

Abstract
Alzheimer’s disease (AD) is a devastating neurodegenerative disease that is characterized by progressive cognitive decline and a prominent loss of hippocampal-dependent memory. Therefore, much focus has been placed on understanding the function and dysfunction of the hippocampus in AD. However, AD is also accompanied by a number of other debilitating cognitive and behavioral alterations including deficits in attention, cognitive processing, and sleep maintenance. The underlying mechanisms that give rise to impairments in such diverse behavioral domains are unknown, and identifying them would shed insight into the multifactorial nature of AD as well as reveal potential new therapeutic targets to improve overall function in AD. We present here several lines of evidence that suggest that dysregulation of the corticothalamic network may be a common denominator that contributes to the diverse cognitive and behavioral alterations in AD. First, we will review the mechanisms by which this network regulates processes that include attention, cognitive processing, learning and memory, and sleep maintenance. Then we will review how these behavioral and cognitive domains are altered in AD. We will also discuss how dysregulation of tightly regulated activity in the corticothalamic network can give rise to non-convulsive seizures and other forms of epileptiform activity that have also been documented in both AD patients and transgenic mouse models of AD. In summary, the corticothalamic network has the potential to be a master regulator of diverse cognitive and behavioral domains that are affected in AD.
Keywords: Thalamic reticular nucleus, sleep maintenance, seizures, hippocampus, thalamus, attention
1. Introduction
Alzheimer’s disease (AD) is a complex and progressive neurodegenerative disease that affects roughly 5 million people in the United States, and over 35 million people worldwide (Ferri et al., 2005; Hebert et al., 2013). Pathological hallmarks of disease include amyloid plaques and neurofibrillary tangles, as well as neuronal loss, and a definitive diagnosis of AD is made at autopsy. However, it is becoming increasingly clear that the disease process begins decades before major pathological hallmarks are established, and well before patients begin to report difficulties due to symptoms.
Clinically, AD typically begins with mild forgetfulness, and early on may be diagnosed as mild cognitive impairment (MCI). As the disease progresses, deficits become more severe and affect day-to-day functioning as they begin to affect other cognitive aspects such as executive and visuospatial function. Other behavioral domains become affected as well, including deficits in both attention and sleep maintenance. Long-term monitoring with electroencephalography (EEG) has also revealed an increase in incidence of epileptiform activity, including epileptic spikes and nonconvulsive seizures that begin to occur near the onset of cognitive decline (Chin and Scharfman, 2013; Vossel et al., 2013; Vossel et al., 2016). Because the characteristic loss of memory is a key feature of AD, much research over the last several decades has focused on understanding (dys)function of the hippocampus in the disease. Molecular, cellular, network, and imaging studies have provided much insight into the complexity of AD, and how AD-related factors like amyloid beta (Aβ), tau, and ApoE alter neuronal and hippocampal function (Huang and Mucke, 2012). Such research has yielded many promising therapeutic targets that have unfortunately led to disappointment in large clinical trials. However it is critical to note that failure to reach particular cognitive endpoint measures that were established a priori does not necessarily mean that the underlying hypothesis was wrong, but perhaps that 1) there are other as yet undefined factors that influence cognition and/or the network mechanisms that underlie it, or 2) treatment was not begun early enough to be able to modulate disease progression, and that identifying other biomarkers or early symptoms of disease is critical to initiating treatment early enough to have beneficial effects.
Therefore it is critical to consider how other brain networks interact with and influence hippocampal function, and to identify the network mechanisms that give rise to the multitude of other symptoms that accompany AD that are not directly subserved by the hippocampus. Are there other brain regions in which dysfunction may occur and exacerbate progression disease even prior to obvious memory loss and cognitive impairment? Are there network mechanisms that could explain how such diverse domains of cognition and behavior are affected in AD? The identification of other vulnerable brain regions and network mechanisms may enable a better understanding of the cellular basis of AD as well as the development of novel therapeutic strategies that could improve several aspects of AD symptomology.
Several lines of evidence suggest that the corticothalamic network may be one such common denominator that underlies many cognitive and behavioral alterations in AD (Figure 1). The corticothalamic network regulates brain processes including sleep and arousal cycles, sleep maintenance, attention, and cognitive processing, as well as influences learning and memory. Moreover, dysregulation of the tightly regulated corticothalamic circuitry generates spike-wave discharges and can give rise to nonconvulsive seizures, which have been described in AD patients and mouse models. Although referred to here as the corticothalamic network, the term cortico-thalamo-cortical network is perhaps more appropriate for reflecting its functions in shaping various behavioral and cognitive domains relevant to understanding the progressive nature of AD. Cortico-thalamo-cortical loops mediate sensory responses, physiologic oscillations during early and deep NREM sleep, attention and perception, as well as absence and nonconvulsive seizures (Beenhakker and Huguenard, 2009; Fogerson and Huguenard, 2016; Jones, 2002; Steriade et al., 1993b; Timofeev et al., 1996). The current review focuses on functional implications of the corticothalamic network and how dysregulation can lead to symptoms in multiple domains observed in AD. We suggest that the corticothalamic network has the potential to be a master regulator of diverse cognitive and behavioral domains that are affected in AD.
Figure 1. Contributions of the corticothalamic network and the hippocampus to particular symptoms that accompany AD.
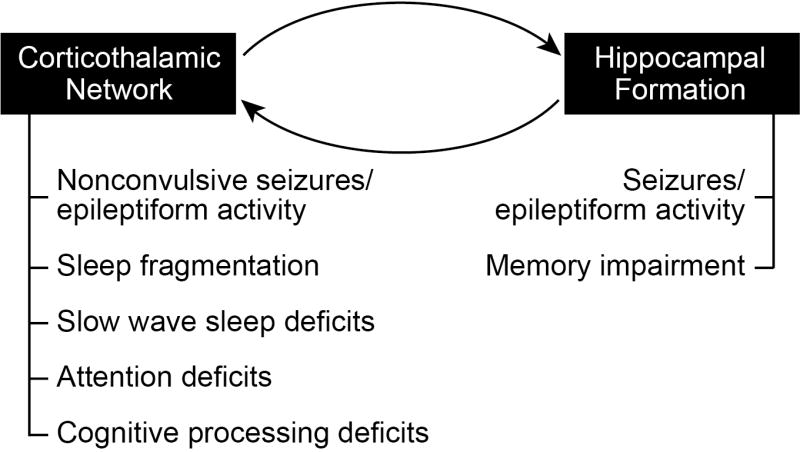
The exquisite vulnerability of the hippocampus in AD is critical for the memory impairments characteristic of AD, and may also contribute to the seizures and epileptiform activity observed in both AD patients and mouse models of the disease. However, alterations in the corticothalamic network are most likely responsible for a number of other deficits that accompany AD such as sleep fragmentation, attention deficits, cognitive processing deficits, and non-convulsive seizures. Notably, many of these other symptoms become evident even prior to memory deficits.
