

Abstract
As a result of the adaptation of life to an aerobic environment, nature has evolved a panoply of metalloproteins for oxidative metabolism and protection against reactive oxygen species. Despite the diverse structures and functions of these proteins, they share common mechanistic grounds. An open-shell transition metal like iron or copper is employed to interact with O2 and its derived intermediates such as hydrogen peroxide to afford a variety of metal–oxygen intermediates. These reactive intermediates, including metal-superoxo, -(hydro)peroxo, and high-valent metal–oxo species, are the basis for the various biological functions of O2-utilizing metalloproteins. Collectively, these processes are called oxygen activation. Much of our understanding of the reactivity of these reactive intermediates has come from the study of heme-containing proteins and related metalloporphyrin compounds. These studies not only have deepened our understanding of various functions of heme proteins, such as O2 storage and transport, degradation of reactive oxygen species, redox signaling, and biological oxygenation, etc., but also have driven the development of bioinorganic chemistry and biomimetic catalysis. In this review, we survey the range of O2 activation processes mediated by heme proteins and model compounds with a focus on recent progress in the characterization and reactivity of important iron–oxygen intermediates. Representative reactions initiated by these reactive intermediates as well as some context from prior decades will also be presented. We will discuss the fundamental mechanistic features of these transformations and delineate the underlying structural and electronic factors that contribute to the spectrum of reactivities that has been observed in nature as well as those that have been invented using these paradigms. Given the recent developments in biocatalysis for non-natural chemistries and the renaissance of radical chemistry in organic synthesis, we envision that new enzymatic and synthetic transformations will emerge based on the radical processes mediated by metalloproteins and their synthetic analogs.
1. Introduction
The highly exergonic four-electron reduction of oxygen to water has an enthalpy change of −80 kcal/mol. The biochemical processes that mediate this reduction provide the energy that sustains all aerobic organisms on earth.1 Paradoxically, however, the utilization of O2-derived energy in aerobic biology represents a significant challenge due to the thermodynamic stability of O2 in its triplet ground state.1−5 The first one-electron reduction of O2 to superoxide ion is endergonic by 7.8 kcal/mol, which makes the stepwise, one-electron processes unfavorable (Figure 1). Direct 2e– reactions between ground-state O2 (S = 1) and many closed-shell organic substrates are spin forbidden unless a stepwise route via diradical intermediates is available. To overcome these difficulties of exploiting O2, nature has evolved families of enzymes to transport O2 and to catalyze, manipulate, and control its reduction. A majority of these enzymes contain transition metals such as iron, copper, and sometimes manganese with unpaired d-electrons.6 These open-shell systems with variable oxidation and electronic states can facilitate the activation of triplet ground-state oxygen and participate at different stages of O2 reduction (Figure 1). The study of these processes, known collectively as oxygen activation, is a central theme in biology and has driven the development of metallobiochemistry and bioinorganic chemistry since their inception.
Figure 1.

Reduction sequence of O2 and representative metalloenzymes related to O2 activation. Reduction potentials shown are versus normal hydrogen electrode (NHE).5
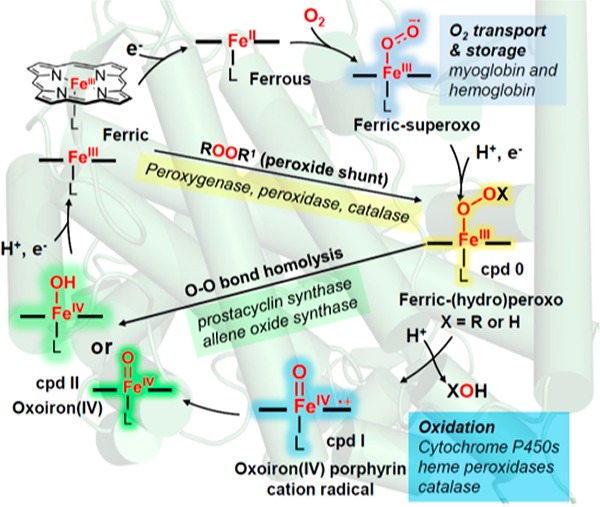
Heme-containing proteins have assumed a key position in our understanding of O2-activation mediated by metalloproteins. The general catalytic cycle for oxygen reduction is shown in Figure 2, which starts with the binding of O2 to a ferrous heme to afford a ferric-superoxo adduct, followed by reduction to ferric–hydroperoxo, and high-valent, iron–oxo (ferryl) intermediates via O–O bond scission. The rich chemistry that occurs between the heme iron and dioxygen provides the basis for the diverse roles of heme proteins in O2 metabolism, including O2 transport (myoglobin and hemoglobin), reduction of O2 to water (cytochrome c oxidases), degradation of reactive oxygen species (catalases and peroxidases), and catalysis of a variety of oxygenation reactions of organic substrates such as steroid hydroxylation, a monooxygenation typical of cytochrome P450s, and dioxygenation reactions (heme dioxygenases).7 The use of metal-cofactors to effect serial reductive activation of O2 is not limited to heme proteins but represents a general strategy employed by nature to harness the oxidation power of dioxygen. The reaction sequence and reactive intermediates similar to that shown in Figure 2a are widely present in a variety of O2-activating metalloenzymes such as nonheme iron oxygenases and copper-containing oxygenases and oxidases (Figure 2b).6,8−11 Accordingly, in spite of their diverse structures and functions, these metalloproteins share many common mechanistic grounds in terms of O2 activation and catalysis.1,6,12
Figure 2.

(a) Interaction of dioxygen with heme and heme–oxygen intermediates in heme proteins. (b) Representative metal–oxygen intermediates in metalloproteins (PHM: peptidylglycine α-hydroxylating monooxygenase).
In this interpretive review, we will survey the range of O2 activation processes mediated by heme proteins and related model compounds with a focus on recent progress in the characterization and reactivity of important iron–oxygen intermediates through mid-2017. Key aspects of background material in the field will also be presented to provide context and perspective. The underlying structural and electronic factors contributing to O2 activation chemistry will be delineated. The topics of O2-activation and oxidative transformations mediated by heme proteins and metalloporphyrins have been extensively reviewed previously.3,4,7,13−32 While most of these reviews focus on either heme proteins or synthetic models, this review will provide a comprehensive perspective of the spectrum of oxidative transformations mediated by the iron–oxygen species in both heme proteins and analogous synthetic model compounds. We will discuss the fundamental mechanistic features of these transformations and how a variety of oxidative pathways are developed from a common mechanistic foundation. For model compounds, we will mainly focus on metalloporphyrins. Other porphyrinoid ligands, such as corrolazines, porphyrazines, and phthalocyanines will not be discussed in detail. Readers can refer to several recent reviews on these families of model compounds.29,33−35
2. Key Intermediates Involved in O2 Activation by Heme Proteins and Model Compounds
2.1. Oxy–Ferrous (Superoxoferric) Intermediates
2.1.1. Structural Features
The first step of O2 activation by heme proteins is the binding of dioxygen to the heme iron, forming an oxy–ferrous intermediate (or ferric-superoxo intermediate, see discussions in section 2.1.2). Heme proteins exhibit varied O2 binding features. For instance, O2 transport proteins like hemoglobin and myoglobin have very high O2 association rate constants (107 M–1 s–1) and moderate to high dissociation rate constants (0.1–20 s–1).36,37 Heme-containing NO or CO sensor proteins are tuned to coordinate these tighter-binding molecules and show little binding of O2 even in O2-saturated solutions.38,39 In P450s, O2 binds to ferrous heme with rate constants around 106 M–1 s–1,40 and the resulting ferric-superoxo intermediate is prone to undergo autoxidation to the ferric state if it is not further reduced to form the ferric hydroperoxo compound 0.41 It is important to note that heme in the ferric state has a very weak oxygen-affinity and will not bind oxygen at ambient temperatures. It is the ease of electron transfer from iron to O2 that is important for the formation of the ferric-superoxo intermediate and thus O2 binding. For instance, highly electron-withdrawing Fe(II) porphyrin such as Fe(TPFPBr8) is very inert toward dioxygen (TPFPBr8 = β-pyrrole brominated 5,10,15,20-tetrakis(pentafluorophenyl)porphyrin).42
The structural and electronic properties of the ferric-superoxo intermediate are crucial for understanding the O2 activation in heme proteins. In the 1930s, Pauling and Coryell found that oxygenated hemoglobin (oxy-Hb) was diamagnetic and proposed that dioxygen was bound to heme in a bent, end-on mode.43,44 This geometric feature was confirmed by an oxy–ferrous model compound synthesized by Collman and by crystal structures of oxy-Mb and oxy-Hb solved by Phillips and Shaanan, respectively.45−47 Following these pioneering efforts, crystal structures of ferric-superoxo intermediates have been solved for a number of heme proteins (Figure 3).48−52 All reported structures reveal an end-on coordination of dioxygen to the iron with a Fe–O bond distance around 1.8 Å, an O–O bond distance ranging from 1.22 to 1.30 Å, and an Fe–O–O angle ranging from 110° to 160°.
Figure 3.

