

Abstract
The functions of the liver are very diverse. From detoxifying blood to storing glucose in the form of glycogen and producing bile to facilitate fat digestion, the liver is a very active and important organ. The liver is comprised of many varied cell types whose functions are equally diverse. Cholangiocytes line the biliary tree and aid in transporting and adjusting the composition of bile as it travels to the gallbladder. Hepatic stellate cells and portal fibroblasts are located in different areas within the liver architecture, but both contribute to the development of fibrosis upon activation after liver injury. Vascular cells, including those that constitute the peribiliary vascular plexus, are involved in functions other than blood delivery to and from the liver, such as supporting the growth of the biliary tree during development. Mast cells are normally found in healthy livers but in very low numbers. However, after injury, mast cell numbers greatly increase as they infiltrate and release factors that exacerbate the fibrotic response. While not an all-inclusive list, these cells have individual roles within the liver, but they are also able to communicate with each other by cellular crosstalk. In this review, we examine some of these pathways that can lead to an increase in the homeostatic dysfunction seen in liver injury.
Keywords: Mast cells, Paracrine, Cholangiocytes, Hepatic stellate cells, Crosstalk
1. Introduction
Cellular crosstalk is an event that has a multitude of effects on liver functions and homeostasis. A number of resident and nonresident cell types are responsible for maintaining proper liver functions and management of the balance between cholangiocyte proliferation and loss. When the biliary tree is damaged, this balance is disrupted and cholangiocytes respond by releasing numerous factors that can activate other cell types. Hepatic stellate cells (HSCs) and portal fibroblasts (PFs) play a prominent role in hepatic fibrosis that can be induced by injury of the biliary tree. These cell types promote collagen formation that can eventually lead to scarring and liver failure. In addition, the vascular bed, including the peribiliary plexus, vascular endothelial cells, and various angiogenic factors, plays a critical role in liver homeostasis and maintenance of the balance between cholangiocyte proliferation and ductopenia. The vascular bed and peribiliary vascular plexus are also important regulators of cholangiocyte damage and repair. Vascular endothelial growth factor (VEGF) is a specific angiogenic factor that has proven to be a therapeutic target of a number of cholangiopathies. Finally, a non-resident liver cell type that has also been demonstrated to be important in maintaining biliary tree homeostasis is the hepatic mast cell (MC). While notoriously known for their role in allergy-mediated diseases, these cells are now more recognized as critical components of tissue remodeling, wound healing, and the progression of hepatic fibrosis. In this review, we will focus on these cell types and how they interact with cholangiocytes during cholestatic liver injury and cholangiopathies such as primary sclerosing cholangitis (PSC) and primary biliary cholangitis (PBC). We have focused our review on studies within the past 5 years, thus striving to provide the reader with the most recent information regarding cellular crosstalk following biliary damage. This review will present a brief overview of the locations and functions of cholangiocytes, HSCs, and PFs, the vascular bed, and MCs, followed by specific studies highlighting these cell types during liver injury.
2. Cholangiocytes
Cholangiocytes are epithelial cells lining the intra and extrahepatic ducts of the three-dimensional network of the biliary tree. Biliary cells comprise approximately 3–5% of cells in the liver and are considered as one of the main cell types in liver epithelia.1–3 General characteristics of cholangiocytes include a multi-lobulated nucleus, several vesicles at the sub-apical region, tight junctions, high microvilli density, lysosomes, and few mitochondria.4 Cholangiocytes contain primary cilia that act as cellular antennae for detection and transmission of signals to influence the function of cholangiocytes. When the normal functions of primary cilia are modified or destroyed, it may result in a chol-angiopathy.5 Cholangiocytes can also be classified into two categories according to their size and function.6
The biliary tree and the functions of cholangiocytes were originally believed to be a passive “plumbing” system whereby the only function was the regulation of bile flow. It is now widely accepted that the biliary tree and cholangiocytes are key regulators of liver homeostasis.7,8 In fact, alterations or damage of the biliary tree can induce a ripple effect of cellular crosstalk and signaling pathways, which can progress into chronic liver disease, fibrosis, scarring, and eventual liver failure. The morphological and functional heterogeneity of cholangiocytes make them unique compared with other epithelial cell types.6,9 Unfortunately, cholangiocytes are also targets of several human diseases including cholangiocarcinoma, PSC, PBC, and vanishing bileduct syndrome.10,11
3. Hepatic stellate cells and portal fibroblasts
HSCs and PFs are two cell types in the liver, which are involved in the development of fibrosis. In normal healthy livers, HSCs are in a quiescent state and represent about 5–8% of the total number of liver cells.12 The function of quiescent HSCs in the liver is not well understood, but there is evidence showing that they play roles as an antigen-presenting cell type and in promoting natural killer cell proliferation.13,14 After liver damage, HSCs convert to an activated state characterized by proliferation, contractility, and chemotaxis.15–18 Once activated, HSCs begin to secrete collagen, leading to fibrosis and potentially cirrhosis of the liver. Some studies have shown that HSCs can begin to enter a senescent state upon activation and accumulate p53, thereby exacerbating the fibrotic reaction and reducing overall cell survival.19 Alternatively, some studies have demonstrated that senescent HSCs limit fibrosis by activating the immune response, causing interactions in natural killer cells.20 This effect can lead to resolution of the fibrotic response seen after acute liver injury.
PFs are also involved in the fibrotic response of the liver and are classified as all fibroblasts in the portal region. First described by Carruthers et al.,21,22 PFs are localized to the portal region, which differs from HSCs that are more distant and found in the peri-sinusoidal region of the liver. A linage tracing study by Asahina et al. provided evidence that HSCs and PFs originate from a common progenitor in early embryonic development.23 PFs have been differentiated from HSCs based on the expression of various markers, although these markers have not been sufficiently examined. Some studies have shown that PF-specific markers include fibulin-2, interleukin (IL)-6, elastin, and ecto-ATPase nucleoside triphosphate diphosphohydrolase-2 (NTPD2). Other markers that have been studied are P100, α2-macroglobulin, and neuronal proteins such as neuronal cell adhesion markers and synaptophysin (SYP).24–26
In cholangiopathies such as PBC, fibrosis begins in the peri-ductular region, which strongly implicates PFs as mediators of biliary fibrosis. PFs have cytoplasmic extensions that extend toward and come very close to the basolateral membrane of cholangiocytes. It has been reported that these dendrite-like extensions increase in number after injury of the liver.27 Following liver injury or when cultured on glass or plastic, PFs undergo myofibroblastic differentiation.28,29 This change causes PFs to produce large amounts of microfilament bundles containing α-SMA and arranged in line with the long axis of the cell.
4. Vascular cells and the peribiliary vascular plexus
The liver is a highly vascularized organ that receives dual blood supplies from the hepatic portal vein (HPV) and hepatic arteries (Fig. 1). The HPV delivers approximately 75% of the total blood supply that the liver receives and carries from the spleen, gastrointestinal (GI), tract organs (i.e. stomach and small intestine), and other GI tract-associated organs. Hepatic arteries supply arterial blood accounting for the remaining 25% of blood received by the liver. Oxygen is brought in by both sources, each providing about half of the total amount of oxygen that the liver requires. Once blood enters the liver, it eventually flows through the liver sinusoids and empties into the central vein of each lobule. These central veins combine into the hepatic veins that exit the liver and drain into the inferior vena cava.
