

Abstract
We report a concise chemical synthesis of kalihinol C via a possible biosynthetic intermediate, ‘protokalihinol,’ which was targeted as a scaffold en route to antiplasmodial analogs. High stereocontrol of the kalihinol framework relies on a heterodendralene cascade to establish the target stereotetrad. Common problems of regio- and chemoselectivity encountered in the kalihinol class are explained and solved.
Graphical Abstract
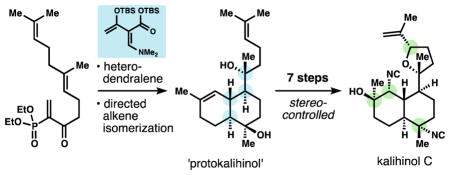
The kalihinols (Figure 1) possess the highest skeletal and functional group complexity of the biologically-enigmatic isocyanoterpene (ICT) class.1,2 Kalihinol A3a also exhibits the highest reported potency of the ICTs against P. falciparum,3b killing with an EC50 of 1 nM, but the mechanism of action has not been rigorously assigned. Proposed mechanisms to explain phenotypic effects of the ICTs include inhibition of heme detoxification4 or copper chelation,5 but these proposals do not fully account for the structure-activity relationships and life-cycle activities reported. For example, our discovery that the amphilectenes and adocianes are cytotoxic against liver-stage parasites militates against heme detoxification inhibition as the exclusive antiplasmodial mechanism.6 Copper chelation is simply not possible for congeners with distant isonitriles. As part of a program to investigate the biological activity of ICTs, we have begun to develop effective chemical syntheses2,6,7 and associated methods8 to produce and modify three main structural classes: amphilectenes, adocianes, and kalihinols.1,2 Prior syntheses9 of the kalihinol class have fought to control stereochemistry in the functionally dense scaffolds, and each contains at least one uncontrolled (ca. 1:1 d.r.) stereogenic step.10 Here we report a short and fully stereocontrolled synthesis of kalihinol C (1) enabled by a new hetereodendralene building block, a directed alkene isomerization and a new method for isonitrile synthesis.
Figure 1.

a. Kalihinol congeners b. hypothetical biosynthesis that informs a proposed chemical synthesis.
The kalihinols appear to derive from a common intermediate, a ‘protokalihinol,’ (2a) where the tetrahydrofurans or -pyrans derive from oxidative cyclization of a pendant prenyl unit. The protokalihinol framework (a dihydroxy-bifloran diterpene)1 would arise from bisabolyl cation intermediate 3 via cation-olefin cyclization and concomitant stereoselective capture of water.11 While such a pathway might globally simplify formation of the kalihinol stereotetrad (in blue), we thought intra-molecular capture of oxygen in synthon 4 might be more realistic than stereoselective carbocation hydration.12
Recently, our lab reported short syntheses of amphilectene7 and adociane6 ICTs that relied on a new class of polarized dendrimeric polyene13 (Danishefsky dendralenes) that forges the stereochemically dense core of these and other terpenes14 in highly diastereoselective Diels-Alder cascades. Given the structural correspondence between amphilectenes and kalihinols, we realized that a dendralene-based approach might emulate the proposed biosynthetic pathway if the oxynucleophile of 4 were embedded in a dendralene.
Short and efficient routes to the heterodendralene and double-dienophile partners are shown in Scheme 1. Preliminary reconnaissance identified two important features of each component. First, the dimethylamine substituent15 in 5 was necessary to offset the electron-withdrawing carboxylate, which rendered the dendralene less reactive with electron-deficient dienophiles. Second, the diethylphosphonate substituent in 6 activated the dienophile for ambient-temperature cycloaddition but was not so destabilizing as to complicate isolation.
Scheme 1.

Routes to building blocks.
