

Abstract
Parthenolide (PTL) has shown great promise as a novel anti-leukemia agent as it selectively eliminates acute myeloid leukemia (AML) blast cells and leukemia stem cells (LSCs) while sparing normal hematopoietic cells. This success has not yet translated to the clinical setting because PTL is rapidly cleared from blood due to its hydrophobicity. To increase the aqueous solubility of PTL, we previously developed micelles formed from predominantly hydrophobic amphiphilic diblock copolymers of poly(styrene-alt-maleic anhydride)-b-poly(styrene) (e.g., PSMA100-b-PS258) that exhibit robust PTL loading (75%efficiency, 11% w/w capacity) and release PTL over 24 h. Here, PTL-loaded PSMA-b-PS micelles were thoroughly characterized in vitro for PTL delivery to MV4-11 AML cells. Additionally, the mechanisms governing micelle-mediated cytotoxicity were examined in comparison to free PTL. PSMA-b-PS micelles were taken up by MV4-11 cells as evidenced by transmission electron microscopy and flow cytometry. Specifically, MV4-11 cells relied on clathrin-mediated endocytosis, rather than caveolae-mediated endocytosis and macropinocytosis. In addition, PTL-loaded PSMA-b-PS micelles exhibited a dose-dependent cytotoxicity towards AML cells and were capable of reducing cell viability by 75% at 10 μM PTL, while unloaded micelles were nontoxic. At 10 μM PTL, the cytotoxicity of PTL-loaded micelles increased gradually over 24 h while free PTL achieved maximal cytotoxicity between 2 and 4 h, demonstrating micelle-mediated delivery of PTL to AML cells and stability of the drug-loaded micelle even in the presence of cells. Both free PTL and PTL-loaded micelles induced NF-κB inhibition at 10 μM PTL doses, demonstrating some mechanistic similarities in cytotoxicity. However, free PTL relied more heavily on exofacial free thiol interactions to induce cytotoxicity than PTL-loaded micelles; free PTL cytotoxicity was reduced by over twofold when cell surface free thiols were depleted, where PTL-loaded micelle doses were unaffected by cell surface thiol modulation. The physical properties, stability, and efficacy of PTL-loaded PSMA-b-PS micelles support further development of a leukemia therapeutic with greater bioavailability and the potential to eliminate LSCs.
Keywords: Nanoparticle, Drug delivery, Endocytosis, Leukemia
INTRODUCTION
In the US, over 50,000 new cases of leukemia are diagnosed each year, and the associated 5-year survivability varies substantially based on the specific sub-type diagnosed, ranging from 25% for acute myeloid leukemia (AML) to nearly 80% for chronic lymphocytic leukemia (CLL).66 For AML, induction therapy results in 60% remission rate, yet 5-year survival remains poor. One possible explanation for this outcome is the inability to eliminate leukemic sub-populations that cause disease recurrence. An infrequent, yet important, sub-population of leukemic cells called leukemia stem cells (LSCs) possess many properties similar to normal hematopoietic cells (HSCs) including multipotency,19 self-renewal,38,45 quiescence,39 and expression of specific cell surface markers.3 These unique properties contribute to LSC evasion of traditional chemotherapeutics (e.g., anthracyclines, nucleoside analogs, alkylating agents, etc.),39 which are more effective towards rapidly dividing cells. Due to insufficiencies of current anti-leukemia agents and importance of LSC elimination, multiple LSC-specific treatments are currently being investigated in clinical trials.22
The sesquiterpene lactone, parthenolide (PTL), has shown potent cytotoxicity in vitro towards both LSCs and malignant progenitor blast cells.24 PTL is known to inhibit the anti-apoptotic transcription factor, nuclear factor-κB (NF-κB),17,18,24,64 which is active amongst AML cells but not normal hematopoietic cells.23 Therefore, PTL therapy offers specific toxicity towards leukemia cells and LSCs, without causing harm to normal hematopoietic cells. Unfortunately, PTL has not shown anti-leukemia activity in vivo due to poor bioavailability.8,25 To increase aqueous solubility and prolong systemic circulation, PTL analogs have been developed43 and have shown enhanced bioavailability and bioactivity in vivo.25 Still, systemic delivery of small molecule drugs has many challenges, including nonspecific cell uptake, off-target organ accumulation and toxicity, clearance by extravasation, inability to overcome multidrug resistant mechanisms, and degradation by enzymes and other cell secreted factors.5,7,28,35 Nanoparticle (NP) drug carriers have emerged as promising tools to overcome these obstacles,36,40 which has led to numerous clinical trials, and approval of a number of NP drug formulations by the Food and Drug Administration (FDA).31 The size (ideally 20–100 nm) and surface characteristics of NPs can prevent premature clearance by the reticuloendothelial system (RES),1,54 and surface functionalization can direct NP delivery to specific cells and tissues.31,63 Thus, successful drug-loaded NPs achieve high therapeutic efficacy while providing favorable circulation and targeting in vivo.
Synthetic polymer micelles are particularly attractive NP drug carriers because they offer vast chemical versatility and highly stable core–shell structures for efficient loading of a variety of therapeutic molecules.21,51 Polymer micelles can also be functionalized with multiple targeting ligands per particle, allowing for NPs with both high potency and cell/tissue-specific affinity.28 Drug-loaded poly(styrene-alt-maleic anhydride) (PSMA) micelles have been extensively studied and show superior circulation time and anti-cancer effects in vivo as compared to free drug.49,50 Drug-loaded poly(styrene-alt-maleic anhydride)-b-poly(styrene) (PSMA-b-PS) micelles have been more recently developed and offer high aqueous stability, as well as robust drug loading and controlled release of a variety of hydrophobic drugs due to the presence of large hydrophobic PS cores.2,26,57 These micelles also exhibit increased drug efficacy towards multidrug resistant ovarian cancer cells compared to free drug, largely due to more robust intracellular accumulation.2 Because PSMA-b-PS micelles combine stable, versatile PS cores with PSMA shells that allow long circulation times, these micelles were studied for their ability to load and deliver PTL to AML cells in vitro. Previously, we demonstrated efficient loading of PTL into highly stable, predominantly hydrophobic PSMA-b-PS micelles. Moreover, PTL release from micelles was quantitative over 24 h.2 Here, PTL-loaded micelles were extensively characterized for PTL delivery to AML cells in vitro while investigating various fundamental mechanisms of micelle-mediated PTL delivery and cytotoxicity versus that of free PTL. In this study, PTL-loaded micelles were tested for uptake kinetics, dose and time-dependent cytotoxicity, and NF-κB inhibition with MV4-11 human AML cells. Importantly, pre-dominant routes of PSMA-b-PS micelle endocytosis utilized by MV4-11 cells were also examined, which has not been characterized previously. The in vitro efficacy and advantageous physiochemical properties of PTL-loaded PSMA-b-PS micelles motivates exploration of this NP-drug formulation for leukemia treatments.
MATERIALS AND METHODS
Materials
Unless otherwise specified, all chemicals were purchased from Sigma Aldrich (St. Louis, Missouri). Styrene (99%, ACS grade) and butyl acrylate (BA, 99% pure ACS grade) were purified by distillation. Maleic anhydride (MA) was recrystalized from chloroform. 2,2′-azo-bis(isobutylnitrile) (AIBN) was recrystalized from methanol. All solvents used were spectroscopic grade. Unless otherwise specified, all water used was distilled with resistivity of 18 MΩ or greater.
Cell Culture
MV4-11 human myelomonocytic leukemia cells (ATCC, CRL-9591) were cultured at 37 °C, 5% carbon dioxide and maintained at 1–5 × 105 cells/mL in Iscove’s modified Dulbecco Media (IMDM) supplemented with 10% v/v heat inactivated fetal bovine serum (FBS) and 1% v/v penicillin–streptomycin.
Synthesis of Poly(styrene-alt-maleic anhydride)-b-Poly(styrene) (PSMA-b-PS) Diblock Copolymers by Reversible Addition-Fragmentation Chain Transfer (RAFT) Polymerization
4-Cyano-4-dodecylsulfanyltrithiocarbonyl sulfanyl pentanoic acid (DCT) was synthesized as previously described and was used as the RAFT chain transfer agent (CTA).52 Amphiphilic PSMA-b-PS copolymers were made in a one-step RAFT polymerization process. Styrene (Sty) was added in excess of maleic anhydride (MA) (4:1 [Sty]:[MA]) in the presence of DCT (100:1 [monomer]:[CTA]) and AIBN thermal initiator (10:1 [CTA]:[Initiator]) in dioxane (50% w/w). The dissolved mixture was kept on ice and purged with nitrogen for 45 min. After purging, the solution was placed in a 60 °C oil bath for polymerization. The polymer molecular weight was monitored throughout the polymerization using gel permeation chromatography (GPC). After the desired molecular weight was attained, the polymer sample was exposed to air and diluted with acetone prior to precipitation in petroleum ether. The final product was dried under vacuum at room temperature.
Gel permeation chromatography (GPC) was performed on a Shimadzu system equipped with a solvent pump (Shimadzu LC-20AD), a differential refractometer (Shimadzu RID-10A), and a light scattering detector (Wyatt Technology DAWNTREOS). A 3-μm linear gel column (Tosoh TSK-Gel Super HM-N, 6.0 mm ID × 15 cm) was used in series with a 3-μm guard column (Tosoh Biosciences) in an oven chamber operating at 60 °C. The mobile phase consisted of spectroscopic grade DMF containing 0.5 M lithium bromide. A flow rate of 0.35 mL/min was used to analyze polymers dissolved in the mobile phase. Polymer molecular weights and polydispersity indices (PDIs) were calculated using ASTRA 6 software (Wyatt Technology). Refractive index increment (dn/dc) of PSMA-b-PS polymers was determined experimentally to be 0.141 mL/g using a Shimadzu refractometer and ASTRA 6 software (Wyatt Technology).
