

Abstract
Objective. To identify muscle gene expression patterns that predict rituximab responses and assess the effects of rituximab on muscle gene expression in PM and DM.
Methods. In an attempt to understand the molecular mechanism of response and non-response to rituximab therapy, we performed Affymetrix gene expression array analyses on muscle biopsy specimens taken before and after rituximab therapy from eight PM and two DM patients in the Rituximab in Myositis study. We also analysed selected muscle-infiltrating cell phenotypes in these biopsies by immunohistochemical staining. Partek and Ingenuity pathway analyses assessed the gene pathways and networks.
Results. Myeloid type I IFN signature genes were expressed at higher levels at baseline in the skeletal muscle of rituximab responders than in non-responders, whereas classic non-myeloid IFN signature genes were expressed at higher levels in non-responders at baseline. Also, rituximab responders have a greater reduction of the myeloid and non-myeloid type I IFN signatures than non-responders. The decrease in the type I IFN signature following administration of rituximab may be associated with the decreases in muscle-infiltrating CD19 + B cells and CD68 + macrophages in responders.
Conclusion. Our findings suggest that high levels of myeloid type I IFN gene expression in skeletal muscle predict responses to rituximab in PM/DM and that rituximab responders also have a greater decrease in the expression of these genes. These data add further evidence to recent studies defining the type I IFN signature as both a predictor of therapeutic responses and a biomarker of myositis disease activity.
Keywords: myositis, rituximab, biomarker, IFN signature
Rheumatology key messages
PM/DM patients who responded to rituximab had higher levels of myeloid type I IFN pathway gene expression in muscle.
Rituximab responders had a greater decrease in the expression of myeloid type I IFN genes.
Type I IFN signalling in skeletal muscle cells may play a role in the pathogenesis of myositis.
Introduction
PM and DM are characterized by chronic inflammation in the muscle and the frequent finding of selected autoantibodies [ 1 ]. Immunosuppressive medications are currently used as therapies for these conditions, however, a large number of patients do not respond completely to the current therapies. A randomized, double-blind, placebo-controlled clinical trial was conducted to assess the efficacy of rituximab in refractory adult and juvenile myositis patients using validated measures of disease activity and a data- and consensus-driven definition of improvement (DOI) [ 2 ]. Although there were no statistically significant differences in the primary or secondary endpoints between the two treatment arms, 83% of refractory adult and juvenile myositis patients met the DOI [ 3 ]. Clinical response to rituximab in myositis (RIM) has been predicted by anti-synthetase and anti-Mi-2 autoantibodies [ 4 ].
Likewise, recent studies indicate that the type I IFN gene and chemokine scores and the levels of pro-inflammatory cytokines (IL-6, IL-8 and TNF-α) may serve as sensitive and responsive longitudinal biomarkers of change in disease activity in juvenile and adult DM [ 5 ]. Type I IFN originates from as well as affects both myeloid and non-myeloid cells. Innate immune myeloid cells rapidly respond to inflammatory stimuli in injured tissues, including skeletal muscle. It is well known that type I IFN signalling significantly alters dynamics of myeloid cells in the injured tissues. For example, Type I IFN initiates arrival of innate myeloid cells in the brains of lymphocytic choriomeningitis virus (LCMV)-infected mice. Over a period of time, the myeloid cell population decreases and returns to a near-normal state in the LCMV-infected brains. This decrease in myeloid cell population coincides with reduced type I IFN production, suggesting type I IFN signalling is responsible for innate myeloid cell dynamics. In fact, LCMV-infected brains of IFNR null mice behave like mock-infected controls, suggesting type I IFN signalling completely controls innate immune activity in injured tissues [ 6 ]. Therefore, we propose that therapies that aim to reduce inflammation should reduce myeloid cells as well as the myeloid type I IFN signature.
To assess the mechanism of response and non-response to rituximab therapy, we performed the first muscle gene expression profiling and analyses before and after treatment in refractory PM and DM patients. Since myeloid cells are known to significantly contribute to type I IFN signature, we specifically investigated the responsiveness of myeloid- and non-myeloid-associated type I IFN signatures in these patients.