Building block 5 was synthesized by condensation of tert-butyl acetoacetate 7 with dimethyl formamide dimethyl acetal (DMF-DMA) to yield vinylogous amide 8, which was doubly silylated to 5 with loss of the tert-butyl group. Geranylphosphonate 6 was also synthesized in two steps by addition of diethyl ethylphosphonate to ethyl geranylacetate (9), followed by in situ selenation and subsequent oxidation/elimination of the selenoxide.16
Cycloaddition of 5 to 6 occurred at 22 °C in CH2Cl2 to yield an inconsequential mixture of diastereomers at the dimethylamino group, which was eliminated to an enone (see synthon 4) by treatment with hydrogen fluoride. The silylester was also cleaved to the corresponding unsaturated carboxylic acid, which engaged in a nearly quantitative intramolecular Diels-Alder cycloaddition17 to provide, after tautomerization, β-ketolactone 10, possessing the targeted stereotetrad of the kalihinols. The stereoisomers (5:1 ratio) corresponded to epimers at the C-P bond, and were converged in the next step.18
Lactone 10 was converted to diol 11 by 1. Krapcho-like dephosphonylation, 2. stereoselective methyl addition to the B-ring ketone, and 3. lactone hydrolysis/decarboxylation; each step deserves some comment. First, the desphonylation is a little-precedented transformation that required the development of a new procedure [LiCl, Py•HCl (aq.), 90→110 °C] to spare the acid-sensitive tert-alkyl lactone and the electrophilic ketones, which underwent retro-Dieckmann reactions under other conditions. Addition of a methyl group prior to decarboxylation preserved the trans-decalin geometry, whereas the corresponding ketone weakly favored the cis-decalin after lactone cleavage. Preferential formation of the disfavored trans-decalin (of 11) has remained unsolved in prior work,2,9,10 and in this case is enabled by the fused lactone, which locks the geometry. Through this short process, multigram quantities of decalone 11 could be generated in a single pass for elaboration to protokalihinol 2a and the metabolite itself (1).
However, establishment of the required Δ3,4 unsaturation was undermined by formation of the Δ4,5 isomer, which predominated upon enolization of ketone 11. Such preference is well-precedented for 3-decalone enolizations19 as well as alkene isomerizations in the heavily-studied amorphane sesquiterpenes.20 Attempts to generate endocyclic alkene 2 by ionization of the tertiary alcohol derived from ketone 11 delivered the isomeric Δ4,5 alkene with unrelenting regularity. Since Brønsted bases can mediate alkene isomerization at high temperatures,21 we wondered if the tertiary alcohol proximal to the C3 methylene of 11 could mediate a selective alkene isomerization as its strongly basic alkoxide. Indeed, we found that the potassium salt 12 could be heated to 140 °C in DMSO to deliver protokalihinol 2a with 7:1 selectivity for Δ3,4 unsaturation (2) over Δ4,5 (14, Table 1). Consistent with this mechanistic model, increased equivalents of base led to increased amounts of 14 (entries 1–3), which would derive from inter-molecular deprotonation. Small amounts (ca. 15%) of isomers derived from chain isomerization (Δ15,16: 2b, 14b) also were observed, but could be removed from 2a (or carried forward and removed from 15). Other alkali metals besides potassium and other solvents besides DMSO performed poorly (entries 4–5; see SI for a list of other variations). Methods like coordinative isomerization22 or HAT isomerization23 (entries 6–7) delivered mixtures of alkenes or the Δ4,5 isomer exclusively.
Table 1.
Alkoxide-directed alkene isomerization.
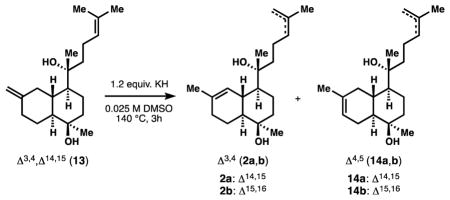
| |||
|---|---|---|---|
| Entry | Variations (& % conversion) | 2:14 | %2a |
| 1 | none (78) | 7:1 | 54 |
| 2a | 4 equiv. KOt-Bu, no KH (82) | 5:1 | 55 |
| 3a | 16 equiv. KOt-Bu, no KH (86) | 1:1 | 28 |
| 4 | 1.2 equiv. n-BuLi, no KH (0) | – | 0 |
| 5 | DMPU instead of DMSO (0) | – | 0 |
|
| |||
| Alternate Conditions | |||
|
| |||
| 6b | 20 mol% RhCl3, EtOH/H2O, 70 °C (56) | 1:1 | 28 |
| 7a | 2 mol% [Co],c 4 mol% PhSiH3, PhH (42) | <1:20 | <5 |
1H NMR;
GCMS;
Co(Salt-Bu,t-Bu)Cl•H2O
A Sharpless-type directed epoxidation24 was identified as the only method capable of controlling the stereochemistry of the targeted tetrahydrofuran. But to our chagrin, the Δ3,4 ring-alkene reacted faster than the Δ14,15 chain-alkene and delivered a C3,4 epoxide opposite to that required for elaboration to the isocyanohydrin of 1 (16, see Figure 2). Fortunately, these same conditions also catalyzed a slower, but stereoselective (93:7 d.r.) epoxidation of the side-chain alkene, as well as concomitant 5-exo-tet cyclization to the targeted tetrahydrofuran, whereas the A-ring epoxide was spared attack. Consequently, a sodium iodide/zinc metal-mediated epoxide deoxygenation selectively removed the unwanted ring epoxide, whereas the C14,15 oxidation was retained as the incipient hydroxy-tetrahydrofuran (see Figure 2).