Micelle Formation and Characterization
Self-assembly of PSMA-b-PS and PSMA-b-PBA Diblock Copolymers into Micelle NPs
Diblock copolymers (200 mg) were dissolved in 30 mL DMF. An equivalent volume of water was added to stirring polymer solutions at a rate of 24 μL/min via syringe pump.58 After addition of water, polymer micelle solutions were dialyzed(MWCO6000–8000 kDa) against water for 3 days, with dialysis water replacement twice daily. Following dialysis, micelle solutions were passed through 0.45 μm hydrophilic PTFE syringe filters. Micelle concentrations were determined following lyophilization. Micelles were stored at 4 °C in distilled water.
Particle Size and Zeta-Potential Measurements by Dynamic Light Scattering (DLS)
Hydrodynamic radii of PSMA-b-PS and PSMA-b-PBA micelles were determined by dynamic light scattering (DLS, Malvern Instruments, Worcestershire, UK) using a 3.0 mm quartz cuvette with 633 nm incident laser source. Micelles were suspended in PBS at 0.1 mg/mL for both particle sizing and zeta-potential measurement.
Particle Size and Morphology by Electron Microscopy
Transmission electron microscopy (TEM) was used to characterize the size and morphology of micelle NPs. Polymer micelles were solvated in PBS at 0.1 mg/mL and incubated 1:1 with 2% (v/v in water) phosphotungstic acid negative stain on 150 mesh formvar/carbon coated grids for 5 min. Excess aqueous solution was wicked away with filter paper.32 The grids were photographed at 80,000–200,000 magnification using a Hitachi 7650 transmission electron microscope operating at 80 with an attached Gatan 11 megapixel Erlangshen digital camera.
Determination of Critical Micelle Concentration (CMC)
The critical micelle concentration (CMC) is a value that describes micelle-solute stability by determining the concentration at which micelles disassociate into polymer unimers.69 CMC measurements were made using a PRODAN assay as previously described.2,73 Briefly, 6-propionyl-2-(dimethylamino)naphthalene (PRODAN) was dissolved in methanol at 24 μM and 10 μL of this solution was placed in each well of a 96-well black plate. The plate was kept covered in a chemical fume hood overnight to allow the methanol to evaporate. Polymer micelle concentrations ranging from 0.1 ng/mL to 0.5 mg/mL were made in PBS and 100 μL of each micelle solution was added to individual wells containing dry PRODAN dye and incubated at 4 °C overnight. Samples were analyzed using a fluorescent plate reader (Tecan). An emission scan ranging from 400 to 600 nm was performed for PRODAN excitation at 360 nm for each micelle dilution. The ratio of the peak hydrophobic emission intensity (436 nm) to the peak hydrophilic emission intensity (518 nm) was plotted against the polymer concentration on a logarithmic scale (see Supplemental Figure 1). The intersection of the hydrophilic and hydrophobic regimes of the dilution curve is designated the CMC.73
PTL Loading into Micelle Nanoparticles
PTL was loaded into micelles as previously described.2 Briefly, 10 mL of micelles dissolved in water at a concentration of 2 mg/mL were added to stirring solutions of PTL (3 mg) dissolved in 1 mL chloroform. The drug-micelle solution was stirred vigorously overnight uncovered in a chemical fume hood shielded from light to allow evaporation of chloroform. Free drug was separated from drug-loaded micelles by centrifugation and centrifugal filtration. Drug-micelle solutions were centrifuged at 2000 RPM for 10 min to remove any insoluble drug aggregates that may have formed during loading, followed by three rounds of centrifugal filtration using 100,000 MWCO Amicon® Ultra-15 centrifugal filter device (Millipore) to remove any unloaded soluble drug. The final concentrate was reconstituted to 5 mL in water, and passed through 0.45 μm PVDF aqueous syringe filters and lyophilized. PTL loading was quantified using high performance liquid chromatography (HPLC) with a mobile phase consisting of HPLC-grade water and methanol. HPLC analysis was completed using a Kromasil C18 column (50 mm × 4.6 mm, 5 μm particle size, 100 A ˚ pore size). The column effluent was monitored with a variable wavelength UV–VIS detector at 210 nm (Shimadzu). Flow conditions were as follows: 0.5 mL/min flow rate, gradient elution (0–3 min 95% water in methanol, 3–10 min 30% water in methanol).
PTL loading efficiency and capacity were calculated based on the following definitions, drug loading efficiency = 100 × (mg drug loaded/mg initial drug mass), and drug loading capacity = 100 × (mg drug loaded/mg micelles).
PTL Release from Polymer Micelles
To analyze drug release, drug-loaded micelles were dialyzed (MWCO 6000–8000 kDa) against 2 L of phosphate buffered saline (PBS) at 37 °C. Release media was replaced once per day and 100 μL samples were taken from the dialysis tubing at 1, 4, 8, and 24 h. PTL concentration was quantified by HPLC as described above.
Analysis of PTL-Loaded Micelle Delivery to MV4-11 Cells In Vitro
Quantitation of PSMA-b-PS Micelle Uptake by MV4-11 Cells Using Flow Cytometry
PSMA-b-PS micelles were functionalized with fluorescein cadaverine using 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDC) chemistry. 16 mL micelles dissolved in water at 2 mg/mL were added to 4 mg fluorescein cadaverine, with 1.2 mg EDC and 20 mg sulfo N-hydroxyl succinimide. The reaction mixture was buffered at pH 7.4 with 0.1 M sodium phosphate, and was stirred for 3 h at room temperature. Unconjugated reactants were removed from solution by extensive dialysis (6000–8000 MWCO) against water.
MV4-11 cells were plated at 1 × 106 cells/well in untreated 24-well plates. Cells were treated with 50 μg/mL fluorescein-labeled micelles (FLM) and incubated at 37 °C for 1, 2, 4, 8, and 24 h. After the designated incubation time, cells were washed thrice with PBS and reconstituted in 0.5 mL PBS containing 0.5% (w/w) bovine serum albumin (BSA). Flow cytometry was performed on an Accuri C6 (BD Biosciences), and FlowJo software was used to analyze 5000 gated cell events. Fluorescein signal from FLM was detected using theFL1 channel (488 nm excitation, 530/30 detector). None of the micelle treatments had adverse effects on cell viability, even at 24 h (data not shown).
Inhibition of PSMA-b-PS Micelle Endocytosis
Specific routes of endocytosis (clathrin-mediated, caveolae-mediated endocytosis, and macropinocytosis) were examined by incubating cells with specific inhibitors of these pathways prior to incubation with FLM. MV4-11 cells were plated at 1 × 106 cells/well in untreated 24-well plates. Cells were incubated with chlorpromazine hydrochloride (10 μg/mL), genistein (250 μM), or rottlerin (5 μg/mL) for 30 min prior to addition of FLM to inhibit uptake by clathrin-mediated endocytosis,34,70 caveolae-mediated endocytosis,56 and macropinocytosis,60 respectively. Cells were incubated with 50 μg/mL FLM in the presence of the inhibitors for an additional hour at 37 °C, before being washed thrice with PBS and reconstituted in 0.5 mL PBS containing 0.5% (w/w) bovine serum albumin (BSA) and 0.01% trypan blue. The addition of trypan blue ensured that all fluorescence was the result of intracellular FLM levels in living cells. Flow cytometry and data analysis were performed as described in “Quantitation of PSMA-b-PS micelle uptake by MV4-11 cells using flow cytometry” section.
Qualitative Assessment of PSMA-b-PS Micelle Uptake by Transmission Electron Microscopy
Circular (12 mm diameter) glass coverslips (VWR) were coated with poly(L-Lysine) (PLL) to promote MV4-11 adhesion to ease imaging. Coverslips were placed in 12-well tissue culture plates and 1 mL solution of PLL in water (0.016 mg/mL) was added to each slide and placed on a rotational shaker for 1 h. PLL-coated slides were transferred to fresh 12 well plates (1 slide/well) and 1 mL of MV4-11 cell suspension (~1.5 × 106 cells/well) was added to each slide. Micelles were added to cell suspensions at a final concentration of 0.2 mg/mL, and incubated for 2 or 24 h. After incubation, media was removed from the well, and slides were fixed with 2.5% glutaraldehyde and 4% paraformaldehyde in 0.1 M Millonig’s buffer. Fixed coverslips were processed into epoxy resin and sectioned (70 nm) onto grids using a “pop-off” technique before imaging under the TEM.13
PTL-Loaded Micelle Toxicity Towards MV4-11 Cells
The toxicity of free PTL, PTL-loaded polymer micelles, and unloaded polymer micelles towards MV4-11 cells was tested in vitro. MV4-11 cells were plated at 5 × 105 cells/mL in 24-well untreated tissue culture plates (1 mL/well) and dosed with 2.5, 5, 7.5, and 10 μM concentrations of PTL. Using the drug loading capacity, the requisite dose of polymer micelles was calculated and used to test the toxicity of unloaded polymer micelles. 24 h after dosing, viable cells were counted using trypan-blue exclusion and an automated cell counter (Bio-Rad TC-20) and compared to the untreated control group. Because of PTL’s limited aqueous solubility, it was necessary to dissolve PTL in DMSO prior to diluting in PBS. These concentrations of DMSO alone did not adversely affect cell viability.
Examination of PTL and PTL-Loaded Micelle Toxicity Towards MV4-11 Cells Over Time (Drug Washout Study)
To determine the necessary longevity of PTL treatment required to realize cytotoxicity, a drug washout study was performed where PTL or PTL-loaded micelle doses were incubated with cells for various amounts of time, and subsequently washed out of solution and replaced with fresh (drug-free) media. MV4-11 cells were plated at 5 × 105 cells/well on a 24-well plate and dosed with 10 μM PTL. At various times (2, 4, and 8 h) after the PTL dose was introduced, cells were washed thrice with PBS and resuspended in fresh (drug-free) culture media. Cell viability was determined 24 h after the PTL dose using an automated cell counter. Washing/resuspending cells did not reduce cell numbers or viability at any time point studied.