Methods
Patients
Treatment-refractory adult PM and adult and juvenile DM patients meeting probable or definite Bohan and Peter criteria and with evidence of moderate disease activity refractory to prednisone and at least one other agent were enrolled into this institutional review board–approved study [ 3 ]. Adult subjects (eight with PM, two with DM) enrolled at the National Institutes of Health and who agreed to have research muscle biopsies underwent surgical thigh muscle biopsies pre- and post-rituximab therapy. This study was approved by the National Institute of Diabetes and Digestive and Kidney Diseases/National Institute of Arthritis and Musculoskeletal and Skin Diseases institutional review board, and all patients signed informed consent in accordance with the Helsinki Declaration.
In this trial, a baseline muscle biopsy was done just prior to rituximab treatment at week 0 and the follow-up biopsy was done at week 16. The randomized placebo-controlled design involved group A receiving rituximab at weeks 0 and 1 and group B receiving it at weeks 8 and 9; both groups were included in our results. Although there were differences in the number of weeks post-therapy before the biopsy, there were no significant differences in the findings between the groups. Muscle biopsy specimens were flash-frozen in liquid nitrogen–cooled isopentane and then stored at −80ºC until use. Clinical response to rituximab was measured by meeting the DOI at week 16. Concomitant therapy with other immunosuppressive agents (prednisone, MTX, AZA or HCQ) was equally distributed between the responder and non-responder groups ( Table 1 ).
T able 1 .
Demographics, response status and other selected features of myositis patients
| Patient no. | Age, years | Gender | DX | Responder a | Anti-Jo-1 or SRP autoantibody present | Therapy |
|---|---|---|---|---|---|---|
| 1 | 75 | F | DM | No | None | PD, PQ, AZA |
| 2 | 59 | M | PM | No | None | PD, AZA |
| 3 | 70 | F | PM | No | None | PD, MTX |
| 4 | 42 | F | PM | No | SRP | PD, MTX |
| 5 | 66 | F | PM | No | None | PD, MTX, AZA |
| 6 | 50 | F | PM | Yes | None | PD, MTX, AZA |
| 7 | 46 | F | PM | Yes | Jo-1 | PD, MTX |
| 8 | 48 | M | DM | Yes | SRP | PD, MTX, AZA |
| 9 | 74 | F | PM | Yes | Jo-1 | PD, MTX |
| 10 | 60 | F | PM | Yes | SRP | PD, MTX, AZA |
a Responders or non-responders to treatment as determined by the primary definition of improvement at week 16. Dx: diagnosis; PD: prednisone; PQ: plaquenil.
RNA extraction and gene expression profiling
Muscle biopsy samples were subjected to total RNA isolation by use of TRIzol reagent (Invitrogen, Carlsbad, CA, USA) followed by the RNeasy MiniElute clean-up kit, and the quality of the RNA samples was assessed with an Agilent 2100 Bioanalyzer (Agilent Technologies., Santa Clara, CA, USA). Gene expression profiling was performed using the GeneChip approach (Affymetrix, Santa Clara, CA, USA).
Partek and Ingenuity pathway analyses
To generate expression values for probe sets, GeneChip-derived CEL files were analysed with the Probe Logarithmic Intensity Error algorithm in Expression Console software (Affymetrix, Santa Clara, CA, USA). Probe Logarithmic Intensity Error algorithm–derived probe sets’ signal intensity values were uploaded directly into the Partek Genomics Suite, version 6.5 (Partek, St Louis, MO, USA) for statistics and data visualization. Differences in gene expression levels between responder and non-responder patients before treatment (at baseline) were analysed using a one-way ANOVA model. A parametric paired-sample t-test was used to test the significance of differences in gene expression levels before and after RIM treatment between patients, who did or did not respond to the treatment. To identify significant molecular networks and pathways, we used an Ingenuity Pathways Analysis software application (Ingenuity Systems, Redwood City, CA, USA). After this analysis, networks generated were ordered by a score denoting significance.