Figure 2.

Stereoselective oxidative cyclization to 15.
The full kalihinol skeleton and correct oxidation state were thus established in eight steps from heterodendralene 5; we next investigated elaboration to a known metabolite. Elimination of the exocyclic tertiary alcohol was effected by selective trifluoroacetylation and syn-elimination via thermolysis since ionizing conditions resulted in hydride shift from the tetrahydrofuran methine. In the same flask, we trifluoroacetylated the remaining alcohol and then installed the A-ring equatorial tert-alkyl isonitrile with our solvolytic stereoinversion.8 The logic leading to this route (Scheme 2) requires some discussion.
Scheme 2.

Synthesis of kalihinol C.
We had originally targeted a tandem epoxide opening/trifluoroacetate stereoinversion of 20 (Figure 3) using our solvolysis conditions to install the bis-isonitrile motif. This approach was reported by Vanderwal to be successful, but low-yielding in his kalihinol B synthesis.9e We similarly found that this tandem reaction yielded only small amounts of the bis-isonitrile 21 (5–6%). The basis of the low yield was not the epoxide opening step, which was efficient and regioselective in substrate 20. Instead, the subsequent B-ring stereoinversion8 was low-yielding by virtue of competitive elimination (22→23).25 Since the epoxide opening occurred faster than trifluoroacetate ionization, and the resulting isocyanohydrin caused elimination in ring B, we concluded that the B-ring C-N bond must be in place prior to epoxidation.
Figure 3.

Tandem isonitrile formation is very low yielding.
We were dismayed to find that the B-ring functional groups influenced the course of epoxide ionization. As shown in Table 2, the B-ring axial trifluoroacetate and alcohol led to A with high selectivity, whereas the equatorial isonitrile or formamide skewed the ratio to favor substantial quantities of regioisomers B and C, as well as semi-pinacol product D.26 So the A and B-ring substituents proved mutually incompatible in the tandem solvolysis sequence reported by Vanderwal.9e
Table 2.
Isocyanohydrin installation.a
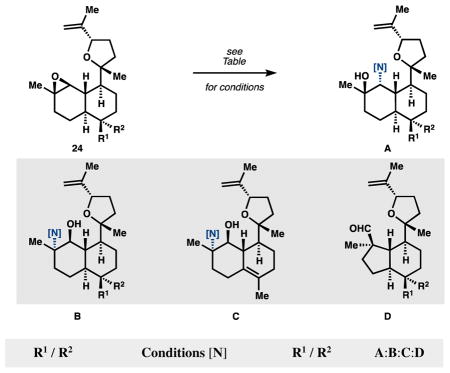
| |||
|---|---|---|---|
| R1 / R2 | Conditions [N] | R1 / R2 | A:B:C:D |
| OTFA / Me | TMSCN, Sc(OTf)3 [NC] | OTFA / Me | 83:0:17:0 |
| OH / Me | TMSCN, Sc(OTf)3 [NC] | OTMS / Me | 100:0:0:0 |
| Me / NC | TMSCN, Sc(OTf)3 [NC] | Me / NC | 61:0:39:0 |
| Me / NHCHO | TMSCN, Sc(OTf)3 [NC] | Me / NHCHO | 54:31:0:15 |
| Me / NC | NH3, MeOH [NH2] | Me / NC | 100:0:0:0 |
Any silylethers were converted to alcohols with TBAF.
Consequently, we relied on a simple but effective aminolysis to regioselectively generate the requisite A-ring functionality (Scheme 2 and Table 2, entry 5). First, chemoselective oxidation of the alkene of 25 occurred in preference to the isonitrile if carried out with dimethyldioxirane (DMDO) in the strongly hydrogen bond-donating solvent HFIP, which we posit deactivates the isonitrile against oxidation (Scheme 2). Aminolysis in methanol cleanly opened the epoxide to deliver a sec-alkyl amino tert-alcohol,27 which was converted to the isocyanohydrin of 1 via difluorocarbene derived from difluoromethyl triflate28 and KOt-Bu. More orthodox methods for amine to isonitrile conversion worked poorly: chloroform/sodium hydroxide29 generated appreciable amounts of a dichlorocyclopropane and an amide; formylation of the hindered amine occurred slowly due to build up of acid (isonitriles are produced by subsequent formamide dehydration).30 Our alternative use of difluorocarbene for isonitrile synthesis therefore offers some advantages over existing methods, especially when multiple functional groups are present or the amine is hindered. This three-step procedure provides an efficient, stereo- and chemoselective strategy to install the kalihinol A-ring isocyanohydrin motif.
