

Abstract
Living cells are complex and dynamic assemblies that carefully sequester and orchestrate multiple diverse processes that enable growth, division, regulation, movement, and communication. Membrane-bound organelles such as the endoplasmic reticulum, mitochondria, plasma membrane, and others are integral to these processes and their functions demand dynamic reorganization in both space and time. Visualizing these dynamics in live cells over long time periods demands probes that label discrete organelles specifically, at high density, and withstand long-term irradiation. Here we describe the evolution of our work on the development of a set of High Density Environmentally sensitive (HIDE) membrane probes that enable long-term, live-cell nanoscopy of the dynamics of multiple organelles in live cells using SMS and STED imaging modalities.
Graphical Abstract
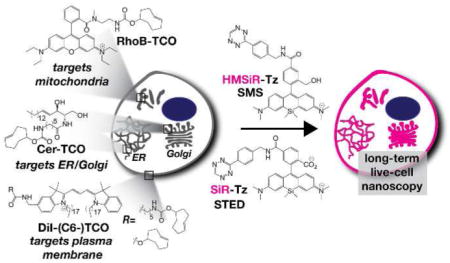
Living cells are the foundational unit of complex organisms. Cells are complex and dynamic assemblies and must carefully sequester and orchestrate a staggering array of biochemistry to perform functions required for growth, division, regulation, movement, and intra- and inter-cellular communication.1 Membrane-bound organelles such as the nucleus, endoplasmic reticulum (ER), Golgi, endosomes, lysosomes, mitochondria, and plasma membrane (PM) are integral to these processes.1 Importantly, organelles are not static assemblies, and their functions demand dynamic reorganization in both space and time. While electron microscopy (EM)2 studies have historically revealed the ultrastructure of cellular organelles at sub-nanometer resolution,3 EM cannot reveal dynamic events in live cells that are evident using traditional light microscopy, albeit, due to the diffraction limit of light,4 at approximately two orders of magnitude lower resolution.
Over the past two decades, a number of super-resolution microscopy techniques have been developed to overcome the resolution barrier posed by the diffraction limit of traditional light microscopy and visualize cellular components and structures at resolutions in the tens of nanometers in living cells.5–11 The development of super-resolution microscopy techniques was recognized by a Nobel Prize in 201412 and many excellent reviews are available to the interested reader.13–15 In brief, super-resolution microscopy or “nanoscopy” techniques that truly break the diffraction limit are embodied by two major modalities that are often referred to as Stimulated Emission Depletion or STED5, 7 and Single Molecule Switching, often referred to as SMS, and which includes techniques known as STORM,9 dSTORM,11 PALM,8 FPALM,6 GSDIM,10 and others. Both STED and SMS rely on targeted or stochastic switching of fluorophores between off (dark) and on (fluorescent) states, and they demand distinct (and sometimes hard to attain) photophysical properties from the requisite chromophores, typically fluorescent proteins and organic fluorophores.13 The STED depletion laser is orders of magnitude more powerful than a traditional excitation laser, which demands exceptionally photostable fluorophores, while SMS requires that the fluorophores “blink” to allow accurate localization of single molecules. These photophysical properties are easy to attain in fixed cells through the use of highly engineered fluorophores,16 anti-fade buffers,17 and “blinking” buffers, 18 but their translation into living cells has been difficult. 15 In this Perspective, we describe the evolution of our work on a unique family of membrane probes that enable long-term live-cell nanoscopy of multiple organelles and their dynamics using both SMS and STED imaging modalities (Figure 1).
Figure 1.

Labeling live cells with HIDE probes derived from the reaction of Cer-TCO (targets ER and/or Golgi), RhoB-TCO (targets mitochondria), or DiI-(C6-)-TCO (targets plasma membrane) with silicon rhodamine dyes HMSiR-Tz or SiR-Tz to enable live-cell nanoscopy using SMS or STED, respectively.
Although membrane-bound organelles have traditionally been visualized at super-resolution by labeling membrane-resident proteins with fluorescent or self-labeling proteins, we reasoned that labeling the organelle membrane itself–the lipid–could offer a genuine advantage because of the exceptionally high density of lipids relative to proteins in a typical membrane (Figure 2). High labeling density is critical for super-resolution methods, because as the resolution or detection volume decreases so does the number of detectable molecules. 15, 19, 20 Excluding polymeric protein assemblies, such as actin and tubulin, the density of lipid in a membrane is naturally over a hundred times higher than that of any protein.21 We further reasoned that by labeling the lipid we could take advantage of known, fast, bioorthogonal22 reactions to perform the labeling reaction in two steps: an initial step in which an orthogonally reactive, but minimally perturbing, lipid is added to cells and a subsequent step in which the dye is appended. This two-component approach provides a high level of flexibility for labelling organelles with dye conjugates across the visible spectrum. Thus, our initial work evaluated whether linking an appropriate dye not to a protein, but to a lipid, would provide a benefit for imaging organelles at super-resolution.
Figure 2.
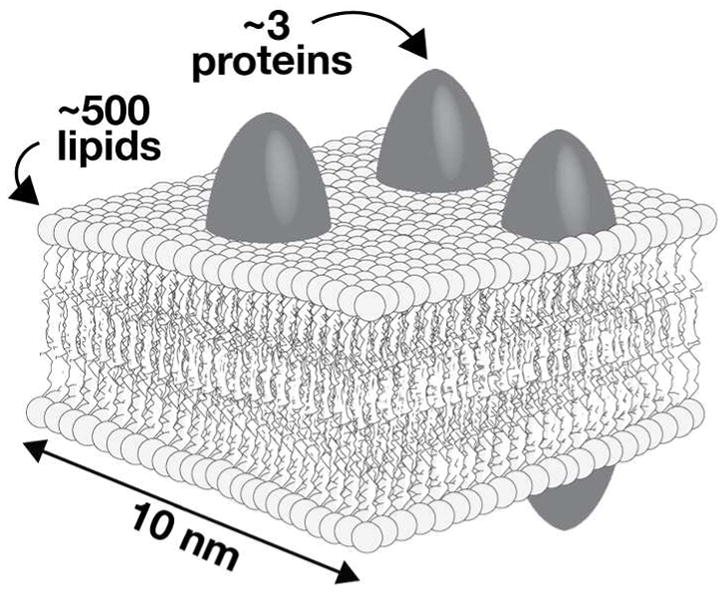
Cartoon illustrating the approximate relative densities of proteins and lipids within the membrane of a typical cellular organelle. There are roughly 3 proteins and 500 lipids in a 10 × 10 nm2 area of a bilayer. Data taken from Quinn et al.21
This two-component membrane probe strategy builds on significant work of many others. Most notable, both Shim et al.19 and Carlini et al.23 recognized the need for high-density labeling and utilized commercially available organelle probes to label live cells for SMS microscopy; these probes unfortunately required non-benign24 405 nm illumination and an oxygen scavenging system. Lukinavičius et al. also recognized the need for high-density labeling and developed a single-component labeling strategy based on small molecules that engage multimeric proteins25, 26 and DNA.27 Our work was also inspired by the unique attributes of PAINT microscopy, which utilizes the reversible binding and fluorescence of classic fluorophores like Nile Red28, 29 or next generation dyes like AzepRh30 in a membrane to achieve high-density stochastic blinking for SMS.
