

Summary
This study describes iterative experiments to define bumped-kinase inhibitor properties needed for clinical efficacy. For these compounds, pharmacokinetics analysis of clinical cryptosporidiosis in mice show that fecal drug levels greater than parasite inhibitory concentrations correlate best with effective therapeutic outcomes.
Keywords: Cryptosporidiosis treatment, calcium-dependent protein kinase 1, bumped kinase inhibitors.
Abstract
Bumped kinase inhibitors (BKIs) of Cryptosporidium parvum calcium-dependent protein kinase 1 (CpCDPK1) are leading candidates for treatment of cryptosporidiosis-associated diarrhea. Potential cardiotoxicity related to anti–human ether-à-go-go potassium channel (hERG) activity of the first-generation anti-Cryptosporidium BKIs triggered further testing for efficacy. A luminescence assay adapted for high-throughput screening was used to measure inhibitory activities of BKIs against C. parvum in vitro. Furthermore, neonatal and interferon γ knockout mouse models of C. parvum infection identified BKIs with in vivo activity. Additional iterative experiments for optimum dosing and selecting BKIs with minimum levels of hERG activity and frequencies of other safety liabilities included those that investigated mammalian cell cytotoxicity, C. parvum proliferation inhibition in vitro, anti–human Src inhibition, hERG activity, in vivo pharmacokinetic data, and efficacy in other mouse models. Findings of this study suggest that fecal concentrations greater than parasite inhibitory concentrations correlate best with effective therapy in the mouse model of cryptosporidiosis, but a more refined model for efficacy is needed.
Cryptosporidiosis is a significant contributor to global diarrheal morbidity and mortality. Cryptosporidium is associated with moderate-to-severe diarrhea, stunting, and death in children <2 years old in resource-limited environments [1–3]. In immunocompromised patients, cryptosporidiosis diarrhea may be life threatening.
Cryptosporidium organisms are obligate intracellular parasites that live in intracellular but extracytosolic parasitophorous vacuoles located beneath the apical plasma membrane of infected intestinal epithelial cells [4]. Large waterborne outbreaks are common, such as the 1993 Milwaukee, Wisconsin, incident in which 403000 cases were reported after the local water supply became contaminated with Cryptosporidium oocysts [5]. The infectious dose for humans ranges from 9 to 132 oocysts, depending on the strain of Cryptosporidium [6, 7]. The incapacitating nature of the diarrhea, high rate of infection, low infectious dose, association with death and stunting, and resistance of parasite to elimination from the environment are all factors that have heightened public health concerns about the disease.
The only Food and Drug Administration–approved therapy for cryptosporidiosis, nitazoxanide, was shown in a randomized trial to reduce the duration of diarrhea by approximately 2 days in immunocompetent individuals [8]. Nitazoxanide also has limited efficacy and a high failure rate in immunocompromised individuals [9]. It is not approved for use in children <12 months of age, whereas studies in malnourished children show poor response rate [10]. With the burden of disease falling disproportionately on young children in regions with a high human immunodeficiency virus prevalence [11] and widespread malnutrition, better drugs are needed for cryptosporidiosis therapy.
Multiple studies have shown that inhibition of apicomplexan calcium-dependent protein kinases (CDPKs) interfere with the parasites’ ability to invade or egress mammalian host cells [12–14]. Calcium regulation in diverse metabolic and functional pathways is essential to apicomplexan parasites’ survival [15–18]. It has previously been demonstrated that C. parvum growth was inhibited by BKIs that target CpCDPK1 [19]. BKI 1294 exhibited anti-Cryptosporidium efficacy in the SCID/beige immunocompromised mouse and the newborn calf C. parvum clinical models [20, 21]. Since Cryptosporidium infections are predominant in the gastrointestinal tract, therapeutic agents with unconventional pharmacokinetic and pharmacodynamic properties may be required for effective treatment. Here we report our progress to identify BKIs that are effective against Cryptosporidium.
MATERIALS AND METHODS
Methods for the propagation of C. parvum (IOWA-II strain) in calves [22, 23], creation of a transgenic C. parvum strain UGA1 transfected to express nanoluciferase (Nluc) [24], purification of oocysts from fecal specimens [23, 25], synthesis of BKIs [19, 26, 27], measurement of in vitro CpCDPK1 activity [19], pharmacokinetic analysis of plasma and fecal BKI concentrations [15, 21], in vitro investigation of BKI plasma protein binding [28], and determination of BKI efficacy in neonatal mice [21] and adult SCID/beige mice [20] have been previously described. Variances from published procedures are included in the Supplementary Materials and Methods.
C. parvum Growth Inhibition by BKIs
Assays to determine growth inhibition and the 50% effective concentration (EC50) were performed using a C. parvum strain UGA1 expressing Nluc in human ileocecal adenocarcinoma (HCT-8) cells (ATCC, Manassas, VA) as previously described [24] with the following modifications. HCT-8 cells were inoculated onto 96-well plates and allowed to grow for 48 hours to 90%–100% confluence. C. parvum oocysts were incubated for 10 minutes in 10% bleach at room temperature and washed with Dulbecco’s phosphate-buffered saline. One thousand oocysts per well were applied to plates with Roswell Park Memorial Institute 1640 (RPMI) medium supplemented with 10% horse serum and 1% penicillin/streptomycin at the same time as compound addition and incubated for 48 hours. The monolayers were lysed for 1 hour before Nano-Glo luciferase reagent (Promega, Madison, WI) was added, and relative luminescence units (RLU) were determined on an EnVision Plate Reader (Perkin Elmer, Waltham, MA). EC50 values were calculated using Prism (GraphPad, La Jolla, CA). Assays were repeated 2–3 times, and representative data are expressed as means ± standard deviation.
Efficacy in Adult Interferon γ (IFN-γ) Knockout (KO) Mice
Female IFN-γ KO mice (B6.129S7-Ifngtm1Ts/J, Jackson Laboratories) aged 8–10 weeks were infected by oral gavage with 1000–10000 C. parvum UGA1 oocysts carrying Nluc in 0.1 mL of Dulbecco’s phosphate-buffered saline. Beginning on day 3 or 6 after infection, mice were dosed orally with BKI suspended in 0.2 mL of oral vehicle (3% ethanol/7% Tween 80/90% saline) or vehicle only once daily for 5 days. Mice were moved to clean cages after each dose and fecal collection. Fecal specimens were collected daily and weighed from each group during dosing and twice weekly out to 20–21 days after infection. Each fecal sample was checked for luminescence on the day of collection [24], and RLU readings were normalized to fecal sample weights. For subcutaneous doses, the vehicle was 60% Phosal 53 MCT/30% PEG/10% ethanol, as oral vehicle can cause inflammation when injected (unpublished data). GraphPad Prism was used to determine the statistical significance of outcomes, using the Student t test and the correlation coefficient (r) for efficacy, with respect to systemic exposure (as measured by the area under curve [AUC]) and fecal concentrations.
