

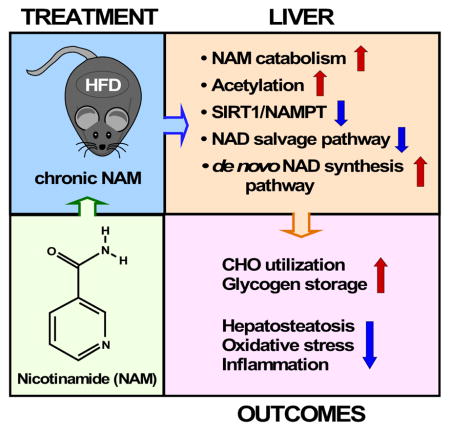
Summary
The role in longevity and healthspan of nicotinamide (NAM), the physiological precursor of NAD+, is elusive. Here, we report that chronic NAM supplementation improves healthspan measures in mice without extending lifespan. Untargeted metabolite profiling of the liver and metabolic flux analysis of liver-derived cells revealed NAM-mediated improvement in glucose homeostasis in mice on high-fat diet (HFD) that was associated with reduced hepatic steatosis and inflammation concomitant with increased glycogen deposition and flux through the pentose phosphate and glycolytic pathways. Targeted NAD metabolome analysis in liver revealed depressed expression of NAM salvage in NAM-treated mice, an effect counteracted by higher expression of de novo NAD biosynthetic enzymes. Though neither hepatic NAD+ nor NADP+ were boosted by NAM, acetylation of some SIRT1 targets was enhanced by NAM supplementation in a diet and NAM dose-dependent manner. Collectively, our results show health improvement in NAM-supplemented HFD-fed mice in the absence of survival effects.
In Brief
Interventions that increase NAD+ bioavailability are of therapeutic interest for the improvement of healthspan and lifespan. Mitchell, Bernier et al. show that chronic treatment with nicotinamide, a NAD+ precursor, is associated with health improvements and lower inflammation in the absence of lifespan extension in high-fat diet-fed mice.
INTRODUCTION
Nicotinamide (NAM) is one of three nicotinamide adenine dinucleotide (NAD) precursor vitamins that is largely made available for NAD salvage via degradation of dietary NAD and NADP (Bogan and Brenner, 2008; Trammell et al., 2016a). In yeast, the NAD precursor nicotinamide riboside (NR) (Bieganowski and Brenner, 2004) increases NAD+ levels and extends replicative lifespan by ~2-fold (Belenky et al., 2007a). Although NAM can also increase NAD biosynthesis, high dose NAM acts as a Sir2 inhibitor that reduces lifespan to about half, similar to the effect of deleting the Sir2 gene (Bitterman et al., 2002; Anderson et al., 2002).
In cells, NAM is formed by NAD+-consuming enzymes, such as sirtuins, CD38, and poly-ADP-ribose polymerases (PARPs) (Belenky et al., 2007b; Cantó et al., 2015; Verdin, 2015). CD38 is a NADase implicated in the age-related decline in NAD and mitochondrial function impairment via its ability to regulate SIRT3 activity (Camacho-Pereira et al., 2016). As a central metabolic regulator, NAD helps maintain mitochondrial fitness and is essential for tissue health including nerves and heart (Scheibye-Knudsen et al., 2014; Kaneko et al., 2006; Swerdlow, 1998; Lee et al., 2016). In aging, maintaining NAD+ levels keeps nuclear-mitochondrial communication (Gomes et al., 2013). Recent studies indicate that dietary supplementation with NR protects against high-fat diet (HFD)-induced obesity in mice (Cantó et al., 2012) while improving hepatic function and protecting against diabetic neuropathy (Trammell et al., 2016b). Similarly, nicotinamide mononucleotide (NMN) enhances insulin sensitivity in HFD-fed mice (Yoshino et al., 2011). Moreover, these NAD precursors are effective at extending lifespan and reducing metabolic disease (Khan et al., 2014; Imai, 2010). For example, NR promotes oxidative metabolism by increasing both the NAD+/NADH ratio in muscle, liver, and brown adipose tissue and the circulating fatty acid levels (Cantó et al., 2012). Repletion of NAD+ stores with NR supplementation improves muscle function and heart defect in a mouse model of muscular dystrophy (Ryu et al., 2016). The similar effects of NMN and NR have been attributed to a common pathway, namely the NR kinase pathway (Ratajczak et al., 2016; Fletcher et al., 2017). In contrast, less is known about the metabolic impact of high NAM dose in rodents, especially with respect to HFD challenge (Yang et al., 2014; Qi et al., 2016). The unanswered questions about NAM are also of interest because the beneficial effects of NR and NMN have been attributed to sirtuin activation in some studies (Yoshino et al., 2011; Cantó et al., 2012; Gomes et al., 2013; Khan et al., 2014) but not others (Trammell et al., 2016b; Zhou et al., 2016).
In the present study, we aimed at characterizing the effects of chronic NAM supplementation on the longevity and healthspan characteristics of male C57BL/6J mice fed a synthetic low-fat diet (SD) and the corresponding HFD. Because of the liver’s importance in maintaining metabolic homeostasis, we carried out histological, biochemical, and untargeted metabolomics surveys to provide an unbiased view of the metabolic impact exerted by 62-week NAM supplementation in liver from SD- and HFD-fed mice. Protein target validation combined with metabolic flux analysis enabled the identification of the underlying mechanisms of enhanced glucose disposal and reduced oxidative stress in response to NAM supplementation. Surprisingly, our data showed that NAM depresses NAD salvage and has complex effects on sirtuin expression and activity. NAM appears to have greater beneficial effects in mice subjected to HFD than SD, which might provide important clues about its therapeutic potential in the fight against obesity and associated comorbidities.
RESULTS AND DISCUSSION
To investigate whether NAM improves mouse healthspan and lifespan, we fed one-year old, male C57BL/6J mice with a SD or HFD supplemented with two doses of NAM (0.5 and 1.0 g/kg of diet) for the remainder of their lives (n=100 mice per experimental group × 6 groups: SD, SDL, SDH, HFD, HFDL, and HFDH). NAM treatment did not affect mean or maximum lifespan of mice on either diet (Figures 1A, 1B). A gross necropsy examination on all mice that died revealed no histopathological differences between cohorts (Table S1), which confirms that NAM was non-toxic at the given doses. SD-fed mice weighed less than mice on HFD, and NAM supplementation did not alter the average body weight (Figures 1C, 1D) or food consumption (data not shown) between experimental groups.
Figure 1. Nicotinamide (NAM) treatment alters whole-body physiology and in vivo metabolism without impacting maximum lifespan.

(A) Kaplan-Meier survival curves for mice fed a standard diet (SD) or SD supplemented with low-dose or high-dose of NAM (n=100/group). (B) Kaplan-Meier survival curves for mice fed a high-fat diet (HFD) or HFD supplemented with either low-dose or high-dose of NAM (n=100/group). (C and D) Body weight trajectories over the course of the study. (E) Timetable for the measure of various outcomes during the treatment protocol. (F and G) Oral glucose tolerance test. Inset, Area under the curve (AUC) (n = 6/group). (H–J) After 49 weeks of treatment, mice were placed into metabolic cages to measure the respiratory exchange ratio (RER) as detailed in Experimental Procedures, n= 6/group. (K) H & E staining depicted steatosis as circular white gaps caused when the dehydration process leaches the fat out of fixed liver tissues. (L) The degree of steatosis was scored and represented as means ± SEM. (M) Periodic acid-Schiff staining (PAS) for the detection of polysaccharides (e.g., glycogen) in fixed liver tissues. (N) Semi-quantification of PAS staining and representation as mean ± SEM. (K, M) Scale bar, 100 μm. 100X final magnification. (O) Dot blot depicting protein carbonylation levels in the liver of the six experimental groups of mice (n=4/group). (P) Quantitative analysis after normalization to protein content. *p<0.05, **p<0.01, ***p<0.001 vs. control. (See also Figure S1).
