

Abstract
Remarkable progress in sequencing technology over the past twenty years has made it possible to comprehensively profile tumors and identify clinically relevant genomic alterations. In breast cancer, the most common malignancy affecting women, we are now increasingly able to use this technology to help specify the use of therapies that target key molecular and genetic dependencies. Large sequencing studies have confirmed the role of well-known cancer-related genes but have also revealed numerous other genes that are recurrently mutated in breast cancer. This growing understanding of patient-to-patient variability at the genomic level in breast cancer is advancing our ability to direct the appropriate treatment to the appropriate patient at the appropriate time – a hallmark of ‘precision cancer medicine.’ This review focuses on the technological advances that have catalyzed these developments, the landscape of mutations in breast cancer, the clinical impact of genomic profiling, and the incorporation of genomic information into clinical care and clinical trials.
Keywords: Breast Cancer, Genomics, Sequencing, Precision Cancer Medicine, Personalized Medicine
Introduction
Breast cancer is the most common malignancy affecting women, with over 230,000 new cases diagnosed annually in the United States alone, affecting approximately one in eight women.1 Outcomes in breast cancer have improved over the past two decades, with a substantial decline in the death rate attributable to breast cancer, from a peak of 33.2 per 100,000 women in 1988 to 21.9 per 100,000 in 2010.1 Over that time, there has been a distinct paradigm shift from treatment based exclusively on anatomic origin of the tumor to the incorporation of key genetic and molecular features to guide therapy. As a field, breast cancer has led many advances in precision cancer medicine, building from immunohistochemical staining to gene expression analysis to, more recently, sequencing of clinical samples.
Remarkable advances over the past two decades in our ability to sequence genes, exomes, and genomes, have made it possible to comprehensively profile tumors to identify clinically relevant mutations and incorporate sequencing into clinical trial design. Our understanding of the patient-to-patient breast cancer variability at the genomic level has begun to advance our ability to direct the appropriate treatment to the appropriate patient at the appropriate time – a hallmark of ‘precision cancer medicine’ – and to develop novel treatments directed at the molecular characteristics of specific breast tumors.2–4 The emerging challenges in the coming years will revolve around on how best to realize the impressive potential benefits of incorporating next-generation sequencing into clinical care in a patient-centered manner.
Historical Perspectives on Precision Medicine in Breast Cancer
Early categorization of breast tumors relied on clinicopathologic features, such as invasive ductal versus invasive lobular carcinomas. This descriptive approach laid the foundation for subsequent advances in the analysis of specific genes. The prognostic significance of the estrogen receptor, detected first through ligand binding assays and later immunohistochemistry, confirmed the value of single-gene interrogation and led to one of the original ‘targeted’ therapies – tamoxifen for estrogen receptor (ER)-positive breast cancer.5–8 Immunohistochemistry also played a critical role in the identification and clinical application of HER2 amplification in breast cancer, which likewise led to powerful targeted therapies for HER2-positive breast cancer.9,10
Analysis of gene expression further delineated breast cancer subsets. Perou and colleagues used gene expression to reveal molecular subtypes that further subdivided the immunohistochemical classification.11 This and subsequent studies not only revealed novel subgroups ultimately shown to have distinct prognoses but also expanded analyses to include expression of hundreds (rather than a few) genes.12,13 Downstream outcomes of these seminal studies include the integration of multi-gene expression assays to guide therapy in breast cancer patients with specific clinical characteristics.14–16
Tumor Genomic Profiling and Targeted Therapies
Over the past 15 years, there has been a significant shift from exclusively anatomic-based treatment strategies toward the use of therapies that target key molecular and genetic dependencies in multiple cancer types. Three key developments are catalyzing this growth: identification and understanding of specific tumor dependencies related to known genomic alterations, a growing repertoire of agents to target these tumor dependencies, and technological advances that allow rapid detection of these genomic alterations. Genomic alterations that have been widely known for several decades, such as BCR-ABL in chronic myelogenous leukemia and HER2 amplification in breast cancer, have become targetable tumor dependencies with impressive improvements in patient outcomes with small molecule or antibody-based therapies.10,17 Individual genomic alterations are also targeted through multiple different agents. For example HER2-amplified breast cancer now has FDA-approved monoclonal antibody (trastuzumab), dimerization inhibiting monoclonal antibody (pertuzumab), small molecule inhibitor (lapatinib), and monoclonal antibody-chemotherapy conjugate (trastuzumab-emtansine).18 As targeted agents increase in number and target diversity, combination or sequential therapy with multiple agents likewise improve outcomes, for example combined BRAF and MEK inhibition in BRAF-mutant melanoma.19–21 More recently, advances in sequencing has allowed the rapid identification of less frequent but targetable alterations, such as ALK- and ROS1-rearrangements in NSCLC.22,23
The majority of genomic profiling to date has been limited to specific single genes that are likely to be altered in a particular cancer. More recently, it has become clear that while certain genomic alterations are particularly common in specific cancers (e.g. HER2 amplification in breast cancer and BRAF mutation in melanoma), these same alterations also occur less commonly in other, unexpected cancer types.24–26 Moreover, there are several emerging instances where these ‘unexpected’ genomic alterations in alternate cancers have responded to targeted therapy, such as response to trastuzumab in HER2 amplified gastric cancer, erlotinib in EGFR-mutant in breast cancer, and vemurafenib in BRAF-mutant lung cancer.27–29 These examples highlight the promise of genomic profiling, which may identify effective therapies that may not have otherwise been considered in a particular tumor type.
The fortuitous intersection of sequencing advances, increasing number of targeted therapies, and greater understanding of the diversity of potential targets supports the potential of incorporating genomically-targeted therapies into clinical practice. However, it is clear that testing for alterations only in an anatomic cancer site-specific approach will miss potentially targetable alterations. Because of this, we have now moved to multiplexed genomic testing – testing cancers for many genomic dependencies simultaneously - including both expected and unexpected alterations. Testing each tumor for the known landscape of potentially actionable alterations to identify the relevant therapies offers a more comprehensive approach to deliver the right therapy to the right patient for the alterations in their specific tumor.
Massively Parallel Sequencing for Precision Cancer Medicine
While tumor genomics is not a novel concept – cancer-related karyotypes have been in use for over four decades – a remarkable leap in our ability to detect, analyze, and correlate genetic changes with outcomes brought genomic analysis from the research world into the clinic.3 Sanger sequencing, though laborious and facing limits with tumor purity and heterogeneity, provided the ability to sequence individual genes and has historically been the gold standard for molecular diagnostics.30 Allele-specific PCR or mass-spectrometric genotyping allows detection of multiple specific mutations in ‘hotspot’ regions of genes such as KRAS, BRAF, EGFR, PIK3CA, and others.4,24,31,32 These technologies could be multiplexed which increased throughput and reduced cost, a significant advance over Sanger sequencing. However, although this high-throughput genotyping approach has represented a remarkable advance in our ability to conduct large-scale tumor genomic profiling, it has the limitation of only detecting pre-specified mutations.24,33,34 Other classes of genomic alterations that are also critical for precision medicine, including chromosomal amplifications or deletions, rearrangements, and most small insertions or deletions (‘indels’), are unable to be detected by this technology.