2. The corticothalamic network and its functions
2.1. Circuitry of the corticothalamic network
The corticothalamic network is an anatomical and functional circuit of reciprocal connectivity between the cerebral cortex and thalamic nuclei. Communication between the cerebral cortex and thalamic relay nuclei is tightly regulated by the thalamic reticular nucleus (TRN), a major control nucleus that is made up exclusively of GABAergic neurons (Figure 2). The TRN resides between the lateral aspect of the thalamus and the internal capsule, spreading across rostrocaudal and dorsoventral axes (Guillery and Harting, 2003; Pinault, 2004). About a century ago, Albert von Kölliker (1896) referred to the thalamic reticular nucleus for the first time in his book, ‘Handbuch der Geweblehre des Menschen’ - as ‘gitterkerne’ (translated from German as ‘core of lattice’) due to the appearance of nucleus, which lies ventral to the anterior thalamic nucleus. The TRN is an organized and complex population of GABAergic neurons that provides strict regulation of the information processing between thalamus and cerebral cortex (Guillery and Harting, 2003; Pinault, 2004). TRN neurons receive dense glutamatergic inputs from dorsal thalamic nuclei and from layer VI of the cerebral cortex, which make up approximately 70% of the synapses that TRN neurons receive (Jones, 2009), as well as cholinergic inputs from pedunculopontine nucleus of the brainstem, the nucleus basalis of Meynert in the basal forebrain, and the amygdala (Clemente-Perez et al., 2017; Cox et al., 1997; Levey et al., 1987; Pinault and Deschenes, 1998; Steriade et al., 1987; Steriade et al., 1988; Warren et al., 1994; Zikopoulos and Barbas, 2012). Corticothalamic projections from cortical layer VI to TRN are collaterals of the primary cortical projections in which the principal axon projects subcortically only to the thalamus (Bourassa and Deschenes, 1995; Deschenes et al., 1998; Pinault, 2004). Corticothalamic projects that begin in layer V of the cortex project directly to the thalamus to activate thalamic relays, without sending collaterals into TRN. However, corticothalamic pathways that originate in layer VI and take indirect pathways through TRN or the perigeniculate nucleus (PGN) are mainly inhibitory due to GABAergic nature of these nuclei (Houser et al., 1980; McCormick, 1992). Thus, Francis Crick’s description of the TRN as the “guardian of the gateway to the cortex”, where “gateway” refers to the thalamus, was aptly stated (Crick, 1984). Notably, although TRN receives excitatory inputs from many regions, the TRN projects solely to thalamic relay neurons wherein an individual TRN neuron innervates a single thalamic nucleus. The projection patterns of TRN neurons therefore make up functional subnetworks within TRN (Halassa et al., 2014). For example, TRN neurons that project to sensory thalamic nuclei are important for sleep spindles, and exhibit elevated activity during sleep. In contrast, TRN neurons that project to limbic nuclei associated with hippocampal processing are not involved in spindles and exhibit less activity during sleep.
Figure 2. Major components and circuitry of the corticothalamic network.

Interconnections between the thalamic reticular nucleus (TRN), thalamic relay nuclei (thalamus), and deep layers of the cortex are the primary components of the corticothalamic network. Cholinergic afferents from the basal forebrain and pedunculopontine nucleus, and glutamatergic afferents from the amygdala as well as other brain regions not shown, also provide input to this network. Projections from the basal forebrain and thalamus to the hippocampus are also illustrated. Red indicates GABA-ergic projections, orange indicates cholinergic projections. All other colors represent glutamatergic projections.
2.2. Corticothalamic circuit function
2.2.1. Sleep
Sleep is made up of 5 stages: Stages 1, 2, 3, and 4 of non-rapid eye movement (NREM) sleep, and REM sleep. The vast majority of time in sleep is spent in NREM sleep, during which brain activity is largely regulated by the thalamocortical circuit. As a person falls asleep, their eyes move very slowly and activity in the muscles slows. This sleep (stage 1) is relatively light and the person can be awakened easily. In stage 2, the eyes stop moving, and there is slowing of EEG brain waves, and occurrence of sleep spindles (7–14 Hz). As sleep deepens into stages 3 and 4 of NREM sleep (deep sleep, or slow wave sleep), delta oscillations (0.5 – 4 Hz) and slow oscillations by cortical neurons (0.3 – 1 Hz) dominate. There is no eye movement or muscle activity. During REM sleep, breathing becomes irregular and the eyes move rapidly in various directions, this is the stage at which dreams occur.
Sleep is an epic phenomenon; certain regions of the brain participate in the generation of rhythmic activities, without input from exterior stimuli, to express dynamic behaviors during sleep. For centuries, it is been a puzzle to elucidate how certain neural networks generate patterns of activity to produce sleep behavior with loss of awareness of the surroundings, but keeping intact memory before sleep (although the few minutes immediately prior to falling asleep are often not remembered) and upon emerging from sleep. Beenhakker and Huguenard (2009) have reviewed the central pattern generators underlying rhythmic oscillations of thalamic neurons that form spindle-like activity even independently of the cerebral cortex. Generalized spindle oscillations in the intact brain are due to a high degree of coherence throughout the corticothalamic, thalamocortical and TRN neuronal networks (Buzsaki, 1991; Steriade et al., 1985; Steriade et al., 1993a). Synchronized, but offset, firing of thalamocortical relay cells and TRN neurons give rise to spindle oscillations at 7 – 14 Hz frequencies. Cortical activation of TRN results in inhibition of downstream thalamocortical relay neurons. However, strong inhibition from TRN transiently evokes regenerative low-threshold Ca2+ spikes due to the deinactivation of T-type Ca2+ channels, together with a hyperpolarization-activated inward rectifier carried by Na+ and K+, leading to post inhibitory rebound responses (Jahnsen and Llinas, 1984a; Jahnsen and Llinas, 1984b; McCormick and Pape, 1990; Pape et al., 1994). Therefore, when TRN activity is particularly high, such as during sleep, its strong inhibitory action on thalamic relay neurons leads to post-inhibitory rebound that re-activates TRN neurons to initiate the next oscillation due to the reciprocal circuitry between TRN and thalamocortical relay nuclei (Golomb et al., 1996). In the intact network, TRN neurons burst with each cycle of spindle oscillation, whereas thalamocortical neurons burst every other or fewer cycles.
As mentioned above, TRN activity is particularly high during sleep, which generally inhibits thalamic relay neurons from transmitting sensory information to and from the cortex (Anderson et al., 2005; Beenhakker and Huguenard, 2009). Therefore, TRN plays a fundamental role in regulating sleep maintenance (the ability to stay asleep when asleep).
Slow wave sleep, which dominates stages 3 and 4 of NREM sleep, is characterized by slow wave activity (SWA) defined by the frequency (0.5 – 4 Hz) and relatively high amplitude power. SWA manifests as a function of interactions between cortex and thalamocortical nuclei that oscillate between UP states (firing of neurons) and silent DOWN states (long-lasting hyperpolarization) (Rechtschaffen, 1998; Vyazovskiy et al., 2009; Vyazovskiy and Harris, 2013). Extended time in the awake state increases SWA during NREM sleep (Hanlon et al., 2011; Lancel et al., 1992; Rodriguez et al., 2016) that has been hypothesized to result from the temporary exhaustion of neurons after a continuous activation state and depletion of energy resources (Dalsgaard and Secher, 2007; O'Donovan and Rinzel, 1997). Therefore, sleep may allow for homeostatic maintenance of synaptic activity and synaptic coupling between neurons. SWA during NREM sleep after an extended wake state or specific learning tasks assists in renormalization of synaptic strength that is beneficial for memory and performance (Hanlon et al., 2011). One mechanism by which SWA can be regulated is through activity in TRN, as it is a major regulator of corticothalamic dynamics. Stimulation of TRN activity induces SWA in cortical regions and sleep in awake mice (Lewis et al., 2015). Stimulation of TRN at the frequency equivalent to spindle activity induces NREM sleep at the expense of wake time (Kim et al., 2012). Taken together, the drive for sleep may be controlled by specified sleep circuits (Mignot, 2008; Steriade and Timofeev, 2003), but the brain activity that occurs during sleep is tightly regulated by the cortico-thalamo-cortical network (Crunelli and Hughes, 2010; Steriade and Timofeev, 2003), which is responsible for also maintaining stable sleep and switching between sleep stages (Bartho et al., 2014; Halassa et al., 2011; Lewis et al., 2015).