Immunohistochemical staining
Frozen human muscle biopsy specimens of myositis (PM and DM patients; n = 8 and 2, respectively) were obtained before and after rituximab treatment. Muscle tissues were sectioned and fixed in ice-cold acetone for 5 min, and immunostaining was performed using the Vectastain Elite ABC kit (Vector Laboratories, Burlingame, CA, USA) using monoclonal mouse anti-human CD19 (Dako, Carpinteria, CA, USA) (1:50 dilution), monoclonal mouse anti-human CD68 (Dako) (1:50 dilution), monoclonal mouse anti-human CD138 (Dako) (1:50 dilution), rabbit anti-IPS1, rabbit IFN-β (Millipore, Billerica, MA, USA), rabbit anti-human MX1 (GeneTex, Irvine, CA, USA) and horseradish peroxidase–conjugated polyclonal rabbit anti-mouse immunoglobulins (Dako) as the primary and secondary antibodies, respectively. A semi-quantitative immunophenotyping assessment of entire stained sections was done in a blinded fashion using a 0–5 scale. Sections with the highest amount of inflammatory infiltrate (>20%) were given a score of 5, 15–20% inflammation a score of 4, 10–15% inflammation a score of 3, 5–10% inflammation a score of 2, <5% inflammation a score of 1 and sections with no inflammatory cells were given a score of 0.
Results
Type I IFN signature genes and their clusters are differentially expressed in muscle biopsies from rituximab non-responder and responder patients
We stratified patients on the basis of the DOI criteria at week 16 [ 2 , 3 ] into rituximab responder and non-responder groups ( Table 1 ). Microarray analysis of gene expression changes in the skeletal muscle of myositis patients before and after rituximab treatment showed differential expression of innate immune and inflammatory genes. Most striking among these genes were type I IFN genes ( Fig. 1 A). These genes are known to have immunomodulatory effects on the infiltrating immune cells as well as skeletal muscle. Since previous reports have also indicated that the type I IFN gene signature score is correlated with disease activity in adult and juvenile myositis patients [ 5 , 7 , 8 ], we selected 37 type I IFN signature genes that represent broad innate immune anti-proliferative functions. Relative expression patterns of these genes varied significantly between responder and non-responder groups. Supervised hierarchical clustering analysis of these genes resulted in five distinct clusters, including myeloid clusters (clusters 1 and 2) and non-myeloid clusters (clusters 3–5) ( Fig. 1 B–D).
F ig . 1 .
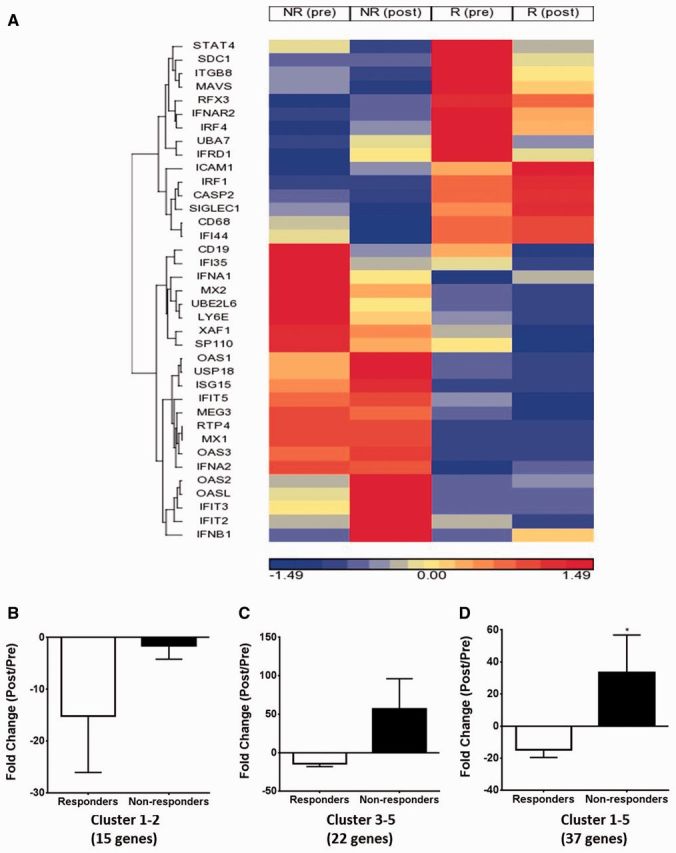
Expression of IFN family genes in rituximab responders and non-responders
( A ) Heat map showing the supervised hierarchical clustering of IFN family gene expression in muscle biopsy specimens in responders and non-responders before and after rituximab treatment. ( B – D ) Fold change in type I IFN genes: ( B ) myeloid clusters 1–2, ( C ) non-myeloid clusters 3–5 and ( D ) all clusters (1–5) as a result of rituximab treatment in responders and non-responders (paired t-test *P < 0.05).
