

Abstract
Cardiovascular disease is the primary cause of mortality for individuals with type 2 diabetes mellitus. During the diabetic condition, cardiovascular dysfunction can be partially attributed to molecular changes in the tissue, including alterations in microRNA (miRNA) interactions. MiRNAs have been reported in the mitochondrion and their presence may influence cellular bioenergetics, creating decrements in functional capacity. In this study, we examined the roles of Argonaute 2 (Ago2), a protein associated with cytosolic and mitochondrial miRNAs, and Polynucleotide Phosphorylase (PNPase), a protein found in the inner membrane space of the mitochondrion, to determine their role in mitochondrial miRNA import. In cardiac tissue from human and mouse models of type 2 diabetes mellitus, Ago2 protein levels were unchanged while PNPase protein expression levels were increased; also, there was an increase in the association between both proteins in the diabetic state. MiRNA-378 was found to be significantly increased in db/db mice, leading to decrements in ATP6 levels and ATP synthase activity, which was also exhibited when overexpressing PNPase in HL-1 cardiomyocytes and in HL-1 cells with stable miRNA-378 overexpression (HL-1-378). To assess potential therapeutic interventions, flow cytometry evaluated the capacity for targeting miRNA-378 species in mitochondria through antimiR treatment, revealing miRNA-378 level-dependent inhibition. Our study establishes PNPase as a contributor to mitochondrial miRNA import through the transport of miRNA-378, which may regulate bioenergetics during type 2 diabetes mellitus. Further, our data provide evidence that manipulation of PNPase levels may enhance the delivery of antimiR therapeutics to mitochondria in physiological and pathological conditions.
Keywords: Mitochondria, microRNA, Bioenergetics, Diabetes mellitus
1. Introduction
Type 2 diabetes mellitus currently affects 29 million Americans, with an inordinate cost to society of $245 billion in medical bills and missed work [1,43]. Further, type 2 diabetes mellitus serves as a major risk factor for cardiovascular disease. During type 2 diabetes mellitus, when cardiac tissue becomes insulin resistant, a proteomic, transcriptomic and epigenomic remodeling occurs as an adaptive mechanism to the pathological insult [2,44,46]. Our laboratory and others have shown that the assault to the transcript can occur in both the cytosol and mitochondria within the cardiomyocyte [44,50]. Cardiac mitochondrial subpopulations have previously been shown to have distinct functional differences [37,47,49,87]. Our laboratory has shown that during type 1 diabetes mellitus, mitochondria located between the myofibrils, interfibrillar mitochondria (IFM), are negatively impacted to a greater extent, while the mitochondria residing directly below the sarcolemmal membrane, subsarcolemmal mitochondria (SSM), are predominantly affected during type 2 diabetes mellitus [8–10,22–25,50,88].
MicroRNAs (miRNAs) are a type of noncoding RNA that have been shown to play a vital role in the regulation of gene expression through degradation of target messenger RNAs (mRNAs) or translational repression [61]. MiRNAs exert their influence through binding to the 3′-untranslated region of the target mRNAs [61]. The association of a miRNA with the RNA-induced silencing complex (RISC) has been shown in the cytoplasm, with the Argonaute (AGO) family proteins functioning to help in miRNA repression through the inhibition of protein synthesis when bound to the 3′-untranslated region of the mRNA [6,39]. A variety of groups, including our own, has also indicated the presence of miRNA in the mitochondria [3,5,13–15,27,28,50,55,83–85, 89,98]. MiRNA species have the ability to target mRNA transcripts produced by the mitochondrial genome in the mitochondrion to control mRNA stability and degradation [27,28,33]. The electron transport chain (ETC) and ATP synthase are housed within the mitochondrial matrix and inner mitochondrial membrane; the differential expression of miRNAs in these mitochondrial locales, with the ability of miRNAs to target these components of cellular bioenergetics, is consequential to the overall functionality of the organelle.
In disease states, miRNA expression in the mitochondria can fluctuate [32,34,68]. Specifically, in diabetes mellitus, miRNA-378 was shown to be increased in the mitochondrion [50]. MiRNA-378 presence was associated with decreases in ATP synthase activity through binding to the ATP6 transcript encoding an essential F0 subunit of ATP synthase. This dynamic led to a decrease in cellular ATP content and bioenergetics [50]. Polynucleotide Phosphorylase (PNPase), situated in the inner mitochondrial membrane and extending into the intermembrane space, is known to facilitate the transport of ribosomal RNA (rRNA) and transfer RNA (tRNA) based on their hairpin-loop structure [3,92,93]. PNPase also functions to edit and degrade RNA, as well as plays a role in controlling cellular senescence and cell cycle [59,76,77,80]. Interestingly, a major determining factor between RNA degradation or transport by PNPase seems to be the length of the RNA species’ 3′ overhang. Wang et al. showed that an 8 nucleotide (nt) 3′ overhang of a mRNA species resulted in degradation while a 0–2 nt 3′ overhang led to transport [57,84]. Other studies suggest that the machinery facilitating protein import into the mitochondria may also contribute to RNA import [3,36,54,75,78,81,82].
PNPase has been established to play an important role in the import of nuclear RNA [75,78,92]; however, evaluating its role in miRNA transport could provide the first mechanistic view of miRNA import into the mitochondria. The current manuscript focuses on PNPase as a key player in miRNA import into the mitochondrion, interacting with Ago2 to shuttle miRNA from the cytosol into the mitochondrion. Specifically, we examine how PNPase expression can control bioenergetics through the regulation of mitochondrial miRNA-378 levels. The present study demonstrates that the overexpression of PNPase in the diseased state may have negative consequences on bioenergetics by facilitating miRNA-378 translocation into the mitochondrion; showing, for the first time, a mitochondrial miRNA import pathway.
2. Materials and methods
2.1. Human tissues
The West Virginia University Institutional Review Board and Institutional Biosafety Committee approved all protocols. Patients undergoing cardiac valve replacement or coronary artery bypass graft surgery at Ruby Memorial Hospital in Morgantown, West Virginia, allowed the release of the right atrial appendage tissue to the West Virginia University School of Medicine. Patients were characterized as non-diabetic or type 2 diabetic based on a previous medical diagnosis of type 2 diabetes mellitus. Pericardial fat was trimmed from right atrial appendage samples and mitochondria isolated as previously described [8,9,22–26,65,67,88,95].
2.2. Experimental animals
The animal experiments performed in this study conform with the National Institutes of Health Eighth Edition Guidelines for the Care and Use of Laboratory Animals and were approved by the West Virginia University Animal Care and Use Committee. Mixed gender db/db mice (strain FVB.BKS(D)-Leprdb/ChuaJ) and wild-type (WT) littermate controls (Jackson Laboratories, Bar Harbor, ME) were housed and bred in the West Virginia University Health Sciences Center animal facility. Microisolator cages were used to maintain animals with ad libitum access to food and water and housed on a 12 h light/dark cycle in a temperature-controlled room. All mice were aged to approximately 20–22 weeks old and then euthanized for experimentation.
2.3. Preparation of individual mitochondrial subpopulations
At approximately 20–22 weeks of age, db/db mice and littermate controls were euthanized, hearts excised and cardiac mitochondrial subpopulations were isolated for analyses as previously described following the methods of Palmer et al. [67] with minor modifications by our laboratory [8,9,22,24–26,65,88]. After isolation of mitochondrial subpopulations from both animal and human cardiac tissue, SSM and IFM were further purified by percoll gradient (23%, 15%, 10% and 3% percoll solution). The samples were centrifuged in a Beckman Optima MAX-XP Ultracentrifuge (Beckman Coulter, Fullerton, CA) at 32,000 ×g for 8 min. Mitochondrial subpopulation pellets were resuspended in the appropriate buffer for each assay. Protein concentrations were determined using the Bradford method with bovine serum albumin as a standard [17].
2.4. Cell culture
For cell culture experiments, the mouse cardiomyocyte cell line (HL-1), which maintains a cardiac-specific phenotype following repeated passages, was used as previously described [10,21,50]. The miRNA-378 HL-1 overexpressing cell line (HL-1-378) generated by our laboratory was also used as previously described [50]; stably overexpressing miRNA-378 through a pre-miRNA-378 miRNA expression plasmid (pCMV-MIRNA) (Product # sc401025; Origene, Rockville, MD). Cells were grown up in a humidified atmosphere of 5% CO2/95% air and maintained at 37 °C in Claycomb media (Sigma Aldrich, St. Louis, MO) with 10% fetal bovine serum and other supplements as previously described [10].
2.5. Overexpression and knockdown of PNPase
Cells were seeded and transfected at 60% to 70% confluence. PNPase transient overexpression, Pnpt1 (BC055826) mouse cDNA clone (Origene, Rockville, MD), and knockdown, Pnpt1 (ID 71701) Trilencer-27 Mouse siRNA (Origene, Rockville, MD), was established through transfection of HL-1 and HL-1-378 cell lines using. Efficiency of the siRNA was determined and transfected at 5 nM. Briefly, FuGENE 6 Transfection Reagent (Promega, Madison, WI) was used per manufacturer’s instructions. Forty-eight hours post transfection, cells were washed with PBS and harvested. In order to account for variations in plasmid uptake by cells, the number of samples in the cohort (n) was determined by the number of independent transfections. Mitochondria were isolated using a mitochondrial isolation kit (Biovision, Milpitas, CA) for protein, qPCR, and enzymatic analyses [50].
2.6. qPCR analyses
Total RNA was isolated from mitochondria in the HL-1 and HL-1-378 overexpression cell lines and FVB-db/db animals and converted to cDNA using the miRNA First Strand cDNA Synthesis kit (Origene) per the manufacturer’s protocol. The cDNA was used with SYBR Green components in a total sample volume of 25 μL: 12.5 μL 2× SYBER Green I qPCR Master Mix (Origene), 9.5 μL RNAse/Nuclease free H2O,1 μL of primer for the control (U6) or experimental group (MP300294 – miRNA-378, Origene), and 2 μL of cDNA (~500 ng/μL). Data are expressed as fold change relative to the difference in Ct expression of the miRNA-378 to U6 log expression. All samples were run in duplicate for both the miRNA-378 and U6 primers. Standard deviation values in either primer pair exceeding a Ct value of 0.5 were excluded from the study. An Applied Biosystems 7900HT Fast Real-Time PCR System (Applied Biosystems, Foster City, CA) was used for analysis, with reaction conditions optimized to Origene’s qSTAR miRNA qPCR Detection System instructions.