Fig. 1. Graphic image of blood flow through the liver and the key cells involved.
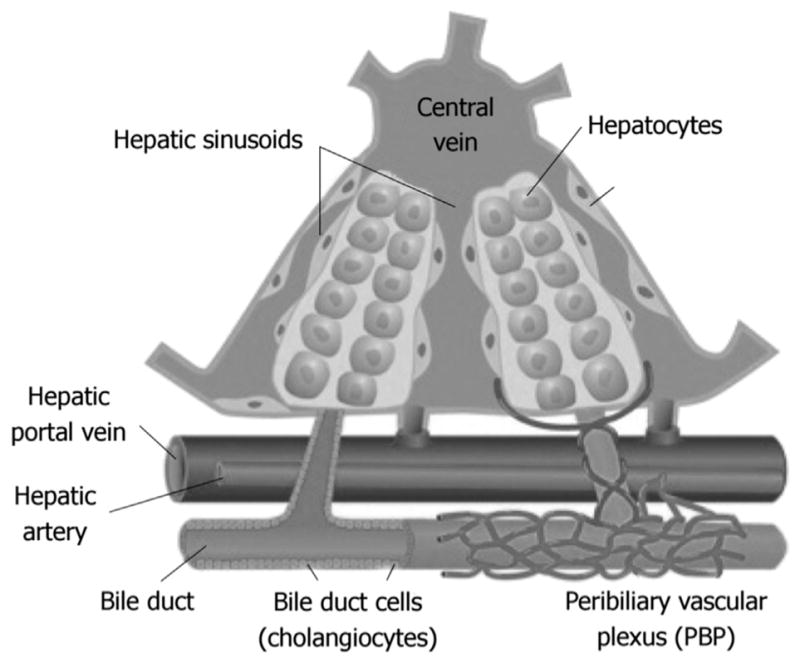
(cite from Onori P1, Gaudio E, Franchitto A, et al. Histamine regulation of hyperplastic and neoplastic cell growth in cholangiocytes. World J Gastrointest Pathophysiol. 2010; 1:38–49.).
The cells that line both blood and lymphatic vessels are called endothelial cells and form a type of simple squamous epithelium. Specifically, vascular endothelial cells form the endothelium that lines the entire circulatory system and come into direct contact with blood. They have unique functions within the cardiovascular system. In highly specialized areas such as the glomerulus in the kidney, they contribute to fluid filtration. The vascular endothelium also influences blood vessel tone by releasing nitric oxide30 and endothelin-1.31 In addition, vascular endothelial cells play a role in hemostasis32 and neutrophil recruitment33 as a part of the global immune response.
A specific portion of the vasculature of the liver is responsible for supporting the needs of cholangiocytes that comprise the intrahepatic biliary tree. This structure is termed as the peribiliary plexus (PBP). The PBP stems from the hepatic artery and is located closely around intrahepatic bile ducts (Fig. 1).34 During development, the formation of bile ducts is closely associated with development of the hepatic artery, which is due to the fact that the hepatic artery branches are in close proximity to the newly formed ductal plates.35 Biliary tree functions are closely linked to the PBP and can be seen when evaluating various models of cholestasis. Changes in the biliary tree (i.e. proliferation due to cholestasis) are associated with changes in the structure of the PBP.34 As cholangiocytes proliferate during cholestasis, they have an increase in metabolic demand. The PBP responds by proliferating and increasing in mass.34 There has also been evidence of a link between new blood vessel formation around bile ducts in response to chronic inflammatory liver diseases such as PBC and PSC. This new vessel growth plays an important role in remodeling the tissue after injury as well as delivering oxygen and other metabolic nutrients to hypoxic areas.34 These newly formed vessels can also provide pathways for the recruitment of inflammatory/immune response cells such as T lymphocytes.36
5. Mast cells
MCs are myeloid cells derived from multipotent CD34-positive hematopoietic stem cells in the bone marrow.37,38 They are ubiquitous in nearly all tissues throughout vertebrates and can be found throughout the body, varying in concentration and size.39 MCs act as sentinel cells and maintain a close proximity in locations where they can rapidly respond to pathogens and antigens. MCs are found in numerous tissues and organs, epithelia, glands, the GI, smooth muscle, blood and lymphatic vessels, and nerves.40
The maturation of MCs is influenced by a diverse group of growth and differentiation factors in the niche where MCs reside.37,41,42 Stem cell factor (SCF) is a crucial signal secreted by tissues and other cells to promote MC migration, adhesion, and de novo proliferation, and can regulate MC development.41 In addition, MCs express c-Kit on their surface, which interacts with SCF and is a vital factor for MC migration and degranulation. It has been demonstrated that c-Kit mutations can result in tumor formation that may also involve MC activation.43
MCs play a key role in the maintaining homeostasis of tissues where they reside by participating in tissue repair, reactions against viral and parasitic infections, and aiding in the regulation of various cell types such as dendritic cells, T cells, and macrophages.44 In addition, MCs are involved in the immune system by secreting proinflammatory mediators such as histamine (HA), proteases, heparin, cytokines, chemokines, and several other molecules that are key players in maintaining the homeostasis of organs and tissues.38,44–46 However, MCs are equally known for their detrimental effects on the body. MCs play a key role in several types of chronic allergic and inflammatory conditions, autoimmune diseases, mastocytosis, cardiovascular diseases, and cancers.39 Consequently, there has been an emphasis on the MC contribution to physiological and pathophysiological processes in recent years with the hopes of better understanding MCs and their roles in disease processes.
5.1. Cholangiocytes →hepatic stellate cells and portal fibroblasts
Cholangiocytes are the main target of injury following cholestatic liver damage. After sustaining damage, they express and secrete a number of factors including, but not limited to, secretin, HA, VEGF, progesterone, and serotonin.9,16,47–49 In addition to the autocrine regulation of cholangiocytes in response to liver injury and repair, there are a number of paracrine actions that require other resident (HSCs, PFs, and vascular cells) and non-resident (MCs) cell types within the liver for successful repair. Cholestatic liver injury damages bile ducts and induces cholangiocyte proliferation that can be associated with hepatic fibrosis.16,50,51 In this section, we will highlight studies that demonstrate interactions between cholangiocytes, HSCs, and PFs during cholestatic liver injury and hepatic fibrosis.
5.1.1. Cholestatic liver injury and fibrosis
A recent study by Kennedy et al. demonstrated that knockout of the microRNA miR-21, which is upregulated in cholangiocytes following injury (Table 1), inhibits biliary hyperplasia, HSC activation, and hepatic fibrosis following bile duct ligation (BDL).17 The authors performed in vitro and in vivo experiments to validate their findings. In miR-21 knockout mice subjected to BDL, the number of proliferating cholangiocytes was significantly reduced along with a decrease in HSC activation. There was a reduction in overall hepatic fibrosis, which was associated with a decrease in portal collagen content and an increase in the expression of Smad-7 that counteracts the fibrotic response.17 In vitro, cholangiocytes and HSCs treated with a miR-21 inhibitor displayed a decrease in proliferation and expression of fibrotic markers, respectively. The link between cholangiocytes and HSCs with regards to this study may be regulation by transforming growth factor-beta 1 (TGF-β1) and more specifically Smad-7 that was found to be increased in their model.17
Table 1.