Glutathione Inhibition of Free and Micelle-Delivered PTL Cytotoxicity
Glutathione (GSH) has been shown to inhibit the exofacial cell binding of PTL and subsequent toxicity to lymphoma cells by depleting the amount free thiol binding targets for PTL on the outside of the cell.65 MV4-11 cells were plated at 5 × 105 cells/well in an untreated 24-well plate. Cells were pretreated with 5 mM GSH in media by incubation at 37 °C for 2 h. Cells were washed 3 times with PBS and resuspended in fresh media containing 10 μM PTL or PTL-loaded micelles. Cell viability was determined 22 h after treatment. GSH treatments had no adverse effect on cell viability.
Quantification of PTL and PTL-Loaded Micelle Induced Nuclear Factor κB (NF-κB) Inhibition by ELISA
The activated, nuclear form of p65 NF-κB subunit was quantified according to Active Motif (Carlsbad, CA) TransAM® p65 ELISA kit. MV4-11 cells were plated at 30 × 106 cells/well in an untreated 6-well plate. Cells were treated with PTL or PTL-loaded micelles at equivalent 10 μM drug concentrations and incubated for 4 or 8 h before cellular nuclear fraction was extracted and quantified for activated p65 NF-κB subunit. Activated protein levels were normalized to total nuclear protein of the sample to account for losses during extraction. Normalized p65 protein levels of treated samples were compared to untreated control groups.
Activated NF-κB levels were also assessed for cells that were treated with GSH prior to PTL dosing. MV4-11 cells were plated at 30 × 106 cells/well in an untreated 6-well plate and incubated with 5 mM GSH for 2 h at 37 °C, 5% CO2. Cells were then washed thrice with PBS and resuspended in 4 mL media containing PTL or PTL-loaded micelles at equivalent 10 μM drug concentrations, and allowed to incubate for 8 h before cellular nuclear fraction was extracted and quantified for activated p65 NF-κB subunit. Activated protein levels were normalized as described above.
Statistical Analysis
Statistical significance was determined using Prism software (GraphPad Version 6.0). Drug loading and release data represent the average of two independently-synthesized micelle batches prepared under identical conditions. For one-variable and two-variable comparisons, one-way or two-way ANOVA with Tukey’s post hoc analysis were used to determine statistical significance (p values are indicated in figure legends).
RESULTS AND DISCUSSION
Predominantly Hydrophobic PSMA-b-PS Micelles are Promising PTL Drug Delivery Systems
Previously, a range of PSMA-b-PS and poly(styrene-alt-maleic anhydride)-b-poly(butyl acrylate) (PSMA-b-PBA) amphiphilic diblock copolymers with a variety of molecular weights and hydrophilic PSMA: hydrophobic PS/PBA ratios were developed.2 In accordance with other studies, the architecture (i.e., chemical composition, overall molecular weight, and relative lengths of hydrophilic/hydrophobic units) of the developed polymers largely influenced the physical properties (e.g., size, aqueous stability) and drug loading capabilities of self-assembled micelle NPs.41,47,69 Specifically for PTL, PS cores load drug in greater capacities than PBA cores, likely due to pi-orbital interaction between PTL and STY units. Additionally, predominantly hydrophobic PSMA-b-PS micelles (i.e., PSMA Mn ≪ PS Mn) are more stable in aqueous media (as evidenced by lower critical micelle concentrations), load PTL more efficiently (up to tenfold greater), and release PTL more gradually (50% release vs. 90–100%release within 4 h in PBS) than other PSMA-b-PS formulations explored (i.e., PSMA Mn ≫ PS Mn or PSMA Mn ~ PS Mn).2
Due to advantageous PTL loading, stability and release rate, predominantly hydrophobic (PS dominant) PSMA-b-PS micelles were explored thoroughly for in vitro PTL delivery herein. Several batches of PSMA-b-PS diblock copolymers were synthesized to duplicate the desirable properties of PSMA100-b-PS258 (shown in Table 1).
TABLE 1.
Characteristics of PSMA-b-PS polymers explored here for PTL delivery to leukemia cells.
| Polymer composition | Diblock Mn (KD)a | Diblock PDI (MW/Mn)a | Micelle hydrodynamic diameter by DLS (nm) | Micelle diameter by TEM (nm) | Micelle zeta potential (mV) | Critical micelle concentration (CMC, μM) |
|---|---|---|---|---|---|---|
| PSMA100-b-PS258 | 46.2 | 1.05 | 40(±10) | 20 | −50(±5) | 0.1 |
Number-average molecular weight and polydispersity index (PDI) of PSMA-b-PS diblocks. Polymers self-assembled into micelles with hydrodynamic diameter and zeta potential listed below, representing the average of three independent experiments (±average peak standard deviation).
Although slight variations in molecular weight and/or relative hydrophobicity were observed, the resulting self-assembled micelles were identical with respect to size, morphology, aqueous stability, PTL loading, rate of PTL release, and cellular interactions (i.e., uptake, cytotoxicity, etc.).
Therefore, predominantly hydrophobic micelles similar to PSMA100-b-PS258, will be generically referred to as PSMA-b-PS micelles, and can be assumed to have properties nearly identical to those listed in Table 1 and shown in Fig. 1.
FIGURE 1.

Size and morphology of self-assembled micelle nanoparticles of predominantly hydrophobic PSMA-b-PS diblocks. (a) TEM images of PSMA100-b-PS258 micelles (×200,000 magnification) with 1% phosphotungstic acid negative stain. (b) Micelle size distribution (number-based) by DLS. PSMA100-b-PS258 micelles dissolved in PBS at 0.05 mg/mL are unimodal with an average diameter of 40 ± 10 nm.
PSMA-b-PS Micelle Nanoparticles are Internalized by MV4-11 Cells
The ability of nanoparticles (NPs) to be efficiently taken up by cells is dependent on both NP and cell characteristics.16 PSMA-b-PS micelle NPs have previously been shown to be taken up by multidrug resistant NCI ADR RES ovarian cancer cells despite negative zeta potentials.2 While many cells readily undergo endocytosis, MV4-11 cells are suspension cells with depleted endocytosis-related gene expression (caveolin-1, caveolin-2, Rab13, and Rab7B), and are typically more difficult cells to access with NPs.27 Nevertheless, the uptake of PSMA-b-PS micelles by MV4-11 cells was characterized by transmission electron microscopy (TEM) and flow cytometry.
TEM has been used previously to qualitatively assess cellular uptake of NPs by cells.2,9–11 MV4-11 cells were adhered to PLL-coated coverslips to ease imaging, incubated with PSMA-b-PS micelles, fixed, sectioned, and imaged by TEM (according to Ref 13). To ensure micelles withstood the fixation, dehydration, and processing of this imaging technique, PSMA-b-PS micelles alone were dried on glass slides and handled exactly as cell samples. Figures 2a and 2b show the dried micelles alone, and although the NPs appeared slightly deformed, they remained stable and distinguishable by TEM. Figures 2c and 2d show MV4-11 cells treated with PSMA-b-PS micelles for 24 h. Specifically, Fig. 2d shows a magnified image of large intracellular compartments (represented by #) that contain multiple PSMA-b-PS micelle NPs (represented by arrows). Although these images allow only qualitative analysis, they confirm that PSMA-b-PS micelles can be taken up by MV4-11 cells despite characteristically poor uptake qualities of these cells. TEM allowed visualization of PSMA-b-PS micelle uptake by MV4-11 cells, showing that multiple micelles could be compartmentalized in intracellular vesicles. Further studies characterizing the specific pathways of endocytosis that were utilized for PSMA-b-PS micelle uptake and sequestration by MV4-11 cells were carried out using flow cytometry (described in “Investigation of endocytic pathways used by MV4-11 cells to take up PSMA-b-PS micelles” section).
FIGURE 2.

Transmission electron micrographs of PSMA-b-PS micelles alone post fixation, dehydration, and sectioning at ×80,000 (a) and ×200,000 (b) magnification, and MV4-11 cells incubated with PSMA-b-PS micelles for 24 h at ×20,000 (c) and ×200,000 magnification (d). PSMA-b-PS micelles were dried on glass slides, fixed and processed identically to MV4-11 cells. Dried micelles aggregated together as the aqueous solvent evaporated. Fixed, processed micelles appear deformed and swollen compared to unprocessed micelles (shown in Fig. 1a). MV4-11 cells show large endocytic compartments (outlined by black box, represented by # and magnified in (d)) containing micelle nanoparticles (representative micelles designated by arrows).
To more closely follow the accumulation of micelles over time, fluorescently labeled PSMA-b-PS micelles (FLM) were incubated with MV4-11 cells for 2, 4, 8, or 24 h and analyzed by flow cytometry. Figure 3a shows the cellular fluorescence of each treatment group normalized to the untreated control groups, and Fig. 3b shows histograms of cellular fluorescence for the different groups. Although there was statistically significant uptake of micelles observed for all time points, there was little change in intracellular fluorescence between 2 and 8 h incubations (1.5-fold increase in intracellular fluorescence over untreated cells at 2 h vs. 1.8-fold increase over untreated at 8 h). However, a significant increase in uptake over these time points was seen at 24 h (2.8-fold increase over untreated cells). Furthermore, trypan blue was used to quench extracellular fluorescence as previously described,29 and no loss of fluorescence was observed for any incubation time (24 h incubation time included in Fig. 3), indicating that fluorescence was intracellular. Through TEM and flow cytometry, MV4-11 cell uptake of PSMA-b-PS micelles was confirmed and characterized as a gradual, yet significant accumulation of NPs over 24 h of incubation.