2.2.2. Attention
The corticothalamic network is also critical for the ability of the brain to switch between sleep and behavioral states of arousal that enable the perception of external stimuli. Several decades of research have established a fundamental understanding of the processing of sensory information, which is filtered through the thalamus before being relayed further to cortex. The interplay between excitatory thalamic nuclei and inhibitory neurons of TRN regulate cortical dynamics that underlie perception, sleep-wake cycles and cognition. Based on functional modalities, TRN can be divided into different sensory sectors which are further divided into sub-sectors (Coleman and Mitrofanis, 1996; Villa, 1990), for review see (Pinault, 2004). Spatial control of the thalamic gateway is regulated by the projection of different subnetworks of TRN to functionally divided regions of thalamus. Inhibition of thalamic relay neurons generally occurs through local circuits. Reduction of stereotypic TRN firing under non-specific modalities allows gating of sensory information through thalamic relays; therefore, suppression of the sensory TRN is a coordinated action that allows perception of the senses. During selective attention, the highly dynamic activity of TRN suppresses gating of unwanted stimuli to allow for specific stimuli to engage selective neural responses. Indeed, visual TRN neurons are modulated by selective attention in behaving primates (McAlonan et al., 2000). When the TRN activity is chronically reduced and therefore its control of downstream thalamic relay nuclei is impaired, attention deficits and hyperactivity arise (Wells et al., 2016). This was demonstrated by TRN-specific knockout of patched domain containing 1 (Ptchd1), which results in attenuation of TRN activity, and models a neurodevelopmental disorder associated with impairment in thalamocortical transmission leading to attention deficit and hyperactivity. Such findings demonstrate that dynamic inhibition of thalamic relay nuclei must occur during the presentation of distractors, or unrelated sensory stimuli, since the reduction of TRN-driven inhibition of thalamic relay nuclei in Ptchd1 knockout mice caused irrelevant stimuli to be particularly distracting during tasks requiring attention (Wells et al., 2016). Therefore, dynamic inhibition of thalamic relay nuclei (primarily accomplished by TRN) is necessary to minimize distraction and focus attention.
2.2.3. Cognitive processing
The interaction between corticothalamic and thalamocortical circuits with feedforward regulation of cortical input to thalamus by TRN provides a common denominator for sleep, arousal and attention. Additionally, the thalamus interacts with a wide range of other brain regions and is implicated in cognitive and behavioral processing. The thalamus plays the role of ‘circuit junction’ in coordinating input signals from brainstem, basal ganglia, cerebellum, cerebral cortex and many other subcortical regions, and relays them primarily to neocortex but also to hippocampus, striatum, and amygdala. Recent studies suggest that functional and anatomical subdivisions of the thalamus, enable its diverse role in relaying information to cortical regions and limbic areas to orchestrate sensory and non-sensory processing.
There are two types of thalamic relays that differ in their sources of afferent input: ‘first order’ relays that receive input from peripheral sensory regions, and ‘higher order’ relays that receive input from cortical regions. Higher order thalamic relays influence communication between different cortical regions (Sherman, 2007; Xiao et al., 2009). The information to the thalamic sub-regions from one region of cortex is modulated and further transmitted to different regions of cortex, thus facilitating and enabling communication between multiple regions of cortex and thalamus, which is critical for in shaping complex cognition and behaviors. The ‘driving’ input (which carries information from cells) and ‘modulatory’ input (which influences how the driving information is interpreted) to thalamic nuclei are communicated through transthalamic pathways and regulated through GABAergic input from TRN before being relaying to cortex (Sherman, 2005; Sherman, 2017).
Communication between the medial prefrontal cortex (mPFC) and medial thalamus is critical to the contribution of the thalamus in cognitive processing and memory. This has been demonstrated in non-human primates, and in rats, by selective lesions of the mediodorsal thalamus (MD), which in monkeys has reported to result in deficits in recognition memory and learning object-in-place (OIP) discrimination tasks. Postoperative lesion of the magnocellular division of the medial thalamus in monkeys resulted in ablation of learning based on new sets of visual cues, however it did not affect preoperatively learned tasks (Mitchell et al., 2007; Mitchell and Gaffan, 2008; Mitchell et al., 2014).
The interaction between mPFC, hippocampus and thalamus is also critical in understanding factors that modulate cognition and behavior. The nucleus of reuniens (Re) of the thalamus is required in establishing interactions between mPFC and dorsal hippocampus, and Re receives reciprocal connections from both structures. Although Re involvement is not crucial for hippocampus-dependent memory retrieval, Remaintained coordination between mPFC and hippocampus is crucial to flexible navigation strategies and goal-directed behavior (Cholvin et al., 2013; Dolleman-van der Weel et al., 2009; Ito et al., 2015). Thus, the recent findings support the notion that communication and interactions between the corticothalamic network and the hippocampus is involved in complex functions.
2.2.4. Absence seizures
The corticothalamic-thalamocortical circuitry is also at the heart of nonconvulsive (absence) seizures. Under normal conditions, reciprocal inhibitory and excitatory connections in this circuitry create oscillatory patterns of activity between cortical and subcortical structures such as the sleep spindles described above. Absence seizures are based on abnormal hypersynchronous activity in the corticothalamic-thalamocortical circuitry. When such hypersynchronous activity occurs it involves both the cortex and thalamus, impairing normal circuit functions and preventing the thalamus from reliably transferring information from peripheral and intrinsic brain afferents to the cortex, contributing to the loss of awareness typical in absence seizures (Hughes, 2009; Jones, 2009).
There are two primary types of alterations, both of which involve TRN, in the corticothalamic-thalamocortical circuitry that have been found to lead to absence seizures. First, many animal models that exhibit absence seizures have been found to exhibit strongly increased activity of TRN. Since such activation of TRN can induce post-inhibitory rebound in its target thalamic nuclei, this leads to pathological oscillations between the cortex and thalamus, and disrupts the thalamus from transferring information to the cerebral cortex (Cope et al., 2009; Fuentealba et al., 2004; Jones, 2009). However, the opposite has also been described. Decreased activity of TRN can lead to disinhibition of target thalamic relay nuclei and result in increase in thalamocortical activity (Ernst et al., 2009; Paz et al., 2011).
Absence epilepsies can be classified into typical and atypical forms. Typical absence epilepsy involves only the corticothalamic network. While typical absence epilepsy can increase synchrony in the hippocampus, it does not generalize into other brain areas, and is not associated with severe memory deficits (Velazquez et al., 2007; Weiergraber et al., 2010). Atypical absence epilepsy is also based in the corticothalamic circuitry, but can spread hypersynchronous activity into vulnerable areas such as the hippocampus, which may underlie the cognitive/memory impairments in atypical absence epilepsy (Chan et al., 2004; Ferlazzo et al., 2010; Velazquez et al., 2007; Wang et al., 2009). The brain regions invaded by hypersynchronous corticothalamic activity may differ depending on patient history/genetics, resulting in the complex presentation of the syndrome (Velazquez et al., 2007; Weiergraber et al., 2010). Because the corticothalamic network underlies typical and atypical absence epilepsies, they are treated with the same pharmacology.
3. Aspects of AD related to the corticothalamic network
3.1. Corticothalamic circuitry and pathology in AD
Many studies have focused on pathology in the hippocampus or cortical areas, both of which are rich in dystrophic neurites, plaques, and tangles. However, there is also Aβ pathology and dystrophic neurites in subcortical areas such as the thalamus (Braak and Braak, 1990; Braak and Braak, 1991). In TRN in particular, there are striking levels of dystrophic neurites (Tourtellotte and Van Hoesen, 1991), which appear to reflect injured axon terminals projecting to the TRN. Imaging studies of AD patients revealed atrophy in several thalamic areas in symptomatic patients (reviewed in Tentolouris-Piperas et al., 2017), but it is not clear whether there are many anatomical or pathological alterations in preclinical stages. Whether functional changes occur in the absence of overt pathology or neuronal loss is unclear.