Two major advances in the last decade have revolutionized genomic profiling, allowing the field to move beyond Sanger sequencing and hotspot genotyping: the advent of massively parallel sequencing (MPS, also known as next generation sequencing) and the completion of the Human Genome Project. The main technological innovation of MPS is a process to sequence genes in a ‘massively parallel’ manner, through which DNA is amplified and fragmented into millions of tiny segments, each of which is sequenced in parallel. This provides a platform to generate nearly a trillion bases per run and led to a rapid decline in the cost to sequence a single genome—from $70 million by the Sanger method in 2007 to less than $5000 using MPS in 2013.2,4 The complete sequencing of the human genome provided a template to interpret the output of MPS - thousands of short sequences in no particular order or organization – allowing any sequence generated from a human sample to be ‘mapped’ to a locus on this draft genome. Advances in the bioinformatic analyses improved our ability to interpret the immense amount of genomic data generated and optimally utilize the human genome data.30 The result of this immense progress is the ability to rapidly obtain high-confidence sequence from human samples in a fraction of the time and at a fraction of the cost of even a few years ago.
Tumor genomes can be interrogated using sequencing to varying degrees. ‘Whole genome’ sequencing interrogates all of the genetic material in the cell, including protein coding regions (exons) as well as non-coding regions (introns). An alternative approach is ‘whole exome’ sequencing, which obtains sequence from only the protein-coding regions – about 25,000 genes or 1% of the whole genome. A third approach is targeted sequencing of a panel of specific – typically ‘actionable’ – genes. In addition to the extent of the genome to be sequenced, sequencing output also varies in the ‘depth’ of the sequencing performed, or the average number of times each basepair is read by the sequencing machinery (Figure 1). Depth of coverage impacts data accuracy and sensitivity, with greater depth sequencing equating to improved detection of mutations present in a small percentage of cells.35 Due to limits in cost, whole genome sequencing typically sacrifices depth for breadth of coverage across the entire genome while whole exome potentially balances these limitations by sequencing only 1% of the genome. Sequencing only a few hundred or thousand genes focuses even further onto genes known to be involved in cancer with direct therapeutic options and can be performed with significant depth for a more reasonable cost.36 Smaller numbers of genes can thus be sequenced to relatively high depth and in a multiplexed approach in a cost-effective manner, optimal characteristics for large-scale clinical applicability.
Figure 1. Tumor Sequencing Approaches: Coverage, Cost, and Depth.
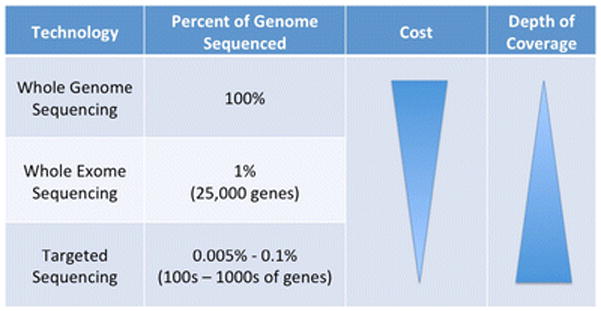
Interrogating tumor genomes via sequencing requires compromise between amount of the genome to be sequenced, cost, and depth. ‘Depth’ refers to the average number of times each basepair is read by the sequencing machinery, which impacts accuracy and sensitivity.
The Landscape of Genomic Alterations in Breast Cancer
Cancer became one of the early targets of our growing ability to rapidly and cost-effectively unravel genomes. Just as the Human Genome Project was a collaborative effort that provided a powerful template for the entire field, several large cooperative efforts to sequence hundreds of breast tumors revolutionized our understanding of the genomic underpinnings of breast cancer. These publicly available data amplified efforts by other institutions and groups to reveal a landscape of mutations and other characteristics including expression, protein, methylation, and miRNA data in breast cancer.
It is becoming increasingly clear that nearly every breast tumor, irrespective of subtype, has multiple genetic abnormalities – not just one or a few driver mutations. In addition, the mutational landscape is not only populated by individual base pair substitutions – the ‘typo’ in genome replication – but by small insertions or deletions (‘indels’) as well as large scale copy number changes involving millions of bases at a time. Early studies provided several key insights: a few genes are recurrently mutated in breast cancer, representing known and likely drivers of tumorigenesis; large copy number changes primarily occur in specific sites within the genome; and immense numbers of genetic changes of unclear significance are present in nearly every breast cancer.
Five large breast cancer sequencing studies, each taking a slightly different approach, have catalogued the landscape of genomic alterations in breast cancers using whole genome or exome sequencing (Table 1).37–41 These studies incorporate nearly 900 largely treatment-naïve primary breast cancers encompassing all breast cancer subtypes. Across all breast cancers, the most commonly mutated genes were TP53 (mutated in 35% of tumors), PIK3CA (34%), GATA3 (9%), MAP3K1 (8%), MLL3 (6%), and CDH1 (6%).42 These findings were remarkably consistent across studies, supporting the validity of these data. Mutations varied by intrinsic breast cancer subtype. For example, in the Cancer Genome Atlas (TCGA) study, TP53 mutation was present in 80% of basal-like and 72% of HER2-enriched but only 12% of luminal A, while GATA3 was mutated in 14% and 15% of luminal A and luminal B, respectively, but only 2% in each of basal-like and HER2-enriched tumors.37
Table 1.
Large-Scale Sequencing Studies in Breast Cancer
| Study | ER+ Breast Tumors | ER- Breast Tumors | TOTAL |
|---|---|---|---|
| Stephens, et al.38 | 79 primary tumors | 21 primary tumors | 100 tumors |
| Banerji, et al.40 | 60 primary tumors | 48 primary tumors | 108 tumors |
| Shah, et al.39 | 0 primary tumors | 104 primary tumors | 104 tumors |
| Ellis, et al.41 | 77 post-neoadjuvant tumors | 0 primary tumors | 77 tumors |
| TCGA37 | 390 primary tumors | 117 primary tumors | 507 tumors |
| TOTAL | 606 tumors | 290 tumors | 896 TOTAL |
The great majority of mutations were point mutations (>90% in all studies) with the remainder small insertions or deletions (‘indels’). Although the commonly mutated genes all have known association with cancer, a large number of lower frequency mutations were also detected. In the TCGA, of the over 28,000 point mutations, more than 10,000 were either nonsense mutations or predicted to be deleterious, yet comparison to existing databases of known cancer genes only identified 619 mutations across 177 previously reported cancer genes.37 This suggests that massive numbers of gene products are potentially affected by mutations in genes not as yet linked to cancer. Interrogation of specific breast cancer subgroups, including triple-negative breast cancer (TNBC),39 BRCA-mutant breast cancer,43 and lobular carcinoma in situ (LCIS)44 likewise revealed multiple mutations in genes previously not related to cancer. In addition, there is evidence that breast fibroadenomas, a non-malignant breast disease, also demonstrate recurrent mutations.45 These mutations present in a small percentage of breast cancers – but recurrent - may be new potential targets for personalizing therapy.
