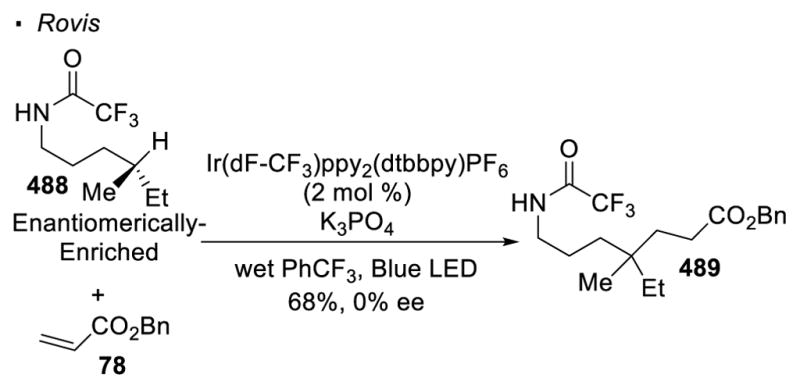
Loss of Stereochemistry in HAT
In addition, a highly diastereoselective reaction with chiral substrates would be non-trivial. The influence of a pre-existing stereogenic center in the substrate is expected to be small on any forming stereogenic center. In contrast to transition metal catalysis that typically involve metalacyclic intermediates, the alkyl radical intermediate, existing in acylic forms, has no conformational constraints. Therefore, the possibility that a pre-existing stereogenic center in the chiral substrate can control the stereochemical outcome of the reaction is slim.
(III) Transfer of Carbenes/Nitrenes
Asymmetric metal-catalyzed carbene transfer is well-established. Doyle developed novel chiral dirhodium catalysts for the enantioselective synthesis of 5-membered lactones through C-H insertion. With Rh2(4S-MPPIM)4 491, γ-lactone 492 can be obtained from acyclic diazo-ester 490 with high enantioselectivity (Scheme 95).194 Doyle also showed that two prochiral alkyl groups can be differentiated with Rh2(4S-MCHIM) 4 494. Thus, desymmetrization of diazo ester 493 provides highly enantiomerically enriched γ-lactone 495.195 The application of asymmetric C-H insertion is demonstrated by Doyle in the synthesis of natural product (+)-isolauricerisinol 498 (Scheme 96).194 Diazo-ester 496 undergoes enantioselective dirhodium-catalyzed intramolecular C-H insertion to yield γ lactone 497, which is further elaborated to afford the natural product. In addition, Taber used chiral dirhodium(II) catalyst 500 to favour the formation of the desired diastereomer 501 through C-H insertion of 499 for the synthesis of (−)-astrogorgiadiol 502.196
Scheme 95.
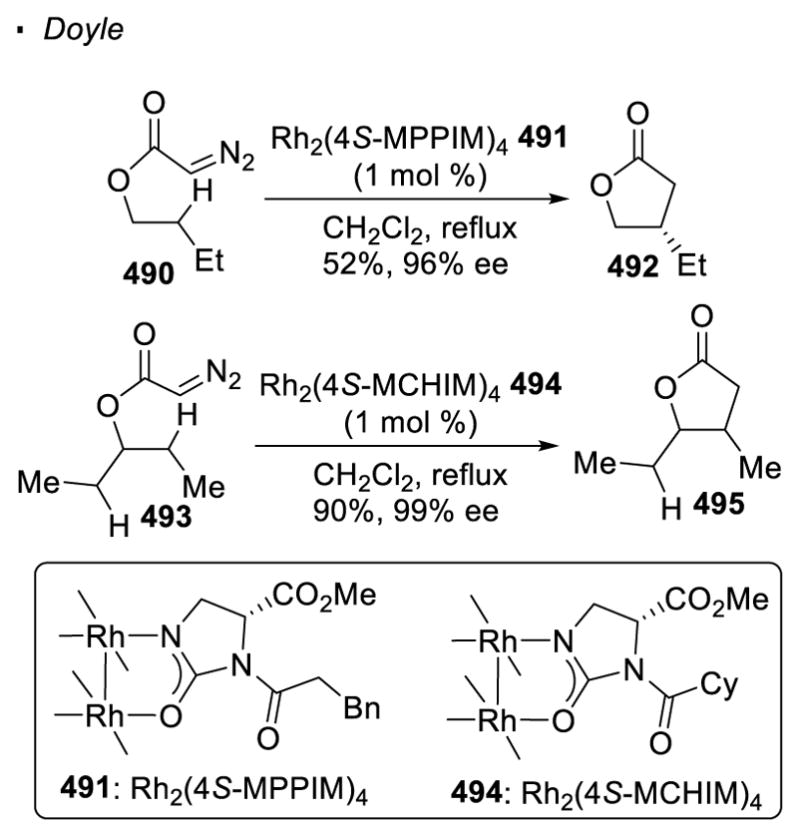
Asymmetric C-H Insertion
Scheme 96.

Applications of Asymmetric C-H Insertion
For enantioselective nitrene transfer, only molecules with activated sp3 C-H bonds (i.e. allylic or benzylic) are competent substrates. This is exemplified by reports from Che,197 Davis198 and Du Bois. 199 Highly enantioselective asymmetric intramolecular nitrene transfer to unactivated sp3 C-H bonds remains to be developed.
Similar to transition-metal catalysis, but in contrast to radical reactions, the cyclic transition state for intramolecular carbene/nitrene transfer provides an opportunity for a pre-existing stereogenic center to communicate effectively with a forming one. Thus, a high level of diastereomeric control is sometimes observed. For example, in the intramolecular C-H insertion of α-diazo ester 503, Taber observed that cyclopentane 504 is obtained with excellent diastereoselectivity (Scheme 97). 200 A simple cyclic 5-membered transition state J1 that places both the phenyl and methyl groups at pseudo-equatorial positions is proposed to account for the observed diastereomer.
Scheme 97.
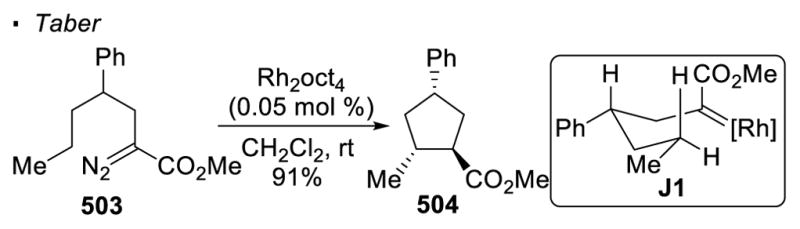
Diastereoselective C-H Insertion
An example of a highly diastereoselective intramolecular nitrene transfer is reported by Du Bois (Scheme 98). The intramolecular C-H amination of sulfamate 505 with Rh2oct4 proceeds with excellent diastereoselectivity. 201 Again, cyclic transition state K1 with all substituents at the pseudo-equatorial positions is invoked. The opposite diastereomer can be obtained when sulfamide 507 is employed as the substrate.202 In this case, the methyl group is placed at the pseudo-axial position to minimize A(1,3) strain with the NBoc group in transition state K2.
Scheme 98.