Structural parameters of ferric-superoxo intermediates of representative heme proteins (oxy-myoglobin, PDB: 1A6M; HRP compound III, PDB: 1H57; P450cam oxygen complex, PDB: 1DZ8). aData presented are for the β subunit of oxyhemoglobin in structure 1HHO.
2.1.2. Nature of Fe–O2 Bonding
The electronic structure of Fe–O2 heme complexes is remarkable because it forms a species with a singlet electronic ground state arising from the combination of high-spin (S = 2) ferrous heme and triplet O2. The nature of Fe–O2 bonding has been a subject of active debate for decades.53 There are three models to explain Fe–O2 bonding (Figure 4).54 In Pauling’s model, an S = 0 state was assigned to both a dioxygen and a ferrous center.43,44 In contrast, Weiss proposed that the Fe–O2 species would be best described as a ferric-superoxide complex in which a low-spin (S = 1/2) ferric center is antiferromagnetically coupled to a doublet superoxide anion O2–.55 In a third model initially suggested by McClure and refined by Goddard, an intermediate-spin (S = 1) ferrous center antiferromagnetically coupled to 3O2 in a 3-center-4-electron bond affords the singlet Fe–O2 species (“Ozone model”).56,57
Figure 4.
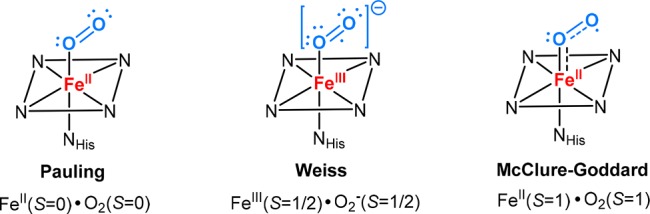
Three models for Fe–O2 bonding in the ferric-superoxo intermediate. Adapted from ref (64). Copyright 2013 American Chemical Society.
Mössbauer studies have lent support to the Weiss model. The Mössbauer parameters of oxyhemoglobin (isomer shift δ = 0.2–0.3 mm/s, quadrupole splitting ΔEQ = 2.00–2.20 mm/s), first measured by Lang and Marshali, are consistent with a low-spin ferric center.58 Sharrock and co-workers have reported similar parameters for oxygenated reduced P450cam (δ = 0.31 mm/s and ΔEQ = 2.15 mm/s at 4.2K).59 The Weiss model is further supported by resonance Raman spectroscopic studies, in which a ferric center was indicated by the oxidation state marker band of oxyHb (ν = 1377 cm–1).60−62
Through density functional theory (DFT) and valence bond (VB) approaches, Shaik has shown that Fe–O2 contains a dative σ(Fe–O) interaction between vacant 3dz2 and π*(O2–) and a rather weak π interaction between dyz(Fe) and π⊥*(O2).53 The H-bonding from a distal histidine residue (His64) in oxy-Mb and the electrostatic polarization from protein backbone stabilizes the negative charges on O2. Although Fe–O2 primarily follows the bonding scheme of the Weiss model, the VB analysis showed a mixing of the other two models, whose contributions depend on protein hosts and active site environments, making Fe–O2 difficult to describe according to canonical oxidation state formalisms. Recent Fe K-edge X-ray absorption spectroscopy (XAS) studies by Sarangi et al. have shown that oxy-Hb is best described as a ferric superoxide species (Weiss model) in solution. However, in crystalline oxy-Hb, the Fe K pre-edge feature indicates Fe2+S = 0 character, consistent with the Pauling model. This surprising change of electronic configuration with environment supports the multiconfigurational description.63 The impact of local environment on the Fe–O2 bonding was also revealed by an iron L-edge XAS study of a ferric-superoxo “picket fence” porphyrin with 1-methylimidazole (1-MeIm) axial ligand, Fe(TpivPP)(1-MeIm)O2 (TpivPP = meso-tetra(α,α,α,α–O-pivalamidophenyl)porphyrin, Figure 5b).64 The results showed a strong π-interaction between dyz(Fe) and π⊥*(O2) as opposed to the weak π-interaction calculated for oxy-Mb. This difference is likely caused by the much weaker hydrogen bonding to peroxo distal oxygen (N···O distance ∼3.9 Å compared to 2.67 Å in oxy-myoglobin) and the lack of active site polarization in this model compound.
Figure 5.

(a) Structure of Im-pyrroheme and Py-mesoheme and their binding affinities to O2 and CO. (b) Structural parameters of Fe(TpivPP)(2-MeHIm)(O2) are from ref (67).
2.1.3. Metal-Dioxygen Intermediates of Metalloporphyrins
Studies of biomimetic synthetic iron porphyrins have greatly aided in the understanding of O2 binding and activation in heme proteins. An early study by Traylor et al. showed that the O2 affinity of imidazole-ligated iron(II) porphyrin (1, Figure 5a) was over 3800 times stronger than that of a pyridine-ligated one (2, Figure 5a), whereas both complexes showed similar CO binding affinities.65 This difference was thought to originate from the large π basicity of imidazole, which could better stabilize the weak O2 ligand. Much progress has been made with sterically protected porphyrin scaffolds like “picket–fence” porphyrins, which prevent the irreversible bimolecular condensation of Fe(II) porphyrins to form μ-oxo-bridged diiron(III) complexes upon reacting with O2. This strategy was first implemented by Collman and led to the characterization of the first Fe–O2 porphyrin compound in the 1970s.45,66 This model binds O2 reversibly with affinity similar to those of Hb and Mb. More importantly, the Fe–O2 intermediate prepared by Collman is diamagnetic and showed a bent end-on binding geometry of dioxygen just as postulated by Pauling. With a similar approach, many synthetic Fe–O2 complexes have been synthesized and characterized with varied O2 binding affinities tuned by ligand environments.22,24 Very recently, the Scheidt and Schulz groups have used multitemperature X-ray crystallography and Mössbauer spectroscopy to study three ferric-superoxo complexes based on “picket fence” porphyrin TpivPP with 1-methyl-, 1-ethyl-, or 2-methylimidazoles as axial ligand, respectively.67 Their results showed a low energy for the rotation of the Fe–O2 unit. An 80K structure of Fe(TpivPP)(O2) (3, Figure 5b) with 2-methylimidazole (2-MeHIm) axial ligand allowed them to accurately determine the position of both oxygen atoms, whose structural details were vailed in previous studies because of the high thermal motion and O2 disorder. They obtained a Fe–O distance of 1.811(5) Å, O–O bond lengths of 1.281(12) Å, Fe–O–O angle of 118.2°, and an off-axis tilt of Fe–O bond of 6.2°.
Heme-containing metal–organic frameworks (MOFs) have recently been used as a platform to study the Fe–O2 adduct because of the spatial separation of heme centers in the MOF scaffold and the gas-absorption properties of MOFs. This approach allows for solvent–free generation of ferric-superoxo intermediates. A rare 5-coordinate heme–O2 complex could be obtained by reacting a Zr-based porphyrinic MOF PCN-224FeII (PCN stands for porous coordination network) with O2 at −78 °C.68 The species revealed a characteristic η1 end-on binding mode of O2 with a Fe–O distance of 1.79(1) Å, O–O distance of 1.15(4), and an Fe–O–O angle of 118(4)o. PCN-224FeII showed a low O2-binding enthalpy of −8 kcal/mol, about half of those of other heme model compounds with imidazole axial ligands. This weak O2 affinity suggests the importance of electron-donation from axial ligands (σ-donation in the case of imidazole) for the formation of Fe–O2 complexes.
Transition-metals other than iron have also been explored for O2 binding in both synthetic metalloporphyrins and heme proteins. A great number of studies have focused on cobalt dioxygen complexes, which have a doublet ground state (S = 1/2) and thus can be probed by EPR spectroscopy.69−73 Co–O2 is best described as a Co(III) superoxo species with the unpaired electron largely on the O2 moiety. Co–O2 also adopts a η1 end-on binding mode as revealed by the structures of both Co-Mb and model compounds (Figure 6).71,74 CoII–O2 binds dioxygen reversibly but with much lower affinity compared to their iron analogs (P1/2 around 60 Torr vs 0.49–1.1 Torr for iron).72 As in iron porphyrins, axial ligands are important for Co–O2 binding. A Co–O2 intermediate without an axial ligand was recently synthesized in a MOF scaffold PCN-224, which showed a much lower O2-binding enthalpy (−3.6 kcal/mol) compared to that of model compound with an imidazole ligand (−13.3 kcal/mol).75 In contrast, cobalt(II) octaethylporphyrin supported on a highly oriented pyrolytic graphite showed a favorable O2 binding (ΔHo = −16.7 ± 3.6 kcal/mol), owing to the partial electron donation from the graphite substrate.76
Figure 6.