3.2. Sleep disturbances in AD patients and animal models
Sleep disturbances are prevalent and impair the quality of life for patients with mild cognitive impairment or Alzheimer’s disease (Ju et al., 2014; Karageorgiou and Vossel, 2017). Common sleep problems reported by caregivers and family members include early awakening and sleeping either longer or shorter than typical hours (McCurry et al., 1999; Tworoger et al., 2006; Westwood et al., 2017). In addition, poor sleep quality due to low sleep efficiency, regulation of breathing during sleep, sleep fragmentation due to frequent awakenings during the sleep period, and an increase in sleep onset latency have been associated with AD (Benedict et al., 2015; Lim et al., 2013; Yaffe et al., 2011). The type of sleep disturbance experienced by the patient may contribute to differing tendencies to sleep longer or shorter than typical hours. In addition, sleep disturbances may complicate the interpretation of findings from different studies that have found that either longer or shorter than typical hours of sleep per night can predict AD, particularly studies that rely on self-reports of sleep patterns. Individuals who sleep poorly may spend increased amounts of time in bed, but are not actually sleeping more, or they may arise early if not sleeping well. Indeed, in some cases, self-reported sleep patterns do not correlate well with simultaneous actigraph data from the same patients (Hita-Yanez et al., 2013).
Sleep disturbances are present early in disease progression. Studies of the underlying sleep physiology have revealed reductions in sleep efficiency, spindle activity, slow wave sleep (SWS) and REM, with an increased arousal index (reviewed in Ju et al., 2014). Individuals with amnestic mild cognitive impairment (aMCI, which often progresses to AD) also exhibit fewer stage 2 spindles, along with prominent reductions in slow wave sleep, and increased time spent awake after sleep onset (Westerberg et al., 2013). Notably, a cross-sectional study of cognitively normal older adults in community-based living found that individuals with more severe sleep fragmentation, measured using actigraphy, have an increased risk for developing Alzheimer’s disease (Lim et al., 2013). A related study of the same population found that the roughly 20% of individuals with one ApoE4 allele that went on to develop AD exhibited poor sleep consolidation, a compound parameter that reflects sleep fragmentation, sleep efficiency, and awake time after sleep onset (Lim et al., 2013). Individuals with one ApoE4 allele that had normal sleep consolidation indices did not develop AD.
Therefore it may not be surprising that there have been many recent efforts to understand the extent to which sleep is impaired in AD, investigate the underlying pathophysiology, and identify the most reliable measures of sleep disturbance. In addition, it is becoming clear that sleep disturbances exacerbate disease pathology, and vice versa. A disruption in NREM slow wave activity (SWA) is observed during the progression of MCI and AD (Hita-Yanez et al., 2013; Prinz et al., 1982; Westerberg et al., 2013). In AD as well as in MCI patients, the quality of early (stage 2) and deep (stage 3 and stage 4) sleep is compromised due to lower spindle power around fast spindle frequencies (13–15 Hz) and slow wave activity (0.5 – 4 Hz) in comparison to healthy controls (Gorgoni et al., 2016; Mander et al., 2015). AD patients also exhibited a roughly 40% decrease in K–complex density (< 1 Hz) in comparison to age–matched controls (De Gennaro et al., 2017).
Perhaps one of the most striking sleep-related differences in AD patients is the reduction in slow wave sleep (SWS), which is severely compromised in AD. Such reductions in SWS are of particular interest because it is the phase of sleep that is critically important for memory consolidation (Born and Wilhelm, 2012) as well as for clearing solutes (including Aβ) from the brain (Ju et al., 2014; Xie et al., 2013). It is also the time during which the default mode network (DMN) is most inactivated. The DMN, which is active when individuals are inattentive, is hyperactive in AD patients and is thought to exacerbate activity-dependent Aβ production. Thus, impairments in SWS might play a critical role, or at least exacerbate, cognitive/memory deficits and Aβ accumulation, the clinical and pathological hallmarks of AD. In subjects with preclinical AD (subjects that were cognitively normal but exhibited low CSF Aβ levels that indicate Aβ deposition in the brain), poor sleep efficiency correlated with lower CSF Aβ42 levels (Ju et al., 2013). In addition, an interaction between sleep efficiency and amyloid deposition in individuals with low Aβ42 deposition was found to be a significant predicator of poor memory performance, whereas there was no correlation between sleep efficiency and memory performance in subjects with no amyloid deposition (Molano 2016). Thus, sleep efficiency modifies the impact of Aβ on memory.
Studies in transgenic mice that overexpress human Aβ have provided mechanistic insight into the relationship between sleep and Aβ. Aβ is produced in an activity-dependent manner in brain slices and in vivo (Bero et al., 2011; Cirrito et al., 2005; Kamenetz et al., 2003), which underlies the diurnal rhythm of Aβ production that is higher during wake phases and lower during sleep phases. However, the relationship between Aβ pathology and sleep disruption is bidirectional. Sleep deprivation elevates the Aβ plaque load in cortex (Kang et al., 2009) whereas during NREM sleep Aβ is cleared (Kang et al., 2009; Xie et al., 2013). On the other hand, in APP transgenic mice, low levels of Aβ induce sleep fragmentation even prior to plaque deposition (Hazra et al., 2016b), as well as after Aβ deposition in cortex (Roh et al., 2012). Human imaging studies and computational models have also implicated sleep disruptions as a key mechanistic pathway by which Aβ impairs hippocampal-dependent cognitive decline (Mander et al., 2015).
3.3. Attention deficits in AD patients
A limited number of studied have reported attention deficits and fluctuation in attention in AD and MCI patients (Alescio-Lautier et al., 2007; Ballard et al., 2001; Bracco et al., 2014; Parasuraman et al., 1992; Rapp and Reischies, 2005; Tales et al., 2002a; Tales et al., 2002b). Focused attention in patients is typically assessed using neuropsychological assessment tasks such as auditory or visual simple-reaction time (RT) and choice reaction time (CRT) tasks. In the RT task, subjects are provided with a single cue; for instance, visual or auditory, where the patient is required to press a button whenever the cue arrives. The reaction time between the arrival of the cue and pressing of the button is recorded. In the CRT task, patients are provided with either of two possible cues, for instance in terms of visual cues, subjects are presented either ‘YES’ or ‘NO’ on the screen. When either word appears on the screen, subjects are expected to press the appropriate designated button for the respective word. The reaction time and accuracy are then analyzed. The SRT task is particularly useful for estimating perception of a visual or auditory cue and the downstream motor response; whereas CRT estimates the perception of sensory stimuli and processing of information.
In both AD and dementia with Lewy bodies patients, the CRT response to a task that consisted of constant inter-stimulus intervals, was found to be slower in comparison to controls (Ballard et al., 2001). Although it has not yet been specifically examined, the role of the corticothalamic network particularly the TRN in regulating sensory information processing and attention as described above, suggests that alterations in this network may contribute to attention deficits observed in AD patients.
3.4. Seizures in AD patients and AD models
AD patients have a 5–10 fold higher risk of developing of seizures compared to reference populations, which has been reviewed elsewhere (Amatniek et al., 2006; Chin and Scharfman, 2013; Larner, 2010; Lozsadi and Larner, 2006; Scarmeas et al., 2009). Here we will discuss the increased seizure incidence with respect to possible alterations in the corticothalamic network that may underlie specific aspects of AD symptoms. It is notable that even when comparing patients with AD versus other types of dementia that can also be associated with seizures, AD patients have a higher risk of seizures (Sherzai et al., 2014). Episodes of seizures in amnestic MCI or AD patients begin near the onset of cognitive deficits and lead to a faster rate of cognitive decline (Irizarry et al., 2012; Vossel et al., 2013; Vossel et al., 2016).
Seizures and epileptiform activity in AD patients may be vastly underestimated due to the occurrence of non-convulsive seizures and other types of subclinical epileptiform activity that are difficult to detect without EEG assessment, or that might not occur frequently enough to be detected in a routine clinical visit. Indeed, with longer-term overnight EEG and magnetoencephalography (MEG), Vossel et al., (2016) recently demonstrated that subclinical epileptiform activity was found in 42% of AD patients. The occurrence of epileptiform activity was associated with a faster rate of cognitive decline. Nearly 90% of all epileptiform activity in AD patients was observed during stage 1 and stage 2 of NREM sleep. Two-thirds of patients with epileptiform activity had epileptic discharges exclusively during sleep. Recently, recordings from bilateral foramen ovale electrodes implanted to target the medial temporal lobe in two patients with AD revealed abundant epileptic spiking and nonconvulsive seizures that were not detected by simultaneous scalp EEG (Lam et al., 2017). Notably, the majority of spiking recorded, and all of the seizures, occurred during sleep.