Our initial work focused on ceramide lipids, which are biosynthesized in the ER before trafficking through the Golgi apparatus for further modification.31 Subsequent trafficking from the Golgi can be halted by a temperature block, which accumulates ceramide within the Golgi/trans-Golgi network.32–34 Indeed, the lipid probe BODIPY-Cer exploits this property and has been used for decades to image Golgi membranes using confocal microscopy. 35 But BODIPY dyes rapidly bleach, especially when exposed to the high-powered illumination required for super-resolution microscopy. We hypothesized that linking a ceramide lipid to a more photostable, STED-compatible dye, such as a member of the silicon rhodamine (SiR) family,36, 37 might allow imaging of the Golgi in live cells at super-resolution.38 To test this hypothesis, we synthesized a modified ceramide lipid equipped with a trans-cyclooctene (Cer-TCO) that could react via a tetrazine ligation reaction22 with SiR-Tz, 37 a silicone rhodamine derivative, to form Cer-SiR in live cells (Figure 1). As expected, we found that Cer-SiR was enriched in the Golgi by a short incubation at 19.5 °C,32–34 as demonstrated by a typical perinuclear labeling pattern and colocalization with GalNaAcT2-GFP, a Golgi-resident protein (Figure 3a–d). Surprisingly, preformed Cer-SiR did not label the Golgi, providing additional support to the utility of our two-componenent strategy.38 The presence of Cer-SiR in the Golgi did not perturb the mobility of Golgi-resident enzymes or the trafficking of cargo from the endoplasmic reticulum through the Golgi and to the plasma membrane.38 Cer-SiR did, however, facilitate the acquisition of STED super-resolution images, and to our surprise, was far more photostable than a conjugate assembled from the same SiR dye joined through a SNAP-tag to the Golgi-resident protein Rab6 (Figure 3e). Notably, we observed the fluorescence of fixed cells labeled with Cer-SiR for more than 29 min (900 images), 4 whereas fixed cells labeled with Rab6-SiR were visible for only 300 frames. This increase in photostability was sufficient to visualize vesicles budding and exiting the Golgi in live cells (Figure 3f).
Figure 3.
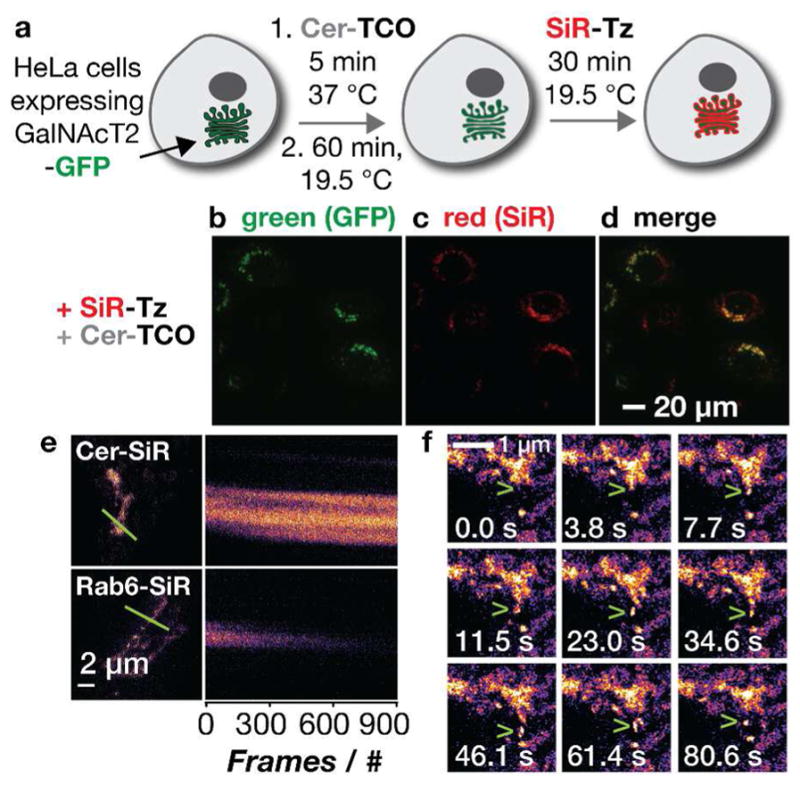
Cer-TCO localizes to the Golgi and reacts with SiR-Tz to enable live cell confocal and STED imaging. a) HeLa cells expressing the Golgi marker Gal-NAcT2-GFP were incubated with Cer-TCO and subjected to a temperature block before treatment with SiR-Tz to label the Golgi with Cer-SiR. b–d) Colocalization with GalNAcT2-GFP was observed only upon addition of Cer-TCO and SiR-Tz. e) Kymographs of fixed cells labelled with Cer-SiR or Rab6-SiR. f) Time lapse STED images of a vesicle budding out of the Golgi in a living cell (green arrows). Figure adapted with permission from Erdmann et al.38
But why was Cer-SiR so much more photostable than Rab6-SiR? An interesting hint was that although Cer-SiR took a longer time to bleach, its initial brightness was not so different than that of Rab6-SiR. We hypothesized that Cer-SiR was more photostable because it resided within a more hydrophobic environment than Rab6-SiR. Like all dyes in the rhodamine family, SiR derivatives exist in an equilibrium between two states: a dark neutral state and a bright charged state (Figure 4a). The position of this equilibrium is affected by both pH and dielectric constant, with the dark state favored at low pH and/or within a lipophilic environment (low dielectric constant).37 Thus, we reasoned that placing the SiR dye in a hydrophobic environment within the membrane could shift the dark/bright equilibrium to favor the dark form, creating a large pool of non-fluorescent molecules that would not absorb excitation light and would be thereby protected from photobleaching (Figure 4b). We reasoned further that this pool could act to slowly replace fluorophores that have been photobleached, leading to a large apparent increase in photostability. By contrast, when appended to a protein (e.g. Rab6), SiR would be in a more aqueous environment, which favors the charged form of the dye, leaving it exposed to photobleaching.