Bonding and electronic properties of various metal-dioxygen porphyrin complexes.
In addition to iron and cobalt, manganese, chromium, and ruthenium dioxygen adducts have also been investigated in porphyrinoid scaffolds.24 We will only briefly summarize their structural and electronic features (Figure 6). Dioxygen adducts of manganese(II) porphyrins have unique features compared to other metal-dioxygen species. Mn(II) binds oxygen with the loss of the axial ligand that usually stabilizes the metal–O2 moiety.77,78 Mn–O2 exhibits an intermediate spin (S = 3/2) and a large zero–field splitting (−2.4 cm–1).79−81 The 900 to 990 cm–1 IR stretching frequencies ν(O2) of Mn–O2 are much lower than those of end-on Fe–O2 and Co–O2 complexes (1100–1200 cm–1).82 These properties indicate that O2 binds to Mn(II) in a symmetric η2 side-on mode, a MnIV–O22– formalism. Oxy-Ru(II) porphyrins are diamagnetic and have O–O stretching frequency of 1103 cm–1, suggesting a low-spin Ru(III) superoxide formulation as described in the Weiss model of Fe–O2.83−85 For CrII–O2 porphyrins, a 1142 cm–1 O–O stretching frequency and a S = 1 ground state also indicates a Weiss model, in which a high-spin S = 3/2 Cr(III) and an S = 1/2 superoxide anion are antiferromagnetically coupled.79,86
2.2. Ferric-(hydro)peroxo Intermediates
2.2.1. Ferric-(hydro)peroxo Intermediates of Heme Proteins
The binding of dioxygen to ferrous heme facilitates its reductive activation. In many hemoproteins such as nitric oxide synthase and cytochrome P450s, an electron and a proton are transferred to the oxy–ferrous complex to afford a ferric–hydroperoxo intermediate [Fe(III)–O–OH], also known as compound 0 (cpd 0). Distinct from most heme proteins, cytochrome c oxidase (CcO) utilizes a copper cofactor complexed with active site residues, including an unusual His-Tyr cross-link, to facilitate the O2 activation. This arrangement leads to the formation of an analogous ferric-peroxo-cupric intermediate. O2 activation by CcO is discussed in detail in section 4.3 of this review.
Cpd 0 species have proven to be short-lived, posing significant challenges for their characterization. Cryogenic radiolytic reduction, as first demonstrated by Davydov and Symons,87−89 has proven to be very useful for the study of cpd 0.90 This method utilizes the hydrated electrons generated through radiolysis to reduce the ferrous-dioxygen intermediate in frozen solvent matrices. The combination of radiolytic cryoreduction with spectroscopic methods, especially EPR and ENDOR spectroscopies employed by Hoffman et al., have afforded critical insight into the electronic and structural properties of cpd 0.91 By controlling the cryogenic conditions, ferric-peroxo anion species [Fe(III)–O–O–] could be obtained and its protonation at the distal oxygen to form the conjugate acid [Fe(III)–O–O–H, cpd 0] upon thermal annealing could be monitored by EPR spectroscopy. It is now known that cpd 0 has a low-spin ferric, S = 1/2 ground state. The g1 value of cpd 0 was found to be indicative of the protonation state of the distal oxygen, with a g1 > 2.30 commonly observed for the ferric–hydroperoxo state.92 The thermal annealing experiments also showed that the ease of proton delivery varied widely across heme proteins. For instance, Mb ferric-peroxo is stable near 100 K, whereas the ferric-peroxo anion of P450cam is readily protonated above 55 K.93−95 This ease of protonation may be related to the presence of the proton relay channel in P450cam. Very recently, Kincaid et al. have characterized ferric-(hydro)peroxo intermediates of lactoperoxidase (LPO) via cryoradiolysis/resonance Raman (rR) spectroscopy.96 Under alkaline conditions (pH 8.2), the ferric-peroxo intermediate is the major form for LPO and no protonation occurred via thermal annealing. Under acidic conditions (pH 5.6), however, a mixture of LPO-peroxo and LPO–hydroperoxo intermediates were formed. Intriguingly, upon thermal annealing to 170 K, LPO-peroxo did not convert to the hydroperoxo species, and its rR intensity decreased substantially, whereas LPO–hydroperoxo was more stable with peak intensities almost unchanged. Radiolytic cryoreduction has also enabled the structural characterization of the fleeting ferric-(hydro)peroxo species with X-ray crystallography. In 2007, Schlichting et al. reported a crystal structure of compound 0 by controlled radiolytic reduction of ferrous-dioxygen intermediate of chloroperoxidase (CPO).97 Cpd 0 of CPO showed an O–O distance of 1.5 Å, a long Fe–O distance of 1.9 Å, and an Fe–O–O angle of 131° (Figure 7a). Soon afterward, Unno and co-workers reported the characterization of a ferric peroxoanion compound of myoglobin, which showed a Fe–O distance of 1.85 Å, Fe–O–O angle of 120°, and a short O–O distance of 1.33 Å (Figure 7b), which distinguishes it from an O-protonated hydroperoxo state.98 Peroxymyoglobin was also characterized by Andersson et al. in 2008 and similar structural features were reported.99
Figure 7.
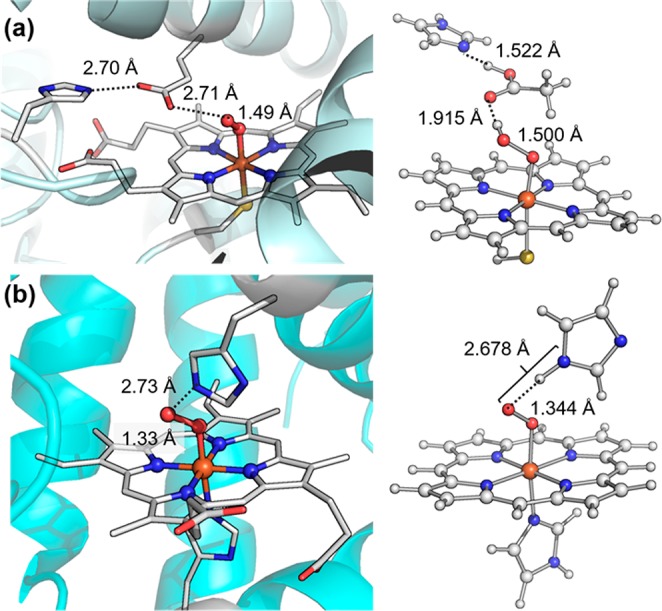
(a) Crystal structure of CPO-0 (PDB: 2J5M) (left) and the QM/MM optimized structure of doublet CPO-0 (generated with molecular coordinates provided in ref (97)). (b) Crystal structure of the ferric-peroxo intermediate of Mb (PDB: 2Z6T) and the corresponding QM/MM optimized structure (doublet ground state) (generated with molecular coordinates provided in ref (98)).
2.2.2. Ferric-(hydro)peroxo Intermediates of Metalloporphyrins
In parallel with the progress with heme proteins, synthesis and characterization of cpd 0 analogs in model compounds have also been actively explored. Valentine and co-workers reported the first preparation of a ferric porphyrin peroxide complex Fe(TPP)O2– (6, Figure 8) by treating FeIII(TPP)Cl (4) with 2 equiv of KO2 or FeII(TPP) (5) with 1 equiv of KO2 in aprotic solvents.100,101 This species exhibited a characteristic EPR spectrum of a rhombic high-spin Fe(III) (S = 5/2) complex with a broad and intense resonance at g ∼ 4.2, which is in contrast to the low-spin S = 1/2 state observed for typical cpd 0 species (g values ∼2). Similar complexes can also be formed by 1e– electrochemical reduction of ferrous dioxygen intermediates.102 The infrared O–O stretching vibration of 6 at 806 cm–1 and the lack of an axial basic ligand suggested that this complex is a side-on η2-peroxo ferric species.101 Several η1 end-on ferric–hydroperoxo models were synthesized by Tajima et al. via addition of H2O2 to ferric porphyrins at low temperatures and subsequently characterized by UV–vis and EPR spectroscopies at 77 K.103−105 The EPR spectra of these complexes showed a low-spin ferric intermediate with g values ∼2, similar to those observed for cpd 0 in heme proteins. In Tajima’s conditions to prepare η1 end-on ferric-peroxo complexes (11), methanol was used as a cosolvent, and hydroxide or imidazole was added as the axial ligand (Figure 8b), in contrast to the conditions of preparing the side-on η2-peroxo ferric species 10, in which an aprotic-solvent was used and no axial ligand was present. Recently, the impact of axial ligands and solvent on ferric-peroxo bonding mode has been extensively studied by Naruta et al. They successfully synthesized both ferric–peroxo and ferric–hydroperoxo complexes of FeII(TMPIm), an iron porphyrin complex bearing a covalently linked imidazole axial ligand (9, Figure 8).106,107 The ferric-peroxo complex 10 was characterized to be a rare seven-coordinate, side-on ferric-peroxo species with axial association of the covalently linked imidazole. An EPR signal for 10 at g ∼ 4.2 and an rR O–O stretching vibration at 807 cm–1, which is characteristic for a high-spin η2-peroxo–ferric heme species, supports this formulation. Addition of methanol to 10 led to the formation of a low spin end-on hydroperoxoferric complex, 12, as judged by changes in the EPR spectra from g ∼ 4.2 to a new set of signals at g = 2.31, 2.19, and 1.95. The identity of 12 was further confirmed by a 4 cm–1 upshift of ν(O–O) in MeOD solvent. The Mössbauer spectrum of 12 showed an isomer shift of 0.25 mm/s and a quadrupole splitting of 2.16 mm/s, which is in agreement with those of the previously characterized low-spin hydroperoxo–heme species.108
Figure 8.
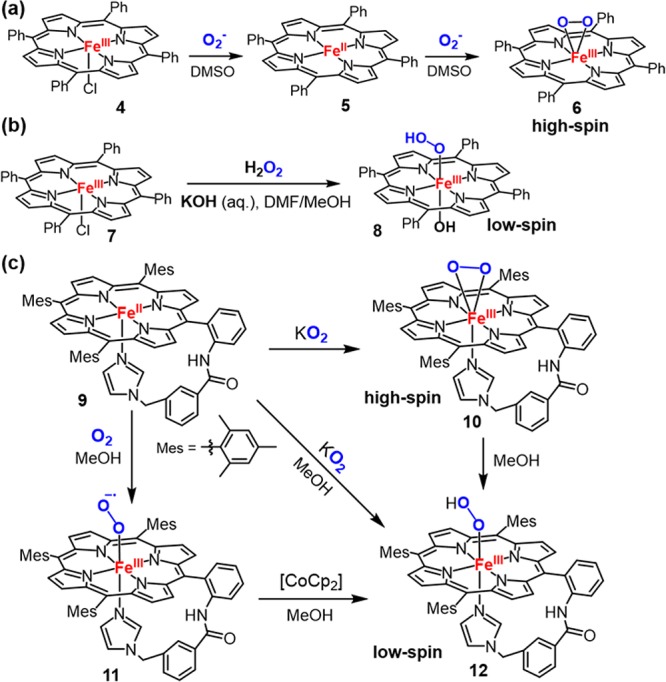
(a) Formation of a high-spin side-on η2-peroxo ferric intermediate (6). (b) Formation of low-spin end-on η1-hydroperoxo ferric intermediate (8). (c) Conversion of a high-spin side-on η2-peroxo ferric intermediate (10) into low-spin end-on η1-hydroperoxo ferric intermediate (12). Adapted with permission from ref (106). Copyright 2009 Wiley-VCH.
Addition of methanol to a 6-coordinated ferric-peroxo complex [(TMP)FeIII(O22–)] (13, Figure 9), however, led to a direct decomposition of 13. This striking difference highlights the importance of the imidazole axial ligand in stabilizing the ferric–hydroperoxo species. Soon afterward, the synthesis of an end-on ferric-peroxo anion intermediate was successfully achieved by the same group with a TMPIm ligand containing a bulky xanthene substituent hanging over the porphyrin (14, Figure 9).109 The EPR spectrum of 14 is characteristic of a low-spin ferric complex with g values of 2.27, 2.16, and 1.96, consistent with those of end-on ferric-peroxo intermediates in heme proteins prepared via radiolytic reduction. Addition of methanol to this species made a new low-spin ferric complex with g values of 2.32, 2.19, and 1.95 and ν(O–O) of 807 cm–1. The small increase in the spread of g values and a g1 > 2.3 are indicative of an end-on ferric–hydroperoxo complex.
Figure 9.
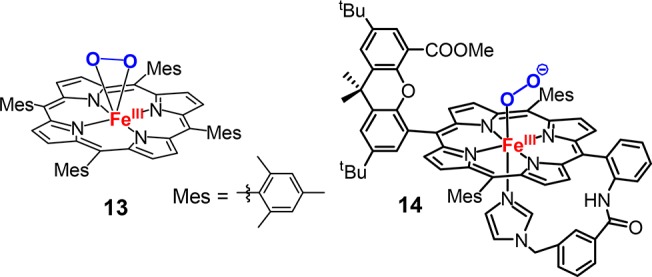
Chemical structure of [(TMP)FeIII(O22–)] (13) and an end-on ferric-peroxo anion species containing a bulky TMPIm ligand (14).
The effect of the second coordination sphere on the formation of ferric hydroperoxo intermediates was recently explored by the same group with an iron(II) porphyrin containing both a covalently appended proton donor and an axial imidazole ligand (15, Figure 10).110 This complex can bind oxygen to afford the ferric-superoxo intermediate 17. Intriguingly, 17 can be reduced by 15 to afford a FeIII-hydroperoxo intermediate 18 presumably via a PCET pathway. This process would be inhibited by converting the carboxylic acid moiety on xanthene to an ester. The corresponding iron(II) porphyrin 16 can still form a ferric–hydroperoxo intermediate 21 upon reacting with cobaltocene (CoCp2) via a PCET pathway. Compared to 21, 18 showed a higher ν(Fe–O) (579 cm–1 vs 576 cm–1) and a lower ν(O–O) (807 cm–1 vs 811 cm–1) compared to 21, indicating that the intramolecular H-bond interaction enhances the Fe–O but weakens the O–O bonding.
Figure 10.
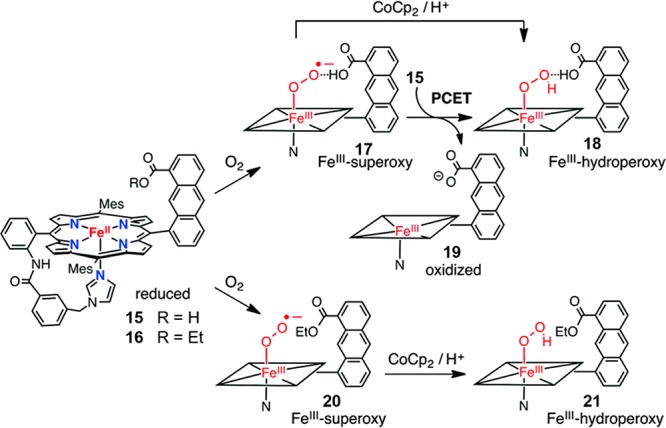
Impact of the second coordination sphere on the formation of a ferric-(hydro)peroxo intermediate. Adapted with permission from ref (110). Copyright 2016 Royal Society of Chemistry.
Ferric hydroperoxo intermediates have also been investigated using electrochemical methods.102,110−114 The use of electrodes modified with iron porphyrins was used to probe for reactive iron porphyrin intermediates under electrocatalytic conditions.111,114 The electrochemical methods also represent a potentially “cleaner” way to generate iron porphyrin intermediates by circumventing the need of reducing agents. A recent example is the preparation of [FeIII(TPFPP)(O2)]− and [(MeIm)FeIII(TPFPP)(OOH)] intermediates via electrochemical reduction of the corresponding ferric-superoxo intermediate [FeIII(TPFPP)(O2•-)] (TPFPP = 5,10,15,20-tetrakis(pentafluorophenyl)porphyrin).112
2.3. Heterolytic O–O Bond Cleavage of Ferric–Hydroperoxo Intermediates
2.3.1. O–O Bond Cleavage of cpd 0 to Generate Cpd I in Heme Proteins
The main role of the ferric–hydroperoxo species (cpd 0) in O2 activation is to generate the key oxoiron(IV) porphyrin cation radical intermediate, which is the canonical compound I (cpd I). Historically, the terms compound I and compound II (the one-electron-reduction product of cpd I) stem from very early spectroscopic studies of the reaction of horseradish peroxidase (HRP) with hydrogen peroxide. In 1937, using an ocular spectroscope, Keilin and Mann observed a red intermediate with a Soret band near 420 nm upon addition of H2O2 to HRP.115 Four year later, Theorell identified a more short-lived green intermediate formed initially in the reaction between HRP and H2O2 (thus termed “cpd I”),116 which then converted into the red species (termed “cpd II”) observed by Keilin and Mann. Similar intermediates were also observed by Stern and Chance for catalase.117−119 Jones and Dunford suggested the presence of an HRP-H2O2 precursor complex in preequilibrium with HRP and H2O2 (termed “cpd 0”) formed prior to the cpd I generation based on exacting kinetic measurements of this reaction.120−123
It is now known that cpd 0 is an Fe(III)–OOH intermediate and is generated via H2O2 binding to the resting ferric heme. The subsequent cpd I formation involves the protonation of Fe(III)–OOH (cpd 0) at the distal oxygen and the subsequent heterolytic scission of the O–O bond to release water. Cpd 0 generation and the ensuing O–O bond cleavage in peroxidases are facilitated by residues at distal pocket acting as acid–base catalysts, as proposed first by Poulos and Kraut.124 This proposal originated from their studies of cytochrome c peroxidase (CcP), in which a highly conserved histidine/arginine (His52/Arg48) diad in the distal pocket was found to be crucial for cpd I generation.124,125 It has been suggested that upon binding of H2O2 to the ferric heme, His52 acts as a general base catalyst to deprotonate the proximal oxygen to form cpd 0 and then act as a general acid catalyst to protonate the distal oxygen of cpd 0, whereas Arg48 stabilized the leaving oxygen through hydrogen bonding. Recently, this mechanistic picture was further elaborated by Raven and Moody using neutron cryo-crystallography to visualize the crucial protons (deuterons) at the active site.126 Surprisingly, their results showed that His52 remained protonated after the formation of CcP cpd I, suggesting that a second proton was delivered to cpd 0, presumably by proton relay through the H-bonded water network, for the O–O bond heterolysis of [Fe(III)–O–OH] (Figure 11).127 In addition to the regulation of proton delivery to the peroxo distal oxygen to create a better leaving group (the “pull” effect), the O–O bond cleavage is further facilitated by the σ-donation (electron push) from an axial histidine ligand by providing electron density to the antibonding O–O orbital.128 The low-spin electronic configuration of Fe–OOH is thought to facilitate O–O bond cleavage via interaction of the filled dxz orbital on iron with the low-lying σ*(O–O) orbital.129 This “push-pull” effect is a general mechanistic feature for the O–O bond to generate cpd I in heme proteins.128,130
Figure 11.