Although somatic mutations and indels account for the majority of alterations in the breast cancer genome, larger shifts in genomic material remain common and can also have major impacts on genes and gene products. These shifts, termed ‘copy number alterations’ (CNAs) are so-called due to an increase (amplification) or decrease (deletion) in the number of copies of a gene or region.35 The most common copy number changes involve well-known breast cancer-related genes via amplification (HER2, PIK3CA, EGFR) or loss (PTEN, MLL3, RB1).37 Copy number analyses in the five large breast cancer sequencing studies described above confirm prior data suggesting that luminal A tumors had significantly fewer CNAs relative to basal-like or HER2-enriched breast cancers. For example, in one study luminal A samples had a median of 30 rearrangements per sample while basal-like and HER2-enriched subtypes had a median of 237 and 246, respectively.40 Although basal-like and HER2-enriched appear less genomically stable, a median of 30 CNAs per sample nonetheless reveals a surprisingly fractured genomic environment within luminal A breast cancers. In the largest copy number analysis to date, Curtis and colleagues evaluated 2,000 breast cancers through copy number and correlative transcriptional analysis.46 Over 10,000 copy number alterations were detected impacting the expression of approximately 40% of the entire genome and could be grouped based on pattern of CNAs.46 A more focused investigation of homozygous deletions reveal that the majority occur in genomic regions that are inherently fragile, or have increased susceptibility to chromosome breakage.47 Copy number alterations clearly play a significant role in cancer but remain difficult to study due to variable CNA size, multiple genes amplified or deleted, and potentially expression of many other genes. Further analyses, particularly of CNA ‘hotspots’ may illuminate novel, targetable breast cancer susceptibilities.
Despite the impressive data available from these and other investigations, several key areas in breast cancer genomics remain under-studied. One pitfall of existing studies is a lack of clinical annotation, such as patient and tumor characteristics, treatment history, and clinical outcomes, among other metrics. Future sequencing approaches that incorporate clinical data will allow us to further interrogate the relationship between specific mutations and clinical outcome. In addition, there are few systematic studies of recurrent and/or metastatic disease, though some studies are now being conducted.48,49 To further understand tumor evolution and therapeutic resistance in breast cancer, it will be imperative to catalogue the mutations present in those tumors that are metastatic at diagnosis or that recur after primary treatment. Beyond the landscape of mutations in metastatic samples, serial biopsies of cancer over time – primary, recurrence, and multiple metastatic sites – will allow us to gain a greater understanding of evolution and resistance in breast cancer, as has been done with other tumor types.50
There are efforts that are beginning to address these issues. Efforts to obtain biopsies are often cited as a challenge yet data suggest that additional or research purposes only biopsies are safe, well-tolerated, and provide a high rate of analyzable tissue.51 Multiple studies involving the prospective collection of clinically annotated research biopsies, both from primary breast tumors and metastases, are now ongoing at many institutions, including our own49,51–55.
Clinical Impact of Genomic Profiling
Our improving knowledge of the genomic landscape of breast cancer has highlighted the potential for identifying clinically relevant genomic alterations for an individual patient with breast cancer. Prospective genomic profiling efforts offer potential utility for multiple aspects of patient care including diagnosis of breast cancer subtype, patient prognosis, prediction of therapeutic response, and markers of resistance. For instance, interrogating ~1000 breast cancer samples recently sequenced as part of the TCGA project (available at cbioportal.org) for genomic alterations in 128 potentially clinically relevant genes56 reveals alterations in numerous genes across the cohort, ranging in frequency from >40% (PIK3CA) to <5% (AKT1, MAP2K2) (Figure 2). Specific alterations in many of these genes may predict sensitivity to several therapies in current use or in clinical development (Table 2).
Figure 2. Genomic Alterations in 128 Potentially Clinically Relevant Genes in 962 TCGA Breast Cancer Samples.
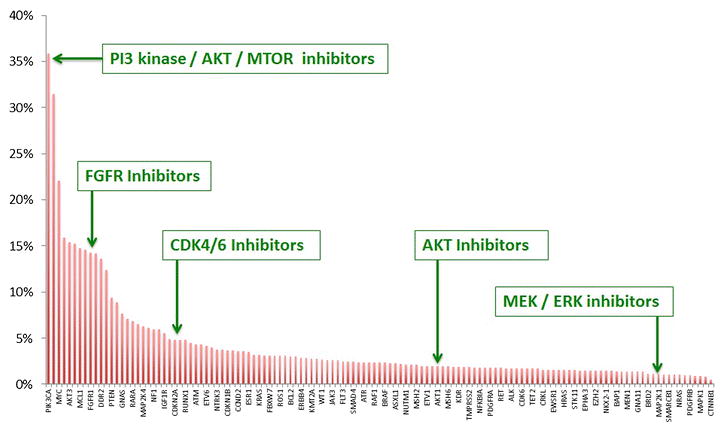
Genomic profiling efforts may identify genomic altertaions that can be used for clinical decision making, such as the choice of therapeutic agent or clinical trials. Interrogating 962 breast cancer samples that have been sequenced as part of the TCGA project (available at cbioportal.org) for genomic alterations in 128 potentially clinically relevant genes56 reveals alterations in numerous genes across the cohort, ranging in frequency from >40% (PIK3CA) to <5% (AKT1, MAP2K2). Examples of therapeutic agents that target several of these genes are highlighted.
Table 2. Common Genomic Alterations in Breast Cancer and Existing Therapeutic Options.