Some of the nonconvulsive seizures in AD share characteristics of absence seizures that arise from dysfunctional corticothalamic network, including a propensity to occur during sleep, and behavioral manifestations such as cessation of movement, staring into space, and loss of consciousness. Not all AD-associated nonconvulsive seizures share such characteristics with absence seizures (see Lam et al., 2017) but some do, and therefore studying whether and/or how corticothalamic network activity may be affected in AD may reveal novel mechanisms of pathophysiology.
Selective changes in corticothalamic network activity can influence thalamocortical oscillations and contribute to the generation of spike-wave discharges. The TRN orchestrates the communication between cerebral cortex and thalamus, however dynamic changes in synaptic strength and excitability in TRN can evoke pathological spike-wave discharges. We recently reported a novel mode of dysfunction in cortico-thalamo-cortical network in the hAPP (J20) mouse line of AD, at an early stage of disease when the onset of epileptiform activities and sleep fragmentation are evident but plaque deposition has not yet begun (Hazra et al., 2016b). We found that TRN activity was markedly reduced whereas downstream thalamic relay nuclei were hyperactive. We therefore hypothesized that reduced activity of TRN disinhibits downstream thalamocortical and thalamolimbic relay nuclei causing hyperactivity in their limbic and cortical targets. Increased cortical and thalamic excitability has also been described in other AD mouse models (Gurevicius et al., 2013). Both convulsive and non-convulsive seizures have been found now in many mouse models (Chin and Scharfman, 2013) however it is not known whether the seizures that occur in those models result also from reductions in TRN activity.
4. Summary and Conclusions
We have reviewed here the basic circuitry and functions of the corticothalamic network, and highlighted key features that are particularly relevant to symptoms associated with Alzheimer’s disease. The striking overlap between the various functions controlled by the corticothalamic network and the dysfunctions that occur in AD suggest that the corticothalamic network may be a master regulator of diverse cognitive and behavioral domains that are affected in AD.
Memory impairment is the most characteristic symptom of AD, and therefore the large body of research on the hippocampus has been and will continue to be a critical area of focus in the field. However, in order to understand the network mechanisms that give rise to the particular pattern of cognitive and behavioral alterations that are associated with AD, it is critical to expand the focus of research to include other brain regions and networks that influence hippocampal function. Such studies have the potential to not only shed light on the network mechanisms that give rise to AD, but may also identify novel therapeutic entry points that can both improve quality of life as well as slow or prevent the progression of disease. It is notable that disturbances in sleep patterns/quality, which is heavily dependent on modulation by the corticothalamic network, can be observed in cognitively normal people many years before they are first diagnosed with cognitive impairments. Given that sleep, particularly slow wave sleep, is critical for both memory consolidation as well as clearance of solutes such as Aβ, it is possible that even slight alterations in activity or function in the corticothalamic network early on, can slowly over many years lead to a tipping point and give rise to manifestation of disease. Future studies that investigate cellular, molecular, and circuit-level aspects of corticothalamic function in AD patients or mouse models may provide critical insight.
Highlights.
Alzheimer’s disease is accompanied by diverse cognitive and behavioral deficits
The corticothalamic network may be a master regulator of multiple domains in AD
Early deficits in sleep may increase Abeta and susceptibility to AD
Investigation of the corticothalamic network in AD may provide new insight to disease
Acknowledgments
This work was supported by a grant from the Ruth K. Broad Biomedical Research Foundation (JC) and NIH grants NS085171 and NS086965 (JC).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alescio-Lautier B, et al. Visual and visuospatial short-term memory in mild cognitive impairment and Alzheimer disease: role of attention. Neuropsychologia. 2007;45:1948–60. doi: 10.1016/j.neuropsychologia.2006.04.033. [DOI] [PubMed] [Google Scholar]
- Amatniek JC, et al. Incidence and predictors of seizures in patients with Alzheimer's disease. Epilepsia. 2006;47:867–72. doi: 10.1111/j.1528-1167.2006.00554.x. [DOI] [PubMed] [Google Scholar]
- Anderson MP, et al. Thalamic Cav3.1 T-type Ca2+ channel plays a crucial role in stabilizing sleep. Proc Natl Acad Sci U S A. 2005;102:1743–8. doi: 10.1073/pnas.0409644102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballard C, et al. Attention and fluctuating attention in patients with dementia with Lewy bodies and Alzheimer disease. Arch Neurol. 2001;58:977–82. doi: 10.1001/archneur.58.6.977. [DOI] [PubMed] [Google Scholar]
- Bartho P, et al. Ongoing network state controls the length of sleep spindles via inhibitory activity. Neuron. 2014;82:1367–79. doi: 10.1016/j.neuron.2014.04.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beenhakker MP, Huguenard JR. Neurons that fire together also conspire together: is normal sleep circuitry hijacked to generate epilepsy? Neuron. 2009;62:612–32. doi: 10.1016/j.neuron.2009.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benedict C, et al. Self-reported sleep disturbance is associated with Alzheimer's disease risk in men. Alzheimers Dement. 2015;11:1090–7. doi: 10.1016/j.jalz.2014.08.104. [DOI] [PubMed] [Google Scholar]
- Bero AW, et al. Neuronal activity regulates the regional vulnerability to amyloid-beta deposition. Nat Neurosci. 2011;14:750–6. doi: 10.1038/nn.2801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Born J, Wilhelm I. System consolidation of memory during sleep. Psychol Res. 2012;76:192–203. doi: 10.1007/s00426-011-0335-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourassa J, Deschenes M. Corticothalamic projections from the primary visual cortex in rats: a single fiber study using biocytin as an anterograde tracer. Neuroscience. 1995;66:253–63. doi: 10.1016/0306-4522(95)00009-8. [DOI] [PubMed] [Google Scholar]
- Braak H, Braak E. Neurofibrillary changes confined to the entorhinal region and an abundance of cortical amyloid in cases of presenile and senile dementia. Acta Neuropathol. 1990;80:479–86. doi: 10.1007/BF00294607. [DOI] [PubMed] [Google Scholar]
- Braak H, Braak E. Alzheimer's disease affects limbic nuclei of the thalamus. Acta Neuropathol. 1991;81:261–8. doi: 10.1007/BF00305867. [DOI] [PubMed] [Google Scholar]
- Bracco L, et al. Do cholinesterase inhibitors act primarily on attention deficit? A naturalistic study in Alzheimer's disease patients. J Alzheimers Dis. 2014;40:737–42. doi: 10.3233/JAD-131154. [DOI] [PubMed] [Google Scholar]
- Buzsaki G. The thalamic clock: emergent network properties. Neuroscience. 1991;41:351–64. doi: 10.1016/0306-4522(91)90332-i. [DOI] [PubMed] [Google Scholar]
- Chan KF, et al. Learning and memory impairment in rats with chronic atypical absence seizures. Exp Neurol. 2004;190:328–36. doi: 10.1016/j.expneurol.2004.08.001. [DOI] [PubMed] [Google Scholar]