Cpd I generation in cytochrome c peroxidase (CcP), PDB: 4CVJ.
The presence of residues acting as acid–base catalysts is common in heme proteins that utilize H2O2 to generate compound I. For instance, the histidine/asparagine residue pair found in catalases functions similarly to the His/Arg pair in peroxidases to promote compound I generation,131 while chloroperoxidase (CPO) and unspecific peroxygenases (UPO, also sometimes termed APO) employ a Glu/His or Glu/Arg acid–base pair. In CPO, as well as the UPO from M. rotula, glutamate hydrogen bonds to a histidine that stabilizes the negative charge built on the glutamate during O–O bond cleavage (Figure 12a),132,133 whereas in UPO from A. aegerita, glutamate hydrogen bonds to an arginine to achieve this stabilization effect (Figure 12c).133,134 CYP152, a family of hydrogen peroxide-utilizing P450 peroxygenases, lack the key histidine or glutamate residue to promote O–O cleavage. Instead, these proteins utilize the carboxyl group from a fatty acid substrate as an acid–base catalyst to rearrange the protons of H2O2 and aid the compound I generation (Figure 12b).135,136 An alternative pathway for cpd I generation involves a O–O homolytic cleavage of the ferric-H2O2 adduct followed by a hydrogen-atom abstraction (HAT) from the Fe(IV)–OH intermediate by the incipient hydroxyl radical.137−139 Recent computational studies of P450SPα (CYP152B1) by Shaik et al. indicated that the carboxyl group of the fatty acid substrate keeps the HO· radical toward the H–O bond of the Fe(IV)–OH intermediate and facilitates the HAT and cpd I generation (dashed bracket in Figure 12b). Inspired by the role of substrate carboxyl group in CYP152, Watanabe et al. have used short-alkyl-chain fatty acids as cocatalysts and decoy molecules to expand the substrate scope of CYP152 to include a variety of substrates that lack the carboxyl acid groups.140,141 In this case, short-alkyl-chain fatty acids served as the proton donor required for O–O bond activation.
Figure 12.