2.7. Immunoprecipitation analyses
Dynabeads® protein G (Life Technologies, Grand Island, NY) were used to determine protein associations between Ago2 and PNPase per the manufacturer’s instructions. Briefly, 50 μL of Dynabeads® protein G were incubated with either anti-PNPT1 (PNPase; OriGene, Rockville, MD) or anti-AGO2 (Abcam, Cambridge, MA) primary antibody overnight. The beads were subsequently washed in NP-40 buffer and 150 μg of mitochondrial protein, as determined by the Bradford method, was resuspended with beads and NP-40 wash/binding buffer; this was incubated overnight. Finally, beads were washed and loading dye added to the samples. Western blots were performed on the eluted protein and subsequently probed with the protein of interest to examine the associations.
2.8. Western blot analyses
SDS-PAGE was run on 4–12% gradient gels, as previously described [8,9,24,56,65,88,95] with modifications. For assessment of protein content, equal amounts of protein were loaded as determined by the Brad-ford method using bovine serum albumin as a standard as previously described [17]. Further, assessment of protein loading control was done by utilizing the COXIV antibody and Ponceau S solution (Sigma, St. Louis, MO). Relative amounts of PNPase, Ago2, ATP6, GAPDH, and COXIV were assessed using the following primary antibodies: anti-PNPT1 (PNPase; OriGene, Rockville, MD); anti-AGO2 (Abcam, Cambridge, MA); anti-ATP6 (Abcam, Cambridge, MA); anti-GAPDH (Abcam, Cambridge, MA); and anti-COXIV (Cell Signaling Technology, Danvers, MA). The secondary antibodies used in the analyses were goat anti-mouse IgG horseradish peroxidase (HRP) conjugate (Pierce Biotechnology, Rockford, IL) for Ago2 and GAPDH and goat anti-rabbit IgG HRP conjugate (Cayman Chemical, Ann Arbor, MI) for the PNPase, ATP6 and CoxIV primary antibodies. Pierce Enhanced Chemiluminescence Western Blotting Substrate (Pierce Biotechnology; Rockford, IL) was used to detect signal per manufacturer’s instructions. A G:Box Bioimaging system (Syngene, Frederick, MD) was used to detect signals and data were captured using GeneSnap/GeneTools software (Syngene, Frederick, MD). Densitometry was analyzed using Image J Software (National Institutes of Health, Bethesda, MD) and expressed as arbitrary optical density units.
2.9. ETC complex activities
ETC complex activities I, III, and IV were measured in mitochondria isolated from the cells spectrophotometrically as previously described [24–26,48,90]. Briefly, by measuring NADH oxidation at 340 nm, complex I activity was determined. Complex III activity was measured by assessing the reduction of cytochrome c at 550 nm in the presence of 50 μM of reduced decylubiquinone, while complex IV activity evaluated cytochrome c oxidation at 550 nm. Further, ATP synthase activity was measured as oligomycin-sensitive ATPase activity using an assay coupled with pyruvate kinase, which converts ADP to ATP and produces pyruvate from phosphoenolpyruvate as described previously [24,38,70,72]. Values for complex activities were expressed as nanomoles substrate converted/min/mg of protein.
2.10. miRCURY LNA™ microRNA transfections
To determine whether a therapeutic designed to competitively bind and repress miRNA-378 in the mitochondria can be enhanced in the presence of increased PNPase levels, we introduced a miRNA-378 antimiR that was fluorescently labeled (product number: 4,100,838-111, sequence: CTTCTGACTCCAAGTCCAG, miRCURY LNA™ Power microRNA inhibitor, 5 nmol, 5′-fluorescein labeled, Exiqon A/S, Vedbaek, Denmark). A scrambled sequence, Negative Control A (product number: 199,006-111, sequence: TAACACGTCTATACGCCCA, miRCURY LNA™ Power microRNA inhibitor control, 5 nmol, 5′-fluorescein labeled, Exiqon A/S, Vedbaek, Denmark) was used as a control. Transfections were performed per manufacture’s guidelines, with some alterations. Briefly, cells were transfected with either the miRNA-378 antimiR or Negative Control A LNA construct in 12-well plates at a 20 nM concentration. Both the HL-1 and HL-1-378 cell lines were used for analysis (described above), with varying expression levels of PNPase (described above). Cells were transfected at 60% confluency and allowed to remain in the transfection media for at least 24 h.
2.11. Fluorescence imaging
Cells were imaged 24–48 post transfection on a Zeiss Axiovert 40 CFL Microscope (Thornwood, NY). The Zeiss AxioCam Icc 1 Rev. 4 Color CCD Camera (Thornwood, NY) was used for both phase contrast (20×/0.30 LD A-Plan Phase Contrast) and epifluorescence GFP imaging (X-Cite 120 Q Light Source (Lumen Dynamics, Ontario, Canada)). Images were captured using the Zen 2011 (Zeiss, Thornwood, NY) software. Images were processed using Adobe’s Photoshop CC 2017.
2.12. Flow cytometry
Total mitochondria were isolated from cells after fluorescence imaging (as described above) and incubated with 100 nM of Mitotracker Deep Red 633 (Product no. M22426, Invitrogen, Carlsbad, CA). Functionality of the mitochondria were maintained during flow cytometric analysis using filtered sucrose. Thesholding, using Mitotracker Deep Red 633, provided the basis for the number of events registered and was used to quantify the percent of GFP fluorescent mitochondria using the FACSDiva 8.0 software (BD Biosciences, San Jose CA) on the LSRFortessa equipped with a FSC PMT (BD Biosciences) in the WVU Flow Cytometry and Single Cell Core Facility. A GFP negative (no stain) cohort in both the HL-1 and HL-1-378 cell lines was used for normalization of GFP expression. MiRNA-378 LNA and scramble LNA (Negative Control A) GFP levels was analyzed using FCS Express 5 Flow Cytometry 32 bit software (De Novo Software, Glendale, CA). Samples were first gated with Mitotracker and FSC-A, followed by SSC-A and FSC-A gating.
2.13. Ingenuity pathway analyses
Conceptual illustration of the PNPase/miRNA-378 mitochondrial import pathway was designed using the Ingenuity Pathway Analysis (IPA) software. The “Mitochondrial Dysfunction Metabolic Pathway” was generated through the use of QIAGEN’s IPA (www.qiagen.com/ingenuity) and used as a structural model for the basic design.
2.14. Statistics
Means ± SEMs were calculated for all data sets. Data were analyzed with a two-tailed Student’s t-test to compare differences between groups (GraphPad Software, La Jolla, CA) where a P ≤ 0.05 was considered significant.
3. Results
3.1. Ago2 expression during type 2 diabetes mellitus
Because Ago2 has been shown to have miRNA binding capacity independent of the RISC complex [16,19,20,31,60,62], we hypothesized that Ago2 may be supplying miRNAs to PNPase at the mitochondrion. We assessed the protein expression of Ago2 in both the mitochondrial and cytosolic fractions of the cell. When assessing the Ago2 protein content in human non-diabetic and type 2 diabetic patients, no change was noted in either cardiac mitochondrial subpopulation (Fig. 1A–B). Similarly in the db/db mouse, SSM and IFM displayed no alterations in Ago2 protein expression levels (Fig. 1D–E). Ago2 protein expression was also not significantly altered in human type 2 diabetic (Fig. 1C) and db/db mouse (Fig. 1F) cytosolic fractions. Though the cytosolic fraction of the human, and to a lesser extent the mouse, appears to have a trend to lower Ago2 levels, lack of an observable change in Ago2 expression in the diabetic state suggests that absolute Ago2 levels are not significantly altered in the heart during type 2 diabetic insult. Nevertheless, these findings do not eliminate the potential of an active role for Ago2 during type 2 diabetic insult, but rather, that a such a role is not associated with changes in its protein content.
Fig. 1.
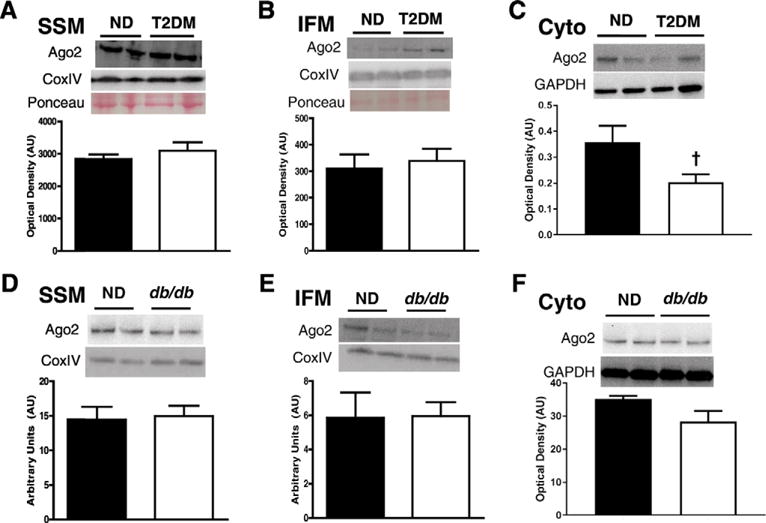
Ago2 expression levels in human and mouse models of type 2 diabetes mellitus. Ago2 expression from non-diabetic (closed bars, n = 5) and diabetic (open bars, n = 5) human atrial tissue in (A) SSM and (B) IFM subpopulations. Ago2 expression levels in non-diabetic and db/db mice in (D) SSM and (E) IFM subpopulations. Ago2 expression levels in cytoplasmic fractions of both (C) human and (F) mouse cardiac tissue. Western blotting quantified to COXIV in the mitochondria. Values are means ± SE. † = P ≤ 0.075. Ago2 = Argonaute 2, SSM = subsarcolemmal mitochondria, IFM = interfibrillar mitochondria, ND = non-diabetic, T2DM = type 2 diabetes mellitus.