Animal Ethics Statement
All animal experiments were approved by the institutional animal care and use committees at the participating universities (University of Washington, University of Georgia, University of Arizona, and University of Texas Medical Branch).
RESULTS
Lead BKIs were selected as potential Cryptosporidium therapies on the basis of favorable in vitro parameters, including c-Src off-target effects, mammalian cytotoxicity, hERG activity, and efficacy in the HCT-8 infection model (Table 1 and Figure 1A). Select lead candidates were evaluated for efficacy in neonatal mice and in plasma and fecal specimens from adult mice after oral dosage. BKIs were also eliminated on the basis of efficacy or exposure findings that approached or exceeded hERG or toxicity levels, as indicated by the in vitro results (Tables 1 and 2 and Figure 1A). Adult mouse efficacies were evaluated for all remaining BKIs, with analyses of efficacious compounds repeated to determine their effect at different treatment times, concentrations, and dose administrations (Figures 1–3).
Table 1.
In Vitro Properties and Neonatal Mouse Efficacy of Bumped-Kinase Inhibitors
| Compound | R1 | R2 | IC50, µM | C. parvum EC50, µM, Mean ± SD | CC50, µM | Percentage of Protein Bound in Plasma, by Species | Percentage Reduction in Parasite Burden in Neonatal Mice, Mean ± SD | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CpCDPK1 | c-SrC | hERG | HepG2 | CRL- 8155 | HCT-8 | Human | Mouse | |||||
| RM-1–22 |
|
|
0.059 | 0.121 | ND | 5.3 ± 0.3 | 30.9 | 10.4 | >20 | ND | ND | ND |
| RM-1–89 |
|
|
0.012 | 0.549 | >10 | 0.7 ± 0.2 | ND | 30 | >20 | ND | ND | ND |
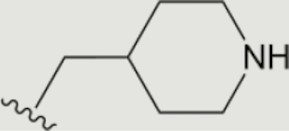
| 1243 |
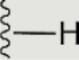
|
|
>5 | >10 | ND | >20 | ND | ND | >20 | ND | ND | ND |
| 1266 |
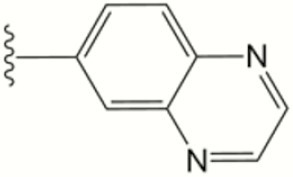
|
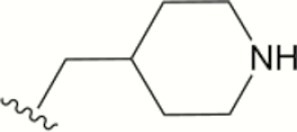
|
0.58 | >10 | ND | >80 | >40 | >40 | >20 | ND | ND | ND |
| 1294 |
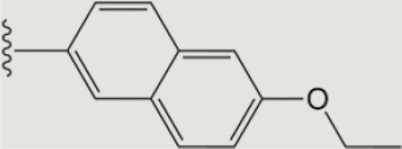
|
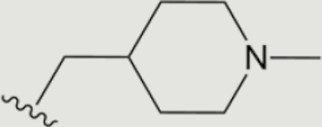
|
0.001 | >10 | 0.76 | 2.7 ± 1 | >40 | >40 | >20 | 85 | 84 | 56 ± 14 |
| 1318 |
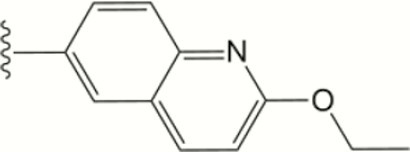
|
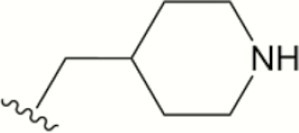
|
0.001 | >10 | 10 | 3.2 ± 0.5 | >40 | >40 | >20 | 77 | 55 | 69 ± 11 |
| 1369 |
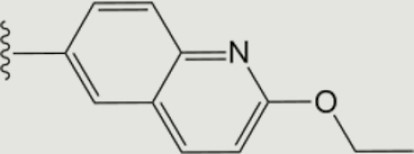
|
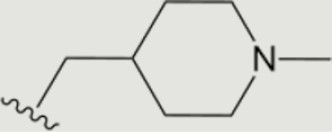
|
0.001 | >10 | 10 | 2.4 ± 0.7 | >40 | >40 | >20 | 77 | 37 | 74 ± 10 |
| 1412 |
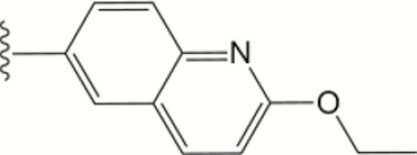
|
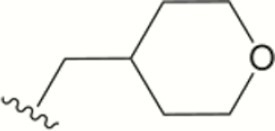
|
0.008 | >10 | >10 | 2.2 ± 0.6 | >40 | >40 | >20 | 94 | 85 | 14 ± 15 |
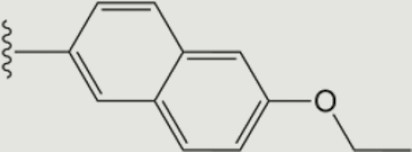
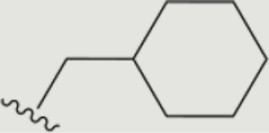
| 1416 |
|
|
0.289 | >10 | ND | 20 ± 2.8 | ND | ND | >20 | ND | ND | ND |
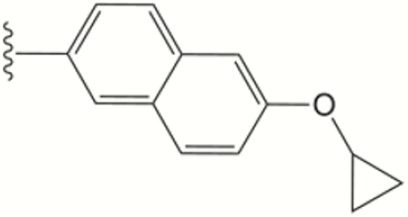
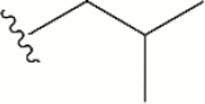
| 1534 |
|
|
0.005 | >10 | 25 | 2.2 ± 0.3 | >40 | 39.9 | >20 | 99.9 | 99.9 | 61 ± 13 |
| 1546 |
|

|
0.001 | >10 | >30 | 1.3 ± 0.3 | >40 | >40 | >20 | 92 | 81 | 33 ± 8 |
| 1550 |
|

|
0.001 | 7.4 | >30 | 5.5 ± 2.4 | >40 | >40 | >20 | 83 | 84 | 50 ± 14 |
| 1553 |
|
|
0.002 | >10 | >30 | 1.6 ± 0.3 | >40 | 39.5 | >20 | 91 | 90 | 36 ± 12 |
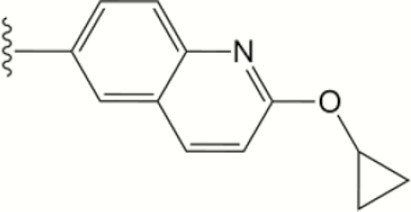
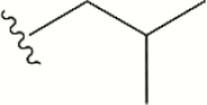
| 1556 |
|
|
0.003 | >10 | 25 | 3.2 ± 0.6 | >40 | 22.7 | >20 | 99.9 | 86 | 61 ± 11 |
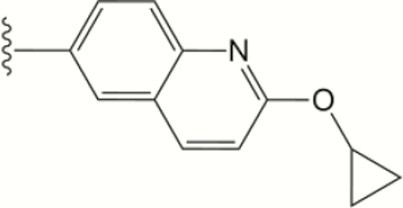
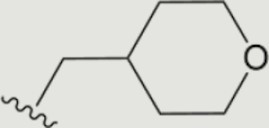
| 1557 |
|
|
0.007 | >10 | 14 | 5.5 ± 0.3 | >40 | 31.1 | >20 | 91 | 84 | 33 ± 11 |
| 1610 |
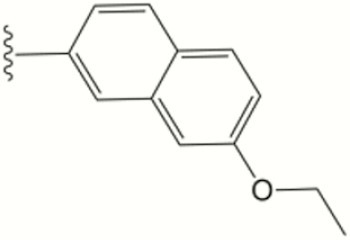
|
|
0.462 | ND | ND | 37.7 ± 5.4 | >40 | >40 | >20 | ND | ND | ND |
| 1624 |
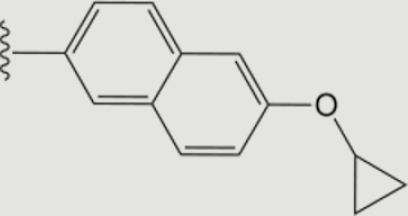
|
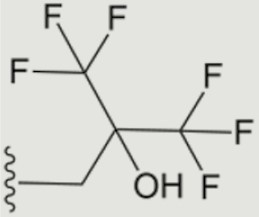
|