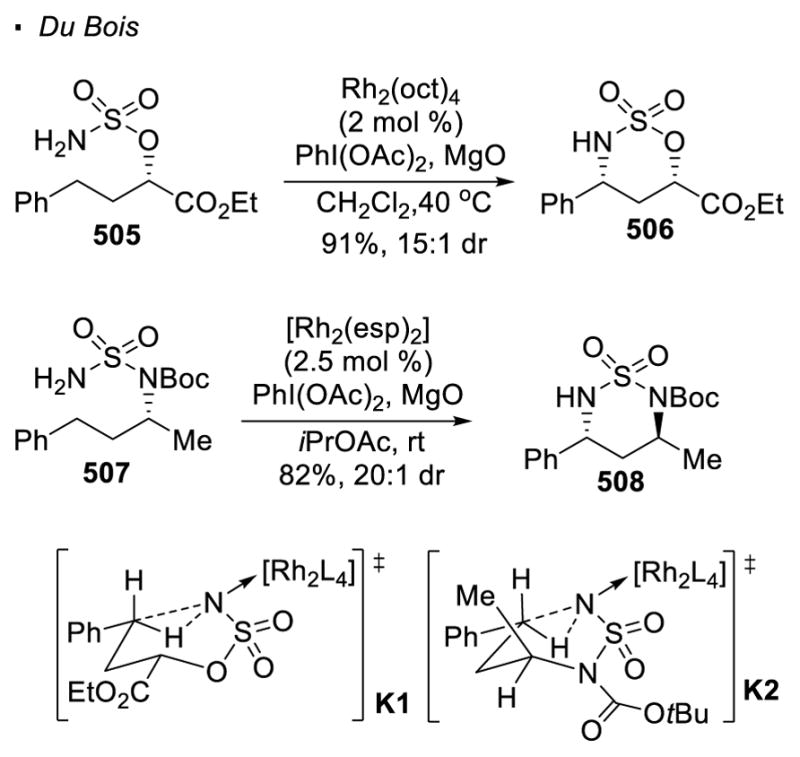
Diastereoselective C-H Amination
With a tertiary stereogenic center, retention of stereochemistry is observed for both dirhodium(II)-catalyzed intramolecular carbene or nitrene transfer (Scheme 99). In Taber’s synthesis of (+)-sulcatine G, a Rh2oct4 catalyzed C-H insertion reaction of enantiomerically enriched α-diazo-β-keto ester 509 gives the desired cyclopentanone 510 with complete retention of stereochemistry.203 Similarly, Du Bois observed that the intramolecular C-H amination of chiral sulfamate ester 512 under dirhodium(II) catalysis proceeds without any sign of racemization in product 513.204 In both dirhodium(II)-catalyzed C-H insertion and amination, the stereochemical outcome can be explained by the aforementioned concerted, yet asynchronous, mechanism.
Scheme 99.
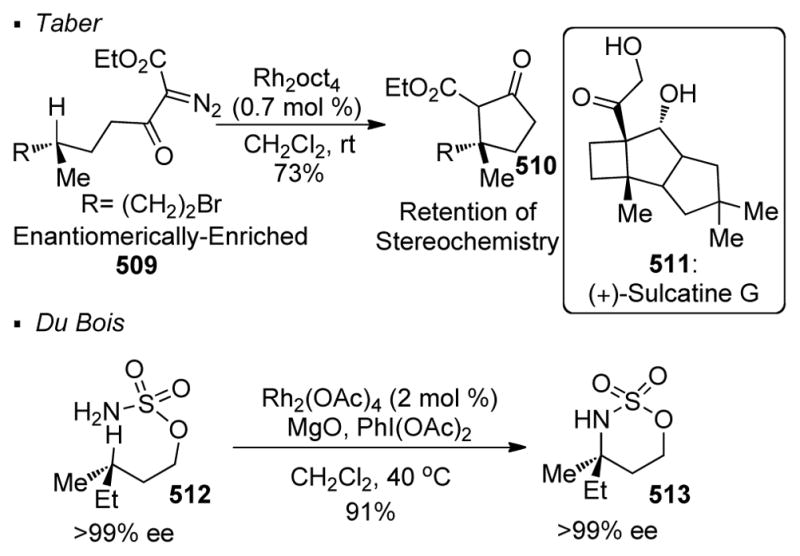
Retention of Stereochemistry in C-H Insertion and Amination
5.4. Scope of Functional Groups that can be Incorporated
The relative breadth of the scope of the three strategies is reflected by the content contribution of this review. 205 As discussed in the following, transition-metal catalyzed C-H activation allows a wide variety of functionalities to be incorporated into sp3 C-H bonds (Scheme 100). For hydrogen atom transfer, the scope is limited by the traditional ways to generate the reactive radicals capable of abstracting hydrogen atoms but the application of photoredox catalysis has shown promising results in extending the scope of this strategy (Scheme 101). On the other hand, for carbene and nitrene transfer, only C-C and C-N bond formation is possible, arguably the two most common and most important types of bonds.
Scheme 100.

Scope of Different Strategies
Scheme 101.
Broadened Scope with Photoredox Catalysis for HAT
(I) Transition-Metal Catalyzed C-H Activation
As illustrated in the representative examples in Section 2.1, transition-metal catalyzed activation provides a means for the formation of various forms at the unactivated sp3 C-H bonds. With the appropriate choice of reaction partners, the formation of C(sp3)-C(sp3), C(sp3)-C(sp2), C(sp3)-C(sp), C(sp3)-O, C(sp3)-N, C(sp3)-F, C(sp3)-S and C(sp3)-B bonds is possible. Currently, transition-metal catalysis demonstrates the broadest scope among all the strategies for C-H functionalization.
(II) Hydrogen Atom Transfer
The strategy of hydrogen atom transfer is traditionally limited in the scope of functional groups that can be incorporated at inert C-H bonds. However, the development of photoredox catalysis has significantly advanced the scope as follows:
For the use of nitrogen radicals, except for one example in which C-H oxygenation is achieved,128 only halogenation or intramolecular amination of unactivated sp3 C-H bonds could be realized with nitrogen radicals before recent reports from Rovis129 and Knowles130 (Scheme 47). The incorporation of other functionalities at C-H bonds is challenging mainly because the trapping of the alkyl radical with a radical couple is often hindered by (1) the presence of other radicals which are formed simultaneously with the nitrogen radicals or (2) the presence of an oxidant that oxidizes the alkyl radical to a carbocation leading to cyclization by the nitrogen. The keys to success of Rovis and Knowles’ photocatalytic systems are (1) the use of an N-H bond as the nitrogen radical precursor that avoids the formation of another radical species that would interfere with the alkyl radical, and (2) the absence of a stoichiometric amount of a strong oxidant that would oxidize the alkyl radical to a carbocation. These features of the reactions allow the alkyl radicals to be trapped by an external electrophilic alkene, allowing for the formation of a carbon-carbon bond.
With oxygen radicals, formation of various bonds at a C-H bond is possible only when the alcohol is pre-activated through the formation of O-O or O-S bonds. This pre-activation step is not trivial and O-O or O-S bonds are difficult to handle, limiting the practicality and feasibility of the use of oxygen radicals. On the other hand, Chen demonstrates that with photoredox catalysis, alkylation of C-H bonds can be accomplished with an easily prepared and stable N-O bond as a precursor of an oxygen radical (Scheme 53).137
Only intramolecular reactions or oxidation of alkyl radicals generated with aryl or vinyl radicals by intermolecular HAT have been demonstrated. Trapping of the alkyl radicals with external reaction partners has not yet been realized. It is not surprising when the method to generate the vinyl or aryl radicals is considered. These radicals are typically generated in the presence of Bu3SnH, which can reduce alkyl radicals. Therefore, the desired reaction between the alkyl radical and an external radical couple must out-compete the reduction of the alkyl radical by Bu3SnH. This criterion might have limited any favorable processes to only intramolecular events. Recently, there appears to be novel protocols to generate aryl or vinyl radicals with photoredox catalysis.206 These methods obviate the use of Bu3SnH and might offer a solution to address the limited scope for sp3 C-H functionalization with aryl radicals.
(III) Transfer of Carbenes/Nitrenes
Metal-catalyzed carbene/nitriene transfer allows intramolecular C-H insertion or amination, resulting in the formation of 5- or 6-membered carbocycles and heterocycles. These products are sometimes hydrolyzable to give acyclic molecules, allowing access to not only cyclic molecules with these C-H functionalization reactions. However, the functionalities that can be incorporated into the substrates are very limited. Only C-C and C-N bond formation can be achieved. This limited scope represents a drawback of metal-catalyzed carbene/nitrene transfer.