Summarization of microRNAs involved in progression of fibrosis in response to hepatic injury; their regulation, targets and effects.
| MicroRNA | Regulation | Targets | Effects |
|---|---|---|---|
| miR-21 | ↑ | SMAD7 | ↑ fibrosis |
| miR-125b | ↓ | VEGF | ↑ biliary proliferation |
| NGF | ↑ fibrosis | ||
| HDC | ↑ inflammation | ||
| miR-let7a | ↓ | VEGF | ↑ biliary proliferation |
| NGF | ↑ fibrosis |
Another analysis of cholangiocyte/HSC interactions was performed by a recent study published in Hepatology. In this study, McDaniel and coauthors reported an interesting finding that mice subjected to BDL and subsequently injected with small cholangiocytes displayed a reduction in hepatic fibrosis, which was associated with decreased activation of HSCs.52 Isolated HSCs were obtained by laser capture microdissection, thus providing direct evidence of the role of HSCs in cholestatic-induced fibrosis. The authors also found that HSC senescence was reduced following small cholangiocyte engraftment, suggesting that factors released from HSCs alter the course of hepatic fibrosis.52 In a study demonstrating that BDL-induced ductular reactions are regulated by integrin αvβ-mediated activation of nuclear factor-kappa B (NF-κB), the authors demonstrated that CCN1 induces Jagged1 expression in HSCs, thereby promoting cholangiocyte proliferation and hepatic fibrosis.53 The authors elegantly showed that CCN1 induction during HSC activation contributes to hepatic progenitor cell differentiation into cholangiocytes, which is regulated via αvβ5/NF-κB/Jagged1 signaling mechanisms.53 Both of these studies demonstrate the capacity for HSCs to interact with cholangiocytes following injury to promote hepatic fibrosis and offer insights into therapeutic avenues for fibrotic therapy.52,53
HSCs and PFs can also be altered following treatment with an antioxidative compound, oltipraz, which is a potential cancer preventative.54 In mice treated with oltipraz at 5 days prior to BDL for 3 days, the authors found that this drug enhances biliary damage, HSC activation, and PF initiation. While this drug has promising effects on cancer, based on this study, it appears that oltipraz should be avoided because it increases all aspects of damage and hepatic fibrosis including induction of extracellular matrix remodeling.54
PFs and HSCs can also affect the response of vascular cells during liver injury. In a study by Lemoinne et al.,55 markers for PFs and endothelial cells, collagen, type XV, alpha 1 (COL15A1) and von Willebrand factor (vWF), respectively, were upregulated in advanced fibrosis. The authors found that COL15A1 was highly expressed around vascular capillaries that were in close proximity to reactive bile ducts. When liver endothelial cells were exposed to PF-conditioned medium in vitro, the cells had increased migration and more readily formed tubular structures. Furthermore, when PFs and endothelial cells were cultured together, intercellular junctions were formed, showing evidence that these cells communicate directly with each other. It was also shown that PFs release microparticles containing VEGF-A, and that these micro-particles can activate VEGF-receptor 2 (VEGF-R2) on endothelial cells, thereby activating their pro-angiogenic effects.55
Zhang et al.56 showed that, in a carbon tetrachloride (CCl4) model of biliary fibrosis in rats, the drug curcumin decreases the fibrotic and angiogenic responses associated with this model. When tested in vitro, curcumin had no effect on the viability or physiological functions of endothelial cells, which led the authors to postulate that this drug exerts its effects on HSCs. Indeed, curcumin decreases the expression of VEGF in HSCs and disrupts platelet-derived growth factor beta receptor (PDGF-βR)/extracellular signal-regulated kinase (ERK) and mechanistic target of rapamycin (mTOR) pathways. Curcumin also inhibits HSC motility and vascularization by blocking the PDGF-βR/focal adhesion kinase/Ras homolog gene family, member A (RhoA) cascade. Curcumin modifies the angiogenic functions of HSCs through activation of peroxisome proliferator-activated receptor-γ.56 These data suggest that PFs and HSCs are key cells in vascular remodeling during liver fibrosis.
5.1.2. Primary sclerosing cholangitis, primary biliary cholangitis, and hepatic fibrosis
Cholangiopathies are a group of autoimmune disorders that target cholangiocytes and include PSC and PBC.57 PSC induces rapid ductular reactions followed by ductopenia as the bile ducts become more constricted. During PBC, there is progressive destruction of bile ducts, leading to cirrhosis and its associated complications. Both PSC and PBC can lead to a buildup of scar tissue and hepatic fibrosis, resulting in liver failure.58–60 Numerous studies have demonstrated the importance of cell–cell communication between cholangiocytes and HSCs in the progression of both PSC and PBC, and we will highlight the most recent findings in this section.
A recent study has found a link between cholangiocytes and HSCs during PSC. Jones et al. used a rodent model [multidrug resistant 2−/− (Mdr2−/−) mice] of PSC and demonstrated that HSC activation increases in Mdr2−/− mice, which is inhibited by blocking MC-derived HA with cromolyn sodium treatment.16 Similarly, in a BDL study,51 1 week of cromolyn sodium treatment decreased biliary mass, liver fibrosis, and HSC activation coupled with a decrease in bile flow, bicarbonate excretion, and total serum and liver bile acid contents.16 It is unclear how cholangiocytes and HSCs interact with one another; however, a potential mechanism may be secretion of TGF-β1. The authors also found that TGF-β1 expression and secretion were upregulated in total liver and cultured HSCs, demonstrating that this factor may be a regulator of communication between cholangiocytes and HSCs.16
In the study mentioned above from McDaniel et al.,52 the authors also analyzed Mdr2−/− mice to evaluate the role of forkhead box A2 (Fox A2) as a regulator of biliary injury during PSC. The authors found that fibrosis was increased in Mdr2−/− mice and HSCs isolated from these mice.52 Similar to the BDL model, when small cholangiocytes were introduced to Mdr2−/− mice, HSC activation and hepatic fibrosis were decreased strongly, indicating a cellular crosstalk mechanism between cholangiocytes and HSCs, which may be linked to FoxA2.52 Further studies are warranted to determine the positive effects of small cholangiocyte therapy on PSC-induced liver injury.
5.2. Cholangiocytes →vascular cells, the peribiliary plexus, and angiogenic factors
The PBP, vascular cells, and angiogenic factors all play a role in cholestatic-induced liver injury. The plexus is an area enriched with vascular cells and secretes large amounts of VEGF into the portal environment, thereby influencing cholangiocytes.34,61,62 Vascular endothelial cells activate following injury such as BDL. Here, we will highlight recent studies that demonstrate the importance of the vascular bed in cholestatic liver injury.