Cluster 1 gene expression, consisting of the STAT4 , SDC1 , ITGB8 , MAVS , RFX3 , IFNAR2 , IRF4 , UBA7 and IFRD1 genes, was elevated in the muscle tissue of the responder group prior to rituximab treatment and was decreased post-treatment, whereas the expression of this gene cluster was low in the non-responder group in both the pre- and post-treatment muscle samples. Cluster 2 gene expression, consisting of the ICAM1 , IRF1 , CASP2 , SIGLEC1 , CD68 and IFI44 genes, was increased in both the pre- and post-treatment muscle biopsies from patients showing a clinical response, whereas the expression of this cluster was low in both the pre- and post-treatment samples from the non-responder group ( Fig. 1 B). Cluster 3 gene expression, consisting of the CD19 , IFI35 , IFNA1 , MX2 , UBE2L6 , LY6E , XAF1 and SP110 genes, was high in the muscle of the non-responder group prior to treatment with rituximab and decreased post-treatment, whereas the expression of this gene cluster was low in the responder group in both the pre- and post-treatment samples. Cluster 4 gene expression, consisting of the OAS1 , USP18 , ISG15 , IFIT5 , MEG3 , RTP4 , MX1 , OAS3 and IFNA2 genes, was increased in pre- and post-treatment non-responders, whereas the expression of this cluster was low in the responder group in both the pre- and post-treatment samples. Cluster 5 gene expression, consisting of the OAS2 , OASL , IFIT3 , IFIT2 and IFNB1 genes, was increased in post-treatment non-responders and low in pre- and post-treatment responders ( Fig. 1 C). Unsupervised comparison of all samples before rituximab treatment indicates that the subjects cluster into two groups (responder vs non-responder) even before treatment ( supplementary Fig. S1 , available at Rheumatology Online).
Type I IFN signature gene myeloid and non-myeloid clusters are differentially altered in rituximab responders compared with non-responders
Comparison of the gene expression between normal and myositis subjects before treatment with rituximab indicate that myositis patients have increased levels of IFN signature genes at baseline ( supplementary Fig. S2 , available at Rheumatology Online). However, in order to assess whether the treatment response alters the type I IFN signature, we calculated the fold change in gene expression by subtracting the pretreatment gene expression levels from the post-treatment gene expression levels in the responder and non-responder groups. We found that rituximab treatment significantly reduced the type I IFN gene expression in clusters 1–2 in responders compared with non-responders ( Fig. 1 B). Likewise, rituximab treatment resulted in relatively increased type I IFN gene expression in clusters 3–5 in non-responders compared with responders ( Fig. 1 C). Overall, rituximab responders have a significantly greater reduction of myeloid and non-myeloid (clusters 1–5) type I IFN signatures than non-responders ( Fig. 1 D).
Muscle-infiltrating B cells and macrophages are also reduced in responders
Since rituximab targets the B cell marker CD20, we semi-quantitatively evaluated the presence of these cells in the muscle biopsy specimens. We found a 20% decrease in CD19 + B cell numbers in responder muscle biopsies. In contrast, non-responder patients showed an ∼53% increase in B cell numbers ( Fig. 2 A and supplementary Fig. S3 , available at Rheumatology Online). Since B cells influence macrophage numbers and function, we also evaluated muscle-infiltrating CD68 + macrophages and found that these cells were similarly decreased in responders, by ∼50%, and increased in non-responders, by ∼31% ( Fig. 2 B and supplementary Fig. S3 , available at Rheumatology Online). However, these changes were not statistically significant.
F ig . 2 .

