

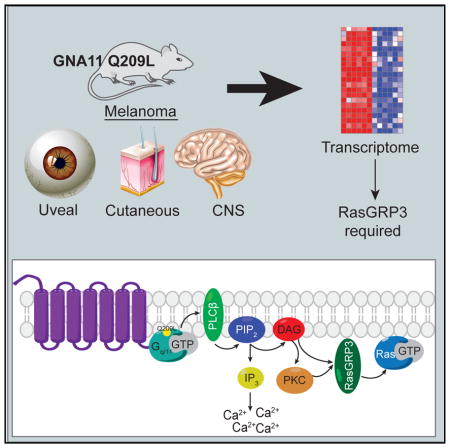
SUMMARY
Uveal melanoma (UM) is characterized by mutually exclusive activating mutations in GNAQ, GNA11, CYSLTR2, and PLCB4, four genes in a linear pathway to activation of PLCβ in almost all tumors and loss of BAP1 in the aggressive subset. We generated mice with melanocyte-specific expression of GNA11Q209L with and without homozygous Bap1 loss. The GNA11Q209L mice recapitulated human Gq-associated melanomas, and they developed pigmented neoplastic lesions from melanocytes of the skin and non-cutaneous organs, including the eye and leptomeninges, as well as at atypical sites, including the lymph nodes and lungs. The addition of Bap1 loss increased tumor proliferation and cutaneous melanoma size. Integrative transcriptome analysis of human and murine melanomas identified RasGRP3 to be specifically expressed in GNAQ/GNA11-driven melanomas. In human UM cell lines and murine models, RasGRP3 is specifically required for GNAQ/GNA11-driven Ras activation and tumorigenesis. This implicates RasGRP3 as a critical node and a potential target in UM.
In Brief
Moore et al. generate a preclinical mouse model of melanoma that recapitulates features of aggressive uveal melanoma. By comparing murine and human melanomas, they identify a dependency on RasGRP3 in uveal melanoma.
INTRODUCTION
Uveal melanomas (UMs) arise from the melanocytes of the eye. While localized disease can be effectively treated surgically, half of all patients develop metastasis, and metastatic UM carries a dismal prognosis with an overall survival of only 6 months (Diener-West et al., 2005). Approximately half of the patients harbor metastases to multiple organs, with liver (93%), lung (24%), bone (16%), and lymph nodes (10%) representing the most common sites (Collaborative Ocular Melanoma Study Group, 2001). Over the past decade, we have gained considerable insight into the genetic basis of UM. This has not yet led to novel therapeutic options and there are still no proven systemic treatments for UM.
UM is highly distinct from cutaneous melanoma (CM) both clinically and molecularly. UM is not associated with sun exposure and has among the lowest mutation rates in cancer, whereas CM has among the highest mutation rates due to UV damage (Furney et al., 2013). A recent comparison of liver metastasis revealed that most CM metastases lacked gross melanin pigmentation while most UM metastases are hyperpigmented and express high levels of melanocyte lineage proteins, such as MART-1 (MLANA) and gp100 (PMEL) (Rothermel et al., 2016). Molecularly, CM is driven by recurrent somatic mutations that activate the mitogen-activated protein kinase (MAPK) pathway, including BRAF, NRAS, NF1, and KIT. Approximately 90% of UMs harbor activating mutations in two homologous G-protein alpha (Gα) subunits, GNA11 (Gα11) and GNAQ (Gαq), at codons Gln209 or Arg183 (Robertson et al., 2017; Van Raamsdonk et al., 2009, 2010). Among the remaining 10% of UMs, most harbor activating mutations in a G-protein-coupled receptor (CYSLTR2 at the Leu129 codon activates Gα11/q) or in phospholipase C β4 (PLCB4) (at the Asp630 codon, a direct downstream effector of Gα11/q cleaves phosphatidylinositol 4,5-bisphosphate [PIP2] to produce the second messengers diacylglycerol [DAG] and inositol triphosphate [IP3] (Johansson et al., 2016; Moore et al., 2016). This indicates a requirement for Gα11/q-coupled signaling and, in particular, the phospholipase Cβ (PLCβ) effector pathway in the initiation of UMs.
While essentially all UMs harbor mutations in the CYSLTR2-Gα11/q-PLCβ pathway, the prognosis is largely determined by the presence of cooperative mutations. Monosomy 3 and an associated poor-prognosis gene expression pattern is the single most negative prognostic factor. Most of these tumors harbor inactivating mutations in BAP1, located at 3q21, and essentially all of these tumors lose expression of the BAP1 protein, implicating BAP1 loss as a critical cooperating lesion driving poor prognosis in UM (Harbour et al., 2010; Robertson et al., 2017). Among tumors with disomy 3, there are mutually exclusive mutations in SF3B1, associated with intermediate prognosis, and in EIF1AX, associated with favorable prognosis (Martin et al., 2013; Robertson et al., 2017).
In addition to UM, CYSLTR2-Gα11/q-PLCβ pathway mutations are found in most leptomeningeal melanocytic neoplasms (LMNs) and blue nevi (Möller et al., 2017; Van Raamsdonk et al., 2009). LMNs are rare neoplasms arising from melanocytes of the leptomeninges. Like UM, in addition to mutations in GNA11 and GNAQ, LMNs harbor mutually exclusive co-mutations in either EIF1AX or SF3B1 in 33% of cases (Küsters-Vandevelde et al., 2016). Blue nevi are common benign neoplasms of dermal melanocytes, which are distinguished from CMs that arise from epidermal melanocytes. Rare malignant melanomas that either arise from blue nevi or show morphologic features of blue nevi are called malignant blue nevi. Recent genetic characterization of a large cohort of blue nevi showed both benign and malignant blue nevi harbored CYSLTR2-Gα11/q-PLCβ pathway mutations (Möller et al., 2017). EIF1AX mutations are found only in benign blue nevi, while SF3B1 and BAP1 mutations are found only in malignant blue nevi (Griewank et al., 2017). Therefore, UM, LMN, and blue nevi represent a molecularly similar spectrum of diseases that commonly harbor CYSLTR2-Gα11/q-PLCβ mutations and whose disease aggressiveness is defined by co-mutations, especially BAP1.
Because the PLCβ pathway is known to activate MAPK, the MEK inhibitor selumetinib has been clinically studied. While a phase 2 trial showed promising improvement in progression-free survival, the phase 3 trial failed to confirm the finding and neither trial showed an improvement in overall survival (Carvajal et al., 2014; Komatsubara et al., 2016). As different MEK inhibitors have distinct properties (Lito et al., 2014), it is still currently unclear whether the MAPK pathway remains a viable therapeutic target in UM.
To identify molecular and lineage events downstream of Gα11 activation, the cooperative role of BAP1 loss, and critical nodes required for Gα11-mediated tumorigenesis, we generated a conditional Rosa26-LSL-GNA11Q209L mouse model and crossed it with conditional Bap1 knockout (KO) mice. These mice recapitulate the features of human Gα11-driven melanomas. They developed neoplastic hyperpigmented melanocytic lesions in the uveal tract, skin, and leptomeninges. These mice developed lesions in the lung and lymph nodes. Deletion of Bap1 accelerated skin tumor growth and mouse mortality. The GNA11Q209L, Bap1 loss tumors were resistant to the MEK inhibitor trametinib. To identify alternative therapeutic targets, we performed integrative analysis comparing BRAF mutant and Gα11/q mutant human and murine cancers, and we identified a critical requirement of a Ras guanine exchange factor (GEF), RasGRP3, for Gα11/q-mediated tumorigenesis.