3.2. PNPase expression during type 2 diabetes mellitus
Because PNPase has RNA binding capacity and because it is has been reported to influence the import or RNA into the mitochondrion, we hypothesized that this role may extend to miRNAs. Interestingly, PNPase protein content was increased in SSM from type 2 diabetic patients, (Fig. 2A); however, no changes in PNPase protein expression in the IFM was noted (Fig. 2B). Similarly, PNPase protein content was increased in SSM in db/db mice (Fig. 2C), while no changes were noted in the IFM (Fig. 2D). PNPase protein expression was also assessed in non-diabetic and type 2 diabetic cytosolic fractions. Unlike Ago2, cytosolic fractions of both the human atrial appendages (Fig. 2E) and db/db whole heart tissue (Fig. 2F) showed no discernable signal, suggesting the localization of PNPase within the mitochondrion.
Fig. 2.
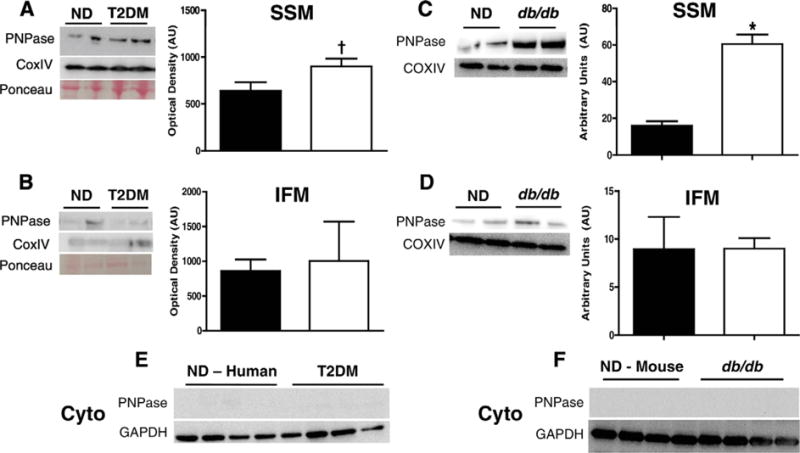
PNPase expression levels in human and mouse models of type 2 diabetes mellitus. PNPase expression from non-diabetic (closed bars, n = 5) and diabetic (open bars, n = 5) human atrial tissue in (A) SSM (P ≤ 0.055) and (B) IFM subpopulations. PNPase expression levels in non-diabetic and db/db mice in (C) SSM and (D) IFM subpopulations. PNPase expression levels in cytoplasmic fractions of both (E) human and (F) mouse cardiac tissue. Western blotting quantified to COXIV in the mitochondria. Values are means ± SE. * = P ≤ 0.05, † = P ≤ 0.075. PNPase = Polynucleotide Phosphorylase, SSM = subsarcolemmal mitochondria, IFM = interfibrillar mitochondria, ND = non-diabetic, T2DM = type 2 diabetes mellitus.
3.3. Ago2 and PNPase association in the mitochondrion
Because both Ago2 and PNPase have been observed in the mitochondrion, we determined whether an association between the two existed in the mitochondrion. We immunoprecipitated Ago2 from the mitochondrial pellet and found that PNPase was co-immunoprecipitated in both the SSM and IFM subpopulations (Fig. 3A–B). Interestingly, the SSM showed an increased association of immunoprecipitated Ago2 with PNPase in db/db mice (Fig. 3A), while the IFM revealed no difference in the association of these proteins (Fig. 3B). We then performed the reciprocal immunoprecipitation with PNPase, immunoprecipitating with PNPase and looking to see if Ago2 co-immunoprecipitated with the protein. Again, we found the association between the 2 proteins (Fig. 3C–D). We observed a trending increase in association between PNPase and Ago2 in the SSM of the type 2 diabetic animals as compared to their controls (Fig. 3C). Because of the much smaller protein quantities immunoprecipitated, protein quantification between groups becomes more difficult to assess [64], potentially attributing to the trending, and not significant, increase observed in the SSM. Though an association between PNPase and Ago2 existed in the IFM, no significant differences were observed between the control and db/db mice (Fig. 3D).
Fig. 3.
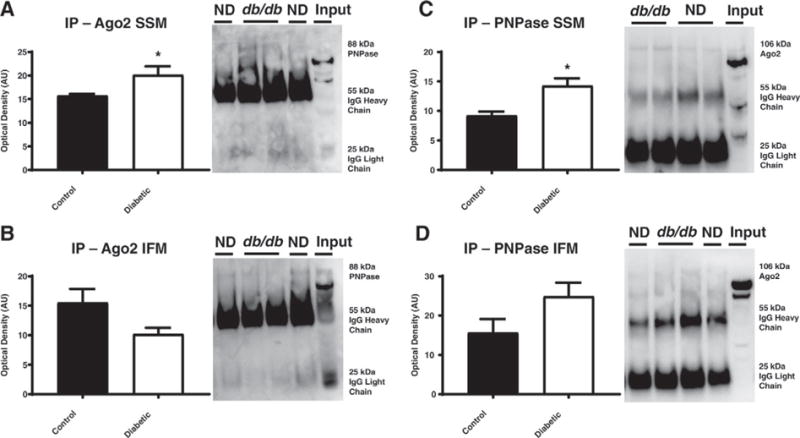
Evaluation of the binding of Ago2 and PNPase through immunoprecipitation in db/db and littermate control mice. Ago2 was immunoprecipitated then transferred and run on a SDS-PAGE gel where the (A) SSM and (B) IFM were probed for PNPase, in both the non-diabetic (closed bars, n = 5) and diabetic (open bars, n = 4) groups. Alternatively, PNPase was immunoprecipitated then transferred and run on a SDS-PAGE gel where the (C) SSM and (D) IFM were probed for Ago2. Values are means ± SE. * = P ≤ 0.05, † = P ≤ 0.075. Ago2 = Argonaute 2, PNPase = Polynucleotide Phosphorylase, SSM = subsarcolemmal mitochondria, IFM = interfibrillar mitochondria, ND = non-diabetic, input = 50% input control.
3.4. miRNA-378 expression in the mitochondrion
To determine whether the type 2 diabetic setting influenced mitochondrial miRNA-378 levels, we used a qPCR approach. We found an increase in miRNA-378 levels in SSM isolated from db/db cardiac tissue, with no changes noted in the IFM (Fig. 4A). Since miRNA-378 is predicted to target ATP6 within the mitochondrion [50], we assessed ATP6 protein levels in the SSM and IFM from control and db/db mice (Fig. 4B). Our data revealed a decrease in ATP6 protein expression in the type 2 diabetic SSM, with no changes in the IFM subpopulation (Fig. 4B). Diminished ATP6 levels in the SSM during type 2 diabetes mellitus should precipitate decrements in ATP synthase activity, which were observed in the type 1 diabetic, but in the IFM [50]. We previously assessed the ATP synthase activity form type 2 diabetic subpopulations and found activity was decreased in the SSM [24]. In order to see if this effect was subpopulation independent, we analyzed total mitochondrial ATP synthase activity, revealing a decrease in activity in db/db mice relative to littermate controls (Fig. 4C). Complexes I, III, and IV activities were also measured in animal and cell culture models. Complex IV was shown to be significantly decreased in cell lines overexpressing miRNA-378, suggestive of the predicted binding affinity of miRNA-378 with mitochondrial cytochrome c oxidase 3 (mtCOX3) (Table 1) [50]. Taken together, these data suggest a coordinated response in which miRNA-378 expression influences mitochondrial genome-encoded ATP6 and ultimately, down-regulation of ATP synthase functionality.
Fig. 4.
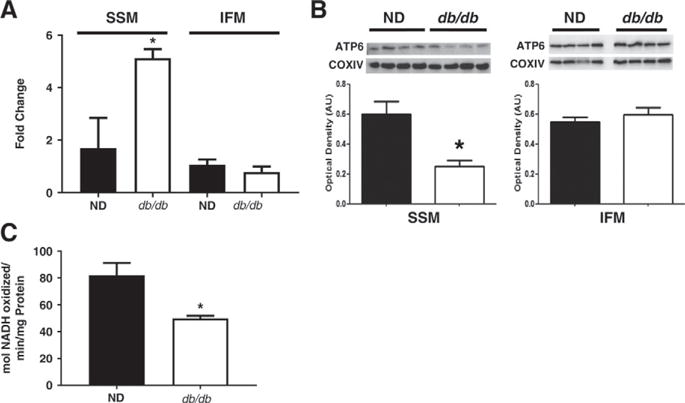
The relationship between miRNA-378 and bioenergetics. (A) MiRNA-378 levels in the SSM and IFM, in both the non-diabetic (closed bars, n = 4) and db/db (open bars, n = 4) groups. (B) ATP6 levels in the SSM and IFM. (C) ATP Synthase activity of mitochondria from control and db/db animals. qPCR Ct values were normalized to U6 expression. Western blotting quantified to COXIV. Values are means ± SE. * = P ≤ 0.05. SSM = subsarcolemmal mitochondria, IFM = interfibrillar mitochondria, ND = non-diabetic.
Table 1.
