

Adeno-associated virus–mediated expression of monoclonal antibodies confers 100% protection in mice when administered intramuscularly as early as 7 days prior to lethal mouse adapted Ebola virus challenge, offering an alternative strategy to traditional vaccination.
Keywords: Ebola virus, hemorrhagic fever, vaccine, neutralizing antibody, ZMapp, adeno-associated virus, vectored immunoprophylaxis
Abstract
The 2013–2016 West Africa outbreak demonstrated the epidemic potential of Ebola virus and highlighted the need for counter strategies. Monoclonal antibody (mAb)–based therapies hold promise as treatment options for Ebola virus infections. However, production of clinical-grade mAbs is labor intensive, and immunity is short lived. Conversely, adeno-associated virus (AAV)–mediated mAb gene transfer provides the host with a genetic blueprint to manufacture mAbs in vivo, leading to steady release of antibody over many months. Here we demonstrate that AAV-mediated expression of nonneutralizing mAb 5D2 or 7C9 confers 100% protection against mouse-adapted Ebola virus infection, while neutralizing mAb 2G4 was 83% protective. A 2-component cocktail, AAV-2G4/AAV-5D2, provided complete protection when administered 7 days prior to challenge and was partially protective with a 3-day lead time. Finally, AAV-mAb therapies provided sustained protection from challenge 5 months following AAV administration. AAV-mAb may be a viable alternative strategy for vaccination against emerging infectious diseases.
Prior to 2012, Ebola virus (EBOV) was responsible for sporadic, well-contained outbreaks primarily localized to central Africa. The 2013–2016 West African epidemic was several magnitudes larger than any previously recorded Ebola outbreak [1]. Despite the overwhelming need for prophylactic and therapeutic options highlighted by the recent West Africa outbreak, there are still no licensed vaccines or therapeutics available. The unusual scale of this outbreak demonstrates the potential for EBOV to cause widespread threat to human life and socioeconomic disruption, justifying further investigation into prevention and treatment strategies.
Monoclonal antibodies (mAbs) are a rapidly expanding set of tools for therapeutic intervention against infectious diseases that pose a significant threat to public health and for which the human population has no preexisting or vaccine-induced immunity [2]. mAb-based therapies are effective at reversing the progression of lethal EBOV infection in mouse, guinea pig, and nonhuman primate models [3–11]. These studies demonstrate that the humoral immune response correlates with survival and plays an important role in protection [12, 13]. Some of the first mAbs shown to confer protection to EBOV were 1H3, 2G4, and 4G7, which formed the mAb cocktail ZMab and subsequently comprised 2 of 3 components of ZMapp, which was used to experimentally treat healthcare workers during the West Africa outbreak [14–16]. Most effective mAbs neutralize EBOV by binding the viral glycoprotein (GP) that studs the exterior of the virion and impede viral entry through inhibition of GP fusion and/or interaction with its receptor, Niemann-Pick C1, as is the case for 2G4 [17]. However, not all effective EBOV mAbs are neutralizing. For example, 5D2 and 7C9 bind the mucin-like domain of the EBOV GP, providing no neutralizing activity but conferring complete protection in mice [18], suggesting that, for some mAbs, immunoglobulin effector functions are critical for protection against EBOV [19, 20].
Adeno-associated virus (AAV) vectors have been used extensively for gene therapy applications and are widely regarded as a safe and effective method of gene transfer [21, 22]. Functional mAbs can be produced directly in vivo through AAV-mediated expression to prevent viral infection [23–26]. Although this process has been termed “vectored immunoprophylaxis” (VIP), suggesting utility as a vaccine administered prior to exposure, ideally this platform could be optimized for use in a postexposure setting, as well. A previous attempt to prevent EBOV infection by AAV9-mediated expression of murine mAbs was unable to confer protection with a 14-day lead time between intramuscular AAV administration and challenge [27]. Here we produced AAV vectors pseudotyped with a novel, rapidly expressing AAV capsid, termed “AAV6.2FF,” encoding murine mAbs (AAV-mAb) 2G4, 5D, and 7C9 and evaluated the protective efficacy of these vectors as monotherapies when delivered intramuscularly. Furthermore, a 2-component cocktail containing AAV-2G4 and AAV-5D2 was administered to the muscle with various lead times prior to EBOV challenge, to elucidate the minimum window required for protection.
METHODS
AAV Vectors
Vector genome plasmids were engineered to contain the muscle-optimized CASI promoter [23] followed by (1) a firefly luciferase (Luc) reporter gene or a murine IgG2a heavy chain linked to a κ light chain by a self-cleaving 2A sequence and (2) a WPRE and a SV40 polyA signal between AAV2 inverted terminal repeats. AAV vectors were produced by cotransfection of human embryonic kidney 293 cells with genome and packaging plasmids as described previously [28]. Vectors pseudotyped with AAV8, AAV9, and AAV-DJ were purified by iodixanol gradient, while AAV6 and AAV6.2FF vectors were purified by use of a heparin column [29]. AAV vector titers were determined by quantitative polymerase chain reaction (qPCR) analysis as described elsewhere [30].
Mice
Mice were purchased from Charles River, and AAV vector administrations were performed on 6-week-old mice. C57BL/6 mice were used in all experiments, with the exception of the comparison of mAb expression in BALB/c mice. In in vivo luciferase imaging experiments, albino C57BL/6 mice were used since their lighter skin pigmentation is more conducive to imaging. All animal experiments were approved by the institutional animal care committees of the Canadian Science Centre for Human and Animal Health and the University of Guelph.