RESULTS
Melanocyte-Specific GNA11Q209L Expression Induces Skin, Uveal, and CNS Neoplasia
To express GNA11Q209L in the melanocyte lineage, we generated a genetically engineered mouse model (GEMM) with a conditional GNA11Q209L allele (R26-LSL-GNA11Q209L) under the control of the endogenous Rosa26 promoter (Figures S1A and S1B). To identify an active Cre-driver for uveal melanocytes, we crossed Tyr-CreERT2 transgenic mice that express tamoxifen-inducible CreERT2 under the melanocyte-specific Tyrosinase (Tyr) promoter with the CAG-LSL-EYFP reporter. One week after tamoxifen injection, there was robust EYFP expression in both uveal and skin (hair follicle) melanocytes (Figure S1C). We thus generated Tyr-CreERT2;GNA11Q209L mice for our studies.
To activate GNA11Q209L expression, we treated 4-week-old mice with a single application of tamoxifen or vehicle by intraperitoneal injection. In tamoxifen-injected, but not vehicle-injected, mice, we observed hyperpigmentation of ears and tail within 2 weeks of treatment and bulging eyes within 1 month of treatment (Figure S2A). In vehicle-treated Tyr-CreERT2-positive or tamoxifen-treated Tyr-CreERT2-negative control mice, there was no discernible pathologic phenotype in the skin or the uveal tract up to 18 months (Figures 1A, 1B, S2B, and S2C). In Tyr-CreERT2;GNA11Q209L mice, pathological analysis of the skin 3 months post-induction showed extensive follicular and dermal melanocytic proliferation (Figure 1C), which progressed to melanomas encompassing the dermis and subcutaneous tissues in 50% of mice by 6 months after injection (Figure 1D). These proliferating melanocytes stained positive for a melanocyte cocktail (Figure S2D). Tumor cells contained abundant pale amphophilic cytoplasm and a small nuclear-to-cytoplasmic ratio (Figure S2D). For comparison, we also treated Tyr-CreERT2;BRafCA/+ mice, which express the conditional BRafV600E allele in melanocytes. We observed melanocytic hyperplasia without progression to melanoma up to 12 months post-induction, similar to previous observations (Figure S2E) (Dankort et al., 2009).
Figure 1. Melanocyte-Specific GNA11Q209L Expression Induces Cutaneous and UM.

(A and B) H&E of skin (A) and eye (B) from Tyr-CreERT2-negative control mice 3 months post-induction.
(C and D) H&E of skin from GNA11Q209L mice 3 months (C) or 6 months (D) post-induction.
(E and F) H&E of eyes from GNA11Q209L mice 3 months (E) or 6 months (F) post-induction.
(G) H&E (i), H&E with melanin bleaching (ii), MITF immunohistochemistry (IHC) using diaminobenzidine (DAB) (brown) (iii), and melanoma cocktail IHC using red chromogen (red) (iv) from a GNA11Q209L mouse 6 months post-induction. High-magnification insets of top panels are shown in bottom panel.
(H) H&E of a selected eye (i) with melanocytic perineural invasion of the optic nerve (ii).
See also Figures S1 and S2.
We next examined the oncogenic role of GNA11Q209L and BRafV600E expression in uveal melanocytes. Within the uveal tract, tamoxifen-injected Tyr-CreERT2;GNA11Q209L mice displayed diffuse hyperplasia, thickening of the choroid and ciliary body that progressed over time to overt UM with intraocular infiltration that distorted the normal architecture of the globe (Figures 1E and 1F). Uveal melanocyte proliferation was evident in mice as early 1 month post-tamoxifen (data not shown). In contrast, the uveal tracts of induced Tyr-CreERT2;BRafCA/+ mice were indistinguishable from those of control mice (Figures S2F and S2G). Further examination revealed tumor cells with pathological characteristics similar to skin melanoma (Figure 1G, i and ii). These cells were positive for MITF and melanocyte cocktail staining (Figure 1G, iii and iv). We further observed perineural spread of malignant melanoma to the optic nerve (Figure 1H).
We next examined the effect of GNA11Q209L in resident melanocytes of other organs, including the heart, harderian gland, and brain (Aoki et al., 2009). Gross examination of the brain revealed focal pigmentation of the leptomeninges in 80% of mice (Figure S3A). Pathological evaluation of the CNS of the Tyr-CreERT2;GNA11Q209L mice showed melanocytic proliferation in the leptomeninges at the base of the brain, around cranial nerve roots, and within the longitudinal fissure (Figure 2A, ii–iv). There was prominent melanocytic hyperplasia within the third ventricle (Figure 2A, i and iv). Clinically, primary melanocytomas occasionally occur within the ventricular system of the CNS (Tandon et al., 2008). There was invasion of melanocytes to the periventricular space, and one mouse exhibited invasion to the olfactory bulb (Figure 2A, iv and v). We observed robust proliferation of resident melanocytes in the harderian gland (Figure 2A, arrow).
Figure 2. Melanocyte-Specific GNA11Q209L Expression Induces Leptomeningeal Melanocytic Neoplasia and Possible Metastasis.

(A) H&E of coronal skull sections (left), magnified images of regions showing melanocytic neoplasia in the (i) choroid plexus of the third ventricle (i), leptomeninges of brain base surrounding cranial nerve roots (ii and iii), longitudinal fissure and ependyma of the third ventricle olfactory bulb (iv and v). Arrow indicates the harderian gland.
(B) H&E of heart and lung. Arrows indicate lesions.
(C) Magnification of heart in (B) showing melanoma in the tricuspid valve, right atrial wall, and interventricular septum indicated by box and gray arrow in (B).
(D and E) Magnification of a selected melanocytic lesion (D) in the lung indicated by box and arrow in (B) and with melanin bleaching (E).
(F and G) H&E (F) and MITF IHC (G) of axillary lymph node melanocytic lesion (G, ii).
See also Figure S3.
Examining the melanocytes of the heart in GNA11Q209L mice, we observed invasive neoplasms that infiltrated and thickened the tricuspid valve and infiltrated the myocardium of the right atrium and interventricular septum (Figures 2B and 2C). We suspect these lesions were primary tumors of the resident melanocytes of the heart. Melanocytic lesions were not evident in Tyr-CreERT2;BRafCA/+ mice in these areas (Figures S3C and S3D).
In GNA11Q209L mice, we observed multi-focal lesions in the lungs that may represent metastases, although we cannot rule out transformation of rare resident lung melanocytes (Figures 2B, 2D, and 2E). However, the morphology of these lesions resembled the primary tumors (Figures 1D, 1G, ii, 2B, and 2C). These lesions occurred early and were observed in mice with less advanced skin and uveal lesions (Figure S3E). We observed melanocytes infiltrating the lymphatic system, as visualized by MITF staining in the axillary lymph nodes (Figures 2F and 2G). In contrast, we did not observe melanocytic lesions in the CNS, heart, lungs, or lymph nodes in Tyr-CreERT2;BRafCA/+ mice (Figures S3B–S3D). Our data are consistent with the clinical absence of BRAF mutations in UMs and LMNs and the sporadic occurrence of GNAQ/11 mutations in CMs.
Recently, a mouse model harboring GNAQQ209L was characterized (Huang et al., 2015). When activated in melanoblasts during embryogenesis with Mitf-Cre, the mice exhibited UM, LMN, neoplastic melanocytic growth in the harderian glands, and rare lesions in the skin, as well as lymph nodes and lung (Huang et al., 2015). When activated in 8-week-old adult mice using Tyr-CreERT2, GNAQQ209L drove melanocyte overgrowth without progression to melanoma. The phenotype of our Tyr-CreERT2; GNA11Q209L mouse model, activated at 4 weeks, appears to be an intermediate between Mitf-Cre activated before birth and Tyr-CreERT2 activated at 8 weeks. The Mitf-Cre;GNAQQ209L mice developed earlier invasive UM and more diffuse LMN, likely due to earlier expression at mid-gestation and prolonged Cre activation (Alizadeh et al., 2008).