6. Summary and Outlook
Each of the three main strategies for the activation of unactivated sp3 bonds, transition-metal catalyzed C-H activation, hydrogen atom transfer and metal-catalyzed transfer of carbenes/nitrenes, has its own strengths and limitations. It is likely that future efforts would be devoted to address the limitations associated with each strategy, adding more tools to the C-H functionalization toolbox.
Transition metal catalyzed C-H activation is a growing research area. Currently, palladium is the most versatile metal catalyst while other noble metals sometimes give novel reactivity that deviates from palladium. Future efforts will certainly be invested to replace these noble metals with earth-abundant metals, as well as to develop new reactivity with earth abundant metals. In addition, the latest research has underscored the possibility of asymmetric catalysis, with more to come. The challenges to activate unactivated tertiary C-H bonds have promising initial results in stoichiometric studies. The development of new directing groups might allow for the activation of the wealth of C-H bonds at positions that are currently not possible.
The research on hydrogen atom transfer has stagnated until the recent development of photoredox catalysis. Initial efforts that incorporated photoredox catalysis into the realm of hydrogen atom transfer have already benefited the functionalization of unactivated sp3 C-H bonds. In particular, this merged strategy has allowed the first C-C bond formation reaction with nitrogen radicals and the use of simple N-H bond as a radical precursor. Photoredox catalysis will probably dominate research in this direction for the foreseeable future and lead to a wider scope of functionalities that can be incorporated into sp3 C-H bonds, obviating/simplifying the pre-functionalization of O-H and N-H for the generation of the radicals. Similar to transition metal catalysis, more enantioselective reactions remain to be developed and the use of new directing groups should lead to functionalization of C-H bonds that cannot be targeted with current methodology.
Current research on the transfer of carbenes/nitrenes in the functionalization of unactivated sp3 C-H bond mainly focuses on intermolecular reactions, 207 the obvious frontier of this chemistry. There have not been many efforts devoted to improving the intramolecular variant, the focus of this review. However, considering the reliability of intramolecular C-H insertion and amination, this strategy would still find applications in the synthesis of complex natural products or other molecules.
While transition-metal catalyzed C-H activation, hydrogen atom transfer and the transfer of carbenes/nitrenes represent three powerful strategies for directed sp3 C-H functionalization, they are distinct in terms of the reactivity of different C-H bonds, positions of reacting C-H bonds relative to the directing groups, stereochemical outcome and the scope of functional groups that can be incorporated. Thus, these strategies are complementary to each other and collaboratively provide powerfule tools in the synthetic arsenal.
Acknowledgments
J. C. K. Chu acknowledges a Croucher Scholarship from the Croucher Foundation (Hong Kong). Our own efforts in this area were supported by the NIGMS (GM80442). We are grateful to Scott M. Thullen (Columbia) for proofreading and revising this review, and to Melissa A. Ashley (Columbia) for creating the cover art.
Biographies

John Chun Kit Chu obtained his BSc in Chemistry at the University of Hong Kong. With the support of a Croucher Scholarship, he pursued his PhD in Chemistry under the supervision of Prof. Tomislav Rovis at Colorado State University. Currently, he is a postdoctoral researcher as a Marie Curie Fellow in the Gaunt Group at the University of Cambridge. His research focuses on the development of novel organic transformations.

Tomislav Rovis was born in Zagreb, Croatia but was raised in Canada. He received his B.Sc. and Ph.D. degrees at the University of Toronto, with Mark Lautens, and then was an NSERC postdoctoral fellow at Harvard with David A. Evans. He began his independent career in 2000 at Colorado State University, was promoted in 2005 and named John K. Stille Chair in 2008. In 2016, he joined Columbia University where he is currently Professor of Chemistry.
Footnotes
Dedicated to Professor Al Padwa on the occasion of his 80th birthday
References
- 1.a) Cernak T, Dykstra KD, Tyagarajan S, Vachael P, Krska SW. Chem Soc Rev. 2016;45:546–576. doi: 10.1039/c5cs00628g. [DOI] [PubMed] [Google Scholar]; Wencel-Delord J, Glorius F. Nature Chem. 2013;5:369–375. doi: 10.1038/nchem.1607. [DOI] [PubMed] [Google Scholar]; b) Chen DYK, Youn SW. Chem Eur J. 2012;18:9452–9474. doi: 10.1002/chem.201201329. [DOI] [PubMed] [Google Scholar]; c) Yamaguchi J, Yamaguchi AD, Itami K. Angew Chem Int Ed. 2012;51:8960–9009. doi: 10.1002/anie.201201666. [DOI] [PubMed] [Google Scholar]; d) Kakiuchi F, Chatani N. Adv Synth Catal. 2003;345:1077–1101. [Google Scholar]
- 2.(a) Siegbahn PEM. J Phys Chem. 1995;99:12723–12729. [Google Scholar]; (b) Martinho Simoes JA, Beauchamp JL. Chem Rev. 1990;90:629–688. [Google Scholar]
- 3.Blanksby SJ, Ellison GB. Acc Chem Res. 2003;36:255–263. doi: 10.1021/ar020230d. [DOI] [PubMed] [Google Scholar]
- 4.a) Cernak T, Dykstra KD, Tyagarajan S, Vachal P, Krska SW. Chem Soc Rev. 2016;45:546–576. doi: 10.1039/c5cs00628g. [DOI] [PubMed] [Google Scholar]; b) Hartwig JF, Larsen MA. ACS Cent Sci. 2016;2:281–292. doi: 10.1021/acscentsci.6b00032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Stoutland PO, Bergman RG, Nolan SP, Hoff CD. Polyhedron. 1988;7:1429–1440. [Google Scholar]
- 6.Wick DD, Jones WD. Organometallics. 1999;18:495–505. [Google Scholar]
- 7.For a recent review on Pd-catalyzed sp3 C-H activation, please see: He J, Wasa M, Chan KSL, Shao Q, Yu J-Q. Chem Rev. doi: 10.1021/acs.chemrev.6b00622.