Figure 4.
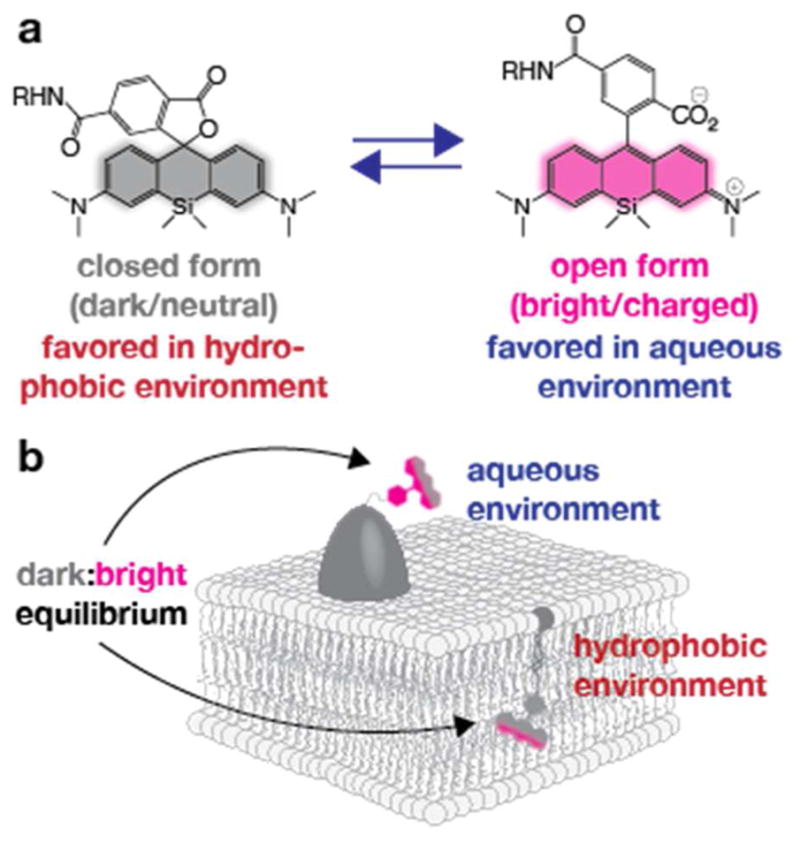
Exploiting the environmental sensitivity of SiR-CO2H to increase apparent photostability. a) SiR-CO2H equilibrates between two forms, a closed, lactone form that is dark, and an open form that is bright. The position of this equilibrium depends on both pH and the polarity of the environment. b) Appending the SiR chromophore to a protein in an aqueous environment shifts this equilibrium towards the open, bright form, while appending it to a lipid in a membrane environment shifts the equilibrium towards the closed, dark form.
Around the time we completed our work with Cer-SiR, the Urano Lab at the University of Tokyo disclosed HMSiR,39 the first fluorophore to blink spontaneously via a ground state mechanism, and demonstrated its utility for SMS nanoscopy. HMSiR is a silicon rhodamine derivative in which the carboxylic acid of SiR-CO2H has been replaced by a hydroxyl group (Figure 5a). This change shifts the intramolecular cyclization equilibrium towards the closed (OFF) form at physiological pH, and endows HMSiR with the ability to “blink” through a ground state mechanism that does not require toxic blinking buffers. At pH 7.4, 1.2–1.3% of HMSiR exists in the open (ON) form, which is sufficient to visualize proteins present at low density by SMS nanoscopy. Unfortunately, an ON-fraction of even 1% is too high to visualize lipids present at high density, as it precludes the localization of single molecules.40
Figure 5.

In vitro evaluation of HMSiR in a membrane environment. a) HMSiR equilibrates between a closed, cyclized form that dark (OFF) and an open (ON)form that is bright. Like SiR-CO2H, the position of the HMSiR equilibrium depends on pH and hydrophobicity. b) Cartoon of an in vitro experiment designed to assess whether the HMSiR ON/OFF fraction is affected by hydrophobicity. HMSiR was immobilized to a glass surface via either a protein (generated upon reaction of HMSiR-CA and Halo-GST) or a liposome (generated upon reaction of Cer-TCO with HMSiR-Tz). c) Super-resolution images of HMSiR immobilized as described in b). Rainbow colored temporal look-up table. Scale bar: 200 nm. d) Plot illustrating the normalized number of localizations observed as a function of time when HMSiR was immobilized to a protein, within an aqueous environment, and within a liposome. τ values were calculated from a single exponential fit (mean ± SEM). Figure adapted with permissionfrom Takakura et al.40
We wondered whether placing HMSiR in a membrane environment would also increase its photostability, in the manner observed for Cer-SiR—this time in the context of SMS nanoscopy. To test this idea, we performed in vitro experiments that compared the apparent photostability of HMSiR within an aqueous (protein) and hydrophobic (liposome) environment assembled on glass slides (Figure 5b). This experiment revealed that the signal from Cer-HMSiR in a hydrophobic liposome environment could be observed for >6-fold longer than that of GST-HMSiR in an aqueous environment (Figure 5c,d).40 We reasoned that placing HMSiR in a hydrophobic membrane environment results in two changes that work together to improve apparent photostability: it shifts the ON/OFF equilibrium to a lower value (0.03% in liposomes vs. 0.2% for GST-HMSiR), preventing the rejection of overlapping single molecules during localization, and simultaneously establishes a dark state reservoir that is protected from photobleaching. Monte Carlo simulations are fully consistent with this model, showing that combining a strongly reduced ON fraction with high labeling density results in a dramatic increase in apparent photostability.40
To test this concept in live cells, we focused first on visualizing the endoplasmic reticulum, whose neutral pH would favor a lower ON/OFF ratio than the more acidic Golgi (pH = 6.4 ± 0.3)41 and is also effectively targeted with ceramide lipids such as Cer-TCO (Figure 6a).40 Subsequent SMS super-resolution imaging with Cer-HMSiR in the ER recapitulated the benefits of combining high-density labeling with a membrane probe: we were able to acquire movies of a highly dynamic ER for >20 mins, imaged at 400 frames/s and combined to achieve a 2 s temporal resolution and a spatial resolution of about 50 nm (Figure 6b). When labeled with Cer-HMSiR, the ER could be imaged for >50-fold longer than when labeled with the protein probe Sec61β-HMSiR (Figure 6c). This increase in imaging time was also accompanied by >30-fold increase in labeling density. By combining high spatial and temporal resolution over an extended time period, we observed ER tubules as well as sheets dynamically interconverting over the course of the >20 min acquisition (Figure 6b). Cer-HMSiR was also amenable to two-color live-cell imaging in combination with mEos3.2. We were able to visualize the ER and the Golgi labeled with Cer-HMSiR and COPβ1-mEos3.2 (Figure 6d). We note that the lower photostability of mEos3.2 precluded the acquisition of long time lapse two-color images, emphasizing the continuing need for orthogonal membrane probe systems and dyes with comparable photostabilities.