Loss of Bap1 Accelerates Skin Melanomas in the Presence of GNA11Q209L
We sought to examine the combinatorial effect of GNA11Q209L and the loss of the tumor suppressor Bap1 in the development of UM. To achieve Bap1 deletion, we crossed Bap1lox/lox mice (LaFave et al., 2015) to the Tyr-CreERT2;GNA11Q209L line. Tamoxifen-treated Tyr-CreERT2;Bap1KO mice had no discernible phenotype and were histologically normal over ~20 months, indicating Bap1 loss alone was insufficient to initiate melanoma (n = 35; Figures S4A and S4B).
We compared the Tyr-CreERT2;GNA11Q209L;Bap1lox/lox mice to the Tyr-CreERT2;GNA11Q209L mice. We observed a stronger ocular phenotype in GNA11Q209L than GNA11Q209L Bap1KO mice (Figures 3A–3C). However, the GNA11Q209L Bap1KO mice succumbed to disease at an accelerated rate compared to GNA11Q209L or GNA11Q209L Bap1lox/+ mice (Figure 3D; p < 0.05), due to increased skin melanoma burden (Figures 3E and 3F). The loss of Bap1 in these mice did not appreciably alter the size or incidence of uveal lesions, but it contributed to an increased progression to skin melanomas originating from the tail and ears (Figures 3E–3G). We confirmed Bap1 KO in uveal melanocytes using PCR (Figure S4C). Unlike in human UM patients, no pigmented liver lesions were observed (Figures S4D and S4E). We observed no significant increase in the size or incidence of lung lesions in the absence of Bap1 (Figures S3E and S4F–S4H). Histologically, GNA11Q209L skin melanomas exhibited slender oval nuclei while GNA11Q209L;Bap1KO had larger euchromatic nuclei (Figure 3H). GNA11Q209L;Bap1KO skin melanomas exhibited a higher proliferation index (Figures 3H and 3I; p < 0.0001).
Figure 3. Bap1 Loss Promotes Aggressive Melanomas.

(A) Photographs of GNA11Q209L (left) and GNA11Q209L Bap1KO (right) mice 3 months post-induction.
(B and C) H&E of eyes from GNA11Q209L (B) and GNA11Q209L Bap1KO (C) mice 12 months post-induction.
(D) Kaplan-Meier curve comparing the survival percentage of GNA11Q209L (blue) or GNA11Q209L Bap1lox/+ (green) to GNA11Q209L Bap1KO mice (red). p = 0.0013.
(E and F) Photograph of large invasive melanoma from the tail dermis (E) and H&E (F) from a GNA11Q209L Bap1KO mouse 12 months post-induction.
(G) H&E of an invasive melanoma from the dermis of the ear from a GNA11Q209L Bap1KO mouse 12 months post-induction.
(H) Ki-67 IHC in GNA11Q209L (top) and GNA11Q209L Bap1KO (bottom) cutaneous tumors.
(I) Quantification of Ki-67-positive cells. Scatter-dot plot: each dot represents the quantification of one field. Error bars represent means ± SEM. p < 0.0001.
(J) GSEA plot using a gene set comprised of genes upregulated in mice (Mouse_Bap1KO_UP) on a profile of genes ranked by correlation to BAP1 expression in the UM TCGA dataset.
(K) GO and SP pathways from leading edge genes (red; J) upregulated in GNA11Q209L Bap1KO tumors and negatively correlated with BAP1 expression in TCGA UM.
(L) GSEA plot using Mouse_Bap1KO_UP gene set (as in J) on a profile of genes ranked by change in UMMP3 cells upon BAP1 wild-type (WT) restoration.
(M) GO and SP pathways from leading edge genes (red; L) upregulated in GNA11Q209L Bap1KO tumors and downregulated upon BAP1 restoration in UMMP3 cells.
See also Figures S4 and S5 and Table S1.
To determine the extent that Bap1 KO molecularly recapitulates human melanoma and to understand the Bap1-regulated transcriptional programs, we performed transcriptome analysis of mouse and human melanomas. RNA sequencing (RNA-seq) revealed the R26-GNA11Q209L transcript level was 4- to 8-fold lower than endogenous murine Gnaq and Gna11 transcript levels, indicating modest expression of mutant GNA11 is required for tumorigenesis (Figure S5A). Analysis of RNA-seq of GNA11Q209L and GNA11Q209L;Bap1KO skin melanomas confirmed deletion of Bap1 (Figure S5B). Using a gene set comprised of genes upregulated in GNA11Q209L;Bap1KO versus GNA11Q209L skin melanomas (Mouse_Bap1KO_UP), we performed gene set enrichment analysis (GSEA) using The Cancer Genome Atlas (TCGA) UM dataset (Robertson et al., 2017). This showed Mouse_Bap1KO_UP genes are significantly enriched among genes negatively correlated with BAP1 expression in UM, suggesting BAP1 deletion in skin melanomas of mice results in the upregulation of similar genes to human UM (Figure 3J; Table S1). To explore the function of the shared genes, we performed functional annotation of the leading edge genes (Figure 3J, red) that drove the GSEA enrichment. We found the most enriched gene ontology (GO) and Swiss-Prot (SP) pathways all involved cell cycle and mitosis (Figure 3K).
In a complementary approach, we identified a UM primary tumor line, UPMM3, contained a frameshift deletion of BAP1 (Figure S5C) (Griewank et al., 2012). We restored wild-type BAP1 (Figure S5D) and generated a gene expression profile using RNA-seq. As controls, we expressed BAP1 with mutations in the deubiquitinase domain (p.Cys91Trp, p.Ala95Pro) found in cancer (Harbour et al., 2010) as well as EGFP. Wild-type BAP1 restoration significantly changed gene expression while the presumably non-functional mutants did not, observed by gene hierarchical clustering (Figure S5E). We next performed GSEA, and we found the Mouse_Bap1KO_UP gene set was significantly enriched among genes downregulated in UPMM3 cells by BAP1 (wild-type [WT]) restoration (Figure 3L). Functional analysis of leading edge genes showed cell cycle pathways were enriched (Figure 3M). GSEA on three BAP1 datasets (mouse model, TCGA, and UPMM3) using the >8,300 gene sets from the Molecular Signatures Database (MSigDB) showed cell cycle and melanoma metastasis signatures are highly enriched in each (Table S1; Figures S5F and S5G). Therefore, in UM, the loss of Bap1 can promote aggressive disease with a propensity to proliferate and metastasize, consistent with clinical data implicating BAP1 loss as a poor prognostic biomarker.
Gα11/q-Driven Cells Have Reduced Sensitivity to MEK Inhibition
Activation of the Gα11/q-PLCβ pathway leads to downstream activation of the MAPK pathway. While preclinical data using UM cell lines suggest MEK inhibition may be a therapeutic strategy (Ambrosini et al., 2012), a recent phase 3 study comparing selumetinib and chemotherapy failed to show significant improvement in progression-free or overall survival (Komatsubara et al., 2016). Prolonged selumetinib treatment induces RAF-MEK dimer formation, leading to reactivation of MAPK signaling, particularly in non-BRAFV600E-driven tumors. The newer MEK inhibitor trametinib uniquely decreases RAF-MEK interaction and MAPK reactivation (Lito et al., 2014).
