

Dilemmas arise concerning how to design studies aimed at preventing or slowing Huntington’s disease (HD) in mutation-positive presymptomatic individuals. HD is an autosomal dominant neurodegenerative disease. Each child of an HD gene expansion carrier (HDGEC) has a 50% chance of inheriting the mutation but the clinical symptoms typically only appear in middle age. In the absence of clinical symptoms or having obtained predictive genetic testing, such a person is considered at risk of HD. While genotyping for the HD mutation approaches 100% accuracy,1 the decision to undergo predictive genetic testing for a debilitating and stigmatized, ultimately fatal disease is highly personal.2 Predictive testing rates in HD are <20% in Europe3 and only 5–7% in the US.
We now know that the underlying neurodegeneration starts at least a decade before clinically overt symptoms appear,4, 5 with the implication that it may be too late to modify the disease once signs and symptoms are apparent. Several potential HD therapeutics are now reaching clinical development,6 raising ethical questions of whether clinical trials should include at risk individuals who choose not to undergo testing. Within this context, there is an important distinction in the way that the genetic status of an individual is identified. Whereas ‘predictive testing’ is a personalized feedback process initiated by a person who wants to know if he or she carries the HD gene expansion,2 ‘research genotyping’ is only done for the purposes of a study; results are not disclosed and are usually only made accessible in the form of aggregate, anonymized databases.
We examine here three possible trial designs for interventions in premanifest HD using the four basic ethical principles (autonomy, beneficence, non-maleficence and justice).7,8
Possible trial designs
Three possible trial designs exist: transparent, concealed-sorted and concealed-unsorted (Figure 1). For each, the results of research genotyping done in the context of the trial is not disclosed to the participants, even to those who choose to have predictive testing.
Figure 1. Possible trial designs.
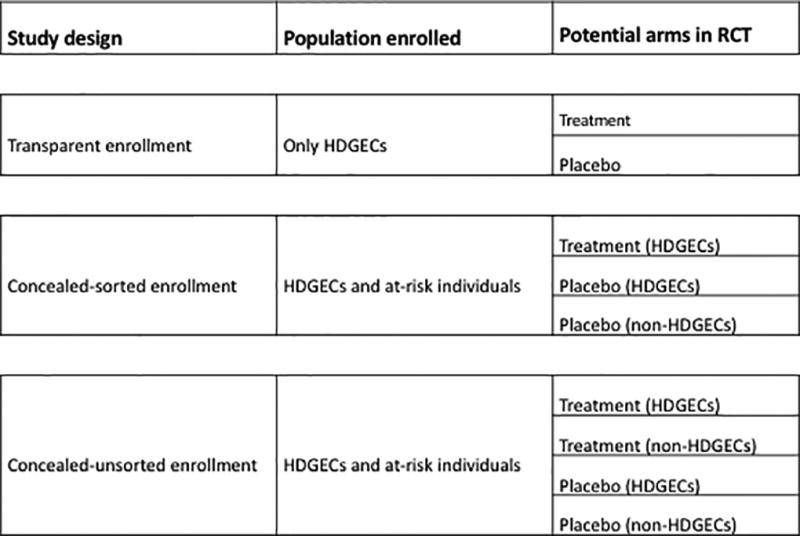
HDGEC: Huntington’s disease gene expansion carriers
We define transparent trials as those in which only individuals who are known beforehand to be HDGECs can participate. Under this model, standard randomized controlled trials can be conducted, with known HDGECs randomized to receive either experimental treatment or placebo. By contrast, trials with concealed enrollment (sorted or unsorted) allow for the participation of both known HDGECs and interested at-risk individuals who are genotyped for the study but not informed of their genetic status and may or may not be mutation-positive. The concealed studies would thus enroll: 1) HDGECs who know their genetic status; 2) HDGECs who choose not to know their genetic status; and 3) non-HDGECs who choose not to know their genetic status. In concealed-sorted designs there are three arms – non HDGECs can only be assigned to placebo/sham treatment. By contrast, the concealed-unsorted design allows all participants (HDGECs and non-HDGECs) to be randomized to receive either treatment or placebo. Unsorted designs are only possible when the safety of test treatment is well-established and does not pose undue risk to the non-HDGECs that are healthy not at risk individuals.
