

Abstract
Progress in understanding the molecular biology of Alzheimer’s disease (AD) has provided a number of plausible therapeutic targets for disease-modifying interventions. To advance these agents towards eventual Food and Drug Administration (FDA) approval and incorporation into clinical practice by physicians and acceptance by patients and caregivers it is necessary to reach consensus on the meaning of disease modification and on what information is needed to provide a compelling factual basis for distinguishing disease modification from symptomatic treatment effects. Disease modification requires that the intervention have an impact of underlying pathology and pathophysiology of AD; “disease course modification”, “illness modification” or “disability sparing” are alternate terminologies that could be applied to symptomatic agents that do not affect the underlying neurobiology of AD. A variety of trial designs have been proposed to provide information supporting disease-modification including change from baseline designs, survival type designs, staggered start designs, and staggered withdrawal designs. Each of these has shortcomings and by themselves trial designs are not likely to provide sufficient information to conclusively prove that disease modification has occurred. Incorporation of a biomarker into clinical trials will support the claim for disease modification. Such a surrogate marker ideally should respond to the intervention, predict the clinical response to the intervention, and be compellingly related to the neurobiology of AD in the pathway affected by the intervention. A third axis of information supportive of disease modification is derived from observation of the effect of treatment in animal models of AD. The triad of a clinical outcome consistent with disease modification, support from a surrogate marker incorporated into the clinical trial, and basic science information indicating the effect of the therapy on a model of AD would combine to make a convincing case for disease modification.
Treatments with the potential to modify the course of Alzheimer’s disease (AD) by affecting the underlying pathophysiology are rapidly evolving. Many such agents are in preclinical phases of testing and some have entered advanced phases of human clinical trials. Disease-modifying treatment strategies are targeted on amyloid beta protein production, accumulation and removal or on enhancing cell viability (“neuroprotection”). Anti-amyloid interventions include vaccination, beta and gamma secretase inhibition or modulation, anti-fibrillization effects, and cholesterol lowering strategies1,2,3,4. A variety of neuroprotective compounds also are under investigation including antioxidants, inflammatory drugs, agents that ameliorate formation of neurofibrillary tangles, caspase inhibitors, drugs to reduce excitotoxicity, nerve growth factor-like drugs, muscarinic agonists, and nicotinic compounds (Table 1)5,6,7.
Table 1.
Principal agents being tested for possible disease-modifying therapies for Alzheimer’s disease.
| Beta-amyloid-related strategies |
| Vaccination/immunization |
| Beta-secretase inhibitors |
| Gamma-secretase modulators or inhibitors |
| Alpha-secretase enhancers |
| Fibrillization inhibitors |
| Statins |
| PPAR-gamma agonists and related agents |
| Zinc and copper chelators |
| Neuroprotective strategies |
| Nerve growth factor-related agents |
| Antioxidants |
| Anti-inflammatory agents |
| NMDA-receptor antagonists |
| AMPA-receptor antagonists |
| Anti-apoptic agents/caspase inhibitors/cell cycle inhibitors |
| Tan-related therapeutics |
| Specific estrogen receptor modulators |
| Monoamine oxidase B inhibitors |
| Nicotinic agonists |
| Muscarinic agonists |
| Calcium channel blocking agents |
| Folic acid and related agents |
Clinical trials of disease-modifying agents strive to demonstrate that the course of AD has been impacted and the rate of progression has been slowed. This objective differs from clinical trials for symptomatic agents in which improvement in cognition, function, and global measures or deferral of decline over a short period of time have been the principal outcomes of interest8,9,10,11,12,13. The differences in objectives raise fundamental questions about optimal clinical trial designs. The issues regarding how best to design clinical trials to demonstrate disease-modifying effects and what additional data may be necessary to convincingly demonstrate an impact on disease course are considered here. The challenge proposed by the need to generate data supportive of disease-modification is of critical importance in drug development and acceptance. The data bear on how best to present a disease-modifying claim for an agent in a new drug application (NDA) to the US Food and Drug Administration (FDA), are critically important to scientists seeking agents that may produce lasting impact on neurodegenerative disorders, and have uncommon significance to patients and families who desperately desire treatments that improve disease prognosis. The basic elements considered in demonstration of disease modification in AD are similar to those discussed when disease-modifying agents are considered for non-Alzheimer neurodegenerative conditions including Parkinson’s disease, dementia with Lewy bodies, frontotemporal lobar degenerations, and amyotrophic lateral sclerosis, and are central to discourse across neurodegenerative disorders.
Terminology
Disease modification has not been rigorously defined and no consensus definition is available. As used here, a disease-modifying agent is a pharmacological treatment that retards the underlying process of AD by intervening in the neurobiological processes that constitute the pathology and pathophysiology of the disease and lead to cell death or dysfunction. The targeted disease process must be in the pathway responsible for producing clinical decline and there must be a clinical response corresponding to the disease intervention. This approach is the same as that of the FDA, which defines disease-modifying treatments as those that exert their effects on the underlying pathology and/or pathophysiology of the disorder14.
“Neuroprotection” refers to a mechanism of action evident for some compounds. Disease-modifying agents can usefully be divided into those that are anti-amyloid and those that are neuroprotective. While anti-amyloid compounds would result in neuroprotection, that outcome would be a secondary “downstream” event and not the primary intent of intervention. Neuroprotective therapies are those that promote cell survival and viability without affecting the primary amyloidogenic process.
Effects on amyloid beta metabolism or neuroprotection are examples of disease-modifying therapies. In contrast, symptomatic agents improve or defer symptoms without affecting the fundamental disease process. The transmitter effects of cholinesterase inhibitors, for example, provide symptomatic improvement in cognition or deferral of decline relative to placebo but appear to have no or limited effects on the underlying pathology and pathophysiology of AD. Clinically, disease-modifying effects would be expected to defer the onset or show the progression of AD. Symptomatic agents defer the progression of AD, but do not change the slope of decline after the initial period of symptomatic response.
Alternative definitions of “disease modification” have been proffered. Sampaio15 suggested a “patient centered” definition of disease-modifying agent as those that “produce a clinically relevant long lasting benefit, sufficiently larger to change relevant milestones in the disease”. As defined, however, it is not required that these effects impact the underlying pathophysiology of the disease and this definition is not consistent with “disease modifying” as used here. When a clinical effect is achieved without modifying the underlying pathophysiology, terms such as “disease-course modifying”, “illness modifying”, or “disability sparing” may be more appropriate.
Can Clinical Trials Distinguish Between Symptomatic and Disease-Modifying Effects?
The classic example of a clinical trial designed to demonstrate symptomatic benefits of an anti-dementia agent is a randomized, double blind, placebo controlled, parallel group study comparing change from baseline in drug and placebo groups at the end of the study. These trials are typically of six months duration. The dual outcomes required by FDA for approval as an anti-dementia agent involve a measure of cognition such as the Alzheimer’s Disease Assessment Scale – cognitive portion (ADAS-cog)16 and a global measure such as the Clinical Global Impression of Change (CGIC) or the Clinician Interview Based Impression of Change with Caregiver Input (CIBIC-plus)17 or (in conjunction with cognitive improvement) a drug benefit compared with placebo on a measure of activities of daily living (ADL)18,14. Drug-placebo differences must be demonstrated in two well-conducted clinical trials. Secondary measures of ADL’s (when not used as a primary outcome), behavior, and caregiver time devoted to care, or pharmacoeconomic impact of treatment may be incorporated into the trial. Currently, instruments used in disease-modifying trials and symptomatic trials are the same.