0.426 | ND | ND | >80 | >40 | >40 | >20 | ND | ND | ND |
| 1649a |
|
|
0.002 | >10 | >30 | 2.3 ± 1.3 | >40 | >40 | >20 | 56 | 90 | 56 ± 10 |
| 1663 |
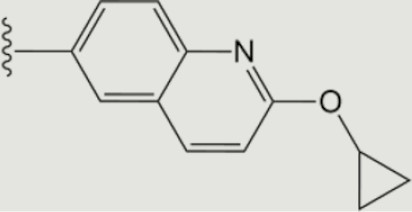
|
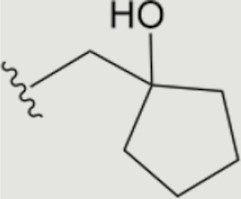
|
0.021 | >10 | 7.6 | 6.7 ± 0.5 | >40 | >40 | >20 | 96 | 86 | 76 ± 13 |
| Paromomycinb | … | … | … | … | … | … | … | … | … | … | … | 18 ± 14 |
The central scaffold of each compounds is , unless otherwise indicated.
Abbreviations: CC50, 50% cytotoxicity concentration; C. parvum, Cryptosporidium parvum; EC50, 50% effective concentration; IC50, 50% inhibitory concentration; ND, not determined.
aThe central scaffold is .
bParomomycin was selected as the standard comparator because it consistently yielded better activity than nitazoxanide in experimental mouse models.
Figure 1.

Criteria for continuing or ceasing additional investigation of bumped-kinase inhibitor (BKIs). A, The colors reflect distinct stages of evaluating BKIs for their suitability as a therapy for cryptosporidiosis. Criteria for advancement (ie, continuing additional investigation) are listed for each assay. B, Summary of the progression of each individual BKI. Colored blocks are filled in when the BKI satisfies the criteria for advancement and left blank when the BKI fails to meet the criteria and was eliminated from additional investigation. Abbreviations: CC50, 50% cytotoxicity concentration; Cmax, maximum concentration; CpCDPK1, Cryptosporidium parvum calcium-dependent protein kinase 1; C. parvum, Cryptosporidium parvum; EC50, 50% effective concentration; IC50, 50% inhibitory concentration; IFN-γ, interferon γ; KO, knockout. aIncluded as a control.
Table 2.
Pharmacokinetic Data of Bumped-Kinase Inhibitors (BKIs) in Uninfected Adult Mice After Oral Dosing
| BKI Compound, by Dosing Route | Dose, mg/kg | Cmax, µM | Tmax, min | AUC, µmol/L min | Half-life, min | Oral Clearance Rate, mL/min | Fecal Concentration, µM |
|---|---|---|---|---|---|---|---|
| Orala | |||||||
| 1294 | 10 | 0.75 | 120 | 430 | 205 | 1.69 | 4.4 |
| 1318 | 10 | 6.5 | 120 | 2209 | 909 | 0.16 | 0.44 |
| 1369 | 10 | 0.26 | 200 | 47 | 197 | 0.78 | 5.20 |
| 1412 | 10 | 0 | 0 | 0 | …b | …b | 1.83 |
| 1534 | 10 | 0.23 | 30 | 58 | 66 | 10.5 | 10.1 |
| 1546 | 10 | 0 | 0 | 0 | …b | …b | 5.29 |
| 1550 | 10 | 3.9 | 30 | 309 | 41 | 2.1 | 0.036 |
| 1553 | 10 | 12.8 | 320 | 13725 | 1188 | 0.02 | 0.52 |
| 1556 | 25 | 7.0 | 50 | 2082 | 320 | 0.56 | … |
| 1557 | 10 | 0.88 | 30 | 67.4 | 49 | 10.9 | 1.67 |
| 1649 | 10 | 4.9 | 400 | 6416 | 895 | 0.01 | 5.76 |
| Subcutaneousc | |||||||
| 1294 | 60 | 12.7 | 280 | 12249 | 6066 | … | 0.92 |
| 1534 | 60 | 4.2 | 180 | 3223 | 1831 | … | 0.44 |
Abbreviations: AUC, area under the curve; Cmax, maximum concentration; Tmax, time at which maximum concentration was observed.
a25 mg/kg dose.
bNo systemic exposure detected.
c60 mg/kg dose.
Enzymatic and Cellular Potency and Off-Target Cytotoxicity
The BKIs described here were tested for the inhibition of recombinant CpCDPK1 activity (Table 1). All BKIs were counter-screened against the mammalian tyrosine kinase c-Src and screened for mammalian cytotoxicity in vitro to eliminate toxic compounds. BKIs with activity against c-Src were eliminated because of concerns about off-target inhibition of mammalian kinases [29]. BKI 1294 had previously been reported to inhibit hERG at concentrations needed for efficacy, and hERG is a cardiotoxicity liability to be avoided [27, 30]. Accordingly, the remaining BKIs were screened for hERG inhibition by the thallium-flux assay for potassium channels [31, 32]. The BKIs listed in (Table 1) have a ≥10 fold higher hERG 50% inhibitory concentration (IC50) than BKI 1294 and thus have less potential for cardiotoxicity.