AAV Vector Administration
Intramuscular administration of AAV was performed in the gastrocnemius muscle, using a 29-gauge needle and a 40-μL injection volume. Injection into the tail vein was conducted on slightly heated mice, using a 100-μL injection volume. Modified intranasal administration of vector was performed as described elsewhere [31]. Single AAV-mAbs were administered intramuscularly or intranasally at a dose of 2 × 1011 vector genomes per mouse, whereas the AAV-2G4+AAV-5D2 cocktail was dosed at 4 × 1011 vector genomes (equal parts AAV-2G4/AAV-5D2). In the cotransduction route of administration experiments, AAV-2G4 and AAV-5D2 were coadministered together, in a single intramuscular injection, or separately, in 2 intramuscular injections (1 per leg); however, the total dose was 4 × 1011 vector genomes regardless of administration method.
In Vivo Luciferase Imaging
A total of 1 × 1011 vector genomes of AAV6-Luc, AAV6.2FF-Luc, AAV8-Luc, AAV9-Luc, or AAV-DJ-Luc were administered intramuscularly to 6-week-old albino C57BL/6 mice in a 40-μL volume. Bioluminescence imaging was performed on days 0, 1, 3, 7, 14, 21, 28, 56, and 112 after vector administration, using the IVIS SpectrumCT instrument (Perkin Elmer, Waltham, MA). Resultant data were analyzed and the signal intensity quantified using Living Image software (Perkin Elmer, Waltham, MA).
AAV-mAb Expression Profiling in Mice
Saphenous vein blood specimen collection was performed on a weekly basis for 1 month and then periodically until day 126 after AAV administration. Serum levels of EBOV-specific antibody were determined by ELISA, as previously described [32].
EBOV Challenge Studies
Mouse challenge studies were performed in the containment level 4 facility at the Canadian Science Centre for Human and Animal Health in Winnipeg, Canada. Groups of 6 mice were challenged intraperitoneally with 1000 times the lethal dose (for 50% of animals) of mouse-adapted EBOV (MA-EBOV) strain Mayinga [33]. Mock control animals received vehicle only (Hank’s balanced salt solution). Clinical signs of infection and body weight were monitored daily for 2 weeks after challenge, and survivors were followed 3 times longer than the death of the last control animal.
Statistical Analysis
GraphPad Prism 7 software was used for statistical analyses. Multiple t tests were used to compare differences in luciferase expression from each AAV capsid and to compare AAV-2G4 and AAV-5D2 mAb expression levels at each time point. Two-way analysis of variance was used to analyze differences in mAb output following different routes of administration of the AAV-2G4/AAV-5D2 cocktail. Challenge survival of AAV-mAb treated groups was compared to the mock group, using the Mantel-Cox log rank test.
RESULTS
AAV Vectors Mediate Early Onset Transgene Expression Following Intramuscular Injection
To identify an AAV vector that promotes robust and rapid transgene expression following intramuscular injection, a panel of AAV capsids representing some of the most commonly used serotypes for muscle transduction, including AAV6, AAV8, and AAV9, were used to pseudotype an AAV-CASI-firefly luciferase (AAV-Luc) genome and monitored for early transgene expression kinetics. In addition to the above-mentioned capsids, a novel variant of AAV6, termed AAV6.2FF, in which we engineered 3 point mutations to enhance muscle transduction [34, 35], and AAV-DJ, which has not previously been evaluated for muscle transduction, were included in the panel. Images illustrating the distribution of firefly luciferase expression from each of the 5 vectors were captured on days 1, 3, 7, 14 (Figure 1A), 21, 28, and 56 after AAV vector administration (Fig. S1A). The flux (in photons/second) generated exclusively at the site of intramuscular injection by animals within each of the AAV vector groups was quantified over time (Figure 1B and Fig. S1B). Since some of the signal migrated from the muscle to the liver, we also calculated the flux value for the muscle plus the liver for each of the AAV capsids (Figure 1C). Luciferase transgene expression was detectable as little as 24 hours after vector administration for all AAV capsids and continued to increase in intensity over the first 14–21 days, before plateauing and remaining relatively stable for at least 112 days. At 24 hours after vector administration, AAV6.2FF expressed luciferase in the muscle at levels that were significantly higher than any of the other capsids investigated, including the gold standards AAV8 and AAV9, with AAV6.2FF flux values that ranged from 10 to 1000 times greater than all other vectors tested (Figure 1B). Since AAV6.2FF promoted the fastest and most robust muscle-specific transgene expression, particularly at early time points, we selected this capsid for AAV-mAb gene transfer experiments.
Figure 1.

Early intramuscular transgene expression kinetics of 5 different adeno-associated virus (AAV) capsids. Albino C57BL/6 mice (n = 4 per group) were injected intramuscularly with 1 × 1011 vector genomes of an AAV vector expressing firefly luciferase (Luc) packaged with either AAV6, AAV6.2FF, AAV8, AAV9, or AAV-DJ capsid. A, In vivo luciferase images were obtained 0, 1, 3, 7, and 14 days after AAV administration to demonstrate vector distribution. B and C, Relative photon emission produced by luciferase from the muscle exclusively (quantified from the lateral view; B) or the muscle plus the liver (quantification of the ventral view; C) of each serotype of AAV was quantified at various time points from 1 to 112 days after injection. Multiple t tests were used to compare each time point. *P < .05 for comparison of AAV6.2FF to all other capsids; #P < .05 for comparison of AAV6.2FF to AAV8, AAV9, and AAV-DJ; †P < .05 for comparison of AAV6.2FF to AAV8 and AAV9; and §P < .05 for comparison of AAV6.2FF to AAV8.