Figure 6.

Long time-lapse SMS imaging of endoplasmic reticulum (ER) dynamics using Cer-HMSiR. a) Schematic illustration of the 2-step procedure employed to label the ER with Cer-HMSiR. b) Single frame from a 25 min movie with 2 s temporal resolution. s: sheet-like ER, f: fenestrated ER sheets, t: tubular ER. Diffraction limited inset shown in grey. Scale bar: 1 μm. c) Comparison of localizations observed between Cer-HMSiR and Sec61β-HMSiR d) Image of COP1 vesicles and the Golgi and ER in HeLa cells expressing the marker COPβ1-mEos3.2 (green) and treated with Cer-TCO and HMSiR-Tz at 37 °C (red). An enlarged view of the image within the yellow box is shown beneath. Figure adapted with permission from Takakura et al.40
Labeling density is especially critical to achieve high quality super-resolution images in three dimensions. We found that the high labeling density and improved photostability provided by Cer-HMSiR allowed the acquisition of a 15 min movie of the ER with 10 s temporal resolution, which represents the longest 3D SMS movie of which we are aware (Figure 7a). This high level of structural detail revealed interconnections between ER tubules at different depths that would not be visible in 2D (Figure 7b,c). Additionally, 3D imaging deeper in the cell exposed ER channel structures traversing the nucleus. 40
Figure 7.
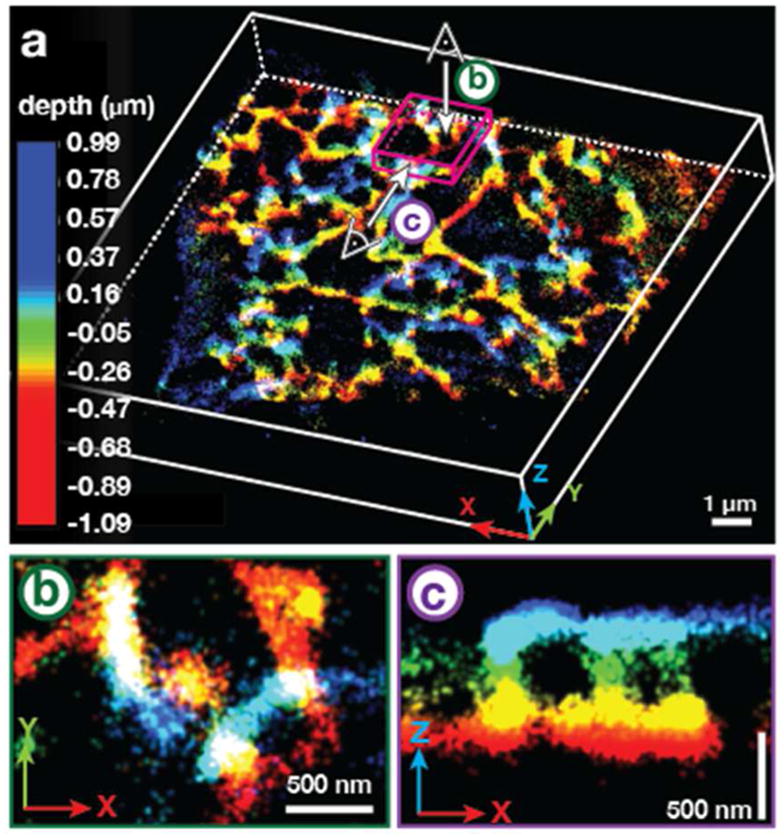
Long time-lapse imaging the ER in 3D with Cer-HMSiR. a) Perspective view of a depth-colored snapshot from a 15 min movie of the ER in HeLa cells treated with Cer-TCO and HMSiR-Tz. b) Top view of the magenta cube shown in (a). c) Front view of the magenta cube shown in (b). Figure adapted with permission from Takakura et al.40
The success of Cer-TCO in STED and SMS nanoscopy encouraged us to apply the HIDE strategy to other organelles. This goal required the identification of new lipid-like molecules known to localize selectively to organelles other than the ER or Golgi. Despite the abundance of information about lipid organization42 in specific organelles, natural lipids can be difficult to synthesize and their localization can be sensitive to even small structural modifications. Instead, we chose to repurpose a pair of known and well-validated organelle-specific probes and modify them with a TCO moiety to enable reaction with a tetrazine-modified silicon rhodamine dye. This approach offered the benefit of synthetic ease and well-established localization protocols, simplifying probe optimization. Furthermore, while the well-validated organelle-specific probes we chose are fluorophores themselves, they are generally not suitable for live cell super-resolution microscopy without additives,19 and because they do not absorb near-IR light, invisible during SMS microscopy. Overall, this strategy furnishes new utility for these canonical organelle probes for live-cell nanoscopy. Two validated organelle-specific probes were chosen: the cationic rhodamine dye RhoB and the family of dialkylindocarbocyanines exemplified by DiI.19
It is well known that cationic rhodamine derivatives localize to mitochondria due to the mitochondrial membrane potential.43 Functionalized Rhodamine B (RhoB) derivatives are no exception.44, 45 After synthesizing RhoB-TCO, we confirmed that it labeled mitochondria (Figure 8a) and proceeded to perform live-cell SMS imaging with HMSiR-Tz.40 Here again, localizing HMSiR to an organelle membrane at high density enabled longterm imaging.40 By targeting HMSiR to the mitochondria membrane with RhoB, we could obtain images for more than 7 min, 13-fold longer than when the mitochondria was visualized using the mitochondrial protein OMP-25 labeled with HMSiR (Figure 8b,c).