In Vitro Inhibition of Cryptosporidium Growth by BKIs
Nluc-expressing C. parvum oocysts were used to infect HCT-8 cells to assay for cellular BKI inhibition [24]. Compounds with an IC50 of >0.10 μM against recombinant CpCDPK1 enzyme showed little or no activity against the parasites in vitro, whereas compounds with a CpCDPK1 IC50 of <0.020 µM inhibited Cryptosporidium proliferation in the low micromolar range (Table 1). BKIs RM-1-22, RM-1-89, 1243, 1266, 1416, 1610, and 1624 were all eliminated from further examination because of poor activity, off-target c-Src inhibition, or mammalian cell cytotoxicity (Figure 1B).
BKI Efficacy Against C. parvum Infection in Neonatal Mice
Twelve BKIs, 1294, 1318, 1369, 1412, 1534, 1546, 1550, 1553, 1556, 1557, 1649, and 1663, have good C. parvum growth inhibition and little cytotoxicity or c-Src inhibition (Table 1). These BKIs were tested in a neonatal mouse model of C. parvum infection at an oral dose of 25 mg/kg given twice daily. For all compounds, infection levels decreased by 14%–76% relative to levels after vehicle treatment (Table 1). The least effective BKI in this series, 1412, reduced oocyst shedding by a level similar to that for paromomycin (Table 1). BKIs 1556, 1649, and 1663 showed signs of toxicity—specifically, protruding sternums—in infected neonatal mice. Although it is not intended to evaluate toxicity, the neonatal mouse model is practical for investigation and interpretation of data for various aspects of cryptosporidiosis in human infants, including potential harm from drug treatment. For this reason and its hERG-associated liability (Table 1), 1663 was eliminated from further consideration (Figure 1B). Compounds 1412, 1546, 1553, and 1557 were eliminated because of suboptimal efficacy in neonatal mice, defined as a <50% reduction of infection levels (Figure 1B).
BKI Levels in Mouse Plasma and Feces
Plasma and stool drug levels were determined after oral dosing in adult mice (Table 2). BKI 1556 was eliminated because its high oral exposure led to a concern that it would reach hERG-inhibiting plasma concentrations (Table 1 and Figure 1B). BKI 1649 was further evaluated at doses of <25 mg/kg twice daily, because 25 mg/kg twice daily was toxic in infected neonatal mice.
BKI Efficacy in the IFN-γ KO Mouse Model of C. parvum Infection
It is not yet clear which mouse model of Cryptosporidium infection predicts effective therapy in humans. In the IFN-γ KO mice model, infection is progressive, whereas immunocompetent neonatal mice are transiently infected for 10 days before the infection is cleared [24]. BKIs 1266, 1294, 1318, 1369, 1534, 1550, and 1649 were delivered orally for 5 days beginning at day 3 after infection (Figure 2A and 2B). Compound 1266 (CpCDPK1 IC50, 0.58 µM; C. parvum EC50, >80 µM) was included as a negative control and showed no clearance of C. parvum, compared with untreated controls (Figure 2A). In contrast, BKIs 1294, 1369, and 1534 significantly reduced parasite shedding (P < .002; Figure 2A and 2B). BKIs 1550 and 1318 (P < .5 for both) only slightly reduced the parasite load and were eliminated from further consideration (Figure 2B). BKI 1649 was delivered at a dose of 6 mg/kg/day, owing to its high oral exposure (Table 2) and neonatal mouse toxicity at 25 mg/kg twice daily, and still significantly reduced infection (P < .01) and was therefore worthy of further evaluation.
Figure 2.

Efficacy of bumped-kinase inhibitor (BKIs) in mouse models of Cryptosporidium parvum infection. A–C, Efficacies against C. parvum excretion in adult interferon γ knockout mice, based on nanoluciferase activity log RLU plotted on y-axis (A–C), and efficacy of BKI 1534 in SCID/beige mice, using reverse transcription–polymerase chain reaction to quantify infection (D). A, BKIs were delivered orally for 5 days, beginning 3 days after infection. In vivo inhibition by BKIs 1266, 1294, 1534, and 1550. BKI 1266, with limited inhibition of the C. parvum calcium-dependent protein kinase 1 target enzyme, was included as a negative control. BKIs 1294 and 1534 significantly reduced parasite shedding as compared to controls. The C. parvum infectious dose was 10000 oocysts. B and C, Significant initial reduction of parasite burden by BKIs, even in the case of higher fecal luciferase levels, which reflect higher parasites load. Parasite excretion rebounds when control animals show a higher infectious burden (B). BKIs 1294 (100 mg/kg once daily), 1369 (100 mg/kg once daily or 50 mg/kg twice daily), and 1649 (10 mg/kg once daily) efficacies (uninfected control mice had the highest recorded levels; C). BKIs substantially reduced Cryptosporidium proliferation, as shown by lower oocyst counts. A once-daily BKI 1369 dose of 100 mg/kg significantly reduced the infection burden (by 100000-fold), relative to that in untreated controls. BKI 1649 was administered at lower doses than other compounds owing to its high oral exposure, but it still significantly reduced infection relative to controls. The C. parvum infectious dose was 1000 oocysts. D, BKI 1534 delivered orally at a dose of 60 mg/kg for 5 days cleared infection with no recurrence. Assays were performed at the University of Georgia (Athens; A), the University of Washington (Seattle; B and C), and the University of Texas Medical Branch (Galveston; D). For assays with results in panels A and B, the percentage inhibition of infection, relative to that in controls, after treatment (day 10) was calculated for each compound as follows: 1294, 99.5%; 1534, 99.4%; and 1550, 59.7% (A); and 1294, 99.9%; 1318, 37.6%; 1369, 99.9%; and 1649, 98.9% (B). There were 3 mice in each experimental group.
No clinical signs of illness were observed in the infected IFN-γ KO mice, except for gradual weight loss and decreased activity in control and unsuccessfully treated mice. Infection intensities varied between the different IFN-γ KO mouse experiments. This can be observed by higher fecal luciferase levels in the second experiment (Figure 2B). The control mice referenced in (Figure 2B) lost ≥20% of their baseline body weight—the threshold at which euthanasia is required—by 20 days after infection, unlike the control mice referenced in (Figure 2A), where this threshold was not reached by the end of the experiment. The increased infection level was associated with relapsed oocyst shedding after the completion of dosing in some experiments. BKI 1294 administration consistently led to a significant reduction of infection, but a recurrent increase in parasite excretion was observed (Figure 2B). This was different than the initial experiment, in which no recurrent increase was noted with the same dose (Figure 2A). The greater relapse in the second experiment as compared to the first may have been due to the increased infection level, judging by the more rapid onset of infection and higher levels of luciferase signal. A recurrent rise in infection level was also observed after BKI 1369 treatment (Figure 2B), again perhaps due to the increased infection level. However, the level of infection after treatment remained at <15% of that for controls, and the mice appeared healthy for the duration of the experiment.