Complex I, III, and IV activities for both animal (FVB and FVB – db/db) and cell culture groups. All groups are represented as an n = 4. Values are means ± SE. HL-1 PNPase = murine cardiomyocyte cell line transiently overexpressing or knocking down Polynucleotide Phosphorylase (PNPase), HL-1-378 PNPase = murine cardiomyocyte cell line stably overexpressing miRNA-378a-3p while transiently overexpressing or knocking down Polynucleotide Phosphorylase (PNPase). AntimiR PNPase = HL-1-378 cell line transfected with antimiR-378 while transiently overexpressing or knocking down Polynucleotide Phosphorylase (PNPase).
| Complex I | Complex III | Complex IV | |
|---|---|---|---|
| FVB | 235.07 ± 34.05 | 63.01 ± 10.82 | 52.75 ± 12.71 |
| FVB - db/db | 339.74 ± 18.16 | 54.90 ± 18.96 | 76.36 ± 18.98 |
| HL-1 | 140.39 ± 3.17 | 38.32 ± 4.55 | 38.31 ± 9.06 |
| HL-1 PNPase OE | 156.66 ± 17.96 | 43.37 ± 3.61 | 42.46 ± 10.83 |
| HL-1 PNPase KD | 131.31 ± 17.83 | 38.19 ± 6.13 | 36.65 ± 7.07 |
| HL-1-378 | 143.48 ± 37.19 | 41.34 ± 1.66 | 12.46 ± 3.31 |
| HL-1-378 PNPase OE | 118.22 ± 40.61 | 36.29 ± 7.41 | 12.38 ± 2.68 |
| HL-1-378 PNPase KD | 131.76 ± 9.88 | 33.14 ± 5.25 | 11.97 ± 2.00 |
| AntimiR | 151.06 ± 13.06 | 45.75 ± 3.06 | 10.88 ± 2.19 |
| AntimiR PNPase OE | 167.07 ± 14.68 | 36.93 ± 3.38 | 14.06 ± 4.82 |
| AntimiR PNPase KD | 157.34 ± 19.57 | 59.56 ± 6.47 | 13.93 ± 1.70 |
3.5. PNPase overexpression and knockdown in cell culture models
We developed a stable HL-1 cell line with enhanced expression of miRNA-378 (HL-1-378) [50]. MiRNA-378 overexpression in the mitochondria isolated from this cell line was substantial (Fig. 5A) and, in previous studies, we found that both ATP6 mRNA and ATP6 protein expression levels were decreased in these cells [50]. Compared to the HL-1 cell line, HL-1-378 cells also had a significantly lower ATP synthase activity, suggesting a change in the basal ATP synthase activity of the cell line (Fig. 5B). HL-1 cardiomyocytes were transfected with either a plasmid encoding PNPase, displaying increased PNPase levels in isolated mitochondria, or siRNA against the PNPT1 gene, displaying decreased PNPase levels in isolated mitochondria (Fig. 5C). Overexpression of PNPase led to a significant increase in miRNA-378 levels in the mitochondrion of HL-1 cells, but knockdown of the protein did not significantly alter mitochondrial miRNA-378 levels (Fig. 5D). In HL-1-378 cells, overexpression of PNPase lead to a significant rise in mitochondrial miRNA-378, while knockdown conversely significantly diminished this effect (Fig. 5E). These findings support the contention that PNPase expression is linked to mitochondrial miRNA-378 import.
Fig. 5.
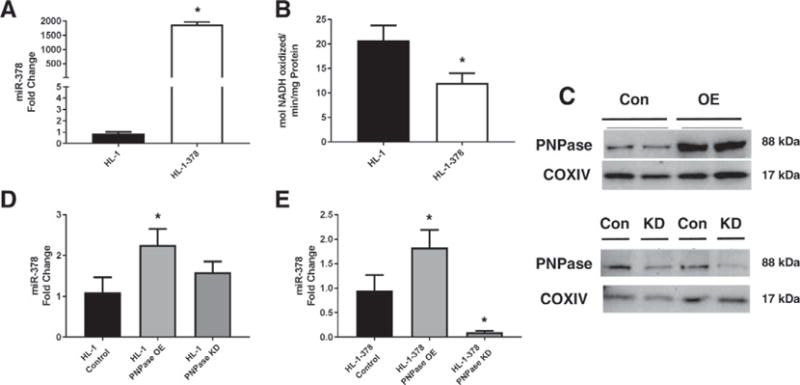
The influence of PNPase on mitochondrial miRNA-378 in cell culture models. (A) HL-1 and HL-1-378 miRNA-378 fold change. (B) ATP synthase activity of HL-1 and HL-1-378 cell lines. (C) Representative western blot of transient PNPase overexpression and knockdown in HL-1 cells. (D) Expression of mitochondrial miRNA-378 in HL-1 cells during normal, overexpressing, and knocked down expression of PNPase. (E) Expression of mitochondrial miRNA-378 in HL-1-378 cells during normal, overexpressing, and knocked down expression of PNPase. qPCR Ct values were normalized to U6 expression. N = 4 for each group. Values are ± SE. * = P ≤ 0.05. HL-1 PNPase = murine cardiomyocyte cell line transiently overexpressing or knocking down Polynucleotide Phosphorylase (PNPase), HL-1-378 PNPase = murine cardiomyocyte cell line stably overexpressing miRNA-378a-3p while transiently overexpressing or knocking down Polynucleotide Phosphorylase (PNPase).
3.6. Effects of PNPase and miRNA-378 expression on cellular function
With changing dynamics of mitochondrial miRNA-378 in the presence of PNPase expression, we wanted to examine how cellular bioenergetics was impacted. ATP synthase activity was measured in HL-1 cells, with PNPase overexpression resulting in a lower ATP synthase activity but with no change during knockdown of the protein (Fig. 6A). In the HL-1-378 cell line, ATP synthase activity revealed a decrease in activity for cells overexpressing PNPase while also showing a significant recovery of activity after knocking down the protein (Fig. 6B). We also wanted to examine how treating cells with an antimiR for miRNA-378 could change the energetic profile. Previously, we have shown that treating with antimiR-378 restored function in HL-1-378 cells [50]. With the overexpression of PNPase, we found that antimiR-378 treatment of HL-1-378 cells restored ATP synthase activity to baseline and knockdown of the protein allowed for a significant increase in ATP synthase activity (Fig. 6C). We also wanted to verify that the change in ATP synthase activity was due to a change in mitochondrial ATP6 levels. Protein levels of HL-1-378 cells revealed a decrease in ATP6 levels when overexpressing PNPase, while no change was observed in the knockdown (Fig. 6D). ATP6 levels were found to increase in antimiR-378 treated HL-1-378 cells when PNPase was overexpressed (Fig. 6E). This data continues to emphasize the role of PNPase in changing bioenergetics through mitochondrial miRNA-378 import.
Fig. 6.
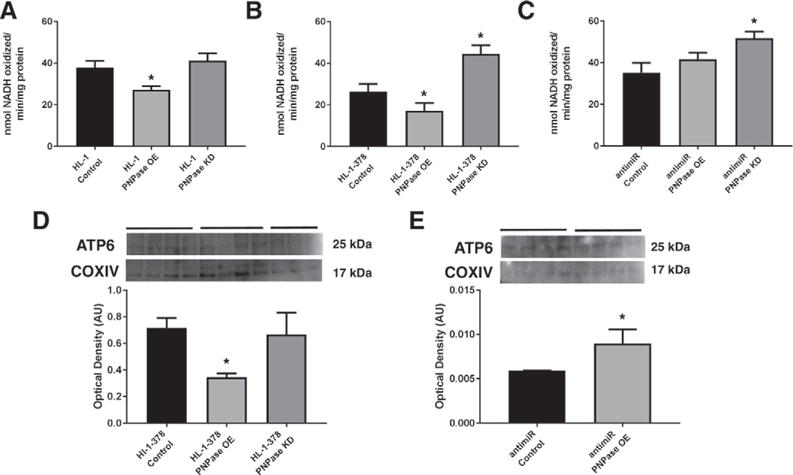
The influence of PNPase and mitochondrial miRNA expression on bioenergetics. ATP synthase activity was measured in (A) HL-1, (B) HL-1-378, and (C) HL-1-378 with antimiR-378 cells with manipulations to the expression of PNPase. (D) Western blot analysis for mitochondrial ATP6 in HL-1-378 control, PNPase overexpressing, and PNPase knockdown cells. (E) Western blot analysis for mitochondrial ATP6 in HL-1-378 cells transfected with antimiR-378 with normal and overexpressing levels of PNPase. Western blotting quantified to COXIV in the mitochondria. Values are means ± SE. * = P ≤ 0.05. HL-1 PNPase = murine cardiomyocyte cell line transiently overexpressing or knocking down Polynucleotide Phosphorylase (PNPase), HL-1-378 PNPase = murine cardiomyocyte cell line stably overexpressing miRNA-378a-3p while transiently overexpressing or knocking down Polynucleotide Phosphorylase (PNPase). AntimiR PNPase = HL-1-378 cell line transfected with antimiR-378 while transiently overexpressing or knocking down Polynucleotide Phosphorylase (PNPase).
3.7. Mitochondrial miRNA-378 antimiR treatment
To explore the potential of therapeutic interventions on controlling mitochondrial miRNA-378 levels, we employed antimiR treatment to competitively inhibit miRNA-378. HL-1 and HL-1-378 cell lines were transfected with miRNA-378a-3p (LNA-378) or a scrambled LNA control (Negative Control A) and fluorometrically examined (Fig. 7A). Representative fluorescent images for the HL-1-378 control, PNPase overexpression, and PNPase knockdown treatments are included, indicating similar transfection capacity across groups. Mitochondria were isolated from cells and examined using flow cytometry, in order to determine the amount of a given LNA that was imported. Mitochondria were gated using Mitotracker Deep Red 633, with the population further filtered using the forward (FSC-A) and side (SSC-A) scattered light (Fig. 7B). The antimiR-378 and scrambled LNA control GFP expression were referenced to the no-GFP baseline (0.22 and 0.26% GFP for the HL-1 and HL-1-378 no stain groups, respectively). Fig. 7C shows a representative expression profile for each of the groups. In the HL-1-378 cell line, transiently overexpressing PNPase increased antimiR-378 incorporation into the mitochondria while knocking down PNPase resulted in a decrease of antimiR-378 import (Fig. 7D). The HL-1 cell line showed a significant decrease in antimiR-378 import during PNPase knockdown, while all other groups, including Scramble controls, showed no significant changes. Demonstrating that antimiR treatment can be differently regulated by PNPase expression provides an initial look into the therapeutic potential of this protein.
Fig. 7.