- 8.Dyker G. Angew Chem Int Ed. 1994;33:103–105. [Google Scholar]
- 9.a) Constable AG, Mcdonald WS, Sawkins LC, Shaw BL. Chem Commun. 1978:1061–1062. [Google Scholar]; b) Baldwin JE, Jones RH, Najera C, Yus M. Tetrahedron. 1985;280:c51–c54. [Google Scholar]
- 10.Dick AR, Hull KL, Sanford MS. J Am Chem Soc. 2004;126:2300–2301. doi: 10.1021/ja031543m. [DOI] [PubMed] [Google Scholar]
- 11.Desai LV, Hull KL, Sanford MS. J Am Chem Soc. 2004;126:9542–9543. doi: 10.1021/ja046831c. [DOI] [PubMed] [Google Scholar]
- 12.Giri R, Chen X, Yu J-Q. Angew Chem Int Ed. 2005;44:2112–2115. doi: 10.1002/anie.200462884. [DOI] [PubMed] [Google Scholar]
- 13.Balavoine G, Clinet JC. J Organomet Chem. 1990;390:c84–c88. [Google Scholar]
- 14.Zaitsev VG, Shabashov D, Daugulis O. J Am Chem Soc. 2005;127:13154–13155. doi: 10.1021/ja054549f. [DOI] [PubMed] [Google Scholar]
- 15.Baudoin O, Herrbach A, Gueritte F. Angew Chem Int Ed. 2003;42:5736–5740. doi: 10.1002/anie.200352461. [DOI] [PubMed] [Google Scholar]
- 16.Barder TE, Walker SD, Marinelli JR, Buchwald SL. J Am Chem Soc. 2005;127:4685–4696. doi: 10.1021/ja042491j. [DOI] [PubMed] [Google Scholar]
- 17.Pan J, Su M, Buchwald SL. Angew Chem Int Ed. 2011;50:8647–8651. doi: 10.1002/anie.201102880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lafrance M, Gorelsky SI, Fagnou K. J Am Chem Soc. 2007;129:14570–14571. doi: 10.1021/ja076588s. [DOI] [PubMed] [Google Scholar]
- 19.Watanabe T, Oishi S, Fujii N, Ohno H. Org Lett. 2008;10:1759–1762. doi: 10.1021/ol800425z. [DOI] [PubMed] [Google Scholar]
- 20.Rousseaux S, Davi M, Sofack-Kreutzer J, Pierre C, Kefalidis CE, Clot E, Fagnou K, Baudoin O. J Am Chem Soc. 2010;132:10706–10716. doi: 10.1021/ja1048847. [DOI] [PubMed] [Google Scholar]
- 21.Sofack-Kreutzer J, Martin N, Renaudat A, Jazzar R, Baudoin O. Angew Chem Int Ed. 2012;51:10399–10402. doi: 10.1002/anie.201205403. [DOI] [PubMed] [Google Scholar]
- 22.Holstein PM, Dailler D, Vantourout J, Shaya J, Millet A, Baudoin O. Angew Chem Int Ed. 2016;55:2805–2809. doi: 10.1002/anie.201511457. [DOI] [PubMed] [Google Scholar]
- 23.Liegault B, Fagnou K. Organometallics. 2008;27:4841–4843. [Google Scholar]
- 24.Minami Y, Noguchi Y, Yamada K, Hiyama T. Chem Lett. 2016;45:1210–1212. [Google Scholar]
- 25.Shen R, Chen T, Zhao Y, Qiu R, Zhou Y, Yin S, Wang X, Goto M, Han L-B. J Am Chem Soc. 2011;133:17037–17044. doi: 10.1021/ja2069246. [DOI] [PubMed] [Google Scholar]
- 26.Thu H-Y, Yu W-Y, Che C-M. J Am Chem Soc. 2006;128:9048–9049. doi: 10.1021/ja062856v. [DOI] [PubMed] [Google Scholar]
- 27.Peng J, Chen C, Xi C. Chem Sci. 2016;7:1383–1387. doi: 10.1039/c5sc03903g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shabashov D, Daugulis O. Org Lett. 2005;7:3657–3659. doi: 10.1021/ol051255q. [DOI] [PubMed] [Google Scholar]
- 29.Chen X, Goodhue CE, Yu J-Q. J Am Chem Soc. 2006;128:12634–12635. doi: 10.1021/ja0646747. [DOI] [PubMed] [Google Scholar]
- 30.Stowers KJ, Fortner KC, Sanford MS. J Am Chem Soc. 2011;133:6541–6544. doi: 10.1021/ja2015586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang S-Y, He G, Nack WA, Zhao Y, Li Q, Chen G. J Am Chem Soc. 2013;135:2124–2127. doi: 10.1021/ja312277g. [DOI] [PubMed] [Google Scholar]
- 32.Nadres ET, Daugulis O. J Am Chem Soc. 2012;134:7–10. doi: 10.1021/ja210959p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.He G, Zhao Y, Zhang S, Lu C, Chen G. J Am Chem Soc. 2012;134:3–6. doi: 10.1021/ja210660g. [DOI] [PubMed] [Google Scholar]
- 34.Zhang S-Y, He G, Zhao Y, Wring K, Nack WA, Chen G. J Am Chem Soc. 2012;134:7313–7316. doi: 10.1021/ja3023972. [DOI] [PubMed] [Google Scholar]
- 35.Zhang L-S, Chen G, Wang X, Guo Q-Y, Zhang X-S, Pan F, Chen K, Shi Z-J. Angew Chem Int Ed. 2014;53:3899–3903. doi: 10.1002/anie.201310000. [DOI] [PubMed] [Google Scholar]
- 36.Xu J-W, Zhang Z-Z, Rao W-H, Shi B-F. J Am Chem Soc. 2016;138:10750–10753. doi: 10.1021/jacs.6b05978. [DOI] [PubMed] [Google Scholar]
- 37.Topczewski JJ, Cabrera PJ, Saper NI, Sanford MS. Nature. 2016;531:220–224. doi: 10.1038/nature16957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chan KSL, Wasa M, Chu L, Laforteza BN, Miura M, Yu J-Q. Nature Chem. 2014;6:146–150. doi: 10.1038/nchem.1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jiang H, He J, Liu T, Yu J-Q. J Am Chem Soc. 2016;138:2055–2059. doi: 10.1021/jacs.5b13462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McNally A, Haffemayer B, Collins BSL, Gaunt MJ. Nature. 2014;510:129–133. doi: 10.1038/nature13389. [DOI] [PubMed] [Google Scholar]
- 41.Smalley AP, Gaunt MJ. J Am Chem Soc. 2015;137:10632–10641. doi: 10.1021/jacs.5b05529. [DOI] [PubMed] [Google Scholar]
- 42.Willcox D, Chappell BGN, Hogg KF, Calleja J, Smalley AP, Gaunt MJ. Science. 2016;354:851–857. doi: 10.1126/science.aaf9621. [DOI] [PubMed] [Google Scholar]
- 43.He C, Gaunt MJ. Angew Chem Int Ed. 2016;54:15840–15844. doi: 10.1002/anie.201508912. [DOI] [PubMed] [Google Scholar]
- 44.Calleja J, Pla D, Gorman TW, Domingo V, Haffemayer B, Gaunt MJ. Nature Chem. 2015;7:1009–1016. doi: 10.1038/nchem.2367. [DOI] [PubMed] [Google Scholar]
- 45.Liu Y, Ge H. Nature Chem. 2017;9:26–32. [Google Scholar]
- 46.Xu Y, Young MC, Wang C, Magness DM, Dong G. Angew Chem Int Ed. 2016;55:9084–9087. doi: 10.1002/anie.201604268. [DOI] [PubMed] [Google Scholar]
- 47.Wu Y, Chen Y-Q, Liu T, Eastgate MD, Yu J-Q. J Am Chem Soc. 2016;138:14554–14557. doi: 10.1021/jacs.6b09653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Huang Z, Wang C, Dong G. Angew Chem Int Ed. 2016;55:5299–5303. doi: 10.1002/anie.201600912. [DOI] [PubMed] [Google Scholar]
- 49.Shabashov D, Daugulis O. J Am Chem Soc. 2014;132:3965–3972. doi: 10.1021/ja910900p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ano Y, Tobisu M, Chatani N. J Am Chem Soc. 2011;133:12984–12986. doi: 10.1021/ja206002m. [DOI] [PubMed] [Google Scholar]