The efficacies of BKIs 1294, 1369, and 1649 were tested at higher oral doses (Figure 2C). BKI 1369 was administered at 100 mg/kg orally once daily or at 50 mg/kg orally twice daily, and BKI 1649 was delivered at 10 mg/kg orally once daily. Compound 1294 was included at 100 mg/kg orally once daily. The infection in this experiment was even more robust, with parasite loads in control mice rising to their highest recorded levels. The control mice had weight loss requiring euthanasia at day 13 after infection, a week earlier than the experiment described in Figure 2B. This infection intensity probably led to BKI 1294 having less apparent efficacy (Figure 2C) than in previous experiments (Figure 2A and 2B), while still significantly reducing infection (P < .05). Nonetheless, all BKIs had a substantial effect on infection (Figure 2C). BKI 1369 at 100 mg/kg once daily significantly reduced the infection (P < .05) to levels >100000 times lower than those in controls. BKI 1369, when administered at 50 mg/kg twice daily, considerably reduced infection levels (P < .05) but with a faster recurrence and greater total oocyst excretion, compared with the same total dose given once daily. BKI 1649 also significantly reduced infection levels (P < .05) at a dose of 10 mg/kg once daily.
SCID/Beige Mouse C. parvum Model
BKI 1534 was tested in the SCID/beige mouse C. parvum infection model. BKI 1534 given orally at 60 mg/kg for 5 days cleared the infection, with no recurrence (Figure 2D).
BKI Therapy of Highly Established Infection in the IFN-γ KO Mouse C. parvum Model
To test the effects of BKIs on higher infection levels, we evaluated the efficacy of our lead inhibitors by delaying dosing to day 6 after infection in IFN-γ KO mice. BKIs 1294, 1369, and 1534 were administered orally at 60 mg/kg once daily and BKI 1649 at 6 mg/kg once daily for 5 days. BKIs 1294 (P < .001), 1369 (P < .001), 1534 (P < .005), and 1649 (P < .001) all showed a significant decrease in the infection level (Figure 3A).
Figure 3.

Efficacy in adult interferon γ knockout mice with dosing starting on day 6 after infection. In all mice, the nanoluciferase-expressing Cryptosporidium parvum infectious dose was 1000 oocysts. A, Efficacy plots of bumped-kinase inhibitors (BKIs) 1294, 1369, 1534, and 1649 used for treatment of highly established infection when administered orally at 60 mg/kg once daily for 5 days. BKI 1649 was administered at 6 mg/kg once daily. All BKIs showed significant decreases in infection. B, Efficacy of subcutaneous versus oral dosing of mice with 1294 at 60 mg/kg, starting on day 6 after infection, for 5 days. BKI 1294 showed a significantly greater reduction in oocyst excretion when delivered orally as compared to an equivalent dose delivered subcutaneously (P < .05). This is probably due to a fecal concentration of orally administered 1294 that was higher than the 50% effective concentration (EC50) early in the treatment period and to the longer presence of the orally administered 1294 in the lumen relative to the subcutaneously administered dose, for which the level after a single dose was below the EC50. C, Efficacy of subcutaneous versus oral dosing with 1534 at 60 mg/kg, starting on day 6 after infection, for 5 days. No significant difference was seen in parasite excretion between the groups receiving the BKI doses.
Comparison of Subcutaneous Versus Oral Administration of BKIs
It is unclear whether the optimal anti-Cryptosporidium agent should be absorbed systemically and/or retained in the gastrointestinal tract [33]. To evaluate subcutaneous administration, a single 60 mg/kg dose of BKI 1294 or 1534 was administered to uninfected mice. Plasma and fecal levels were measured and compared to oral dose pharmacokinetics (Tables 2 and 3). Subcutaneous delivery of BKI 1294 and 1534 resulted in higher systemic exposure and lower fecal concentrations than oral delivery, even when considering the different doses administered (10 mg/kg orally vs 60 mg/kg subcutaneously in single-dose pharmacokinetics analyses). To compare the efficacy of subcutaneous versus oral dosing, mice were given BKI 1294 or 1534 at 60 mg/kg once daily orally or subcutaneously for 5 days, starting on day 6 after infection (Figure 3B and 3C). Fecal specimens were collected 24 hours after the fifth dose, to measure BKI concentrations (Table 3). The fecal concentration of BKI 1294 was only marginally higher after the fifth dose delivered subcutaneously, compared with the pharmacokinetics of the subcutaneously delivered single dose, while the fecal concentration of BKI 1534 was >10-fold higher after subcutaneous delivery of the fifth dose, compared with pharmacokinetics of the single dose administered subcutaneously (Table 3). BKI 1294 showed a significantly greater reduction in infection when delivered orally, compared with an equivalent dose delivered subcutaneously (P < .05; Figure 3B). However, BKI 1534 did not show a significant difference in parasite excretion between groups dosed orally and subcutaneously (Figure 3C).
Table 3.
Single-Dose Fecal Concentrations of Bumped-Kinase Inhibitors 1294 and 1534 in Uninfected Adult Mice, Compared With the Fifth-Dose Fecal Concentrations in Infected Adult Mice
| Infection Status, Dose | Concentration, µM | |
|---|---|---|
| 1294 | 1534 | |
| Uninfected, single dosea | ||
| 25 mg/kg orally | 4.4 | 10.1 |
| 60 mg/kg subcutaneously | 0.92 | 0.44 |
| Infected, fifth doseb | ||
| 60 mg/kg orally | 4.1 | 17.8 |
| 60 mg/kg subcutaneously | 1.1 | 4.7 |
aConcentration (weight/volume) in pooled fecal specimens from adult uninfected mice given a single oral or subcutaneous dose.
bConcentration (weight/volume) in pooled fecal specimens from interferon γ knockout mice infected with Cryptosporidium parvum after the fifth oral or subcutaneous dose.
DISCUSSION
A challenge to the development of effective therapy for Cryptosporidium infection is the differential efficiency of parasite clearance in various in vitro and in vivo models [37, 38]. A number of drug candidates have shown good inhibitory activity in vitro but have demonstrated low potency in clinical models and were not further considered [39–41]. In addition to C. parvum’s natural resistance to many drug therapies, its characteristic subcellular location on the apical-surface of mammalian intestinal epithelium cells may constitute a barrier to effective drug delivery, preventing drugs from reaching therapeutic concentrations in the parasite [42, 43]. This provides the rationale for targeting enzymes such as CpCDPK1 that phenotypically affect extracellular stages.