Proposed mechanism for generation of cpd I from cpd 0. The following structures are used: (a) Cpd 0 of CPO (CPO-0) from Leptoxyphium fumago (PDB: 2J5M) and the active site structure of MroUPO (PDB: 5FUJ); (b) P450BSβ from Bacillus subtilis complexed with a fatty acid substrate (PDB: 1IZO); (c) crystal structure of peroxygenase from Agrocybe aegerita (PDB: 2YP1); and (d) oxygen complex of P450cam from Pseudomonas putida (PDB: 1DZ8).
Unlike peroxidases, most P450s generate their compound 0 through reduction of iron-bound dioxygen, initially generating a ferric-peroxo anion species. Therefore, two protons are needed to produce cpd I, one for protonating the distal oxygen to generate cpd 0 and the other for facilitating O–O bond cleavage to generate water.25 As revealed by the crystal structure of P450cam, these protons are delivered through a hydrogen bonding network of a water channel involving the key residue Thr252 that hydrogen bonds to the distal oxygen and forms an extended hydrogen bonding network with water molecules in the active site (Figure 12d).49,50 This threonine is highly conserved in most P450 enzymes.142 Mutation of this site to alanine does not affect the formation of cpd 0 but diminishes the hydroxylation reactivity, affording H2O2 and the ferric heme, a so-called uncoupling process.21,94
The conversion of O2-dependent P450s into peroxygenases that can utilize hydrogen peroxide as a cosubstrate and terminal oxidant has generated considerable current interest. The low cost and high step-economy of using H2O2 as well as the increased process efficiency derived from not having to recycle reducing cofactors (e.g., NAD(P)H) make such peroxygenase-P450s highly attractive as biocatalysts.140 Protein engineering techniques such as directed evolution have been extensively used to obtain variants that can efficiently utilize H2O2 for monooxygenation reactions.143,144 The mutation of a conserved phenylalanine (F87 in P450BM3, Figure 13) within a substrate recognition site to a smaller residue (alanine, glycine, or valine) has been shown to be crucial for the introduction of peroxygenation activity into O2-dependent P450s.144,145 Recently, Watanabe et al. found that the mutation of the highly conserved threonine (T268 in P450BM3, Figure 13) into glutamic acid substantially enhanced the peroxygenase activity of a number of P450s, including P450BM3, P450cam, and CYP119.146 The glutamic acid mutation is thought to mimic the role of the substrate carboxyl group in CYP152 for O–O bond activation.
Figure 13.
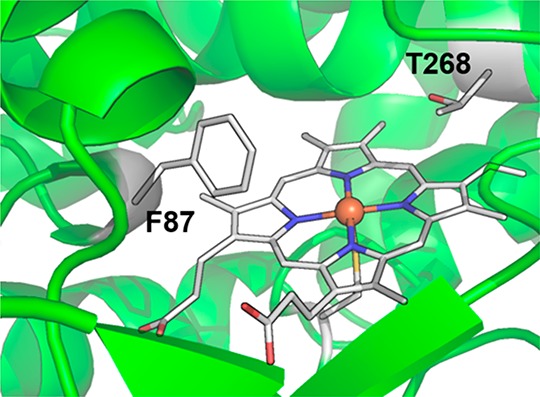
Key mutation sites (F87 and T268) for the induction of peroxygenase activity in P450BM3 (PDB: 1BVY).
In addition to O–O bond heterolytic cleavage of iron-peroxo intermediates to generate cpd I, some heme proteins can interact with alkyl hydroperoxides to form alkoxyl radicals and ferryl intermediates (cpd II). A representative example is allene oxide synthase, which converts fatty acid hydroperoxides into allene oxides. This reaction is discussed in detail in section 4.1.
2.3.2. O–O Bond Cleavage in Metal-Peroxo Porphyrin Complexes
Metal-mediated O–O bond cleavage was extensively explored in model metalloporphyrin compounds.15 In particular, acylperoxy-iron(III) porphyrins have afforded many insights to the mechanistic details of this step.147−151 It has been shown that the addition of m-chloroperbenzoic acid (mCPBA) to the solution of HO–FeIII(TMP) (22, Figure 14a) led to the formation of acylperoxy-iron(III) complex 23. This species undergoes facile acid-catalyzed O–O bond heterolysis to afford cpd I 24 with a first-order rate constant (khetero) of 6.1 × 10–3 s–1 (−48 °C) and an Ea = 4 ± 0.4 kcal/mol, ΔH‡ = 3.6 ± 0.4 kcal/mol, and ΔS‡ > −25 cal mol–1 K–1.149,151 In nonpolar solvents and the absence of acids, however, O–O bond homolysis occurred to afford an unusual iron(III) porphyrin N-oxide species, 26, and a diacylperoxide as products, whereas a ferryl species, 25, was formed for alkyl acylperoxy-iron(III) complexes due to more rapid decarboxylation of the alkyl acyl radicals. The impact of the porphyrin meso-substituents and axial ligands on O–O bond cleavage have been investigated by Watanabe et al.152 They found that both electron-donating meso-substituents and imidazole axial ligands enhanced the O–O heterolytic cleavage (Figure 14b). For instance, the khetero of the most electron-donating [(mCPB)FeIII(TTMPP)] was found to be ∼55–fold faster than that of (mCPB)FeIII(TDMPP) (mCPB = m-chloroperbenzoate). The presence of 1-methylimidazole (1-MeIm) axial ligand increased khetero by over 50-fold for [(mCPB)FeIII(TDMPP)]. The effects of axial ligands were also investigated for O–O bond homolysis in toluene, in which imidazole ligation showed a much less rate enhancement (2.5-fold for 1-MeIm).
Figure 14.

(a) O–O bond heterolysis and homolysis of acylperoxy-iron(III) porphyrins. (b) Effect of axial ligands and meso-substituents on O–O bond heterolytic cleavage of acylperoxy-iron(III) porphyrins.
More recently, acylperoxy-iron(III) porphyrin intermediates have been further studied with different porphyrin ligand scaffolds and axial ligands under both stoichiometric and catalytic conditions.14,153−157 It was found that the rates of oxygen atom transfer reactions mediated by acylperoxy-iron(III) porphyrins were orders of magnitude slower than those of corresponding cpd I species,153,156−158 demonstrating that cpd I is the active intermediate for the oxygenation reactivity. Woggon and van Eldik et al. synthesized an iron(III) porphyrin with covalently appended sulfonate axial ligand (27, Figure 15).153 Upon reacting with mCPBA, this complex generated cpd I with khetero of 2.4 ± 0.1 s–1 (238 K). The faster khetero determined in this case was attributed to the high polarity of acetonitrile solvent. Nocera et al. studied the impact of the secondary coordinating environment on O–O bond cleavage with so-called “hangman” iron porphyrins containing either a carboxylic acid (HPX-CO2H) or a methyl ester (HPX-CO2Me) functional group, respectively (Figure 15).155 Both complexes afforded cpd I upon mCPBA oxidation in CH2Cl2 with similar rates, whereas in toluene, the O–O homolysis was inhibited for FeIII(HPX-CO2H), and only FeIII(HPX-CO2Me) afforded the O–O homolysis product porphyrin N-oxide. This result suggests that the presence of the acidic group facilitates the 2e– pathway for O–O bond cleavage in the same manner as peroxidases. The nature of the leaving group O–X within the peroxo unit [Fe–O–OX] also affected the preference between 2e– and 1e– pathways. Alkyl hydroperoxides such as tBuOOH and cumene hydroperoxide, which have weaker alkoxy leaving groups, were shown to follow a homolytic O–O bond scission pathway upon reacting with Fe(III) porphyrins.159,160
Figure 15.
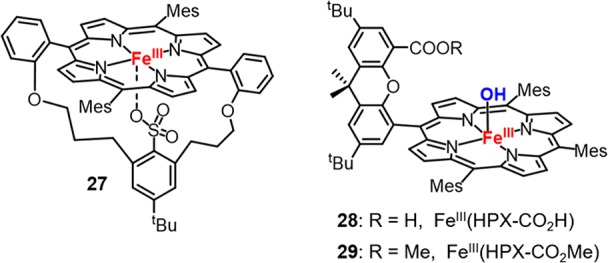
Chemical structures of compound 27, FeIII(HPX-CO2H) (28), and FeIII(HPX-CO2Me) (29).
The O–O bond cleavage of ferric–hydroperoxo intermediates are mainly studied indirectly in oxidation reactions.148,150,161−163 It has been shown that iron(III) porphyrins could react with H2O2 in protic solvents to afford porphyrin cation radical intermediate.164,165 Recently, van Eldik studied the reaction between H2O2 and an iron(III) octa-anionic porphyrin complex (30, Figure 16).166 They found that cpd I was formed at pH < 9, whereas one-electron oxidized oxoiron(IV) intermediate was observed at pH > 9. At pH = 8, the rate constants for the coordination and dissociation of H2O2 was determined to be k1 = 2259 ± 149 M–1 s–1, k–1 = 1.1 ± 0.1 s–1, and O–O heterolytic cleavage rate constant was measured to be ∼0.18 s–1. In another study, the same group also synthesized two different ferric–hydroperoxo intermediates: five-coordinate high-spin [FeIII(TPFPP)(OOH)] and six-coordinate low-spin [FeIII(TPFPP)(OH)(OOH)]− (31 and 32, Figure 16).167 These two species displayed very different modes of O–O bond cleavage in which the former underwent O–O homolysis to afford an oxoiron(IV) porphyrin complex and the latter formed cpd I via O–O heterolysis.
Figure 16.