5.2.1. Cholestatic liver injury and fibrosis
VEGF has been demonstrated to be important for the regulation of biliary proliferation and hepatic fibrosis, and cholangiocytes secrete VEGF in response to damage.34,61,62 A study by Meng et al.63 revealed that miR-125b regulates the HA/VEGF axis in cholestatic mice subjected to BDL. The authors found significant down-regulation of biliary miR-125b in mice subjected to BDL (Table 1). When the BDL mice were treated with an inhibitor of HDC, there was a significant decrease in biliary proliferation, hepatic fibrosis, and VEGF secretion and expression, demonstrating that HA can also partially regulate angiogenic factors. In vitro, overexpression of miR-125b or knockdown of HDC decreases both HDC and VEGF expression in cholangiocytes.63 These studies demonstrated an interaction between cholangiocytes and angiogenic factors, which may be regulated by HA.
Further confirmation of the importance of the HDC/HA/VEGF axis was obtained using global HDC−/− mice subjected to sham or BDL surgeries.50 In this study, the authors demonstrated that knockdown of HDC decreases BDL-induced biliary proliferation, hepatic fibrosis, and VEGF secretion and expression. Again, these data indicate a role of cholangiocytes and VEGF (most of which is secreted from the PBP) in regulation of the balance between cholangiocyte proliferation and loss.50 Furthermore, this study demonstrated the regulatory effects of HDC and HA on angiogenic factors, offering targets for potential therapeutic intervention. The study was expanded in vitro to reveal that decreased biliary proliferation following HDC knockdown is regulated via protein kinase A (PKA)/ERK1/2 signaling.50
In addition to HA, numerous factors have been found to play a role in the vascular bed during cholestatic liver injury. In a recent study by Glaser et al., the authors found that secretin is a major regulator of biliary proliferation and VEGF expression.9 In secretin knockout mice subjected to sham or BDL to induce cholestatic liver injury, the authors found reduced bile duct mass, cholangiocyte proliferation, and VEGF-A, but not VEGF-C, compared with wild-type controls.9 Secretin regulation of the biliary response and VEGF expression are also mediated by miR-125b and miR-let7a (Table 1), which also demonstrates the importance of microRNAs in cholestatic induced liver injury. There have been numerous studies demonstrating the role of secretin in autocrine regulation of biliary injury and responses.6,9,64 A current study included evaluation of S cells of the duodenum, which also express the secretin receptor and secrete secretin. The authors found that secretin release from both cholangiocytes and S cells altered miR-125b and miR-let7a levels, thereby increasing VEGF expression.9 These important findings highlight the importance of both resident and non-resident liver cells in maintaining biliary functions and responses to injury.
Because VEGF is required for non-pathological angiogenesis, one study attempted to elucidate a method to inhibit pathological production of VEGF without disturbing any normal processes. The authors used H5V cells (a mouse endothelial cell line) and showed that activation of cytoplasmic polyadenylation element binding protein (CPEB) 1 resulted in aberrant nuclear processing of VEGF and CPEB4, leading to the deletion of certain translation repressor elements and increasing CPEB4 expression.65 This increase in CPEB4 expression promoted an increase in VEGF mRNA translation, which eventually led to the formation of tubular structures. In addition, the authors found increasing expression of CPEB1 and CPEB4 in patients with cirrhotic livers and in the BDL rat model. Interestingly, mice lacking CPEB1 and 4 did not have any alterations in VEGF levels or any sign of increases in angiogenesis after liver injury.65 Another study by Coch et al. showed upregulation of VEGF and vasohibin-1 in cirrhotic rats and patients. This upregulation drives a pathological angiogenesis event through a negative feedback loop. However, if vasohibin-1 is upregulated further through adenoviral gene transfer, the negative feedback loop is disrupted and the authors observed a decrease in pathological angiogenesis, reduction of fibrogenesis, inhibition of HSC activation, and a decrease in portal pressure and portohepatic resistance in the liver vasculature.66
Pharmacological inhibition of purinergic receptor subtype P2X7 has been shown to prevent splanchnic hyperemia, new blood vessel formation in mesenteries, and decrease liver fibrosis in the BDL rat model.67 The authors of this study used brilliant blue G and oxidized ATP as P2X7 antagonists, and found that both drugs induced a significant reduction in the fibrogenic response seen in the BDL model. Another drug that has been used to attenuate hepatic fibrosis is Brivanib.68 This drug is an inhibitor of VEGF-R and fibroblast growth factor receptor (FGFR) tyrosine kinases. In this study, Nakamura et al. used three models of fibrosis: BDL, chronic CCl4 administration, and chronic thioacetamide (TTA) treatment. In all three models, fibrosis was significantly decreased after Brivanib treatment, and collagen type-1a and α-SMA gene expression was downregulated. The authors also treated HSCs in vitro with Brivanib and observed decreases in HSC proliferation and cell viability. These data suggest that blocking aberrant angiogenesis could be a therapeutic route for the treatment of chronic liver diseases.68
Alternatively, some studies show that angiogenesis can actually play a beneficial role in promoting the resolution of liver fibrosis. Yang et al.69 developed a model of fibrosis resolution by performing a cholecystojejunostomy in BDL C57BL/6 mice, which facilitates reestablishment of bile flow after BDL surgery. The mice were then administered VEGF-neutralizing antibodies, resulting in a reduction of the fibrogenic events normally seen in BDL mice. However, these antibodies also disrupted fibrosis resolution. The authors found that inhibition of VEGF disrupts liver sinusoidal permeability during the resolution of fibrosis, which is associated with reductions in monocyte migration, adhesion, and infiltration into the cirrhotic liver. They also showed that scar-associated macrophages contribute to this event by producing C-X-C motif ligand 9 (CXCL9) and matrix metalloproteinase (MMP) 13. As an extension of this study, Yang et al.69 performed a cholecystojejunostomy in macrophage Fas-induced apoptosis mice in which macrophages can be selectively depleted. Fibrosis resolution was impaired in these mice. However, CXCL9 overexpression led to an increase in the resolution of fibrosis.69 Another group also obtained a similar finding that the resolution of fibrosis is dependent on sinusoidal angiogenesis.70 These authors produced a VEGF-specific knockout in myeloid cells and used these mice to establish BDL and CCl4 fibrotic models. It was shown that suppressing a rise in myeloid-specific VEGF prevented fibrosis resolution. Subsequently, VEGF-expressing myeloid cells were reintroduced by bone marrow transplantation, and the authors found that collagen breakdown continued and fibrosis resolution occurred.70 These data suggest that VEGF promotes fibrosis production, but it is also required for fibrosis resolution.
Another important factor that may influence damage of the biliary tree and progression of hepatic fibrosis is extrahepatic platelet-derived growth factor-β (PDGF-β) that promotes activation of HSCs.71 In Mdr2−/− mice with advanced biliary fibrosis, there is an increase in PDGF-β, and the source of PDGF-β was found to be platelet clusters. Furthermore, the authors demonstrated that serum PDGF-β levels increase in patients with liver fibrosis. When Mdr2−/− mice were depleted of platelets, there was a dramatic decrease in PDGF-β and HSC activation along with reduced fibrosis.71 Interestingly, platelet reintroduction induced activation of isolated HSCs in vitro through PDGF-β signaling, demonstrating an additional cellular mechanism for hepatic fibrosis induction via activation of HSCs.