Figure 8.

Long time-lapse, SMS imaging of mitochondria using RhoB-HMSiR. a) Schematic illustration of the 2-step procedure employed to label mitochondria with RhoB-HMSiR. b,c) Comparison between time-colored super-resolution images of mitochondria labeled with the protein probe OMP25-HMSiR and RhoB-HMSiR. The diffraction-limited image obtained from the RhoB channel is marked by the dashed white line in (c). Kymographs of both images, derived from the time-dependent signal along each brown line vs. time, are shown on the right side of each panel. Scale bar: 1 μm. Figure adapted with permission from Takakura et al.40
It is also well known that dialkylindocarbocyanines such as DiI localize to the plasma membrane due to their long lipophilic C18 chains.35 We initially synthesized DiI-TCO (Figure 1), which labeled the plasma membrane upon reaction with HMSiR-Tz (Figure 9a), and was 14-fold more photostable than the plasma membrane marker Smoothened labeled with HMSiR,40 and using DiI-HMSiR we could visualize filopodia dynamics over long time periods by SMS (Figure 9b,c). Wondering whether we could improve photostability further, we reasoned that an analog of DiI-TCO that contained a longer alkyl chain between HMSiR and the DiI chromophore would allow for enhanced interaction with the membrane, leading to a further decrease in the ON/OFF ratio of HMSiR and a concomitant increase in apparent photostability. We therefore synthesized DiI-C6-TCO (Figure 1) and performed SMS nanoscopy after its reaction with HMSiR-Tz. Indeed, the increased linker length yielded a probe that could be imaged for 2.4-fold longer than DiI-HMSiR alone.40 The simplicity of this rational design represents yet another advantage of two component lipid probes for high-density labeling of cellular organelles.
Figure 9.

Long time-lapse, SMS imaging of the plasma membrane using DiI-HMSiR and DiI-C6-HMSiR. a) Schematic diagram of the 2-step procedure used to label the plasma membrane with DiI-HMSiR or DiI-C6-HMSiR. b) Super-resolution image of the plasma membrane visualized with DiI-HMSiR. Scale bar: 1 μm. c) Time-lapse images of filopodia dynamics from purple box in (b). Yellow arrows highlight major changes. Scale bar: 1 μm. Figure adapted with permission from Takakura et al.40
Drawing from the observation that our membrane-labelling strategy improved apparent photostability for Cer-SiR and Cer-HMSiR during both STED and SMS nanoscopy, respectively, we next applied our membrane probe DiI-TCO to live-cell STED imaging by pairing it with SiR-Tz.46 As expected, DiI-TCO reacts with SiR-Tz and labels the plasma membrane of live HeLa cells (Figure 10a). Additionally, we were able to observe HeLa cell dynamics for more than 25 minutes with minimal photobleaching—in stark contrast to the fluorescence of cells labeled with a plasma membrane resident protein labeled with the same SiR core and observed under STED conditions (Figure 10a).
Figure 10.
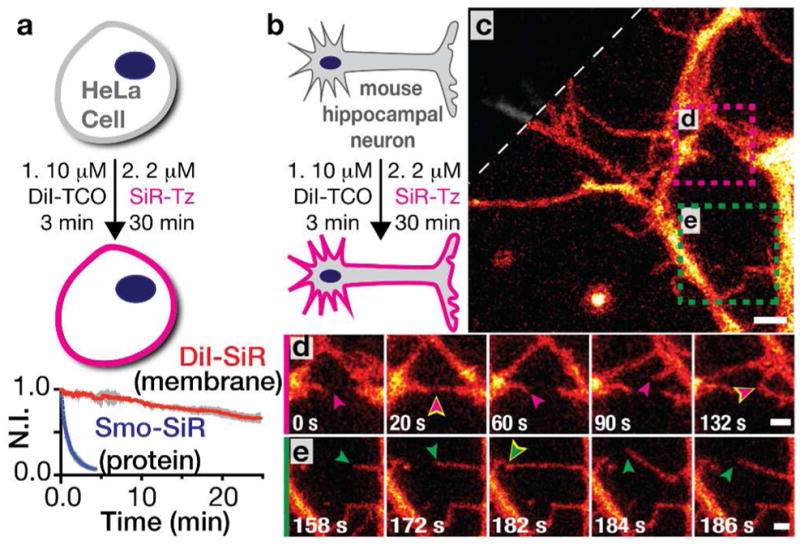
Long time-lapse STED imaging of live HeLa cells and mouse hippocampal neurons labeled with DiI-SiR. a) HeLa cells treated with DiI-TCO and SiR-Tz were compared with cells transiently expressing Smo-Halo and labelled with SiR-CA. A plot of the normalized fluorescence intensity of Smo-SiR (blue) and DiI-SiR (red) over time (mean ± SD) is shown below. b) DIV 4 neurons were labeled with DiI-TCO followed by SiR-Tz. c) Snapshot of a STED movie with a confocal cutaway in gray. d) Pink, and e) green square outline from c; time courses with corresponding pink and green arrows denoting filopodia dynamics; yellow outlines designate contact events. Scale bars: 2 μm in c, 1 μm in d,e. Figure adapted with permission from Thompson et al.46
Like all membrane probes, DiI-TCO does not require transfection. This advantage is significant when one wants to visualize primary cell lines that are difficult to transfect. Our standard DiI-TCO labeling conditions adapted easily to label live mouse hippocampal neurons, and facilitated the dynamic STED imaging of early development events for more than 9 min at a 2 sec temporal resolution (Figure 10b,c).46 Using DiI-SiR, we could visualize the dynamics of neuronal development, including self/nonself discrimination,47–49 (Figure 10d,e) which is essential for the control of synaptic partner selection. These observations underscore the utility of HIDE probes for applications in primary cell lines.