Recently, a lack of correlation between anti-Cryptosporidium and CpCDPK1 enzyme inhibition for some pyrazolo[2,3-d]pyrimidine (PP) compounds was reported, suggesting potential alternative-target activity [34]. The observed c-Src activity for some BKIs supports this potential for off-target kinase activity. BKI 1243 does not have a bumped R1 aryl substituents at the C-3 position. For all other BKIs used in this study, especially with bumped R1, which project into CpCDPK1’s enlarged glycine gatekeeper hydrophobic pocket, there is a statistically significant correlation between the CpCDPK1 IC50 and the C. parvum cellular EC50 (r16 = 0.93; P < .0001). This suggests that the R1 groups are important in keeping these PP BKIs on target (Figure 4). Additionally, antisense methods were recently used to reduce CpCDPK1 expression [35, 36]. These experiments demonstrated that knocking down expression of CDPK1 significantly decreased parasite invasion and growth, and there was an additive effect with BKIs in reducing the proliferation of C. parvum parasites for BKIs 1294 (PP) and 1517 (an aminopyrazole-carboxamide compound) [36]. This potentiation of BKI activity when CpCDPK1 expression is reduced is further evidence that CpCDPK1 is a target of our BKIs. Further chemical-genetic validation experiments are needed to test the hypothesis that CpCDPK1 is the primary target of our selective BKIs.
Figure 4.

Comparison of the in vitro Cryptosporidium parvum calcium-dependent protein kinase 1 (CpCDPK1) 50% inhibitory concentration (IC50) to the C. parvum 50% effective concentration (EC50). A, Graphed cellular C. parvum EC50 versus CpCDPK1 IC50 from Table 1. Bumped-kinase inhibitor 1243 was excluded because of its lack of an R1 group. Values show a statistically significant correlation (r16 = 0.93; P < .0001). B, Values graphed on logarithmic scale to better show the separation of IC50 values <0.1 mM.
There is an observed difference between EC50 values against C. parvum in vitro determined using a reverse transcription–quantitative polymerase chain reaction (RT-qPCR) assay [20, 36] and those determined by the Nluc assay reported here. This could be attributed to the end point readout of the RT-qPCR assay: RNA may degrade faster than the luciferase protein used in the Nluc assay. Thus, delayed degradation of luciferase protein could underestimate compound potency, compared with that determined by RT-qPCR. Nevertheless, luminescence readouts from Nluc-expressing C. parvum have been previously demonstrated to correlate with the level of oocyst shedding determined by real-time qPCR using 18S ribosomal RNA primers and DNA extracted from fecal material [24].
The hERG IC50 of 10 µM for BKI 1369 was initially a reason for concern in using the compound in humans. Previous studies of hERG suggests that the risk of arrhythmia is reduced for most drugs if the hERG IC50 is >30-fold higher than the effective free therapeutic plasma concentration [47]. BKI 1369 displayed a maximum concentration of 0.26 μM at 10 mg/kg, which was 40-fold less than its hERG IC50. Additionally, BKI 1369 is 76% protein bound in human plasma (Table 1). The low maximum concentration and unbound fraction of BKI 1369 should mitigate the risks of hERG inhibition [47]. BKI 1294 was efficacious in SCID/beige mice [20] and neonatal mice (Table 1) and had significant therapeutic effects in the calf clinical model of cryptosporidiosis [21]. The efficacy of BKI 1294 was also demonstrated using IFN-γ KO mice infected with Nluc-expressing C. parvum. BKI 1294 was associated with relapses after treatment in both SCID/beige [20] and IFN-γ KO mouse models. In contrast, no relapses were detected with BKI 1534. The congruence of results across models supports the validity of the IFN-γ KO model for drug development purposes. A likely explanation for recrudescence, especially after high levels of infection are cleared, is the limited immunological resistance of this mouse strain. They will readily reinfect themselves, owing to factors such as oocysts on fur or cage bedding. It is therefore impossible to determine whether infections are recurring because of incomplete clearance or reinfection from grooming, coprophagy, or exposure to fecal matter from cohabitating mice. Future exploration into whether these BKIs will maintain their highly efficacious results in larger mammals is warranted.
Differences in efficacy between the neonatal and immunocompromised adult mice were observed for some compounds, including BKIs 1318 and 1550, which could be due to a number of reasons. These include the transient neonatal versus chronic IFN-γ KO infection and differential physiological characteristics relating to insufficiently developed neonatal organ-specific drug transporters and metabolic systems that may affect drug absorption, distribution, and elimination [44–46]. Furthermore, BKIs tested showed a lack of association between neonatal efficacy and plasma or fecal exposure in adult mice. Nonetheless, intraspecies metabolic barriers may have limited impact on the therapeutic outcome for treatment of cryptosporidiosis with BKIs, based on successful results of recent calf and SCID/beige mouse investigations [20, 21, 35].
In IFN-γ KO mice, there is a lack of correlation between BKI systemic exposure (AUC) and Cryptosporidium efficacy (r5 = –0.02; P < 1). In contrast, BKIs with high fecal concentrations (Table 2) were efficacious. BKIs 1318, 1550, 1553, and 1649 display the largest plasma AUCs of all compounds tested, yet BKI 1649 proved to be the only compound that was efficacious. BKIs 1318 and 1550 failed to significantly lower infection levels in IFN-γ KO mice, and BKI 1553 was the least effective compound of those tested in neonatal calves [21]. All 3 BKIs showed fecal concentrations in mice below their respective 50% effective concentrations (Table 1). However, the fecal concentration of BKI 1649 was much higher at 5.76 µM (Table 2). All other BKIs tested that had higher fecal concentrations, including BKIs 1294 (4.4 µM), 1369 (5.2 µM), and 1534 (10.1 µM), were efficacious in mice despite low systemic exposures. This demonstrates that PP-BKI fecal levels correlate with efficacy against Cryptosporidium organisms (r5 = 0.78; P < .05), in line with the fact that infections localize at the lumen interface within the gut [42, 48]. Since fecal levels are imperfect estimators of exposure in the gastrointestinal tract, future studies are necessary to examine properties beyond plasma and fecal levels to thoroughly explain the pharmacodynamics of successful therapies for cryptosporidiosis.
For efficacy comparison between oral and subcutaneous doses, the BKI 1294 fecal concentration after the fifth dose was similar to its value after a single dose (Table 3). Fecal exposure of BKI 1294 given subcutaneously was around 1 µM, well below the 50% effective concentration for this compound. This offers a possible explanation for the subcutaneous dose being less efficacious than the equivalent oral dose. For BKI 1534, the fecal concentration appeared to rise, with pharmacokinetic analysis of the subcutaneously delivered single dose yielding a concentration of 0.44 µM, compared with 4.76 µM after the fifth dose (Table 3). This potentially explains the lack of significant difference between subcutaneous and oral delivery of BKI 1534 in reducing infection, as subcutaneous administration showed that the BKI concentration in feces increased to therapeutic levels by the end of dosing. This could provide an advantage over BKI 1294 in species for which oral dosing is difficult. Also, the hERG-associated liability is significantly reduced for BKI 1534 (25 µM) as compared to BKI 1294 (0.76 µM). Overall, the 2 compounds are equally efficacious in this mouse model. Given the localization of Cryptosporidium infection in the gastrointestinal tract in immunocompetent hosts but biliary and respiratory sites in immunocompromised hosts [49, 50], some systemic absorption is important for optimal efficacy. Further investigations of different dosing routes and formulations are needed, especially in terms of systemic as compared to gastrointestinal exposure.