5.2.2. Primary sclerosing cholangitis, primary biliary cholangitis, and hepatic fibrosis
Studies have demonstrated an alteration of the vascular bed, and that it regulates biliary responses during PSC, PBC, and/or hepatic fibrosis. In fact, angiogenic factors have been found to also regulate cellular crosstalk with HSCs. In a study by Luo et al.,72 the authors found that VEGF is a key regulator of HSC activation in a model of chronic fibrosis. In their model, there was an increase of endothelial cell proliferation and VEGF-A production, which subsequently activated HSCs and increased collagen-1a and α-SMA expression.72 This study is also supported by findings demonstrating that HSC activation can be inhibited by a specific collagen-binding domain (CBD) fused to the N-terminal of VEGF that induces anti-fibrotic effects.73 Following treatment with CBD-VEGF, CCl4-induced fibrosis was rescued and HSC activation was reduced.73 Taken together, these studies provide evidence that targeting the vascular bed may be a therapeutic option for patients with liver fibrosis.
During sclerosing cholangitis, altered vascular endothelial cells and peribiliary glands contribute to the scarring associated with PSC along with an increase in chemokine expression, primarily chemokine (C-C motif) ligand 1 (CCL1).74 In human PSC, it was found that IL-4, IL-10, and CCL1 were increased compared with controls, and CCL1 was upregulated in peribiliary glands and vascular endothelial cells. These findings suggest a recruitment mechanism triggered by these cells, which induces inflammation via CCL1 and promotes sclerosing cholangitis.74
In the study mentioned above by Jones and Hargrove, the authors also found that vascular endothelial cells play a role in regulating PSC-induced fibrosis. Using vWF as a marker of vascular cells and measuring VEGF-A expression, the study showed an increase of both markers in Mdr2−/− mice. Furthermore, these parameters were decreased in Mdr2−/− mice treated with cromolyn sodium, which was coupled with decreases in biliary proliferation and the bile duct mass, demonstrating that MCs may also work with the vascular bed to maintain cholangiocyte homeostasis.16
5.3. Cholangiocytes →mast cells
In addition to HSCs and vascular cells, both resident liver cells, MCs, which are non-resident cells, play a role in the regulation of the cholangiocyte response and hepatic fibrosis following liver injury. MCs are immune cells that migrate into tissue following injury. While primarily known for their role in mediating allergic responses, MCs are becoming more recognized for their pathological role in a number of disease states including cholestatic liver injury.16,39,41,51
5.3.1. Cholestatic liver injury and fibrosis
MC infiltration begins at day 2 post-BDL, which is coupled with the appearance of bile duct proliferation and an increase in the bile duct mass. Hargrove et al. have shown that infiltrated MCs settle in close proximity to bile ducts, but not within the hepatocyte parenchyma, demonstrating that they are in close contact to interact with proliferating cholangiocytes.75 Furthermore, MCs have been isolated from BDL rats at day 14, which were treated with saline or the MC stabilizer cromolyn sodium. Isolated MC culture supernatants were collected and placed on cultured cholangiocytes or HSCs. The authors found that treatment with BDL saline-treated MC culture supernatants increased biliary proliferation and HSC activation, demonstrating a direct interaction between MCs, cholangiocytes, and HSCs.75 More interestingly, when cultured cholangiocytes or HSCs were stimulated with culture supernatants of MCs collected from cromolyn sodium-treated BDL rats, biliary proliferation and HSC activation were reduced significantly. These studies support a strong relationship and regulation between MCs and HSCs during biliary damage and repair.
In another study, BDL rats were treated with saline or cromolyn sodium for 1 week, and there was a decrease in biliary damage, hepatic fibrosis, and the MC number.51 These studies are the first to demonstrate a synergistic interaction between MCs, cholangiocytes, and HSCs during cholestatic liver injury.51,75 In addition, these studies indicate that MCs and MC-derived HA are key regulators of the biliary response in addition to the activation of HSCs during cholestatic-induced hepatic fibrosis.
Protease-activated receptor (PAR) 2 is a Gprotein-coupled receptor involved in fibrosis formation through activation of extra-cellular proteins, which is activated upon cleavage by serine proteases. One MC-specific serine protease is tryptase. In a study by Lu et al.,76 PAR-2, which is expressed in HSCs, was inhibited using APC 366 in a BDL rat model. The authors observed a reduction in fibrosis after treatment, which was associated with decreases in PAR-2 and α-SMA expression.76
5.3.2. Primary sclerosing cholangitis, primary biliary cholangitis, and hepatic fibrosis
In human PSC, Jones et al.16 demonstrated an increase in the liver MC number and MC marker expression. In this study, Mdr2−/− mice were used as the PSC model to evaluate the effect of MCs on disease progression. The mice were treated with cromolyn sodium and, following treatment, biliary proliferation and fibrosis as well as HA serum levels were all decreased, showing that MC-derived HA plays a direct role in the progression of PSC.16 The authors also knocked down HDC in MCs in vitro and co-cultured these cells with human HSCs. The HSCs co-cultured with MCs containing depleted levels of HDC exhibited decreases in proliferation and activation compared with HSCs co-cultured with MCs containing normal levels of HDC.16
In another study, the effect of chymase, another MC protease, was observed on the activation and proliferation of HSCs. Yin et al. isolated and treated HSCs from rats with various concentrations of chymase. As a result, the HSCs showed increases in their proliferative rates, α-SMA and TGF-β1 protein expression, and TGF-β1 and collagen type-1a gene expression.77 Finally, Knight et al.78 used a PAR-2 knockout mouse for the CCl4 fibrosis model. As mentioned earlier, PAR-2 is involved in the progression of fibrosis upon activation. After CCl4 administration, PAR-2 knockout mice showed reductions in fibrosis progression, collagen gene expression, and hydroxyproline content. The authors also found a reduction in TGF-β gene and protein expression as well as a decrease in MMP2. The authors also showed in vitro activation of human HSCs treated with PAR-1 or PAR-2 agonists (hexapeptide SFLLRN-NH2 and hexapeptide SLIGKV, respectively), as well as increases in proliferation and collagen production, and TGF-β protein upregulation.78 Taken together, these studies suggest that blocking MC migration into the liver and/or blocking the release of MC mediators such as HA could be potential therapeutic strategies for cholangiopathies including PSC and PBC.
6. Conclusion
Cellular crosstalk is an important event that occurs during disease progression and is critical to better understand how cells, tissues, and organs respond to injury and repair. While specific cells are the targets of certain diseases (e.g., cholangiocytes in cholangiopathies), this does not underscore the importance of cellular crosstalk between both resident and non-resident liver cells during disease processes and progression. It appears that there is great interest in evaluating the full events of liver injury and repair with regards to cellular crosstalk as evidenced by the studies highlighted in this review. Based on these studies, cholangiocytes, HSCs/PFs, vascular cells, and MCs may all interact with each other to promote liver damage as summarized in Fig. 2, which may facilitate targeting these cells to induce liver repair. The hope would be that these studies of cellular crosstalk allow for the identification of potential therapeutic targets as well as understanding disease processes more thoroughly.
Fig. 2.

The cellular crosstalk pathways.