This work provides additional support for the hypothesis that the apparent photostability of silicon rhodamine dyes can be enhanced during STED and SMS nanoscopy by positioning them within the hydrophobic membrane environment that characterizes the boundaries of many cellular organelles. This enhancement is possible because the dyes exist in two forms, one that fluoresces and one that does not. This increase in apparent photostability arises not through fluorophore engineering, but through environmental engineering. Indeed, we think of our membrane probes as if they are “hiding” the appended fluorophore in the membrane to protect it from photobleaching. We therefore find it fitting to describe our strategy as High Density Environmentally sensitive labeling, or HIDE.
We hope that this Perspective paints a bright future for long-term, live-cell nanoscopy of membrane-bound organelles. We foresee the application of HIDE probes across multiple cell types and organelles, into three dimensions, and using new imaging modalities, and especially in multicolor mode to probe lipid and organelle interactions and dynamics. Fluorophore design will likely be an important contributor in enabling even longer term studies. Modifications to the fluorophores appended to membrane probes to endow them with useful properties such as fluorogenicity50, 51 or membrane-specific 30 utility will also augment imaging capabilities. Phototoxicity remains a major hurdle for long-term imaging with high laser intensities, but brighter dyes50–52 illuminated with near infrared light will enable the use of lower laser intensities that are less phototoxic.24 Additionally, the adoption of cell-friendly super-resolution microscopes outfitted with incubators will likely improve cell viability. In order to truly leverage these modalities for studying biological processes, it will be important to apply these strategies to 2- and 3-color imaging with orthogonal labeling chemistries22 and dye combinations. Additionally, imaging for long periods with high temporal resolution generates extremely large datasets (many of the SMS single movies have several TB (!) of raw data), which can make handling, analysis, and storage difficult. The application of small-molecule based approaches to solving problems in microscopy is only in its infancy. We expect that as the field continues to push the boundaries of live-cell imaging, the focus will turn more and more towards the probes.
Acknowledgments
Funding Sources
This work was supported by the Wellcome Trust (095927/A/11/Z) and in part by the NIH (GM 83257 to A.S., P30 DK45735 to J.B., R01GM118486 to J.B. and D.T.) A.D.T. was supported by an NIH Ruth L. Kirschstein NRSA F31GM119259.
We thank the numerous collaborators who made possible the work described in this Perspective.
Footnotes
A.D.T., D.T., and A.S. disclose a pending patent for novel lipid probes. J. B. discloses significant financial interest in Bruker Corp. and Hamamatsu Photonics.
Author Contributions
The manuscript was written with contributions from all authors.
References
- 1.Alberts B. Molecular biology of the cell. 6. Garland Science, Taylor and Francis Group; New York, NY: 2015. [Google Scholar]
- 2.Kourkoutis LF, Plitzko JM, Baumeister W. Electron Microscopy of Biological Materials at the Nanometer Scale. Annu Rev Mater Res. 2012;42:33–58. [Google Scholar]
- 3.Palade G. Intracellular aspects of the process of protein synthesis. Science. 1975;189:867. doi: 10.1126/science.189.4206.867-b. [DOI] [PubMed] [Google Scholar]
- 4.Abbe E. Beitrage zur Theorie des Mikroskops und der mikroskopischen Wahrnehmung. Arch Mikroskop Anat. 1873;9:413–420. [Google Scholar]
- 5.Hell SW, Wichmann J. Breaking the Diffraction Resolution Limit by Stimulated-Emission - Stimulated-Emission-Depletion Fluorescence Microscopy. Opt Lett. 1994;19:780–782. doi: 10.1364/ol.19.000780. [DOI] [PubMed] [Google Scholar]
- 6.Hess ST, Girirajan TPK, Mason MD. Ultra-high resolution imaging by fluorescence photoactivation localization microscopy. Biophys J. 2006;91:4258–4272. doi: 10.1529/biophysj.106.091116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Klar TA, Hell SW. Subdiffraction resolution in far-field fluorescence microscopy. Opt Lett. 1999;24:954–956. doi: 10.1364/ol.24.000954. [DOI] [PubMed] [Google Scholar]
- 8.Betzig E, Patterson GH, Sougrat R, Lindwasser OW, Olenych S, Bonifacino JS, Davidson MW, Lippincott-Schwartz J, Hess HF. Imaging intracellular fluorescent proteins at nanometer resolution. Science. 2006;313:1642–1645. doi: 10.1126/science.1127344. [DOI] [PubMed] [Google Scholar]
- 9.Rust MJ, Bates M, Zhuang XW. Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM) Nat Methods. 2006;3:793–795. doi: 10.1038/nmeth929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Folling J, Bossi M, Bock H, Medda R, Wurm CA, Hein B, Jakobs S, Eggeling C, Hell SW. Fluorescence nanoscopy by ground-state depletion and single-molecule return. Nat Methods. 2008;5:943–945. doi: 10.1038/nmeth.1257. [DOI] [PubMed] [Google Scholar]
- 11.Heilemann M, van de Linde S, Schuttpelz M, Kasper R, Seefeldt B, Mukherjee A, Tinnefeld P, Sauer M. Subdiffraction-resolution fluorescence imaging with conventional fluorescent probes. Angew Chem Int Edit. 2008;47:6172–6176. doi: 10.1002/anie.200802376. [DOI] [PubMed] [Google Scholar]