Figure 1 shows the design of a typical trial constructed to capture symptomatic effects of an anti-dementia agent. Using the Mini Mental State Examination (MMSE)19, there is approximately a one-point improvement in MMSE followed by decline to baseline after one year of therapy. Double blind studies extending over one and two years suggest that this degree of improvement above placebo is maintained20,21, but the slope of decline is not changed or changed to only a minor extent.
Figure 1.
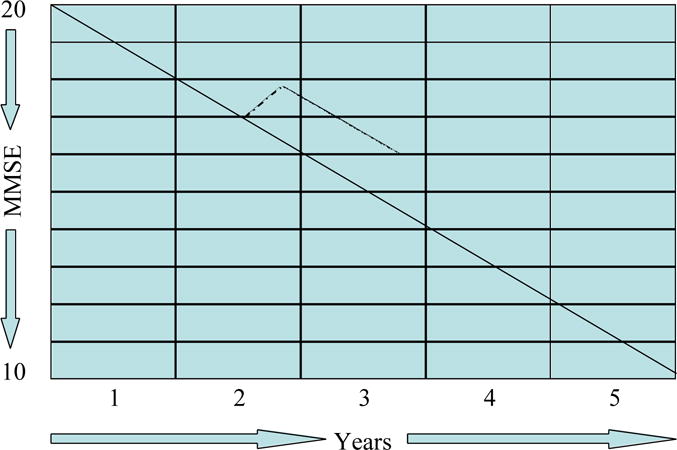
Graph of the typical effects of a symptomatic agent on the course of Alzheimer’s disease (MMSE = Mini Mental State Examination).
The objective of a clinical trial designed to demonstrate a disease-modifying effect is to show that the underlying disease process has been affected. Figure 2 shows the outcome of a hypothetical disease-modifying trial using the same parallel group design. In this model, the rate of disease progression is reduced by 50%; there is gradual divergence of disease trajectories representing approximately five MMSE points after 3.5 years of therapy. The difference in slopes of decline or increasing divergence of treatment and placebo groups provide support for a disease-modifying effect.
Figure 2.
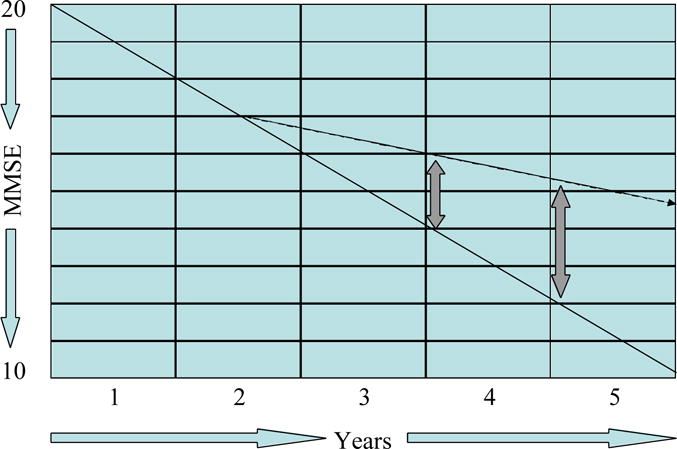
Graph, showing the anticipated effects of a disease-modifying agent on the course of Alzheimer’s disease in a parallel group design (MMSE = Mini Mental State Examination).
Figure 3 superimposes the projected clinical trial outcomes of a symptomatic agent and a disease-modifying agent. For the first 12 months of such a comparison, patients on symptomatic agents will perform at higher levels than those on disease-modifying agents; between 12 and 18 months they would have cognitive and global function of comparable levels; after 18 months there would be an increasing divergence of trajectories in favor of the disease-modifying agent. These observations indicate that trials of 12-18 months duration will be required to support disease modification using a parallel group design. Longer trials have greater attrition as well as greater measurement variability over time. These challenges must be anticipated in the trial design. If approved, agents requiring long term administration to achieve their effects will need to have benign side effect profiles and formulations that encourage chronic treatment adherence.
Figure 3.
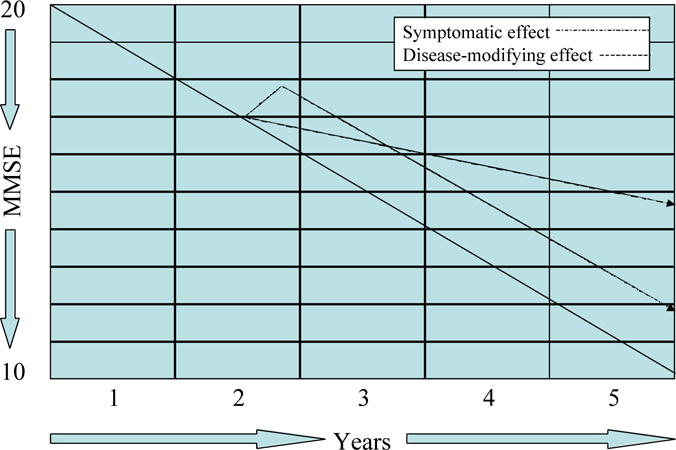
Graph, showing a comparison of the effects of disease-modifying and symptomatic effects on the course of AD (MMSE = Mini Mental State Examination).
A variety of strategies have been considered to allow clinical trials to provide more insight into the underlying mechanisms of action of agents tested. Several trials assessing putative disease-modifying agents have used survival type designs comparing time taken for patients on placebo compared to patients on active agents to meet specific disease milestones such as progression from Mild Cognitive Impairment (MCI) to AD22 or progression to death, nursing home placement, or decline to pre-specified cognitive and functional levels in patients with moderately severe AD (Figure 4)23. The longitudinal design by itself, however, cannot distinguish symptomatic from disease-modifying effects since symptomatic agents may delay progression to pre-specified levels of decline24.
Figure 4.
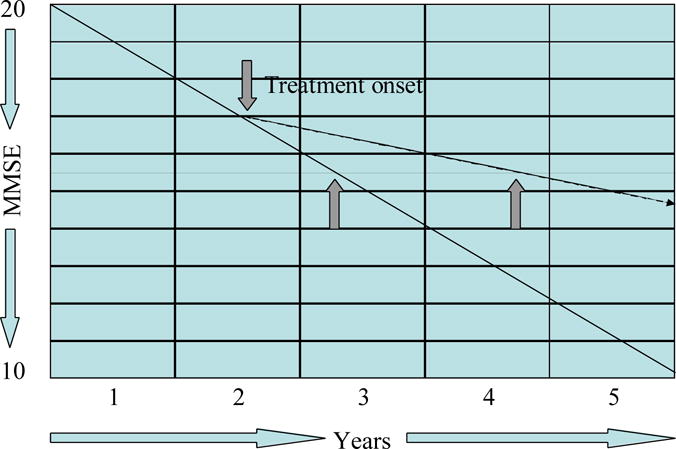
Graph, demonstrating a “delay to milestone” survival type of clinical trial design.