Acknowledgments
Portions of this work were supported by (i) a VA Merit Award (1I01BX003031) from the United States Department of Veteran’s affairs, Biomedical Laboratory Research and Development Service and an RO1 from NIH NIDDK (DK108959) and (ii) the Dr. Nicholas C. Hightower Centennial Chair of Gastroenterology from Baylor Scott & White Health. This material is the result of work supported with resources and the use of facilities at the Central Texas Veterans Health Care System, Temple, Texas. The content is the responsibility of the author(s) alone and does not necessarily reflect the views or policies of the Department of Veterans Affairs or the United States Government.
Footnotes
Conflict of interest
The authors declare that they have no conflict of interest.
References
- 1.Carpino G, Renzi A, Franchitto A, et al. Stem/progenitor cell niches involved in hepatic and biliary regeneration. Stem Cells Int. 2016;2016:3658013. doi: 10.1155/2016/3658013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tabibian JH, Masyuk AI, Masyuk TV, et al. Physiology of cholangiocytes. Compr Physiol. 2013;3:541–565. doi: 10.1002/cphy.c120019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Venter J, Francis H, Meng F, et al. Development and functional characterization of extrahepatic cholangiocyte lines from normal rats. Dig Liver Dis. 2015;47:964–972. doi: 10.1016/j.dld.2015.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Han Y, Glaser S, Meng F, et al. Recent advances in the morphological and functional heterogeneity of the biliary epithelium. Exp Biol Med (Maywood) 2013;238:549–565. doi: 10.1177/1535370213489926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Larusso NF, Masyuk TV. The role of cilia in the regulation of bile flow. Dig Dis. 2011;29:6–12. doi: 10.1159/000324121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Glaser S, Francis H, Demorrow S, et al. Heterogeneity of the intrahepatic biliary epithelium. World J Gastroenterol. 2006;12:3523–3536. doi: 10.3748/wjg.v12.i22.3523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Alpini G, Roberts S, Kuntz SM, et al. Morphological, molecular, and functional heterogeneity of cholangiocytes from normal rat liver. Gastroenterology. 1996;110:1636–1643. doi: 10.1053/gast.1996.v110.pm8613073. [DOI] [PubMed] [Google Scholar]
- 8.Alpini G, Ulrich C, Roberts S, et al. Molecular and functional heterogeneity of cholangiocytes from rat liver after bile duct ligation. Am J Physiol. 1997;272:G289–G297. doi: 10.1152/ajpgi.1997.272.2.G289. [DOI] [PubMed] [Google Scholar]
- 9.Glaser S, Meng F, Han Y, et al. Secretin stimulates biliary cell proliferation by regulating expression of microRNA 125b and microRNA let7a in mice. Gastroenterology. 2014;146:1795–1808. e1712. doi: 10.1053/j.gastro.2014.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee SO. Physiologic and pathologic experimental models for studying cholangiocytes. Korean J Hepatol. 2008;14:139–149. doi: 10.3350/kjhep.2008.14.2.139. [DOI] [PubMed] [Google Scholar]
- 11.Yoo KS, Lim WT, Choi HS. Biology of cholangiocytes: from bench to bedside. Gut Liver. 2016;10:687–698. doi: 10.5009/gnl16033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Geerts A. History, heterogeneity, developmental biology, and functions of quiescent hepatic stellate cells. Semin Liver Dis. 2001;21:311–335. doi: 10.1055/s-2001-17550. [DOI] [PubMed] [Google Scholar]
- 13.Scholzel K, Schildberg FA, Welz M, et al. Transfer of MHC-class-I molecules among liver sinusoidal cells facilitates hepatic immune surveillance. J Hepatol. 2014;61:600–608. doi: 10.1016/j.jhep.2014.04.028. [DOI] [PubMed] [Google Scholar]
- 14.Winau F, Hegasy G, Weiskirchen R, et al. Ito cells are liver-resident antigen-presenting cells for activating T cell responses. Immunity. 2007;26:117–129. doi: 10.1016/j.immuni.2006.11.011. [DOI] [PubMed] [Google Scholar]
- 15.Bataller R, Gines P, Nicolas JM, et al. Angiotensin II induces contraction and proliferation of human hepatic stellate cells. Gastroenterology. 2000;118:1149–1156. doi: 10.1016/s0016-5085(00)70368-4. [DOI] [PubMed] [Google Scholar]
- 16.Jones H, Hargrove L, Kennedy L, et al. Inhibition of mast cell-secreted histamine decreases biliary proliferation and fibrosis in primary sclerosing cholangitis Mdr2(−/−) mice. Hepatology. 2016;64:1202–1216. doi: 10.1002/hep.28704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kennedy LL, Meng F, Venter JK, et al. Knockout of microRNA-21 reduces biliary hyperplasia and liver fibrosis in cholestatic bile duct ligated mice. Lab Invest. 2016;96:1256–1267. doi: 10.1038/labinvest.2016.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Reifart J, Rentsch M, Mende K, et al. Modulating CD4+ T cell migration in the postischemic liver: hepatic stellate cells as new therapeutic target? Transplantation. 2015;99:41–47. doi: 10.1097/TP.0000000000000461. [DOI] [PubMed] [Google Scholar]
- 19.Lujambio A, Akkari L, Simon J, et al. Non-cell-autonomous tumor suppression by p53. Cell. 2013;153:449–460. doi: 10.1016/j.cell.2013.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Krizhanovsky V, Yon M, Dickins RA, et al. Senescence of activated stellate cells limits liver fibrosis. Cell. 2008;134:657–667. doi: 10.1016/j.cell.2008.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Carruthers JS, Kalifat SR, Steiner JW. The ductular cell reaction of rat liver in extrahepatic cholestasis. II. The proliferation of connective tissue. Exp Mol Pathol. 1962;1:377–396. doi: 10.1016/0014-4800(62)90032-1. [DOI] [PubMed] [Google Scholar]
- 22.Steiner JW, Carruthers JS. Studies on the fine structure of proliferated bile ductules. II. Changes of the ductule-connective tissue envelope relationship. Can Med Assoc J. 1961;85:1275–1287. [PMC free article] [PubMed] [Google Scholar]
- 23.Asahina K, Tsai SY, Li P, et al. Mesenchymal origin of hepatic stellate cells, submesothelial cells, and perivascular mesenchymal cells during mouse liver development. Hepatology. 2009;49:998–1011. doi: 10.1002/hep.22721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cassiman D, Libbrecht L, Desmet V, et al. Hepatic stellate cell/myofibroblast subpopulations in fibrotic human and rat livers. J Hepatol. 2002;36:200–209. doi: 10.1016/s0168-8278(01)00260-4. [DOI] [PubMed] [Google Scholar]