To address whether improved MEK inhibition can lead to therapeutic efficacy in UM, we utilized the GEMMs to perform in vivo trametinib treatment. We needed relevant control tumors that formed nodules of a similar size and were responsive to trametinib treatment. We observed Bap1KO also accelerated BRafV600E-driven tumors to form nodules amenable for treatment (unpublished data). We subcutaneously grafted skin melanomas, isolated from BRafV600E;Bap1KO and GNA11Q209L;Bap1KO mice, into severe combined immunodeficiency (SCID) mice. The grafts retained features of the in situ tumors, where GNA11Q209L;Bap1KO tumors retained hyper-pigmentation whereas BRafV600E;Bap1KO tumors were hypopigmented and exhibited elevated MAPK output (Figures 4A and 4B). In BRafV600E Bap1KO tumors, short-term trametinib treatment decreased proliferation and MAPK output, whereas in GNA11Q209L Bap1KO tumors, the effects were modest (Figures 4A–4C). Long-term treatment resulted in initial tumor shrinkage followed by stabilization in BRafV600E Bap1KO tumors (Figure 4D). However, GNA11Q209L Bap1KO tumors were resistant to trametinib treatment (Figure 4D). To determine if relative resistance to trametinib treatment was more generalized, we treated human BRAFV600E CM and Gαq/11 mutant UM cell lines with a clinically achievable concentration of trametinib (10 nM) (Infante et al., 2012). Trametinib sustainably inhibited MAPK in CM cells. In UM cells, MEK phosphorylation was stable or increased over time and ERK phosphorylation was variably inhibited but rebounded by 24 hr (Figure 4E). This is consistent with known hypersensitivity of BRAFV600E melanoma to MEK inhibition (Solit et al., 2006). Together, activating mutations in the Gα11/q pathway exhibit in vivo and in vitro resistance to MEK inhibition. This highlights the need for novel therapeutic targets in UM.
Figure 4. Reduced MEK Sensitivity in Gα11/q-Driven Tumors.

(A) H&E and Ki-67 IHC of GNA11Q209L Bap1KO and BRAFV600E Bap1KO tumors treated with trametinib or vehicle.
(B) Immunoblots for MAPK in explanted control and trametinib-treated GNA11Q209L Bap1KO and BRAFV600E Bap1KO tumors.
(C) Quantification of Ki67 in explanted control and trametinib-treated GNA11Q209L Bap1KO and BRAFV600E Bap1KO tumors, shown as Tukey box-and-whisker plots. Outliers are shown as dots. p < 0.0001.
(D) Tumor growth of grafted GNA11Q209LBap1KO or BRAFV600E Bap1KO tumors in SCID mice with treatment as indicated. n = tumors per group. Error bars, SEM. p < 0.001 for BRAFV600E Bap1KO treatment.
(E) Immunoblot of Gα11/q mutant UM and BRAF mutant CM cell lines treated with 10 nM trametinib.
Cross-Species Analysis Shows Gα11/q-Driven Tumors Enforce a Melanocyte Lineage Program and Express High Levels of RASGRP3
To identify critical nodes in Gα11/q-mediated tumorigenesis, we utilized a cross-species transcriptome analysis approach of human and murine melanoma. We sought to generate a transcriptional signature of Gα11-driven GEMM melanoma. We generated GEMM Gα11 and BRaf signatures with differentially expressed genes between GNA11Q209L Bap1KO and BRafV600E Bap1KO melanomas (>3-fold, false discovery rate [FDR] < 0.01). In human melanoma, we combined and curated TCGA skin CM (SKCM) (Cancer Genome Atlas, 2015) and UM datasets, and we compared tumors with hotspot mutations in Gα11/q (74/80 UM and 5/333 SKCM) with BRAFV600E (0/80 UM and 121/333 SKCM). GSEA using the GEMM signatures on the TCGA transcriptomes showed significant enrichment of the Gα11 and Braf signatures in human Gα11/q-mutated and BRAFV600E tumors, respectively (Figure 5A). Therefore, the oncogenic signaling driver, in addition to the location of the tumor, contributes to the oncogenic transcriptome. Functional annotation of the leading edge Gα11/q signature genes showed upregulation of pigmentation and melanocyte differentiation pathways (Figures 5B–5D), consistent with the observation of highly pigmented melanomas in the GNA11Q209L GEMM (Figures 1, 2, and 4). This is also consistent with the clinicopathological observations that metastatic UMs retain greater pigmentation than CMs (Rothermel et al., 2016) and with our previous observation that CYSLTR2L129Q enforces a melanocyte lineage (Moore et al., 2016).
Figure 5. Gα11/q-Driven Tumors Enforce a Melanocyte Lineage Program and Express High Levels of RASGRP3.

(A) GSEA profile using the GEMM gene sets (Mouse BRaf UP and Mouse Gα11/q UP) on a profile ranked by expression difference between Gα11/q mutant and BRAFV600E mutant melanomas in the combined TCGA SKCM and UM datasets.
(B) GO and SP pathways from leading edge genes (left; red; A) overexpressed in Gα11/q GEMM tumors and human Gα11/q mutant.
(C and D) Melanocyte-lineage genes shown as Tukey box-and-whisker plots from BRafV600E and GNA11Q209L GEMM (C) and TCGA SKCM melanomas (D). Outliers are shown as dots. p < 10−6.
(E) RASGRP3 expression from pan-cancer TCGA shown as Tukey box-and-whisker plots. Outliers are shown as red dots. UM is highlighted in red. SKCM is highlighted in blue. Outliers with GNAQ, GNA11, BAP1, and SF3B1 mutations are circled and detailed.
(F) RasGRP3 expression shown as Tukey box-and-whisker plots from GEMM tumors. p < 10−6.
(G) Immunoblot of RASGRP3 in human UM and CM cell lines.
See also Figure S6.
Further examination of top-ranked genes identified RASGRP3 as highly expressed in both Gα11/q-mutated human and GEMM melanomas (Figures 5E and 5F). Pan-cancer analysis of RNA-seq datasets from TCGA (Figure 5E) and Affymetrix U133Plus2 datasets curated by gene expression across normal and tumor tissue (GENT) (Figure S6) showed RASGRP3 is expressed at significantly higher levels in UMs than in CMs and other cancer types. Notably, all TCGA UMs expressed high RASGRP3, and the five TCGA SKCMs with the highest RASGRP3 expression harbored either GNAQ or GNA11 mutations and either BAP1 loss (mutation or monosomy 3) or SF3B1 mutations, suggesting they may be malignant blue nevi (Griewank et al., 2017) (Figure 5E, circled). RASGRP3 encodes Ras guanyl-releasing protein 3 (RasGRP3), a GEF that promotes the release of GDP-bound Ras in order to bind GTP, yielding active Ras (Ras-GTP) (Rebhun et al., 2000). RasGRP3 activation is dependent on both DAG binding and phosphorylation on Thr 133 by protein kinase C (PKC) (Aiba et al., 2004; Zheng et al., 2005). Together, these signaling events place the activation of RasGRP3 downstream of UM-activating mutations (CYSLTR2, GNAQ, GNA11, and PLCB4), and they suggest RasGRP3 may be a key signaling node that can integrate the UM-activating mutations into the MAPK pathway.
To determine if RasGRP3 expression is retained in cell lines, we screened a panel of UM and CM cells, and we found RasGRP3 to be expressed exclusively in UM cells (Figure 5G).
RasGRP3 Is Required for Ras-MAPK Activation and Growth in UM Cells
To determine if RasGRP3 is required for Gα11/q-mediated activation of the MAPK pathway and for growth in UM, we generated two short hairpin RNAs (shRNAs) to mediate knockdown of RASGRP3 (shRasGRP3-1 and shRasGRP3-2) and a control (shSCR). Depletion of RasGRP3 significantly reduced cell proliferation in GNA11 or GNAQ mutant cells (Figures 6A and S7A). In contrast, knockdown of RasGRP3 in CM cells did not (Figures 6B and S7A).
Figure 6. RASGRP3 Is Required for Growth and ras Activation in UM Cells.

(A and B) Growth curves of UM (A) or CM (B) cells with shSCR, shRASGRP3-1, or shRASGRP3-2, shown as relative luminescence units (RLUs). Error bars, means ± SEM from six technical replicates. p < 0.001 (A), p = not significant (ns), and p = 0.016 (B).