“Staggered start” and “staggered withdrawal” designs have been proposed in an effort to derive information regarding disease-modifying effects from clinical trials25,26. In the staggered start approach, one trial group receives placebo for a defined period of time while the other is started on placebo. After a delay, those receiving placebo are administered the active agent. Failure of this group to achieve a symptomatic benefit that places them at the same level as the group receiving treatment from onset of the trial, suggests that the agent has exerted a disease-modifying effect (Figure 5, A and B). Re-randomization at the point of treatment of the placebo group has been proposed as a means of maintaining the blind through the second phase of the study27.
Figure 5.
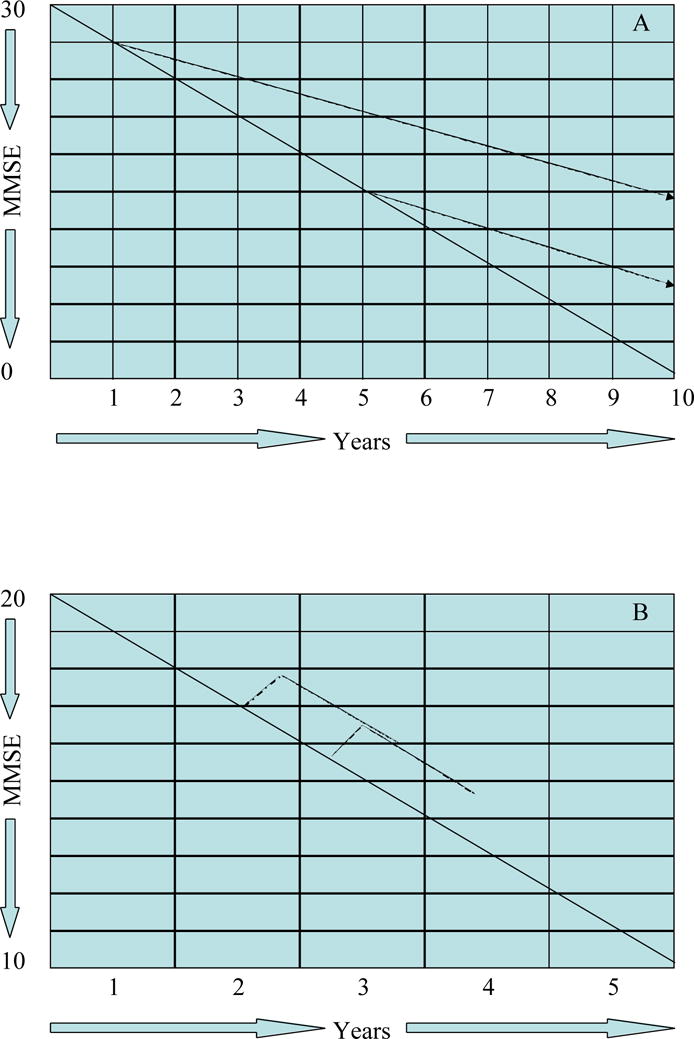
Staggered start design. A – disease modifying; B – symptomatic (MMSE = Mini Mental State Examination).
In the staggered withdrawal design, patients are withdrawn from therapy after a defined period of treatment to determine if they decline to the level of function of patients maintained on placebo and predicted on the basis of uninterrupted disease progression. If patients do not decline to the predicted level, the trial suggests that a disease-modifying effect has occurred (Figure 6, A and B). Again re-randomization at the point of withdrawal could be used to maintain blinding throughout the trial27.
Figure 6.
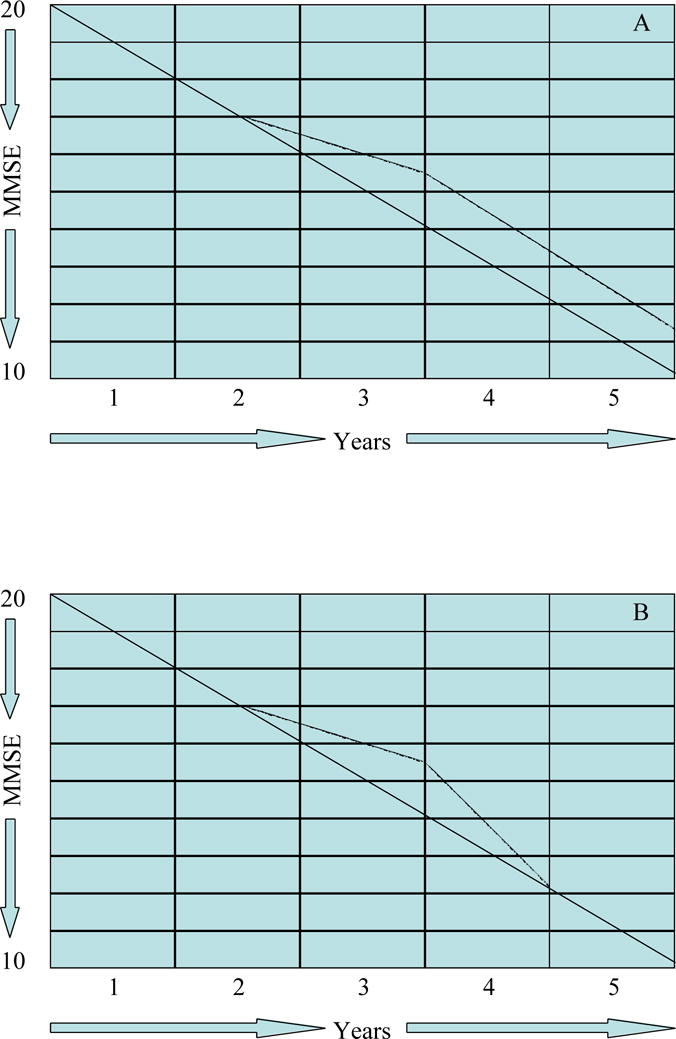
Staggered withdrawal design. A – disease modifying; B – symptomatic (MMSE = Mini Mental State Examination).
Staggered start and staggered withdrawal designs have rarely been used in clinical trials. There are uncertainties about the required length of therapy before a disease-modifying effect could be demonstrated in the staggered start design. Similarly, it is unclear how long a treatment period is needed and how long a withdrawal period would have to extend before the possibility of continuing symptomatic effects could be excluded in the staggered withdrawal design. The effect of beginning at two different baselines for the intervention groups in the staggered start design would also complicate interpretation of the results. There are ethical challenges to the withdrawal design28. While theoretically providing an additional avenue for gaining information on disease modification, the staggered start and staggered withdrawal designs are fraught with uncertainty and difficult to implement. No approved anti-dementia agent has utilized one of these approaches.
The situation is made more complex by the possibility that disease-modifying agents may have modest symptomatic benefit. Anti-amyloid or neuroprotective interventions may rescue populations of dysfunctional but still viable neurons. Correction of dysfunction would produce a beneficial symptomatic effect in addition to the long-term effects on disease progression. This possibility is supported by the observation that transgenic mice fated to develop the amyloid-related changes of AD and exhibiting cognitive impairment, have improved test performance when treated with some anti-amyloid therapies29,30,31. In addition, cholinesterase inhibitors previously considered as purely symptomatic agents may have at least mild disease-modifying effects. Clinical, imaging, and basic science studies support a limited disease-modifying role for cholinesterase inhibitors32,33,34,35.