We assessed several physiological parameters displayed by mice during the experiment (Figure 1E). Body-fat percentage was significantly reduced in SDL mice after 25 and 51 weeks of treatment (81 and 107 weeks of age, respectively) (Figure S1A, S1B) with respect to SD. Though NAM-treated mice on SD exhibited unaltered oral glucose tolerance (OGTT, Figure 1F), the SDL mice had significantly lower fasting blood glucose without effect on circulating insulin levels (Figures S1C, S1D). Mice on HFD supplemented with NAM exhibited a significant decrease in glucose at the OGTT peak (15-min) (Figure 1G), indicating a higher rate of glucose clearance that was associated with a trend toward lower AUC values (inset Figure 1G, p=0.08). Insulin levels and homeostatic model assessment of insulin resistance (HOMA-IR) remained unchanged with NAM treatment, although both readouts exhibited a downward trend in HFDL and HFDH mice (Figures S1D, S1E).
In vivo metabolism in mice showed a greater reliance of HFD-fed mice on fatty acid (FA) oxidation as their predominant fuel source (RER ~0.75) than SD-fed animals, which consumed a mix of fat and carbohydrates (RER ~0.83) (Figure 1I vs. 1H). NAM supplementation provoked predominant carbohydrate fueling in SD-fed mice (RER ~0.9–0.95) (Figure 1H), with a similar trend in HFDH mice (RER ~ 0.78) (Figure 1I). The flattening of the RER amplitude upon consumption of HFD vs. SD was consistent with an earlier report (Kohsaka et al., 2007). Although HFD-fed mice were not as active as their SD counterparts, the HFDH group was associated with greater VO2 and VCO2 values and locomotor activity than HFD controls (Figure S1F, S1G). Note the occurrence of circadian rhythmicity in ambulation (tabulated as activity counts) in all groups of mice. It would appear, therefore, that NAM supplementation partly corrects the diet-associated dampening of in vivo metabolic/activity pattern.
We then set out to assess healthspan using behavioral and locomotor tests, and the results indicated an improvement in motor coordination and locomotor activity when HFD-fed mice were on chronic NAM supplementation (Figures S1H–S1K).
Histological staining revealed that NAM treatment reduced morphological alterations in the liver that occurs as part of normal aging and in response to HFD. Significant improvement in steatosis was observed in HFD-fed mice treated with NAM (Figures 1K, 1L) and, in agreement with the heightened glucose metabolism bestowed by NAM supplementation, periodic acid-Schiff staining indicated a marked recovery in hepatic glycogen deposition of non-fasted mice on HFD and NAM (Figures 1M, 1N). Hepatocytic lipid accumulation predisposes to reactive oxygen species excess, endoplasmic reticulum stress, and lipotoxicity (Bechmann et al., 2012). Here, NAM supplementation reduced the formation of carbonylated proteins in liver lysates (Figures 1O, 1P).
Together, these results suggested that dietary supplementation with NAM improves glucose metabolism and effectively inhibits liver pathology and oxidative stress while enhancing physical performance of HFD-fed mice.
Liver metabolite profiling in NAM-treated mice
To better understand the liver’s metabolic response to a chronic (62-week) NAM supplementation we performed untargeted metabolomics in 118 weeks-old mice subjected to SD or HFD diet.
Both diet and NAM treatment influenced the relative abundance of several metabolites, as evidenced by significant differences between control and treated groups (Figures 2A, S2A–S2C). Both, two-way ANOVA and Principal Component analyses revealed combined diet-NAM dose-dependent effects underscoring a higher impact of NAM supplementation on liver metabolism from HFD-fed mice.
Figure 2. Impact of NAM on hepatic metabolites profile in mice fed SD or HFD.

(A) Metabolomics analysis illustrating the relative levels of glucose, fructose, glucose 6-phosphate, glucose 1-phosphate, glycerol 3-phosphate, and citrate (n=6/group; metabolites key: red, accumulated; green, depleted) (B) Diagram depicting the alteration in glycolytic metabolite concentrations in HFD livers in response to low NAM supplementation. The blockade of phosphofructokinase, liver type (PFKL) by citrate may account for the reduced formation of glyceraldehyde 3-phosphate, which is central in several metabolic pathways. The depletion in glucose 1-phosphate suggests active glycogen synthesis in response to NAM supplementation. All box plots represent 6 mice/group. *, p<0.05; **, p<0.01; ***, p<0.001; ****, p<0.0001. (See also Figure S2).
Remarkably, NAM supplementation influenced the levels of biochemical markers of glucose metabolism from the metabolomes of SD and HFD groups. Unlike in SD-fed mice, glucose 6-phosphate (G6P) and glucose 1-phosphate (G1P) were less abundant whereas the tricarboxylic acid cycle intermediate citrate increased significantly in HFDL compared to HFD control liver (Figure 2A). The polyol pathway intermediate sorbitol (Brownlee, 2005) exhibited lower levels in mice on HFD and NAM (Figure S2D) concomitantly with a significant decrease in fructose (Figure 2A), suggesting a return path to glycolysis for glyceraldehyde 3-phosphate via fructose 1-phosphate, thus bypassing the potential block of phosphofructokinase-1 by citrate (Hue and Taegtmeyer, 2009) (Figure 2B). However, the flux through the polyol pathway appears to be negligible as suggested by metabolic flux balance analysis (see below).
Together, the data presented suggest that, in HFD-fed mice, NAM supplementation favors liver glucose catabolism concomitantly through glycolysis and glycogen storage.
Metabolic flux analysis and insulin responsiveness in HepG2 cells in the presence of NAM
Physiological (HOMA-IR, glucose tolerance) and metabolomics readouts suggested that NAM exerts a salutary effect on liver function, apparently mediated by upstream modifications of key steps in glucose metabolism, especially in mice subjected to HFD.
To investigate the NAM’s ability to confer protection against fatty acid-induced metabolic impairment, we examined quantitatively the impact of this compound on glucose and lipid catabolism using a combined experimental-computational approach in liver-derived HepG2 cells. In the absence (control) or the presence of 1 or 2 mM NAM, direct measurements of substrate uptake (glucose, palmitate, O2), efflux (pH, lactate), and ATP demand were performed using high throughput Seahorse oxygen consumption (OCR) and extracellular acidification (ECAR) rate measurements (Figures 3A, 3B). Comparatively to controls, ECAR rather than OCR measurements exhibited the more consistent and significant effect of NAM.
Figure 3. Metabolic measurements and metabolic flux analysis in HepG2 cells in the presence or absence of NAM.

(A) Baseline oxygen consumption rate (OCR) and (B) extracellular acidification rate (ECAR) were monitored in the presence of 1 mM glucose and 100 nM insulin along with the NAM levels that were used during the overnight preincubation period (0, 1, or 2 mM). The values shown were determined after addition of either 4 mM glucose (Glc, final 5 mM), 200 μM palmitate (Palm, in a 4:1 ratio with fatty acid-free BSA), or the combination of both substrates (Glc+Palm). Two-way ANOVA tests examined the influence of each independent variable (NAM concentration and type of substrate utilization), and provided evidence of interaction between them. *, **, *** P < 0.05, 0.01, and 0.001, respectively. (C) Effect of NAM on experimental and estimated fluxes through main glucose and lipid catabolic pathways. Depicted is an aggregate version of the metabolic pathways involved in the catabolism of glucose and palmitate that accounts only for stoichiometric information and includes glucose uptake and glycogen degradation (omitted in the scheme for the sake of simplicity), lactate generation and efflux, and pyruvate (Pyr) uptake into mitochondria. Mitochondrial acetyl CoA (AcCoA) may be derived from either pyruvate dehydrogenase (PDH) activity or from β-oxidation of palmitoyl-CoA whereas pyruvate may be carboxylated into oxaloacetate (Oaa) by pyruvate carboxylase (PCb). Because at steady state this reaction replenishes the TCA cycle, the PCb flux is equivalent to the citrate efflux from mitochondria. On the other hand, the flux of Pyr decarboxylation through PDH is equivalent to that through the TCA cycle from citrate to Oaa, while the flux through citrate synthase (CS) is the sum of the two, i.e. PDH + PCb. See Experimental Procedures for additional information. Red bars, measured values; blue bars, estimated values; all fluxes in nmol/min or pmol/min depicted in panels A–C were normalized to 1.6 × 104 cells, i.e., nmol/min/1.6 × 104 cells or pmol/min/1.6 × 104 cells.