- Chin J, Scharfman HE. Shared cognitive and behavioral impairments in epilepsy and Alzheimer's disease and potential underlying mechanisms. Epilepsy Behav. 2013;26:343–51. doi: 10.1016/j.yebeh.2012.11.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cholvin T, et al. The ventral midline thalamus contributes to strategy shifting in a memory task requiring both prefrontal cortical and hippocampal functions. J Neurosci. 2013;33:8772–83. doi: 10.1523/JNEUROSCI.0771-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirrito JR, et al. Synaptic activity regulates interstitial fluid amyloid-beta levels in vivo. Neuron. 2005;48:913–22. doi: 10.1016/j.neuron.2005.10.028. [DOI] [PubMed] [Google Scholar]
- Clemente-Perez A, et al. Distinct Thalamic Reticular Cell Types Differentially Modulate Normal and Pathological Cortical Rhythms. Cell Rep. 2017;19:2130–2142. doi: 10.1016/j.celrep.2017.05.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman KA, Mitrofanis J. Organization of the visual reticular thalamic nucleus of the rat. Eur J Neurosci. 1996;8:388–404. doi: 10.1111/j.1460-9568.1996.tb01222.x. [DOI] [PubMed] [Google Scholar]
- Cope DW, et al. Enhanced tonic GABAA inhibition in typical absence epilepsy. Nat Med. 2009;15:1392–8. doi: 10.1038/nm.2058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox CL, Huguenard JR, Prince DA. Nucleus reticularis neurons mediate diverse inhibitory effects in thalamus. Proc Natl Acad Sci U S A. 1997;94:8854–9. doi: 10.1073/pnas.94.16.8854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crick F. Function of the thalamic reticular complex: the searchlight hypothesis. Proc Natl Acad Sci U S A. 1984;81:4586–90. doi: 10.1073/pnas.81.14.4586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crunelli V, Hughes SW. The slow (<1 Hz) rhythm of non-REM sleep: a dialogue between three cardinal oscillators. Nat Neurosci. 2010;13:9–17. doi: 10.1038/nn.2445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalsgaard MK, Secher NH. The brain at work: a cerebral metabolic manifestation of central fatigue? J Neurosci Res. 2007;85:3334–9. doi: 10.1002/jnr.21274. [DOI] [PubMed] [Google Scholar]
- De Gennaro L, et al. The Fall of Sleep K-Complex in Alzheimer Disease. Sci Rep. 2017;7:39688. doi: 10.1038/srep39688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deschenes M, Veinante P, Zhang ZW. The organization of corticothalamic projections: reciprocity versus parity. Brain Res Brain Res Rev. 1998;28:286–308. doi: 10.1016/s0165-0173(98)00017-4. [DOI] [PubMed] [Google Scholar]
- Dolleman-van der Weel MJ, Morris RG, Witter MP. Neurotoxic lesions of the thalamic reuniens or mediodorsal nucleus in rats affect non-mnemonic aspects of watermaze learning. Brain Struct Funct. 2009;213:329–42. doi: 10.1007/s00429-008-0200-6. [DOI] [PubMed] [Google Scholar]
- Ernst WL, et al. Genetic enhancement of thalamocortical network activity by elevating alpha 1g-mediated low-voltage-activated calcium current induces pure absence epilepsy. J Neurosci. 2009;29:1615–25. doi: 10.1523/JNEUROSCI.2081-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferlazzo E, et al. Lennox-Gastaut syndrome in adulthood: clinical and EEG features. Epilepsy Res. 2010;89:271–7. doi: 10.1016/j.eplepsyres.2010.01.012. [DOI] [PubMed] [Google Scholar]
- Ferri CP, et al. Global prevalence of dementia: a Delphi consensus study. Lancet. 2005;366:2112–7. doi: 10.1016/S0140-6736(05)67889-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fogerson PM, Huguenard JR. Tapping the Brakes: Cellular and Synaptic Mechanisms that Regulate Thalamic Oscillations. Neuron. 2016;92:687–704. doi: 10.1016/j.neuron.2016.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuentealba P, et al. Synaptic interactions between thalamic and cortical inputs onto cortical neurons in vivo. J Neurophysiol. 2004;91:1990–8. doi: 10.1152/jn.01105.2003. [DOI] [PubMed] [Google Scholar]
- Golomb D, Wang XJ, Rinzel J. Propagation of spindle waves in a thalamic slice model. J Neurophysiol. 1996;75:750–69. doi: 10.1152/jn.1996.75.2.750. [DOI] [PubMed] [Google Scholar]
- Gorgoni M, et al. Parietal Fast Sleep Spindle Density Decrease in Alzheimer's Disease and Amnesic Mild Cognitive Impairment. Neural Plast. 2016;2016:8376108. doi: 10.1155/2016/8376108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillery RW, Harting JK. Structure and connections of the thalamic reticular nucleus: Advancing views over half a century. J Comp Neurol. 2003;463:360–71. doi: 10.1002/cne.10738. [DOI] [PubMed] [Google Scholar]
- Gurevicius K, Lipponen A, Tanila H. Increased cortical and thalamic excitability in freely moving APPswe/PS1dE9 mice modeling epileptic activity associated with Alzheimer's disease. Cereb Cortex. 2013;23:1148–58. doi: 10.1093/cercor/bhs105. [DOI] [PubMed] [Google Scholar]
- Halassa MM, et al. Selective optical drive of thalamic reticular nucleus generates thalamic bursts and cortical spindles. Nat Neurosci. 2011;14:1118–20. doi: 10.1038/nn.2880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halassa MM, et al. State-dependent architecture of thalamic reticular subnetworks. Cell. 2014;158:808–21. doi: 10.1016/j.cell.2014.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanlon EC, et al. Synaptic potentiation and sleep need: clues from molecular and electrophysiological studies. Curr Top Med Chem. 2011;11:2472–82. doi: 10.2174/156802611797470312. [DOI] [PubMed] [Google Scholar]
- Hazra A, et al. Corticothalamic network dysfunction and behavioral deficits in a mouse model of Alzheimer's disease. Neurobiol Aging. 2016;44:96–107. doi: 10.1016/j.neurobiolaging.2016.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebert LE, et al. Alzheimer disease in the United States (2010–2050) estimated using the 2010 census. Neurology. 2013;80:1778–83. doi: 10.1212/WNL.0b013e31828726f5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hita-Yanez E, Atienza M, Cantero JL. Polysomnographic and subjective sleep markers of mild cognitive impairment. Sleep. 2013;36:1327–34. doi: 10.5665/sleep.2956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houser CR, et al. GABA neurons are the major cell type of the nucleus reticularis thalami. Brain Res. 1980;200:341–54. doi: 10.1016/0006-8993(80)90925-7. [DOI] [PubMed] [Google Scholar]
- Huang Y, Mucke L. Alzheimer mechanisms and therapeutic strategies. Cell. 2012;148:1204–22. doi: 10.1016/j.cell.2012.02.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes JR. Absence seizures: a review of recent reports with new concepts. Epilepsy Behav. 2009;15:404–12. doi: 10.1016/j.yebeh.2009.06.007. [DOI] [PubMed] [Google Scholar]
- Irizarry MC, et al. Incidence of new-onset seizures in mild to moderate Alzheimer disease. Arch Neurol. 2012;69:368–72. doi: 10.1001/archneurol.2011.830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito HT, et al. A prefrontal-thalamo-hippocampal circuit for goal-directed spatial navigation. Nature. 2015;522:50–5. doi: 10.1038/nature14396. [DOI] [PubMed] [Google Scholar]
- Jahnsen H, Llinas R. Electrophysiological properties of guinea-pig thalamic neurones: an in vitro study. J Physiol. 1984a;349:205–26. doi: 10.1113/jphysiol.1984.sp015153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahnsen H, Llinas R. Ionic basis for the electro-responsiveness and oscillatory properties of guinea-pig thalamic neurones in vitro. J Physiol. 1984b;349:227–47. doi: 10.1113/jphysiol.1984.sp015154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones EG. Thalamic circuitry and thalamocortical synchrony. Philos Trans R Soc Lond B Biol Sci. 2002;357:1659–73. doi: 10.1098/rstb.2002.1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones EG. Synchrony in the interconnected circuitry of the thalamus and cerebral cortex. Ann N Y Acad Sci. 2009;1157:10–23. doi: 10.1111/j.1749-6632.2009.04534.x. [DOI] [PubMed] [Google Scholar]