Given the complex issues surrounding clinical trial design, implementation, and analysis it is unlikely that data derived from clinical trials alone will be sufficient to conclusively demonstrate disease modification. The most convincing evidence would be derived from a well conducted positive staggered start study14. A long-term parallel group design demonstrating increasing drug-placebo divergence will provide supportive information and has greater feasibility for most agents of interest but will not produce invincible evidence of disease-modification.
Can Biomarkers Help?
A biological marker is defined as a characteristic that is objectively measured and evaluated as an indicator of normal biological processes, pathogenic processes or pharmacologic responses to a therapeutic intervention36. A surrogate marker or surrogate endpoint is a biomarker that is intended to substitute for a clinical endpoint and is expected to predict clinical benefit based on epidemiologic, therapeutic, physiologic, or other scientific evidence36,37. Thus, all surrogate markers are biological markers, but not all biological markers will serve as surrogate endpoints. To support the concept of disease-modification, surrogate markers must reflect the disease process itself (not merely a correlate of the disease); the intervention’s effect on the surrogate must reliably predict the clinical outcome; and the treatment effect should be explained by the effect measured by the surrogate marker14,38.
The concept of disease modification in AD, entails the idea that the agent has an effect on the fundamental disease processes such as amyloid production, accumulation or removal or on the protection of neurons exposed to the adversities of the cerebral environment created by amyloid beta toxicity. A worthy surrogate marker must be linked to the neurobiological alterations underlying the clinical changes exhibited by the patients. Surrogate measures of the effects of the disease-modifying agent on the neurobiological processes would add validity to the claim that an agent has disease-modifying effects.
Magnetic resonance imaging (MRI) is currently most advanced as a candidate biomarker for inclusion in clinical trials of putative disease-modifying agents. However, brain atrophy is a gross measure of cerebral disease and provides little insight into the precise mechanisms by which a disease-modifying agent might affect AD pathophysiology. Reduction in disease activity would be expected to be reflected in a diminished rate of cerebral cortical volume loss and cerebral ventricular enlargement, although results from at least one study appear to contradict this assumption39. The National Institute on Aging (NIA) is currently sponsoring a national multi-center study of serial volume changes on MRI in the course of normal aging, MCI and AD. The Alzheimer’s Disease Neuroimaging Initiative (ADNI) will establish a data bank of serial imaging and other biomarkers that will assist in developing research designs and performing power calculations necessary to know what sample size is needed to establish a drug-placebo difference for disease-modifying therapies of varying efficacy40. The archival images also can be used as historical controls in futility studies41,42. Reproducibility of serial MRI measures lend themselves to use in clinical trials; substantially fewer patients are required to show a reduction in rate of change using MRI than using established clinical measures43,44,45. The relative lack of specificity of atrophy and the distant link between atrophy and specific effects on the underlying disease of anticipated interventions make MRI less than ideal as a candidate surrogate for clinical trials in AD.
Brain metabolism as measured by fluorodeoxyglucose positron emission tomography (FDG PET) provides a global measure of brain glucose metabolism and could be used as an outcome measure in clinical trials46. However, symptomatic agents also have effects on brain glucose metabolism47, 48 and FDG PET would provide relatively limited insight into the underlying neurobiological effects of a putative treatment.
Recent advances in molecular imaging promise to provide more specific information on the neurobiology of AD. Pittsburgh Compound-B (PIB) is an amyloid beta-specific radioligand that allows in vivo intracerebral visualization of beta-amyloid deposited in the brains of AD patients49, 50. Very early intervention with an anti-amyloid agent might be expected to limit the amyloid beta-related signal compared to placebo groups over the course of a clinical trial. The utility of PIB in trials will depend on how early amyloid is deposited in AD and how dynamic the amyloid signal on PIB imaging is found to be.
Other types of molecular imaging are evolving. PET imaging with 2-(1-{6-[(2-[18F]fluoroethyl)(methyl)amino]-2-naphthyl}ethylidene)malononitrile([18F]FDDNP) provides molecular labeling and imaging of both amyloid plaques and neurofibrillary tangles51. Imaging markers of activated microglia in dementia have been developed and could represent a measure of the inflammatory response as a downstream consequence of amyloid beta toxicity and as a direct measure of efficacy of an anti inflammatory agent52.
Scans useful as biological markers for agents with neuroprotective effects would need to measure some feature reflective of cell viability. Magnetic resonance spectroscopy (MRS) is one such candidate measure53. Molecular imaging of serotonin 1A receptors — concentrated in the hippocampus on cells lost early in the course of AD — provides another possible window on cell survival that might be incorporated into clinical trials54. A variety of imaging measures are evolving that may be used as biological markers of disease progression and possibly as surrogate measures to determine the effects of disease-modifying agents on the course of AD. Among imaging modalities, molecular imaging using ligands that label disease process intermediaries have the greatest promise for meeting the rigorous requirements of a surrogate marker.
Cerebrospinal fluid (CSF) measures may serve as biomarkers in clinical trials. Cerebrospinal fluid levels of total tau and phosphorylated tau protein are increased early in the course of AD and remain elevated55,56, 57,58. Tau measures can serve as a surrogate marker for the effect of a disease-modifying agent directed at tau metabolism and phosphorylation or could possibly serve as a measure of the effect of an agent on amyloid beta metabolism if tau hypophosphorylation is a product of amyloid toxicity. Measures of amyloid beta protein in the CSF are problematic as surrogate measures in clinical trials; levels increase early in AD and then decline, making the effect of anti-amyloid agents difficult to predict59,60. Other CSF measures that may have potential utility in clinical trials depending on the mechanism of the intervention include neurofilament protein, alpha-antichymotrypsin, and measures of oxidative metabolism61,62,63,64.
Plasma levels of amyloid beta protein may evolve as surrogate measures potentially useful in clinical trials65,66. Serum measures of inflammation61,67 and measures of oxidation such as isoprostanes and lipoxygenase products68,69,70 are elevated in the serum of patients with AD and could represent surrogate measures of the effects of therapeutic agents on targeted disease pathways. CSF and serum markers promise to perform better as surrogate markers than nonspecific imaging approaches since they have a closer relationship to the disease pathways targeted by emerging therapies.
Combinations of biomarkers may increase the power to detect the effect of disease-modifying agents. Such combinations should reflect different aspects of the effect of the treatment. Agents targeting tau hyperphosphorylation, for example, would be expected to be associated with decreased levels of CSF tau as well as less atrophy on MRI.
Surrogate measures might be included in proof of concept (phase IIb) studies, incorporated in all patients participating in a clinical trial, or collected from nested groups of patients in specific trial centers.
To be most convincing, a significant correlation would be expected between the effect on the surrogate endpoint and the effect on clinical measures if the two are mechanistically related. The combination of clinical trial outcomes consistent with disease modification, a drug-placebo difference in a relevant biomarker, an evident relationship between the surrogate marker and the disease process targeted by the therapeutic compound, and a significant relationship between the clinical outcome and the surrogate marker would represent substantial evidence of disease-modification.