Next, we performed flux balance analysis (Savinell and Palsson, 1992; Cortassa et al., 1995) to evaluate steady-state metabolic fluxes under the conditions in which OCR and ECAR were measured. OCR and ECAR fluxes were employed as a reference for an aggregated computational model of central catabolism, which includes carbohydrate (glycolysis, pentose phosphate [PP], glycogen) and lipid (β-oxidation) degradation pathways in cytoplasmic and mitochondrial compartments. In the presence of 5 mM glucose (Glc), 200 μM palmitate (Palm, bound to fatty acid-free bovine serum albumin 4:1) and 100 nM insulin, 1 or 2 mM NAM increased ~50% the uptake of glucose (VGlc) and, only modestly, palmitate (VPalm_Ox) (Figure 3C). Concomitantly, O2 consumption (VO2) and lactate efflux (VLact) rates augmented ~15% and ~25%, respectively, accompanied by a slight increase in the cellular ATP demand (VATPdemand, assessed from the flux of protein synthesis as a function of HepG2 cells doubling time under similar conditions). Using our computational model, we could estimate the flux increase through glycolytic (VGlycolysis) and PP (VPPP) pathways as well as the mitochondrial flux distribution via pyruvate dehydrogenase (VPDH), pyruvate carboxylase (VPCb), and citrate synthase (VCS) (Figure 3C). Remarkably, low or high NAM concentrations activated 8- to 9-fold the PP pathway flux, accompanied by a relatively much lower but consistent glycolytic flux enhancement. In mitochondria, 2 mM NAM produced a modest increase in VO2, an effect mediated by flux enhancement through the TCA cycle via acetyl CoA (AcCoA) supplied from β-oxidation of Palm, and glycolytic pyruvate (Pyr) through pyruvate dehydrogenase, complemented by anaplerotic replenishment of oxaloacetate (Oaa) in the TCA cycle through pyruvate carboxylase (Figure 3C). Enzymatically assessed via the levels of fructose in the extracellular medium, the flux through the polyol pathway was negligible (fructose level < 20 pmol at time zero and throughout the assay).
Together, the results obtained show that, in HepG2 cells in the presence of lipids (mimicking HFD), NAM increases glucose catabolism from enhanced glucose uptake and degradation of internal stores of glycogen (more abundant in the liver of HFD-fed mice treated with NAM, see Figures 1M, 1N) processed through glycolytic and PP pathways. In HepG2 cells, ~50% of glucose is oxidized via oxidative phosphorylation and the remaining is excreted as lactate. The remarkable stimulation of the glucose flux redirection through the PP pathway suggests that the favorable effect of NAM on glucose homeostasis might be linked to improved cytoplasmic redox homeostasis via augmented PP pathway-derived NADPH, the main electron donor for cytoplasmic and mitochondrial antioxidant systems (Kembro et al., 2013). Thus, the improvement in glucose homeostasis may be linked with redox balance in HFD-fed mice treated with NAM.
NAM-mediated changes in NAD metabolism
Next, we performed quantitative targeted metabolomics of the NAMPT-NAD-SIRT1 pathway to further assess the NAM impact on liver redox metabolism. The abundance of nicotinamide phosphoribosyl transferase (NAMPT), that catalyzes NAM conversion into NMN, was markedly lower in HFD compared to SD livers, and NAM treatment caused a significant reduction in NAMPT levels from SD-fed mice (Figures 4A right panel, S3A). In contrast, the expression of SIRT1 protein was significantly higher in HFD vs. SD livers and in response to NAM supplementation irrespective of diet (Figures 4A left panel, S3A). Figure 4B depicts the strong negative correlation observed between the NAMPT and SIRT1 protein levels (r2=0.761, F=74.14, p<0.001). Comparatively, NAMPT levels were sharply reduced in liver and white adipose tissue (WAT), but not in skeletal muscle (Figures S3B, S3C). Likewise, alterations in SIRT1 protein levels were detected in WAT but not in skeletal muscle upon NAM supplementation (Figures S3B, S3C).
Figure 4. Impact of NAM supplementation on hepatic NAD+ metabolome and the NAMPT-NAD-SIRT1 pathway.

(A) Scatter plots depicting densitometric analysis after normalization of SIRT1 and NAMPT immunoblots to Ponceau S staining of the membrane, n=4/group. (B) Scatter plot displaying the association between NAMPT and SIRT1 protein expression. (C) Densitometric analysis after normalization of IDO, NMNAT1 and NADS immunoblots to Ponceau S staining of the membrane. Bars represent the average ± SEM (n=6). (D) Histograms show the relative tryptophan levels in serum metabolome. *, p<0.05; **, p<0.01; ***, p<0.001. (E) Heatmap illustrates the log2(fold change) values of 13 metabolites from the NAD-related pathway analysis that was evaluated after data normalization with median fold change in each group. (F) Diagram depicting the effect of dietary supplementation of NAM on the liver NAD+ biosynthetic, salvage, and catabolic pathways. Abbreviations: AOX, aldehyde oxidase 1; IDO, indoleamine-pyrrole 2,3-dioxygenase 1; Me-2-py, N-methyl-2-pyridone-5-carboxamide; Me-4-py, N-methyl-4-pyridone-5-carboxamide; MeNAM, methylnicotinamide; NAAD, nicotinic acid adenine dinucleotide; NAD+/NADH, oxidized/reduced nicotinamide adenine dinucleotide; NADP+/NADPH, oxidized/reduced nicotinamide adenine dinucleotide phosphate; NADS, nicotinamide adenine dinucleotide synthase; NAM, nicotinamide; NAMN, nicotinic acid mononucleotide; NAMPT, nicotinamide phosphoribosyltransferase; NMN, nicotinamide mononucleotide; NMNAT, nicotinamide/nicotinic acid mononucleotide adenyltransferase; NNMT, NAM N-methyltransferase; NR, nicotinamide riboside; QA, quinolinic acid; SIRT1, sirtuin 1; Trp, tryptophan. (See also Figures S3, S4).
NAM is also known to alter protein acetylation (Luo et al., 2001) through its concentration-dependent action on sirtuins (Bitterman et al., 2002; Guan et al., 2014) which, in turn, might act as NAM and NAD sensor. While increased NAD bioavailability and greater SIRT1 expression might promote sirtuin activity, increased NAM might also depress sirtuin activity (Bitterman et al., 2002). Immunoblotting of total liver lysates with acetyl-lysine antibody revealed a clear increase in global protein acetylation in response to NAM treatment, especially in HFD-fed mice (Figures S3D, S3E), suggesting overall sirtuin inhibition. Focusing on known SIRT1 downstream targets, we observed a significant NAM-dependent deacetylation of the transcription factors p53 and p65Rel in SD liver, while the acetylated forms of p53 and p65Rel were found elevated in HFDH liver (Figures S3F, S3G). Unlike SIRT1 targets, acetylation of tubulin (SIRT2 target) was significantly reduced in HFDH liver, whereas SOD2 acetylation (SIRT3 target) was unresponsive to NAM (Figures S3F, S3G). These results underscore the complex, dose-dependent, effects of NAM on protein acetylation.
Diminished NAMPT levels due to NAM supplementation was surprising, despite previous evidence showing the down-modulation of this enzyme with age and HFD (Yoshino et al., 2011). The involvement in the NAM’s effect of the de novo pathway that converts tryptophan to kynurenine to generate NAD+ via NAMN and NAAD intermediates (Oxenkrug, 2013) was investigated. In HFDL mice liver, we found a significant induction of tryptophan 2,3-dioxygenase (IDO). Immunoblotting of total liver lysates showed significant increase in IDO, NMNAT1, and NADS levels under NAM (Figures 4C, S4A). Circulating tryptophan was significantly reduced with NAM supplementation (Figure 4D) without altering hepatic tryptophan levels (data not shown), suggesting that NAM leads to activation of the de novo NAD+ biosynthetic pathway at the expense of NAD salvage.