Supplementary Data
Supplementary materials are available at The Journal of Infectious Diseases online. Consisting of data provided by the authors to benefit the reader, the posted materials are not copyedited and are the sole responsibility of the authors, so questions or comments should be addressed to the corresponding author.
Supplementary Material
Notes
Acknowledgments. We thank Geno De Hostos and Robert Choy (PATH Drug Solutions), for their consultation in the drug development pathway, and Gail M. Freiberg (AbbVie) for establishing hERG inhibition of the BKIs.
Disclaimer. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health or the US Department of Agriculture National Institute of Food and Agriculture.
Financial support. This work was supported by the National Institute of Allergy and Infectious Diseases, National Institutes of Health (awards R01AI089441, R01AI111341, R01A1112427 and R01HD080670), BMGF OPP 1134302, and the US Department of Agriculture National Institute of Food and Agriculture (award 2014–06183).
Potential conflicts of interest. W. C. V. V. is the president of ParaTheraTech, a company involved in developing BKIs for use in animal health. All other authors report no potential conflicts. The author has submitted the ICMJE Form for Disclosure of Potential Conflicts of Interest. Conflicts that the editors consider relevant to the content of the manuscript have been disclosed.
References
- 1. Kotloff KL, Nataro JP, Blackwelder WC, et al. Burden and aetiology of diarrhoeal disease in infants and young children in developing countries (the Global Enteric Multicenter Study, GEMS): a prospective, case-control study. Lancet 2013; 382:209–22. [DOI] [PubMed] [Google Scholar]
- 2. Checkley W, White AC, Jr, Jaganath D, et al. A review of the global burden, novel diagnostics, therapeutics, and vaccine targets for cryptosporidium. Lancet Infect Dis 2015; 15:85–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Platts-Mills JA, Babji S, Bodhidatta L, et al. Pathogen-specific burdens of community diarrhoea in developing countries: a multisite birth cohort study (MAL-ED). Lancet Glob Health 2015; 3:e564–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. White AC. Cryptosporidium species. In: Bennett JE, Dolin R, Blaser MJ, eds. Mandell, Douglas, and Bennett’s principles and practice of infectious diseases. 8th ed Philadelphia, PA: Elsevier/Saunders, 2015. [Google Scholar]
- 5. Mac Kenzie WR, Hoxie NJ, Proctor ME, et al. A massive outbreak in Milwaukee of cryptosporidium infection transmitted through the public water supply. N Engl J Med 1994; 331:161–7. [DOI] [PubMed] [Google Scholar]
- 6. Okhuysen PC, Chappell CL, Crabb JH, Sterling CR, DuPont HL. Virulence of three distinct Cryptosporidium parvum isolates for healthy adults. J Infect Dis 1999; 180:1275–81. [DOI] [PubMed] [Google Scholar]
- 7. DuPont HL, Chappell CL, Sterling CR, Okhuysen PC, Rose JB, Jakubowski W. The infectivity of Cryptosporidium parvum in healthy volunteers. N Engl J Med 1995; 332:855–9. [DOI] [PubMed] [Google Scholar]
- 8. Rossignol JF, Ayoub A, Ayers MS. Treatment of diarrhea caused by Cryptosporidium parvum: a prospective randomized, double-blind, placebo-controlled study of nitazoxanide. J Infect Dis 2001; 184:103–6. [DOI] [PubMed] [Google Scholar]
- 9. Sparks H, Nair G, Castellanos-Gonzalez A, White AC., Jr Treatment of cryptosporidium: what we know, gaps, and the way forward. Curr Trop Med Rep 2015; 2:181–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Amadi B, Mwiya M, Musuku J, et al. Effect of nitazoxanide on morbidity and mortality in Zambian children with cryptosporidiosis: a randomised controlled trial. Lancet 2002; 360:1375–80. [DOI] [PubMed] [Google Scholar]
- 11. Pavlinac PB, John-Stewart GC, Naulikha JM, et al. High-risk enteric pathogens associated with HIV infection and HIV exposure in Kenyan children with acute diarrhoea. AIDS 2014; 28:2287–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lourido S, Tang K, Sibley LD. Distinct signalling pathways control Toxoplasma egress and host-cell invasion. EMBO J 2012; 31:4524–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ojo KK, Larson ET, Keyloun KR, et al. Toxoplasma gondii calcium-dependent protein kinase 1 is a target for selective kinase inhibitors. Nat Struct Mol Biol 2010; 17:602–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kieschnick H, Wakefield T, Narducci CA, Beckers C. Toxoplasma gondii attachment to host cells is regulated by a calmodulin-like domain protein kinase. J Biol Chem 2001; 276:12369–77. [DOI] [PubMed] [Google Scholar]
- 15. Ojo KK, Pfander C, Mueller NR, et al. Transmission of malaria to mosquitoes blocked by bumped kinase inhibitors. J Clin Invest 2012; 122:2301–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Billker O, Dechamps S, Tewari R, Wenig G, Franke-Fayard B, Brinkmann V. Calcium and a calcium-dependent protein kinase regulate gamete formation and mosquito transmission in a malaria parasite. Cell 2004; 117:503–14. [DOI] [PubMed] [Google Scholar]
- 17. Billker O, Lourido S, Sibley LD. Calcium-dependent signaling and kinases in apicomplexan parasites. Cell Host Microbe 2009; 5:612–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lourido S, Shuman J, Zhang C, Shokat KM, Hui R, Sibley LD. Calcium-dependent protein kinase 1 is an essential regulator of exocytosis in Toxoplasma. Nature 2010; 465:359–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Murphy RC, Ojo KK, Larson ET, et al. Discovery of potent and selective inhibitors of calcium-dependent protein kinase 1 (CDPK1) from C. parvum and T. gondii. ACS Med Chem Lett 2010; 1:331–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Castellanos-Gonzalez A, White AC, Jr, Ojo KK, et al. A novel calcium-dependent protein kinase inhibitor as a lead compound for treating cryptosporidiosis. J Infect Dis 2013; 208:1342–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Schaefer DA, Betzer DP, Smith KD, et al. Novel bumped kinase inhibitors are safe and effective therapeutics in the calf clinical model for cryptosporidiosis. J Infect Dis 2016; 214:1856–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Riggs MW, McGuire TC, Mason PH, Perryman LE. Neutralization-sensitive epitopes are exposed on the surface of infectious Cryptosporidium parvum sporozoites. J Immunol 1989; 143:1340–5. [PubMed] [Google Scholar]