Chemical structures of compound 30, Fe(TPFPP)(OOH) (31), and [Fe(TPFPP)(OH)(OOH)]− (32).
Manganese-peroxo porphyrin complexes have afforded a particularly informative example of metal-mediated O–O bond scission. Groves and co-workers have prepared an acylperoxomanganese(III) porphyrin [(mCPB)MnIII(TMP)] (33, Figure 17a) by both direct acylation of manganese-dioxygen intermediate and direct addition of peroxyacid salts to manganese(III) porphyrins.168 Warming a CH2Cl2 solution of 33 from −78 °C to −30 °C led to the formation of an oxoMn(V) intermediate 34. Weiss et al. prepared a manganese(III)-peroxycarbonate intermediate 35 by treating manganese-dioxygen complex with CO2 at −70 °C, which readily converted to oxoMn(V) species 36 by elevating the temperature to −35 °C (Figure 17b).169 Bruice et al. studied the effect of axial ligands on oxygen transfer from various percarboxylic acids and alkyl hydroperoxides to manganese(III) porphyrins. They found that the nitrogen base ligation lead to more than a 100-fold increase in O–O bond cleavage rate and subsequent oxygen transfer.170 Recently, Groves et al. have determined the activation parameters of O–O bond heterolysis for a water-soluble manganese porphyrin cpd 0, [MnIII(OH)(OOH)(TDMImP)] (37, Figure 17c).171 That work determined that the optimal pH was in the range of 8.5–11.5 for the reaction between H2O2 and MnIII(TDMImP). The H2O2 oxidation was significantly slowed outside this range. Significantly, O–O bond cleavage was pH-independent over that range, proceeding with a first-order rate constant of 66 ± 12 s–1. The activation parameters, ΔH‡ = 4.2 ± 0.2 kcal/mol and ΔS‡ = −36 ± 1 cal mol–1 K–1, revealed a remarkably low enthalpic barrier for this reaction. The pH independence suggests a concerted “push-pull” mechanism for O–O cleavage, in which the axial OH ligand is partially deprotonated to become more electron-donating (“push”) and the terminal oxygen of hydroperoxo is partially protonated to release a water molecule to the medium (“pull” effect) to afford the product trans-dioxomanganese(V) species (Figure 17c).
Figure 17.

(a) Conversion of [(mCPB)MnIII(TMP)] into oxoMn(V) intermediate 34. (b) Conversion of manganese(III)-peroxycarbonate 35 into oxoMn(V) intermediate 36. (c) “Push-pull” mechanism for the O–O bond heterolysis of [MnIII(OH)(OOH)(TDMImP)].
2.4. Compound I: Oxoiron(IV) Porphyrin Cation Radicals
2.4.1. Compounds I of Heme Proteins
Compound I (cpd I) is the primary intermediate that gives rise to the diverse oxidative reactivities of most heme proteins. The electronic structure of cpd I is best described as an S = 1 ferryl coupled to an S = 1/2 porphyrin π cation radical, which can afford either doublet or quartet states.172,173 The net spin of cpd I is thus determined by the exchange coupling parameter (J) between the ferryl and the porphyrin cation radical (Table 1). Weak ferromagnetic coupling (J = 0–10 cm–1) and a net spin S = 3/2 have been observed for cpd I of several peroxidases and catalase,173−178 whereas for heme-thiolate proteins like CPO and P450s strong antiferromagnetic coupling (|J| > 40 cm–1) was seen and a total spin of S = 1/2 was observed using EPR and Mössbauer spectroscopy.18,179−182 The two-oxidizing-equivalent intermediate in cytochrome c peroxidase (CcP) obtained after H2O2 oxidation, known as compound ES, has unusual features compared to those of cpd I of most heme proteins. The UV spectrum of CcP-ES lacks the characteristic 690 nm absorbance of the porphyrin cation radical, and its EPR spectrum exhibited a unique free radical signal (g⊥ = 2.006, g∥ = 2.034).183 It is recognized now that CcP compound ES is a ferryl porphyrin intermediate like compound II with an uncoupled protein radical at a tryptophan residue near the axial histidine (Figure 11).184
Table 1. Structural and Spectroscopic Parameters of Representative Compound I Intermediates of Heme Proteins.
| entry | Stotal | J (cm–1) | D (cm–1) | |J|/D | δ (mm/s) | ΔEQ (mm/s) | d(FeIV=O) (Å) | ref |
|---|---|---|---|---|---|---|---|---|
| HRP-I | 3/2 | |J| ≤ 2 | 26 | ∼0.1 | 0.08 | 1.25 | 1.64a | (173) |
| Catalase-I | 3/2 | ∼10 | ∼25 | 0.4 | – | – | – | (176) |
| LiP-Ib | 3/2 | ∼10 | ∼33 | 0.29 | – | – | – | (174) |
| APX-Ic | 3/2 | – | – | 0.28 | – | – | – | (175) |
| CPO-I | 1/2 | –37 | 36 | 1.02 | 0.15 | 1.02 | 1.661d | (179) |
| CYP119A1-I | 1/2 | –52d | 40d | 1.30 | 0.11 | 0.90 | 1.670d | (182) |
| SeCYP119–1 | 1/2 | – | – | 1.43 | 0.07 | 1.49 | – | (201) |
| CYP158A2*-Ie | 1/2 | – | – | – | 0.13 | 0.90 | 1.669d | (260) |
| P450cam-I | 1/2 | – | – | 1.38 | 0.11 | 0.90 | – | (180) |
While cpd I of horseradish peroxidase and chloroperoxidase have been well characterized for decades,179,185−187 the direct observation and characterization of a cytochrome P450 cpd I (P450-I) was much more challenging. Best known for their roles in drug metabolism and oxidative tailoring in biosynthetic pathways, P450s catalyze remarkably selective aliphatic C–H functionalization reactions.16 The oxygen rebound mechanism (Figure 18), first proposed by Groves in the 1970s,188−190 invoked the oxoiron(IV) porphyrin cation radical, compound I, as the “active oxygen species.” Compound I was proposed to activate aliphatic C–H substrates via hydrogen atom transfer (HAT) to the ferryl oxygen. Subsequently, the incipient substrate radical recombines with the Fe(IV)–OH of compound II (oxygen rebound) to afford hydroxylation products. Because of the exceptional reactivity of P450 enzymes, efforts to understand P450-I have attracted sustained attention for four decades. Further, these pursuits have provided valuable insights for the development of new catalysts for selective aliphatic C–H functionalization.
Figure 18.
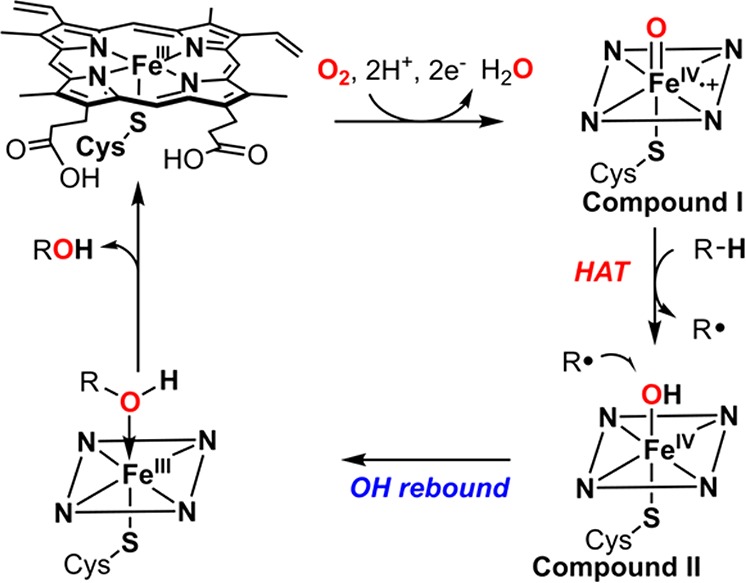
Oxygen rebound mechanism for C–H hydroxylation catalyzed by cytochrome P450s.
The main obstacles for studying P450-I included its very short lifetime and low yield.18,191−193 Chloroperoxidase cpd I (CPO-I) has long been used as an analog of P450-I because it also contains an axial cysteine thiolate. However, CPO-I can only hydroxylate weak benzylic C–H bonds.194 This lack of reactivity for C–H activation led to proposals of other possible intermediates such as an oxoFe(V) electromer or Fe(III)–OOH for P450 reactivity.195−197 The long-sought P450-I was finally elucidated in 2010.182 Rittle and Green greatly improved the yield of cpd I of a thermophilic cytochrome P450, CYP119. Spectroscopic studies of CYP119-I revealed a broad UV absorbance around 690 nm typical of a porphyrin cation radical (Figure 19a) and a Mössbauer isomer shift of 0.11 mm/s and quadrupole splitting of 0.90 mm/s signaling iron(IV). An antiferromagnetic coupling between S = 1 ferryl and porphyrin cation radical with a |J/D| of 1.3 was determined by EPR spectroscopy. These data clearly showed that CYP119-I is an antiferromagnetically coupled (S = 1/2) oxoiron(IV) porphyrin cation radical. Unpaired electron density of the axial thiolate was also suggested. More importantly, CYP119-I was found to be highly reactive for C–H hydroxylation with apparent second-order rate constants ranging from 104 to 107 M–1 s–1 (Figure 19b). Following the characterization of CYP119-I, compounds I of CYP158A2 Y352F variant and P450cam have also been characterized.180,181 All these P450 compounds I have similar spectroscopic features and exhibit high reactivity toward alkyl C–H bonds.
Figure 19.