Commonly mutated genes in breast cancer are grouped by pathway and listed with estimated mutation frequency. Agents currently in clinical trials targeting each pathway are listed. Adapted from Metzger-Filho & Polyak.42
| PI3-Kinase Pathway | ERBB2/ HER2 |
Fibroblast Growth Factor |
Insulin-Like Growth Factor |
Estrogen Receptor |
BRCA Mutation |
c-MET | ||||
|---|---|---|---|---|---|---|---|---|---|---|
|
| ||||||||||
| PIK3CA (34%), PIK3R1 (2%), AKT1 (2%) | ERBB2 (14%) | FGFR1 (13%), FGFR2 (3%), FGFR3 (2%) |
IGF1R (6%), IGF2R (3%), IGF1 (1%) |
ESR1 (4%) | BRCA1 (4%), BRCA2 (4%) |
MET (2%) | ||||
|
| ||||||||||
| Inhibitors | PI3K Inhibitors | mTOR inhibitors | AKT inhibitors | PI3K/mTOR Inhibitors | PARP Inhibitors | |||||
| Buparlisib (BKM120) | Everolimus | GDC-0068 | BGT226 | Trastuzumab | AZD4547 | BMS-754807 | Tamoxifen | Olaparib | SGX523 | |
| Pictilisib (GDC-0941) | Temsirolimus | MK2206 | GDC-0980 | T-DM1 | BGJ398 | Cixutumumab | Toremifene | Veliparib | INC280 | |
| BYL120 | AZD8055 | GSK2141795 | PKI-587 | Pertuzumab | Dovitinib | Dalotuzumab | Anastrazole | Rucaparib | Tivantinib | |
| GDC-0032 | INK128 | XL765 | Lapatinib | Lucitinib (E-3810)** | Figitumumab | Letrozole | BMN-673 | PF-02341066* | ||
| INK1117 | Afatinib | HGS1036 | Ganitumab | Exemestane | CEP-9722 | Cabozantinib** | ||||
| PF-04691502 | Canertinib | BAY1163877 | Linsitinib | Fulvestrant | E7016 | GSK1363089 | ||||
| PX-866 | Dacomitinib | GSK3052230 | MEDI-573 | INO-1001 | ARQ197 | |||||
| XL147 | Neratinib | MK4827 | ||||||||
| MM-121 | ||||||||||
Also inhibits ALK
Also inhibits VEGFR
Numerous institutional and inter-institutional efforts have now begun to utilize sequencing initiatives to examine the effect of genomic profiling on clinical decision-making and, ultimately, clinical outcomes in patients with advanced breast cancer. So-called umbrella trials, which utilize genomic profiling in a single cancer type, in breast cancer include both the SAFIR and AURORA trials, as well as additional initiatives taking place at individual institutions. The SAFIR-01 and SAFIR-02 trials in France leverage array CGH and targeted sequencing to identify somatic mutations and CNAs in breast cancer samples and then direct single-agent or combination therapy based on those results.52 SAFIR-01 included over 400 patients with metastatic breast cancer, 12% of whom were treated with matched therapies based on their genomic data with 3% clinical benefit rate from this approach.53 In Europe, the AURORA initiative will enroll patients with metastatic breast cancer and no more than one line of systemic therapy for metastatic disease, sequencing both primary and metastasis for a panel of cancer-related genes.55 Those patients with actionable mutations will be directed to innovative clinical trials assessing molecularly targeted agents while all patients will be followed for 10 years. 55
Similarly, basket trials utilize genomic profiling to identify specific “actionable” genomic alterations in multiple tumor types, matching these alterations to particular therapies, based either on predetermined rules or decisions make by a genomics/molecular tumor board. The National Cancer Institute’s (NCI) ‘Precision Medicine Initiatives’ under its National Clinical Trials Network includes several efforts to measure the impact of genomic profiling. The NCI-MPACT Trial (Molecular Profiling based Assignment of Cancer Therapeutics), utilizes a randomized design to determine if assigning treatment based on genomic profiling can improve response in approimarly 180 patients with advanced solid tumors. The NCI MATCH (“Molecular Analysis for Therapy Choice”) trial will enroll 3,000 patients with solid tumors or lymphoma who have progressed on standard therapy, using genomic profiling to match at least 1,000 patients to a treatment with a targeted drug or drug combination57.
Additional institution-specific efforts using sequencing-based genomic profiling include the PROFILE and CanSeq initiatives at Dana-Farber Cancer Institute34, the MiOncoSeq program at the University of Michigan58, a targeted sequencing effort at Vanderbilt-Ingram Cancer Center33; the Integrated Molecular Profiling in Advanced Cancers Trial (IMPACT) at Princess Margaret Cancer Center59, the MSK-IMPACT at Memorial Sloan Kettering Cancer Center, the Signature Trial from Novartis60, the My Pathway trial from Genentech, and many others.
In addition to guiding therapy, detecting mutations that may impact resistance to standard therapies could guide therapeutic choices up-front or upon recurrence. To date, multiple mutations have been associated with resistance, including MCL1 in chemoresistance among triple-negative breast cancers,61 ESR1 mutations in resistance to endocrine therapy in ER+ breast cancers,62–67 and activating mutations of downstream signaling molecules in HER2+ breast cancers49,68 (reviewed in 69). The complementary approach - sequencing patients who demonstrate extraordinary response to particular therapies – is also now being used to reveal potential susceptibilities within cancers.55,70–74
Future Challenges in Breast Cancer Sequencing
Patient-Centered Genomic Medicine
The potential positive impact of clinical sequencing in breast cancer is immense. However, we face numerous challenges to incorporate sequencing into clinical practice. As sequencing transitions from an academic endeavor to a clinical tool, the medical community will have to address several potential challenges. One major challenge is the clinical analysis, interpretation, and communication of clinically relevant mutations to clinicians and patients. With the growing amount of sequencing data, we will require massive computational power and storage as well as databases to catalogue mutations with associated clinical annotation. For ‘actionable’ mutations, we face logistical challenges to link patients with appropriate therapies and ensure that therapeutics are available. We also now have the capability to detect germline variants or mutations that may or may not be related to a patient’s cancer, such as unexpected germline p53 mutations in patients with HER2+ breast cancer.75 In addition, we are increasingly detecting mutations of unknown significance – both in genes known to be related to cancer and in other genes.76 How to deal with these unknown alterations in the clinical setting remains unclear. All of these findings require care in how they are communicated to patients, making physician and patient education increasingly important. The growth of both sequencing and commercial genetic screening tools are putting increasing pressure on geneticists and genetic counselors, an area that will need to grow to meet anticipated need.
Detecting and Addressing Intra-tumor Heterogeneity
An additional outcome of the progress with MPS and associated computational analyses is the ability to detect subpopulations within individual tumors. Increasingly, sequencing data suggest that all cells within a tumor are not identical but instead that there are multiple distinct subpopulations that can be identified by the unique collection of mutations in each individual subpopulation, so-called ‘intra-tumor heterogeneity. By treating tumors as a single, homogenous entity we may be ignoring very small, pre-existing resistant populations that may be undetectable by standard methods.77 Several early studies evaluated multiple samples from the same patient – such as a lobular primary and metastasis78 as well as primary tumor, metastasis, blood, and patient-derived xenograft from a basal-like breast cancer.79 These and studies in other tumor types provided important information about the diversity of subpopulations within individual tumors.’50,80 Two key observations revealed by these studies were 1) metastasis likely derived from a small number of initiating cells and 2) metastases demonstrated subsequent evolution from the primary after establishment of the metastasis.
An evaluation of 100 triple-negative breast cancers with coverage of recurrent somatic mutations to median 20,000× suggested that most TNBCs include 2–10 clonal clusters.39 A more detailed analysis of 21 breast cancers suggested that an early ‘driver’ event led to the expansion of a dominant clone over an extended period of time during which many hundreds or thousands of mutations collect.81 Ultimately, it appears that one of these mutations facilitates the expansion of a subpopulation (or ‘subclone’) of the dominant clone, shifting the dynamic of the developing tumor.81 Another approach that uses sequencing at the single-cell level reveal separate clonal populations and implicated punctuated clonal expansion.82,83 Although complex, these approaches have revealed many insights into the diversity of subpopulations, often termed ‘clones’ or ‘subclones,’ within tumors. These populations are not static but instead evolve over time - certain subpopulations expand while others decline or disappear and new mutations develop allowing identification of novel subpopulations. The presence of multiple subpopulations within individual tumors and their ongoing evolution will challenge our ability to optimally use sequencing data.