Concealed designs allow for placebo comparisons. However, non-HDGECs would be long-term study participants experiencing study burden (e.g., risks of trial procedures) While non-HDGECs will contribute to the safety data, their outcomes will never meaningfully contribute to the understanding of efficacy. The non-HDGEC contribution to the understanding of safety is also limited because in the sorted design non-HDGECs are only exposed to placebo, and unsorted studies can only be conducted when the safety of test treatment is already well-established. As autonomous individuals, participants can choose this path. They are helping to advance science, but the informed consent should disclose that they will be research-genotyped, and that those who are HDGEC would be randomized to receive the experimental treatment or placebo, while those who are non-HDGEC would all receive placebo with the risks associated, including the possibility of nocebo effects.
Ethical analysis of trial designs
Autonomy
Participants must be free to choose if they want to be tested and/or know their genetic status.2, 9, 10 Transparent trials only enroll participants who know they are HDGECs, because of prior predictive testing. The inclusion/exclusion criteria establish the conditions of trial participation. The requirement for a confirmed genetic diagnosis is not different from that in standard clinical trials where the inclusion criteria explicitly list constraints to enrollment (e.g., trials may require a change/discontinuation of medications). However, because these criteria are made public, it could be argued that the existence of a trial itself could constitute undue influence, attracting patients who want the possibility of taking a new experimental treatment. These individuals may want to avoid predictive testing but participate in a clinical trial. At-risk individuals could believe they will miss a therapeutic opportunity if they do not undergo predictive testing to meet eligibility criteria. They may thus have a ‘therapeutic misconception’ believing that they can individually benefit from the trial, rather than having a main goal of contributing to scientific knowledge.11
Participants may also wish to enter the trial to please the investigator who may be the patient’s physician’ as well. The relationship between ‘asymptomatic’ premanifest participants and researchers is very different from doctor-patient relationships in a therapeutic situation, but can blur, especially since the premanifest phase evolves with the progression of disease. Even without genetic testing, the accumulation of clinical symptoms will become obvious to the physician (and participant), changing the doctor-participant relationship. Participants may thus think they are receiving direct therapeutic benefit from the trial, when they are not.
In amyotrophic lateral sclerosis, the phrasing of informed consent affects perceptions of the existence of personal benefit and willingness to participate in clinical trials.11 Accurately conveying the trial aims requires careful education which can reduce therapeutic misconception among potential participants, and help reduce some of these tensions. We argue that the risk of therapeutic misconception is outweighed by a properly executed informed consent process that emphasizes that the individual benefit in trials is unknown. Nevertheless, we acknowledge that our argument depends on the effectiveness of informed consent, which has recognized weaknesses.12 The informed consent process should emphasize and clearly convey that a clinical trial is not a form of personal medical therapy.
Concealed-sorted designs (like concealed-unsorted designs) respect autonomy because they give both HDGECs and non-HDGECs a choice, but can strain researchers who must withhold the information from individuals who wish to remain unaware of their genetic results. Such concealment of information might be difficult if the medication has distinctive side-effects that reveal intervention assignment (and the possibilities of side effects revealing this assignment should be disclosed as part of the informed consent), thus indirectly disclosing the participants’ genetic status.
Beneficence/Nonmaleficence
Clinical research can benefit participants, providing helpful knowledge, but can also cause unintended harms. Transparent, but not concealed, enrollment might induce increases in predictive testing since in order to enroll in a study, HD family members might decide to undergo testing when they otherwise would not do so – introducing possible risks. Evidence suggests that predictive testing for personal reasons (properly done with genetic counseling and sufficient time and steps for individuals to change their mind2, 9, 10) is not associated with negative psychological consequences (e.g., suicidality),13, 14 and can even enhance relationships.15, 16 However, no empirical data exist regarding the impact of predictive testing when the motive is to participate in research; studies should explore this question.