- 23. Riggs MW, Perryman LE. Infectivity and neutralization of Cryptosporidium parvum sporozoites. Infect Immun 1987; 55:2081–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Vinayak S, Pawlowic MC, Sateriale A, et al. Genetic modification of the diarrhoeal pathogen Cryptosporidium parvum. Nature 2015; 523:477–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Arrowood MJ, Donaldson K. Improved purification methods for calf-derived Cryptosporidium parvum oocysts using discontinuous sucrose and cesium chloride gradients. J Eukaryot Microbiol 1996; 43:89S. [DOI] [PubMed] [Google Scholar]
- 26. Johnson SM, Murphy RC, Geiger JA, et al. Development of Toxoplasma gondii calcium-dependent protein kinase 1 (TgCDPK1) inhibitors with potent anti-toxoplasma activity. J Med Chem 2012; 55:2416–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Vidadala RS, Rivas KL, Ojo KK, et al. Development of an orally available and central nervous system (CNS) penetrant Toxoplasma gondii calcium-dependent protein kinase 1 (TgCDPK1) inhibitor with minimal human ether-a-go-go-related gene (hERG) activity for the treatment of toxoplasmosis. J Med Chem 2016; 59:6531–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tatipaka HB, Gillespie JR, Chatterjee AK, et al. Substituted 2-phenylimidazopyridines: a new class of drug leads for human African trypanosomiasis. J Med Chem 2014; 57:828–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Keyloun KR, Reid MC, Choi R, et al. The gatekeeper residue and beyond: homologous calcium-dependent protein kinases as drug development targets for veterinarian Apicomplexa parasites. Parasitology 2014; 141:1499–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ojo KK, Eastman RT, Vidadala R, et al. A specific inhibitor of PfCDPK4 blocks malaria transmission: chemical-genetic validation. J Infect Dis 2014; 209:275–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bowes J, Brown AJ, Hamon J, et al. Reducing safety-related drug attrition: the use of in vitro pharmacological profiling. Nat Rev Drug Discov 2012; 11:909–22. [DOI] [PubMed] [Google Scholar]
- 32. Bridal TR, Margulis M, Wang X, Donio M, Sorota S. Comparison of human Ether-à-go-go related gene screening assays based on IonWorks Quattro and thallium flux. Assay Drug Dev Technol 2010; 8:755–65. [DOI] [PubMed] [Google Scholar]
- 33. Bessoff K, Spangenberg T, Foderaro JE, Jumani RS, Ward GE, Huston CD. Identification of Cryptosporidium parvum active chemical series by Repurposing the open access malaria box. Antimicrob Agents Chemother 2014; 58:2731–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kuhlenschmidt TB, Rutaganira FU, Long S, et al. Inhibition of calcium-dependent protein kinase 1 (CDPK1) in vitro by pyrazolopyrimidine derivatives does not correlate with sensitivity of Cryptosporidium parvum growth in cell culture. Antimicrob Agents Chemother 2015; 60:570–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Castellanos-Gonzalez A, Perry N, Nava S, White AC., Jr Preassembled single-stranded RNA-argonaute complexes: a novel method to silence genes in cryptosporidium. J Infect Dis 2016; 213:1307–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Castellanos-Gonzalez A, Sparks H, Nava S, et al. A novel calcium-dependent kinase inhibitor, bumped kinase inhibitor 1517, cures cryptosporidiosis in immunosuppressed mice. J Infect Dis 2016; 214:1850–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Woods KM, Nesterenko MV, Upton SJ. Efficacy of 101 antimicrobials and other agents on the development of Cryptosporidium parvum in vitro. Ann Trop Med Parasitol 1996; 90:603–15. [DOI] [PubMed] [Google Scholar]
- 38. Rehg JE. Anti-cryptosporidial activity of macrolides in immunosuppressed rats. J Protozool 1991; 38:228–30S. [PubMed] [Google Scholar]
- 39. Soave R. Treatment strategies for cryptosporidiosis. Ann N Y Acad Sci 1990; 616:442–51. [DOI] [PubMed] [Google Scholar]
- 40. Portnoy D, Whiteside ME, Buckley E, 3rd, MacLeod CL. Treatment of intestinal cryptosporidiosis with spiramycin. Ann Intern Med 1984; 101:202–4. [DOI] [PubMed] [Google Scholar]
- 41. Wittenberg DF, Miller NM, van den Ende J. Spiramycin is not effective in treating cryptosporidium diarrhea in infants: results of a double-blind randomized trial. J Infect Dis 1989; 159:131–2. [DOI] [PubMed] [Google Scholar]
- 42. Mead JR. Cryptosporidiosis and the challenges of chemotherapy. Drug Resist Updat 2002; 5:47–57. [DOI] [PubMed] [Google Scholar]
- 43. Griffiths JK, Balakrishnan R, Widmer G, Tzipori S. Paromomycin and geneticin inhibit intracellular Cryptosporidium parvum without trafficking through the host cell cytoplasm: implications for drug delivery. Infect Immun 1998; 66:3874–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. van den Anker JN, Schwab M, Kearns GL. Developmental pharmacokinetics. Handb Exp Pharmacol 2011; 205:51–75. [DOI] [PubMed] [Google Scholar]
- 45. Mooij MG, Schwarz UI, de Koning BA, et al. Ontogeny of human hepatic and intestinal transporter gene expression during childhood: age matters. Drug Metab Dispos 2014; 42:1268–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Smits A, Annaert P, Allegaert K. Drug disposition and clinical practice in neonates: cross talk between developmental physiology and pharmacology. Int J Pharm 2013; 452:8–13. [DOI] [PubMed] [Google Scholar]
- 47. Redfern WS, Carlsson L, Davis AS, et al. Relationships between preclinical cardiac electrophysiology, clinical QT interval prolongation and torsade de pointes for a broad range of drugs: evidence for a provisional safety margin in drug development. Cardiovasc Res 2003; 58:32–45. [DOI] [PubMed] [Google Scholar]
- 48. Marcial MA, Madara JL. Cryptosporidium: cellular localization, structural analysis of absorptive cell-parasite membrane-membrane interactions in guinea pigs, and suggestion of protozoan transport by M cells. Gastroenterology 1986; 90:583–94. [DOI] [PubMed] [Google Scholar]
- 49. Clifford CP, Crook DW, Conlon CP, Fraise AP, Day DG, Peto TE. Impact of waterborne outbreak of cryptosporidiosis on AIDS and renal transplant patients. Lancet 1990; 335:1455–6. [DOI] [PubMed] [Google Scholar]
- 50. Hunter PR, Nichols G. Epidemiology and clinical features of Cryptosporidium infection in immunocompromised patients. Clin Microbiol Rev 2002; 15:145–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.