Somatic Mutational Processes in Breast Cancer
As whole genomes were decoded, it became clear that somatic mutation events were not random. Instead, these somatic mutations could be grouped into specific mutational processes. Early studies relied on mutational signatures from known carcinogens, such as tobacco carcinogens, UV light, and alkylating chemotherapy while more recent studies have implicated novel processes that contribute to mutations in tumors, including the APOBEC cytidine deaminases.38,84–86 These APOBEC-related mutations did not occur randomly across the genome but in specific regions of hypermutation, termed ‘kataegis,’ a phenomenon that was particularly frequent in breast cancer.86 A more recent analysis that incorporated sequence information from over 7,000 tumors suggested that the majority of the nearly 5 million mutations fell into 20 distinct mutational signatures.87 As these processes become better understood, we will need to consider how to incorporate mutational signatures into clinical care.
Additional Applications of Clinical Sequencing
To date, clinical sequencing primarily provides prognostic information and, as described, identification of potentially targetable genomic alterations. However, the potential applications of clinical sequencing are immense. Circulating tumor cells and circulating free DNA are both promising technologies in breast and other cancers to improve prognostication, track tumor dynamics over time, assess drug sensitivity, and potentially detect mutations non-invasively.66,88–93 Additionally, immunotherapy is become a promising therapeutic approach for cancer and personalized approaches using neoantigens and personalized vaccines may have a role in precision medicine in breast cancer in the future.94
Conclusions
Over the last two decades, we have witnessed a revolution in sequencing - technology, bioinformatics, and cost – making this an exciting time in clinical and translational cancer research, specifically in breast cancer. Large sequencing studies have confirmed the role of well-known cancer-related genes – TP53, PIK3CA, ERBB2, PTEN – but have also revealed numerous other genes that are recurrently mutated in breast cancer. These data demonstrate that comprehensive genomic profiling can reveal the clinically relevant mutations present in most breast cancers. This suggests that breast cancers could potentially be subdivided into smaller and smaller groupings for which therapies may be targeted to the specific mutational profile of individual tumors. Beyond guiding therapy as part of trials, clinical sequencing has also revealed novel mechanisms underlying both resistance and extraordinary response to therapy. At the same time, there remain contexts within breast cancer where additional sequencing information is still needed – metastatic disease, rare subtypes such as inflammatory breast cancer, and serial samples over time to evaluate tumor evolution and therapeutic resistance.
Some clinical trials have begun to incorporate clinical sequencing and early evidence of clinical benefit in a subset of patients with advanced cancer is promising. In the coming years, we will need to expand novel clinical trials that incorporate sequencing and establish shared databases to centralize genomic data. Along with significant promise, precision medicine in breast cancer also faces a number of challenges – social (ethical implications and patient education), biological (annotation and investigation of novel mutations), technical (cost and widespread implementation), and infrastructure (data storage and management). Careful attention will also need to be given to these areas as we usher in an era of genomics-driven precision medicine in breast cancer.
Footnotes
Compliance with Ethics Guidelines
Conflict of Interest
Daniel G. Stover has received a grant from The Susan G. Komen Breast Cancer Foundation.
Nikhil Wagle has received consultancy fees from and has stock options in Foundation Medicine.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
References
- 1.SEER Cancer Statistics Factsheets. Breast Cancer. National Cancer Institute; 2014. [Accessed July 3, 2014]. at http://seer.cancer.gov/statfacts/html/breast.html. [Google Scholar]
- 2.Garraway LA, Lander ES. Lessons from the cancer genome. Cell. 2013;153:17–37. doi: 10.1016/j.cell.2013.03.002. [DOI] [PubMed] [Google Scholar]
- 3.Garraway LA. Genomics-driven oncology: framework for an emerging paradigm. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2013;31:1806–14. doi: 10.1200/JCO.2012.46.8934. [DOI] [PubMed] [Google Scholar]
- 4.MacConaill LE. Existing and emerging technologies for tumor genomic profiling. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2013;31:1815–24. doi: 10.1200/JCO.2012.46.5948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Knight WA, Livingston RB, Gregory EJ, McGuire WL. Estrogen receptor as an independent prognostic factor for early recurrence in breast cancer. Cancer research. 1977;37:4669–71. [PubMed] [Google Scholar]
- 6.Fisher B, Redmond C, Brown A, et al. Treatment of primary breast cancer with chemotherapy and tamoxifen. The New England journal of medicine. 1981;305:1–6. doi: 10.1056/NEJM198107023050101. [DOI] [PubMed] [Google Scholar]
- 7.Ingle JN, Ahmann DL, Green SJ, et al. Randomized clinical trial of diethylstilbestrol versus tamoxifen in postmenopausal women with advanced breast cancer. The New England journal of medicine. 1981;304:16–21. doi: 10.1056/NEJM198101013040104. [DOI] [PubMed] [Google Scholar]
- 8.Harvey JM, Clark GM, Osborne CK, Allred DC. Estrogen receptor status by immunohistochemistry is superior to the ligand-binding assay for predicting response to adjuvant endocrine therapy in breast cancer. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 1999;17:1474–81. doi: 10.1200/JCO.1999.17.5.1474. [DOI] [PubMed] [Google Scholar]
- 9.van de Vijver MJ, Peterse JL, Mooi WJ, et al. Neu-protein overexpression in breast cancer. Association with comedo-type ductal carcinoma in situ and limited prognostic value in stage II breast cancer The New England journal of medicine. 1988;319:1239–45. doi: 10.1056/NEJM198811103191902. [DOI] [PubMed] [Google Scholar]
- 10.Slamon DJ, Leyland-Jones B, Shak S, et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. The New England journal of medicine. 2001;344:783–92. doi: 10.1056/NEJM200103153441101. [DOI] [PubMed] [Google Scholar]
- 11.Perou CM, Sorlie T, Eisen MB, et al. Molecular portraits of human breast tumours. Nature. 2000;406:747–52. doi: 10.1038/35021093. [DOI] [PubMed] [Google Scholar]
- 12.Sorlie T, Perou CM, Tibshirani R, et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:10869–74. doi: 10.1073/pnas.191367098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sorlie T, Tibshirani R, Parker J, et al. Repeated observation of breast tumor subtypes in independent gene expression data sets. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:8418–23. doi: 10.1073/pnas.0932692100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Paik S, Shak S, Tang G, et al. A multigene assay to predict recurrence of tamoxifen-treated, node-negative breast cancer. The New England journal of medicine. 2004;351:2817–26. doi: 10.1056/NEJMoa041588. [DOI] [PubMed] [Google Scholar]