Concealed enrollment designs can cause several potential harms that are absent in transparent enrollment. Non-HDGEC participants will be exposed to trial risks, including potential side-effects of the active intervention in the unsorted design. The concealed-unsorted design (where non-HDGECs receive active treatment) is only acceptable if the risks of such treatment are minimal and well-established. Studies that meet those criteria will presumably be few. Hence, this design is only feasible when the test product is, for instance, a food supplement or exercise practice. The PRECREST trial was only able to include a mixed population (HDGECs and at-risk individuals with an affected first-degree relative) because research ethics committees considered the expected toxicities from creatine “modest and reversible”.17 Unlike creatine, most disease-modifying therapeutics now entering clinical development for HD may pose not fully understood risks (such as those inherent in the suppression of htt) or utilize invasive delivery techniques. These limitations exclude these therapeutics from both unsorted and sorted designs because intrathecal sham procedures carry considerable burden.
There is also a possibility that an at-risk participant’s mutation-positive status will be disclosed during the trial because of error, symptom onset, or a serious adverse event. Such disclosures would occur in the context of crisis management, rather than under the optimized conditions of planned genetic testing. Concealed enrollment also requires a much larger sample size, raising trial costs, and is unlikely to be offset by faster recruitment. In general, less costly trials, if they provide equal or better results, as with transparent enrollment, are in patients’ best interests.
Justice
Justice concerns might arise if access to studies are imbalanced.18 Transparent enrollment needs to be conducted in countries with higher rates of predictive testing (e.g., Europe). However, clinical development centered in Europe does not necessarily violate principles of justice. Therapeutic interventions, if proven efficacious and safe, will presumably be approvable by all regulatory authorities (e.g., the FDA) and with the right provisos become licensed. However, the asymmetry in clinical trial activity goes far beyond the specialized topic of predictive testing. Transparent enrollment is more likely to occur in countries with more clinical trials, and thus unlikely to alter this existing asymmetry in clinical trial access.
Logistical considerations
Compared to a transparent enrollment design, the main advantages of both types of concealed enrollment are: 1) wide engagement of affected communities; 2) potentially faster recruitment, given the larger population base; and 3) extension of indirect benefits of research participation to the at-risk population. Key disadvantages of concealed versus transparent enrollment include the complexity of design and data interpretation. Concealed designs also require more participants than a transparent design for adequate study powering. The final sample size will be a result of the ratio between HDGECs and non-HDGEC at the enrollment screening. Since non-HDGECs outnumber HDGECs, enrollment must be drawn from a pre-existing registry with enrollment of non-HDGEC limited to a preset number. One could expect the number of non-HDGECs in concealed-sorted designs to equal, the other two HDGEC groups (equating to a 33% trial size increase). However, the trial will never be able to accept all-comers if the proportion of non-HDGECs in the initial population is too high – thus detracting from the argument of higher inclusivity.
Feasibility
HD research studies have successfully recruited large cohorts of premanifest participants with a prerequisite of predictive testing (PREDICT,5 TRACK4 and PREQUEL19). To further evaluate the potential of transparent enrollment, we examined published US and European HD prevalence figures20 and the census population numbers to estimate the number of potential participants. We estimate (see Table 1) that at the current rates of uptake of predictive testing, approximately 30,499–52,284 tested HDGECs reside in potential North American and European study populations. Putative transparent design trials with samples of 1,000–2,000, which are in the range of published predictions, will need to enroll only 3–5% of these individuals to provide sufficient participant numbers to evaluate efficacy and safety, without the risks of unintended genetic disclosure. Given the current HD clinical experience,21 this recruitment yield seems feasible.
Table 1.