Preclinical Evidence of Mechanism of Action
A plausible mechanism of action established by preclinical studies of the candidate therapeutic intervention is necessary to guide the choice of agent to study as a disease-modifying therapy. This basic evidence would increase the confidence of clinicians and patients when such drugs come to market, and effects on compelling disease models would assist in justifying the resource commitments by pharmaceutical and biotechnology companies to pursue development of a drug candidate.
Drug discovery in academic medical centers, biotechnology companies and the pharmaceutical industry employ a variety of screening techniques to document the potential effects of an agent on relevant neurobiological processes. Paradigms vary from basic molecular and biochemical investigations71 through cellular models, non-vertebrate models of AD such as transgenic drosophila72,73, to transgenic mouse and rat models that mimic most of the pathologic and some of the clinical features of AD74. Demonstration of the effect in mammalian models imitating human AD as closely as possible provides the most credible evidence to support the potential for disease-modifying activity in human neurotherapeutics.
The case for a disease-modifying effect of a candidate therapy would be most firmly supported by the concomitant development of surrogate markers applicable in both the animal model and human patient; the use of related doses, formulations, and administration approaches in both settings; and the use of behavioral measures in animals and humans that are conceptually related (Figure 7).
Figure 7.
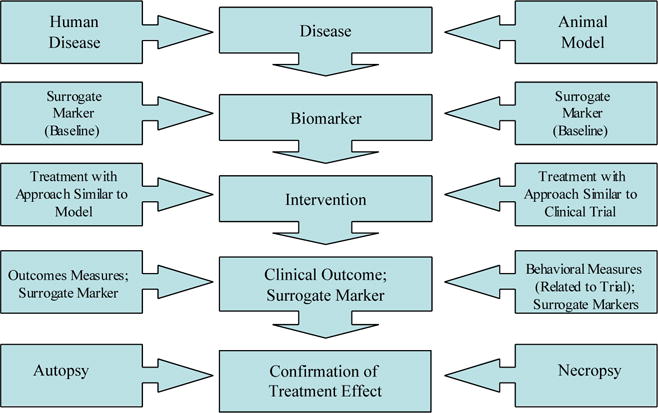
Model of harmonization of drug testing in animal models and clinical trials including surrogate markers in human populations with Alzheimer’s disease.
Comment
There are three critical elements to developing the case for a disease modifying effect of a therapeutic agent: 1) a plausible preclinical model supporting the effects of the agent on a fundamental aspect of the posited disease process; 2) results from clinical trials consistent with an effect on disease progression; 3) inclusion in the clinical trial of a surrogate biological marker reflecting the effect of the agent on the disease process and correlating with the clinical outcome. All three of these elements are necessary to establish disease-modifying activity; no element or combination of two elements is sufficient to support the claim that an agent is disease-modifying (Table 2).
Table 2.
Requirements for demonstrating disease modification by a candidate agent for the treatment of Alzheimer’s disease (AD).
| Preclinical evidence of disease modification |
| The candidate treatment impacts the pathologic and clinical aspects of an animal model that closely mimics AD |
| Supportive clinical trial observations |
| Randomized double-blinded placebo controlled trials establish a drug-placebo difference; parallel group designs with increasing drug-placebo divergence or staggered start designs are the most feasible approaches to conducting such trials |
| Supportive biomarker data |
| A biomarker that reflects the basic disease mechanism is incorporated into the clinical trial |
| There is a drug-placebo difference between the groups at the end of the trial on the surrogate measures |
| There is a correlation between the effects on the surrogate marker and the clinical effects of the compound in the trial |
Disease-modifying agents will be most efficacious and most meaningful in preserving quality of life for AD patients if they are implemented very early in AD or in MCI patients considered to be in the pre-dementia phase of AD. Figure 8 shows the trajectories of patients, whose therapy was begun when the patient had an MMSE score of 27, and contrasts the outcome with a patient, whose treatment was begun when the MMSE score was 20. After five years, the first patient has a MMSE score above 20 and is in the mild stage of AD; the patient began when disease was more severe would be predicted to have a MMSE score of 15 compatible with moderate-to-severe AD.
Figure 8.
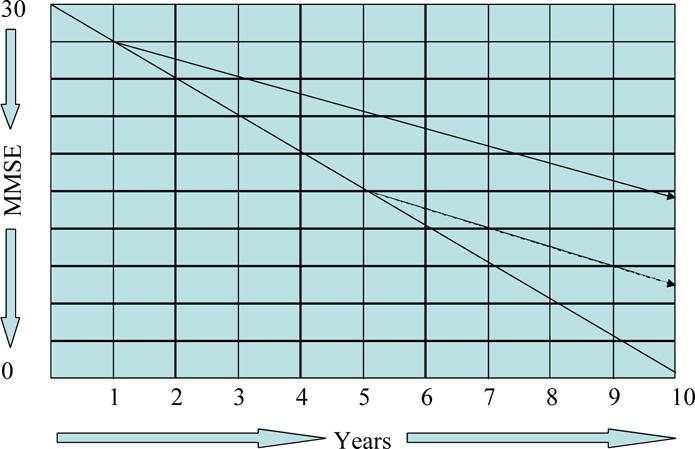
Graph, showing the effect of early versus late intervention with a disease-modifying agent in Alzheimer’s disease (MMSE = Mini Mental State Examination).
Trials with MCI must be larger and their methodology is less well established75, but development of effective therapeutics for this population is critical to prevent patients from developing Alzheimer’s dementia or remaining in the most mild stages of the disease. Whether current measurement methods developed for AD trials and adopted in recent trials of MCI75 are optimal for assessing outcomes in this phase of the disease warrants greater study.
Death from age-related competitive mortality is likely in this elderly population if AD progression is minimized. Greater longevity is not necessarily a desirable outcome in the therapy of AD: longer periods with no or few symptoms is the outcome of choice. Death from age-related disease in a patient with mild cognitive symptoms would prevent patients from entering the severe stages of AD and is the principal intent of disease-modifying therapy implemented in the early phases of AD.
Patients and caregivers are most concerned with symptom control and reducing clinical decline regardless of how it is achieved. Amelioration of illness or progressive disability is the most important outcome regardless of drug mechanism. Likewise clinicians are most interested in optimizing patient function and quality of life whatever the therapeutic mechanism. This emphasizes the importance of continuing to optimize the use of currently available symptomatic agents, as well as to encourage the development of new potentially more efficacious symptomatic interventions. On the other hand, it seems unlikely that symptomatic agents alone can compensate for the ever more severe decline in cognition, function and behavior manifested over the course of AD. Disease-modifying agents will be required to preserve sufficient cerebral integrity to permit symptomatic agents to create synergies that come closer to normalizing cognition. Combinations of symptomatic cognitive-enhancing and psychotropic agents76 with disease-modifying interventions are anticipated to achieve optimal patient quality of life and preservation of maximal function.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosure Statement. Dr. Cummings has provided consultation to the following pharmaceutical companies: Avanir, Eisai, EnVivo, Forest, Janssen, Lilly, Lundbeck, Merz, Novartis, Ono Pharma, Pfizer, Sanofi-Aventis, and Sepracor and Taketa.