(C) Immunoblots of RASGRP3 and MAPK pathway. UM and CM cells stably expressing dox-shSCR, dox-shRASGRP3-1, or dox-shRASGRP3-2 in the presence of dox are shown.
(D) Percentage GFP-positive cells over time expressing KRASG12V-IRES-GFP with shSCR, shRASGRP3-1, or shRASGRP3-2. Change of percentage GFP-positive indicates relative growth of KRASG12V-expressing cells to non-expressing cells.
(E) Immunoblot of bulk cells (D) against the indicated proteins.
See also Figure S7.
We next sought to characterize the response of RasGRP3 depletion in the context of Ras activation and subsequent downstream MAPK signaling. We stably expressed two doxycycline (dox)-inducible RasGRP3 shRNAs (dox-shRasGRP3-1 and dox-shRasGRP3-2) and a dox-inducible control (dox-shSCR) in the panel of UM and CM cells. Depletion of RasGRP3 with dox significantly reduced the proportion of Ras-GTP and phosphorylation of ERK1/2 and P90RSK in UM cells (Figure 6C). Consistent with the cellular growth data, there was no change in Ras activation or MAPK signaling upon depletion of RasGRP3 in CM cells (Figure 6C).
To elucidate the requirement of the Ras GEF activity of RasGRP3, we examined if ectopic expression of KRASG12V could rescue the proliferation and MAPK signaling in three UM cell lines depleted of RasGRP3. We performed growth competition assays in which we first generated cells where 20%–50% expressed KRASG12V-IRES-GFP and empty vector at low MOI. We next infected these cells with shSCR or shRasGRP3, and we tracked the percentage of GFP-positive cells over time using fluorescence-activated cell sorting (FACS). The percentage of empty vector-expressing GFP-positive cells remained stable over time regardless of RasGRP3 depletion in both CM and UM cells (Figure S7B). In contrast, the percentage of KRASG12V-expressing GFP-positive cells increased after RasGRP3 depletion compared to shSCR in all three UM lines (Figure 6D), indicating KRASG12V conveys a growth advantage specifically after RasGRP3 depletion. Expression of KRASG12V rescued the reduction in ERK phosphorylation observed upon depletion of RasGRP3 (Figure 6E). Ectopic expression of KRASG12V in a CM cell line (A375) provided no changes in proliferation or ERK phosphorylation (Figures 6D and 6E). Therefore, UM cells require RasGRP3 for Ras activation and cellular proliferation.
RasGRP3 Is Required for Gα11/q-Mediated Growth
Human UM cells harboring Gα11/q mutations selectively require RasGRP3 for growth and MAPK activation, suggesting Gα11/q-mediated oncogenesis might require RasGRP3. To explore this hypothesis, we determined the requirement of RasGRP3 in an immortalized mouse melanocytic cell line, melan-a. Melan-a cells require phorbol esters, such as the DAG analog TPA (12-O-tetradecanoylphorbol-13-acetate), that can activate PKC for growth, and they can become TPA independent upon the expression of oncogenic mutations (Wellbrock et al., 2004). We transduced melan-a cells with GNAQQ209L, BRAFV600E, and KRASG12V, and we cultured the cells in the absence of TPA to establish oncogene-dependent growth. At baseline, melan-a cells with TPA-dependent growth expressed endogenous Rasgrp3. While GNAQQ209L-dependent cells retained Rasgrp3, BRAFV600E-and KRASG12V-dependent cells lost Rasgrp3 (Figure S7C). BRAFV600E- and KRASG12V-dependent cells lost pigmentation and expression of melanocyte lineage proteins, while GNAQQ209L-dependent cells retained pigmentation and melanocyte lineage proteins (Figure S7C). Re-introduction of TPA to the media of BRAFV600E- and KRASG12V-dependent cells failed to re-establish Rasgrp3 expression (Figure S7D). The difference in melanocyte lineage commitment between Gα11/q and RAS/RAF-driven transformed melanocytes is consistent with observations in our GEMMs and patient tumors.
To determine the potential role of RasGRP3 in Gα11/q-, BRAF-, and KRAS-mediated tumorigenesis, we performed shRNA-mediated knockdown of Rasgrp3 (shRasgrp3-1 and shRasgrp3-2). Knockdown of Rasgrp3 significantly reduced cell growth in GNAQQ209L-dependent, but not in BRAFV600E- or KRASG12V-dependent, melan-a cells (Figures 7A and S7E). Depletion of Rasgrp3 in GNAQQ209L melan-a cells reduced Ras-GTP and the phosphorylation of ERK and P90RSK (Figure 7B). Therefore, RasGRP3 is specifically required for Gα11/q-mediated oncogenic growth.
Figure 7. Rasgrp3 Is Required for Gα11/q-Mediated Growth and MAPK Activation in Melan-a Cells.

(A) Growth curves of GNAQQ209L, BRAFV600E, or KRASG12V melan-a cells, grown in the absence of TPA and expressing shSCR, shRasgrp3-1, or shRasgrp3-2, shown as RLUs. Error bars, means ± SEM from six technical replicates. p < 0.001 (GNAQQ209L) and p = ns (BRAFV600E and KRASG12V) for reduction in growth.
(B) Immunoblots of Rasgrp3 and MAPK pathway following Rasgrp3 depletion.
See also Figure S7.
DISCUSSION
UMs, LMNs, and blue nevi harbor activating mutations along the CYSLTR2-Gα11/q-PLCβ pathway, and they can have mutually exclusive cooperating mutations in BAP1, SF3B1, and EIF1AX that convey poor, intermediate, and favorable risk, respectively (de la Fouchardière et al., 2015; Goldman-Lévy et al., 2016; Küsters-Vandevelde et al., 2016). This distinct molecular profile is observed in a small subset of CMs and ~10% of mucosal melanomas (Sheng et al., 2016). Pathologically, UMs are characterized by their retention of the melanocyte lineage program, including pigmentation (Rothermel et al., 2016).
No proven effective therapies exist for UM. As with Ras, it is difficult to target G-proteins with competitive inhibitors to the nucleotide-binding site due to the high cellular concentrations of GTP. Since Gα11/q signaling activates PKC and the MAPK pathway, via PLCβ, many groups have studied the role of PKC and MAPK in UM. Cells with Gα11/q mutations were modestly sensitive to MEK inhibition and combination treatment of PKC and MEK inhibitors (Chen et al., 2014). Unfortunately, PKC targeting is limited by toxicity, and a completed phase 3 trial with selumetinib showed no clinical benefit (Komatsubara et al., 2016). In addition to PLCβ, Gαq directly interacts with the Trio family of Rho-GEFs (Trio, p63-RhoGEF, and Kalirin) to activate Rac and Rho and downstream YAP, and this pathway may represent a therapeutic target (Feng et al., 2014; Yu et al., 2014). Another promising target is ARF6, a GTPase involved in vesicle trafficking and required for proper shuttling of activated Gαq to cytoplasmic vesicles, where downstream signaling to both PLCβ and Rho were localized (Yoo et al., 2016). In addition to Gα11/q signaling, another therapeutic strategy is targeting the melanocyte lineage. IMCgp100 is a bispecific antibody that binds gp100 (PMEL) on tumor cells and CD3 on T cells (Carvajal et al., 2014, J. Stem Cell Res. Ther., abstract). A phase 1 trial in melanoma showed a disease control rate of 21% and 57% in CM and UM, respectively, and an expanded study in UM showed a similar disease control rate with some durable responses (Iams et al., 2017).