FIGURE 3.

Uptake of fluorescein-labeled PSMA-b-PS micelles by MV4-11 cells, measured by flow cytometry. Fluorescent micelles were incubated with cells for 2, 4, 8, or 24 h before being analyzed by flow cytometry. Micelles show gradual increases in intracellular fluorescence, with significant increases at 24 h compared to the first 8 h. The presence of trypan blue in the flow assay buffer did not impact the cellular fluorescence indicating fluorescence was intracellular. n = 3 ± standard deviation, **p<0.01 (one-way ANOVA, Tukey’s post hoc).
Investigation of Endocytic Pathways Used by MV4-11 Cells to Take Up PSMA-b-PS Micelles
While some nanoparticle drug delivery systems have been evaluated for drug delivery to MV4-11 AML cells,33,53,74 to our knowledge, no publications have investigated the routes of endocytosis utilized by nanoparticle formulations with this cell type. Endocytic processing is dependent on both the cell type and the composition of the nanoparticle,16 making this characterization material and application specific. Chlorpromazine, genistein, and rottlerin were chosen as chemical inhibitors because they have shown specific inhibition of clathrin-mediated endocytosis,34,70 caveloae- mediated endocytosis,56 and macropinocytosis,60 respectively. Still, these inhibitors can affect cell viability and compromise cell membrane integrity over long incubation times, so experiments were designed to investigate the initial cellular response upon inhibition of common endocytic pathways without influencing cell viability.
Treatment of MV4-11 cells with 10 μg/mL chlorpromazine for 30 min prior to and throughout FLM treatments resulted in a 35% reduction in FLM uptake compared to FLM only controls (see Fig. 4). Genistein and rottlerin treatment during FLM dosing did not significantly affect the intracellular fluorescence of MV4-11 cells. Chlorpromazine, which depletes clathrin levels at the cell surface, was the only inhibitor to reduce uptake of PSMA-b-PS micelles in MV4-11 cells, suggesting that clathrin-mediated endocytosis is an important route for PSMA-b-PS uptake. Specific routes of endocytosis have signature endosomal/lysosomal pathways with unique physiological characteristics, and therefore largely impact degradation and subsequent efficacy of NP-mediated nucleic acid delivery.16,56 Because PTL activity relies heavily on exofacial interactions with AML cells, which is described more thoroughly later in this section, the endocytic route taken by micelles may have less of an effect on PTL cytotoxicity, provided PTL successfully reaches the cytoplasm. Still, efficient uptake of PSMA-b-PS micelles by clathrin-mediated endocytosis was observed with MV4-11 cells, which allowed for a comparison between micelle-mediated PTL and free PTL delivery.
FIGURE 4.

Examination of endocytic pathways used by MV4-11 cells to internalize PSMA-b-PS micelles by flow cytometry. Uptake of FLMs via clathrin-mediated endocytosis, caveolae-mediated endocytosis, and macropinocytosis was inhibited by chlorpromazine hydrochloride (10 μg/mL), genistein (250 μM), or rottlerin (5 μg/mL), and intracellular fluorescence was quantified by flow cytometry. All fluorescence levels were normalized to intracellular fluorescence of MV4-11 cells treated with FLMs in the absence of any inhibitor of endocytosis (FLM alone). n = 3 ± standard deviation ***p<0.001 vs. FLM alone control (1-way ANOVA, Tukey’s post hoc).
In Vitro Toxicity of PTL and PTL-Loaded Micelles Towards MV4-11 Cells
PTL has been extensively studied in vitro for therapeutic potential towards numerous cancer cells, including leukemia cells.18,24,30,44,55,67 Specifically, PTL induces apoptosis in human primary acute myeloid leukemia (AML) cells with a half-maximal inhibitory concentration (IC50) between 2.5 and 7.5 μM, while sparing normal hematopoietic cells.24 The specific toxicity observed for isolated human primary cells is attributed to inhibition of the anti-apoptotic transcription factor, NF-κB, as NF-κB is constitutively active in AML but not normal hematopoietic cells.23 PTL is known to bind to IκB kinase (IKK),30,44 as well as NF-κB directly,17,18,24 which inhibits degradation of IκB, subsequent translocation of the activated form of NF-κB to the nucleus, and NF-κB binding to target DNA in the nucleus. In addition, PTL induces cytotoxicity through proapoptotic p53 activation and generation of cellular oxidative stress.24,55,64,71 Unfortunately, the low aqueous solubility of PTL (1 mg/mL) has limited its bioavailability and therapeutic effects in vivo.24,25 Therefore a NP drug formulation, which we previously demonstrated to exhibit robust PTL loading, was assessed for efficacy towards the MV4-11 human myelomonocytic leukemia cell line in vitro.
In agreement with previous studies, free PTL exhibited potent cytotoxicity towards MV4-11 cells with an IC50 ~ 5 μM. Figure 5 shows the dose dependent cytotoxicity of free PTL and PTL-loaded PSMA-b-PS micelles at 24 and 48 h. Free PTL was significantly more effective than PTL-loaded micelles between 2.5 and 7.5 μM PTL (by as much as twofold), however, no statistical difference was apparent between the two treatments at 10 μM. Importantly, no significant cytotoxicity was observed for cells treated with unloaded PSMA-b-PS micelles, at doses equivalent to the 10 μM PTL-loaded micelles.
FIGURE 5.

Dose-dependent cytotoxicity of PTL and PTL-loaded PSMA-b-PS micelles in vitro. MV4-11 leukemia cell counts were taken after 24 (a) and 48 h (b) incubations with 0–10 μM PTL (either free or loaded within PSMA-b-PS micelles). All cell counts were normalized to the untreated control groups (media only). Unloaded micelles, at the highest delivered dose, showed no significant toxicity towards MV4-11 cells. n = 3 ± standard deviation *p<0.05, **p <0.01, ****p <0.0001 free PTL vs. micelle PTL (2-way ANOVA, Tukey’s post hoc).
Given that PTL releases nearly quantitatively from micelles over the first 24 h,2 the observed differences in cytotoxicity in the intermediate doses was somewhat surprising. However, PTL release from PSMA-b-PS was not immediate, with retention of ~30% of the drug still within micelles as late as 8 h after the start of treatments, as reported previously.2 Interestingly, the controlled release of PTL from micelles did not result in a delay in cytotoxicity, as a sizeable gap in toxicity remained between free PTL and PTL-loaded micelles at intermediate doses even after 48 h (see Fig. 5b). Rather, the initial shortage of free PTL caused an overall inhibition of activity at doses below 10 μM. The cytotoxicity of PTL towards leukemia cells is directly influenced by the ability of PTL to bind free thiols on the surface of the cell.64 Thus, it is possible that within the intermediate dosing regime, a significant amount of PTL was restricted from the initial cell surface binding event, and the majority of PTL was trafficked intracellularly via endocytosis, resulting in significant inhibition of cytotoxicity by PTL. In support of this hypothesis, a sub-optimal combinatory dose of 2.5 μM free PTL with 2.5 μM PTL-loaded micelles resulted in the same cytotoxicity as 5 μM free PTL (~50% reduction in cell viability over 24 h, see Fig. 6), and significantly higher cytotoxicity than 2.5 μM free or micelle-loaded PTL individually (25 and 10% reduction in cell viability, respectively), as well as 5 μM micelle-loaded PTL (25%reduction in cell viability).
FIGURE 6.
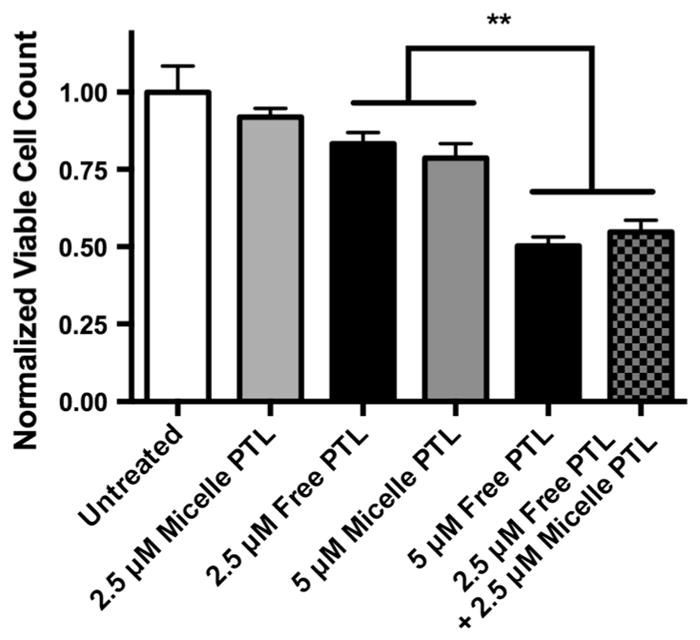
Cytotoxicity of 24 h treatments of free PTL and PTL-loaded PSMA-b-PS micelles. MV4-11 cells were simultaneously dosed with a combination of 2.5 μM free PTL and 2.5 μM PTL-loaded micelles (checkered gray/black bar) and incubated for 24 h. This combination dose was significantly more effective than 2.5 (not shown) and 5 μM PTL-loaded micelles (grey bar) and 2.5 μM free PTL (black bar) alone. Additionally, the combination dose rescued full cytotoxicity of 5 μM free PTL. n = 3 ± standard deviation, **p<0.01 (one-way ANOVA, Tukey’s post hoc).
In previous studies, PTL toxicity towards primary AML cells becomes irreversible after 6 h of treatment. 25,64 Similar toxicity kinetics were confirmed in the present study for free PTL with MV4-11 leukemia cells, as shown in Fig. 7. Cell viability was reduced by only 50% when incubated with 10 μM free PTL for 2 h prior to drug washout. However, cell viability was reduced by 75% when incubated with 10 μM free PTL for 4 or 8 h prior to drug washout (WO), which was the same effect as 24 h PTL treatments (no WO).