Substantial Extramuscular Luciferase Expression Detected Following Intramuscular Administration of AAV8 and AAV9 Vectors
Intramuscular administration of AAV8 and AAV9 vectors expressing firefly luciferase was found to mediate substantial transgene expression in tissues other than the muscle (Figure 2A and B, Fig. S2A and B). This inadvertent transduction was particularly striking for AAV9, where luciferase expression in extramuscular tissues, particularly in the abdomen (liver), represented 70%–90% of the total flux 24 hours after vector administration (Figure 2D). This value was somewhat lower and more variable for AAV8, with peak extramuscular transgene expression occurring 3 days after vector administration and representing 0%–85% of the total flux (Figure 2C). In both cases, extramuscular transgene expression waned such that it was no longer detectable 21 and 56 days after AAV8 and AAV9 administration, respectively.
Figure 2.

Substantial extramuscular transgene expression mediated by adeno-associated virus 8 (AAV8) and AAV9 vectors following intramuscular administration. A and B, Albino C57BL/6 mice (n = 4 per group) injected intramuscularly with 1 × 1011 vector genomes of AAV8 (A) or AAV9 (B) vectors expressing firefly luciferase were imaged ventrally 1, 3, 7, 14, 21, 28, and 56 days after injection. C and D, The relative photon emission produced by luciferase from AAV8 (C) and AAV9 (D) was quantified at various time points from 1 to 56 days after injection, and the percentage of extramuscular transgene expression relative to the total transgene expression was graphed.
Sustained AAV6.2FF-Mediated mAb Expression Levels in the Serum >18 Weeks After Intramuscular Vector Administration
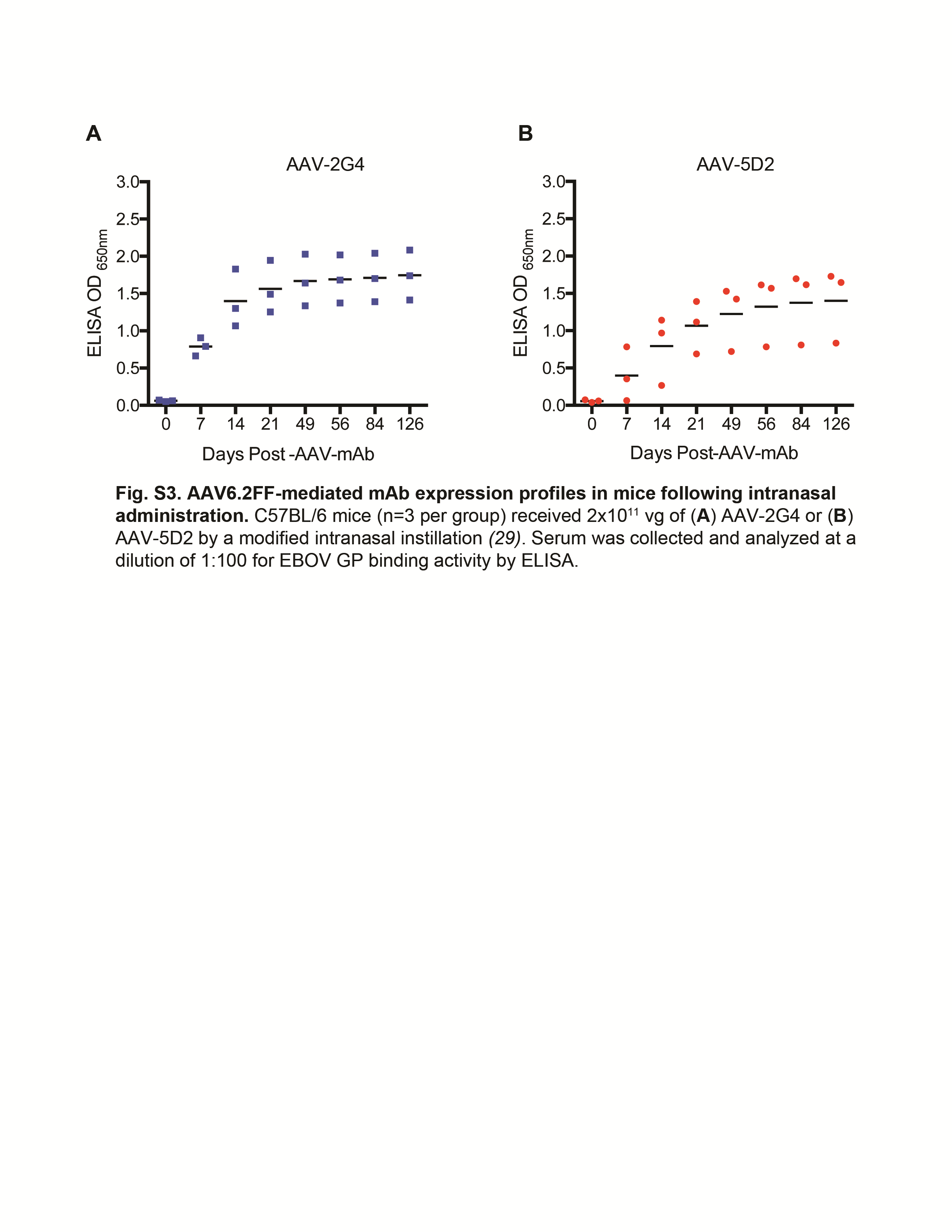
Antibody expression levels in mice transduced with AAV6.2FF-pseudotyped AAV-2G4 and AAV-5D2 were monitored 0–126 days following intramuscular administration (Figure 3). Serum 2G4 and 5D2 antibody levels rose steeply during the first 14 days and subsequently slowed but had yet to decline by day 126 after AAV administration. While serum containing 2G4 tended to yield somewhat higher ELISA ODs than 5D2, there was no statistically significant difference in serum EBOV GP binding capacity at any time point between the AAV-2G4 and AAV-5D2 groups. Moreover, levels of AAV-2G4 and AAV-5D2 mAb expression were comparable following intranasal administration; however, this mode of vector administration resulted in lower overall serum antibody levels than intramuscular administration (Fig. S3) [31]. Finally, differences in AAV-mAb expression levels were detected in mice with different genetic backgrounds, such that mAb expression levels were considerably higher in BALB/c mice than in C57BL/6 mice regardless of route of administration, and this was statistically significant 14 and 21 days after intramuscular and intranasal AAV administration, respectively (Fig. S4A and B). Taken together, these results indicate that intramuscular delivery of EBOV mAbs by use of AAV6.2FF can promote robust and sustained serum mAb expression levels for >4 months and that AAV-mediated mAb expression differs depending on the genetic background of the mouse.