Here we sought to generate a clinically relevant GEMM of aggressive Gα11-driven melanoma, combining GNA11Q209L and BAP1 loss, and we compared it to an isogenic BRafV600E model to identify Gα11-driven phenotypes and vulnerabilities. We found GNA11Q209L drove neoplastic growth in cutaneous and many non-cutaneous sites whereas BRafV600E only promotes CM. While the lung and lymph nodes are the preferential sites of metastasis in the Tyr-CreERT2;BRafCA/+;Ptenflox/flox mouse (Dankort et al., 2009) and in a transgenic mouse of Tyr-driven SV40 T-antigen (Bradl et al., 1991), one limitation in our mouse model was the inability to specifically activate GNA11Q209L in defined melanocytic subsets, and this hampers the ability to definitively assign metastasis (Gibson et al., 2010; Klein-Szanto et al., 1991).
GNA11Q209L-driven tumors were highly pigmented compared to BRafV600E, consistent with clinical observation that Gα11/q-driven primary blue nevi, UM, and UM metastases retain pigmentation (Emley et al., 2011; Rothermel et al., 2016). Therefore, Gαq/11 signaling drives lineage commitment, and targeting the lineage, such as IMCgp100, is a promising therapeutic strategy (Carvajal et al., 2014, J. Stem Cell Res. Ther., abstract). Bap1 loss in our GEMM accelerated skin melanoma growth, consistent with the clinical observation that BAP1 loss is found in transformed, but not benign, blue nevi (Griewank et al., 2017). Yet, there was no significant change of uveal pathology, highlighting a limitation of our model.
Cross-species comparison between representative GEMM models with human disease can identify critical mediators of tumorigenesis (Johnson et al., 2010). By cross-referencing the RNA-seq data from the GEMM and human disease data, we identified a Ras-GEF, RasGRP3, as a required signaling node for UM. Consistent with a recently published study (Chen et al., 2017), we observed RasGRP3 is highly upregulated in UM and is required for proliferation. We additionally showed engineered cells driven by mutant Gα11/q specifically require RasGRP3 for Ras activation and growth. RASGPR3 expression is tissue specific and, among cancers, constrained to Gα11/q-driven melanomas, leukemias, and lymphomas, suggesting RasGRP3 is a specific vulnerability in Gα11/q-driven tumors and potentially a therapeutic target. The interaction between G-proteins and their GEFs is a viable drug target, as exemplified by the antibiotic brefeldin A, which blocks interaction between ARF1 and its GEF Sec7 (Mossessova et al., 2003), and RasGRP3 may be similarly targeted.
EXPERIMENTAL PROCEDURES
Further details and an outline of the resources used in this work can be found in the Supplemental Experimental Procedures.
Mouse Experiments
All animal studies were performed in accordance with the MSKCC IACUC (11-12-029). For GEMM studies, three cohorts of mice, Tyr-CreERT2;GNA11Q209L, Tyr-CreERT2;GNA11Q209L;Bap1lox/lox, and Tyr-CreERT2;BRafCA/+;Bap1lox/lox, were administered with intraperitoneal tamoxifen at 4 weeks of age with no regard to the sex of the animals, and histology was similar between males and females. Mice developed tumors in situ after tamoxifen injection. Mice were euthanized in response but not limited to the following: tumors larger than 1 cm3, tumor ulceration, tumors located too close to the trunk of the mice to impede movement and blood flow, and tumor burden, and time of euthanization was used for Kaplan-Meier survival analysis. For allograft studies, GEMM-derived tumors were grafted into 6- to 8-week-old female CB17-SCID mice and treated with vehicle or trametinib via oral gavage.
Histology, Immunohistochemistry, and Immunofluorescence
All tissues were fixed at 4°C overnight in 4% paraformaldehyde. Tissue processing, embedding, sectioning, H&E staining, and H&E staining with melanin bleaching were performed by Histoserv. Skull sections were performed following decalcification.
RNA-Seq
Total RNA was extracted from fresh-frozen tissue or cell lines using QIAGEN’s RNeasy Mini Kit. The isolated RNA was processed for RNA-seq by the Integrated Genomics Core Facility at MSKCC.
Cell Lines
Melan-a cells were provided by D. Bennett (Bennett et al., 1987); MEL202, MEL270, OMM1.3, COLO800, UPMM3, A375, and A2058 cells were submitted for short tandem repeat (STR) profiling and MSK-IMPACT (integration mutation profiling of actionable cancer targets) for mutational status at MSKCC to confirm their authenticity.
Statistics
Boxplots represent 25th and 75th percentiles with midline indicating the median; whiskers extend to the lowest/highest value within 1.5 times the inter-quartile range. Outliers are shown as dots. Comparisons for growth curves and xenograft experiments between two groups were performed using a two-tailed parametric unpaired t test. All statistics were performed using GraphPad Prism 6.0 software.
Supplementary Material
Highlights.
GNA11 Q209L mouse model induces uveal, cutaneous, and leptomeningeal melanoma
Loss of Bap1 promotes aggressive melanomas
RasGRP3 links GNA11/GNAQ activation to RAS activation
RasGRP3 is required for GNA11/GNAQ-driven tumorigenesis
Acknowledgments
We gratefully acknowledge David Abramson, Brian Marr, Irina Belinsky, and Taha Merghoub for their intellectual input. Next-generation sequencing and gene expression arrays were done at the MSKCC Integrated Genomics Operation. Gene targeting was performed by the Rockefeller University Gene Targeting Facility (Chingweng Yang), and blastocyst injection was performed by the MSKCC Mouse Genetic Facility (Willie Marks). FACS was performed at the MSKCC Flow Cytometry Core. Mouse pathology was reviewed by the MSKCC Laboratory of Comparative Pathology Core Facility. This work was supported by MSKCC Support Grant/Core Grant (P30 CA008748) and grants from the NCI (K08CA140946, Y.C.; R01CA193837, Y.C.; P50CA092629, Y.C.; P50CA140146, P.C.; K08CA151660, P.C.; and DP2 CA174499, P.C.), US DOD (W81XWH-10-1-0197, P.C.), the Prostate Cancer Foundation (16CHAL03, Y.C.), the Starr Cancer Consortium (I7-A722, Y.C. and P.C.), the Geoffrey Beene Cancer Research Center (Y.C. and P.C.), the Gerstner Family Foundation (Y.C.), Bressler Scholars Fund (Y.C.), and Cycle for Survival (Y.C.).
Footnotes
DATA AND SOFTWARE AVAILABILITY
The accession number for the data reported in this paper is GEO: GSE97225.
Supplemental Information includes Supplemental Experimental Procedures, seven figures, and three tables and can be found with this article online at https://doi.org/10.1016/j.celrep.2018.01.081.
AUTHOR CONTRIBUTIONS
Project Planning and Experimental Design, P.C., Y.C., A.R.M., L.R., K.G.G., and A.N.S.; Pathology Review, R.M. and S.M.; Bioinformatics, Y.C. and A.R.M.; Cellular Assays, A.R.M., L.R., Y.G., and T.D.H.; Mice, A.R.M., J.J.S., and E.G.W.; Expression Vectors, A.R.M., Y.G., J.Q.Z., C.H., and T.W.; Manuscript Writing, A.R.M., P.C., and Y.C.; Review of the Final Manuscript, all authors.
DECLARATION OF INTERESTS
The authors declare no competing interests.