FIGURE 7.

Time-dependent induction of cytotoxicity by PTL-loaded PSMA-b-PS micelles. 10 μM PTL (black) and PTL-loaded micelles (grey) were incubated with MV4-11 cells for 2, 4, or 8 h before cells were washed with PBS, resuspended in drug-free media, and returned to the incubator. Cell counts were taken 24 h after the initial dose was administered. n = 3 ± standard deviation **p<0.01, ***p <0.001 free PTL vs. micelle PTL (2-way ANOVA, Tukey’s post hoc).
Interestingly, 10 μM PTL-loaded micelle treatments decreased cell viability more gradually with respect to incubation time. For 2 and 4 h PTL-loaded micelle incubations, cell viability was reduced by 30 and 40%, respectively, and only after 8 h did toxicity become equivalent to 24 h PTL-loaded micelle treatments (no WO). This behavior further supports a gradual release of PTL rather than an immediate burst release of drug that can affect cells extracellularly and intracellularly. Finally, although 10 μM PTL-loaded micelles achieved high toxicity over 24 h, the initial release of PTL by the micelles did not produce this effect, as further accumulation of PTL-loaded micelles was required to match cytotoxicity of free PTL.
To confirm that cytotoxicity was the result of NF-κB inhibition, as shown previously,24,64 MV4-11 cells were treated with either 10 μM free PTL or PTL-loaded micelles for 4 and 8 h before cell nuclear fractions were isolated and quantified for activated p65 NF-κB by ELISA. Figures 8a and 8b show a twofold reduction in activated NF-κB cells treated with free PTL for 4 and 8 h. In addition, Figs. 8a and 8b show a trend of increasing NF-κB inhibition over time for PTL-loaded micelles with a 1.2- and 2.5-fold reduction in activated NF-κB compared to untreated cells at 4 and 8 h. Overall, PTL-loaded micelles remained stable over treatment times, gradually released PTL, and resulted in MV4-11 cytotoxicity and NF-κB inhibition.
FIGURE 8.

NF-κB inhibition by free PTL and PTL-loaded PSMA-b-PS micelles. Activated NF-κB levels in MV4-11 cell nuclear extracts were quantified after 4 (a) and 8 h (b) incubations with free PTL (black) or PTL-loaded micelles (grey) by ELISA. There is a trend towards greater NF-κB inhibition with longer incubation time for PTL-loaded micelles, although this trend is not statistically significant. n = 3 ± standard deviation.
PTL-Loaded Micelles Overcome Glutathione-Induced Inhibition of Exofacial PTL-Thiol Binding to Recover Cytotoxicity In Vitro
As previously mentioned, PTL is known to bind efficiently to cell surface (exofacial) free thiols via Michael-like addition reactions.64 Furthermore, this extracellular binding event is essential to the apoptotic cascade that follows, including NF-κB inhibition.65 Skalska et al. used glutathione (GSH) to attenuate exofacial free thiols on lymphoma cell lines, which inhibited cell surface binding of PTL, diminished PTL inhibition of NF-κB, and reduced cytotoxicity of PTL treatment. Importantly, GSH pretreatment did not inhibit intracellular reactive oxygen species (ROS) generation by PTL, indicating that GSH activity was limited to the extracellular space. In this study, MV4-11 cells were pretreated with 5 mM GSH for 2 h prior to 10 μM PTL or PTL-loaded micelle treatments. Figure 9 shows the normalized cell counts 22 h after PTL dosing. A twofold loss in free PTL efficacy was observed when MV4-11 cells were pretreated with GSH. Interestingly, no loss in efficacy was seen for PTL-loaded micelles following GSH pretreatment. This result may indicate that micelle-mediated delivery and cytotoxicity of PTL is independent of exofacial PTL binding at optimal doses (10 μM). Also, Fig. 9 shows efficient micelle-mediated PTL delivery by endocytic pathways, and the resulting cytotoxicity suggests that PTL is not altered by the physiological conditions of endosomal/lysosomal pathways upon intracellular PTL release.
FIGURE 9.
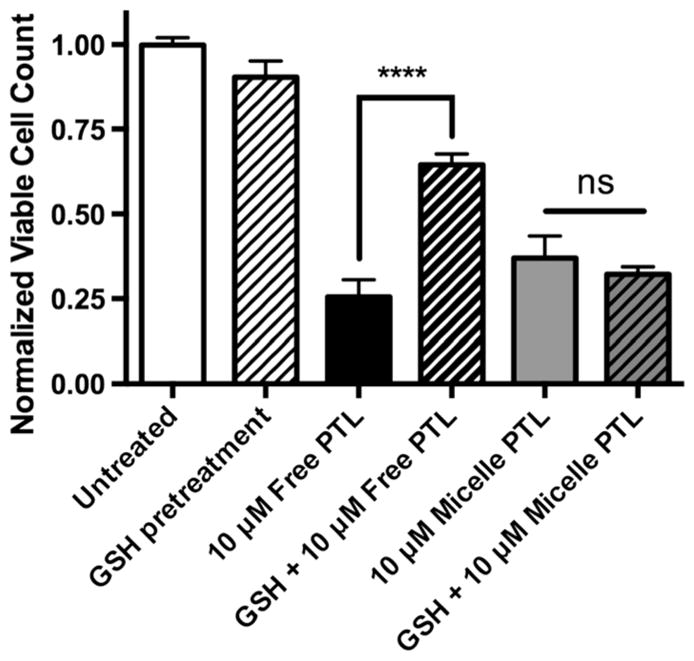
Inhibition of free PTL cytotoxicity towards MV4-11 cells by pre-incubation with glutathione (GSH). Cells were incubated with 5 mM GSH for 2 h prior to incubation with 10 μM free PTL (black) or PTL-loaded loaded micelles (grey). GSH surface binding decreased the efficacy of free PTL by over twofold, but had no effect on the ability of PTL-loaded micelles to induce cell death over 22 h incubations. Cell counts normalized to untreated control. n = 3 ± standard deviation, ****p<0.0001 (2-way ANOVA, Tukey’s post hoc testing).
When examining the effect of GSH pretreatment on PTL cytotoxicity, care was taken to remove GSH after the 2 h pretreatment because the reactive methylene group of PTL binds directly to sulfhydrl groups present on GSH molecules. Thus, by removing excess GSH that has not participated in cell surface thiol depletion, there is no reduction in the effective PTL dose delivered. As expected, when GSH was not washed out after pretreatment, free PTL did not affect MV4-11 viable cell counts over 48 h treatment times (see Supplemental Figure 2). Interestingly, under the same conditions, PTL-loaded micelles significantly reduced the viable cell count by 35% compared to untreated control groups over 48 h incubations. This is further evidence that PSMA-b-PS NPs sequester PTL in the micelle core, protecting PTL from both cells and deactivating chemicals (such as GSH here). Additionally, micelles remain stable in cell suspensions and retain a significant amount of loaded PTL prior to uptake by AML cells, and PTL that is released intracellularly remains bioactive.
The cascade of events following exofacial binding of PTL has not been fully characterized. However, it has been hypothesized that the interaction of PTL with extracellular thioredoxin activates ion channels on the cell membrane and influences lipid raft-mediated internalization, causing changes in intracellular redox states.64 In this respect, micelle-mediated delivery of PTL may induce intracellular oxidative stress more efficiently than free PTL, as PTL-loaded micelles were able to overcome inhibition of exofacial thiol binding without loss of efficacy towards AML cells. It is not known how GSH pretreatment influences the ability of PTL to enter cells quantitatively, although it has been shown that PTL, with or without GSH pretreatment, increases intracellular reactive oxygen species, which is an indication of PTL internalization. Similarly, PSMA-b-PS micelle internalization was not affected by GSH pretreatment, as no difference in intracellular fluorescence was observed for FL-PSMA-b-PS micelle treated cells with or without GSH pretreatment (data not shown).
As stated previously, GSH pretreatments inhibit the ability of free PTL to inhibit NF-κB activation. To confirm that this mechanism is true of micelle-mediated delivery of PTL, activated NF-κB protein was quantified for MV4-11 cells that were pretreated with 5 mM GSH for 2 h prior to PTL and PTL-loaded micelle doses. Activated NF-κB levels were not decreased by either free PTL or PTL-loaded micelles when cells were pretreated with GSH (see Supplemental Figure 3). Therefore, PTL must interact with exofacial free thiols to inhibit NF-κB activation regardless of the mode of entry into the cell. However, unlike free PTL, PTL-loaded micelles showed no loss in cytotoxicity when exofacial interaction between free thiols and PTL was restricted. For such cases, PTL-loaded micelles may induce apoptosis by other mechanisms described for PTL, such as increased oxidative stress or proapoptotic p53 activation.
PTL-Loaded PSMA-b-PS Micelles as an Improved Anti-leukemia Chemotherapeutic
PTL has great advantages over typical anti-leukemia agents because of its ability to eliminate LSCs, while sparing normal hematopoietic cells. However, like many hydrophobic small molecule drugs, PTL is plagued by poor bioavailability and inefficacy in vivo due to rapid blood clearance. PSMA-b-PS micelles possess many desirable characteristics for avoidance of obstacles presented by biological systems that limit drug efficacy in vivo. In this study, it has been demonstrated that PTL can be efficiently loaded within PSMA-b-PS micelles. Further, PTL-loaded PSMA-b-PS micelles are taken up by MV4-11 cells via endocytosis and release PTL gradually over time. Finally 10 μM PTL-loaded micelles are cytotoxic towards AML cells even in the absence of exofacial interaction between free thiols and PTL. Therefore, the favorable physiochemical properties of PTL-loaded PSMA-b-PS NPs, along with demonstrated therapeutic promise in vitro, may lead to development of a viable anti-leukemia agent in vivo.