- Ju YE, et al. Sleep quality and preclinical Alzheimer disease. JAMA Neurol. 2013;70:587–93. doi: 10.1001/jamaneurol.2013.2334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju YE, Lucey BP, Holtzman DM. Sleep and Alzheimer disease pathology--a bidirectional relationship. Nat Rev Neurol. 2014;10:115–9. doi: 10.1038/nrneurol.2013.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamenetz F, et al. APP processing and synaptic function. Neuron. 2003;37:925–37. doi: 10.1016/s0896-6273(03)00124-7. [DOI] [PubMed] [Google Scholar]
- Kang JE, et al. Amyloid-beta dynamics are regulated by orexin and the sleep-wake cycle. Science. 2009;326:1005–7. doi: 10.1126/science.1180962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karageorgiou E, Vossel KA. Brain rhythm attractor breakdown in Alzheimer's disease: Functional and pathologic implications. Alzheimers Dement. 2017 doi: 10.1016/j.jalz.2017.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim A, et al. Optogenetically induced sleep spindle rhythms alter sleep architectures in mice. Proc Natl Acad Sci U S A. 2012;109:20673–8. doi: 10.1073/pnas.1217897109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolliker RAV. Handbuch der Gewebelehre des Menschen. Vol. 2. W. Engelmann; 1896. [Google Scholar]
- Lam AD, et al. Silent hippocampal seizures and spikes identified by foramen ovale electrodes in Alzheimer's disease. Nat Med. 2017 doi: 10.1038/nm.4330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lancel M, van Riezen H, Glatt A. Enhanced slow-wave activity within NREM sleep in the cortical and subcortical EEG of the cat after sleep deprivation. Sleep. 1992;15:102–18. doi: 10.1093/sleep/15.2.102. [DOI] [PubMed] [Google Scholar]
- Larner AJ. Epileptic seizures in AD patients. Neuromolecular Med. 2010;12:71–7. doi: 10.1007/s12017-009-8076-z. [DOI] [PubMed] [Google Scholar]
- Levey AI, Hallanger AE, Wainer BH. Cholinergic nucleus basalis neurons may influence the cortex via the thalamus. Neurosci Lett. 1987;74:7–13. doi: 10.1016/0304-3940(87)90042-5. [DOI] [PubMed] [Google Scholar]
- Lewis LD, et al. Thalamic reticular nucleus induces fast and local modulation of arousal state. Elife. 2015;4:e08760. doi: 10.7554/eLife.08760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim AS, et al. Sleep Fragmentation and the Risk of Incident Alzheimer's Disease and Cognitive Decline in Older Persons. Sleep. 2013;36:1027–1032. doi: 10.5665/sleep.2802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lozsadi DA, Larner AJ. Prevalence and causes of seizures at the time of diagnosis of probable Alzheimer's disease. Dement Geriatr Cogn Disord. 2006;22:121–4. doi: 10.1159/000093664. [DOI] [PubMed] [Google Scholar]
- Mander BA, et al. beta-amyloid disrupts human NREM slow waves and related hippocampus-dependent memory consolidation. Nat Neurosci. 2015;18:1051–7. doi: 10.1038/nn.4035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McAlonan K, Brown VJ, Bowman EM. Thalamic reticular nucleus activation reflects attentional gating during classical conditioning. J Neurosci. 2000;20:8897–901. doi: 10.1523/JNEUROSCI.20-23-08897.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormick DA, Pape HC. Properties of a hyperpolarization-activated cation current and its role in rhythmic oscillation in thalamic relay neurones. J Physiol. 1990;431:291–318. doi: 10.1113/jphysiol.1990.sp018331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormick DA. Neurotransmitter actions in the thalamus and cerebral cortex and their role in neuromodulation of thalamocortical activity. Prog Neurobiol. 1992;39:337–88. doi: 10.1016/0301-0082(92)90012-4. [DOI] [PubMed] [Google Scholar]
- McCurry SM, et al. Characteristics of sleep disturbance in community-dwelling Alzheimer's disease patients. J Geriatr Psychiatry Neurol. 1999;12:53–9. doi: 10.1177/089198879901200203. [DOI] [PubMed] [Google Scholar]
- Mignot E. Why we sleep: the temporal organization of recovery. PLoS Biol. 2008;6:e106. doi: 10.1371/journal.pbio.0060106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell AS, Baxter MG, Gaffan D. Dissociable performance on scene learning and strategy implementation after lesions to magnocellular mediodorsal thalamic nucleus. J Neurosci. 2007;27:11888–95. doi: 10.1523/JNEUROSCI.1835-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell AS, Gaffan D. The magnocellular mediodorsal thalamus is necessary for memory acquisition, but not retrieval. J Neurosci. 2008;28:258–63. doi: 10.1523/JNEUROSCI.4922-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell AS, et al. Advances in understanding mechanisms of thalamic relays in cognition and behavior. J Neurosci. 2014;34:15340–6. doi: 10.1523/JNEUROSCI.3289-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molano JR, Roe CM, Ju YS. The interaction of sleep and amyloid deposition on cognitive performance. J Sleep Res. 2016 doi: 10.1111/jsr.12474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Donovan MJ, Rinzel J. Synaptic depression: a dynamic regulator of synaptic communication with varied functional roles. Trends Neurosci. 1997;20:431–3. doi: 10.1016/s0166-2236(97)01124-7. [DOI] [PubMed] [Google Scholar]
- Pape HC, et al. Prevention of Ca(2+)-mediated action potentials in GABAergic local circuit neurones of rat thalamus by a transient K+ current. J Physiol. 1994;478(Pt 3):403–22. doi: 10.1113/jphysiol.1994.sp020261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parasuraman R, et al. Visuospatial attention in dementia of the Alzheimer type. Brain. 1992;115(Pt 3):711–33. doi: 10.1093/brain/115.3.711. [DOI] [PubMed] [Google Scholar]
- Paz JT, et al. A new mode of corticothalamic transmission revealed in the Gria4(−/−) model of absence epilepsy. Nat Neurosci. 2011;14:1167–73. doi: 10.1038/nn.2896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinault D, Deschenes M. Projection and innervation patterns of individual thalamic reticular axons in the thalamus of the adult rat: a three-dimensional, graphic, and morphometric analysis. J Comp Neurol. 1998;391:180–203. doi: 10.1002/(sici)1096-9861(19980209)391:2<180::aid-cne3>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- Pinault D. The thalamic reticular nucleus: structure, function and concept. Brain Res Brain Res Rev. 2004;46:1–31. doi: 10.1016/j.brainresrev.2004.04.008. [DOI] [PubMed] [Google Scholar]
- Prinz PN, et al. Sleep, EEG and mental function changes in senile dementia of the Alzheimer's type. Neurobiol Aging. 1982;3:361–70. doi: 10.1016/0197-4580(82)90024-0. [DOI] [PubMed] [Google Scholar]
- Rapp MA, Reischies FM. Attention and Executive Control Predict Alzheimer Disease in Late Life: Results From the Berlin Aging Study (BASE) The American Journal of Geriatric Psychiatry. 2005;13:134–141. doi: 10.1176/appi.ajgp.13.2.134. [DOI] [PubMed] [Google Scholar]
- Rechtschaffen A. Current perspectives on the function of sleep. Perspect Biol Med. 1998;41:359–90. doi: 10.1353/pbm.1998.0051. [DOI] [PubMed] [Google Scholar]