Figure 3.

Adeno-associated virus 6.2FF (AAV6.2FF)–monoclonal antibody (mAb) expression levels in mice following intramuscular administration. C57BL/6 mice (n = 4) were injected intramuscularly with 2 × 1011 vector genomes of AAV-2G4 (A) or AAV-5D2 (B). Serum was collected from 1 to 126 days after administration and analyzed at a 1:100 dilution for Ebola virus glycoprotein binding capacity by enzyme-linked immunosorbent assay (ELISA).
Optimizing AAV Vector–Mediated Delivery of mAb Cocktails
Since therapeutic antibodies against EBOV are often given in the form of a mAb cocktail, we investigated the feasibility of coadministering 2 different AAV-mAb vectors in the same dose while still producing functional antibodies. We administered an equal dose of the AAV-2G4/AAV-5D2 cocktail via a single tail vein injection or a single intramuscular injection. A third group received 2 separate intramuscular injections—AAV-2G4 in the left leg and AAV-5D2 in the right leg—to investigate whether heterodimer formation due to AAV cotransduction would influence production of functional mAb titers. We found no significant difference in mAb expression levels between intravenous injection, 1 intramuscular injection, or 2 intramuscular injections at 7, 49 or 126 days after AAV administration; however, there was a trend toward slightly better serum mAb expression levels in the mice that received AAV-mAb vectors in 2 separate intramuscular injections (Figure 4). For this reason, we administered the AAV-mAb cocktail as 2 intramuscular injections for the challenge studies.
Figure 4.

Comparison of adeno-associated virus 6.2FF (AAV6.2FF)–mediated monoclonal antibody (mAb) cocktail (2G4 + 5D2) expression levels in mice following intravenous, single, or separate intramuscular injections. Serum mAb expression levels in C57BL/6 mice (n = 4) following AAV6.2FF-2G4/AAV-5D2 cocktail (4 × 1011 vector genomes total) administration either combined and administered intramuscularly (1xIM) or intravenously (IV) or separated and administered in 2 intramuscular injections (2xIM) on either leg were evaluated by enzyme-linked immunosorbent assay (ELISA) on 7, 49, and 126 days after administration. No significant differences in mAb expression levels were detected by 2-way analysis of variance when comparing routes of administration or combined versus separate injections of AAV vectors. NS, not significant.
AAV-Mediated mAb Expression Provides Complete Protection Against EBOV Challenge
Previous studies using AAV vectors to express mAbs in a prophylactic setting allowed 14–28 days for antibody to accumulate in the serum before animals were challenged [27, 36]. Given the speed with which transgene expression was detectable after AAV6.2FF muscle transduction (Figure 1A and B), mice were initially challenged with MA-EBOV 14 days after AAV-mAb administration. For this experiment, all mice receiving AAV-5D2 or AAV-7C9 (expressing nonneutralizing mAbs) were protected from lethal MA-EBOV challenge, whereas 83% of the group that received AAV-2G4 survived (Figure 5A). Moreover, mice that received the AAV-mAb monotherapies experienced negligible weight loss (Figure 5B).
Figure 5.

Adeno-associated virus 6.2FF (AAV6.2FF)–mediated expression of monoclonal antibodies provides complete protection against mouse-adapted Ebola virus (MA-EBOV) challenge. C57BL/6 mice (n = 6 per group) received an intramuscular injection of 2 × 1011 vector genomes of single AAV6.2FF-mAbs or a cocktail of 2 × 1011 vector genomes of AAV6.2FF-2G4 and 2 × 1011 vector genomes of AAV6.2FF-5D2 for a total dose of 4 × 1011 vg. All AAV monotherapies were given 14 days prior to intraperitoneal challenge with 1000 times the lethal dose (50%) of MA-EBOV. A and B, Kaplan-Meyer survival plots of AAV6.2FF-2G4, AAV6.2FF-5D2, and AAV6.2FF-7C9 monotherapies (A) and averaged mouse group weights (B). Survival of treated groups was compared to that of the mock group by using the Mantel-Cox log rank test. P = .0009 for 2G4 and P = .0005 for 5D2 and 7C9. C and D, Kaplan-Meyer survival plots of AAV6.2FF-2G4/AAV6.2FF-5D2 cocktail survival plots at various lead times between AAV administration and MA-EBOV challenge (C) and averaged mouse group weights (D). Survival of treated groups was compared to the mock group using the Mantel-Cox log rank test. P = .005 for 14 and 7 days, P = .2801 for 3 days, and P>0.9999 for 1 day and 0 days.