References
- Aiba Y, Oh-hora M, Kiyonaka S, Kimura Y, Hijikata A, Mori Y, Kurosaki T. Activation of RasGRP3 by phosphorylation of Thr-133 is required for B cell receptor-mediated Ras activation. Proc Natl Acad Sci USA. 2004;101:16612–16617. doi: 10.1073/pnas.0407468101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alizadeh A, Fitch KR, Niswender CM, McKnight GS, Barsh GS. Melanocyte-lineage expression of Cre recombinase using Mitf regulatory elements. Pigment Cell Melanoma Res. 2008;21:63–69. doi: 10.1111/j.1755-148X.2007.00425.x. [DOI] [PubMed] [Google Scholar]
- Ambrosini G, Pratilas CA, Qin LX, Tadi M, Surriga O, Carvajal RD, Schwartz GK. Identification of unique MEK-dependent genes in GNAQ mutant uveal melanoma involved in cell growth, tumor cell invasion, and MEK resistance. Clin Cancer Res. 2012;18:3552–3561. doi: 10.1158/1078-0432.CCR-11-3086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoki H, Yamada Y, Hara A, Kunisada T. Two distinct types of mouse melanocyte: differential signaling requirement for the maintenance of non-cutaneous and dermal versus epidermal melanocytes. Development. 2009;136:2511–2521. doi: 10.1242/dev.037168. [DOI] [PubMed] [Google Scholar]
- Bennett DC, Cooper PJ, Hart IR. A line of non-tumorigenic mouse melanocytes, syngeneic with the B16 melanoma and requiring a tumour promoter for growth. Int J Cancer. 1987;39:414–418. doi: 10.1002/ijc.2910390324. [DOI] [PubMed] [Google Scholar]
- Bradl M, Klein-Szanto A, Porter S, Mintz B. Malignant melanoma in transgenic mice. Proc Natl Acad Sci USA. 1991;88:164–168. doi: 10.1073/pnas.88.1.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas N.; Cancer Genome Atlas Network. Genomic Classification of Cutaneous Melanoma. Cell. 2015;161:1681–1696. doi: 10.1016/j.cell.2015.05.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvajal RD, Sosman JA, Quevedo JF, Milhem MM, Joshua AM, Kudchadkar RR, Linette GP, Gajewski TF, Lutzky J, Lawson DH, et al. Effect of selumetinib vs chemotherapy on progression-free survival in uveal melanoma: a randomized clinical trial. JAMA. 2014;311:2397–2405. doi: 10.1001/jama.2014.6096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Wu Q, Tan L, Porter D, Jager MJ, Emery C, Bastian BC. Combined PKC and MEK inhibition in uveal melanoma with GNAQ and GNA11 mutations. Oncogene. 2014;33:4724–4734. doi: 10.1038/onc.2013.418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X, Wu Q, Depeille P, Chen P, Thornton S, Kalirai H, Coupland SE, Roose JP, Bastian BC. RasGRP3 Mediates MAPK Pathway Activation in GNAQ Mutant Uveal Melanoma. Cancer Cell. 2017;31:685–696. e6. doi: 10.1016/j.ccell.2017.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collaborative Ocular Melanoma Study Group. Assessment of metastatic disease status at death in 435 patients with large choroidal melanoma in the Collaborative Ocular Melanoma Study (COMS): COMS report no. 15. Arch Ophthalmol. 2001;119:670–676. doi: 10.1001/archopht.119.5.670. [DOI] [PubMed] [Google Scholar]
- Dankort D, Curley DP, Cartlidge RA, Nelson B, Karnezis AN, Damsky WE, Jr, You MJ, DePinho RA, McMahon M, Bosenberg M. Braf(V600E) cooperates with Pten loss to induce metastatic melanoma. Nat Genet. 2009;41:544–552. doi: 10.1038/ng.356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Fouchardière A, Cabaret O, Pètre J, Aydin S, Leroy A, de Potter P, Pissaloux D, Haddad V, Bressacde Paillerets B, Janin N. Primary leptomeningeal melanoma is part of the BAP1-related cancer syndrome. Acta Neuropathol. 2015;129:921–923. doi: 10.1007/s00401-015-1423-2. [DOI] [PubMed] [Google Scholar]
- Diener-West M, Reynolds SM, Agugliaro DJ, Caldwell R, Cumming K, Earle JD, Hawkins BS, Hayman JA, Jaiyesimi I, Jampol LM, et al. Collaborative Ocular Melanoma Study Group. Development of metastatic disease after enrollment in the COMS trials for treatment of choroidal melanoma: Collaborative Ocular Melanoma Study Group Report No. 26. Arch Ophthalmol. 2005;123:1639–1643. doi: 10.1001/archopht.123.12.1639. [DOI] [PubMed] [Google Scholar]
- Emley A, Nguyen LP, Yang S, Mahalingam M. Somatic mutations in GNAQ in amelanotic/hypomelanotic blue nevi. Hum Pathol. 2011;42:136–140. doi: 10.1016/j.humpath.2010.05.027. [DOI] [PubMed] [Google Scholar]
- Feng X, Degese MS, Iglesias-Bartolome R, Vaque JP, Molinolo AA, Rodrigues M, Zaidi MR, Ksander BR, Merlino G, Sodhi A, et al. Hippo-independent activation of YAP by the GNAQ uveal melanoma oncogene through a trio-regulated rho GTPase signaling circuitry. Cancer Cell. 2014;25:831–845. doi: 10.1016/j.ccr.2014.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furney SJ, Pedersen M, Gentien D, Dumont AG, Rapinat A, Desjardins L, Turajlic S, Piperno-Neumann S, de la Grange P, Roman-Roman S, et al. SF3B1 mutations are associated with alternative splicing in uveal melanoma. Cancer Discov. 2013;3:1122–1129. doi: 10.1158/2159-8290.CD-13-0330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson P, Tong Y, Robinson G, Thompson MC, Currle DS, Eden C, Kranenburg TA, Hogg T, Poppleton H, Martin J, et al. Subtypes of medulloblastoma have distinct developmental origins. Nature. 2010;468:1095–1099. doi: 10.1038/nature09587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman-Lévy G, Rigau V, Blèchet C, Bens G, Muckensturm B, Delage M, Labrousse F, Haddad V, Attignon V, Pissaloux D, de la Fouchardière A. Primary Melanoma of the Leptomeninges with BAP1 Expression-Loss in the Setting of a Nevus of Ota: A Clinical, Morphological and Genetic Study of 2 Cases. Brain Pathol. 2016;26:547–550. doi: 10.1111/bpa.12363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griewank KG, Yu X, Khalili J, Sozen MM, Stempke-Hale K, Bernatchez C, Wardell S, Bastian BC, Woodman SE. Genetic and molecular characterization of uveal melanoma cell lines. Pigment Cell Melanoma Res. 2012;25:182–187. doi: 10.1111/j.1755-148X.2012.00971.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griewank KG, Müller H, Jackett LA, Emberger M, Möller I, van de Nes JA, Zimmer L, Livingstone E, Wiesner T, Scholz SL, et al. SF3B1 and BAP1 mutations in blue nevus-like melanoma. Mod Pathol. 2017;30:928–939. doi: 10.1038/modpathol.2017.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harbour JW, Onken MD, Roberson ED, Duan S, Cao L, Worley LA, Council ML, Matatall KA, Helms C, Bowcock AM. Frequent mutation of BAP1 in metastasizing uveal melanomas. Science. 2010;330:1410–1413. doi: 10.1126/science.1194472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang JL, Urtatiz O, Van Raamsdonk CD. Oncogenic G Protein GNAQ Induces Uveal Melanoma and Intravasation in Mice. Cancer Res. 2015;75:3384–3397. doi: 10.1158/0008-5472.CAN-14-3229. [DOI] [PubMed] [Google Scholar]
- Iams WT, Sosman JA, Chandra S. Novel Targeted Therapies for Metastatic Melanoma. Cancer J. 2017;23:54–58. doi: 10.1097/PPO.0000000000000242. [DOI] [PubMed] [Google Scholar]