- 15.van’t Veer LJ, Dai H, van de Vijver MJ, et al. Gene expression profiling predicts clinical outcome of breast cancer. Nature. 2002;415:530–6. doi: 10.1038/415530a. [DOI] [PubMed] [Google Scholar]
- 16.van’t Veer LJ, Bernards R. Enabling personalized cancer medicine through analysis of gene-expression patterns. Nature. 2008;452:564–70. doi: 10.1038/nature06915. [DOI] [PubMed] [Google Scholar]
- 17.Druker BJ, Sawyers CL, Kantarjian H, et al. Activity of a specific inhibitor of the BCR-ABL tyrosine kinase in the blast crisis of chronic myeloid leukemia and acute lymphoblastic leukemia with the Philadelphia chromosome. The New England journal of medicine. 2001;344:1038–42. doi: 10.1056/NEJM200104053441402. [DOI] [PubMed] [Google Scholar]
- 18.Krop I, Winer EP. Further progress in HER2-directed therapy. Lancet Oncol. 2012;13:2–3. doi: 10.1016/S1470-2045(11)70388-6. [DOI] [PubMed] [Google Scholar]
- 19.Wagle N, Van Allen EM, Treacy DJ, et al. MAP kinase pathway alterations in BRAF-mutant melanoma patients with acquired resistance to combined RAF/MEK inhibition. Cancer discovery. 2014;4:61–8. doi: 10.1158/2159-8290.CD-13-0631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Long GV, Stroyakovskiy D, Gogas H, et al. Combined BRAF and MEK Inhibition versus BRAF Inhibition Alone in Melanoma. The New England journal of medicine. 2014 doi: 10.1056/NEJMoa1406037. [DOI] [PubMed] [Google Scholar]
- 21.Flaherty KT, Infante JR, Daud A, et al. Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. The New England journal of medicine. 2012;367:1694–703. doi: 10.1056/NEJMoa1210093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shaw AT, Kim DW, Mehra R, et al. Ceritinib in ALK-rearranged non-small-cell lung cancer. The New England journal of medicine. 2014;370:1189–97. doi: 10.1056/NEJMoa1311107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shaw AT, Ou SH, Bang YJ, et al. Crizotinib in ROS1-Rearranged Non-Small-Cell Lung Cancer. The New England journal of medicine. 2014 doi: 10.1056/NEJMoa1406766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Thomas RK, Baker AC, Debiasi RM, et al. High-throughput oncogene mutation profiling in human cancer. Nat Genet. 2007;39:347–51. doi: 10.1038/ng1975. [DOI] [PubMed] [Google Scholar]
- 25.Lawrence MS, Stojanov P, Polak P, et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature. 2013;499:214–8. doi: 10.1038/nature12213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kan Z, Jaiswal BS, Stinson J, et al. Diverse somatic mutation patterns and pathway alterations in human cancers. Nature. 2010;466:869–73. doi: 10.1038/nature09208. [DOI] [PubMed] [Google Scholar]
- 27.Bang YJ, Van Cutsem E, Feyereislova A, et al. Trastuzumab in combination with chemotherapy versus chemotherapy alone for treatment of HER2-positive advanced gastric or gastro-oesophageal junction cancer (ToGA): a phase 3, open-label, randomised controlled trial. Lancet. 2010;376:687–97. doi: 10.1016/S0140-6736(10)61121-X. [DOI] [PubMed] [Google Scholar]
- 28.Oxnard GR, Binder A, Janne PA. New targetable oncogenes in non-small-cell lung cancer. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2013;31:1097–104. doi: 10.1200/JCO.2012.42.9829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29*.Ali SM, Alpaugh RK, Buell JK, et al. Antitumor response of an ERBB2 amplified inflammatory breast carcinoma with EGFR mutation to the EGFR-TKI erlotinib. Clinical breast cancer. 2014;14:e14–6. doi: 10.1016/j.clbc.2013.09.010. In this case report, a patient with advanced HER2+ inflammatory breast cancer, that had developed resistance to HER2-targeted therapy was found by genomic profiling to have an L858R base substitution in EGFR, a well-known activating mutation in non-small cell lung cancer. Treatment with erlotinib, an EGFR-targeted therapy approved in NSCLC, was effective, with an 8-month period of cancer regression as well as symptomatic improvement. This case report is an example of how genomic profiling can identify targeted therapies that otherwise might not be considered for patients with breast cancer. [DOI] [PubMed] [Google Scholar]
- 30.Meyerson M, Gabriel S, Getz G. Advances in understanding cancer genomes through second-generation sequencing. Nat Rev Genet. 2010;11:685–96. doi: 10.1038/nrg2841. [DOI] [PubMed] [Google Scholar]
- 31.Dias-Santagata D, Akhavanfard S, David SS, et al. Rapid targeted mutational analysis of human tumours: a clinical platform to guide personalized cancer medicine. EMBO molecular medicine. 2010;2:146–58. doi: 10.1002/emmm.201000070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.MacConaill LE, Campbell CD, Kehoe SM, et al. Profiling critical cancer gene mutations in clinical tumor samples. PloS one. 2009;4:e7887. doi: 10.1371/journal.pone.0007887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Johnson DB, Dahlman KH, Knol J, et al. Enabling a genetically informed approach to cancer medicine: a retrospective evaluation of the impact of comprehensive tumor profiling using a targeted next-generation sequencing panel. Oncologist. 2014;19:616–22. doi: 10.1634/theoncologist.2014-0011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.MacConaill LE, Garcia E, Shivdasani P, et al. Prospective enterprise-level molecular genotyping of a cohort of cancer patients. The Journal of molecular diagnostics: JMD. 2014;16:660–72. doi: 10.1016/j.jmoldx.2014.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Van Allen EM, Wagle N, Levy MA. Clinical analysis and interpretation of cancer genome data. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2013;31:1825–33. doi: 10.1200/JCO.2013.48.7215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wagle N, Berger MF, Davis MJ, et al. High-throughput detection of actionable genomic alterations in clinical tumor samples by targeted, massively parallel sequencing. Cancer discovery. 2012;2:82–93. doi: 10.1158/2159-8290.CD-11-0184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cancer Genome Atlas N. Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stephens PJ, Tarpey PS, Davies H, et al. The landscape of cancer genes and mutational processes in breast cancer. Nature. 2012;486:400–4. doi: 10.1038/nature11017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shah SP, Roth A, Goya R, et al. The clonal and mutational evolution spectrum of primary triple-negative breast cancers. Nature. 2012;486:395–9. doi: 10.1038/nature10933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Banerji S, Cibulskis K, Rangel-Escareno C, et al. Sequence analysis of mutations and translocations across breast cancer subtypes. Nature. 2012;486:405–9. doi: 10.1038/nature11154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ellis MJ, Ding L, Shen D, et al. Whole-genome analysis informs breast cancer response to aromatase inhibition. Nature. 2012;486:353–60. doi: 10.1038/nature11143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Polyak K, Metzger Filho O. SnapShot: breast cancer. Cancer Cell. 2012;22:562–e1. doi: 10.1016/j.ccr.2012.06.021. [DOI] [PubMed] [Google Scholar]