- 25.Guyot C, Lepreux S, Combe C, et al. Hepatic fibrosis and cirrhosis: the (myo) fibroblastic cell subpopulations involved. Int J Biochem Cell Biol. 2006;38:135–151. doi: 10.1016/j.biocel.2005.08.021. [DOI] [PubMed] [Google Scholar]
- 26.Ramadori G, Saile B. Portal tract fibrogenesis in the liver. Lab Invest. 2004;84:153–159. doi: 10.1038/labinvest.3700030. [DOI] [PubMed] [Google Scholar]
- 27.Tuchweber B, Desmouliere A, Bochaton-Piallat ML, et al. Proliferation and phenotypic modulation of portal fibroblasts in the early stages of cholestatic fibrosis in the rat. Lab Invest. 1996;74:265–278. [PubMed] [Google Scholar]
- 28.Kruglov EA, Nathanson RA, Nguyen T, et al. Secretion of MCP-1/CCL2 by bile duct epithelia induces myofibroblastic transdifferentiation of portal fibroblasts. Am J Physiol Gastrointest Liver Physiol. 2006;290:G765–G771. doi: 10.1152/ajpgi.00308.2005. [DOI] [PubMed] [Google Scholar]
- 29.Li Z, Dranoff JA, Chan EP, Uemura M, Sevigny J, Wells RG. Transforming growth factor-beta and substrate stiffness regulate portal fibroblast activation in culture. Hepatology. 2007;46:1246–1256. doi: 10.1002/hep.21792. [DOI] [PubMed] [Google Scholar]
- 30.Shu X, Keller TCt, Begandt D, et al. Endothelial nitric oxide synthase in the microcirculation. Cell Mol Life Sci. 2015;72:4561–4575. doi: 10.1007/s00018-015-2021-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gao Y, Chen T, Raj JU. Endothelial and smooth muscle cell interactions in the pathobiology of pulmonary hypertension. Am J Respir Cell Mol Biol. 2016;54:451–460. doi: 10.1165/rcmb.2015-0323TR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Roumenina LT, Rayes J, Frimat M, Fremeaux-Bacchi V. Endothelial cells: source, barrier, and target of defensive mediators. Immunol Rev. 2016;274:307–329. doi: 10.1111/imr.12479. [DOI] [PubMed] [Google Scholar]
- 33.Marki A, Esko JD, Pries AR, et al. Role of the endothelial surface layer in neutrophil recruitment. J Leukoc Biol. 2015;98:503–515. doi: 10.1189/jlb.3MR0115-011R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gaudio E, Onori P, Pannarale L, et al. Hepatic microcirculation and peribiliary plexus in experimental biliary cirrhosis: a morphological study. Gastroenterology. 1996;111:1118–1124. doi: 10.1016/s0016-5085(96)70081-1. [DOI] [PubMed] [Google Scholar]
- 35.Terada T, Nakanuma Y. Development of human peribiliary capillary plexus: a lectin-histochemical and immunohistochemical study. Hepatology. 1993;18:529–536. [PubMed] [Google Scholar]
- 36.Medina J, Sanz-Cameno P, Garcia-Buey L, Martin-Vilchez S, Lopez-Cabrera M, Moreno-Otero R. Evidence of angiogenesis in primary biliary cirrhosis: an immunohistochemical descriptive study. J Hepatol. 2005;42:124–131. doi: 10.1016/j.jhep.2004.09.024. [DOI] [PubMed] [Google Scholar]
- 37.Huang H, Li Y. Mechanisms controlling mast cell and basophil lineage decisions. Curr Allergy Asthma Rep. 2014;14:457. doi: 10.1007/s11882-014-0457-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Theoharides TC, Alysandratos KD, Angelidou A, et al. Mast cells and inflammation. Biochim Biophys Acta. 2012;1822:21–33. doi: 10.1016/j.bbadis.2010.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.da Silva EZ, Jamur MC, Oliver C. Mast cell function: a new vision of an old cell. J Histochem Cytochem. 2014;62:698–738. doi: 10.1369/0022155414545334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Galli SJ, Tsai M, Marichal T, et al. Approaches for analyzing the roles of mast cells and their proteases in vivo. Adv Immunol. 2015;126:45–127. doi: 10.1016/bs.ai.2014.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ammendola M, Sacco R, Sammarco G, et al. Mast cell-targeted strategies in cancer therapy. Transfus Med Hemother. 2016;43:109–113. doi: 10.1159/000444942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Galli SJ. Mast cells and basophils. Curr Opin Hematol. 2000;7:32–39. doi: 10.1097/00062752-200001000-00007. [DOI] [PubMed] [Google Scholar]
- 43.Wedemeyer J, Tsai M, Galli SJ. Roles of mast cells and basophils in innate and acquired immunity. Curr Opin Immunol. 2000;12:624–631. doi: 10.1016/s0952-7915(00)00154-0. [DOI] [PubMed] [Google Scholar]
- 44.Krystel-Whittemore M, Dileepan KN, Wood JG. Mast cell: a multi-functional master cell. Front Immunol. 2015;6:620. doi: 10.3389/fimmu.2015.00620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Abraham SN, St John AL. Mast cell-orchestrated immunity to pathogens. Nat Rev Immunol. 2010;10:440–452. doi: 10.1038/nri2782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jonsson F, Daeron M. Mast cells and company. Front Immunol. 2012;3:16. doi: 10.3389/fimmu.2012.00016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Glaser S, DeMorrow S, Francis H, et al. Progesterone stimulates the proliferation of female and male cholangiocytes via autocrine/paracrine mechanisms. Am J Physiol Gastrointest Liver Physiol. 2008;295:G124–G136. doi: 10.1152/ajpgi.00536.2007. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 48.Mancinelli R, Franchitto A, Gaudio E, et al. After damage of large bile ducts by gamma-aminobutyric acid, small ducts replenish the biliary tree by amplification of calcium-dependent signaling and de novo acquisition of large cholangiocyte phenotypes. Am J Pathol. 2010;176:1790–1800. doi: 10.2353/ajpath.2010.090677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Marzioni M, Glaser S, Francis H, et al. Autocrine/paracrine regulation of the growth of the biliary tree by the neuroendocrine hormone serotonin. Gastroenterology. 2005;128:121–137. doi: 10.1053/j.gastro.2004.10.002. [DOI] [PubMed] [Google Scholar]
- 50.Graf A, Meng F, Hargrove L, et al. Knockout of histidine decarboxylase decreases bile duct ligation-induced biliary hyperplasia via downregulation of the histidine decarboxylase/VEGF axis through PKA-ERK1/2 signaling. Am J Physiol Gastrointest Liver Physiol. 2014;307:G813–G823. doi: 10.1152/ajpgi.00188.2014. [DOI] [PubMed] [Google Scholar]
- 51.Kennedy LL, Hargrove LA, Graf AB, et al. Inhibition of mast cell-derived histamine secretion by cromolyn sodium treatment decreases biliary hyperplasia in cholestatic rodents. Lab Invest. 2014;94:1406–1418. doi: 10.1038/labinvest.2014.129. [DOI] [PubMed] [Google Scholar]