Expression of B cell macrophage and plasma cell markers in rituximab responders and non-responders
Quantification of ( A ) CD19, ( B ) CD68, ( C ) CD138 and ( D ) IPS-1 staining patterns in responders and non-responders both before and after treatment with rituximab.
CD138 + plasma cells are increased in non-responder patients
CD138 is a recognized plasma cell marker. There was no difference in the number of CD138 + cells before and after treatment in responder patients, but non-responders showed an increase of 71% post-treatment ( Fig. 2 C). However, these changes were not statistically significant. We also stained tissues with an antibody that recognizes IPS-1, an adaptor triggering RIG-I- and MDA5-mediated induction of type I IFN. Nuclei of both muscle and infiltrating cells stained for IPS1, but there were no significant differences in either group between the pre- and post-treatment levels ( Fig. 2 D). Likewise, we stained the tissues for Mx1 and IFN-β, both of which stained muscle fibres and infiltrating cells, but these levels did not differ between responders and non-responders (data not shown).
Discussion
In the present study we have shown that (i) rituximab responders have higher muscle type I IFN signature genes before treatment than non-responders, (ii) expression of type I IFN myeloid signature genes are significantly decreased in muscle after treatment with rituximab in responders, (iii) muscle-infiltrating CD19 + B cells and CD68 + macrophages are reduced after treatment with rituximab in responders and (iv) muscle-infiltrating CD138 + plasma cells are increased in non-responder patients.
The recently completed RIM clinical trial demonstrated that although there were no significant differences in primary and secondary endpoints in the two treatment arms, 83% of the refractory adult and juvenile myositis patients met the DOI. We divided patients in the present study into responders and non-responders based on DOI criteria and assessed muscle gene expression and cellular infiltrates in an attempt to identify correlations with the response to rituximab.
In the RIM study, peripheral B cells were fully depleted in all patients [ 3 ]. We found that muscle-infiltrating B cells decreased in the responder group but not in the non-responder group, suggesting that there is either incomplete depletion of B cells in the target tissues or increased repopulation of B cells in the non-responder group. Failure of B cell depletion in the peripheral pool is associated with a poor clinical response in RA patients [ 9 ].
We recently showed that depletion of peripheral blood B cells did not correlate with clinical response at week 16, in that responders and non-responders (based on the DOI) both reduced CD20 + B cells to a similar extent. Similar trends were observed for CD20 + CD27 + B cells, except that one non-responder had an increase in memory B cells at week 16, suggesting that B cell markers and IFN may have distinct roles in the therapeutic response to rituximab [ 10 ]. We also found a decrease in CD20 + cells at week 16 in skeletal muscle, but this did not reach statistical significance (data not shown). Because of the small sample size, we could not draw a meaningful conclusion between CD20 + B cells in the skeletal muscle and peripheral blood. Since B cells affect macrophage numbers, we evaluated CD68 + macrophages and found a similar pattern to that for B cells, suggesting that B cell depletion affects the macrophage number in muscle tissue.
Several groups have independently shown a marked increase in type I IFN-inducible transcripts and proteins in muscle biopsies of clinically active adult and juvenile myositis patients [ 5 , 11–13 ]. While most studies see a more dramatic increase in type I INF signatures in juvenile and adult DM patients, an increase in some PM patients has also been identified [ 6 , 14 ].
Walsh et al. [ 15 ] also showed that type I IFN-inducible gene expression in blood reflects disease activity in DM and PM. Previous studies indicated that there is correlation between rituximab treatment and both the type I IFN signature and clinical outcome in RA patients and that clinical response can be predicted by the type I IFN signature score [ 16 , 17 ]. Our study further demonstrates that non-responders have a higher overall expression of myeloid type I IFN signature genes and responders have lower myeloid type I IFN signatures after treatment with rituximab. Clusters containing myeloid cell–specific genes such as CD68 , ICAM-1 , ITGB4 , SIGLEC1 , etc. are grouped as myeloid clusters. Rituximab treatment results in a greater decrease in myeloid clusters (clusters 1 and 2) in responders than non-responders, whereas non-myeloid clusters (clusters 3–5) are decreased in responders and increased in non-responders. The molecular basis for this differential response in these two cell types in responders and non-responders needs further investigation. Recent studies suggest that type I IFN and myeloid signatures are candidate markers of disease activity in myositis [ 18 ]. We identified five clusters of type I IFN-related genes that are coordinately regulated, although it is unclear how genes within each cluster are related to each other functionally. Overall, clusters that represented the classical IFN-stimulated genes (clusters 3–5) were high in non-responders after treatment with rituximab. This result in non-responders is consistent with the presence of higher levels of muscle-infiltrating CD138 + plasma cells in this group. The decrease in myeloid type I IFN signature along with the decrease in CD68 + cells in rituximab responders suggests that reduced activity of innate immune cells is beneficial to myositis patients, therefore therapeutic interventions aimed directly at reducing activity of myeloid cells are likely to be beneficial to myositis patients.