NPs have been useful in circumventing multidrug resistant (MDR) efflux pumps that limit the intracellular concentration and subsequent therapeutic efficacy of hydrophobic small molecule drugs.14,72 For cells that utilize p-glycoprotien (PGP) and other ATP-binding cassette (ABC) transporters, NP-mediated endocytosis and intracellular trafficking has been beneficial for hydrophobic drug accumulation and cytotoxicity by circumventing transporter efflux mechanisms.15,62 For example, delivery of doxorubicin (DOX) via PSMA-b-PS micelles enhanced drug retention by MDR ovarian cancer cells and ultimately led to increased cytotoxicity compared to free DOX treatments.2 Several types of MDR mechanisms have been reported in addition to cell membrane transporters, including production of intracellular detoxifying agents,61 mutations to apoptotic pathways,48 and alterations to DNA replication mechanisms.4 Both transport-based and apoptosis related MDR mechanisms have been reported in multiple leukemia subtypes, including AML.20,59,68 In particular, MV4-11 cells have exhibited resistance to DOX,6 although the exact mechanism of this resistance has not been characterized. To determine whether micelle-mediated intracellular delivery of DOX could overcome this apparent resistance, DOX-loaded PSMA-b-PS micelles were prepared as previously described.2 Subsequently, MV4-11 cells were treated with free and micelle-loaded DOX formulations to examine potential benefits of micelle-mediated drug delivery with regard to MDR. However, no significant differences in cytotoxicity were observed between free DOX and DOX-loaded micelles over 48 h with a range of DOX concentrations (0.1–50 μM, data not shown). These data may indicate that MV-411 cells possess resistances to drug-induced DNA damage instead of, or in addition to, MDR membrane transporters. Interestingly, both DOX and fludarabine are less toxic towardsMV4-11 cells compared to other AML cell lines,6 which offers some support that MDR efflux pumps are not solely responsible for the observed drug resistance, as DOX is a PGP substrate (i.e., is effluxed by PGP) while fludarabine is not.20 While micelle-mediated delivery of hydrophobic anticancer agents can improve intracellular accumulation of drugs, other MDR mechanisms can still limit the therapeutic efficacy of the drug by altering cell-cycle machinery and apoptotic pathways.
Aside from evasion of MDR efflux mechanisms, PSMA-b-PS micelles offer advantages for systemic drug delivery. They are an optimal size (~40 nm) to avoid renal and hepatic filtration as NPs smaller than 10 nm can pass through the glomerular capillary wall resulting in elimination by the kidneys.12 In addition, NPs larger than 100 nm show greater clearance by opsonization, phagocytic cell recognition, macrophage activation, and hepatic uptake.1,54 Thus, PSMA-b-PS NPs, which possess a hydrodynamic diameter of ~40 nm, are an ideal size for avoidance of clearance by the RES. In addition to size, NP surface charge is an important property that can affect circulation time. Generally, greater charge (positive or negative) increases opsonization54 and macrophage scavenging.12 However, negatively charged micelles have shown less non-specific internalization and accumulation within organs like the liver and spleen when compared to neutral and positively charged micelles.75 Furthermore, PSMA macromolecules are known to bind serum albumin non-covalently,42 and although binding of serum proteins usually correlates with opsonization and clearance, albumin binding does not result in faster clearance of micelles,1 and has actually increased the circulation time and stability of drug-loaded PSMA NPs.46,49,50 With the pharmacokinetics primarily depending on the physical dimensions and surface characteristics of the NP, PSMA-b-PS micelles present a high likelihood for long circulation times in vivo but future studies supporting this claim are necessary.
With drug efficacy in the forefront, there are characteristics of PTL-loaded PSMA-b-PS micelles that can be optimized for success as a leukemia therapeutic. Although loading of PTL within PSMA-b-PS micelles markedly enhanced the aqueous solubility of PTL (over fivefold), release of PTL was quite rapid and uptake of PSMA-b-PS micelles by AML cells was gradual. Thus, efficacy may be lost due to premature PTL release before reaching target cells. Fortunately, the PTL-loaded micelles that were taken up by cells were efficacious without exofacial binding, proving feasibility of PTL-loaded micelle therapy in the absence of free extracellular PTL. Still, functionalization of PTL-loaded micelles with cell-specific targeting molecules can further increase the likelihood of successful therapy by ensuring loaded NPs reach cells of interest. In this respect, active targeting can balance extended NP circulation promoted by PSMA coronas, and achieve more robust therapeutic effects of PTL-loaded PSMA-b-PS micelles in vivo. In addition, incorporation of LSC-specific targeting molecules such as antibodies or peptides may allow for selective treatment of a traditionally evasive leukemia cell sub-type that is responsible for poor long-term survivability. Of the known LSC specific surface markers,3 decoration of PSMA-b-PS micelles with CD123 antibodies is a particularly attractive option because the 7G3 clone of anti-CD123 alone inhibits proliferation, engraftment, and function of LSCs.37 Thus, incorporation of anti-CD123 antibodies on the surface of PSMA-b-PS micelles can combine two LSC-targeted treatment options that are currently in FDA clinical trials22 by localizing an NF-κB inhibitor with anti-CD123 antibodies. Towards this goal, PSMA-b-PS micelle functionalization can be achieved using the carboxylic acid moieties of the preserved RAFT CTA or PSMA units of the micelle corona. Various bioconjugation techniques are currently being explored to realize this targeting strategy for PTL-loaded micelles to make this therapeutic approach more effective and specific to LSCs.
CONCLUSIONS
PSMA-b-PS micelles were utilized to increase the aqueous solubility of PTL by over fivefold, and were stable in solution allowing extended release of PTL over 24 h. NPs controlled drug-cell interactions and promoted intracellular accumulation of PTL-loaded NPs through clathrin-mediated endocytosis, resulting in potent cytotoxicity to AML cells in vitro. Although free PTL was more potent than PTL-loaded micelles, the feasibility of the more bioavailable PTL-loaded micelle formulation was demonstrated. In addition to robust PTL loading, PSMA-b-PS micelles possess attractive physiochemical properties and readily modifiable coronas, making these NPs a promising PTL delivery system. The ability to eliminate LSCs in vivo may significantly enhance survivability for AML, which is currently one of the most lethal and prevalent forms of leukemia.
Supplementary Material
Acknowledgments
The authors gratefully acknowledge Cheryl Corbett for helpful advice on in vitro assays and Karen Bentley, M.S., of the University of Rochester Medical Center Electron Microscopy Shared Laboratory for sample preparation and EM image acquisition. This work was supported by Alex’s Lemonade Stand Foundation for Childhood Cancer, the I Care I Cure Foundation, and the Leukemia Research Foundation (D.S.W.B.), and the National Science Foundation Graduate Research Fellowship Program (M.P.B.).
Biography
Danielle Benoit is the James P. Wilmot Distinguished Assistant Professor within the Department of Biomedical Engineering with appointments also in Chemical Engineering and the Center for Musculoskeletal Research at the University of Rochester. She directs the Therapeutic Biomaterials Laboratory, which specializes in the rational design of polymeric materials for regenerative medicine and drug delivery applications. Her work has provided insights into the translation of tissue engineering strategies for bone allograft repair, development of pH-responsive nanoparticles for nucleic acid and small molecule drug delivery, and novel targeting strategies for bone-specific delivery of therapeutics. Prof. Benoit has received numerous awards for her research program including the 2015 Young Innovator Award in Cellular and Molecular Bioengineering, an NSF CAREER Award, and Alex’s Lemonade Stand Young Investigator Award. Prof. Benoit received her undergraduate degree in Biological Engineering from the University of Maine and M.S. and Ph.D. in Chemical Engineering from the University of Colorado, where she was mentored by Dr. Kristi Anseth. She then trained at the University of Washington where she was a Damon Runyon Cancer Research Foundation Postdoctoral Fellow, working with Drs. Patrick Stayton and Allan Hoffman. Prof. Benoit joined the faculty at the University of Rochester in 2010.

Footnotes
ELECTRONIC SUPPLEMENTARY MATERIAL
The online version of this article (doi:10.1007/s12195-015-0391-x) contains supplementary material, which is available to authorized users.
CONFLICT OF INTEREST
Michael P. Baranello, Louisa Bauer, Craig T. Jordan, Ph.D., and Danielle S.W. Benoit, Ph.D. each declare no conflicts of interest associated with this work.
ETHICAL STANDARDS
Neither animal studies nor human studies were carried out by the authors for data reported herein.