In addition to the use of AAV-mAb expression in a prophylactic setting, we are interested in the use of this strategy in a postexposure or therapeutic setting. Using a 2-component mAb cocktail comprising AAV-2G4 (a neutralizing mAb) and AAV-5D2 (a nonneutralizing mAb), we characterized the minimum lead time required to confer protection against lethal MA-EBOV challenge in mice. Similar to the AAV-mAb monotherapies, the cocktail provided protection to 100% of recipients when administered 14 days prior to challenge (Figure 5C). Full protection was also observed with a 7-day lead time, without apparent morbidity (Figure 5D). Remarkably, 1 of 6 mice in the cocktail treatment group survived after only a 3-day lead time for mAb accumulation; however, this mouse experienced weight loss and displayed clinical signs of infection prior to recovery. Interestingly, we did not observe any extension of life in the other mice in the 3-day group, and the sole mouse to die of infection in the AAV-2G4 monotherapy group died only 1 day after the control mice. MA-EBOV challenge the same day or 24 hours after AAV-mAb delivery did not extend survival, and these mice died on day 6 after infection along with control mice.
Protection From EBOV Challenge Extends 5 Months After a Single Intramuscular Injection of AAV-mAb
Next we investigated the protective efficacy of AAV-mediated mAb expression from a single administration given 5 months prior to challenge. Groups of mice received AAV-2G4, AAV-5D2, or an AAV-2G4/AAV-5D2 cocktail intramuscularly (administered as 2 separate injections) and were subsequently monitored for serum mAb levels by ELISA (Figure 3A and 3B and Figure 4). At day 126 after AAV-mAb administration, blood specimens were obtained from mice, and serum mAb levels were evaluated by ELISA. As shown in Table 1, ELISA ODs ranged from 0.8 to 2.9. At 140 days after AAV-mAb administration, mice were challenged intraperitoneally with 1000 times the LD50 of MA-EBOV and monitored for clinical signs of disease and weight loss. Age-matched control mice died on day 7, whereas all of the AAV-mAb recipients survived challenge (Figure 6). Interestingly, in this second study, in which older mice were challenged, the survivors lost weight for a longer period than the 6–8-week-old mice challenged 7–14 days after AAV-mAb administration (Figure 5B and 5D). These results demonstrate that AAV VIP mediates stable, long-term mAb expression in the serum of mice and can confer protection against lethal MA-EBOV challenge >5 months after a single intramuscular injection.
Table 1.
Results of Mouse-Adapted Ebola Virus (MA-EBOV) Challenge 140 Days Following Intramuscular Delivery of Adeno-Associated Virus (AAV) Monoclonal Antibody Vectors
| AAV Vector, Mouse | ELISA OD Before Challengea | Outcome |
|---|---|---|
| AAV-2G4 | ||
| 1 | 2.8 | Survived |
| 2 | 2.2 | Survived |
| 3 | 2.1 | Survived |
| 4 | 2.8 | Survived |
| AAV-5D2 | ||
| 1 | 1.5 | Survived |
| 2 | 2.0 | Survived |
| 3 | 2.4 | Survived |
| 4 | 2.3 | Survived |
| AAV-2G4 + AAV-5D2 | ||
| 1 | 2.1 | Survived |
| 2 | 2.5 | Survived |
| 3 | 2.9 | Survived |
| 4 | 0.8 | Survived |
Abbreviation: ELISA, enzyme-linked immunosorbent assay.
aSerum specimens were collected from mice 14 days before intraperitoneal challenge with 1000 times the lethal dose (50%) of MA-EBOV.
Figure 6.

Sustained adeno-associated virus 6.2FF (AAV6.2FF)–mediated monoclonal antibody (mAb) expression protects mice from mouse-adapted Ebola virus (MA-EBOV) challenge 5 months after a single intramuscular injection. C57BL/6 mice received an intramuscular injection of 2 × 1011 vector genomes of single AAV6.2FF-mAbs (n = 4 per group) or a cocktail of 2 × 1011 vector genomes of AAV6.2FF-2G4 and 2 × 1011 vector genomes of AAV6.2FF-5D2 (n = 4 per group) for a total dose of 4 × 1011 vector genomes. AAV vectors were administered 140 days prior to intraperitoneal challenge with 1000 times the lethal dose (50%) of MA-EBOV. A, Kaplan-Meyer survival plots of AAV6.2FF-2G4, AAV6.2FF-5D2, and AAV6.2FF-2G4/AAV6.2FF-5D2 cocktail (A) and averaged mouse group weights (B).
DISCUSSION
VIP offers a novel approach for preexposure and postexposure prophylaxis against pathogens of public health importance for which no vaccines or therapies are available. VIP has been shown to be highly effective at protecting mice, ferrets, and nonhuman primates from a variety of infectious agents, including human immunodeficiency virus [23, 24], influenza virus [25, 26] and Plasmodium falciparum sporozoites [37]. Recently, AAV9-mediated delivery of 2 of the antibody components of the ZMapp cocktail protected mice against systemic and airway challenge with MA-EBOV when delivered 14 days prior to challenge [27]. The aim of the current study was to investigate whether AAV-mediated antibody gene transfer of a single neutralizing or nonneutralizing antibody could protect mice from systemic EBOV challenge and to determine the minimum therapeutic window between AAV-mediated antibody transfer and challenge.