References
- 1.Gilman S, Koller M, Black RS, et al. Clinical effects of Aβ immunization (AN1792) in patients with AD in an interrupted trial. Neurology. 2005;64:1553–1562. doi: 10.1212/01.WNL.0000159740.16984.3C. [DOI] [PubMed] [Google Scholar]
- 2.Gandy S, Martins RN, Buxbaum J. Molecular and cellular basis for anti-amyloid therapy in Alzheimer disease. Alzheimer Dis Assoc Disord. 2003;17:259–266. doi: 10.1097/00002093-200310000-00011. [DOI] [PubMed] [Google Scholar]
- 3.Citron M. Strategies for disease modification in Alzheimer’s disease. Nat Rev Neurosci. 2004;5:677–685. doi: 10.1038/nrn1495. [DOI] [PubMed] [Google Scholar]
- 4.Wolfe MS. Therapeutic strategies for Alzheimer’s disease. Nat Rev Drug Discov. 2002;1:859–866. doi: 10.1038/nrd938. [DOI] [PubMed] [Google Scholar]
- 5.Akwa Y, Allain H, Bentue-Ferrer D, et al. Neuroprotection and neurodegenerative diseases: from biology to clinical practice. Alzheimer Dis Assoc Disord. 2005;19:226–239. doi: 10.1097/01.wad.0000189053.25817.d6. [DOI] [PubMed] [Google Scholar]
- 6.Scarpini E, Scheltens P, Feldman H. Treatment of Alzheimer’s disease: current status and new perspectives. Lancet Neurol. 2003;2:539–547. doi: 10.1016/s1474-4422(03)00502-7. [DOI] [PubMed] [Google Scholar]
- 7.Fischer F, Matthisson M, Herrling P. List of Drugs in Development for Neurodegenerative Diseases. Neurodegenerative Diseases. 2004;1:50–70. doi: 10.1159/000077879. [DOI] [PubMed] [Google Scholar]
- 8.Corey-Bloom J, Anand R, Veach J, ENA 713 B352 Study Group A randomized trial evaluating the efficacy and safety of ENA 713 (rivastigmine tartrate), a new acetylcholinesterase inhibitor, in patients with mild to moderately severe Alzheimer’s disease. Int J Geriatr Psychopharmacology. 1998;1:55–65. [Google Scholar]
- 9.Feldman H, Gauthier S, Hecker J, et al. A 24-week, randomized, double-blind study of donepezil in moderate to severe Alzheimer’s disease. Neurology. 2001;57:613–620. doi: 10.1212/wnl.57.4.613. [DOI] [PubMed] [Google Scholar]
- 10.Rosler M, Anand R, Cicin-Sain A, et al. Efficacy and safety of rivastigmine in patients with Alzheimer’s disease: international randomized controlled trial. BMJ. 1999;318:633–638. doi: 10.1136/bmj.318.7184.633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tariot PN, Solomon PR, Morris JC, et al. A 5-month, randomized, placebo-controlled trial of galantamine in AD. Neurology. 2000;54:2269–2276. doi: 10.1212/wnl.54.12.2269. [DOI] [PubMed] [Google Scholar]
- 12.Wilkinson D, Murray J, Galantamine Research Group Galantamine: a randomized, double-blind, dose comparison in patients with Alzheimer’s disease. Int J Geriatr Psychiatry. 2001;16:852–857. doi: 10.1002/gps.409. [DOI] [PubMed] [Google Scholar]
- 13.Rogers SL, Farlow MR, Doody RS, et al. A 24-week, double-blind, placebo-controlled trial of donepezil in patients with Alzheimer’s disease. Neurology. 1998;50:136–145. doi: 10.1212/wnl.50.1.136. [DOI] [PubMed] [Google Scholar]
- 14.Mani RB. The evaluation of disease modifying therapies in Alzheimer’s disease: a regulatory viewpoint. Stat Med. 2004;23:305–314. doi: 10.1002/sim.1718. [DOI] [PubMed] [Google Scholar]
- 15.Sampaio C. Alzheimer Disease: Disease Modifying TrialsWhere are We? Where do We Need to Go? A Reflective Paper. J Nutr Health Aging. 2006;10:113–115. [PubMed] [Google Scholar]
- 16.Rosen WG, Mohs RC, Davis KL. A new rating scale for Alzheimer’s disease. Am J Psychiatry. 1984;141:1356–1364. doi: 10.1176/ajp.141.11.1356. [DOI] [PubMed] [Google Scholar]
- 17.Schneider LS, Olin JT, Doody RS, et al. Validity and reliability of the Alzheimer’s Disease Cooperative Study-Clinical Global Impression of Change. Alzheimer Dis Assoc Disord. 1997;11(Suppl 2):S22–S32. doi: 10.1097/00002093-199700112-00004. [DOI] [PubMed] [Google Scholar]
- 18.Leber P. Food and Drug Administration; 1990. Washington, D C: 1990. Guidelines for the Clinical Evaluation of Antidementia Drugs. [Google Scholar]
- 19.Folstein MF, Folstein SE, McHugh PR. “Mini-Mental State”: a practical method for grading the cognitive state of patients for the clinician. J Psychiatr Res. 1975;12:189–198. doi: 10.1016/0022-3956(75)90026-6. [DOI] [PubMed] [Google Scholar]
- 20.Winblad B, Engedal K, Soininen H, et al. A 1-year, randomized, placebo-controlled study of donepezil in patients with mild to moderate AD. Neurology. 2001;57:489–495. doi: 10.1212/wnl.57.3.489. [DOI] [PubMed] [Google Scholar]
- 21.AD2000 Collaborative Group. Long-term donepezil treatment in 565 patients with Alzheimer’s disease (AD2000): randomised double-blind trial. Lancet. 2004;363:2105–2115. doi: 10.1016/S0140-6736(04)16499-4. [DOI] [PubMed] [Google Scholar]
- 22.Petersen RC, Thomas RG, Grundman M, et al. Vitamin E and donepezil for the treatment of mild cognitive impairment. N Engl J Med. 2005;352:2379–2388. doi: 10.1056/NEJMoa050151. [DOI] [PubMed] [Google Scholar]
- 23.Sano M, Ernesto C, Thomas RG, et al. A controlled trial of selegiline, α-tocopherol, or both as treatment for Alzheimer’s disease. N Engl J Med. 1997;336:1216–1222. doi: 10.1056/NEJM199704243361704. [DOI] [PubMed] [Google Scholar]
- 24.Mohs RC, Doody RS, Morris JC, et al. A 1-year, placebo-controlled preservation of function survival study of donepezil in AD patients. Neurology. 2001;57:481–488. doi: 10.1212/wnl.57.3.481. [DOI] [PubMed] [Google Scholar]
- 25.Leber P. Slowing the progression of Alzheimer disease: methodologic issues. Alzheimer Dis Assoc Disord. 1997;11(Suppl 5):S10–21. [PubMed] [Google Scholar]
- 26.Leber P. Observations and suggestions on antidementia drug development. Alzheimer Dis Assoc Disord. 1996;10(Suppl 1):31–35. doi: 10.1097/00002093-199601031-00009. [DOI] [PubMed] [Google Scholar]
- 27.Andrieu S, Rascol O, Lang T, et al. Methodological Issues and Statistical Analyses. The Journal of Nutrition, Health & Aging. 2006;10:116–117. [PubMed] [Google Scholar]