- 51.Wang B, Lu C, Zhang S-Y, He G, Nack WA, Chen G. Org Lett. 2014;16:6260–6263. doi: 10.1021/ol503248f. [DOI] [PubMed] [Google Scholar]
- 52.Shan G, Huang G, Rao Y. Org Biomol Chem. 2015;13:697–701. doi: 10.1039/c4ob02389g. [DOI] [PubMed] [Google Scholar]
- 53.Reddy BVS, Reddy LR, Corey EJ. Org Lett. 2006;8:3391–3394. doi: 10.1021/ol061389j. [DOI] [PubMed] [Google Scholar]
- 54.Xiong H-Y, Besset T, Cahard D, Pannecoucke X. J Org Chem. 2015;80:4204–4212. doi: 10.1021/acs.joc.5b00505. [DOI] [PubMed] [Google Scholar]
- 55.Rao W-H, Zhan B-B, Chen K, Ling P-X, Zhang Z-Z, Shi B-F. Org Lett. 2015;17:3552–3555. doi: 10.1021/acs.orglett.5b01634. [DOI] [PubMed] [Google Scholar]
- 56.Kanyiva KS, Kuninobu Y, Kanai M. Org Lett. 2014;16:1968–1971. doi: 10.1021/ol500519y. [DOI] [PubMed] [Google Scholar]
- 57.Zhu Q, Ji D, Liang T, Wang X, Xu Y. Org Lett. 2015;17:3798–3801. doi: 10.1021/acs.orglett.5b01774. [DOI] [PubMed] [Google Scholar]
- 58.Ying X, Sun Y, Sun T-Y, Rao Y. Chem Commun. 2016;52:6423–6426. doi: 10.1039/c6cc00234j. [DOI] [PubMed] [Google Scholar]
- 59.a) Wasa M, Engle KM, Yu JQ. J Am Chem Soc. 2009;131:9886–9887. doi: 10.1021/ja903573p. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Wasa M, Chan KSL, Zhang XG, He J, Miura M, Yu JQ. J Am Chem Soc. 2012;134:18570–18572. doi: 10.1021/ja309325e. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) He J, Li S, Deng Y, Fu H, Laforteza BN, Spangler JE, Homs A, Yu JQ. Science. 2014;343:1216–1220. doi: 10.1126/science.1249198. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) He J, Takise R, Fu H, Yu JQ. J Am Chem Soc. 2015;137:4618–4621. doi: 10.1021/jacs.5b00890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhu R-Y, He J, Wang X-C, Yu J-Q. J Am Chem Soc. 2014;136:13194–13197. doi: 10.1021/ja508165a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.a) He J, Wasa M, Chan KSL, Yu JQ. J Am Chem Soc. 2013;135:3387–3390. doi: 10.1021/ja400648w. [DOI] [PubMed] [Google Scholar]; b) Fu H, Shen PX, He J, Zhang F, Li S, Wang P, Liu T, Yu JQ. Angew Chem Int Ed. 2017;129:1899–1902. doi: 10.1002/anie.201610426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wasa M, Engle KM, Yu J-Q. J Am Chem Soc. 2010;132:3680–3681. doi: 10.1021/ja1010866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Yoo EJ, Wasa M, Yu J-Q. J Am Chem Soc. 2010;132:17378–17380. doi: 10.1021/ja108754f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.He J, Shigenari T, Yu J-Q. Angew Chem Int Ed. 2015;54:6545–6549. doi: 10.1002/anie.201502075. [DOI] [PubMed] [Google Scholar]
- 65.Zhu R-Y, Tanaka K, Li G-C, He J, Fu H-Y, Li S-H, Yu J-Q. J Am Chem Soc. 2015;137:7067–7070. doi: 10.1021/jacs.5b04088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.a) He J, Jiang H, Takise R, Zhu RY, Chen G, Dai HX, Dhar TGM, Shi J, Zhang H, Cheng PTW, Yu JQ. Angew Chem Int Ed. 2016;55:785–789. doi: 10.1002/anie.201509996. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) He J, Shao Q, Wu Q, Yu JQ. J Am Chem Soc. 2017;139:3344–3347. doi: 10.1021/jacs.6b13389. [DOI] [PubMed] [Google Scholar]
- 67.Wang D-H, Wasa M, Giri R, Yu J-Q. J Am Chem Soc. 2008;130:7190–7191. doi: 10.1021/ja801355s. [DOI] [PubMed] [Google Scholar]
- 68.Giri R, Maugel N, Li J-J, Wang D-H, Breazzano SP, Saunders LB, Yu J-Q. J Am Chem Soc. 2007;129:3510–3511. doi: 10.1021/ja0701614. [DOI] [PubMed] [Google Scholar]
- 69.Chen G, Shigenari T, Jian P, Zhang Z, Jin Z, He J, Li S, Mapelli C, Miller MM, Poss MA, Yeung PMSK-S, Yu J-Q. J Am Chem Soc. 2015;137:3338–3351. doi: 10.1021/ja512690x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Chen G, Chuang Z, Li G-C, Saint-Denis TG, Hsiao Y, Joe CL, Yu J-Q. Angew Chem Int. 2017;56:1506–1509. doi: 10.1002/anie.201610580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Gong W, Zhang G, Liu T, Giri R, Yu J-Q. J Am Chem Soc. 2014;136:16940–16946. doi: 10.1021/ja510233h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zhang F-L, Hong K, Li T-J, Park H, Yu J-Q. Science. 2016;351:252–256. doi: 10.1126/science.aad7893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Yang K, Li Q, Liu Y, Li G, Ge H. J Am Chem Soc. 2016;138:12775–12778. doi: 10.1021/jacs.6b08478. [DOI] [PubMed] [Google Scholar]
- 74.Li S, Chen G, Feng CG, Gong W, Yu J-Q. J Am Chem Soc. 2014;136:5267–5270. doi: 10.1021/ja501689j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Li S, Zhu R-Y, Xiao K-J, Yu J-Q. Angew Chem Int Ed. 2016;55:4317–4321. doi: 10.1002/anie.201512020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.a) Ren Z, Mo F, Dong G. J Am Chem Soc. 2012;134:16991–16994. doi: 10.1021/ja3082186. [DOI] [PubMed] [Google Scholar]; b) Xu Y, Yan G, Ren Z, Dong G. Nature Chem. 2015;7:829–834. doi: 10.1038/nchem.2326. [DOI] [PubMed] [Google Scholar]
- 77.Thimpson SJ, Thach DQ, Dong G. J Am Chem Soc. 2015;137:11586–11589. doi: 10.1021/jacs.5b07384. [DOI] [PubMed] [Google Scholar]
- 78.Yang W, Ye S, Fanning D, Coon T, Schmidt Y, Krenitsky P, Stamos D, Yu J-Q. Angew Chem Int Ed. 2015;54:2501–2504. doi: 10.1002/anie.201410462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hasegawa N, Charra V, Inoue S, Fukumoto Y, Chatani N. J Am Chem Soc. 2011;133:8070–8073. doi: 10.1021/ja2001709. [DOI] [PubMed] [Google Scholar]
- 80.Simmons EM, Hartwig JF. Nature. 2012;483:70–73. doi: 10.1038/nature10785. [DOI] [PubMed] [Google Scholar]
- 81.Simmons EM, Hartwig JF. J Am Chem Soc. 2010;132:17092–17095. doi: 10.1021/ja1086547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Li B, Matthias D, Hartwig JF. J Am Chem Soc. 2014;136:6586–6589. doi: 10.1021/ja5026479. [DOI] [PubMed] [Google Scholar]