- Infante JR, Fecher LA, Falchook GS, Nallapareddy S, Gordon MS, Becerra C, DeMarini DJ, Cox DS, Xu Y, Morris SR, et al. Safety, pharmacokinetic, pharmacodynamic, and efficacy data for the oral MEK inhibitor trametinib: a phase 1 dose-escalation trial. Lancet Oncol. 2012;13:773–781. doi: 10.1016/S1470-2045(12)70270-X. [DOI] [PubMed] [Google Scholar]
- Johansson P, Aoude LG, Wadt K, Glasson WJ, Warrier SK, Hewitt AW, Kiilgaard JF, Heegaard S, Isaacs T, Franchina M, et al. Deep sequencing of uveal melanoma identifies a recurrent mutation in PLCB4. Oncotarget. 2016;7:4624–4631. doi: 10.18632/oncotarget.6614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson RA, Wright KD, Poppleton H, Mohankumar KM, Finkelstein D, Pounds SB, Rand V, Leary SE, White E, Eden C, et al. Cross-species genomics matches driver mutations and cell compartments to model ependymoma. Nature. 2010;466:632–636. doi: 10.1038/nature09173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein-Szanto A, Bradl M, Porter S, Mintz B. Melanosis and associated tumors in transgenic mice. Proc Natl Acad Sci USA. 1991;88:169–173. doi: 10.1073/pnas.88.1.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komatsubara KM, Manson DK, Carvajal RD. Selumetinib for the treatment of metastatic uveal melanoma: past and future perspectives. Future Oncol. 2016;12:1331–1344. doi: 10.2217/fon-2015-0075. [DOI] [PubMed] [Google Scholar]
- Küsters-Vandevelde HV, Creytens D, van Engen-van Grunsven AC, Jeu- nink M, Winnepenninckx V, Groenen PJ, Küsters B, Wesseling P, Blokx WA, Prinsen CF. SF3B1 and EIF1AX mutations occur in primary leptomeningeal melanocytic neoplasms; yet another similarity to uveal melanomas. Acta Neuropathol Commun. 2016;4:5. doi: 10.1186/s40478-016-0272-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaFave LM, Béguelin W, Koche R, Teater M, Spitzer B, Chramiec A, Papalexi E, Keller MD, Hricik T, Konstantinoff K, et al. Loss of BAP1 function leads to EZH2-dependent transformation. Nat Med. 2015;21:1344–1349. doi: 10.1038/nm.3947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lito P, Saborowski A, Yue J, Solomon M, Joseph E, Gadal S, Saborowski M, Kastenhuber E, Fellmann C, Ohara K, et al. Disruption of CRAF-mediated MEK activation is required for effective MEK inhibition in KRAS mutant tumors. Cancer Cell. 2014;25:697–710. doi: 10.1016/j.ccr.2014.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M, Maßhöfer L, Temming P, Rahmann S, Metz C, Bornfeld N, van de Nes J, Klein-Hitpass L, Hinnebusch AG, Horsthemke B, et al. Exome sequencing identifies recurrent somatic mutations in EIF1AX and SF3B1 in uveal melanoma with disomy 3. Nat Genet. 2013;45:933–936. doi: 10.1038/ng.2674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Möller I, Murali R, Müller H, Wiesner T, Jackett LA, Scholz SL, Cosgarea I, van de Nes JA, Sucker A, Hillen U, et al. Activating cysteinyl leukotriene receptor 2 (CYSLTR2) mutations in blue nevi. Mod Pathol. 2017;30:350–356. doi: 10.1038/modpathol.2016.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore AR, Ceraudo E, Sher JJ, Guan Y, Shoushtari AN, Chang MT, Zhang JQ, Walczak EG, Kazmi MA, Taylor BS, et al. Recurrent activating mutations of G-protein-coupled receptor CYSLTR2 in uveal melanoma. Nat Genet. 2016;48:675–680. doi: 10.1038/ng.3549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mossessova E, Corpina RA, Goldberg J. Crystal structure of ARF1*Sec7 complexed with Brefeldin A and its implications for the guanine nucleotide exchange mechanism. Mol Cell. 2003;12:1403–1411. doi: 10.1016/s1097-2765(03)00475-1. [DOI] [PubMed] [Google Scholar]
- Rebhun JF, Castro AF, Quilliam LA. Identification of guanine nucleotide exchange factors (GEFs) for the Rap1 GTPase. Regulation of MR-GEF by M-Ras-GTP interaction. J Biol Chem. 2000;275:34901–34908. doi: 10.1074/jbc.M005327200. [DOI] [PubMed] [Google Scholar]
- Robertson AG, Shih J, Yau C, Gibb EA, Oba J, Mungall KL, Hess JM, Uzunangelov V, Walter V, Danilova L, et al. TCGA Research Network. Integrative Analysis Identifies Four Molecular and Clinical Subsets in Uveal Melanoma. Cancer Cell. 2017;32:204–220. e15. doi: 10.1016/j.ccell.2017.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothermel LD, Sabesan AC, Stephens DJ, Chandran SS, Paria BC, Srivastava AK, Somerville R, Wunderlich JR, Lee CC, Xi L, et al. Identification of an Immunogenic Subset of Metastatic Uveal Melanoma. Clin Cancer Res. 2016;22:2237–2249. doi: 10.1158/1078-0432.CCR-15-2294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng X, Kong Y, Li Y, Zhang Q, Si L, Cui C, Chi Z, Tang B, Mao L, Lian B, et al. GNAQ and GNA11 mutations occur in 9.5% of mucosal melanoma and are associated with poor prognosis. Eur J Cancer. 2016;65:156–163. doi: 10.1016/j.ejca.2016.06.019. [DOI] [PubMed] [Google Scholar]
- Solit DB, Garraway LA, Pratilas CA, Sawai A, Getz G, Basso A, Ye Q, Lobo JM, She Y, Osman I, et al. BRAF mutation predicts sensitivity to MEK inhibition. Nature. 2006;439:358–362. doi: 10.1038/nature04304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tandon N, O’Neill TJ, Vollmer DG, Wang M. Intraventricular occurrence of a melanocytoma. J Neurosurg. 2008;109:480–485. doi: 10.3171/JNS/2008/109/9/0480. [DOI] [PubMed] [Google Scholar]
- Van Raamsdonk CD, Bezrookove V, Green G, Bauer J, Gaugler L, O’Brien JM, Simpson EM, Barsh GS, Bastian BC. Frequent somatic mutations of GNAQ in uveal melanoma and blue naevi. Nature. 2009;457:599–602. doi: 10.1038/nature07586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Raamsdonk CD, Griewank KG, Crosby MB, Garrido MC, Vemula S, Wiesner T, Obenauf AC, Wackernagel W, Green G, Bouvier N, et al. Mutations in GNA11 in uveal melanoma. N Engl J Med. 2010;363:2191–2199. doi: 10.1056/NEJMoa1000584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wellbrock C, Ogilvie L, Hedley D, Karasarides M, Martin J, Niculescu-Duvaz D, Springer CJ, Marais R. V599EB-RAF is an oncogene in melanocytes. Cancer Res. 2004;64:2338–2342. doi: 10.1158/0008-5472.can-03-3433. [DOI] [PubMed] [Google Scholar]
- Yoo JH, Shi DS, Grossmann AH, Sorensen LK, Tong Z, Mleynek TM, Rogers A, Zhu W, Richards JR, Winter JM, et al. ARF6 Is an Actionable Node that Orchestrates Oncogenic GNAQ Signaling in Uveal Melanoma. Cancer Cell. 2016;29:889–904. doi: 10.1016/j.ccell.2016.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu FX, Luo J, Mo JS, Liu G, Kim YC, Meng Z, Zhao L, Peyman G, Ouyang H, Jiang W, et al. Mutant Gq/11 promote uveal melanoma tumorigenesis by activating YAP. Cancer Cell. 2014;25:822–830. doi: 10.1016/j.ccr.2014.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y, Liu H, Coughlin J, Zheng J, Li L, Stone JC. Phosphorylation of RasGRP3 on threonine 133 provides a mechanistic link between PKC and Ras signaling systems in B cells. Blood. 2005;105:3648–3654. doi: 10.1182/blood-2004-10-3916. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.