- 43.Natrajan R, Mackay A, Lambros MB, et al. A whole-genome massively parallel sequencing analysis of BRCA1 mutant oestrogen receptor-negative and -positive breast cancers. J Pathol. 2012;227:29–41. doi: 10.1002/path.4003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.De Brot M, Andrade V, Morrogh M, et al. Novel mutations in lobular carcinoma in situ (LCIS) as uncovered by targeted parallel sequencing. Cancer research. 2012:72. [Google Scholar]
- 45.Lim WK, Ong CK, Tan J, et al. Exome sequencing identifies highly recurrent MED12 somatic mutations in breast fibroadenoma. Nature genetics. 2014;46:877–80. doi: 10.1038/ng.3037. [DOI] [PubMed] [Google Scholar]
- 46.Curtis C, Shah SP, Chin SF, et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature. 2012;486:346–52. doi: 10.1038/nature10983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bignell GR, Greenman CD, Davies H, et al. Signatures of mutation and selection in the cancer genome. Nature. 2010;463:893–8. doi: 10.1038/nature08768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Blackwell K, Hamilton E, Marcom P, et al. Abstract S4-03: Exome sequencing reveals clinically actionable mutations in the pathogenesis and metastasis of triple negative breast cancer. Cancer research. 2013;73:S4–03. [Google Scholar]
- 49.Wagle N, Lin NU, Richardson AL, et al. Whole-exome sequencing (WES) of HER2+ metastatic breast cancer (MBC) from patients (pts) treated with prior trastuzumab (T): A correlative analysis of TBCRC003. ASCO Meeting Abstracts. 2014;32:536. [Google Scholar]
- 50.Gerlinger M, Rowan AJ, Horswell S, et al. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. The New England journal of medicine. 2012;366:883–92. doi: 10.1056/NEJMoa1113205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Vaz-Luis I, Zeghibe CA, Frank ES, et al. Prospective clinical experience with research biopsies in breast cancer patients. Breast Cancer Res Treat. 2013;142:203–9. doi: 10.1007/s10549-013-2717-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Andre F, Bachelot TD, Campone M, et al. Array CGH and DNA sequencing to personalize targeted treatment of metastatic breast cancer (MBC) patients (pts): A prospective multicentric trial (SAFIR01) ASCO Meeting Abstracts. 2013;31:511. [Google Scholar]
- 53*.Andre F, Bachelot T, Commo F, et al. Comparative genomic hybridisation array and DNA sequencing to direct treatment of metastatic breast cancer: a multicentre, prospective trial (SAFIR01/UNICANCER) The Lancet Oncology. 2014;15:267–74. doi: 10.1016/S1470-2045(13)70611-9. The SAFIR01 study evaluated 423 patients with metastatic breast cancer, identifying a targetable genomic alteration in 195 patients (46%). While only a small subset – 55 patients (13%) received ‘personalized’ therapy, this trial provides a proof of concept for incorporating genomic analyses into clinical studies. [DOI] [PubMed] [Google Scholar]
- 54.Roychowdhury S, Iyer MK, Robinson DR, et al. Personalized oncology through integrative high-throughput sequencing: a pilot study. Science translational medicine. 2011;3:111ra21. doi: 10.1126/scitranslmed.3003161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55*.Zardavas D, Maetens M, Irrthum A, et al. The AURORA initiative for metastatic breast cancer. Br J Cancer. 2014 doi: 10.1038/bjc.2014.341. The AURORA initiative is a prospective study that will perform next generation sequencing for a panel of cancer-related genes on tumour tissue and blood samples from patients with meastatic breast cancer, then follow them longitudinally. This multi-national study hopes to direct patients into clinical trials based on their genomic characteristics, while also collecting information about long-term outcomes for this cohort. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Van Allen EM, Wagle N, Stojanov P, et al. Whole-exome sequencing and clinical interpretation of formalin-fixed, paraffin-embedded tumor samples to guide precision cancer medicine. Nature medicine. 2014;20:682–8. doi: 10.1038/nm.3559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Conley BA, Doroshow JH. Molecular Analysis for Therapy Choice: NCI MATCH. Semin Oncol. 2014;41:297–9. doi: 10.1053/j.seminoncol.2014.05.002. [DOI] [PubMed] [Google Scholar]
- 58.Everett JN, Gustafson SL, Raymond VM. Traditional roles in a non-traditional setting: genetic counseling in precision oncology. J Genet Couns. 2014;23:655–60. doi: 10.1007/s10897-014-9698-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sleijfer S, Bogaerts J, Siu LL. Designing transformative clinical trials in the cancer genome era. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2013;31:1834–41. doi: 10.1200/JCO.2012.45.3639. [DOI] [PubMed] [Google Scholar]
- 60.Slosberg ED, Kang B, Beck JT, et al. The signature program, a series of tissue-agnostic, mutation-specific signal finding trials. ASCO Meeting Abstracts. 2014;32:TPS2646. [Google Scholar]
- 61.Balko JM, Giltnane JM, Wang K, et al. Molecular profiling of the residual disease of triple-negative breast cancers after neoadjuvant chemotherapy identifies actionable therapeutic targets. Cancer discovery. 2014;4:232–45. doi: 10.1158/2159-8290.CD-13-0286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Jeselsohn R, Yelensky R, Buchwalter G, et al. Emergence of constitutively active estrogen receptor-alpha mutations in pretreated advanced estrogen receptor-positive breast cancer. Clinical cancer research: an official journal of the American Association for Cancer Research. 2014;20:1757–67. doi: 10.1158/1078-0432.CCR-13-2332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Li S, Shen D, Shao J, et al. Endocrine-therapy-resistant ESR1 variants revealed by genomic characterization of breast-cancer-derived xenografts. Cell Rep. 2013;4:1116–30. doi: 10.1016/j.celrep.2013.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64*.Robinson DR, Wu YM, Vats P, et al. Activating ESR1 mutations in hormone-resistant metastatic breast cancer. Nat Genet. 2013;45:1446–51. doi: 10.1038/ng.2823. These two studies demonstrated mutations in the estrogen receptor gene, ESR1, as a novel mechanism of resistance to hormonal therapy in breast cancer. This observation illuminates the importance of genomic characterization not just for potential susceptibilities but also resistance to therapy. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65*.Toy W, Shen Y, Won H, et al. ESR1 ligand-binding domain mutations in hormone-resistant breast cancer. Nat Genet. 2013;45:1439–45. doi: 10.1038/ng.2822. These two studies demonstrated mutations in the estrogen receptor gene, ESR1, as a novel mechanism of resistance to hormonal therapy in breast cancer. This observation illuminates the importance of genomic characterization not just for potential susceptibilities but also resistance to therapy. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yu M, Bardia A, Aceto N, et al. Cancer therapy. Ex vivo culture of circulating breast tumor cells for individualized testing of drug susceptibility Science. 2014;345:216–20. doi: 10.1126/science.1253533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Merenbakh-Lamin K, Ben-Baruch N, Yeheskel A, et al. D538G mutation in estrogen receptor-alpha: A novel mechanism for acquired endocrine resistance in breast cancer. Cancer research. 2013;73:6856–64. doi: 10.1158/0008-5472.CAN-13-1197. [DOI] [PubMed] [Google Scholar]