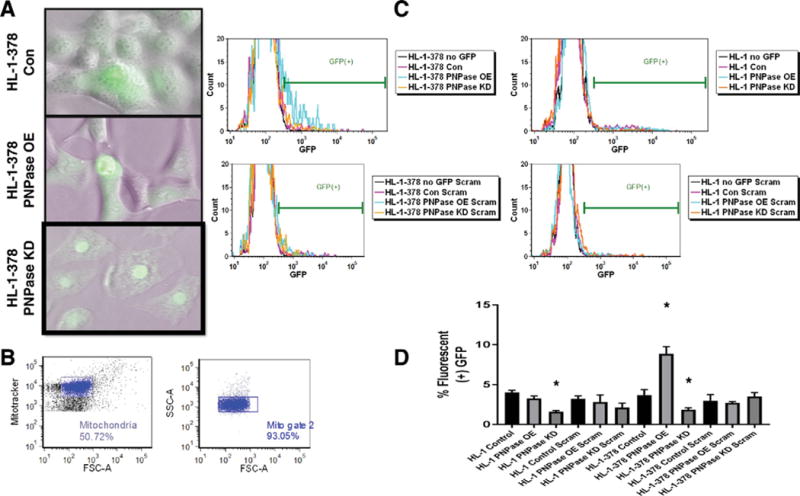
MiRNA-378 antimiR import into the mitochondria visualized through fluorescence imaging and flow cytometry. Cells were transfected with (A) miRNA-378 antimiR LNA or scramble LNA in both HL-1 and HL-1-378 cell lines and visualized qualitatively for transfection efficiency. (B) Flow cytometry was used to measure mean fluorescence of mitochondria, gating to Mitotracker Deep Red 633. (C) Samples were compared to respective no-stain GFP controls (Mean fluorescence ≤ 0.25). Control HL-1 and HL-1-378 cells, (n = 4) and PNPase overexpression and knockdown in each cell line (n = 4) were evaluated. (D) Observed changes during flow cytometric analysis. Values are means ± SE. * = P ≤ 0.05. LNA = locked nucleic acid for miRNA-378 antimiR LNA, scram = locked nucleic acid for Negative Control A, HL-1 PNPase = murine cardiomyocyte cell line transiently overexpressing or knocking down Polynucleotide Phosphorylase (PNPase), HL-1-378 PNPase = murine cardiomyocyte cell line stably overexpressing miRNA-378a-3p while transiently overexpressing or knocking down Polynucleotide Phosphorylase (PNPase).
4. Discussion
Mitochondrial dysfunction is central to the etiology of cardiovascular complications seen in diabetic patients [8,9,23–25,40,73,79,88,95]. Within the mitochondrion, complex functional processes occur, such as oxidative phosphorylation and ATP synthesis, which are critical in an appropriately functioning organelle. With the mitochondrion playing a crucial role in providing energy and maintaining homeostasis in the cell, it is important to evaluate the impact that miRNAs have on mRNA and protein expression in the mitochondrion. MiRNAs localized to the mitochondrion have been found to target essential processes directly, such as energy metabolism, and indirectly such as apoptosis [27,28,50, 52,55,68]. The recent characterization of miRNA localization to various organelles within the cell, along with changes in miRNA species and levels during pathological states suggests a role in miRNA-mediated pathologic phenotypes [3,41,69].
Das et al. found an increase in miRNA-181c in cardiomyocyte mitochondria during heart failure, which targets and represses mitochondrial cytochrome c oxidase 1 (mtCOX1), an important component of ETC complex IV [27,28,30]. Further, the role of miRNA-378 in controlling metabolic processes through targeting mediator complex subunit 13 (Med13) in the liver and insulin-like growth factor 1 (IGF-1) and ATP6 in the heart has been demonstrated [18,50,53]. Our laboratory previously found an increase in miRNA-378 in cardiac mitochondria during type 1 diabetic insult, which resulted in decreased ATP6 protein content and ATP synthase activity in IFM [50]. A more complete understanding of miRNAs on metabolic processes and translocation dynamics within the mitochondrion during disease states could prove imperative in fully understanding the pathology. The mechanisms of miRNA intercellular transport via exosomes and intracellular transport into the nucleus have been elucidated; including nuclear translocation of miRNAs through co-localization of the Ago2/miRNA complex with Importin 8 on the nuclear membrane [94]. The process of miRNA import into the mitochondrion, however, has proven elusive [7,29,35,94]. To date, there are no mechanistic pathways described for the complete mitochondrial miRNA import process, and it is unknown how pathological states can alter the mitochondrial miRNA import dynamic [66,71].
Utilizing PNPase for RNA import into the mitochondrion requires the presence of a specific stem-loop secondary structure in the target RNA [57,92]. Interestingly, Barrey et al. found that pre-miRNA-302a and pre-let-7b are predicted to fold into these stem-loop configurations, leading to the assumption that PNPase could be critical for the import of these pre-miRNAs into the mitochondrion [5]. Additionally, we propose in this study that Ago2 as part of the RISC serves as a mediator in the localization of miRNAs to the mitochondria and potentially facilitates miRNA movement into the mitochondrial matrix via PNPase. In this manuscript, we have shown the association of PNPase with Ago2, which is increased during type 2 diabetic insult, potentially allowing for additional miRNA entry into the mitochondrion. Interestingly, Ago2 has been shown to associate with mitochondrial transcripts mtCOX1 and mtCOX3, along with miRNA-181c, indicating that this protein and the RISC may play a key role in the association of miRNAs being imported into the mitochondrion [4,12,28]. However, at current, the mechanism allowing Ago2 to migrate into the inner mitochondrial membrane to interact with PNPase, or any other protein constituent, is unknown.
Structurally, PNPase is composed of four to five primary domains with the KH and S1 domain located at the C-terminal end of the polypeptide [57,77,99]. The KH and S1 domain are conserved motifs known to have RNA-binding specificity [86,96] and are likely the key regulators controlling recognition of miRNAs by PNPase. It has been shown that altering amino acids within the KH and S1 domains of PNPase can change how the protein interacts with RNA, decreasing the RNA binding efficiency and disrupting the synergistic role of the two domains [96]. In mitochondrial miRNA import, the regulation of amino acids within the KH and S1 domains may contribute to the diversity of miRNAs imported.
The current study shows that PNPase expression influences mitochondrial miRNA-378 levels, but evaluating how the protein is regulated within the nuclear genome could prove vital in understanding the onset/progression of a pathology. In the diabetic condition, it is now beginning to be understood how cardiovascular complications can be linked to changes in the epigenome, both through DNA methylation [91,97] and histone modifications [42]. Studies in diabetic skeletal muscle have also demonstrated how nuclear-encoded mitochondrial protein expression can be regulated through DNA methylation [58,74]. Assessing the epigenetic regulation of PNPase during diabetes mellitus could provide a more complete picture of the transcriptional control of the protein and how therapeutic approaches can be used to alter its expression.
Other therapeutic approaches could include miRNA mimics, increasing miRNA species levels, or supplementation with antimiRs or antagomirs, decreasing miRNA species levels; this may have the potential to attenuate or reverse decrements due to pathological states. MiRNA therapeutic strategies are currently being used in the clinic to treat hepatitis C virus, with clinical trials of miRNA mimics and antagomirs underway [51]. A variety of miRNA-based therapies to address type 2 diabetes mellitus and heart disease are in the preclinical trial stage, providing the need for comprehensive study on miRNA effects on different cellular organelles. One such biologic is antagomir-208a, which has been shown to increase systemic metabolism, display a lean phenotype when provided a high-fat diet, and improve overall cardiac function [11,45,63]. This study has begun to examine the potential use of antimiRs in targeting miRNA-378 within mitochondria, but it is of interest whether inhibition of miRNA-378 using an antimiR during type 2 diabetes mellitus will provide benefits to cardiac mitochondrial function and cardiac contractile function. Further, our data provide evidence that manipulation of PNPase levels may enhance our potential for delivering antimiR therapeutics to mitochondria in physiological and pathological conditions.
In the current study we acknowledge some limitations to the design and interpretations of results. The knockdown of PNPase, though shown to express significantly lower protein levels than control, may not be sufficient in some cases to completely diminish the effects of miRNA-378. Though complete knockout of PNPase is embryonic lethal [93], a conditional animal knockout or cell culture knockout of PNPase should be examined in the future to observe how this impacts miRNA import into the mitochondria.
In conclusion, our data support the concept that PNPase participates in the translocation of miRNA-378 into the mitochondrion (Fig. 8). In the diabetic condition, PNPase levels were shown to increase, correlating with increased mitochondrial miRNA-378. With transient overexpression of PNPase in cell culture models, the diabetic condition was mimicked, showing increased mitochondrial miRNA-378 and decreased ATP synthase activity. Additionally, we have established the capacity of antimiR therapy to be used in targeting mitochondrial miRNA-378 as a potential therapeutic for restoring bioenergetic function. Together, these findings suggest that during type 2 diabetes mellitus, PNPase may be a constituent in the import mechanism of miRNAs into cardiac mitochondria. This work begins to elucidate the potential of PNPase as a mitochondrial miRNA import protein, revealing new questions about the regulation and specificity, and how to target this process in the future.
Fig. 8.
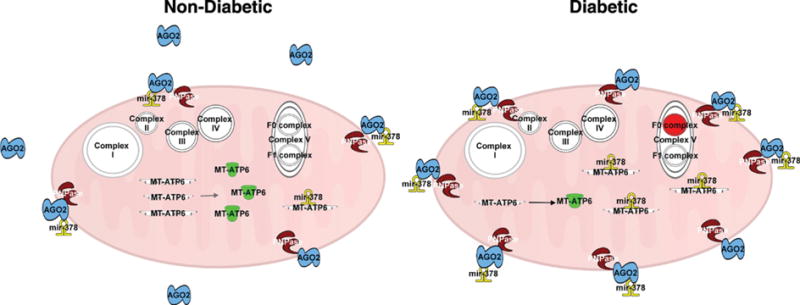
Graphical illustration of PNPase and the changing dynamics in the mitochondrion during type 2 diabetes mellitus. In the non-diabetic state, fewer PNPase proteins are located within the intermembrane space of the mitochondria, resulting in fewer miRNA-378 transcripts being imported. During type 2 diabetes mellitus, an increase in PNPase expression results in increased associations with Ago2, which provide for additional influx of miRNA-378 into the mitochondrion. These miRNA-378 transcripts target mt-ATP6 mRNA, resulting in fewer catalytically active proteins. The lack of mt-ATP6 protein expression contributes to a decrease in bioenergetics through disrupting the F0 complex of ATP synthase. Ago2 = Argonaute 2, mt-ATP6 = mitochondrial ATP synthase F0 subunit 6, PNPase = Polynucleotide Phosphorylase.