To assess whether long-term dietary NAM supplementation modifies hepatic NAD+ metabolism, targeted metabolomics was performed using mass spectrometry analysis as described (Trammell et al., 2016a). The normalized fold-change in 13 metabolites from the NAD-related pathways’ metabolome in the six experimental groups are displayed as a heatmap (Figure 4E), and the expression level of different enzymes indicated (Figure 4F, red and blue dots). We found no significant differences between the NAD metabolome of old mice on SD vs. HFD (Figure S4B). Likely, this may be explained by the high carbohydrate content of the SD diet and the effect of age, which appears to dominate the impact of high fat. Though NAM supplementation did not elevate the steady-state concentration of hepatic NAM, it increased in a dose-dependent manner the levels of methylated and oxidized NAM metabolites. Increased Me-Nam upon NAM supplementation (Figures 4F, S4B) correlated perfectly with higher SIRT1 protein accumulation (Figure 4A), consistent with reports that Me-Nam blocks hepatic SIRT1 proteolysis (Hong et al., 2015; Trammell and Brenner, 2015).
Prior work with younger male C57BL/6J mice on a chow diet, HFD or HFD with low streptozotocin showed mildly and greatly depressed hepatic NAD+ and NADP+, respectively, as a function of obesity and diabetes—these dysregulated NAD+ metabolomes were largely corrected by NR supplementation at 3 g/kg of diet (Trammell et al., 2016b). In agreement with previous work, we show a NAM-elicited global alteration in the hepatic acetylation profile, which may underlie differential catabolism in HFD- vs. SD-fed mice. Acetylation of more than 2,000 non-histone proteins regulates key intermediary metabolic processes involved in glycolysis, TCA cycle, and fatty acid metabolism (Wang et al., 2010; Zhao et al., 2010; Choudhary et al., 2009; Kim et al., 2006). The dysregulation in acetylation is linked to various human diseases, such as neurodegeneration, cancer, and metabolic disorders (Iyer 2012; Marks, 2010; Kazantsev and Thompson, 2008). Here, we observed an increase in global acetylation in response to a high dose of NAM, especially in HFD-fed mice. The dual action of NAM, acting both as a SIRT1 inhibitor and NAD+ precursor, illustrates the complex pharmacological profile of this compound.
We describe beneficial effects of chronic NAM supplementation on the healthspan of HFD-fed mice as revealed by liver metabolism and physical activity measurements. Two of the biochemical effects of NAM, namely increased PP activity and reduced protein carbonylation, are processes that are potentially limited by NADPH. However, in the old mice in this study, NAM depressed NAM salvage and did not produce a net boost in the NAD metabolome. While the increased SIRT1 accumulation might have been anticipated based on Me-Nam-dependent stabilization mechanism (Hong et al., 2015; Trammell and Brenner, 2015), the ability of NAM to depress NAMPT expression was not anticipated. Quantitative metabolomic analysis has made it clear that oral NR boosts hepatic NAD metabolism and sirtuin activity independent of NAMPT (Ratajczak et al., 2016; Fletcher et al., 2017), thereby leading to a wave of hepatic NAM production (Trammell et al., 2016a). Whether this metabolite would undermine or amplify the effect of the boosted NAD metabolome remained in question. Further work is needed to determine whether alteration of the control of NAM catabolism in the liver can promote pro-longevity effects like those elicited by the NAD precursors NR and NMN (de Picciotto et al., 2016; Mills et al., 2016). The data in this study support salutary effects of NAM.
Limitations of the study
This long-term study on the effect of NAM supplementation was conducted in male mice under two dietary conditions. Our work provides quantifiable evidence that NAM exerts beneficial effects on hepatic glucose metabolism regulation and oxidative stress reduction in the liver of HFD-fed mice, coincident with improved behavioral/physical and metabolic performance without extending longevity. It remains unclear whether similar effects can be observed in other strains of mice and in females. Longitudinal evaluation of phenotypes associated with healthspan would have allowed a better assessment of the impact of NAM on behavioral and motor function tests.
Concluding Remarks
We show for the first time that chronic NAM supplementation can be well-tolerated and prevents diet-induced hepatosteatosis while improving glucose metabolism and redox status in livers of HFD-fed mice at 118 weeks of age, independently from food intake, body weight or body composition. In HFD-fed mice, NAM restored glycogen deposition to the levels observed in the SD-fed group, and in liver-derived HepG2 cells augmented flux through glucose uptake, glycolysis and PP pathways in the presence of the fatty acid palmitate (to mimic HFD). The remarkable stimulation of the glucose flux redirection through the PP pathway suggests that the favorable effect of NAM on glucose homeostasis might be linked to improved cytoplasmic redox balance via augmented PP pathway-derived NADPH, the main electron donor for cytoplasmic and mitochondrial antioxidant systems. The salutary effects of NAM on reducing oxidative stress and inflammation represent potentially counteracting factors against DNA damage elicited by age and HFD.
STAR METHODS
KEY RESOURCES TABLE
The table has been sent as a separate document entitled: “key resource Table_NAM_011318”.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| rabbit anti-acetyl-lysine | ImmunoChem Pharmaceuticals Inc. | cat#ICP0380 |
| mouse anti-tubulin | Santa Cruz Biotechnology | cat#sc-5286; RRID: AB_628411 |
| rabbit anti-acetylated tubulin | Cell Signaling Technology | cat#5335S; RRID:AB_10544694 |
| mouse anti-SIRT1 | Sigma-Aldrich | cat#S5196; RRID:AB_532284 |
| mouse anti-p53 | Cell Signaling Technology | cat#2524; RRID:AB_331743 |
| rabbit anti-acetylated p53 | Cell Signaling Technology | cat# 2570S; RRID:AB_823591 |
| rabbit anti-p65 | Abcam | cat#ab32536; RRID:AB_776751 |
| rabbit anti-acetylated p65 | Abcam | cat#ab19870; RRID:AB_776753 |
| rabbit anti-SOD2 | Abcam | cat#ab13533; RRID:AB_300434 |
| rabbit anti-acetylated SOD2 | Abcam | cat#ab137037 |
| rabbit anti-NAMPT | Bethyl Laboratories | cat#A300-372A-M |
| goat anti-IDO | Abcam | cat#ab134197 |
| rabbit anti-NMNAT-1 | Santa Cruz | cat#sc-98249; RRID:AB_2153131 |
| rabbit anti-NADS | Abcam | cat#ab139561 |
| Bacterial and Virus Strains | ||
| Biological Samples | ||
| Chemicals, Peptides, and Recombinant Proteins | ||
| Critical Commercial Assays | ||
| Mouse insulin ELISA | Crystal Chem | 90080 |
| OxiSelect Protein Carbonyl Immunoblot kit | Cell Biolabs, Inc. | STA-308 |
| Deposited Data | ||
| Experimental Models: Cell Lines | ||
| Human: HepG2 | ATCC | HB-8065 |
| Experimental Models: Organisms/Strains | ||
| Mouse: C57BL/6J | The Jackson Laboratory | JAX 000664 |
| Oligonucleotides | ||
| Recombinant DNA | ||
| Software and Algorithms | ||
| Prism 6.0 | GraphPad | http://www.graphpad.com/scientific-software/prism; RRID:SCR_015807 |
| Excel 2010 | Microsoft Corp. | |
| SigmaStat 3.0 | Aspire Software Int. | http://www.sigmaplot.com/products/sigmaplot/sigmaplot-details.php RRID:SCR_010285 |
| ImageJ | National Institutes of Health (NIH) | https://imagej.nih.gov/ij/; RRID:SCR_003070 |
| Other | ||
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by Rafael de Cabo (decabora@mail.nih.gov).