To investigate the potential usefulness of VIP in a postexposure scenario, it was important to select an AAV capsid that promoted rapid transgene expression. While a number of studies have quantified luciferase expression from various AAV capsids following different routes of administration [38–41], the focus has largely been on quantification of signal longevity, as opposed to characterizing the kinetics of transgene expression at early time points. Unexpectedly, all of the AAV capsids evaluated in this study generated a robust luciferase signal that was detectable 24 hours after a single intramuscular injection. This is the first report evaluating transgene expression from AAV vectors 24 hours after transduction in vivo. AAV6.2FF outperformed AAV6 and all other capsids evaluated, potentially because of the removal of surface-exposed tyrosine residues, which are known to mitigate capsid ubiquitination and degradation [42]. The remarkable speed of transgene expression from AAV6.2FF strongly suggests that AAV-VIP could potentially be used in a postexposure scenario.
AAV8 and AAV9 are popular choices for AAV-antibody gene transfer, owing to their ability to mediate high-level transgene expression [23, 26]; however, migration of transgene expression from the site of intramuscular administration has been reported for AAV8 [43], similar to what we observed. Here, we are the first to demonstrate that a large proportion (70%–90%) of AAV9-mediated transgene expression is detected outside the muscle, in the liver, within the first 24 hours after vector administration. The mechanism of AAV8/9 migration to other tissues is not yet known. Interestingly, although systemic administration of AAV-DJ almost exclusively targets the liver [44], transgene expression from AAV-DJ remained intramuscular, unlike that from AAV8/9. Delivery of AAV-mAbs intramuscularly is more practical and safer than systemic administration; therefore, selecting a capsid that does not migrate away from the muscle is optimal.
We characterized the expression profiles of 2 distinct AAV-mAbs and demonstrate that both are capable of high-level expression in mice. Despite the lack of statistical significance, AAV-2G4 consistently produced higher ELISA values in both intramuscularly and intranasally treated groups. However, since the mAb transgenes were murine IgG2a molecules, it was not possible to quantify the amount of mAb in the serum, and higher ELISA ODs do not necessarily indicate higher expression.
Cotransfection had a severe impact on functional mAb output in vitro (data not shown); however, coadministration of AAV-2G4 and AAV-5D2 did not significantly alter mAb expression in vivo. It is possible that, in vitro, a higher proportion of cells were cotransfected, leading to the production of improperly paired antibody chains and therefore poorly functional mAbs. A greater number of available cells in vivo may reduce the likelihood of cotransduction; therefore, although erroneous heterodimers are still likely to be generated, they are not the primary product and do not seem to impede overall serum concentrations of functional mAbs.
The AAV-2G4/AAV-5D2 cocktail conferred 100% protection with only a 7-day lead time before EBOV challenge. To our knowledge this is the shortest prophylactic window in which AAV-mAb expression has demonstrated full protection against EBOV challenge and the first time that a nonneutralizing mAb has been shown to protect against EBOV via VIP. Interestingly, we observed minimal extension of life in groups with partial protection. Although there was a sole survivor in the group with a 3-day lead time, all of its cage mates died the same day as the untreated controls, and the mouse that died of infection in the AAV-2G4 monotherapy group did so only 1 day after the controls. With minimal protection conferred by the AAV-2G4/AAV-5D2 cocktail 3 days after AAV administration, it appears that we missed the minimum lead time required for full protection, which would be between 7 and 3 days after AAV administration. These challenge experiments were conducted using C57BL/6 mice prior to the observation that BALB/c mice displayed significantly greater 5D2 mAb levels; therefore, it is possible that we would have observed better efficacy with BALB/c mice. In addition to generating protection with minimal lead time, groups treated with AAV-2G4, AAV-5D2, or an AAV-2G4/AAV-5D2 cocktail 5 months prior to challenge experienced 100% survival. Long-term expression of mAbs at protective levels demonstrates the potential of this platform to be used as an alternative vaccination strategy for various infectious diseases.
Both of the nonneutralizing AAV-mAb monotherapies provided 100% protection with a 14-day lead time, while the only neutralizing mAb we examined was partially effective. These results are similar to what Qiu et al observed when mice were treated with these same mAbs intraperitoneally and challenged with MA-EBOV [18]. Since 5D2 and 7C9 were engineered as murine IgG2a antibodies with functional Fc domains, they may induce complement-dependent cytotoxicity or antibody-dependent cell-mediated cytotoxicity in place of viral neutralization.
Despite preclinical success in postsymptomatic treatment of nonhuman primates with ZMapp [45], a clinical trial treating humans with the mAb cocktail failed to significantly improve patient outcome [46]. Limited doses of ZMapp were available during the West Africa outbreak, and it was not feasible to scale up production quickly enough, since the mAbs were manufactured in Nicotiana benthamiana. In the context of epidemic response, AAV-mediated mAb production in vivo offers an alternative strategy because vectors can be lyophilized and stockpiled for future use [47]. Furthermore, a single dose of AAV-mAb provides circulating mAbs for much longer periods than passively administered mAbs. Since AAV-mAb expression is independent of the host immune response, it can provide protection to older or immunocompromised individuals, which offers an advantage over traditional vaccines. Alternatively, in healthy individuals, AAV-mAb expression offers a stopgap, slowing viral replication to allow the immune response time to catch up to the infection and generate natural immunity [25]. While postexposure AAV-mAb therapy is not yet feasible, the mAbs used in ZMapp and in this study are of a first generation, and recently countless, more potent mAbs have been characterized that may facilitate this advancement [48–50].
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Notes
Acknowledgments. We thank those who contributed to the care and health of the animals involved at the University of Guelph and the National Microbiology Laboratory; and David Russell, for providing the pDGM6 plasmid.
Financial support. This work was supported by the Canadian Institutes for Health Research (grant N351310 to S. K. W. and G. P. K.), the Ontario Lung Association/Ontario Thoracic Society (grant 052919), the Ontario Veterinary College (PhD scholarship to L. P. V.), and the Ontario Graduate Scholarship Program (to L. P. V.).