Acknowledgments
Grants
This work was supported by the NIH/NHLBI (R56 HL128485 and R01 HL128485), awarded to J. M. Hollander; the NIH/NIGMS (U54 GM104942) as a Pilot Grant to I. Martinez; the NSF Integrative Graduate Education and Research Traineeship: Research and Education in Nanotoxicology at West Virginia University Fellowship (1144676), awarded to Q. A. Hathaway; an American Heart Association Predoctoral Fellowship (13PRE16850066), awarded to C. E. Nichols; and a WV-INBRE NIH Grant P20GM103434 awarded to S. Sreekumar for summer research. Flow Cytometry experiments were performed in the West Virginia University Flow Cytometry & Single Cell Core Facility, which is supported by the NIH (RR020866) and the Institutional Development Award (IDeA) from the NIH/NIGMS CoBRE (P30GM103488) and INBRE (P20GM103434).
Footnotes
Disclosures
No conflicts of interest, financial or otherwise, are declared by the author(s).
References
- 1.Prevention CfDCa, editor. National Diabetes Statistics Report: Estimates of Diabetes and its Burden in the United States. 2014. [Google Scholar]
- 2.Asrih M, Steffens S. Emerging role of epigenetics and miRNA in diabetic cardiomyopathy, Cardiovasc. Pathol. 2013;22:117–125. doi: 10.1016/j.carpath.2012.07.004. [DOI] [PubMed] [Google Scholar]
- 3.Bandiera S, Mategot R, Girard M, Demongeot J, Henrion-Caude A. MitomiRs delineating the intracellular localization of microRNAs at mitochondria. Free Radic Biol Med. 2013;64:12–19. doi: 10.1016/j.freeradbiomed.2013.06.013. [DOI] [PubMed] [Google Scholar]
- 4.Bandiera S, Ruberg S, Girard M, Cagnard N, Hanein S, Chretien D, Munnich A, Lyonnet S, Henrion-Caude A. Nuclear outsourcing of RNA interference components to human mitochondria. PLoS One. 2011;6:e20746. doi: 10.1371/journal.pone.0020746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Barrey E, Saint-Auret G, Bonnamy B, Damas D, Boyer O, Gidrol X. Pre-microRNA and mature microRNA in human mitochondria. PLoS One. 2011;6:e20220. doi: 10.1371/journal.pone.0020220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 7.Baseler WA, Croston TL, Hollander JM. Functional characteristics of mortalin. In: Kaul SC, Wadhwa R, editors. Mortalin Biology: Life, Stress and Death. Springer; New York: 2012. pp. 55–80. [Google Scholar]
- 8.Baseler WA, Dabkowski ER, Jagannathan R, Thapa D, Nichols CE, Shepherd DL, Croston TL, Powell M, Razunguzwa TT, Lewis SE, Schnell DM, Hollander JM. Reversal of mitochondrial proteomic loss in Type 1 diabetic heart with overexpression of phospholipid hydroperoxide glutathione peroxidase. Am J Phys Regul Integr Comp Phys. 2013;304:R553–565. doi: 10.1152/ajpregu.00249.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Baseler WA, Dabkowski ER, Williamson CL, Croston TL, Thapa D, Powell MJ, Razunguzwa TT, Hollander JM. Proteomic alterations of distinct mitochondrial subpopulations in the type 1 diabetic heart: contribution of protein import dysfunction. Am J Phys Regul Integr Comp Phys. 2011;300:R186–200. doi: 10.1152/ajpregu.00423.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Baseler WA, Thapa D, Jagannathan R, Dabkowski ER, Croston TL, Hollander JM. miR-141 as a regulator of the mitochondrial phosphate carrier (Slc25a3) in the type 1 diabetic heart. Am J Phys Cell Physiol. 2012;303:C1244–1251. doi: 10.1152/ajpcell.00137.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Baskin KK, Grueter CE, Kusminski CM, Holland WL, Bookout AL, Satapati S, Kong YM, Burgess SC, Malloy CR, Scherer PE, Newgard CB, Bassel-Duby R, Olson EN. MED13-dependent signaling from the heart confers leanness by enhancing metabolism in adipose tissue and liver. EMBO Mol Med. 2014;6:1610–1621. doi: 10.15252/emmm.201404218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Beitzinger M, Peters L, Zhu JY, Kremmer E, Meister G. Identification of human microRNA targets from isolated argonaute protein complexes. RNA Biol. 2007;4:76–84. doi: 10.4161/rna.4.2.4640. [DOI] [PubMed] [Google Scholar]
- 13.Bienertova-Vasku J, Sana J, Slaby O. The role of microRNAs in mitochondria in cancer. Cancer Lett. 2013;336:1–7. doi: 10.1016/j.canlet.2013.05.001. [DOI] [PubMed] [Google Scholar]
- 14.Borralho PM, Rodrigues CM, Steer CJ. microRNAs in mitochondria: an unexplored niche. Adv Exp Med Biol. 2015;887:31–51. doi: 10.1007/978-3-319-22380-3_3. [DOI] [PubMed] [Google Scholar]
- 15.Borralho PM, Rodrigues CMP, Steer CJ. Mitochondrial microRNAs and their potential role in cell function. Current Pathobiology Reports. 2014;2:123–132. [Google Scholar]
- 16.Bouasker S, Simard MJ. The slicing activity of miRNA-specific Argonautes is essential for the miRNA pathway in C. elegans. Nucleic Acids Res. 2012;40:10452–10462. doi: 10.1093/nar/gks748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 18.Carrer M, Liu N, Grueter CE, Williams AH, Frisard MI, Hulver MW, Bassel-Duby R, Olson EN. Control of mitochondrial metabolism and systemic energy homeostasis by microRNAs 378 and 378. Proc Natl Acad Sci U S A. 2012;109:15330–15335. doi: 10.1073/pnas.1207605109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cheloufi S, Dos Santos CO, Chong MM, Hannon GJ. A dicer-independent miRNA biogenesis pathway that requires Ago catalysis. Nature. 2010;465:584–589. doi: 10.1038/nature09092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cifuentes D, Xue H, Taylor DW, Patnode H, Mishima Y, Cheloufi S, Ma E, Mane S, Hannon GJ, Lawson ND, Wolfe SA, Giraldez AJ. A novel miRNA processing pathway independent of Dicer requires Argonaute2 catalytic activity. Science. 2010;328:1694–1698. doi: 10.1126/science.1190809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Claycomb WC, Lanson NA, Jr, Stallworth BS, Egeland DB, Delcarpio JB, Bahinski A, Izzo NJ., Jr HL-1 cells: a cardiac muscle cell line that contracts and retains phenotypic characteristics of the adult cardiomyocyte. Proc Natl Acad Sci U S A. 1998;95:2979–2984. doi: 10.1073/pnas.95.6.2979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Croston TL, Shepherd DL, Thapa D, Nichols CE, Lewis SE, Dabkowski ER, Jagannathan R, Baseler WA, Hollander JM. Evaluation of the cardiolipin biosynthetic pathway and its interactions in the diabetic heart. Life Sci. 2013;93:313–322. doi: 10.1016/j.lfs.2013.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Croston TL, Thapa D, Holden AA, Tveter KJ, Lewis SE, Shepherd DL, Nichols CE, Long DM, Olfert IM, Jagannathan R, Hollander JM. Functional deficiencies of subsarcolemmal mitochondria in the type 2 diabetic human heart. Am J Physiol Heart Circ Physiol. 2014;307:H54–65. doi: 10.1152/ajpheart.00845.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dabkowski ER, Baseler WA, Williamson CL, Powell M, Razunguzwa TT, Frisbee JC, Hollander JM. Mitochondrial dysfunction in the type 2 diabetic heart is associated with alterations in spatially distinct mitochondrial proteomes. Am J Physiol Heart Circ Physiol. 2010;299:H529–540. doi: 10.1152/ajpheart.00267.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dabkowski ER, Williamson CL, Bukowski VC, Chapman RS, Leonard SS, Peer CJ, Callery PS, Hollander JM. Diabetic cardiomyopathy-associated dysfunction in spatially distinct mitochondrial subpopulations. Am J Physiol Heart Circ Physiol. 2009;296:H359–369. doi: 10.1152/ajpheart.00467.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dabkowski ER, Williamson CL, Hollander JM. Mitochondria-specific transgenic overexpression of phospholipid hydroperoxide glutathione peroxidase (GPx4) attenuates ischemia/reperfusion-associated cardiac dysfunction. Free Radic Biol Med. 2008;45:855–865. doi: 10.1016/j.freeradbiomed.2008.06.021. [DOI] [PubMed] [Google Scholar]
- 27.Das S, Bedja D, Campbell N, Dunkerly B, Chenna V, Maitra A, Steenbergen C. miR-181c regulates the mitochondrial genome, bioenergetics, and propensity for heart failure in vivo. PLoS One. 2014;9:e96820. doi: 10.1371/journal.pone.0096820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Das S, Ferlito M, Kent OA, Fox-Talbot K, Wang R, Liu D, Raghavachari N, Yang Y, Wheelan SJ, Murphy E, Steenbergen C. Nuclear miRNA regulates the mitochondrial genome in the heart. Circ Res. 2012;110:1596–1603. doi: 10.1161/CIRCRESAHA.112.267732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Das S, Halushka MK. Extracellular vesicle microRNA transfer in cardiovascular disease, Cardiovasc. Pathol. 2015;24:199–206. doi: 10.1016/j.carpath.2015.04.007. [DOI] [PubMed] [Google Scholar]
- 30.Dennerlein S, Rehling P. Human mitochondrial COX1 assembly into cytochrome c oxidase at a glance. J Cell Sci. 2015;128:833–837. doi: 10.1242/jcs.161729. [DOI] [PubMed] [Google Scholar]
- 31.Diederichs S, Haber DA. Dual role for argonautes in microRNA processing and post-transcriptional regulation of microRNA expression. Cell. 2007;131:1097–1108. doi: 10.1016/j.cell.2007.10.032. [DOI] [PubMed] [Google Scholar]