(a) UV–vis transients observed upon 1:1 mixing of 20 μM ferric CYP119 with 40 μM mCPBA. Maximum yield of CYP119-I was ∼70% at 35 ms. (b) Observed first-order decay rates vs camphor concentration. (c) UV–vis transients observed upon 1:1 mixing of 13 μM ferric AaeUPO with 25 μM mCPBA. Maximum yield of AaeAPO-I was ∼70% at 30 ms. (d) Observed first-order decay rates vs p-ethylbenzoic acid concentration. (a) and (b) were adapted with permission from ref (182). Copyright 2010 American Association for the Advancement of Science. (c) and (d) were adapted from ref (200). Copyright 2012 American Chemical Society.
Unspecific peroxygenases (UPO, initially termed APO for aromatic peroxygenase) comprise a large and newly discovered family of P450-like, heme thiolate hydroxylases of fungal origin.198 These enzymes are showing considerable promise as biocatalysts with high reactivity toward even aliphatic hydrocarbons.199 Compound I of an unspecific peroxygenase from the common edible mushroom Agrocybe aegerita (AaeUPO) was recently characterized by Wang and Groves et al.200 UPOs are very significant and unusual because they can oxidize strong C–H bonds with H2O2 as oxidant and do not require any cofactors. AaeUPO-I displays similar UV–vis features as CYP119-I and fast rate constants for the hydroxylation of strong C–H bonds (up to 100 kcal/mol) in the range of 10–105 M–1 s–1 (Figure 19c and 19d).200
Very recently, Green et al. reported the characterization of a selenocysteine-ligated cpd I of CYP119 (SeCYP119-I).201 Given the structural similarity and the difference in electronic properties between selenocysteine and cysteine, SeCYP119-I provides a good opportunity to study the effect of electron-donation of axial ligands on cpd I reactivity. Compared to CPO-I and CYP119-I, SeCYP119-I exhibits a larger |J/D| value (|J/D| = 1.43) and a smaller isomer shift δ = 0.07 mm/s, suggesting a stronger electron-donation from selenolate [ R-Se–] axial ligand. Reactivity studies showed that CYP119-I and SeCYP119-I were formed at similar rates (5 × 106 M–1 s–1 and 7 × 106 M–1 s–1 for CYP119 and SeCY119, respectively) upon treating corresponding ferric enzymes with mCPBA. But SeCYP119-I was more reactive toward C–H bonds than CYP119-I, leading to a significantly less accumulation of SeCYP119-I during reactions with substrates.
2.4.2. Compounds I of Iron Porphyrins
The first synthesis and characterization of an oxoiron(IV) porphyrin cation radical model compound was achieved by our group in 1981.202 Treatment of a sterically hindered iron porphyrin, FeIII(TMP)Cl (39, Figure 20a) with mCPBA in CH2Cl2/MeOH (4:1) at −78 °C produced a green compound formulated as [FeIV(O)TMP•+]L (40, Figure 20a). This species had a Soret band at 406 nm and a broad absorption at 645 nm, typical of a porphyrin cation radical. EXAFS data showed a short Fe–O bond length of 1.6 Å. A ferryl iron–oxygen stretch, ν(Fe=O) of 801 cm–1 (L = Cl) and 828 cm–1 (L = MeOH), was obtained by resonance Raman spectroscopy.203 Magnetic susceptibility and EPR studies showed a total spin of 3/2 with strong ferromagnetic coupling between the ferryl and porphyrin cation radical (J > 40 cm–1). These features stand in sharp contrast to the antiferromagnetic coupling observed in P450 cpd I.204 This green species [O=FeIVTMP•+] was found to be reactive in oxygen atom transfer reactions.205 The direct oxidation of norbornene by [O=FeIVTMP•+] in the presence of H218O led to the formation of an epoxide product with 99% 18O incorporation, indicating facile ferryl–oxygen exchange with water.
Figure 20.

(a) The synthesis of the first model cpd I (TMP•+)FeIV=O and its structural and electronic parameters. (b) Depiction of the C–H activation mode of (TMP•+)FeIV=O and its Mössbauer spectrum, which indicates high-valent iron. Adapted from ref (205). Copyright 1983 American Chemical Society.
A number of compound I analogues were synthesized with varied substitutions at meso- and pyrrole-β positions of the porphyrin ligand. These studies have been summarized in a number of reviews.15,26,27,31,32,206−208 In general, both meso- and pyrrole-β substituents affect cpd I electronic structures via altering the energy of porphyrin SOMO/HOMO orbitals a1u and a2u (Figure 21a). In unsubstituted porphyrins, a2u is the higher-energy orbital and is singly occupied in cpd I. Since a2u has a large spin density at meso-positions (as well as the pyrrole nitrogens), the increase of electron-deficiency of meso-substituents will substantially stabilize a2u.27 For instance, the most electron-withdrawing pentafluorophenyl derivative, FeIV(O)(TPFPP•+), has an a1u porphyrin radical configuration (Figure 21b). Fujii et al. showed that compounds I with pyrrole β-substituents are generally a1u porphyrin radicals, and the electronic properties of pyrrole β-substituents were shown to have similar influences on a1u and a2u (Figure 21c). The nature of the porphyrin radical (a1u or a2u) has a large impact on its exchange interaction with iron center, in which large ferromagnetic coupling (J > 30 cm–1) was observed for a2u radicals due to large pyrrole nitrogen orbital coefficients and, by contrast, weak antiferromagnetic coupling (J < 10 cm–1) for a1u radicals.209,210
Figure 21.