- 28.Whitehouse PJ, Kittner B, Roessner M, et al. Clinical trial designs for demonstrating disease-course-altering effects in dementia. Alzheimer Dis Assoc Disord. 1998;12:281–294. doi: 10.1097/00002093-199812000-00007. [DOI] [PubMed] [Google Scholar]
- 29.Dodart JC, Bales KR, Gannon KS, et al. Immunization reverses memory deficits without reducing brain Aβ burden in Alzheimer’s disease model. Nat Neurosci. 2002;5:452–457. doi: 10.1038/nn842. [DOI] [PubMed] [Google Scholar]
- 30.Janus C, Pearson J, McLaurin J, et al. Aβ peptide immunization reduces behavioural impairment and plaques in a model of Alzheimer’s disease. Nature. 2000;408:979–982. doi: 10.1038/35050110. [DOI] [PubMed] [Google Scholar]
- 31.Wilcock DM, Rojiani A, Rosenthal A, et al. Passive immunotherapy against Aβ in aged APP-transgenic mice reverses cognitive deficits and depletes parenchymal amyloid deposits in spite of increased vascular amyloid and microhemorrhage. J Neuroinflammation. 2004;1:24. doi: 10.1186/1742-2094-1-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Doody RS, Dunn JK, Clark CM, et al. Chronic donepezil treatment is associated with slowed cognitive decline in Alzheimer’s disease. Dement Geriatr Cogn Disord. 2001;12:295–300. doi: 10.1159/000051272. [DOI] [PubMed] [Google Scholar]
- 33.Francis PT, Nordberg A, Arnold SE. A preclinical view of cholinesterase inhibitors in neuroprotection: do they provide more than symptomatic benefits in Alzheimer’s disease? Trends Pharmacol Sci. 2005;26:104–111. doi: 10.1016/j.tips.2004.12.010. [DOI] [PubMed] [Google Scholar]
- 34.Krishnan KR, Charles HC, Doraiswamy PM, et al. Randomized, placebo-controlled trial of the effects of donepezil on neuronal markers and hippocampal volumes in Alzheimer’s disease. Am J Psychiatry. 2003;160:2003–2011. doi: 10.1176/appi.ajp.160.11.2003. [DOI] [PubMed] [Google Scholar]
- 35.Sabbagh MN, Farlow MR, Relkin N, et al. Do cholinergic therapies have disease-modifying effects in Alzheimer’s disease? Alzheimer’s & Dementia. 2006;2:118–125. doi: 10.1016/j.jalz.2006.02.001. [DOI] [PubMed] [Google Scholar]
- 36.Biomarkers Definitions Working Group. Biomarkers and surrogate endpoints: preferred definitions and conceptual framework. Clin Pharmacol Ther. 2001;69:89–95. doi: 10.1067/mcp.2001.113989. [DOI] [PubMed] [Google Scholar]
- 37.Feigin A. Evidence from biomarkers and surrogate endpoints. NeuroRx. 2004;1:323–330. doi: 10.1602/neurorx.1.3.323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fleming TR, DeMets DL. Surrogate end points in clinical trials: are we being misled? Ann Intern Med. 1996;125:605–613. doi: 10.7326/0003-4819-125-7-199610010-00011. [DOI] [PubMed] [Google Scholar]
- 39.Fox NC, Black RS, Gilman S, et al. Effects of Aβ immunization (AN1792) on MRI measures of cerebral volume in Alzheimer disease. Neurology. 2005;64:1563–1572. doi: 10.1212/01.WNL.0000159743.08996.99. [DOI] [PubMed] [Google Scholar]
- 40.Mueller SG, Weiner MW, Thal LJ, et al. Ways toward an early diagnosis in Alzheimer’s disease: The Alzheimer’s Disease Neuroimaging Initiative (ADNI) Alzheimer’s & Dementia. 2005;1:55–66. doi: 10.1016/j.jalz.2005.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tilley BC, Palesch YY, Kieburtz K, et al. Optimizing the ongoing search for new treatments for Parkinson disease: using futility designs. Neurology. 2006;66:628–633. doi: 10.1212/01.wnl.0000201251.33253.fb. [DOI] [PubMed] [Google Scholar]
- 42.Levin B. The utility of futility. Stroke. 2005;36:2331–2332. doi: 10.1161/01.STR.0000185722.99167.56. [DOI] [PubMed] [Google Scholar]
- 43.Jack CR, Slomkowski M, Gracon S, et al. MRI as a biomarker of disease progression in a therapeutic trial of milameline for AD. Neurology. 2003;60:253–260. doi: 10.1212/01.wnl.0000042480.86872.03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fox NC, Cousens S, Scahill R, et al. Using serial registered brain magnetic resonance imaging to measure disease progression in Alzheimer disease. Arch Neurol. 2000;57:339–344. doi: 10.1001/archneur.57.3.339. [DOI] [PubMed] [Google Scholar]
- 45.Schott JM, Price SL, Frost C, et al. Measuring atrophy in Alzheimer disease: a serial MRI study over 6 and 12 months. Neurology. 2005;65:119–124. doi: 10.1212/01.wnl.0000167542.89697.0f. [DOI] [PubMed] [Google Scholar]
- 46.Alexander GE, Chen K, Pietrini P, Rapoport SI, Reiman EM. Longitudinal PET evaluation of cerebral metabolic decline in dementia: a potential outcome measure in Alzheimer’s disease treatment studies. Am J Psychiatry. 2002;159:738–745. doi: 10.1176/appi.ajp.159.5.738. [DOI] [PubMed] [Google Scholar]
- 47.Mega MS, Cummings JL, O’Connor SM, et al. Cognitive and metabolic responses to metrifonate therapy in Alzheimer disease. Neuropsychiatry, Neuropsychol, Behav Neurol. 2001;14:63–68. [PubMed] [Google Scholar]
- 48.Mega M, Dinov ID, Porter V, et al. Metabolic patterns associated with the clinical response to galantamine therapy. Arch Neurol. 2005;62:721–728. doi: 10.1001/archneur.62.5.721. [DOI] [PubMed] [Google Scholar]
- 49.Klunk WE, Engler H, Nordberg A, et al. Imaging brain amyloid in Alzheimer’s disease with Pittsburgh Compound-B. Ann Neurol. 2004;55:306–319. doi: 10.1002/ana.20009. [DOI] [PubMed] [Google Scholar]
- 50.Mathis CA, Klunk WE, Price JC, DeKosky ST. Imaging technology for neurodegenerative diseases: progress toward detection of specific pathologies. Arch Neurol. 2005;62:196–200. doi: 10.1001/archneur.62.2.196. [DOI] [PubMed] [Google Scholar]
- 51.Shoghi-Jadid K, Small GW, Agdeppa ED, et al. Localization of neurofibrillary tangles and β-amyloid plaques in the brains of living patients with Alzheimer disease. Am J Geriatr Psychiatry. 2002;10:24–35. [PubMed] [Google Scholar]
- 52.Cagnin A, Brooks DJ, Kennedy AM, et al. In-vivo measurement of activated microglia in dementia. Lancet. 2001;358:461–467. doi: 10.1016/S0140-6736(01)05625-2. [DOI] [PubMed] [Google Scholar]