- 32.Divakaran V, Mann DL. The emerging role of microRNAs in cardiac remodeling and heart failure. Circ Res. 2008;103:1072–1083. doi: 10.1161/CIRCRESAHA.108.183087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Duarte FV, Palmeira CM, Rolo AP. The emerging role of mitomiRs in the pathophysiology of human disease. Adv Exp Med Biol. 2015;888:123–154. doi: 10.1007/978-3-319-22671-2_8. http://dx.doi.org/10.1007/978-3-319-22671-2_8. [DOI] [PubMed] [Google Scholar]
- 34.Duarte FV, Palmeira CM, Rolo AP. The role of microRNAs in mitochondria: small players acting wide. Genes (Basel) 2014;5:865–886. doi: 10.3390/genes5040865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dudek J, Rehling P, van der Laan M. Mitochondrial protein import: common principles and physiological networks. Biochim Biophys Acta. 2013;1833:274–285. doi: 10.1016/j.bbamcr.2012.05.028. [DOI] [PubMed] [Google Scholar]
- 36.Entelis NS, Kolesnikova OA, Dogan S, Martin RP, Tarassov IA. 5 S rRNA and tRNA import into human mitochondria. Comparison of in vitro requirements. J Biol Chem. 2001;276:45642–45653. doi: 10.1074/jbc.M103906200. [DOI] [PubMed] [Google Scholar]
- 37.Fancher IS, Dick GM, Hollander JM. Diabetes mellitus reduces the function and expression of ATP-dependent K(+) channels in cardiac mitochondria. Life Sci. 2013;92:664–668. doi: 10.1016/j.lfs.2012.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Feniouk BA, Suzuki T, Yoshida M. Regulatory interplay between proton motive force, ADP, phosphate, and subunit epsilon in bacterial ATP synthase. J Biol Chem. 2007;282:764–772. doi: 10.1074/jbc.M606321200. [DOI] [PubMed] [Google Scholar]
- 39.Filipowicz W, Bhattacharyya SN, Sonenberg N. Mechanisms of post-transcriptional regulation by microRNAs: are the answers in sight? Nat Rev Genet. 2008;9:102–114. doi: 10.1038/nrg2290. [DOI] [PubMed] [Google Scholar]
- 40.Flarsheim CE, Grupp IL, Matlib MA. Mitochondrial dysfunction accompanies diastolic dysfunction in diabetic rat heart. Am J Phys. 1996;271:H192–202. doi: 10.1152/ajpheart.1996.271.1.H192. [DOI] [PubMed] [Google Scholar]
- 41.Foldes-Papp Z, Konig K, Studier H, Buckle R, Breunig HG, Uchugonova A, Kostner GM. Trafficking of mature miRNA-122 into the nucleus of live liver cells. Curr Pharm Biotechnol. 2009;10:569–578. doi: 10.2174/138920109789069332. [DOI] [PubMed] [Google Scholar]
- 42.Gaikwad AB, Sayyed SG, Lichtnekert J, Tikoo K, Anders HJ. Renal failure increases cardiac histone h3 acetylation, dimethylation, and phosphorylation and the induction of cardiomyopathy-related genes in type 2 diabetes. Am J Pathol. 2010;176:1079–1083. doi: 10.2353/ajpath.2010.090528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Goldberg RB. Type 2 diabetes. In: Gotto AMaTPP, editor. Comprehensive Management of High Risk Cardiovascular Patients. Taylor & Francis Group; Boca Raton, FL: 2007. [Google Scholar]
- 44.Greco S, Fasanaro P, Castelvecchio S, D’Alessandra Y, Arcelli D, Di Donato M, Malavazos A, Capogrossi MC, Menicanti L, Martelli F. MicroRNA dysregulation in diabetic ischemic heart failure patients. Diabetes. 2012;61:1633–1641. doi: 10.2337/db11-0952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Grueter CE, van Rooij E, Johnson BA, DeLeon SM, Sutherland LB, Qi X, Gautron L, Elmquist JK, Bassel-Duby R, Olson EN. A cardiac microRNA governs systemic energy homeostasis by regulation of MED13. Cell. 2012;149:671–683. doi: 10.1016/j.cell.2012.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hamblin M, Friedman DB, Hill S, Caprioli RM, Smith HM, Hill MF. Alterations in the diabetic myocardial proteome coupled with increased myocardial oxidative stress underlies diabetic cardiomyopathy. J Mol Cell Cardiol. 2007;42:884–895. doi: 10.1016/j.yjmcc.2006.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hatano A, Okada J, Washio T, Hisada T, Sugiura S. Distinct functional roles of cardiac mitochondrial subpopulations revealed by a 3D simulation model. Biophys J. 2015;108:2732–2739. doi: 10.1016/j.bpj.2015.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hathaway QA, Nichols CE, Shepherd DL, Stapleton PA, McLaughlin SL, Stricker JC, Rellick SL, Pinti MV, Abukabda AB, McBride CR, Yi J, Stine SM, Nurkiewicz TR, Hollander JM. Maternal engineered nanomaterial exposure disrupts progeny cardiac function and bioenergetics. Am J Physiol Heart Circ Physiol. 2016 doi: 10.1152/ajpheart.00634.2016. 00634 02016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Holmuhamedov EL, Oberlin A, Short K, Terzic A, Jahangir A. Cardiac subsarcolemmal and interfibrillar mitochondria display distinct responsiveness to protection by diazoxide. PLoS One. 2012;7:e44667. doi: 10.1371/journal.pone.0044667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jagannathan R, Thapa D, Nichols CE, Shepherd DL, Stricker JC, Croston TL, Baseler WA, Lewis SE, Martinez I, Hollander JM. Translational regulation of the mitochondrial genome following redistribution of mitochondrial microRNA in the diabetic heart. Circ Cardiovasc Genet. 2015;8:785–802. doi: 10.1161/CIRCGENETICS.115.001067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Janssen HL, Reesink HW, Lawitz EJ, Zeuzem S, Rodriguez-Torres M, Patel K, van der Meer AJ, Patick AK, Chen A, Zhou Y, Persson R, King BD, Kauppinen S, Levin AA, Hodges MR. Treatment of HCV infection by targeting microRNA. N Engl J Med. 2013;368:1685–1694. doi: 10.1056/NEJMoa1209026. [DOI] [PubMed] [Google Scholar]
- 52.Keane J, Tajouri L, Gray B. The effect of growth hormone administration on the regulation of mitochondrial apoptosis in-vivo. Int J Mol Sci. 2015;16:12753–12772. doi: 10.3390/ijms160612753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Knezevic I, Patel A, Sundaresan NR, Gupta MP, Solaro RJ, Nagalingam RS, Gupta M. A novel cardiomyocyte-enriched microRNA, miR-378, targets insulin-like growth factor 1 receptor: implications in postnatal cardiac remodeling and cell survival. J Biol Chem. 2012;287:12913–12926. doi: 10.1074/jbc.M111.331751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kolesnikova OA, Entelis NS, Mireau H, Fox TD, Martin RP, Tarassov IA. Suppression of mutations in mitochondrial DNA by tRNAs imported from the cytoplasm. Science. 2000;289:1931–1933. doi: 10.1126/science.289.5486.1931. [DOI] [PubMed] [Google Scholar]
- 55.Kren BT, Wong PY, Sarver A, Zhang X, Zeng Y, Steer CJ. MicroRNAs identified in highly purified liver-derived mitochondria may play a role in apoptosis. RNA Biol. 2009;6:65–72. doi: 10.4161/rna.6.1.7534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 57.Lin CL, Wang YT, Yang WZ, Hsiao YY, Yuan HS. Crystal structure of human polynucleotide phosphorylase: insights into its domain function in RNA binding and degradation. Nucleic Acids Res. 2012;40:4146–4157. doi: 10.1093/nar/gkr1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ling C, Poulsen P, Simonsson S, Ronn T, Holmkvist J, Almgren P, Hagert P, Nilsson E, Mabey AG, Nilsson P, Vaag A, Groop L. Genetic and epigenetic factors are associated with expression of respiratory chain component NDUFB6 in human skeletal muscle. J Clin Invest. 2007;117:3427–3435. doi: 10.1172/JCI30938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Liou GG, Chang HY, Lin CS, Lin-Chao S. DEAD box RhlB RNA helicase physically associates with exoribonuclease PNPase to degrade double-stranded RNA independent of the degradosome-assembling region of RNase E. J Biol Chem. 2002;277:41157–41162. doi: 10.1074/jbc.M206618200. [DOI] [PubMed] [Google Scholar]
- 60.Liu X, Jin DY, McManus MT, Mourelatos Z. Precursor microRNA-programmed silencing complex assembly pathways in mammals. Mol Cell. 2012;46:507–517. doi: 10.1016/j.molcel.2012.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Macfarlane LA, Murphy PR. MicroRNA: biogenesis, function and role in cancer. Curr Genomics. 2010;11:537–561. doi: 10.2174/138920210793175895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Meister G. Argonaute proteins: functional insights and emerging roles. Nat Rev Genet. 2013;14:447–459. doi: 10.1038/nrg3462. [DOI] [PubMed] [Google Scholar]
- 63.Montgomery RL, Hullinger TG, Semus HM, Dickinson BA, Seto AG, Lynch JM, Stack C, Latimer PA, Olson EN, van Rooij E. Therapeutic inhibition of miR-208a improves cardiac function and survival during heart failure. Circulation. 2011;124:1537–1547. doi: 10.1161/CIRCULATIONAHA.111.030932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Murphy RM, Lamb GD. Important considerations for protein analyses using antibody based techniques: down-sizing Western blotting up-sizes outcomes. J Physiol. 2013;591:5823–5831. doi: 10.1113/jphysiol.2013.263251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Nichols CE, Shepherd DL, Knuckles TL, Thapa D, Stricker JC, Stapleton PA, Minarchick VC, Erdely A, Zeidler-Erdely PC, Alway SE, Nurkiewicz TR, Hollander JM. Cardiac and mitochondrial dysfunction following acute pulmonary exposure to mountaintop removal mining particulate matter. Am J Physiol Heart Circ Physiol. 2015;309:H2017–2030. doi: 10.1152/ajpheart.00353.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Nouws J, Shadel GS. microManaging mitochondrial translation. Cell. 2014;158:477–478. doi: 10.1016/j.cell.2014.07.010. [DOI] [PubMed] [Google Scholar]