- 52.McDaniel K, Meng F, Wu N, et al. Forkhead box A2 regulated biliary heterogeneity and senescence during cholestatic liver injury. Hepatology. 2016;66:544–559. doi: 10.1002/hep.28831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kim KH, Chen CC, Alpini G, et al. CCN1 induces hepatic ductular reaction through integrin alphavbeta(5)-mediated activation of NF-kappaB. J Clin Invest. 2015;125:1886–1900. doi: 10.1172/JCI79327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Weerachayaphorn J, Luo Y, Mennone A, et al. Deleterious effect of oltipraz on extrahepatic cholestasis in bile duct-ligated mice. J Hepatol. 2014;60:160–166. doi: 10.1016/j.jhep.2013.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lemoinne S, Cadoret A, Rautou PE, et al. Portal myofibroblasts promote vascular remodeling underlying cirrhosis formation through the release of microparticles. Hepatology. 2015;61:1041–1055. doi: 10.1002/hep.27318. [DOI] [PubMed] [Google Scholar]
- 56.Zhang F, Zhang Z, Chen L, et al. Curcumin attenuates angiogenesis in liver fibrosis and inhibits angiogenic properties of hepatic stellate cells. J Cell Mol Med. 2014;18:1392–1406. doi: 10.1111/jcmm.12286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Trauner M, Fickert P, Baghdasaryan A, et al. New insights into autoimmune cholangitis through animal models. Dig Dis. 2010;28:99–104. doi: 10.1159/000282072. [DOI] [PubMed] [Google Scholar]
- 58.Corpechot C, Chazouilleres O, Poupon R. Early primary biliary cirrhosis: biochemical response to treatment and prediction of long-term outcome. J Hepatol. 2011;55:1361–1367. doi: 10.1016/j.jhep.2011.02.031. [DOI] [PubMed] [Google Scholar]
- 59.Kaplan MM, Gershwin ME. Primary biliary cirrhosis. N Engl J Med. 2005;353:1261–1273. doi: 10.1056/NEJMra043898. [DOI] [PubMed] [Google Scholar]
- 60.Tischendorf JJ, Hecker H, Kruger M, et al. Characterization, outcome, and prognosis in 273 patients with primary sclerosing cholangitis: a single center study. Am J Gastroenterol. 2007;102:107–114. doi: 10.1111/j.1572-0241.2006.00872.x. [DOI] [PubMed] [Google Scholar]
- 61.Gaudio E, Barbaro B, Alvaro D, et al. Vascular endothelial growth factor stimulates rat cholangiocyte proliferation via an autocrine mechanism. Gastroenterology. 2006;130:1270–1282. doi: 10.1053/j.gastro.2005.12.034. [DOI] [PubMed] [Google Scholar]
- 62.Gaudio E, Onori P, Franchitto A, et al. Hepatic microcirculation and cholangiocyte physiopathology. Ital J Anat Embryol. 2005;110:71–75. [PubMed] [Google Scholar]
- 63.Meng F, Onori P, Hargrove L, et al. Regulation of the histamine/VEGF axis by miR-125b during cholestatic liver injury in mice. Am J Pathol. 2014;184:662–673. doi: 10.1016/j.ajpath.2013.11.008. [DOI] [PubMed] [Google Scholar]
- 64.Alpini G, Ulrich CD, 2nd, Phillips JO, et al. Upregulation of secretin receptor gene expression in rat cholangiocytes after bile duct ligation. Am J Physiol. 1994;266:G922–G928. doi: 10.1152/ajpgi.1994.266.5.G922. [DOI] [PubMed] [Google Scholar]
- 65.Calderone V, Gallego J, Fernandez-Miranda G, et al. Sequential functions of CPEB1 and CPEB4 regulate pathologic expression of vascular endothelial growth factor and angiogenesis in chronic liver disease. Gastroenterology. 2016;150:982–997. e930. doi: 10.1053/j.gastro.2015.11.038. [DOI] [PubMed] [Google Scholar]
- 66.Coch L, Mejias M, Berzigotti A, et al. Disruption of negative feedback loop between vasohibin-1 and vascular endothelial growth factor decreases portal pressure, angiogenesis, and fibrosis in cirrhotic rats. Hepatology. 2014;60:633–647. doi: 10.1002/hep.26995. [DOI] [PubMed] [Google Scholar]
- 67.Tung HC, Lee FY, Wang SS, et al. The beneficial effects of P2X7 antagonism in rats with bile duct ligation-induced cirrhosis. PLoS One. 2015;10:e0124654. doi: 10.1371/journal.pone.0124654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Nakamura I, Zakharia K, Banini BA, et al. Brivanib attenuates hepatic fibrosis in vivo and stellate cell activation in vitro by inhibition of FGF, VEGF and PDGF signaling. PLoS One. 2014;9:e92273. doi: 10.1371/journal.pone.0092273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yang L, Kwon J, Popov Y, et al. Vascular endothelial growth factor promotes fibrosis resolution and repair in mice. Gastroenterology. 2014;146:1339–1350. e1331. doi: 10.1053/j.gastro.2014.01.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kantari-Mimoun C, Castells M, Klose R, et al. Resolution of liver fibrosis requires myeloid cell-driven sinusoidal angiogenesis. Hepatology. 2015;61:2042–2055. doi: 10.1002/hep.27635. [DOI] [PubMed] [Google Scholar]
- 71.Yoshida S, Ikenaga N, Liu SB, et al. Extrahepatic platelet-derived growth factor-beta, delivered by platelets, promotes activation of hepatic stellate cells and biliary fibrosis in mice. Gastroenterology. 2014;147:1378–1392. doi: 10.1053/j.gastro.2014.08.038. [DOI] [PubMed] [Google Scholar]
- 72.Luo J, Liang Y, Kong F, Qiu J, Liu X, Chen A, et al. Vascular endothelial growth factor promotes the activation of hepatic stellate cells in chronic schistosomiasis. Immunol Cell Biol. 2017;95(4):399–407. doi: 10.1038/icb.2016.109. [DOI] [PubMed] [Google Scholar]
- 73.Wu K, Huang R, Wu H, et al. Collagen-binding vascular endothelial growth factor attenuates CCl4-induced liver fibrosis in mice. Mol Med Rep. 2016;14:4680–4686. doi: 10.3892/mmr.2016.5826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zen Y, Liberal R, Nakanuma Y, et al. Possible involvement of CCL1-CCR8 interaction in lymphocytic recruitment in IgG4-related sclerosing cholangitis. J Hepatol. 2013;59:1059–1064. doi: 10.1016/j.jhep.2013.06.016. [DOI] [PubMed] [Google Scholar]
- 75.Hargrove L, Graf-Eaton A, Kennedy L, et al. Isolation and characterization of hepatic mast cells from cholestatic rats. Lab Invest. 2016;96:1198–1210. doi: 10.1038/labinvest.2016.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lu J, Chen B, Li S, et al. Tryptase inhibitor APC 366 prevents hepatic fibrosis by inhibiting collagen synthesis induced by tryptase/protease-activated receptor 2 interactions in hepatic stellate cells. Int Immunopharmacol. 2014;20:352–357. doi: 10.1016/j.intimp.2014.04.001. [DOI] [PubMed] [Google Scholar]
- 77.Yin M, Wu L. Effect of mast cell chymase on activation, proliferation and transdifferentiation of hepatic stellate cells. Hepatogastroenterology. 2015;62:1007–1010. [PubMed] [Google Scholar]
- 78.Knight V, Tchongue J, Lourensz D, et al. Protease-activated receptor 2 promotes experimental liver fibrosis in mice and activates human hepatic stellate cells. Hepatology. 2012;55:879–887. doi: 10.1002/hep.24784. [DOI] [PubMed] [Google Scholar]