- 53.Lin A, Ross BD, Harris K, et al. Efficacy of proton magnetic resonance spectroscopy in neurological diagnosis and neurotherapeutic decision making. NeuroRx. 2005;2:197–214. doi: 10.1602/neurorx.2.2.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kepe V, Barrio JR, Huang S-C, et al. Serotonin 1A receptors in the living brain of Alzheimer’s disease patients. Proc Natl Acad Sci USA. 2006;103:702–707. doi: 10.1073/pnas.0510237103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Andreasen N, Minthon L, Clarberg A, et al. Sensitivity, specificity, and stability of CSF-tau in AD in a community-based patient sample. Neurology. 1999;53:1488–1494. doi: 10.1212/wnl.53.7.1488. [DOI] [PubMed] [Google Scholar]
- 56.Itoh N, Arai H, Urakami K, et al. Large-scale, multicenter study of cerebrospinal fluid tau protein phosphorylated at Serine 199 for the antemortem diagnosis of Alzheimer’s disease. Ann Neurol. 2001;50:150–156. doi: 10.1002/ana.1054. [DOI] [PubMed] [Google Scholar]
- 57.Maddalena A, Papassotiropoulos A, Muller-Tillmanns B, et al. Biochemical diagnosis of Alzheimer disease by measuring the cerebrospinal fluid ratio of phosphorylated tau protein to β-amyloid peptide42. Arch Neurol. 2003;60:1202–1206. doi: 10.1001/archneur.60.9.1202. [DOI] [PubMed] [Google Scholar]
- 58.Sunderland T, Linker G, Mirza N, et al. Decreased β-amyloid1-42 and increased tau levels in cerebrospinal fluid of patients with Alzheimer disease. JAMA. 2003;289:2094–2103. doi: 10.1001/jama.289.16.2094. [DOI] [PubMed] [Google Scholar]
- 59.Jensen M, Schroder J, Blomberg M, et al. Cerebrospinal fluid Aβ42 is increased early in sporadic Alzheimer’s disease and declines with disease progression. Ann Neurol. 1999;45:504–511. doi: 10.1002/1531-8249(199904)45:4<504::aid-ana12>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 60.Kanai M, Matsubara E, Isoe K, et al. Longitudinal study of cerebrospinal fluid levels of tau, Aβ1-40, and Aβ1-42(43) in Alzheimer’s disease: a study in Japan. Ann Neurol. 1998;44:17–26. doi: 10.1002/ana.410440108. [DOI] [PubMed] [Google Scholar]
- 61.DeKosky ST, Ikonomovic MD, Wang X, et al. Plasma and cerebrospinal fluid α1-antichymotrypsin levels in Alzheimer’s disease: correlation with cognitive impairment. Ann Neurol. 2003;53:81–90. doi: 10.1002/ana.10414. [DOI] [PubMed] [Google Scholar]
- 62.Kahle PJ, Jakowec M, Teipel SJ, et al. Combined assessment of tau and neuronal thread protein in Alzheimer’s disease CSF. Neurology. 2000;54:1498–1504. doi: 10.1212/wnl.54.7.1498. [DOI] [PubMed] [Google Scholar]
- 63.Rosengren LE, Karlsson JE, Sjogren M, Blennow K, Wallin A. Neurofilament protein levels in CSF are increased in dementia. Neurology. 1999;52:1090–1093. doi: 10.1212/wnl.52.5.1090. [DOI] [PubMed] [Google Scholar]
- 64.Sun Y-X, Minthon L, Wallmark A, Warkentin S, Blennow K, Janciauskiene S. Inflammatory markers in matched plasma and cerebrospinal fluid from patients with Alzheimer’s disease. Dement Geriatr Cogn Disord. 2003;16:136–144. doi: 10.1159/000071001. [DOI] [PubMed] [Google Scholar]
- 65.Fukumoto H, Tennis M, Locascio JJ, et al. Age but not diagnosis is the main predictor of plasma amyloid β-protein levels. Arch Neurol. 2003;60:958–964. doi: 10.1001/archneur.60.7.958. [DOI] [PubMed] [Google Scholar]
- 66.Mayeux R, Honig LS, Tang M-X, et al. Plasma Aβ40 and Aβ42 and Alzheimer’s disease: relation to age, mortality, and risk. Neurology. 2003;61:1185–1190. doi: 10.1212/01.wnl.0000091890.32140.8f. [DOI] [PubMed] [Google Scholar]
- 67.Sala G, Galimberti G, Canevari C, et al. Peripheral cytokine release in Alzheimer patients: correlation with disease severity. Neurobiol Aging. 2003;24:909–914. doi: 10.1016/s0197-4580(03)00010-1. [DOI] [PubMed] [Google Scholar]
- 68.Montine TJ, Beal MF, Cudkowicz ME, et al. Increased CSF F2-isoprostane concentration in probable AD. Neurology. 1999;52:562–565. doi: 10.1212/wnl.52.3.562. [DOI] [PubMed] [Google Scholar]
- 69.Pratico D, Clark CM, Lee VM-Y, et al. Increased 8,12-iso-iPF2α-VI in Alzheimer’s disease: correlation of a noninvasive index of lipid peroxidation with disease severity. Ann Neurol. 2000;48:809–812. [PubMed] [Google Scholar]
- 70.Yao Y, Clark CM, Trojanowski JQ, Lee VM-Y, Pratico D. Elevation of 12/15 lipoxygenase products in AD and mild cognitive impairment. Ann Neurol. 2005;58:623–626. doi: 10.1002/ana.20558. [DOI] [PubMed] [Google Scholar]
- 71.Vetrivel KS, Thinakaran G. Amyloidogenic processing of β-amyloid precursor protein in intracellular compartments. Neurology. 2006;66:S69–73. doi: 10.1212/01.wnl.0000192107.17175.39. [DOI] [PubMed] [Google Scholar]
- 72.Greeve I, Kretzschmar D, Tschape J-A, et al. Age-dependent neurodegeneration and Alzheimer-amyloid plaque formation in transgenic Drosophila. J Neurosci. 2004;24:3899–3906. doi: 10.1523/JNEUROSCI.0283-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Jackson G, Wiedau-Pazos M, Sang T-K, et al. Human wild-type tau interacts with wingless pathway components and produces neurofibrillary pathologyin Drosophila. Neuron. 2002;34:509–519. doi: 10.1016/s0896-6273(02)00706-7. [DOI] [PubMed] [Google Scholar]
- 74.Oddo S, Caccamo A, Shepherd JD, et al. Triple-transgenic model of Alzheimer’s disease with plaques and tangles: intracellular Aβ and synaptic dysfunction. Neuron. 2003;39:409–421. doi: 10.1016/s0896-6273(03)00434-3. [DOI] [PubMed] [Google Scholar]
- 75.Jelic V, Kivipelto M, Winblad B. Clinical trials in mild cognitive impairment: lessons for the future. J Neurol Neurosurg Psychiatry. 2006;77:429–438. doi: 10.1136/jnnp.2005.072926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Cummings JL, Zhong K. Treatments for behavioral disorders in neurodegenerative diseases: drug development strategies. Nature Drug Discovery. 2006;5:64–74. doi: 10.1038/nrd1928. [DOI] [PubMed] [Google Scholar]