- 83.Larsen MA, Cho SH, Hartwig J. J Am Chem Soc. 2016;138:762–765. doi: 10.1021/jacs.5b12153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.a) Kang T, Kim Y, Lee D, Wang Z, Chang S. J Am Chem Soc. 2014;136:4141–4144. doi: 10.1021/ja501014b. [DOI] [PubMed] [Google Scholar]; Kang T, Kim H, Kim JG, Chang S. Chem Commun. 2014;50:12073–12075. doi: 10.1039/c4cc05655h. [DOI] [PubMed] [Google Scholar]
- 85.Xiao X, Hou C, Zhang Z, Ke Z, Lan J, Jiang H, Zeng W. Angew Chem Int Ed. 2016;55:11897–11901. doi: 10.1002/anie.201606531. [DOI] [PubMed] [Google Scholar]
- 86.Gao P, Guo W, Xue J, Zhao Y, Yuan Y, Xia Y, Shi Z. J Am Chem Soc. 2015;137:12231–12240. doi: 10.1021/jacs.5b06758. [DOI] [PubMed] [Google Scholar]
- 87.Huang X, Wang Y, Lan J, You J. Angew Chem Int Ed. 2015;54:9404–9408. doi: 10.1002/anie.201504507. [DOI] [PubMed] [Google Scholar]
- 88.Wang X, Yu D-G, Glorius F. Angew Chem Int Ed. 2015;54:10280–10283. doi: 10.1002/anie.201503888. [DOI] [PubMed] [Google Scholar]
- 89.a) Wang H, Tang G, Li X. Angew Chem Int Ed. 2015;54:13049–13052. doi: 10.1002/anie.201506323. [DOI] [PubMed] [Google Scholar]; b) Yu S, Tang G, Li Y, Zhou X, Lan Y, Li X. Angew Chem Int Ed. 2016;55:8696–8700. doi: 10.1002/anie.201602224. [DOI] [PubMed] [Google Scholar]; c) Hu XH, Yang XF, Loh TP. ACS Catal. 2016;6:5930–5934. [Google Scholar]
- 90.Nakao Y, Morita E, Idei H, Hiyama T. J Am Chem Soc. 2011;133:3264–3267. doi: 10.1021/ja1102037. [DOI] [PubMed] [Google Scholar]
- 91.Aihara Y, Chatani N. J Am Chem Soc. 2014;136:898–901. doi: 10.1021/ja411715v. [DOI] [PubMed] [Google Scholar]
- 92.Wu X, Zhao Y, Ge H. J Am Chem Soc. 2014;136:1789–1792. doi: 10.1021/ja413131m. [DOI] [PubMed] [Google Scholar]
- 93.Lin Y-J, Zhang Z-Z, Yan S-Y, Liu Y-H, Shi B-F. Chem Commun. 2015;51:7899–7902. doi: 10.1039/c5cc02254a. [DOI] [PubMed] [Google Scholar]
- 94.Lin C, Chen Z, Liu Z, Zhang Y. Org Lett. 2017;19:850–853. doi: 10.1021/acs.orglett.6b03856. [DOI] [PubMed] [Google Scholar]
- 95.a) Lin C, Xu W, Yao J, Wang B, Liu Z, Zhang Y. Org Lett. 2015;17:1340–1343. doi: 10.1021/acs.orglett.5b00471. [DOI] [PubMed] [Google Scholar]; b) Yan SY, Lin YJ, Liu B, Liu YH, Zhang ZZ, Shi BF. Chem Commun. 2015;51:7341–7344. doi: 10.1039/c5cc01436k. [DOI] [PubMed] [Google Scholar]; c) Ye X, Petersen JL, Shi X. Chem Commun. 2015;51:7863–7866. doi: 10.1039/c5cc01970b. [DOI] [PubMed] [Google Scholar]; d) Wang X, Qiu R, Yan C, Reddy VP, Zhu L, Xu X, Yin S-F. Org Lett. 2015;17:1970–1973. doi: 10.1021/acs.orglett.5b00706. [DOI] [PubMed] [Google Scholar]
- 96.Wu X, Zhao Y, Ge H. Chem Eur J. 2014;20:9530–9533. doi: 10.1002/chem.201403356. [DOI] [PubMed] [Google Scholar]
- 97.Aihara Y, Chatani N. ACS Catal. 2016;6:4323–4329. [Google Scholar]
- 98.Wu X, Zhao Y, Ge H. J Am Chem Soc. 2015;137:4924–4927. doi: 10.1021/jacs.5b01671. [DOI] [PubMed] [Google Scholar]
- 99.Shang R, Ilies L, Matsumoto A, Nakamura E. J Am Chem Soc. 2013;135:6030–6032. doi: 10.1021/ja402806f. [DOI] [PubMed] [Google Scholar]
- 100.Shang R, Ilies L, Matsumoto A, Nakamura E. J Am Chem Soc. 2015;137:7660–7663. doi: 10.1021/jacs.5b04818. [DOI] [PubMed] [Google Scholar]
- 101.Ilies L, Itabashi Y, Shang R, Nakamura E. ACS Catal. 2017;7:89–92. [Google Scholar]
- 102.Zhang J, Chen H, Lin C, Liu Z, Wang C, Zhang Y. J Am Chem Soc. 2015;137:12990–12996. doi: 10.1021/jacs.5b07424. [DOI] [PubMed] [Google Scholar]
- 103.Wu X, Yang K, Zhao Y, Sun H, Li G, Ge H. Nature Commun. 2015;6:6462. doi: 10.1038/ncomms7462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Barsu N, Bolli SK, Sundararaju B. Chem Sci. 2017;8:2431–2435. doi: 10.1039/c6sc05026c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Willamson P, Galvan A, Gaunt MJ. Chem Sci. doi: 10.1039/c6sc05581h. [DOI] [Google Scholar]
- 106.Wu X, Zhao Y, Zhang G, Ge H. Angew Chem Int Ed. 2014;53:3706–3710. doi: 10.1002/anie.201311263. [DOI] [PubMed] [Google Scholar]
- 107.Wang C, Yang Y, Qin D, He Z, You J. J Org Chem. 2015;80:5424–8429. doi: 10.1021/acs.joc.5b01302. [DOI] [PubMed] [Google Scholar]
- 108.Wu X, Miao J, Li Y, Li G, Ge H. Chem Sci. 2016;7:5260–5264. doi: 10.1039/c6sc01087c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.a) Gou Q, Yang YW, Liu ZN, Qin J. Chem Eur J. 2016;22:16057–16061. doi: 10.1002/chem.201603370. [DOI] [PubMed] [Google Scholar]; b) Wang C, Yang Y. Tetrahedron Lett. 2017;58:935–940. [Google Scholar]
- 110.Robertson J, Pillai J, Lush RK. Chem Soc Rev. 2001;30:94–103. [Google Scholar]
- 111.Cekovic Z. J Serb Chem Soc. 2005;70:287–318. [Google Scholar]
- 112.Zard SZ. Chem Soc Rev. 2008;37:1603–1618. doi: 10.1039/b613443m. [DOI] [PubMed] [Google Scholar]
- 113.Lalevee J, Allonas X, Fouassier J-P. J Am Chem Soc. 2002;124:9613–9621. doi: 10.1021/ja0204168. [DOI] [PubMed] [Google Scholar]
- 114.Hofmann AW. Ber Dtsch, Chem Ges. 1879;12:984–990. [Google Scholar]
- 115.a) Hernandez R, Rivera A, Salazar JA, Suarez E. J Chem Soc Chem Commun. 1980:958–959. [Google Scholar]; b) Carrau R, Hernandez R, Suarez E. J Chem Soc Perkin Trans 1. 1987:937–943. [Google Scholar]; c) de Armas P, Francisco CG, Hernandez R, Salazar JA, Suarez E. J Chem Soc Perkin Trans. 1;1988:3255–3265. [Google Scholar]; d) Betancor C, Concepcion JI, Hernandez R, Salazar JA, Suarez E. J Org Chem. 1983;48:4430–4432. [Google Scholar]; e) de Armas P, Carrau R, Concepcion JI, Francisco CG, Hernandez R, Suarez E. Tetrahedron Lett. 1985;26:2493–2496. [Google Scholar]
- 116.Togo H, Hoshina Y, Yokoyama M. Tetrahedron Lett. 1996;37:6129–6132. [Google Scholar]
- 117.Fan R, Pu D, Wen F, Wu J. J Org Chem. 2007;72:8994–8997. doi: 10.1021/jo7016982. [DOI] [PubMed] [Google Scholar]
- 118.Martinez C, Muniz K. Angew Chem Int Ed. 2015;54:8287–8291. doi: 10.1002/anie.201501122. [DOI] [PubMed] [Google Scholar]
- 119.Liu T, Mei T-S, Yu J-Q. J Am Chem Soc. 2015;137:5871–5874. doi: 10.1021/jacs.5b02065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Minozzi M, Nanni D, Spagnolo P. Chem Eur J. 2009;15:7830–7840. doi: 10.1002/chem.200802710. [DOI] [PubMed] [Google Scholar]