In summary, we have demonstrated a concise route to access the kalihinol (bifloran) ICTs via a putative biosynthetic inter-mediate, protokalihinol (2a) that we anticipate can be divergently advanced to the natural series of metabolites. The synthesis compares favorably to the current best approach to the kalihinols by Vanderwal: it is longer in total step count (17 vs. 12), but higher in yield by an order of magnitude (1.3% vs. 0.13%). The higher efficiency derives from solutions to stereochemical and chemoselectivity problems raised by prior work but left unsolved. Some of these solutions include 1. a method to synthesize the kalihinol stereotetrad using an iterative cycloaddition of the new building block, ‘heterodendralene’ 5; 2. an alkoxide-directed isomerization method to access the thermodynamically-disfavored Δ3,4 unsaturated trans-bifloran skeleton found throughout the diterpene class, and 3. a short, high-yielding, regio- and stereoselective strategy for installing the A-ring isocyanohydrin motif, including difluorocarbene-mediated isonitrile synthesis. This short and divergent route from protokalihinol 2a allowed us to generate several analogs related to the metabolite series. We are currently using these compounds to interrogate the antiplasmodial activity and mechanism(s) of the kalihinol class.
Supplementary Material
Acknowledgments
Funding Sources
Financial support for this work was provided by the NIH (GM105766) and the NSF (GRFP to C. A. R.). Additional support was provided by Eli Lilly, Novartis, Bristol-Myers Squibb, Amgen, Boehringer-Ingelheim, the Sloan Foundation and the Baxter Foundation.
We thank Dr. Milan Gembicky, Dr. Curtis Moore, and Professor Arnold L. Rheingold for X-ray crystallographic analysis. We thank Chris Vanderwal (UC Irvine) for open communication about his ongoing work on the ICTs.
Footnotes
Supporting Information. Detailed experimental procedures, spectral data, chromatograms and x-ray crystallography. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Emsermann J, Kauhl U, Opatz T. Mar Drugs. 2016;14:16. doi: 10.3390/md14010016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shenvi RA, Schnermann MJ. Nat Prod Rep. 2015;32:543. doi: 10.1039/c4np00109e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Isolation: Chang CWJ, Patra A, Roll DM, Scheuer PJ. J Am Chem Soc. 1984;106:4644.kalihinol antimalarial activity: Miyaoka H, Shimomura M, Kimura H, Yamada Y. Tetrahedron. 1998;54:13467.
- 4.(a) Wright AD, Wang HQ, Gurrath M, Konig GM, Kocak G, Neumann G, Loria P, Foley M, Tilley L. J Med Chem. 2001;44:873. doi: 10.1021/jm0010724. [DOI] [PubMed] [Google Scholar]; (b) Wright AD, McCluskey A, Robertson MJ, MacGregor KA, Gordon CP, Guenther J. Org Biomol Chem. 2011;9:400. doi: 10.1039/c0ob00326c. [DOI] [PubMed] [Google Scholar]; (c) Young RM, Adendorff MR, Wright AD, Davies-Coleman MT. Eur J Med Chem. 2015;93:373. doi: 10.1016/j.ejmech.2015.02.011. [DOI] [PubMed] [Google Scholar]
- 5.Sandoval IT, Manos EJ, Van Wagoner RM, Delacruz RGC, Edes K, Winge DR, Ireland CM, Jones DA. Chem Biol. 2013;20:753. doi: 10.1016/j.chembiol.2013.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lu H-H, Pronin SV, Antonova-Koch Y, Meister S, Winzeler EA, Shenvi RA. J Am Chem Soc. 2016;138:7268. doi: 10.1021/jacs.6b03899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pronin SV, Shenvi RA. J Am Chem Soc. 2012;134:19604. doi: 10.1021/ja310129b. [DOI] [PubMed] [Google Scholar]