- Rodriguez AV, et al. Why Does Sleep Slow-Wave Activity Increase After Extended Wake? Assessing the Effects of Increased Cortical Firing During Wake and Sleep. J Neurosci. 2016;36:12436–12447. doi: 10.1523/JNEUROSCI.1614-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roh JH, et al. Disruption of the sleep-wake cycle and diurnal fluctuation of beta-amyloid in mice with Alzheimer's disease pathology. Sci Transl Med. 2012;4:150ra122. doi: 10.1126/scitranslmed.3004291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scarmeas N, et al. Seizures in Alzheimer disease: who, when, and how common? Arch Neurol. 2009;66:992–7. doi: 10.1001/archneurol.2009.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherman SM. Thalamic relays and cortical functioning. Prog Brain Res. 2005;149:107–26. doi: 10.1016/S0079-6123(05)49009-3. [DOI] [PubMed] [Google Scholar]
- Sherman SM. The thalamus is more than just a relay. Curr Opin Neurobiol. 2007;17:417–22. doi: 10.1016/j.conb.2007.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherman SM. Functioning of Circuits Connecting Thalamus and Cortex. Compr Physiol. 2017;7:713–739. doi: 10.1002/cphy.c160032. [DOI] [PubMed] [Google Scholar]
- Sherzai D, et al. Seizures and dementia in the elderly: Nationwide Inpatient Sample 1999–2008. Epilepsy Behav. 2014;36:53–6. doi: 10.1016/j.yebeh.2014.04.015. [DOI] [PubMed] [Google Scholar]
- Steriade M, et al. Abolition of spindle oscillations in thalamic neurons disconnected from nucleus reticularis thalami. J Neurophysiol. 1985;54:1473–97. doi: 10.1152/jn.1985.54.6.1473. [DOI] [PubMed] [Google Scholar]
- Steriade M, et al. Cholinergic and non-cholinergic neurons of cat basal forebrain project to reticular and mediodorsal thalamic nuclei. Brain Res. 1987;408:372–6. doi: 10.1016/0006-8993(87)90408-2. [DOI] [PubMed] [Google Scholar]
- Steriade M, et al. Projections of cholinergic and non-cholinergic neurons of the brainstem core to relay and associational thalamic nuclei in the cat and macaque monkey. Neuroscience. 1988;25:47–67. doi: 10.1016/0306-4522(88)90006-1. [DOI] [PubMed] [Google Scholar]
- Steriade M, McCormick DA, Sejnowski TJ. Thalamocortical oscillations in the sleeping and aroused brain. Science. 1993a;262:679–85. doi: 10.1126/science.8235588. [DOI] [PubMed] [Google Scholar]
- Steriade M, Nunez A, Amzica F. A novel slow (< 1 Hz) oscillation of neocortical neurons in vivo: depolarizing and hyperpolarizing components. J Neurosci. 1993b;13:3252–65. doi: 10.1523/JNEUROSCI.13-08-03252.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steriade M, Timofeev I. Neuronal plasticity in thalamocortical networks during sleep and waking oscillations. Neuron. 2003;37:563–76. doi: 10.1016/s0896-6273(03)00065-5. [DOI] [PubMed] [Google Scholar]
- Tales A, et al. Spatial shifts in visual attention in normal ageing and dementia of the Alzheimer type. Neuropsychologia. 2002a;40:2000–12. doi: 10.1016/s0028-3932(02)00057-x. [DOI] [PubMed] [Google Scholar]
- Tales A, et al. Age-related changes in the preattentional detection of visual change. Neuroreport. 2002b;13:969–72. doi: 10.1097/00001756-200205240-00014. [DOI] [PubMed] [Google Scholar]
- Tentolouris-Piperas V, et al. Brain imaging evidence of early involvement of subcortical regions in familial and sporadic Alzheimer's disease. Brain Res. 2017;1655:23–32. doi: 10.1016/j.brainres.2016.11.011. [DOI] [PubMed] [Google Scholar]
- Timofeev I, Contreras D, Steriade M. Synaptic responsiveness of cortical and thalamic neurones during various phases of slow sleep oscillation in cat. J Physiol. 1996;494(Pt 1):265–78. doi: 10.1113/jphysiol.1996.sp021489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tourtellotte WG, Van Hoesen GW. The axonal origin of a subpopulation of dystrophic neurites in Alzheimer's disease. Neurosci Lett. 1991;129:11–6. doi: 10.1016/0304-3940(91)90709-3. [DOI] [PubMed] [Google Scholar]
- Tworoger SS, et al. The association of self-reported sleep duration, difficulty sleeping, and snoring with cognitive function in older women. Alzheimer Dis Assoc Disord. 2006;20:41–8. doi: 10.1097/01.wad.0000201850.52707.80. [DOI] [PubMed] [Google Scholar]
- Velazquez JL, et al. Typical versus atypical absence seizures: network mechanisms of the spread of paroxysms. Epilepsia. 2007;48:1585–93. doi: 10.1111/j.1528-1167.2007.01120.x. [DOI] [PubMed] [Google Scholar]
- Villa AE. Physiological differentiation within the auditory part of the thalamic reticular nucleus of the cat. Brain Res Brain Res Rev. 1990;15:25–40. doi: 10.1016/0165-0173(90)90010-l. [DOI] [PubMed] [Google Scholar]
- Vossel KA, et al. Seizures and epileptiform activity in the early stages of Alzheimer disease. JAMA Neurol. 2013;70:1158–66. doi: 10.1001/jamaneurol.2013.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vossel KA, et al. Incidence and impact of subclinical epileptiform activity in Alzheimer's disease. Ann Neurol. 2016;80:858–870. doi: 10.1002/ana.24794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vyazovskiy VV, et al. Cortical firing and sleep homeostasis. Neuron. 2009;63:865–78. doi: 10.1016/j.neuron.2009.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vyazovskiy VV, Harris KD. Sleep and the single neuron: the role of global slow oscillations in individual cell rest. Nat Rev Neurosci. 2013;14:443–51. doi: 10.1038/nrn3494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, et al. The circuitry of atypical absence seizures in GABA(B)R1a transgenic mice. Pharmacol Biochem Behav. 2009;94:124–30. doi: 10.1016/j.pbb.2009.07.017. [DOI] [PubMed] [Google Scholar]
- Warren RA, Agmon A, Jones EG. Oscillatory synaptic interactions between ventroposterior and reticular neurons in mouse thalamus in vitro. J Neurophysiol. 1994;72:1993–2003. doi: 10.1152/jn.1994.72.4.1993. [DOI] [PubMed] [Google Scholar]
- Weiergraber M, Stephani U, Kohling R. Voltage-gated calcium channels in the etiopathogenesis and treatment of absence epilepsy. Brain Res Rev. 2010;62:245–71. doi: 10.1016/j.brainresrev.2009.12.005. [DOI] [PubMed] [Google Scholar]
- Wells MF, et al. Thalamic reticular impairment underlies attention deficit in Ptchd1(Y/-) mice. Nature. 2016;532:58–63. doi: 10.1038/nature17427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westerberg C, et al. Distinct medial temporal contributions to different forms of recognition in amnestic mild cognitive impairment and Alzheimer's disease. Neuropsychologia. 2013;51:2450–61. doi: 10.1016/j.neuropsychologia.2013.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westwood AJ, et al. Prolonged sleep duration as a marker of early neurodegeneration predicting incident dementia. Neurology. 2017;88:1172–1179. doi: 10.1212/WNL.0000000000003732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao D, Zikopoulos B, Barbas H. Laminar and modular organization of prefrontal projections to multiple thalamic nuclei. Neuroscience. 2009;161:1067–81. doi: 10.1016/j.neuroscience.2009.04.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie L, et al. Sleep drives metabolite clearance from the adult brain. Science. 2013;342:373–7. doi: 10.1126/science.1241224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaffe K, et al. Sleep-disordered breathing, hypoxia, and risk of mild cognitive impairment and dementia in older women. JAMA. 2011;306:613–9. doi: 10.1001/jama.2011.1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zikopoulos B, Barbas H. Pathways for emotions and attention converge on the thalamic reticular nucleus in primates. J Neurosci. 2012;32:5338–50. doi: 10.1523/JNEUROSCI.4793-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]