References
- 1.Alexis F, Pridgen E, Molnar LK, Farokhzad OC. Factors affecting the clearance and biodistribution of polymeric nanoparticles. Mol Pharm. 2008;5:505–515. doi: 10.1021/mp800051m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Baranello MP, Bauer L, Benoit DSW. Poly (styrene-alt-maleic anhydride)-based diblock copolymer micelles exhibit versatile hydrophobic drug loading, drug-dependent release, and internalization by multidrug resistant ovarian cancer cells. Biomacromolecules. 2014;15:2629–2641. doi: 10.1021/bm500468d. [DOI] [PubMed] [Google Scholar]
- 3.Becker MW, Jordan CT. Leukemia stem cells in 2010: current understanding and future directions. Blood Rev. 2011;25:75–81. doi: 10.1016/j.blre.2010.11.001. [DOI] [PubMed] [Google Scholar]
- 4.Boshoff HIM, Reed MB, Barry CE, Mizrahi V. Dnae2 polymerase contributes to in vivo survival and the emergence of drug resistance in mycobacterium tuberculosis. Cell. 2003;113:183–193. doi: 10.1016/s0092-8674(03)00270-8. [DOI] [PubMed] [Google Scholar]
- 5.Chidambaram M, Manavalan R, Kathiresan K. Nanotherapeutics to overcome conventional cancer chemotherapy limitations. J Pharm Pharm Sci. 2011;14:67–77. doi: 10.18433/j30c7d. [DOI] [PubMed] [Google Scholar]
- 6.Colado E, Alvarez-Fernandez S, Maiso P, et al. The effect of the proteasome inhibitor bortezomib on acute myeloid leukemia cells and drug resistance associated with the cd34+ immature phenotype. Haematologica. 2008;93:57–66. doi: 10.3324/haematol.11666. [DOI] [PubMed] [Google Scholar]
- 7.Corrie PG. Cytotoxic chemotherapy: clinical aspects. Medicine. 2011;39:717–722. [Google Scholar]
- 8.Curry EA, III, Murry DJ, Yoder C, et al. Phase i dose escalation trial of feverfew with standardized doses of parthenolide in patients with cancer. Invest New Drugs. 2004;22:299–305. doi: 10.1023/B:DRUG.0000026256.38560.be. [DOI] [PubMed] [Google Scholar]
- 9.Dausend J, Musyanovych A, Dass M, et al. Uptake mechanism of oppositely charged fluorescent nanoparticles in hela cells. Macromol Biosci. 2008;8:1135–1143. doi: 10.1002/mabi.200800123. [DOI] [PubMed] [Google Scholar]
- 10.Dausend J, Musyanovych A, Dass M, et al. Uptake mechanism of oppositely charged fluorescent nanoparticles in hela cells. Macromol Biosci. 2008;8:1135–1143. doi: 10.1002/mabi.200800123. [DOI] [PubMed] [Google Scholar]
- 11.Davis ME, Chen Z, Shin DM. Nanoparticle therapeutics: an emerging treatment modality for cancer. Nat Rev Drug Discov. 2008;7:771–782. doi: 10.1038/nrd2614. [DOI] [PubMed] [Google Scholar]
- 12.Davis ME, Chen ZG, Shin DM. Nanoparticle therapeutics: an emerging treatment modality for cancer. Nat Rev Drug Discov. 2008;7:771–782. doi: 10.1038/nrd2614. [DOI] [PubMed] [Google Scholar]
- 13.de Mesy Jensen KL. Tech Sample CY-1. American Society of Clinical Pathologists; 1987. “Pop-off” Technique for FNA Smears for Diagnostic Electron Microscopy. [Google Scholar]
- 14.Dong X, Mattingly CA, Tseng MT, et al. Doxorubicin and paclitaxel-loaded lipid-based nanoparticles overcome multidrug resistance by inhibiting p-glycoprotein and depleting atp. Cancer Res. 2009;69:3918–3926. doi: 10.1158/0008-5472.CAN-08-2747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dong XW, Mattingly CA, Tseng MT, et al. Doxorubicin and paclitaxel-loaded lipid-based nanoparticles overcome multidrug resistance by inhibiting p-glycoprotein and depleting atp. Cancer Res. 2009;69:3918–3926. doi: 10.1158/0008-5472.CAN-08-2747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Douglas KL, Piccirillo CA, Tabrizian M. Cell line-dependent internalization pathways and intracellular trafficking determine transfection efficiency of nanoparticle vectors. Eur J Pharm Biopharm. 2008;68:676–687. doi: 10.1016/j.ejpb.2007.09.002. [DOI] [PubMed] [Google Scholar]
- 17.Garcia-Pineres AJ, Castro V, Mora G, et al. Cysteine 38 in p65/nf-kappa b plays a crucial role in DNA binding inhibition by sesquiterpene lactones. J Biol Chem. 2001;276:39713–39720. doi: 10.1074/jbc.M101985200. [DOI] [PubMed] [Google Scholar]
- 18.Garcia-Pineres AJ, Lindenmeyer MT, Merfort I. Role of cysteine residues of p65/nf-kappa b on the inhibition by the sesquiterpene lactone parthenolide and-n-ethyl maleimide, and on its transactivating potential. Life Sci. 2004;75:841–856. doi: 10.1016/j.lfs.2004.01.024. [DOI] [PubMed] [Google Scholar]
- 19.Gereige LM, Mikkola HK. DNA methylation is a guardian of stem cell self-renewal and multipotency. Nat Genet. 2009;41:1164–1166. doi: 10.1038/ng1109-1164. [DOI] [PubMed] [Google Scholar]
- 20.Gottesman MM, Fojo T, Bates SE. Multidrug resistance in cancer: role of atp-dependent transporters. Nat Rev Cancer. 2002;2:48–58. doi: 10.1038/nrc706. [DOI] [PubMed] [Google Scholar]
- 21.Guo S, Huang L. Nanoparticles containing insoluble drug for cancer therapy. Biotechnol Adv. 2014;32:778–788. doi: 10.1016/j.biotechadv.2013.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Guzman ML, Allan JN. Concise review: leukemia stem cells in personalized medicine. Stem Cells. 2014;32:844–851. doi: 10.1002/stem.1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Guzman ML, Neering SJ, Upchurch D, et al. Nuclear factor-kappab is constitutively activated in primitive human acute myelogenous leukemia cells. Blood. 2001;98:2301–2307. doi: 10.1182/blood.v98.8.2301. [DOI] [PubMed] [Google Scholar]
- 24.Guzman ML, Rossi RM, Karnischky L, et al. The sesquiterpene lactone parthenolide induces apoptosis of human acute myelogenous leukemia stem and progenitor cells. Blood. 2005;105:4163–4169. doi: 10.1182/blood-2004-10-4135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Guzman ML, Rossi RM, Neelakantan S, et al. An orally bioavailable parthenolide analog selectively eradicates acute myelogenous leukemia stem and progenitor cells. Blood. 2007;110:4427–4435. doi: 10.1182/blood-2007-05-090621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Han JT, Silcock P, McQuillan AJ, Bremer P. Preparation and characterization of poly(styrene-alt-maleic acid)-b-polystyrene block copolymer self-assembled nanoparticles. Colloid Polym Sci. 2008;286:1605–1612. [Google Scholar]
- 27.He W, Bennett MJ, Luistro L, et al. Discovery of siRNA lipid nanoparticles to transfect suspension leukemia cells and provide in vivo delivery capability. Mol Ther. 2013;22:359–370. doi: 10.1038/mt.2013.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Heath JR, Davis ME. Nanotechnology and cancer. Annu Rev Med. 2008;59:251–265. doi: 10.1146/annurev.med.59.061506.185523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hed J, Hallden G, Johansson SGO, Larsson P. The use of fluorescence quenching in flow cytofluorometry to measure the attachment and ingestion phases in phagocytosis in peripheral blood without prior cell separation. J Immunol Methods. 1987;101:119–125. doi: 10.1016/0022-1759(87)90224-9. [DOI] [PubMed] [Google Scholar]
- 30.Hehner SP, Hofmann TG, Droge W, Schmitz ML. The antiinflammatory sesquiterpene lactone parthenolide inhibits nf-kappa b by targeting the i kappa b kinase complex. J Immunol. 1999;163:5617–5623. [PubMed] [Google Scholar]
- 31.Heidel JD, Davis ME. Clinical developments in nanotechnology for cancer therapy. Pharm Res. 2011;28:187–199. doi: 10.1007/s11095-010-0178-7. [DOI] [PubMed] [Google Scholar]
- 32.Horne RW, Wildy P. Virus structure revealed by negative staining. Adv Virus Res. 1963;10:101–170. doi: 10.1016/s0065-3527(08)60698-3. [DOI] [PubMed] [Google Scholar]
- 33.Huang X, Schwind S, Yu B, et al. Targeted delivery of microrna-29b by transferrin-conjugated anionic lipopolyplex nanoparticles: a novel therapeutic strategy in acute myeloid leukemia. Clin Cancer Res. 2013;19:2355–2367. doi: 10.1158/1078-0432.CCR-12-3191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Huth US, Schubert R, Peschka-Suss R. Investigating the uptake and intracellular fate of ph-sensitive liposomes by flow cytometry and spectral bio-imaging. J Control Release. 2006;110:490–504. doi: 10.1016/j.jconrel.2005.10.018. [DOI] [PubMed] [Google Scholar]
- 35.Jaggi AS, Singh N. Mechanisms in cancer-chemotherapeutic drugs-induced peripheral neuropathy. Toxicology. 2012;291:1–9. doi: 10.1016/j.tox.2011.10.019. [DOI] [PubMed] [Google Scholar]
- 36.Jain KK. Nanomedicine: application of nanobiotechnology in medical practice. Med Princ Pract. 2008;17:89–101. doi: 10.1159/000112961. [DOI] [PubMed] [Google Scholar]
- 37.Jin LQ, Lee EM, Ramshaw HS, et al. Monoclonal antibody-mediated targeting of cd123, il-3 receptor alpha chain, eliminates human acute myeloid leukemic stem cells. Cell Stem Cell. 2009;5:31–42. doi: 10.1016/j.stem.2009.04.018. [DOI] [PubMed] [Google Scholar]
- 38.Jordan CT. Unique molecular and cellular features of acute myelogenous leukemia stem cells. Leukemia. 2002;16:559–562. doi: 10.1038/sj.leu.2402446. [DOI] [PubMed] [Google Scholar]