Potential conflicts of interest. All authors: No reported conflicts of interest. All authors have submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed.
References
- 1. Spengler JR, Ervin ED, Towner JS, Rollin PE, Nichol ST. Perspectives on West Africa Ebola virus disease outbreak, 2013–2016. Emerg Infect Dis 2016; 22:956–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Sparrow E, Friede M, Sheikh M, Torvaldsen S. Therapeutic antibodies for infectious diseases. Bull World Health Organ 2017; 95:235–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Qiu X, Kobinger GP. Antibody therapy for Ebola: Is the tide turning around?Hum Vaccin Immunother 2014; 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Mupapa K, Massamba M, Kibadi K et al. . Treatment of Ebola hemorrhagic fever with blood transfusions from convalescent patients. International Scientific and Technical Committee. J Infect Dis 1999; 179(Suppl 1):S18–23. [DOI] [PubMed] [Google Scholar]
- 5. Qiu X, Wong G, Fernando L et al. . mAbs and Ad-Vectored IFN-alpha therapy rescue Ebola-infected nonhuman primates when administered after the detection of viremia and symptoms. Sci Transl Med 2013; 5:207ra143. [DOI] [PubMed] [Google Scholar]
- 6. Dye JM, Herbert AS, Kuehne AI et al. . Postexposure antibody prophylaxis protects nonhuman primates from filovirus disease. Proc Natl Acad Sci U S A 2012; 109:5034–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Pettitt J, Zeitlin L, Kim do H et al. . Therapeutic intervention of Ebola virus infection in rhesus macaques with the MB-003 monoclonal antibody cocktail. Sci Transl Med 2013; 5:199ra13. [DOI] [PubMed] [Google Scholar]
- 8. Qiu X, Audet J, Wong G et al. . Successful treatment of Ebola virus-infected cynomolgus macaques with monoclonal antibodies. Sci Transl Med 2012; 4:138ra81. [DOI] [PubMed] [Google Scholar]
- 9. Marzi A, Yoshida R, Miyamoto H et al. . Protective efficacy of neutralizing monoclonal antibodies in a nonhuman primate model of Ebola hemorrhagic fever. PLoS One 2012; 7:e36192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Olinger GG Jr, Pettitt J, Kim D et al. . Delayed treatment of Ebola virus infection with plant-derived monoclonal antibodies provides protection in rhesus macaques. Proc Natl Acad Sci U S A 2012; 109:18030–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Qiu X, Wong G, Fernando L et al. . Monoclonal antibodies combined with adenovirus-vectored interferon significantly extend the treatment window in Ebola virus-infected guinea pigs. J Virol 2013; 87:7754–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Marzi A, Engelmann F, Feldmann F et al. . Antibodies are necessary for rVSV/ZEBOV-GP-mediated protection against lethal Ebola virus challenge in nonhuman primates. Proc Natl Acad Sci U S A 2013; 110:1893–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wong G, Richardson JS, Pillet S et al. . Immune parameters correlate with protection against Ebola virus infection in rodents and nonhuman primates. Sci Transl Med 2012; 4:158ra46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Qiu X, Alimonti JB, Melito PL, Fernando L, Ströher U, Jones SM. Characterization of Zaire ebolavirus glycoprotein-specific monoclonal antibodies. Clin Immunol 2011; 141:218–27. [DOI] [PubMed] [Google Scholar]
- 15. Audet J, Wong G, Wang H et al. . Molecular characterization of the monoclonal antibodies composing ZMAb: a protective cocktail against Ebola virus. Sci Rep 2014; 4:6881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fauci AS. Ebola–underscoring the global disparities in health care resources. N Engl J Med 2014; 371:1084–6. [DOI] [PubMed] [Google Scholar]
- 17. Tran EE, Nelson EA, Bonagiri P et al. . Mapping of Ebolavirus neutralization by monoclonal antibodies in the zmapp cocktail using cryo-electron tomography and studies of cellular entry. J Virol 2016; 90:7618–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Qiu X, Fernando L, Melito PL et al. . Ebola GP-specific monoclonal antibodies protect mice and guinea pigs from lethal Ebola virus infection. PLoS Negl Trop Dis 2012; 6:e1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hessell AJ, Hangartner L, Hunter M et al. . Fc receptor but not complement binding is important in antibody protection against HIV. Nature 2007; 449:101–4. [DOI] [PubMed] [Google Scholar]
- 20. Cadogan M, Dalgleish AG. HIV immunopathogenesis and strategies for intervention. Lancet Infect Dis 2008; 8:675–84. [DOI] [PubMed] [Google Scholar]
- 21. Naso MF, Tomkowicz B, Perry WL 3rd, Strohl WR. Adeno-associated virus (AAV) as a vector for gene therapy. BioDrugs 2017; 31:317–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mingozzi F, High KA. Therapeutic in vivo gene transfer for genetic disease using AAV: progress and challenges. Nat Rev Genet 2011; 12:341–55. [DOI] [PubMed] [Google Scholar]
- 23. Balazs AB, Chen J, Hong CM, Rao DS, Yang L, Baltimore D. Antibody-based protection against HIV infection by vectored immunoprophylaxis. Nature 2012; 481:81–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Balazs AB, Ouyang Y, Hong CM et al. . Vectored immunoprophylaxis protects humanized mice from mucosal HIV transmission. Nat Med 2014; 20:296–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Balazs AB, Bloom JD, Hong CM, Rao DS, Baltimore D. Broad protection against influenza infection by vectored immunoprophylaxis in mice. Nat Biotechnol 2013; 31:647–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Limberis MP, Adam VS, Wong G et al. . Intranasal antibody gene transfer in mice and ferrets elicits broad protection against pandemic influenza. Sci Transl Med 2013; 5:187ra72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Limberis MP, Tretiakova A, Nambiar K et al. . Adeno-associated virus serotype 9-expressed ZMapp in mice confers protection against systemic and airway-acquired Ebola virus infection. J Infect Dis 2016; 214:1975–9. [DOI] [PubMed] [Google Scholar]