- 121.Chen H, Chiba S. Org Biomol Chem. 2014;12:42–46. doi: 10.1039/c3ob41871e. [DOI] [PubMed] [Google Scholar]
- 122.Qin Q, Yu S. Org Lett. 2015;17:1894–1897. doi: 10.1021/acs.orglett.5b00582. [DOI] [PubMed] [Google Scholar]
- 123.Zhu X, Wang Y-F, Ren W, Zhang F-L, Chiba S. Org Lett. 2013;15:3214–3217. doi: 10.1021/ol4014969. [DOI] [PubMed] [Google Scholar]
- 124.Liu T, Myers MC, Yu J-Q. Angew Chem Int Ed. 2017;56:306–309. doi: 10.1002/anie.201608210. [DOI] [PubMed] [Google Scholar]
- 125.Nikishin GI, Troyansky EI, Lazareva MI. Tetrahedron. 1985;41:4279–4288. [Google Scholar]
- 126.Yang M, Su B, Wang Y, Chen K, Jiang X, Zhang Y-F, Zhang X-S, Chen G, Cheng Y, Cao Z, Guo Q-Y, Wang L, Shi Z-J. Nature Commun. 2014;5:4707. doi: 10.1038/ncomms5707. [DOI] [PubMed] [Google Scholar]
- 127.Zhou T, Luo F-X, Yang M-Y, Shi Z-J. J Am Chem Soc. 2015;137:14586–14589. doi: 10.1021/jacs.5b10267. [DOI] [PubMed] [Google Scholar]
- 128.Wang Y-F, Chen H, Zhu X, Chiba S. J Am Chem Soc. 2012;134:11980–11983. doi: 10.1021/ja305833a. [DOI] [PubMed] [Google Scholar]
- 129.Chu JCK, Rovis T. Nature. 2016;539:272–275. doi: 10.1038/nature19810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Choi GJ, Zhu Q, Miller DC, Gu CJ, Knowles RR. Nature. 2016;539:268–271. doi: 10.1038/nature19811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Cekovic Z. Tetrahedron. 2003;59:8073–8090. [Google Scholar]
- 132.Barton DHR, Beaton JM. J Am Chem Soc. 1960;82:2640–2641. [Google Scholar]
- 133.Walling C, Padwa A. J Am Chem Soc. 1963;85:1597–1601. [Google Scholar]
- 134.Petrovic G, Saicic RN, Cekovic Z. Tetrahedron Lett. 1997;38:7107–7110. [Google Scholar]
- 135.Petrovic G, Cekovic Z. Tetrahedron. 1999;55:1377–1390. [Google Scholar]
- 136.Cekovic Z, Cvetkovic M. Tetrahedron Lett. 1982;23:3791–3794. [Google Scholar]
- 137.Zhang J, Li Y, Zhang F, Hu C, Chen Y. Angew Chem Int Ed. 2016;55:1872–1875. doi: 10.1002/anie.201510014. [DOI] [PubMed] [Google Scholar]
- 138.Mihailovic ML, Cekovic Z, Maksimovic Z, Jememic D, Lorenc L, Mamuzic RI. Tetrahedron. 1965;21:2799–2812. [Google Scholar]
- 139.Concepcion JI, Francisco CG, Hernandez R, Salazar JA, Suarez E. Tetrahedron Lett. 1984;25:1953–1956. [Google Scholar]
- 140.a) Tsunoi S, Ryu I, Sonoda N. J Am Chem Soc. 1994;116:5473–5474. [Google Scholar]; b) Tsunoi S, Ryu I, Okuda T, Tanaka M, Komastsu M, Sonoda N. J Am Chem Soc. 1998;120:8692–8701. [Google Scholar]
- 141.Rawal VH, Newton RC, Krishnamurthy V. J Org Chem. 1990;55:5181–5183. [Google Scholar]
- 142.Kim S, Lee S, Koh JS. J Am Chem Soc. 1991;113:5106–5107. [Google Scholar]
- 143.Barton DHR, Motherwell RSH, Motherwell WB. J Chem Soc Perkin Trans 1. 1981:2363–2367. [Google Scholar]
- 144.Yang NC, Yang D-DH. J Am Chem Soc. 1958;80:2913–2914. [Google Scholar]
- 145.Lathbury DC, Parsons PJ, Pinto I. J Chem Soc Chem Commun. 1988:81–82. [Google Scholar]
- 146.Curran DP, Shen W. J Am Chem Soc. 1993;115:6051–6059. [Google Scholar]
- 147.Curran DP, Xu J. J Am Chem Soc. 1996;118:3142–3147. [Google Scholar]
- 148.Bogen S, Gulea M, Fensterbank L, Malacria M. J Org Chem. 1999;64:4920–4925. doi: 10.1021/jo9904260. [DOI] [PubMed] [Google Scholar]
- 149.Devin P, Fensterbank L, Malacria M. J Org Chem. 1998;63:6764–6765. doi: 10.1021/jo9809783. [DOI] [PubMed] [Google Scholar]
- 150.Voica A-F, Mendoza A, Gutekunst WR, Fraga JO, Baran PS. Nature Chem. 2012;4:629–635. doi: 10.1038/nchem.1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Davies HML, Manning JR. Nature. 2008;451:471–424. doi: 10.1038/nature06485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.For C-H Insertion: Davies HML, Beckwith REJ. Chem Rev. 2003;103:2861–2904. doi: 10.1021/cr0200217.Merlic CA, Zechman AL. Synthesis. 2003:1137–1156.Slattery CN, Ford A, Maguire AR. Tetehradeon. 2010;66:6681–6705.
- 153.For C-H Amination: Dauban P, Dodd RH. Synlett. 2003:1571–1586.Collet F, Dodd RH, Dauban P. Chem Commun. 2009:5061–5074. doi: 10.1039/b905820f.Collet F, Lescot C, Dauban P. Chem Soc Rev. 2011;40:1926–1936. doi: 10.1039/c0cs00095g.Starkov P, Jamison TF, Marek I. Chem Eur J. 2015;21:5278–5300. doi: 10.1002/chem.201405779.
- 154.Ford A, Miel H, Ring A, Slattery CN, Maguire AR, McKervery MA. Chem Rev. 2015;115:9981–10080. doi: 10.1021/acs.chemrev.5b00121. [DOI] [PubMed] [Google Scholar]
- 155.Demonceau A, Noels AF, Hubert AJ, Teyssie P. J Chem Soc Chem Commun. 1981:688–689. [Google Scholar]
- 156.Wenkert E, Davis LL, Mylari BL, Solomon MF, da Silva RR, Shulman S, Warnet RJ. J Org Chem. 1982;47:3242–3247. [Google Scholar]
- 157.Taber DF, Petty EH. J Org Chem. 1982;47:4808–4809. [Google Scholar]
- 158.a) Breslow R, Gellman SH. J Chem Soc Chem Commun. 1982:1400–1401. [Google Scholar]; b) Yu X-Q, Huang J-S, Zhou X-G, Che C-M. Org Lett. 2000;2:2233–2236. doi: 10.1021/ol000107r. [DOI] [PubMed] [Google Scholar]
- 159.Roizen J, Harvey ME, Du Bois J. Acc Chem Res. 2012;45:911–922. doi: 10.1021/ar200318q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Taber DF, Schuchardt JL. J Am Chem Soc. 1985;107:5289–5290. [Google Scholar]
- 161.Taber DF, You KK. J Am Chem Soc. 1995;117:5757–5762. [Google Scholar]
- 162.Cane DE, Thomas PJ. J Am Chem Soc. 1984;106:5295–5303. [Google Scholar]
- 163.Wehn PM, Du Bois J. J Am Chem Soc. 2002;124:12950–12951. doi: 10.1021/ja028139s. [DOI] [PubMed] [Google Scholar]
- 164.Hinman A, Du Bois J. J Am Chem Soc. 2003;125:11510–11511. doi: 10.1021/ja0368305. [DOI] [PubMed] [Google Scholar]
- 165.Reger DL, Garza DG, Baxter JC. Organometallics. 1990;9:873–874. [Google Scholar]
- 166.Ren Z, Dong G. Organometallics. 2016;35:1057–1059. [Google Scholar]