Previous studies have shown that IFN gene expression and antibody status might be linked to disease activity. It has been previously shown that the presence of autoantibodies (e.g. anti-synthetase and anti-Mi-2 autoantibodies) predicts clinical improvement in patients with refractory myositis [ 19 ]. Further, in a recent study, Reed et al. [ 20 ] showed IFN chemokine (IFNCK) scores were higher at baseline in subjects with autoantibodies and autoantibody-positive subjects had a greater improvement in IFNCK scores at 16 weeks after rituximab, suggesting that both IFNCK high scores and autoantibodies predict clinical improvement in these patients. In our study, most of the autoantibody-positive patients were responders and showed greater improvements in overall IFN signature genes, suggesting that the presence of autoantibodies and improvements in type I IFN signature may predict clinical improvement. However, the small sample size in our study precludes any meaningful correlations with autoantibody status.
In summary, our study confirms that myeloid and type I IFN signatures are important in myositis pathogenesis and rituximab treatment alters these signatures. Rituximab responders have a greater reduction of the myeloid signature and non-myeloid type I IFN signature than non-responders. Some of the limitations of our study include small sample size, considerable heterogeneity across all patients and a high degree of variation in the histological evaluations. Future studies are needed to validate these findings in independent patient cohorts treated with rituximab.
Supplementary Material
Acknowledgements
K.N. is supported by the National Institutes of Health (RO1-AR050478, 5U54HD053177 and K26OD011171), the Myositis Association and the US Department of Defense (W81XWH-11-1-0809). S.R. is supported by a pre-doctoral fellowship from the Association Francaise Contre les Myopathies. This study was supported by intramural funds from the National Institute of Environmental Health Sciences, National Institutes of Health (ES101074) to F.W. M. and L.G.R.
We thank Drs Robert Colbert and Yi-Wen Chen for their useful comments after reviewing the manuscript, the Rituximab in Myositis investigators for their clinical assistance in organizing and conducting the study, Dr Terrance O’Hanlon for technical assistance and Dr Deborah McClellan for editorial assistance. Authors’ contributions: K.N. planned experiments, analysed data and wrote the manuscript. S.G. performed gene expression profiling and analysed the data, S.R. performed confirmatory experiments and analysed data. A.P. performed immunohistochemistry experiments and analysed data. L.G.R. analysed the data and wrote the manuscript. E.P.H. analysed data and contributed the manuscript. F.W.M. planned experiments, analysed data and wrote the manuscript.
Funding : This study was supported in part by the Intramural Research Program of the National Institute of Environmental Health Sciences, National Institutes of Health.
Disclosure statement : The authors have declared no conflicts of interest.