Potential study population estimates
| Populations | USA | Europe | Total |
|---|---|---|---|
|
| |||
| Manifest HD | |||
| (High estimate: prevalence 12:100,000)* | 38,280 | 89,160 | 127,440 |
| (Low estimate: prevalence 7:100,000)* | 22,330 | 52,010 | 74,340 |
|
| |||
| Estimates of premanifest HDGEC | |||
| (High estimate: prevalence 12:100,000**) | 114,840 | 267,480 | 383,320 |
| (Low estimate: prevalence 7:100,000**) | 66,990 | 156,030 | 223,020 |
|
| |||
| Current uptake of predictive genetic testing of 5% | |||
| (High-Low prevalence estimates) | 5,742-3,350 | ||
|
| |||
| Current uptake of predictive genetic testing of 17.4%*** | 46,542–27,149 | ||
| (High-Low prevalence estimates) | |||
|
| |||
| Current uptake of predictive genetic testing | |||
| (High-Low prevalence estimates) | 52,284 – 30,499 | ||
The disease prevalence is a crucial factor in our analyses. In a similar ethical analysis, regarding Familial Alzheimer disease (FAD), Kim et al. argued that transparent enrollment does not unfairly exploit vulnerable participants or limit generalizability of scientific findings of prevention trials.21 However, since FAD is very rare, they also suggested that if a community’s preferences might affect the rigor or feasibility of a prevention trial using transparent enrollment, the investigators may be required, for scientific validity, to use blinded enrollment.22 Indeed, in one ongoing trial, an extended family at risk for FAD has opted to remain unaware of their genetic status.23 While we agree that social and cultural circumstances might alter the ethical calculus, we performed our analyses regarding HD, which, although considered rare (~10/100,000 in Caucasians), is more prevalent than FAD (rare clusters). HD clinical trials do not need to be conducted in high prevalence clusters that might not be generalizable to the wider population.
Conclusions
In summary, transparent enrollment appears to be the most ethically and methodologically appropriate design, and should be adopted as the preferred approach for premanifest HD studies. Future research should evaluate patients’ and professionals’ views, and further probe the feasibility of transparent enrollment in different cultural settings. Ultimately, clear guidelines should be developed for such clinical trials.
Acknowledgments
All authors take responsibility for the integrity of the analysis. CHDI Foundation provided funding for editorial assistance in preparing the final paper for submission from Anita Chadha-Patel PhD (ACP Clinical Communications Ltd) under direction of the authors. Robert Klitzman has been supported by The National Human Genome Research Institute: #3 P50 HG007257 04S1, The Center for Research on the Ethical, Legal and Social Implications of Psychiatric, Neurologic and Behavioral Genetics, (PI: Paul S. Appelbaum).
Footnotes
Authors’ Roles: 1. Research Project: A. Conception, B. Organization, C. Execution; 2. Statistical Analysis: A. Design, B. Execution, C. Review and Critique; 3. Manuscript Preparation: A. Writing the First Draft, B. Review and Critique.
C.S. 1A, 1B, 1C, 3A, 3B
J.L. 1A, 1B, 1C, 3A, 3B
R.K. 1A, 1B, 1C, 3A, 3B
Financial Disclosures:
Cristina Sampaio has management functions within CHDI. Jamie Levey has management functions within CHDI and the European Huntington’s Disease Network. Robert Klitzman has no conflicts of interest to report.
References
- 1.Levin BC, Richie KL, Jakupciak JP. Advances in Huntington's disease diagnostics: development of a standard reference material. Expert Rev Mol Diagn. 2006;6(4):587–596. doi: 10.1586/14737159.6.4.587. [DOI] [PubMed] [Google Scholar]
- 2.MacLeod R, Tibben A, Frontali M, et al. Recommendations for the predictive genetic test in Huntington's disease. Clin Genet. 2013;83(3):221–231. doi: 10.1111/j.1399-0004.2012.01900.x. [DOI] [PubMed] [Google Scholar]
- 3.Baig SS, Strong M, Rosser E, et al. 22 Years of predictive testing for Huntington's disease: the experience of the UK Huntington's Prediction Consortium. Eur J Hum Genet. 2016;24(10):1396–1402. doi: 10.1038/ejhg.2016.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tabrizi SJ, Scahill RI, Owen G, et al. Predictors of phenotypic progression and disease onset in premanifest and early-stage Huntington's disease in the TRACK-HD study: analysis of 36-month observational data. Lancet Neurol. 2013;12(7):637–649. doi: 10.1016/S1474-4422(13)70088-7. [DOI] [PubMed] [Google Scholar]