- 167.a) Saget T, Perez D, Cramer N. Org Lett. 2013;15:1354–1357. doi: 10.1021/ol400380y. [DOI] [PubMed] [Google Scholar]; b) Hoshiya N, Kobayashi T, Arisawa M, Shuto S. Org Lett. 2013;15:6202–6205. doi: 10.1021/ol4030452. [DOI] [PubMed] [Google Scholar]; c) Hoshiya N, Takenaka K, Shuto S, Jun’ichi Uenishi. Org Lett. 2016;18:48–51. doi: 10.1021/acs.orglett.5b03229. [DOI] [PubMed] [Google Scholar]
- 168.Yang X, Shan G, Yang Z, Huang G, Dong G, Sheng C, Rao Y. Chem Commun. 2017;53:1534–1537. doi: 10.1039/c6cc06897a. [DOI] [PubMed] [Google Scholar]
- 169.Giese B. Angew Chem Int Ed. 1983;22:753–764. [Google Scholar]
- 170.Taber DF, Ruckle RE., Jr J Am Chem Soc. 1986;108:7686–7693. doi: 10.1021/ja00284a037. [DOI] [PubMed] [Google Scholar]
- 171.Doyle MP, Westrum LJ, Wolthuis WNE, See MM, Boone WP, Bagheri V, Pearson MM. J Am Chem Soc. 1993;115:958–964. [Google Scholar]
- 172.Hashimoto S-i, Watanabe N, Anada M, Ikegami S. J Synth Org Chem Jpn. 1996;54:988–999. [Google Scholar]
- 173.Fiori KW, Espino CG, Brodsky BH, Du Bois J. Tetrahedron. 2009;65:3042–3051. [Google Scholar]
- 174.Micovic VM, Mamuzic RI, Jeremic DD, Mihailovic ML. Tetrahedron. 1964;20:2279–2287. [Google Scholar]
- 175.Nishio T, Koyama H, Sasaki D, Sakamoto M. Helv Chim Acta. 2005;88:996–1003. [Google Scholar]
- 176.Breslow R, Winnik MA. J Am Chem Soc. 1969;91:3083–3084. [Google Scholar]
- 177.a) Breslow R, Baldwin S, Flechtner T, Kalicky P, Liu S, Washburn W. J Am Chem Soc. 1973;95:3251–3262. doi: 10.1021/ja00791a031. [DOI] [PubMed] [Google Scholar]; b) Breslow R, Baldwin S. J Am Chem Soc. 1970;92:732–734. [Google Scholar]; c) Breslow R, Kalicky P. J Am Chem Soc. 1971;93:3540–3541. [Google Scholar]
- 178.Wille U, Plath C. Liebigs Ann. 1977:111–119. [Google Scholar]
- 179.Chen K, Richter JM, Baran PS. J Am Chem Soc. 2008;130:7247–7249. doi: 10.1021/ja802491q. [DOI] [PubMed] [Google Scholar]
- 180.Rey V, Pierini AB, Penenory AB. J Org Chem. 2009;74:1223–1230. doi: 10.1021/jo801892c. [DOI] [PubMed] [Google Scholar]
- 181.Nakanishi M, Katayev D, Besnard C, Kundig EP. Angew Chem Int Ed. 2011;50:7438–7441. doi: 10.1002/anie.201102639. [DOI] [PubMed] [Google Scholar]
- 182.Saget T, Lemouzy SJ, Cramer N. Angew Chem Int Ed. 2012;51:2238–2242. doi: 10.1002/anie.201108511. [DOI] [PubMed] [Google Scholar]
- 183.Anas S, Cordi A, Kagan HB. Chem Commun. 2011;47:11483–11485. doi: 10.1039/c1cc14292e. [DOI] [PubMed] [Google Scholar]
- 184.a) Martin N, Pierre C, Davi M, Jazzar R, Baudoin O. Chem Eur J. 2012;18:4480–4484. doi: 10.1002/chem.201200018. [DOI] [PubMed] [Google Scholar]; b) Holstein PM, Vogler M, Larini P, Pilet G, Clot E, Baudoin O. ACS Catal. 2015;5:4300–4308. [Google Scholar]
- 185.Xiao K-J, Lin DW, Miura M, Zhu R-Y, Gong W, Wasa M, Yu J-Q. J Am Chem Soc. 2014;136:8138–8142. doi: 10.1021/ja504196j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Chen G, Gong W, Zhuang Z, Andra MS, Chen Y-Q, Hong X, Yang Y-F, Liu T, Houk KN, Yu J-Q. Science. 2016;353:1023–1027. doi: 10.1126/science.aaf4434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Wu Q-F, Shen P-X, He J, Wang X-B, Zhang F, Shao Q, Zhu R-Y, Mapelli C, Qiao JX, Poss MA, Yu J-Q. Science. 2017;355:499–503. doi: 10.1126/science.aal5175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Smalley AP, Cuthbertson JD, Gaunt MJ. J Am Chem Soc. 2017;139:1412–1415. doi: 10.1021/jacs.6b12234. [DOI] [PubMed] [Google Scholar]
- 189.Wang C-W, Derosa J, Biscoe MR. Chem Sci. 2015;6:5105–5113. doi: 10.1039/c5sc01710f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Porter NA, Scott DM, Lacher B, Giese B, Zeitz HG, Lindner HJ. J Am Chem Soc. 1989;111:8311–8312. [Google Scholar]
- 191.a) Sibi MP, Ji J. J Am Chem Soc. 1996;118:9200–9201. [Google Scholar]; b) Sibi MP, Ji J. J Org Chem. 1997;62:3800–3801. [Google Scholar]
- 192.Sibi MP, Nad S. Angew Chem Int Ed. 2007;46:9231–9234. doi: 10.1002/anie.200702976. [DOI] [PubMed] [Google Scholar]
- 193.Wong C, Harms K, Meggers E. Angew Chem Int Ed. 2016;55:13495–13498. doi: 10.1002/anie.201607305. [DOI] [PubMed] [Google Scholar]
- 194.Bode JW, Doyle MP, Protopopova MN, Zhou Q-L. J Org Chem. 1996;61:9146–9155. [Google Scholar]
- 195.Doyle MP, Zhou Q-L, Dyatkin AB, Ruppar DA. Tetrahedron Lett. 1995;36:7579–7582. [Google Scholar]
- 196.Taber DF, Malcolm SC. J Org Chem. 2001;66:944–953. doi: 10.1021/jo001519g. [DOI] [PubMed] [Google Scholar]
- 197.Liang J-L, Yuan S-X, Huang J-S, Yu W-Y, Che C-M. Angew Chem Int Ed. 2002;41:3465–3468. doi: 10.1002/1521-3773(20020916)41:18<3465::AID-ANIE3465>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- 198.Reddy RP, Davies HML. Org Lett. 2006;8:5013–5016. doi: 10.1021/ol061742l. [DOI] [PubMed] [Google Scholar]
- 199.Zakatan DN, Du Bois J. J Am Chem Soc. 2008;130:9220–9221. doi: 10.1021/ja8031955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Taber DF, You KK, Rheingold AL. J Am Chem Soc. 1996;118:547–556. [Google Scholar]
- 201.Wehn PM, Lee J, Du Bois J. Org Lett. 2003;5:4823–4826. doi: 10.1021/ol035776u. [DOI] [PubMed] [Google Scholar]
- 202.Kurokawa T, Kim M, Du Bois J. Angew Chem Int Ed. 2009;48:2777–2779. doi: 10.1002/anie.200806192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Taber DF, Frankowski KJ. J Org Chem. 2005;70:6417–6421. doi: 10.1021/jo0508752. [DOI] [PubMed] [Google Scholar]
- 204.Espino CG, When PM, Chow J, Du Bois J. J Am Chem Soc. 2001;123:6935–6936. [Google Scholar]
- 205.A Scifinder search conducted on June 4, 2017 underscores this point: “C-H Activation”: 7788 hits, “Hydrogen Atom Transfer”: 2819 hits, “Carbene Transfer”: 464 hits, “Nitrene Transfer”: 299 hits.
- 206.Nguyen JD, D’Amato EM, Narayanam JMR, Stephenson CRJ. Nature Chem. 2012;4:854–859. doi: 10.1038/nchem.1452. [DOI] [PubMed] [Google Scholar]
- 207.Liao K, Negretti S, Musaev DG, Bacsa J, Davis HML. Nature. 2016;533:230–234. doi: 10.1038/nature17651. [DOI] [PubMed] [Google Scholar]