- 8.Pronin SV, Reiher CA, Shenvi RA. Nature. 2013;501:195. doi: 10.1038/nature12472. [DOI] [PubMed] [Google Scholar]
- 9.(a) White RD, Wood JL. Org Lett. 2001;3:1825. doi: 10.1021/ol015828k. [DOI] [PubMed] [Google Scholar]; (b) White RD, Keaney GF, Slown CD, Wood JL. Org Lett. 2004;6:1123. doi: 10.1021/ol049918b. [DOI] [PubMed] [Google Scholar]; (c) Miyaoka H, Abe Y, Sekiya N, Mitome H, Kawashima E. Chem Commun. 2012;48:901. doi: 10.1039/c1cc16468f. [DOI] [PubMed] [Google Scholar]; (d) Miyaoka H, Abe Y, Kawashima E. Chem Pharm Bull. 2012;60:1224. doi: 10.1248/cpb.c12-00485. [DOI] [PubMed] [Google Scholar]; (e) Daub ME, Prudhomme J, Le Roch K, Vanderwal CD. J Am Chem Soc. 2015;137:4912. doi: 10.1021/jacs.5b01152. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Daub ME, Prudhomme J, Mamoun CB, Le Roch KG, Vanderwal CD. ACS Med Chem Lett. doi: 10.1021/acsmedchemlett.7b00013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kalihinene X was synthesized with stereocontrol in 35-steps, but lacks the A-ring isocyanohydrin and characteristic trans-ring fusion of the kalihinols. See: Miyaoka H, Shida H, Yamada N, Mitome H, Yamada Y. Tetrahedron Lett. 2002;43:2227.
- 11.Dewick PM. Medicinal Natural Products: A Biosynthetic Approach. Wiley; 2009. [Google Scholar]
- 12.For an exception, see: Zhao C, Toste FD, Raymond KN, Bergman RG. J Am Chem Soc. 2014;136:14409. doi: 10.1021/ja508799p.
- 13.For an excellent review of dendralenes, see: Hopf H, Sherburn MS. Angew Chem Int Ed. 2012;51:2298. doi: 10.1002/anie.201102987.
- 14.Newton CG, Drew SL, Lawrence AL, Willis AC, Paddon-Row MN, Sherburn MS. Nat Chem. 2015;7:82. doi: 10.1038/nchem.2112. [DOI] [PubMed] [Google Scholar]
- 15.Kozmin SA, Rawal VH. J Org Chem. 1997;62:5252–5253. [Google Scholar]
- 16.Reich HJ, Renga JM, Reich IL. J Org Chem. 1975;97:5434. [Google Scholar]
- 17.For the closest precedent to this stereoselective cycloaddition, see: Tietze LF, Beifuss U, Ruther M. J Org Chem. 1989;54:3120.
- 18.The diastereomers of 10 could also be separated and shown to converge to the same product upon dephosphonylation.
- 19.Huffman JW, Balke WH. J Org Chem. 1988;53:3828. [Google Scholar]
- 20.Ngo K, Brown GD. Tetrahedron. 1999;55:15109. [Google Scholar]
- 21.Schriesheim A, Muller RJ, Rowe CA. J Am Chem Soc. 1962;84:3164. [Google Scholar]
- 22.Harrod JF, Chalk AJ. J Am Chem Soc. 1964;86:1776. [Google Scholar]
- 23.Crossley SWM, Barabé F, Shenvi RA. J Am Chem Soc. 2014;136:16788. doi: 10.1021/ja5105602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nicolaou KC, Harrison ST. Angew Chem Int Ed. 2006;45:3256. doi: 10.1002/anie.200601116. [DOI] [PubMed] [Google Scholar]
- 25.Stereoselectivity was also poor and the remaining material could not be identified. See Ref ixf.
- 26.Stereochemistry depicted assumes a thermally-allowed 2-electron suprafacial Wagner-Meerwein shift.
- 27.Wood accesses a related amino alcohol (Ref. ixb) by epoxide-opening with NaN3, followed by Na0 reduction, conditions which are reported to reduce isonitriles.
- 28.(a) Fier PS, Hartwig JF. Angew Chem Int Ed. 2013;52:2092. doi: 10.1002/anie.201209250. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Levin VV, Dilman AD, Belyakov PA, Struchkova MI, Tartakovsky VA. J Fluorine Chem. 2009;130:667. [Google Scholar]
- 29.(a) Hofmann AW. Ann. 1868;298:202. [Google Scholar]; (b) Sasaki T, Eguchi S, Katada T. J Org Chem. 1974;39:1239. [Google Scholar]
- 30.Hertler WR, Corey EJ. J Org Chem. 1958;23:1221. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.