- 12.Mockl L, Lamb DC, Brauchle C. Super-resolved fluorescence microscopy: Nobel Prize in Chemistry 2014 for Eric Betzig, Stefan Hell, and William E Moerner. Angew Chem Int Ed Engl. 2014;53:13972–13977. doi: 10.1002/anie.201410265. [DOI] [PubMed] [Google Scholar]
- 13.Hell SW. Far-field optical nanoscopy. Science. 2007;316:1153–1158. doi: 10.1126/science.1137395. [DOI] [PubMed] [Google Scholar]
- 14.Huang B, Babcock H, Zhuang XW. Breaking the Diffraction Barrier: Super-Resolution Imaging of Cells. Cell. 2010;143:1047–1058. doi: 10.1016/j.cell.2010.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Toomre D, Bewersdorf J. A New Wave of Cellular Imaging. Annu Rev Cell Dev Bi. 2010;26:285–314. doi: 10.1146/annurev-cellbio-100109-104048. [DOI] [PubMed] [Google Scholar]
- 16.Wurm CA, Kolmakov K, Gottfert F, Ta H, Bossi M, Schill H, Berning S, Jakobs S, Donnert G, Belov VN, Hell SW. Novel red fluorophores with superior performance in STED microscopy. Optical Nanoscopy. 2012;1:7. [Google Scholar]
- 17.Cordes T, Maiser A, Steinhauer C, Schermelleh L, Tinnefeld P. Mechanisms and advancement of antifading agents for fluorescence microscopy and single-molecule spectroscopy. Phys Chem Chem Phys. 2011;13:6699–6709. doi: 10.1039/c0cp01919d. [DOI] [PubMed] [Google Scholar]
- 18.Dempsey GT, Vaughan JC, Chen KH, Bates M, Zhuang XW. Evaluation of fluorophores for optimal performance in localization-based super-resolution imaging. Nat Methods. 2011;8:1027–1036. doi: 10.1038/nmeth.1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shim SH, Xia CL, Zhong GS, Babcock HP, Vaughan JC, Huang B, Wang X, Xu C, Bi GQ, Zhuang XW. Super-resolution fluorescence imaging of organelles in live cells with photoswitchable membrane probes. P Natl Acad Sci USA. 2012;109:13978–13983. doi: 10.1073/pnas.1201882109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shroff H, Galbraith CG, Galbraith JA, Betzig E. Live-cell photoactivated localization microscopy of nanoscale adhesion dynamics. Nat Methods. 2008;5:417–423. doi: 10.1038/nmeth.1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Quinn P, Griffiths G, Warren G. Density of Newly Synthesized Plasma-Membrane Proteins in Intracellular Membranes .2. Biochemical-Studies. J Cell Biol. 1984;98:2142–2147. doi: 10.1083/jcb.98.6.2142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lang K, Chin JW. Cellular Incorporation of Unnatural Amino Acids and Bioorthogonal Labeling of Proteins. Chem Rev. 2014;114:4764–4806. doi: 10.1021/cr400355w. [DOI] [PubMed] [Google Scholar]
- 23.Carlini L, Manley S. Live Intracellular Super-Resolution Imaging Using Site-Specific Stains. Acs Chem Biol. 2013;8:2643–2648. doi: 10.1021/cb400467x. [DOI] [PubMed] [Google Scholar]
- 24.Waldchen S, Lehmann J, Klein T, van de Linde S, Sauer M. Light-induced cell damage in live-cell super-resolution microscopy. Scientific Reports. 2015;5:15348. doi: 10.1038/srep15348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lukinavicius G, Reymond L, D’Este E, Masharina A, Gutfert F, Ta H, Guether A, Fournier M, Rizzo S, Waldmann H, Blaukopf C, Sommer C, Gerlich DW, Arndt HD, Hell SW, Johnsson K. Fluorogenic probes for live-cell imaging of the cytoskeleton. Nat Methods. 2014;11:731–733. doi: 10.1038/nmeth.2972. [DOI] [PubMed] [Google Scholar]
- 26.Lukinavicius G, Reymond L, Umezawa K, Sallin O, D’Este E, Gottfert F, Ta H, Hell SW, Urano Y, Johnsson K. Fluorogenic Probes for Multicolor Imaging in Living Cells. J Am Chem Soc. 2016;138:9365–9368. doi: 10.1021/jacs.6b04782. [DOI] [PubMed] [Google Scholar]
- 27.Lukinavicius G, Blaukopf C, Pershagen E, Schena A, Reymond L, Derivery E, Gonzalez-Gaitan M, D’Este E, Hell SW, Gerlich DW, Johnsson K. SiR-Hoechst is a far-red DNA stain for live-cell nanoscopy. Nat Commun. 2015;6:8497. doi: 10.1038/ncomms9497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Giannone G, Hosy E, Levet F, Constals A, Schulze K, Sobolevsky AI, Rosconi MP, Gouaux E, Tampe R, Choquet D, Cognet L. Dynamic superresolution imaging of endogenous proteins on living cells at ultra-high density. Biophys J. 2010;99:1303–1310. doi: 10.1016/j.bpj.2010.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sharonov A, Hochstrasser RM. Wide-field subdiffraction imaging by accumulated binding of diffusing probes. Proc Natl Acad Sci U S A. 2006;103:18911–18916. doi: 10.1073/pnas.0609643104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Legant WR, Shao L, Grimm JB, Brown TA, Milkie DE, Avants BB, Lavis LD, Betzig E. High-density three-dimensional localization microscopy across large volumes. Nat Methods. 2016;13:359–365. doi: 10.1038/nmeth.3797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Maceyka M, Spiegel S. Sphingolipid metabolites in inflammatory disease. Nature. 2014;510:58–67. doi: 10.1038/nature13475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Griffiths G, Pfeiffer S, Simons K, Matlin K. Exit of Newly Synthesized Membrane-Proteins from the Trans Cisterna of the Golgi-Complex to the Plasma-Membrane. J Cell Biol. 1985;101:949–964. doi: 10.1083/jcb.101.3.949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kuliawat R, Arvan P. Protein Targeting Via the Constitutive-Like Secretory Pathway in Isolated Pancreatic-Islets-Passive Sorting in the Immature Granule Compartment. J Cell Biol. 1992;118:521–529. doi: 10.1083/jcb.118.3.521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Simon JP, Ivanov IE, Bo SS, Hersh D, Adesnik M, Sabatini DD. The in vitro generation of post-Golgi vesicles carrying viral envelope glycoproteins requires an ARF-like GTP-binding protein and a protein kinase C associated with the Golgi apparatus. J Biol Chem. 1996;271:16952–16961. doi: 10.1074/jbc.271.28.16952. [DOI] [PubMed] [Google Scholar]