EXPERIMENTAL MODEL DETAILS
Animals and Diets
Male C57BL/6J mice were purchased from the Jackson Laboratory (Bar Harbor, ME) (stock number: 000664) and housed at the Gerontology Research Center and Biomedical Research Center (Baltimore, MD). Mice were housed in cages of four with ad libitum access to diet and tap water. Mice were electronically tagged for identification (Biomedic Data System Inc., Maywood, NJ), and bodyweight and food intake were monitored every two weeks from the start of the study for the duration of their lifespan. Animal rooms were maintained at 20–22°C with 30–70% relative humidity and a 12-hour light/dark cycle. Diets were started at 56 weeks of age after randomization into six groups of 100 mice per group. Mice were fed one of six study diets: (a) standard AIN-93G diet (SD; carbohydrate:protein:fat ratio of 64:19:17 percent of kcal), (b) SD + 0.5g nicotinamide (NAM)/kg chow (SDL), (c) SD+1.0g NAM/kg chow (SDH), (d) a high fat diet (HFD; carbohydrate:protein:fat ratio of 16:23:61 percent of kcal), (e) HFD+0.5g NAM/kg chow (HFDL) or (f) HFD + 1.0g NAM/kg chow (HFDH). These levels of NAM corresponded to 37.5 (SHD, HDFL) and 75 (SDH, HFDH) mg/g BW/day. Study diets were purchased from Dyets, Inc. (Bethlehem, PA). NAM was obtained from Sigma-Aldrich (St Louis, MO). No inclusion or exclusion criteria were used. All animal protocols were approved by the Animal Care and Use Committee (352-TGB-2013, 352-TGB-2016) of the National Institute on Aging.
Survival Study
Animals were inspected twice daily for health issues and deaths were recorded for each animal. Moribund animals were euthanized and every animal found dead or euthanized was necropsied. Criteria for euthanasia were based on an independent assessment by a veterinarian according to the AAALAC guidelines. Only cases where the condition of the animal was considered incompatible with continued survival are represented as deaths in the curves. Animals removed at sacrifice or euthanized due to reasons unrelated to incompatible survival were considered as censored deaths and these included animals that died due to flooded cages or sacrificed for tissue collection. For euthanasia and subsequent tissue collection, mice were on their normal feeding cycle (study diet adlibitum) and not fasted.
Cell Culture
The HepG2 human hepatocellular carcinoma cell line (ATCC, Manassas, VA) was derived from the liver tissue of a 15-year-old Causasian male. ATCC uses short tandem repeat (STR) analysis to authenticate cell lines. Upon receipt from ATCC, HepG2 cells were expanded for a few passages to generate new frozen stocks. Cells were resuscitated as needed and used for no more than 10–12 passages or no more than 8–10 weeks after resuscitation. Cells were maintained in MEM supplemented with 2 mM Glutamax, 1% sodium pyruvate, 100 units/ml penicillin, 100 μg/ml streptomycin, and 10% fetal bovine serum (Hyclone, Logan, UT). Cells were maintained in culture at 37 °C in a humidified incubator with 5% CO2 and the medium was replaced every 2–3 days. Details about metabolic flux analysis in these cells can be found in a sub-section below.
METHOD DETAILS
Body Composition
Measurements of lean, fat and fluid mass in live mice were acquired by nuclear magnetic resonance (NMR) using the Minispec LF90 (Bruker Optics, Billerica, MA). Measurements were made in 81- and 107-week old mice, which correspond to 25 and 51 weeks on diet, respectively.
Metabolic Assessment
Mouse metabolic rate was assessed by indirect calorimetry in open-circuit oxymax chambers using the Comprehensive Lab Animal Monitoring System (CLAMS; Columbus Instruments, Columbus, OH) as previously described (Minor et al., 2011). Mice (105-week old, 49 weeks on diet) were housed singly with water and food available ad libitum and maintained at ~24°C under a 12:12-h light-dark cycle (light period 0600-1800). All mice were acclimatized to monitoring cages for 3–6 h before recording began. Sample air was passed through an oxygen (O2) sensor for determination of O2 content. O2 consumption was determined by measuring oxygen concentration in air entering the chamber compared with air leaving the chamber. The sensor was calibrated against a standard gas mix containing defined quantities of O2, carbon dioxide (CO2), and nitrogen (N2). Constant airflow (0.6 L/min) was drawn through the chamber and monitored by a mass-sensitive flow meter. The concentrations of O2 and CO2 were monitored at the inlet and outlet of the sealed chambers to calculate oxygen consumption. Each chamber was measured for 30 s at 30-min intervals and data were recorded for 60 h total. Movement (both horizontal and vertical) was also monitored. The system has beams 0.5-inch apart on the horizontal plane providing a high-resolution grid covering the XY-planes and the software provides counts of beam breaks by the mouse in 30-s epochs.
Behavioral Assessment
Mice were tested at the same time of day over a five-day period. On the testing day, mice were brought to the testing room and allowed to acclimatize for 2–3 h. Rotarod. Mice were given a habituation trial on day 1 where they were placed on the rotarod at a constant speed (4 r.p.m.) and had to remain on the rotarod for 5 min. On the testing day, mice were placed on the accelerating rotarod (4–40 r.p.m. over 5 min) and given three trials (30-min rest period in between trials). Results shown are the average of three trials per mouse. The maximum trial length was 5 min. Cage top. A metal cage top was turned upside down, with its sides covered in duct tape, and the experimenter placed the mouse on top of cage top. The cage top was then turned until the mouse was hanging from the cage top with all four limbs grasping the bars. Each mouse was given three trials (maximum latency of 60 s) with a 30-min rest period in between trials. Data is shown as the average of three tests. Open field. Mice were placed individually into the center of a plexiglass square measuring 23in × 23in. The open field was evenly illuminated and exploratory behavior was measured for 300 s using AnyMaze software (Stoelting Co., Wood dale, IL). The apparatus was cleaned with 70% alcohol before testing the next mouse. This was repeated over a five-day period and data are presented as the average over this period. Mice were 115-week old, 59 weeks on diet.
Oral Glucose Tolerance Test (OGTT)
Following an overnight fast, mice (97-week old, 41 weeks on diet, n=6 per group) received a 30% glucose solution (1.5 g/kg glucose by gavage). Blood glucose was measured using an Breeze2 glucose meter (Bayer, Mishawaka, IN) at 0, 15, 30, 60 and 120 min following gavage.
Serum Markers and HOMA-IR Calculation
Fasting blood glucose (16h fast) was determined using an Breeze2 glucose meter and fasting serum insulin (16h fast) was measured using an enzyme-linked immunosorbent assay (Crystal Chem, Downers Grove, IL) according to the manufacturer’s instructions. Insulin resistance was calculated from fasted glucose and insulin values using the HOMA2 Calculator software available from the Oxford Centre for Diabetes, Endocrinology and Metabolism, Diabetes Trials Unit website (www.dtu.ox.ac.uk).
Histology
Fresh frozen liver sections from non-fasted mice were cut on a cryostat (Leica Microsystems, Wetzlar, Germany) at 10 μm thickness and mounted on glass coverslips. H & E staining was performed to evaluate liver architecture while Periodic-Acid Schiff (PAS) reagent was used to stain for glycogen according to well-established protocols. Scoring of the glycogen content (PAS staining) was performed by 3 independent observers blinded to the treatment groups. This was performed on at least 10 fields/animal (minimum 60 images/treatment group) with the data representing the average of three scores. PAS staining was determined by assigning an equally weighted arbitrary score of 0 (no visible glycogen staining) to 3 (normal glycogen deposition) for each image and averaging the score per treatment group.