- 68.Bose R, Kavuri SM, Searleman AC, et al. Activating HER2 mutations in HER2 gene amplification negative breast cancer. Cancer Discov. 2013;3:224–37. doi: 10.1158/2159-8290.CD-12-0349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Arteaga CL, Sliwkowski MX, Osborne CK, Perez EA, Puglisi F, Gianni L. Treatment of HER2-positive breast cancer: current status and future perspectives. Nat Rev Clin Oncol. 2012;9:16–32. doi: 10.1038/nrclinonc.2011.177. [DOI] [PubMed] [Google Scholar]
- 70.Wagle N, Grabiner BC, Van Allen EM, et al. Activating mTOR mutations in a patient with an extraordinary response on a phase I trial of everolimus and pazopanib. Cancer discovery. 2014;4:546–53. doi: 10.1158/2159-8290.CD-13-0353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wagle N, Grabiner BC, Van Allen EM, et al. Response and acquired resistance to everolimus in anaplastic thyroid cancer. The New England journal of medicine. 2014;371:1426–33. doi: 10.1056/NEJMoa1403352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Abrams J, Conley B, Mooney M, et al. National Cancer Institute’s Precision Medicine Initiatives for the new National Clinical Trials Network. American Society of Clinical Oncology educational book/ASCO American Society of Clinical Oncology Meeting; 2014; pp. 71–6. [DOI] [PubMed] [Google Scholar]
- 73.Al-Ahmadie H, Iyer G, Hohl M, et al. Synthetic lethality in ATM-deficient RAD50-mutant tumors underlies outlier response to cancer therapy. Cancer discovery. 2014;4:1014–21. doi: 10.1158/2159-8290.CD-14-0380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Iyer G, Hanrahan AJ, Milowsky MI, et al. Genome sequencing identifies a basis for everolimus sensitivity. Science. 2012;338:221. doi: 10.1126/science.1226344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Rath MG, Masciari S, Gelman R, et al. Prevalence of germline TP53 mutations in HER2+ breast cancer patients. Breast Cancer Res Treat. 2013;139:193–8. doi: 10.1007/s10549-012-2375-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76*.Dewey FE, Grove ME, Pan C, et al. Clinical interpretation and implications of whole-genome sequencing. Jama. 2014;311:1035–45. doi: 10.1001/jama.2014.1717. An evaluation of whole genome sequencing on 12 patients reveals the challenges of sequencing the whole genome - incomplete coverage and low reproducibility of known disease-associated genetic variants with difficulty determining what findings to convey to patients. This supports a multiplexed, targeted sequencing approach of actionable genes for tumors rather than whole genome sequencing. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Swanton C, Burrell RA, Futreal PA. Breast cancer genome heterogeneity: a challenge to personalised medicine? Breast Cancer Res. 2011;13:104. doi: 10.1186/bcr2807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Shah SP, Morin RD, Khattra J, et al. Mutational evolution in a lobular breast tumour profiled at single nucleotide resolution. Nature. 2009;461:809–13. doi: 10.1038/nature08489. [DOI] [PubMed] [Google Scholar]
- 79.Ding L, Ellis MJ, Li S, et al. Genome remodelling in a basal-like breast cancer metastasis and xenograft. Nature. 2010;464:999–1005. doi: 10.1038/nature08989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Gerlinger M, Horswell S, Larkin J, et al. Genomic architecture and evolution of clear cell renal cell carcinomas defined by multiregion sequencing. Nat Genet. 2014;46:225–33. doi: 10.1038/ng.2891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Nik-Zainal S, Van Loo P, Wedge DC, et al. The life history of 21 breast cancers. Cell. 2012;149:994–1007. doi: 10.1016/j.cell.2012.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Navin N, Kendall J, Troge J, et al. Tumour evolution inferred by single-cell sequencing. Nature. 2011;472:90–4. doi: 10.1038/nature09807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Wang Y, Waters J, Leung ML, et al. Clonal evolution in breast cancer revealed by single nucleus genome sequencing. Nature. 2014;512:155–60. doi: 10.1038/nature13600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Greenman C, Stephens P, Smith R, et al. Patterns of somatic mutation in human cancer genomes. Nature. 2007;446:153–8. doi: 10.1038/nature05610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Pleasance ED, Cheetham RK, Stephens PJ, et al. A comprehensive catalogue of somatic mutations from a human cancer genome. Nature. 2010;463:191–6. doi: 10.1038/nature08658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Nik-Zainal S, Alexandrov LB, Wedge DC, et al. Mutational processes molding the genomes of 21 breast cancers. Cell. 2012;149:979–93. doi: 10.1016/j.cell.2012.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Alexandrov LB, Nik-Zainal S, Wedge DC, et al. Signatures of mutational processes in human cancer. Nature. 2013;500:415–21. doi: 10.1038/nature12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Lohr JG, Adalsteinsson VA, Cibulskis K, et al. Whole-exome sequencing of circulating tumor cells provides a window into metastatic prostate cancer. Nature biotechnology. 2014;32:479–84. doi: 10.1038/nbt.2892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Aceto N, Bardia A, Miyamoto DT, et al. Circulating tumor cell clusters are oligoclonal precursors of breast cancer metastasis. Cell. 2014;158:1110–22. doi: 10.1016/j.cell.2014.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Yu M, Stott S, Toner M, Maheswaran S, Haber DA. Circulating tumor cells: approaches to isolation and characterization. J Cell Biol. 2011;192:373–82. doi: 10.1083/jcb.201010021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Dawson SJ, Rosenfeld N, Caldas C. Circulating tumor DNA to monitor metastatic breast cancer. The New England journal of medicine. 2013;369:93–4. doi: 10.1056/NEJMc1306040. [DOI] [PubMed] [Google Scholar]
- 92.Dawson SJ, Tsui DW, Murtaza M, et al. Analysis of circulating tumor DNA to monitor metastatic breast cancer. The New England journal of medicine. 2013;368:1199–209. doi: 10.1056/NEJMoa1213261. [DOI] [PubMed] [Google Scholar]
- 93.Smerage JB, Barlow WE, Hortobagyi GN, et al. Circulating Tumor Cells and Response to Chemotherapy in Metastatic Breast Cancer: SWOG S0500. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2014 doi: 10.1200/JCO.2014.56.2561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.van Rooij N, van Buuren MM, Philips D, et al. Tumor exome analysis reveals neoantigen-specific T-cell reactivity in an ipilimumab-responsive melanoma. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2013;31:e439–42. doi: 10.1200/JCO.2012.47.7521. [DOI] [PMC free article] [PubMed] [Google Scholar]