References
- 1. Rider LG, Miller FW. Deciphering the clinical presentations, pathogenesis, and treatment of the idiopathic inflammatory myopathies . JAMA 2011. ; 305 : 183 – 90 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Rider LG, Giannini EH, Brunner HI. et al. . International consensus on preliminary definitions of improvement in adult and juvenile myositis . Arthritis Rheum 2004. ; 50 : 2281 – 90 . [DOI] [PubMed] [Google Scholar]
- 3. Oddis CV, Reed AM, Aggarwal R. et al. . Rituximab in the treatment of refractory adult and juvenile dermatomyositis and adult polymyositis: a randomized, placebo-phase trial . Arthritis Rheum 2013. ; 65 : 314 – 24 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Aggarwal R, Bandos A, Reed AM. et al. . Predictors of clinical improvement in rituximab-treated refractory adult and juvenile dermatomyositis and adult polymyositis . Arthritis Rheum 2013. ; 66 : 740 – 9 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Reed AM, Peterson E, Bilgic H. et al. . Changes in novel biomarkers of disease activity in juvenile and adult dermatomyositis are sensitive biomarkers of disease course . Arthritis Rheum 2012. ; 64 : 4078 – 86 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Nayak D, Johnson KR, Heydari S. et al. . Type I interferon programs innate myeloid dynamics and gene expression in the virally infected nervous system . PLoS Pathog 2013. ; 9 : e1003395.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bilgic H, Ytterberg SR, Amin S. et al. . Interleukin-6 and type I interferon-regulated genes and chemokines mark disease activity in dermatomyositis . Arthritis Rheum 2009. ; 60 : 3436 – 46 . [DOI] [PubMed] [Google Scholar]
- 8. O’Connor KA, Abbott KA, Sabin B, Kuroda M, Pachman LM. MxA gene expression in juvenile dermatomyositis peripheral blood mononuclear cells: association with muscle involvement . Clin Immunol 2006. ; 120 : 319 – 25 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Dass S, Rawstron AC, Vital EM. et al. . Highly sensitive B cell analysis predicts response to rituximab therapy in rheumatoid arthritis . Arthritis Rheum 2008. ; 58 : 2993 – 9 . [DOI] [PubMed] [Google Scholar]
- 10. Rider LG, Yip AL, Horkayne-Szakaly I. et al. . Novel assessment tools to evaluate clinical and laboratory responses in a subset of patients enrolled in the Rituximab in Myositis trial . Clin Exp Rheumatol 2014. ; 32 : 689 – 96 . [PMC free article] [PubMed] [Google Scholar]
- 11. Salajegheh M, Kong SW, Pinkus JL. et al. . Interferon-stimulated gene 15 (ISG15) conjugates proteins in dermatomyositis muscle with perifascicular atrophy . Ann Neurol 2010. ; 67 : 53 – 63 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Baechler EC, Bauer JW, Slattery CA. et al. . An interferon signature in the peripheral blood of dermatomyositis patients is associated with disease activity . Mol Med 2007. ; 13 : 59 – 68 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Niewold TB, Kariuki SN, Morgan GA, Shrestha S, Pachman LM. Elevated serum interferon-alpha activity in juvenile dermatomyositis: associations with disease activity at diagnosis and after thirty-six months of therapy. Arthritis R heum 2009. ; 60 : 1815 – 24 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Greenberg SA. Type 1 interferons and myositis . Arthritis Res Ther 2010. ; 12(Suppl 1) : S4.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Walsh RJ, Kong SW, Yao Y. et al. . Type I interferon-inducible gene expression in blood is present and reflects disease activity in dermatomyositis and polymyositis . Arthritis Rheum 2007. ; 56 : 3784 – 92 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Raterman HG, Vosslamber S, de Ridder S. et al. . The interferon type I signature towards prediction of non-response to rituximab in rheumatoid arthritis patients . Arthritis Res Ther 2012. ; 14 : R95.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Verweij CL, Vosslamber S. New insight in the mechanism of action of rituximab: the interferon signature towards personalized medicine . Discov Med 2011. ; 12 : 229 – 36 . [PubMed] [Google Scholar]
- 18. Nasr R, Reed AM, Peterson EJ. Update: biomarkers for idiopathic inflammatory myopathies . Curr Opin Rheumatol 2012. ; 24 : 609 – 15 . [DOI] [PubMed] [Google Scholar]
- 19. Aggarwal R, Bandos A, Reed AM. et al. . Predictors of clinical improvement in rituximab-treated refractory adult and juvenile dermatomyositis and adult polymyositis . Arthritis Rheumatol 2014. ; 66 : 740 – 9 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Reed AM, Crowson CS, Hein M. et al. . Biologic predictors of clinical improvement in rituximab-treated refractory myositis . BMC Musculoskelet Disord 2015. ; 16 : 257.. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.