Gel Electrophoresis and Western Blotting
Separation of mouse liver extracts was carried out according to standard procedures. N=6 mice per group, 118-week old, 62 weeks on dietary intervention. Tissues were lysed either in radioimmunoprecipitation buffer (RIPA) or urea lysis buffer supplemented with ethylenediaminetetraacetic acid and ethylene glycol tetraacetic acid (Boston BioProducts, Ashland, MA) along with KDAC inhibitors [10 μM trichostatin A, 10 mM NAM, and 50 mM butyric acid, all from Sigma-Aldrich], and protease and phosphatase inhibitors (Roche, Indianapolis, IN). Following homogenization with a polytron homogenizer, samples were centrifuged (14,000 rpm, 30 min at 4°C) and protein concentration in the clarified lysates was then quantified using the Bradford assay method (Bio-Rad). Proteins were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis under reducing conditions and then transferred to nitrocellulose membranes. Western blots were performed according to standard methods unless otherwise specified. Membranes were blocked for 1 h in 5% milk, and then incubated overnight at 4°C with the antibody of interest, followed by incubation with a secondary antibody. Antibodies used were anti-acetyl-lysine antibody (cat#: ICP0380; ImmunoChem Pharmaceuticals Inc., Burnaby, BC, Canada); tubulin (cat#: sc-5286; Santa Cruz Biotechnology, Santa Cruz, CA); acetylated tubulin (cat#: cs-5335; Cell Signaling Technology (CST), Denvers, MA); SIRT1 (cat#: s5196, Sigma-Aldrich; and cat#: ab110304, Abcam, Cambridge, MA); p53 (cat#: cs-2524; CST); acetylated p53 (cat#: cs-2570; CST); p65 (cat#: ab32536; Abcam); acetylated p65 (cat#: ab19870; Abcam); SOD2 (cat#: ab13533; Abcam), acetylated SOD2 (cat#: ab137037; Abcam); NAMPT (cat.# A300-372A-M, Bethyl Laboratories, Montgomery, TX); IDO (cat.# ab134197, Abcam); NMNAT-1 (cat# sc98249, Santa Cruz); NADS (cat# ab139561, Abcam). The visualization of immunoreactive bands was performed using the ECL Plus Western blotting detection system (GE Healthcare, Pascataway, NJ). The quantification was performed by volume densitometry using ImageJ software (National Institutes of Health, Bethesda, MD) and normalization to Ponceau S staining (Sigma-Aldrich).
Metabolomics
Metabolomic analysis was performed by the West Coast Metabolomics Center at UC Davis (Davis, CA) in livers and serum of non-fasted animals as previously described (Mitchell et al., 2016). In brief, liver tissue (4 mg) was homogeneized in extraction solution (acetonitrile:isopropanol:water, 3:3:2), then vortexed for 45 s and incubated for 5 min at 4 °C. Following centrifugation at 14,000 rcf for 2 min, two aliquots of the supernatant (500 μl each aliquot) were made for analysis and one for backup. One aliquot was dried via evaporation overnight in the Labconco Centrivap cold trap concentrator (Labconco, Kansas City, MO). The dried aliquot was then resuspended with 500 μl 50% acetonitrile (degassed) and centrifuged at 14,000 rcf for 2 min. The supernatant was transferred to a clean Eppendorf tube and evaporated again to dryness. Internal standards (C8–C30 fatty acid methyl esters) were then added and the sample was derivatized by methoxyamine. HCl in pyridine and subsequently by N-methyl-N-trimethylsilyltrifluoro acetemide for trimethylsilylation of acidic protons. Data were acquired using the method as described (Fiehn et al., 2008), which is briefly summarized in Mitchell et al. (2016). Data are presented as a ratio of the metabolite to the total metabolites returned. N=6 mice per group, 118-week old, 62 weeks on dietary intervention.
Determination of Metabolic Fluxes in HepG2 cells
HepG2 cells were seeded at 1.6 × 104 cells on a 24-multiwell Seahorse plate in low-glucose DMEM supplemented with 5% FBS, 100 nM insulin, and in presence of NAM (0, 1 or 2 mM) overnight. Cells were washed twice and then maintained in Seahorse glucose-free DMEM in the absence of FBS, insulin, and NAM for 45 min in the temperature controlled instrument set at 37 °C before the addition of glucose (1 mM final concentration), insulin (100 nM), and NAM (0, 1 or 2 mM) for 1 h, as indicated. After three baseline measurements of the oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) on the Seahorse XF24 instrument (North Billerica, MA), aliquots of stock solutions containing glucose (4 mM final concentration + 1 mM already with cells = 5 mM), palmitate (200 μM in a 4:1 ratio with fatty acid-free BSA), or the combination glucose + palmitate were added. For the next hour, the metabolic phenotype of these cells —mitochondrial respiration and glycolysis— was measured through the relative utilization of the two substrates. Preliminary experiments were carried out to demonstrate the linearity of the OCR and ECAR response as a function of the cell number and the expected behavior in the presence of the mitochondrial inhibitors, oligomycin (10 μM), FCCP (1.23 μM), and antimycin A/rotenone (10 μM each) (Reily et al., 2013).
Metabolic flux balance analysis (Savinell and Palsson, 1992; Cortassa et al., 1995) was used to estimate the fluxes through the network of metabolic pathways involved in the degradation of glucose and palmitate (Palm) consumption. The algebraic calculation of metabolic fluxes requires the input of several fluxes: glucose uptake (VGlc), lactate efflux (VLact), oxygen consumption (VO2), Palm oxidation (VPalm_Ox) and ATP demand (VATPdemand, estimated from the rate of protein synthesis during growth under similar conditions to those used in the Seahorse experiments under the assumption that protein synthesis is ~50% of the total ATP demand). Those five fluxes were used as a reference in the algebraic solution corresponding to the estimated fluxes through the metabolic network considered. These estimated fluxes include VPPP, which stands for the glucose flux through the pentose phosphate (PP) pathway that in the presence of NAM is derived from degradation of glycogen stores, glycolysis (VGlycolysis), citrate synthase (VCit), pyruvate dehydrogenase (VPDH), and pyruvate carboxylase (VPCb).
Quantitation of the NAD+ Metabolome
Sample preparation and targeted quantitative metabolomics were carried out for complete analysis of the NAD+ metabolome as described (Trammell et al., 2016a). Heatmaps of the average intensity of metabolites in each group and Log2(fold change) calculation were performed using Microsoft Excel (Microsoft, Redmond, WA). Mice were 118-week old, and 62 weeks on dietary intervention. SD, n=6; SDL, n=5; SDH, n=5; HFD, n=6; HFDL, n=6; HFDH, n=5.
QUANTIFICATION AND STATISTICAL ANALYSIS
Mortality during the survival study was assessed using the log rank test to compare the differences in Kaplan-Meier survival curves. Maximal lifespan was defined as the 10th percentile of mice still alive. Two-way analysis of variance (ANOVA) was performed for mRNA, protein expression, or metabolomics analysis, and the two independent factors were diet (SD or HFD) and NAM concentrations (none, low and high). Data are expressed as means ± standard error of the mean (SEM), with p value of ≤0.05 considered statistically significant. The statistical parameters (n, mean, SEM) can be found within the figure legends. No statistical method was used to determine whether the data met assumptions of the statistical approach. Analyses were performed using Graph Pad Prism 6.0 (San Diego, CA), Excel 2010 (Microsoft Corp., Redmond, WA), and SigmaStat 3.0 (Aspire Software International, Ashburn, VA).
Supplementary Material
Highlights.
Nicotinamide (NAM) supplementation does not extend lifespan in mice
NAM prevents hepatosteatosis in obese mice while improving glucose metabolism
NAM reduces oxidative stress and inflammation
NAM depresses NAM salvage and does not produce a net boost in tissue NAD levels
Acknowledgments
This research was conducted under a Cooperative Research and Development Agreement (CRADA) between Glaxo Smith-Kline and the National Institute on Aging, National Institutes of Health (NIA/NIH), and supported in part by the Intramural Research Program of the NIA/NIH. E.Y.K. was supported by a grant from the KRIBB Research Initiative Program (Korean Biomedical Scientist Fellowship Program), Korea Research Institute of Bioscience and Biotechnology, Republic of Korea. D.A.S. was supported by The Paul F. Glenn Foundation for Medical Research, NIH/NIA MERIT award R01 AG028730, and a gift from Edward Schulak. Special thanks to the members of the Translational Gerontology Branch and the Comparative Medicine Section of the NIA. We acknowledge Dawn Phillips, Dawn Nines and Justice Lucas for animal care; Olga Carlson for insulin measurements; Elin Lehrmann and Paul Bastian for their expertise in microarray analysis; Theresa Ward, Robin K. Minor, Michael A. Petr, Irene Alfaras, and Joseph H. Garcia for their contributions to this work.