- 67.Palmer JW, Tandler B, Hoppel CL. Biochemical properties of subsarcolemmal and interfibrillar mitochondria isolated from rat cardiac muscle. J Biol Chem. 1977;252:8731–8739. [PubMed] [Google Scholar]
- 68.Pinti MV, Hathaway QA, Hollander JM. Role of microRNA in metabolic shift during heart failure. Am J Physiol Heart Circ Physiol. 2017;312:H33–H45. doi: 10.1152/ajpheart.00341.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Politz JC, Zhang F, Pederson T. MicroRNA-206 colocalizes with ribosome-rich regions in both the nucleolus and cytoplasm of rat myogenic cells. Proc Natl Acad Sci U S A. 2006;103:18957–18962. doi: 10.1073/pnas.0609466103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Pullman ME, Penefsky HS, Datta A, Racker E. Partial resolution of the enzymes catalyzing oxidative phosphorylation. I. Purification and properties of soluble dinitrophenol-stimulated adenosine triphosphatase. J Biol Chem. 1960;235:3322–3329. [PubMed] [Google Scholar]
- 71.Richter-Dennerlein R, Dennerlein S, Rehling P. Integrating mitochondrial translation into the cellular context. Nat Rev Mol Cell Biol. 2015;16:586–592. doi: 10.1038/nrm4051. [DOI] [PubMed] [Google Scholar]
- 72.Ritov VB, Menshikova EV, He J, Ferrell RE, Goodpaster BH, Kelley DE. Deficiency of subsarcolemmal mitochondria in obesity and type 2 diabetes. Diabetes. 2005;54:8–14. doi: 10.2337/diabetes.54.1.8. [DOI] [PubMed] [Google Scholar]
- 73.Rolo AP, Palmeira CM. Diabetes and mitochondrial function: role of hyperglycemia and oxidative stress. Toxicol Appl Pharmacol. 2006;212:167–178. doi: 10.1016/j.taap.2006.01.003. [DOI] [PubMed] [Google Scholar]
- 74.Ronn T, Poulsen P, Hansson O, Holmkvist J, Almgren P, Nilsson P, Tuomi T, Isomaa B, Groop L, Vaag A, Ling C. Age influences DNA methylation and gene expression of COX7A1 in human skeletal muscle. Diabetologia. 2008;51:1159–1168. doi: 10.1007/s00125-008-1018-8. [DOI] [PubMed] [Google Scholar]
- 75.Rubio MA, Rinehart JJ, Krett B, Duvezin-Caubet S, Reichert AS, Soll D, Alfonzo JD. Mammalian mitochondria have the innate ability to import tRNAs by a mechanism distinct from protein import. Proc Natl Acad Sci U S A. 2008;105:9186–9191. doi: 10.1073/pnas.0804283105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sarkar D, Leszczyniecka M, Kang DC, Lebedeva IV, Valerie K, Dhar S, Pandita TK, Fisher PB. Down-regulation of Myc as a potential target for growth arrest induced by human polynucleotide phosphorylase (hPNPaseold-35) in human melanoma cells. J Biol Chem. 2003;278:24542–24551. doi: 10.1074/jbc.M302421200. [DOI] [PubMed] [Google Scholar]
- 77.Sarkar D, Park ES, Emdad L, Randolph A, Valerie K, Fisher PB. Defining the domains of human polynucleotide phosphorylase (hPNPaseOLD-35) mediating cellular senescence. Mol Cell Biol. 2005;25:7333–7343. doi: 10.1128/MCB.25.16.7333-7343.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Schneider A. Mitochondrial tRNA import and its consequences for mitochondrial translation. Annu Rev Biochem. 2011;80:1033–1053. doi: 10.1146/annurev-biochem-060109-092838. [DOI] [PubMed] [Google Scholar]
- 79.Shen X, Zheng S, Thongboonkerd V, Xu M, Pierce WM, Jr, Klein JB, Epstein PN. Cardiac mitochondrial damage and biogenesis in a chronic model of type 1 diabetes. Am J Physiol Endocrinol Metab. 2004;287:E896–905. doi: 10.1152/ajpendo.00047.2004. [DOI] [PubMed] [Google Scholar]
- 80.Slomovic S, Schuster G. Stable PNPase RNAi silencing: its effect on the processing and adenylation of human mitochondrial RNA. RNA. 2008;14:310–323. doi: 10.1261/rna.697308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Smirnov A, Comte C, Mager-Heckel AM, Addis V, Krasheninnikov IA, Martin RP, Entelis N, Tarassov I. Mitochondrial enzyme rhodanese is essential for 5 S ribosomal RNA import into human mitochondria. J Biol Chem. 2010;285:30792–30803. doi: 10.1074/jbc.M110.151183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Smirnov A, Entelis N, Martin RP, Tarassov I. Biological significance of 5S rRNA import into human mitochondria: role of ribosomal protein MRP-L18. Genes Dev. 2011;25:1289–1305. doi: 10.1101/gad.624711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Srinivasan H, Das S. Mitochondrial miRNA (MitomiR): a new player in cardiovascular health. Can J Physiol Pharmacol. 2015;93:855–861. doi: 10.1139/cjpp-2014-0500. [DOI] [PubMed] [Google Scholar]
- 84.Sripada L, Tomar D, Prajapati P, Singh R, Singh AK, Singh R. Systematic analysis of small RNAs associated with human mitochondria by deep sequencing: detailed analysis of mitochondrial associated miRNA. PLoS One. 2012;7:e44873. doi: 10.1371/journal.pone.0044873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Sripada L, Tomar D, Singh R. Mitochondria: one of the destinations of miRNAs. Mitochondrion. 2012;12:593–599. doi: 10.1016/j.mito.2012.10.009. [DOI] [PubMed] [Google Scholar]
- 86.Stickney LM, Hankins JS, Miao X, Mackie GA. Function of the conserved S1 and KH domains in polynucleotide phosphorylase. J Bacteriol. 2005;187:7214–7221. doi: 10.1128/JB.187.21.7214-7221.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Suh JH, Heath SH, Hagen TM. Two subpopulations of mitochondria in the aging rat heart display heterogenous levels of oxidative stress. Free Radic Biol Med. 2003;35:1064–1072. doi: 10.1016/s0891-5849(03)00468-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Thapa D, Nichols CE, Lewis SE, Shepherd DL, Jagannathan R, Croston TL, Tveter KJ, Holden AA, Baseler WA, Hollander JM. Transgenic overexpression of mitofilin attenuates diabetes mellitus-associated cardiac and mitochondria dysfunction. J Mol Cell Cardiol. 2015;79:212–223. doi: 10.1016/j.yjmcc.2014.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Towheed A, Markantone DM, Crain AT, Celotto AM, Palladino MJ. Small mitochondrial-targeted RNAs modulate endogenous mitochondrial protein expression in vivo. Neurobiol Dis. 2014;69:15–22. doi: 10.1016/j.nbd.2014.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Trounce IA, Kim YL, Jun AS, Wallace DC. Assessment of mitochondrial oxidative phosphorylation in patient muscle biopsies, lymphoblasts, and transmitochondrial cell lines. Methods Enzymol. 1996;264:484–509. doi: 10.1016/s0076-6879(96)64044-0. [DOI] [PubMed] [Google Scholar]
- 91.Valencia-Morales Mdel P, Zaina S, Heyn H, Carmona FJ, Varol N, Sayols S, Condom E, Ramirez-Ruz J, Gomez A, Moran S, Lund G, Rodriguez-Rios D, Lopez-Gonzalez G, Ramirez-Nava M, Rocha Cdela, Sanchez-Flores A, Esteller M. The DNA methylation drift of the atherosclerotic aorta increases with lesion progression. BMC Med Genet. 2015;8(7) doi: 10.1186/s12920-015-0085-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Wang G, Chen HW, Oktay Y, Zhang J, Allen EL, Smith GM, Fan KC, Hong JS, French SW, McCaffery JM, Lightowlers RN, Morse HC, III, Koehler CM, Teitell MA. PNPASE regulates RNA import into mitochondria. Cell. 2010;142:456–467. doi: 10.1016/j.cell.2010.06.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Wang G, Shimada E, Koehler CM, Teitell MA. PNPASE and RNA trafficking into mitochondria. Biochim Biophys Acta. 2012;1819:998–1007. doi: 10.1016/j.bbagrm.2011.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Wei Y, Li L, Wang D, Zhang CY, Zen K. Importin 8 regulates the transport of mature microRNAs into the cell nucleus. J Biol Chem. 2014;289:10270–10275. doi: 10.1074/jbc.C113.541417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Williamson CL, Dabkowski ER, Baseler WA, Croston TL, Alway SE, Hollander JM. Enhanced apoptotic propensity in diabetic cardiac mitochondria: influence of subcellular spatial location. Am J Physiol Heart Circ Physiol. 2010;298:H633–642. doi: 10.1152/ajpheart.00668.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Wong AG, McBurney KL, Thompson KJ, Stickney LM, Mackie GA. S1 and KH domains of polynucleotide phosphorylase determine the efficiency of RNA binding and autoregulation. J Bacteriol. 2013;195:2021–2031. doi: 10.1128/JB.00062-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Zaina S, Heyn H, Carmona FJ, Varol N, Sayols S, Condom E, Ramirez-Ruz J, Gomez A, Goncalves I, Moran S, Esteller M. DNA methylation map of human atherosclerosis. Circ Cardiovasc Genet. 2014;7:692–700. doi: 10.1161/CIRCGENETICS.113.000441. [DOI] [PubMed] [Google Scholar]
- 98.Zhang X, Zuo X, Yang B, Li Z, Xue Y, Zhou Y, Huang J, Zhao X, Zhou J, Yan Y, Zhang H, Guo P, Sun H, Guo L, Zhang Y, Fu XD. MicroRNA directly enhances mitochondrial translation during muscle differentiation. Cell. 2014;158:607–619. doi: 10.1016/j.cell.2014.05.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Zuo Y, Deutscher MP. Exoribonuclease superfamilies: structural analysis and phylogenetic distribution. Nucleic Acids Res. 2001;29:1017–1026. doi: 10.1093/nar/29.5.1017. [DOI] [PMC free article] [PubMed] [Google Scholar]