- 39.Jordan CT. The leukemic stem cell. Best Pract Res Clin Haematol. 2007;20:13–18. doi: 10.1016/j.beha.2006.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kateb B, Chiu K, Black KL, et al. Nanoplatforms for constructing new approaches to cancer treatment, imaging, and drug delivery: what should be the policy? Neuroimage. 2011;54(Suppl 1):S106–S124. doi: 10.1016/j.neuroimage.2010.01.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kim S, Shi Y, Kim JY, Park K, Cheng JX. Overcoming the barriers in micellar drug delivery: loading efficiency, in vivo stability, and micelle-cell interaction. Expert Opin Drug Deliv. 2010;7:49–62. doi: 10.1517/17425240903380446. [DOI] [PubMed] [Google Scholar]
- 42.Kobayashi A, Oda T, Maeda H. Protein binding of macromolecular anticancer agent SMANCS: characterization of poly(styrene-co-maleic acid) derivatives as an albumin binding ligand. J Bioact Compat Polym Biomed Appl. 1988;3:319–333. [Google Scholar]
- 43.Kolev JN, O’Dwyer KM, Jordan CT, Fasan R. Discovery of potent parthenolide-based antileukemic agents enabled by late-stage p450-mediated c-h functionalization. ACS Chem Biol. 2014;9:164–173. doi: 10.1021/cb400626w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kwok BH, Koh B, Ndubuisi MI, Elofsson M, Crews CM. The anti-inflammatory natural product parthenolide from the medicinal herb feverfew directly binds to and inhibits ikappab kinase. Chem Biol. 2001;8:759–766. doi: 10.1016/s1074-5521(01)00049-7. [DOI] [PubMed] [Google Scholar]
- 45.Lapidot T, Sirard C, Vormoor J, et al. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature. 1994;367:645–648. doi: 10.1038/367645a0. [DOI] [PubMed] [Google Scholar]
- 46.Larson N, Greish K, Bauer H, Maeda H, Ghandehari H. Synthesis and evaluation of poly(styrene-co-maleic acid) micellar nanocarriers for the delivery of tanespimycin. Int J Pharm. 2011;420:111–117. doi: 10.1016/j.ijpharm.2011.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lin LY, Lee NS, Zhu JH, et al. Tuning core vs. shell dimensions to adjust the performance of nanoscopic containers for the loading and release of doxorubicin. J Control Release. 2011;152:37–48. doi: 10.1016/j.jconrel.2011.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Liu YY, Han TY, Giuliano AE, Cabot MC. Ceramide glycosylation potentiates cellular multidrug resistance. FASEB J. 2001;15:719–730. doi: 10.1096/fj.00-0223com. [DOI] [PubMed] [Google Scholar]
- 49.Maeda H. Smancs and polymer-conjugated macromolecular drugs: advantages in cancer chemotherapy. Adv Drug Deliv Rev. 2001;46:169–185. doi: 10.1016/s0169-409x(00)00134-4. [DOI] [PubMed] [Google Scholar]
- 50.Maeda H, Bharate GY, Daruwalla J. Polymeric drugs for efficient tumor-targeted drug delivery based on EPR-effect. Eur J Pharm Biopharm. 2009;71:409–419. doi: 10.1016/j.ejpb.2008.11.010. [DOI] [PubMed] [Google Scholar]
- 51.Miyata K, Christie RJ, Kataoka K. Polymeric micelles for nano-scale drug delivery. React Funct Polym. 2011;71:227–234. [Google Scholar]
- 52.Moad G, Rizzardo E, Thang SH. Living radical polymerization by the raft process. Aust J Chem. 2005;58:379–410. [Google Scholar]
- 53.Myhren L, Nilssen IM, Nicolas V, Doskeland SO, Barratt G, Herfindal L. Efficacy of multi-functional liposomes containing daunorubicin and emetine for treatment of acute myeloid leukaemia. Eur J Pharm Biopharm. 2014;88:186–193. doi: 10.1016/j.ejpb.2014.04.002. [DOI] [PubMed] [Google Scholar]
- 54.Owens DE, III, Peppas NA. Opsonization, biodistribution, and pharmacokinetics of polymeric nanoparticles. Int J Pharm. 2006;307:93–102. doi: 10.1016/j.ijpharm.2005.10.010. [DOI] [PubMed] [Google Scholar]
- 55.Pei S, Minhajuddin M, Callahan KP, et al. Targeting aberrant glutathione metabolism to eradicate human acute myelogenous leukemia cells. J Biol Chem. 2013;288:33542–33558. doi: 10.1074/jbc.M113.511170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rejman J, Bragonzi A, Conese M. Role of clathrinand caveolae-mediated endocytosis in gene transfer mediated by lipo- and polyplexes. Mol Ther. 2005;12:468–474. doi: 10.1016/j.ymthe.2005.03.038. [DOI] [PubMed] [Google Scholar]
- 57.Rodriguez VB, Henry SM, Hoffman AS, Stayton PS, Li XD, Pun SH. Encapsulation and stabilization of indocyanine green within poly(styrene-altmaleic anhydride) block-poly(styrene) micelles for near-infrared imaging. J Biomed Opt. 2008;13:014025. doi: 10.1117/1.2834296. [DOI] [PubMed] [Google Scholar]
- 58.Rodriguez VB, Henry SM, Hoffman AS, Stayton PS, Li XD, Pun SH. Encapsulation and stabilization of indocyanine green within poly(styrene-altmaleic anhydride) block-poly(styrene) micelles for near-infrared imaging. J Biomed Opt. 2008;13:014025. doi: 10.1117/1.2834296. [DOI] [PubMed] [Google Scholar]
- 59.Ross DD. Novel mechanisms of drug resistance in leukemia. Leukemia. 2000;14:467–473. doi: 10.1038/sj.leu.2401694. [DOI] [PubMed] [Google Scholar]
- 60.Sarkar K, Kruhlak MJ, Erlandsen SL, Shaw S. Selective inhibition by rottlerin of macropinocytosis in monocyte-derived dendritic cells. Immunology. 2005;116:513–524. doi: 10.1111/j.1365-2567.2005.02253.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Schuetz EG, Beck WT, Schuetz JD. Modulators and substrates of p-glycoprotein and cytochrome p4503a coordinately up-regulate these proteins in human colon carcinoma cells. Mol Pharmacol. 1996;49:311–318. [PubMed] [Google Scholar]
- 62.Siddiqui A, Gupta V, Liu YY, Nazzal S. Doxorubicin and MBO-asGCS oligonucleotide loaded lipid nanoparticles overcome multidrug resistance in adriamycin resistant ovarian cancer cells (NCI/ADR-RES) Int J Pharm. 2012;431:222–229. doi: 10.1016/j.ijpharm.2012.04.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Singh R, Lillard JW., Jr Nanoparticle-based targeted drug delivery. Exp Mol Pathol. 2009;86:215–223. doi: 10.1016/j.yexmp.2008.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Skalska J, Brookes PS, Nadtochiy SM, et al. Modulation of cell surface protein free thiols: a potential novel mechanism of action of the sesquiterpene lactone parthenolide. PLoS One. 2009;4:e8115. doi: 10.1371/journal.pone.0008115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Skalska J, Brookes PS, Nadtochiy SM, et al. Modulation of cell surface protein free thiols: a potential novel mechanism of action of the sesquiterpene lactone parthenolide. PLoS One. 2009;4:e8115. doi: 10.1371/journal.pone.0008115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Society, A.C. Cancer Facts & Figures 2014. Atlanta: American Cancer Society; 2014. [Google Scholar]
- 67.Sweeney CJ, Mehrotra S, Sadaria MR, et al. The sesquiterpene lactone parthenolide in combination with docetaxel reduces metastasis and improves survival in a xenograft model of breast cancer. Mol Cancer Ther. 2005;4:1004–1012. doi: 10.1158/1535-7163.MCT-05-0030. [DOI] [PubMed] [Google Scholar]
- 68.Tallman MS. Drug therapy for acute myeloid leukemia. Blood. 2005;106:2243. doi: 10.1182/blood-2005-01-0178. [DOI] [PubMed] [Google Scholar]
- 69.Torchilin VP. Structure and design of polymeric surfactant-based drug delivery systems. J Control Release. 2001;73:137–172. doi: 10.1016/s0168-3659(01)00299-1. [DOI] [PubMed] [Google Scholar]
- 70.Wang LH, Rothberg KG, Anderson RG. Misassembly of clathrin lattices on endosomes reveals a regulatory switch for coated pit formation. J Cell Biol. 1993;123:1107–1117. doi: 10.1083/jcb.123.5.1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wen J, You KR, Lee SY, Song CH, Kim DG. Oxidative stress-mediated apoptosis—the anticancer effect of the sesquiterpene lactone parthenolide. J Biol Chem. 2002;277:38954–38964. doi: 10.1074/jbc.M203842200. [DOI] [PubMed] [Google Scholar]
- 72.Wong HL, Bendayan R, Rauth AM, Xue HY, Babakhanian K, Wu XY. A mechanistic study of enhanced doxorubicin uptake and retention in multidrug resistant breast cancer cells using a polymer-lipid hybrid nanoparticle system. J Pharmacol Exp Ther. 2006;317:1372–1381. doi: 10.1124/jpet.106.101154. [DOI] [PubMed] [Google Scholar]
- 73.Wong JE, Duchscherer TM, Pietraru G, Cramb DT. Novel fluorescence spectral deconvolution method for determination of critical micelle concentrations using the fluorescence probe prodan. Langmuir. 1999;15:6181–6186. [Google Scholar]
- 74.Wu Y, Ihme S, Feuring-Buske M, et al. A core-shell albumin copolymer nanotransporter for high capacity loading and two-step release of doxorubicin with enhanced anti-leukemia activity. Adv Healthc Mater. 2013;2:884–894. doi: 10.1002/adhm.201200296. [DOI] [PubMed] [Google Scholar]
- 75.Yamamoto Y, Nagasaki Y, Kato Y, Sugiyama Y, Kataoka K. Long-circulating poly(ethylene glycol)-poly(d, l-lactide) block copolymer micelles with modulated surface charge. J Control Release. 2001;77:27–38. doi: 10.1016/s0168-3659(01)00451-5. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.