Footnotes
AUTHOR CONTRIBUTIONS
R.d.C., J.L.E., P.J.E., D.A.S., and M.B. conceived and designed the study. S.J.M. and H.H.P. performed in vivo experiments, gross necropsies, histopathological analyses as well as mouse lifespan analysis. S.J.M. and T.K. performed histology staining. E.F.F., T.B.W., and V.A.B. contributed to and analyed some of the in vivo studies. H.P.P., A.A., I.N.E., A.D.F., and E.Y.K. contributed to Western blot analysis and E.Y.K. assisted in analyzing the metabolomics data. M.A.A. and S.C. performed metabolic flux analyses and contributed to analyzing the metabolomics data. D.W.F., J.A.B., N.Z., A.D.F., A.A.S., M.S.S. and C.B. contributed to analyzing the results from targeted metabolomics and quantitation of NAD+-related metabolites. M.B and M.A.A. took the leading role in writing the manuscript and creating the figures. Most authors contributed to the editing and proofreading of the final draft.
DECLARATION of INTERESTS
J.L.E. is a former employee and a stockholder in GlaxoSmithKline (GSK), and a current SAB member of Metro Biotech. P.J.E. is a former employee of Sirtris, a GSK company. V.A.B. has a CRADA with ChromaDex. C.B. owns stock in ChromaDex and serves as a consultant to ChromaDex and Cytokinetics. AAS receives royalties on commercial sales of nicotinamide riboside from ChromaDex. AAS is also a consultant and co-founder of Metro MidAtlantic Biotech LLC. D.A.S. is a consultant, equity owner, and inventor on patents licensed to Life Biosciences, Metro International Biotech, Jumpstart Fertility, Liberty Biosecurity, Ovascience and GSK. The remaining authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bogan KL, Brenner C. Nicotinic acid, nicotinamide, and nicotinamide riboside: a molecular evaluation of NAD+ precursor vitamins in human nutrition. Annu Rev Nutr. 2008;28:115–130. doi: 10.1146/annurev.nutr.28.061807.155443. [DOI] [PubMed] [Google Scholar]
- Trammell SA, Schmidt MS, Weidemann BJ, Redpath P, Jaksch F, Dellinger RW, Li Z, Abel ED, Migaud ME, Brenner C. Nicotinamide riboside is uniquely and orally bioavailable in mice and humans. Nat Commun. 2016a;7:12948. doi: 10.1038/ncomms12948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bieganowski P, Brenner C. Discoveries of nicotinamide riboside as a nutrient and conserved NRK genes establish a Preiss-Handler independent route to NAD+ in fungi and humans. Cell. 2004;117:495–502. doi: 10.1016/s0092-8674(04)00416-7. [DOI] [PubMed] [Google Scholar]
- Belenky P, Racette FG, Bogan KL, McClure JM, Smith JS, Brenner C. Nicotinamide riboside promotes Sir2 silencing and extends lifespan via Nrk and Urh1/Pnp1/Meu1 pathways to NAD+ Cell. 2007a;129:473–484. doi: 10.1016/j.cell.2007.03.024. [DOI] [PubMed] [Google Scholar]
- Bitterman KJ, Anderson RM, Cohen HY, Latorre-Esteves M, Sinclair DA. Inhibition of silencing and accelerated aging by nicotinamide, a putative negative regulator of yeast sir2 and human SIRT1. J Biol Chem. 2002;277:45099–45107. doi: 10.1074/jbc.M205670200. [DOI] [PubMed] [Google Scholar]
- Anderson RM, Bitterman KJ, Wood JG, Medvedik O, Cohen H, Lin SS, Manchester JK, Gordon JI, Sinclair DA. Manipulation of a nuclear NAD+ salvage pathway delays aging without altering steady-state NAD+ levels. J Biol Chem. 2002;277:18881–18890. doi: 10.1074/jbc.M111773200. [DOI] [PubMed] [Google Scholar]
- Belenky P, Bogan KL, Brenner C. NAD+ metabolism in health and disease. Trends Biochem Sci. 2007b;32:12–19. doi: 10.1016/j.tibs.2006.11.006. [DOI] [PubMed] [Google Scholar]
- Cantó C, Menzies KJ, Auwerx J. NAD(+) Metabolism and the Control of Energy Homeostasis: A Balancing Act between Mitochondria and the Nucleus. Cell Metab. 2015;22:31–53. doi: 10.1016/j.cmet.2015.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verdin E. NAD+ in aging, metabolism, and neurodegeneration. Science. 2015;350:1208–1213. doi: 10.1126/science.aac4854. [DOI] [PubMed] [Google Scholar]
- Camacho-Pereira J, Tarragó MG, Chini CC, Nin V, Escande C, Warner GM, Puranik AS, Schoon RA, Reid JM, Galina A, et al. CD38 Dictates Age-Related NAD Decline and Mitochondrial Dysfunction through an SIRT3-Dependent Mechanism. Cell Metab. 2016;23:1127–1139. doi: 10.1016/j.cmet.2016.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheibye-Knudsen M, Mitchell SJ, Fang EF, Iyama T, Ward T, Wang J, Dunn CA, Singh N, Veith S, Hasan-Olive MM, et al. A high-fat diet and NAD(+) activate Sirt1 to rescue premature aging in cockayne syndrome. Cell Metab. 2014;20:840–855. doi: 10.1016/j.cmet.2014.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneko S, Wang J, Kaneko M, Yiu G, Hurrell JM, Chitnis T, Khoury SJ, He Z. Protecting axonal degeneration by increasing nicotinamide adenine dinucleotide levels in experimental autoimmune encephalomyelitis models. J Neurosci. 2006;26:9794–9804. doi: 10.1523/JNEUROSCI.2116-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swerdlow RH. Is NADH effective in the treatment of Parkinson’s disease? Drugs Aging. 1998;13:263–268. doi: 10.2165/00002512-199813040-00002. [DOI] [PubMed] [Google Scholar]
- Lee CF, Chavez JD, Garcia-Menendez L, Choi Y, Roe ND, Chiao YA, Edgar JS, Goo YA, Goodlett DR, Bruce JE, et al. Normalization of NAD+ Redox Balance as a Therapy for Heart Failure. Circulation. 2016;134:883–894. doi: 10.1161/CIRCULATIONAHA.116.022495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes AP, Price NL, Ling AJ, Moslehi JJ, Montgomery MK, Rajman L, White JP, Teodoro JS, Wrann CD, Hubbard BP, et al. Declining NAD(+) induces a pseudohypoxic state disrupting nuclear-mitochondrial communication during aging. Cell. 2013;155:1624–1638. doi: 10.1016/j.cell.2013.11.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantó C, Houtkooper RH, Pirinen E, Youn DY, Oosterveer MH, Cen Y, Fernandez-Marcos PJ, Yamamoto H, Andreux PA, Cettour-Rose P, et al. The NAD(+) precursor nicotinamide riboside enhances oxidative metabolism and protects against high-fat diet-induced obesity. Cell Metab. 2012;15:838–847. doi: 10.1016/j.cmet.2012.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trammell SA, Weidemann BJ, Chadda A, Yorek MS, Holmes A, Coppey LJ, Obrosov A, Kardon RH, Yorek MA, Brenner C. Nicotinamide Riboside Opposes Type 2 Diabetes and Neuropathy in Mice. Sci Rep. 2016b;6:26933. doi: 10.1038/srep26933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshino J, Mills KF, Yoon MJ, Imai S. Nicotinamide mononucleotide, a key NAD(+) intermediate, treats the pathophysiology of diet- and age-induced diabetes in mice. Cell Metab. 2011;14:528–536. doi: 10.1016/j.cmet.2011.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan NA, Auranen M, Paetau I, Pirinen E, Euro L, Forsström S, Pasila L, Velagapudi V, Carroll CJ, Auwerx J, et al. Effective treatment of mitochondrial myopathy by nicotinamide riboside, a vitamin B3. EMBO Mol Med. 2014;6:721–731. doi: 10.1002/emmm.201403943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imai S. A possibility of nutriceuticals as an anti-aging intervention: activation of sirtuins by promoting mammalian NAD biosynthesis. Pharmacol Res. 2010;6:242–247. doi: 10.1016/j.phrs.2010.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu D, Zhang H, Ropelle ER, Sorrentino V, Mázala DA, Mouchiroud L, Marshall PL, Campbell MD, Ali AS, Knowels GM, et al. NAD+ repletion improves muscle function in muscular dystrophy and counters global PARylation. Sci Transl Med. 2016;8:361ra139. doi: 10.1126/scitranslmed.aaf5504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratajczak J, Joffraud M, Trammell SA, Ras R, Canela N, Boutant M, Kulkarni SS, Rodrigues M, Redpath P, Migaud ME, et al. NRK1 controls nicotinamide mononucleotide and nicotinamide riboside metabolism in mammalian cells. Nat Commun. 2016;7:13103. doi: 10.1038/ncomms13103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang SJ, Choi JM, Kim L, Park SE, Rhee EJ, Lee WY, Oh KW, Park SW, Park CY. Nicotinamide improves glucose metabolism and affects the hepatic NAD-sirtuin pathway in a rodent model of obesity and type 2 diabetes. J Nutr Biochem. 2014;25:66–72. doi: 10.1016/j.jnutbio.2013.09.004. [DOI] [PubMed] [Google Scholar]