- 28. Halbert CL, Allen JM, Miller AD. Efficient mouse airway transduction following recombination between AAV vectors carrying parts of a larger gene. Nat Biotechnol 2002; 20:697–701. [DOI] [PubMed] [Google Scholar]
- 29. Halbert CL, Allen JM, Chamberlain JS. AAV6 vector production and purification for muscle gene therapy. Methods Mol Biol 2018; 1687:257–66. [DOI] [PubMed] [Google Scholar]
- 30. Aurnhammer C, Haase M, Muether N et al. . Universal real-time PCR for the detection and quantification of adeno-associated virus serotype 2-derived inverted terminal repeat sequences. Hum Gene Ther Methods 2012; 23:18–28. [DOI] [PubMed] [Google Scholar]
- 31. Santry LA, Ingrao JC, Yu DL et al. . AAV vector distribution in the mouse respiratory tract following four different methods of administration. BMC Biotechnol 2017; 17:43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hevey M, Negley D, Geisbert J, Jahrling P, Schmaljohn A. Antigenicity and vaccine potential of Marburg virus glycoprotein expressed by baculovirus recombinants. Virology 1997; 239:206–16. [DOI] [PubMed] [Google Scholar]
- 33. Bray M, Davis K, Geisbert T, Schmaljohn C, Huggins J. A mouse model for evaluation of prophylaxis and therapy of Ebola hemorrhagic fever. J Infect Dis 1998; 178:651–61. [DOI] [PubMed] [Google Scholar]
- 34. Qiao C, Zhang W, Yuan Z et al. . Adeno-associated virus serotype 6 capsid tyrosine-to-phenylalanine mutations improve gene transfer to skeletal muscle. Hum Gene Ther 2010; 21:1343–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Limberis MP, Vandenberghe LH, Zhang L, Pickles RJ, Wilson JM. Transduction efficiencies of novel AAV vectors in mouse airway epithelium in vivo and human ciliated airway epithelium in vitro. Mol Ther 2009; 17:294–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Limberis MP, Racine T, Kobasa D et al. . Vectored expression of the broadly neutralizing antibody FI6 in mouse airway provides partial protection against a new avian influenza A virus, H7N9. Clin Vaccine Immunol 2013; 20:1836–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Deal C, Balazs AB, Espinosa DA, Zavala F, Baltimore D, Ketner G. Vectored antibody gene delivery protects against Plasmodium falciparum sporozoite challenge in mice. Proc Natl Acad Sci U S A 2014; 111:12528–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Prasad KM, Smith RS, Xu Y, French BA. A single direct injection into the left ventricular wall of an adeno-associated virus 9 (AAV9) vector expressing extracellular superoxide dismutase from the cardiac troponin-T promoter protects mice against myocardial infarction. J Gene Med 2011; 13:333–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hu C, Lipshutz GS. AAV-based neonatal gene therapy for hemophilia A: long-term correction and avoidance of immune responses in mice. Gene Ther 2012; 19:1166–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Körbelin J, Dogbevia G, Michelfelder S et al. . A brain microvasculature endothelial cell-specific viral vector with the potential to treat neurovascular and neurological diseases. EMBO Mol Med 2016; 8:609–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Michelfelder S, Varadi K, Raupp C et al. . Peptide ligands incorporated into the threefold spike capsid domain to re-direct gene transduction of AAV8 and AAV9 in vivo. PLoS One 2011; 6:e23101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zhong L, Li B, Mah CS et al. . Next generation of adeno-associated virus 2 vectors: point mutations in tyrosines lead to high-efficiency transduction at lower doses. Proc Natl Acad Sci U S A 2008; 105:7827–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Greig JA, Peng H, Ohlstein J et al. . Intramuscular injection of AAV8 in mice and macaques is associated with substantial hepatic targeting and transgene expression. PLoS One 2014; 9:e112268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Grimm D, Lee JS, Wang L et al. . In vitro and in vivo gene therapy vector evolution via multispecies interbreeding and retargeting of adeno-associated viruses. J Virol 2008; 82:5887–911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Qiu X, Wong G, Audet J et al. . Reversion of advanced Ebola virus disease in nonhuman primates with ZMapp. Nature 2014; 514:47–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Davey RT, Dodd L, Proschan MA et al. . a randomized, controlled trial of ZMapp for Ebola virus infection. N Engl J Med 2016; 375:1448–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Croyle MA, Cheng X, Wilson JM. Development of formulations that enhance physical stability of viral vectors for gene therapy. Gene Ther 2001; 8:1281–90. [DOI] [PubMed] [Google Scholar]
- 48. Flyak AI, Shen X, Murin CD et al. . Cross-reactive and potent neutralizing antibody responses in human survivors of natural Ebolavirus infection. Cell 2016; 164:392–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zhang Q, Gui M, Niu X et al. . Potent neutralizing monoclonal antibodies against Ebola virus infection. Sci Rep 2016; 6:25856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Corti D, Misasi J, Mulangu S et al. . Protective monotherapy against lethal Ebola virus infection by a potently neutralizing antibody. Science 2016; 351:1339–42. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.