- 5.Paulsen JS, Langbehn DR, Stout JC, et al. Detection of Huntington's disease decades before diagnosis: the Predict-HD study. J Neurol Neurosurg Psychiatry. 2008;79(8):874–880. doi: 10.1136/jnnp.2007.128728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Borowsky B, Sampaio C. Experimental therapeutics in huntington's disease : moving forward in clinical trials. In: Bates G, Tabrizi S, Jones L, editors. Huntington's disease. 4. Oxford: Oxford University Press; 2014. [Google Scholar]
- 7.The Department of Health, Education and Welfare. The Belmont Report 1979. [Date of last access March 16, 2017]; Available from: https://www.hhs.gov/ohrp/regulations-and-policy/belmont-report/
- 8.Beauchamp and Childress. Principles of Biomedical Ethics. Seventh. Oxford: Oxford University Press; 2013. [Google Scholar]
- 9.Guidelines for the molecular genetics predictive test in Huntington's disease. International Huntington Association (IHA) and the World Federation of Neurology (WFN) Research Group on Huntington's Chorea. Neurology. 1994;44(8):1533–1536. [PubMed] [Google Scholar]
- 10.The Huntington’s Disease Society of America. Guidelines for genetic testing. New York: The Huntington’s Disease Society of America; 1994. [Google Scholar]
- 11.Kim SY, Wilson R, De Vries R, Ryan KA, Holloway RG, Kieburtz K. Are patients with amyotrophic lateral sclerosis at risk of a therapeutic misconception? J Med Ethics. 2016;42(8):514–518. doi: 10.1136/medethics-2015-103319. [DOI] [PubMed] [Google Scholar]
- 12.Manson NC, O'Neill O. Rethinking Informed Consent in Bioethics. Cambridge University Press; 2007. [Google Scholar]
- 13.Dufrasne S, Roy M, Galvez M, Rosenblatt DS. Experience over fifteen years with a protocol for predictive testing for Huntington disease. Mol Genet Metab. 2011;102(4):494–504. doi: 10.1016/j.ymgme.2010.12.001. [DOI] [PubMed] [Google Scholar]
- 14.Mandich P, Jacopini G, Di Maria E, et al. Predictive testing for Huntington's disease: ten years' experience in two Italian centres. Ital J Neurol Sci. 1998;19(2):68–74. doi: 10.1007/BF02427559. [DOI] [PubMed] [Google Scholar]
- 15.Decruyenaere M, Evers-Kiebooms G, Cloostermans T, et al. Predictive testing for Huntington's disease: relationship with partners after testing. Clin Genet. 2004;65(1):24–31. doi: 10.1111/j..2004.00168.x. [DOI] [PubMed] [Google Scholar]
- 16.Richards F. Couples' experiences of predictive testing and living with the risk or reality of Huntington disease: a qualitative study. Am J Med Genet A. 2004;126A(2):170–182. doi: 10.1002/ajmg.a.20583. [DOI] [PubMed] [Google Scholar]
- 17.Rosas HD, Doros G, Gevorkian S, et al. PRECREST: a phase II prevention and biomarker trial of creatine in at-risk Huntington disease. Neurology. 2014;82(10):850–857. doi: 10.1212/WNL.0000000000000187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Velasquex M, Andre C, Shanks T, Meyer MJ. Justice and Fairness. Ethics V3 N2 Markkula Center for Applied Ethics. Santa Clara University. [Last accessed 24th March 2017];1990 https://www.scu.edu/ethics/ethics-resources/ethical-decision-making/justice-and-fairness/
- 19.Chandra A, Johri A, Beal MF. Prospects for neuroprotective therapies in prodromal Huntington's disease. Mov Disord. 2014;29(3):285–293. doi: 10.1002/mds.25835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rawlins MD, Wexler NS, Wexler AR, et al. The Prevalence of Huntington's Disease. Neuroepidemiology. 2016;46(2):144–153. doi: 10.1159/000443738. [DOI] [PubMed] [Google Scholar]
- 21.Landwehrmeyer GB, Fitzer-Attas CJ, Giuliano JD, et al. Data Analytics from Enroll-HD, a Global Clinical Research Platform for Huntington's Disease. Movement Disorders Clinical Practice. 2016:n/a–n/a. doi: 10.1002/mdc3.12388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim SY, Karlawish J, Berkman BE. Ethics of genetic and biomarker test disclosures in neurodegenerative disease prevention trials. Neurology. 2015;84(14):1488–1494. doi: 10.1212/WNL.0000000000001451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. [Date of last access 16th March 2017]; http://www.alzforum.org/news/conference-coverage/nih-director-announces-100m-prevention-trial-genentech-antibody.