- Qi Z, Xia J, Xue X, He Q, Ji L, Ding S. Long-term treatment with nicotinamide induces glucose intolerance and skeletal muscle lipotoxicity in normal chow-fed mice: compared to diet-induced obesity. J Nutr Biochem. 2016;36:31–41. doi: 10.1016/j.jnutbio.2016.07.005. [DOI] [PubMed] [Google Scholar]
- Fletcher RS, Ratajczak J, Doig CL, Oakey LA, Callingham R, Da Silva Xavier G, Garten A, Elhassan YS, Redpath P, Migaud ME, et al. Nicotinamide riboside kinases display redundancy in mediating nicotinamide mononucleotide and nicotinamide riboside metabolism in skeletal muscle cells. Mol Metab. 2017;6:819–832. doi: 10.1016/j.molmet.2017.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou CC, Yang X, Hua X, Liu J, Fan MB, Li GQ, Song J, Xu TY, Li ZY, Guan YF, et al. Hepatic NAD(+) deficiency as a therapeutic target for non-alcoholic fatty liver disease in ageing. Br J Pharmacol. 2016;173:2352–2368. doi: 10.1111/bph.13513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohsaka A, Laposky AD, Ramsey KM, Estrada C, Joshu C, Kobayashi Y, Turek FW, Bass J. High-fat diet disrupts behavioral and molecular circadian rhythms in mice. Cell Metab. 2007;6:414–421. doi: 10.1016/j.cmet.2007.09.006. [DOI] [PubMed] [Google Scholar]
- Bechmann LP, Hannivoort RA, Gerken G, Hotamisligil GS, Trauner M, Canbay A. The interaction of hepatic lipid and glucose metabolism in liver diseases. J Hepatol. 2012;56:952–964. doi: 10.1016/j.jhep.2011.08.025. [DOI] [PubMed] [Google Scholar]
- Brownlee M. The pathobiology of diabetic complications: a unifying mechanism. Diabetes. 2005;54:1615–1625. doi: 10.2337/diabetes.54.6.1615. [DOI] [PubMed] [Google Scholar]
- Hue L, Taegtmeyer H. The Randle cycle revisited: a new head for an old hat. Am J Physiol Endocrinol Metab. 2009;297:E578–E591. doi: 10.1152/ajpendo.00093.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savinell JM, Palsson BO. Optimal selection of metabolic fluxes for in vivo measurements. I. Development of Mathematical Methods. J Theor Biol. 1992;155:201–214. doi: 10.1016/s0022-5193(05)80595-8. [DOI] [PubMed] [Google Scholar]
- Cortassa S, Aon JC, Aon MA. Fluxes of carbon, phosphorylation, and redox intermediates during growth of Saccharomyces cerevisiae on different carbon sources. Biotechnol Bioeng. 1995;47:193–208. doi: 10.1002/bit.260470211. [DOI] [PubMed] [Google Scholar]
- Kembro JM, Aon MA, Winslow RL, O’Rourke B, Cortassa S. Integrating mitochondrial energetics, redox and ROS metabolic networks: a two-compartment model. Biophys J 1. 2013;04:332–343. doi: 10.1016/j.bpj.2012.11.3808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo J, Nikolaev AY, Imai S, Chen D, Su F, Shiloh A, Guarante L, Gu W. Negative control of p53 by Sir2alpha promotes cell survival under stress. Cell. 2001;107:137–148. doi: 10.1016/s0092-8674(01)00524-4. [DOI] [PubMed] [Google Scholar]
- Guan X, Lin P, Knoll E, Chakrabarti R. Mechanism of inhibition of the human sirtuin enzyme SIRT3 by nicotinamide: computational and experimental studies. PLoS One. 2014;9:e107729. doi: 10.1371/journal.pone.0107729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oxenkrug G. Insulin resistance and dysregulation of tryptophan-kynurenine and kynurenine-nicotinamide adenine dinucleotide metabolic pathways. Mol Neurobiol. 2013;48:294–301. doi: 10.1007/s12035-013-8497-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong S, Moreno-Navarrete JM, Wei X, Kikukawa Y, Tzameli I, Prasad D, Lee Y, Asara JM, Fernandez-Real JM, Maratos-Flier E, et al. Nicotinamide N-methyltransferase regulates hepatic nutrient metabolism through Sirt1 protein stabilization. Nat Med. 2015;21:887–894. doi: 10.1038/nm.3882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trammell SA, Brenner C. NNMT: A Bad Actor in Fat Makes Good in Liver. Cell Metab. 2015;22:200–201. doi: 10.1016/j.cmet.2015.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Zhang Y, Yang C, Xiong H, Lin Y, Yao J, Li H, Xie L, Zhao W, Yao Y, et al. Acetylation of metabolic enzymes coordinates carbon source utilization and metabolic flux. Science. 2010;327:1004–1007. doi: 10.1126/science.1179687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao S, Xu W, Jiang W, Yu W, Lin Y, Zhang T, Yao J, Zhou L, Zeng Y, Li H, et al. Regulation of cellular metabolism by protein lysine acetylation. Science. 2010;327:1000–1004. doi: 10.1126/science.1179689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhary C, Kumar C, Gnad F, Nielsen ML, Rehman M, Walther TC, Olsen JV, Mann M. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science. 2009;325:834–840. doi: 10.1126/science.1175371. [DOI] [PubMed] [Google Scholar]
- Kim SC, Sprung R, Chen Y, Xu Y, Ball H, Pei J, Cheng T, Kho Y, Xiao H, Xiao L, et al. Substrate and functional diversity of lysine acetylation revealed by a proteomics survey. Mol Cell. 2006;23:607–618. doi: 10.1016/j.molcel.2006.06.026. [DOI] [PubMed] [Google Scholar]
- Iyer A, Fairlie DP, Brown L. Lysine acetylation in obesity, diabetes and metabolic disease. Immunol Cell Biol. 2012;90:39–46. doi: 10.1038/icb.2011.99. [DOI] [PubMed] [Google Scholar]
- Marks PA. The clinical development of histone deacetylase inhibitors as targeted anticancer drugs. Expert Opin Investig Drugs. 2010;19:1049–1066. doi: 10.1517/13543784.2010.510514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazantsev AG, Thompson LM. Therapeutic application of histone deacetylase inhibitors for central nervous system disorders. Nat Rev Drug Discov. 2008;7:854–868. doi: 10.1038/nrd2681. [DOI] [PubMed] [Google Scholar]
- de Picciotto NE, Gano LB, Johnson LC, Martens CR, Sindler AL, Mills KF, Imai S, Seals DR. Nicotinamide mononucleotide supplementation reverses vascular dysfunction and oxidative stress with aging in mice. Aging Cell. 2016;15:522–530. doi: 10.1111/acel.12461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills KF, Yoshida S, Stein LR, Grozio A, Kubota S, Sasaki Y, Redpath P, Migaud ME, Apte RS, Uchida K, et al. Long-term administration of nicotinamide mononucleotide mitigates age-associated physiological decline in mice. Cell Metab. 2016;24:1–12. doi: 10.1016/j.cmet.2016.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.