- 35.Michelle TZ, Spence IDJ. The molecular probes handbook: a guide to fluorescent probes and labeling technologies. Life Technologies Corporation; 2010. [Google Scholar]
- 36.Koide Y, Urano Y, Hanaoka K, Terai T, Nagano T. Evolution of Group 14 Rhodamines as Platforms for Near-Infrared Fluorescence Probes Utilizing Photoinduced Electron Transfer. Acs Chem Biol. 2011;6:600–608. doi: 10.1021/cb1002416. [DOI] [PubMed] [Google Scholar]
- 37.Lukinavicius G, Umezawa K, Olivier N, Honigmann A, Yang G, Plass T, Mueller V, Reymond L, Correa IR, Jr, Luo ZG, Schultz C, Lemke EA, Heppenstall P, Eggeling C, Manley S, Johnsson K. A near-infrared fluorophore for live-cell super-resolution microscopy of cellular proteins. Nat Chem. 2013;5:132–139. doi: 10.1038/nchem.1546. [DOI] [PubMed] [Google Scholar]
- 38.Erdmann RS, Takakura H, Thompson AD, Rivera-Molina F, Allgeyer ES, Bewersdorf J, Toomre D, Schepartz A. Super-Resolution Imaging of the Golgi in Live Cells with a Bioorthogonal Ceramide Probe. Angew Chem Int Edit. 2014;53:10242–10246. doi: 10.1002/anie.201403349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Uno SN, Kamiya M, Yoshihara T, Sugawara K, Okabe K, Tarhan MC, Fujita H, Funatsu T, Okada Y, Tobita S, Urano Y. A spontaneously blinking fluorophore based on intramolecular spirocyclization for live-cell super-resolution imaging. Nat Chem. 2014;6:681–689. doi: 10.1038/nchem.2002. [DOI] [PubMed] [Google Scholar]
- 40.Takakura H, Zhang Y, Erdmann RS, Thompson AD, Lin Y, McNellis B, Rivera-Molina F, Uno S-n, Kamiya M, Urano Y, Rothman JE, Bewersdorf J, Schepartz A, Toomre D. Long time-lapse nanoscopy with spontaneously blinking membrane probes. Nat Biotech advance online publication. 2017 doi: 10.1038/nbt.3876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wu MM, Llopis J, Adams S, McCaffery JM, Kulomaa MS, Machen TE, Moore HPH, Tsien RY. Organelle pH studies using targeted avidin and fluorescein-biotin. Chem Biol. 2000;7:197–209. doi: 10.1016/s1074-5521(00)00088-0. [DOI] [PubMed] [Google Scholar]
- 42.van Meer G, Voelker DR, Feigenson GW. Membrane lipids: where they are and how they behave. Nat Rev Mol Cell Bio. 2008;9:112–124. doi: 10.1038/nrm2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Xu W, Zeng ZB, Jiang JH, Chang YT, Yuan L. Discerning the Chemistry in Individual Organelles with Small-Molecule Fluorescent Probes. Angew Chem Int Edit. 2016;55:13658–13699. doi: 10.1002/anie.201510721. [DOI] [PubMed] [Google Scholar]
- 44.Wu SQ, Song YL, Li Z, Wu ZS, Han JH, Han SF. Covalent labeling of mitochondria with a photostable fluorescent thiol-reactive rhodamine-based probe. Anal Methods-Uk. 2012;4:1699–1703. [Google Scholar]
- 45.Yasueda Y, Tamura T, Fujisawa A, Kuwata K, Tsukiji S, Kiyonaka S, Hamachi I. A Set of Organelle-Localizable Reactive Molecules for Mitochondrial Chemical Proteomics in Living Cells and Brain Tissues. J Am Chem Soc. 2016;138:7592–7602. doi: 10.1021/jacs.6b02254. [DOI] [PubMed] [Google Scholar]
- 46.Thompson AD, Omar MH, Rivera-Molina F, Xi Z, Koleske AJ, Toomre D, Schepartz A. Long-Term Live-Cell STED Nanoscopy of Primary and Cultured Cells with the Plasma Membrane HIDE Probe DiI-SiR. Angew Chem Int Edit advance online publication. 2017 doi: 10.1002/anie.201704783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lefebvre JL, Kostadinov D, Chen WSV, Maniatis T, Sanes JR. Protocadherins mediate dendritic self-avoidance in the mammalian nervous system. Nature. 2012;488:517–521. doi: 10.1038/nature11305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lefebvre JL, Sanes JR, Kay JN. Development of Dendritic Form and Function. Annu Rev Cell Dev Bio. 2015;31:741–777. doi: 10.1146/annurev-cellbio-100913-013020. [DOI] [PubMed] [Google Scholar]
- 49.Zipursky SL, Grueber WB. The Molecular Basis of Self-Avoidance. Annu Rev Neurosci. 2013;36:547–568. doi: 10.1146/annurev-neuro-062111-150414. [DOI] [PubMed] [Google Scholar]
- 50.Grimm JB, English BP, Chen JJ, Slaughter JP, Zhang ZJ, Revyakin A, Patel R, Macklin JJ, Normanno D, Singer RH, Lionnet T, Lavis LD. A general method to improve fluorophores for live-cell and single-molecule microscopy. Nat Methods. 2015;12:244–249. doi: 10.1038/nmeth.3256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Butkevich AN, Mitronova GY, Sidenstein SC, Klocke JL, Kamin D, Meineke DN, D’Este E, Kraemer PT, Danzl JG, Belov VN, Hell SW. Fluorescent Rhodamines and Fluorogenic Carbopyronines for Super-Resolution STED Microscopy in Living Cells. Angew Chem Int Ed Engl. 2016;55:3290–3294. doi: 10.1002/anie.201511018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Butkevich AN, Belov VN, Kolmakov K, Sokolov VV, Shojaei H, Sidenstein SC, Kamin D, Matthias J, Vlijm R, Engelhardt J, Hell SW. Hydroxylated Fluorescent Dyes for Live-Cell Labeling: Synthesis, Spectra and Super-Resolution STED. Chem Eur J. 2017:1–7. doi: 10.1002/chem.201701216. [DOI] [PMC free article] [PubMed] [Google Scholar]