(a) Orbital diagrams of porphyrin a1u and a2u orbitals. (b) Effect of meso-substituents on the electronic structure of cpd I. (c) Effect of pyrrole-β substituents on the electronic structure of cpd I. (b and c) were adapted from ref (27). Copyright 2002 Elsevier B. V.
Although these cpd I analogs display a range of oxygen transfer reactions, their second-order rate constants for C–H activation (k) are generally less than 10 M–1 s–1.211 These values for the model compounds are 3 to 6 orders of magnitude slower than those recently measured for P450-I or UPO-I. A highly reactive synthetic compound I was reported by our group (Figure 22).212 This species was generated by reacting a cationic iron(III) porphyrin complex FeIII(4-TMPyP) with mCPBA under rapid-mixing conditions. The resulting [FeIV(O)(4-TMPyP•+)] could oxidize a range of C–H bonds with second-order rate constants, ranging from (2.85 ± 0.3) × 106 M–1 s–1 for xanthene to (7.5 ± 0.1) × 103 M–1 s–1 for ethylbenzoic acid. The rate constant of ethylbenzoic acid oxidation is comparable to those of CYP119 and CPO cpd I for ethylbenzene oxidation.213 It was suggested that stabilization of the porphyrin a2u HOMO and the increased electron deficit at the metal core due to the cationic porphyrin contribute to the very high activity of [FeIV(O)(4-TMPyP•+)]. The significance of these findings is that no unusual properties of the protein architecture need to be invoked to explain high rates of C–H bond cleavage by heme proteins.
Figure 22.
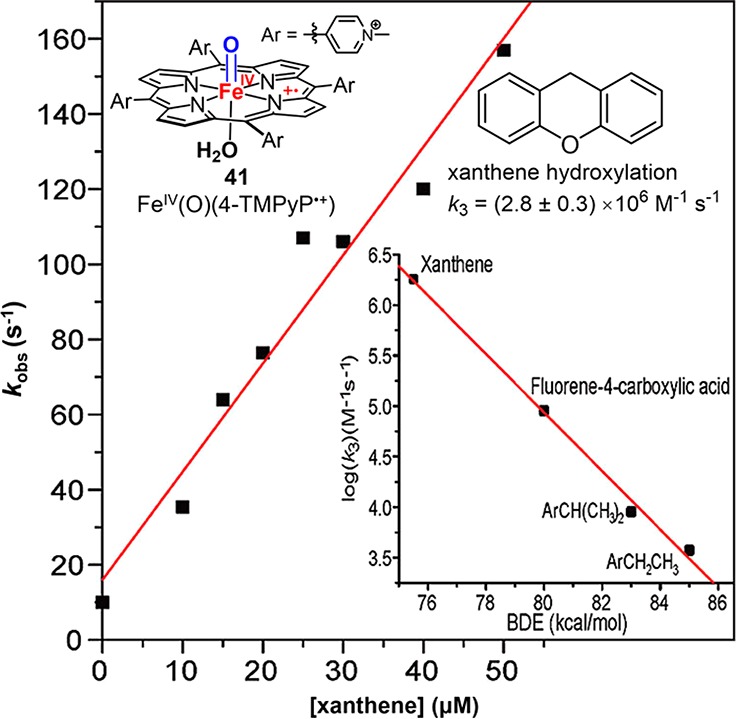
Observed rate constant kobs at 673 nm vs xanthene concentration for its oxidation by 41 at pH 4.7, 14.5 °C, yielding second-order rate constant k3 = (2.8 ± 0.3) × 106 M–1 s–1. Inset: correlation of k3 with C–H bond dissociation energy. Adapted from ref (212). Copyright 2009 American Chemical Society.
Very recently, Sorokin and co-workers have reported the synthesis of a series of dimeric oxoiron(IV) cation radical species [(L)FeIV=N–FeIV(L•+)=O] by treating μ-nitrido diiron porphyrins or phthalocyanines (Pc) with peroxides or mCPBA (42 and 43, Figure 23).214,215 A high-valent μ-nitrido diiron–oxo porphyrin cation radical intermediate [(TPP)FeIV=N–FeIV(TPP•+)=O] was successfully isolated and characterized (44, Figure 23).216 This species has a green color similar to other porphyrin cation radicals and has a half-life of ∼20 min at −80 °C. The single narrow EPR signal at g = 2.001 suggested an S = 1/2 state with the unpaired electron localized on the porphyrin macrocycle. In line with the EPR results, magnetic susceptibility measurements revealed a strong antiferromagnetic coupling between two iron centers with −JFe–Fe > 600 cm–1, which leads to a SFeFe = 0. The Mössbauer spectrum showed a very small isomer shift (0.00(1) mm/s) and a moderate quadrupole splitting (0.75(2) mm/s), also consistent with iron(IV). These high-valent μ-nitrido diiron–oxo complexes have exceptional activity for C–H activation.217 Under catalytic conditions, they were shown to activate C–H bonds of methane (BDE = 104 kcal/mol) to afford formic acid with total turnover numbers (TTN) up to ∼220.218 Recent computational studies showed that the activation barriers of [(L)FeIV=N–FeIV(L•+)=O] type intermediates for methane oxidation are over 10 kcal/mol lower than that of a model P450 cpd I.219 The calculation also suggests that the donor properties of the bridged nitrido group dramatically increased the basicity of the oxo group, which is crucial for the high reactivity of this class of compounds.
Figure 23.
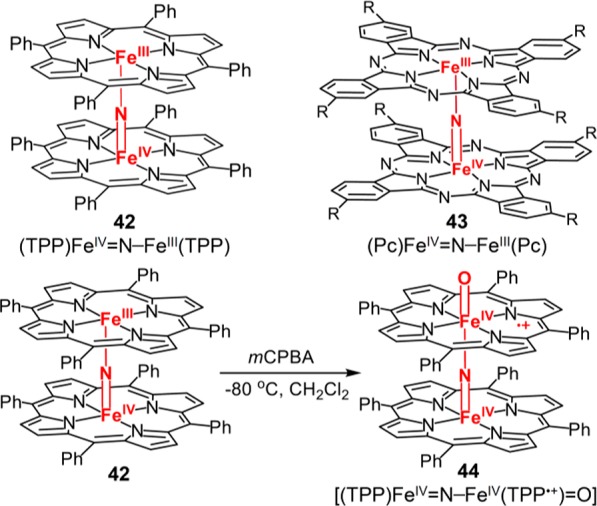
Structures of μ-nitrido diiron porphyrins and phthalocyanines (Pc) and formation of cpd I 44 from 42 and mCPBA.
2.4.3. Manganese Porphyrin Analogues of Compound I
Synthetic manganese porphyrin analogues of compound I have long been known to have high reactivity toward C–H bonds.220 While relatively stable oxoMn(IV) porphyrin species have been isolated and well-characterized,221,222 the direct characterization of oxoMn(V) porphyrin complexes has been very challenging due to their high reactivity. The presence of reactive oxoMn(V) porphyrin complexes has been usually implicated from reactivity patterns and 18O-exchange into products from water.223−225 The first Mn porphyrin compound I (MnPor-I) to be kinetically evaluated was generated by treating MnIIITMPyP with a peroxyacid such as mCPBA using rapid-mixing stopped-flow techniques (45 to 46, Figure 24).226 MnPor-I 46 rapidly converted to previously characterized oxoMn(IV) via 1e– reduction with a first-order rate constant of 5.7 s–1. Unlike compound I of iron porphyrins, MnPor-I was diamagnetic, displaying a well-resolved and unshifted proton NMR spectrum and lacked the characteristic broad absorbance between 650–700 nm of porphyrin cation radical, suggesting MnPor-I was an oxoMn(V) species.22746 catalyzed very fast olefin epoxidation with a second-order rate constant of 6.5 × 105 M–1 s–1. A 35% 18O incorporation into the epoxidation products was also observed when carrying out the reaction in the presence of H218O, which is in accordance with an aqua–oxo interconversion via prototropy as suggested by Meunier (47 to 48, Figure 24).223,225 By contrast, manganese-substituted heme proteins, such as Mn-HRP, form only manganese(IV) species and a protein radical.228 Apparently, the reduction potential of oxoMn(V)protoporphyrin IX is too high to be tolerated by the more redox sensitive protein.
Figure 24.

Generation of a cpd I manganese porphyrin analogue and the oxo-aqua interconversion of oxoMn(V) intermediate.
A related oxoMn(V) porphyrin was synthesized by treating cationic [MnIII(TF4TMAP)](CF3SO3)5 (49, Figure 25) with two equivalents of alkaline hydrogen peroxide.229 This species transformed olefins to epoxides with good yields (∼50%). In line with the 18O exchange experiment of 46, the epoxidation mediated by 49 with H216O2 in H218O water led to a 45% 18O incorporation. Newcomb et al. prepared a series of oxoMnV porphyrins (Figure 25b) in organic solvents via laser flash photolysis (LFP) of corresponding MnIII perchlorate porphyrin complexes.230 The second-order rate constants for olefin epoxidation by these complexes were in the range from 104 to 106 M–1 s–1, consistent with that of water-soluble oxoMnV(TMPyP). For oxoMnV(TPFPP), the second-order rate constant for ethylbenzene oxidation was determined to be 1.2 × 105 M–1 s–1, more than 105 times faster than the corresponding iron complex.211 A dinuclear oxoMnV porphyrin complex has been described by Naruta et al. This species was prepared from the oxidation of a manganese(III) porphyrin dimer with mCPBA in the presence of excess tetrabutylammonium hydroxide (nBu4OH) (Figure 25c).231 Interestingly, decomposition of this oxoMnV dimer produced oxygen. A number of oxoMnV complexes have been prepared with other porphyrinoid ligands such as corroles and corrolazines, as recently reviewed by Goldberg et al.29
Figure 25.
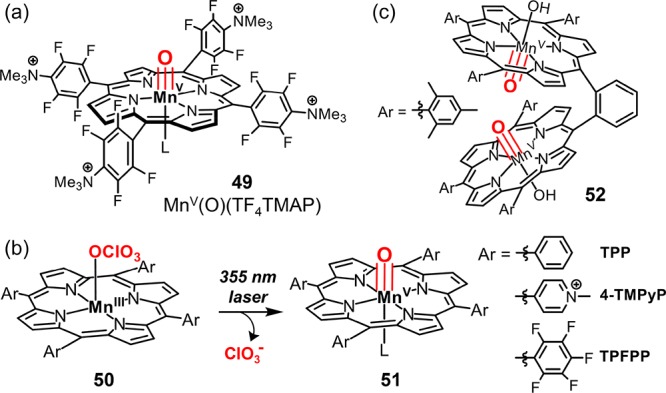
(a) Chemical structure of MnV(O)(TF4TMAP). (b) Generation of oxoMnV intermediates via laser flash photolysis (LFP). (c) A dinuclear oxoMnV intermediate 52.
In addition to these oxoMnV complexes, an unusual trans-dioxoMn(V) porphyrin structure was deduced from the pH-dependent halide oxidation mediated by oxoMnV porphyrins. Apparent second-order rate constants spanned a range of 5 orders of magnitude from ∼107 M–1 s–1 at pH 5.2 to ∼102 M–1 s–1 at pH 9.232 This profound pH dependence was analyzed to result from two acid–base equilibria between the trans-dioxoMn(V), oxo–hydroxo Mn(V), and oxo-aqua Mn(V) species, which displayed increasing oxygen-atom-transfer activities along the sequence. Proton ionizations with pKa1 < 5 and pKa2 = 7.7 were estimated for oxo-aqua species from the Nernst eq (Figure 26a). The synthesis and characterization of such trans-dioxoMn(V) porphyrin complexes were reported by Groves and Spiro. These complexes were synthesized by treating Mn(III) porphyrins with H2O2 in the presence of excess nBu4OH (Figure 26b).233,234 These compounds exhibited Raman νsym(O=MnV=O) values of 741–744 cm–1, which were decreased by 21–26 cm–1 and 39–44 cm–1, respectively, upon single and double 18O substitution on O=MnV=O moiety. Significantly, νassym(O=MnV=O) values detected by IR were predictably different, confirming the linear, triatomic structure of the dioxoMn(V) unit (Figure 26c). A similar MnV(O) intermediate was synthesized by Nam et al. and was assigned as a six coordinate trans-oxohydroxo Mn(V) complex.235 However, a Mn–O distance of 1.68 Å determined by EXAFS indicates a double rather than a triple bond between Mn and oxygen. It is very likely that this species is also a trans-dioxoMn(V) complex.
Figure 26.

(a) pH-dependent reactivity of Mn(III) porphyrins for halide oxidation and the acid-base equilibria between the trans-dioxoMn(V), oxo–hydroxo Mn(V), and oxo-aqua Mn(V) species. (b) Synthesis of a trans-dioxoMn(V) porphyrin 54. (c) Characterization of 54 via rR and IR spectroscopies. (d) Mn–O bond length vs 1/F1/3(Mn–O). (c) and (d) were adapted from ref (233). Copyright 2007 American Chemical Society.
On the basis of the νsym and νassym data obtained for compound 54, the Mn–O bond force constant (F) and stretch–stretch constant (k) were determined to be 454 and 67.2 N/m, respectively. Using the recently reported EXAFS bond length of 54 and available data of various terminal monooxo-manganese complexes,235 a linear correlation was found between F–1/3 and Mn–O bond lengths (Figure 26d), in line with predictions of Badger’s rule.236,237 A similar correlation between F–1/3 and Fe–O bond lengths has previously been reported by Green et al. for oxoiron(IV) porphyrins.236 Although trans-dioxo metal porphyrins have been characterized for second- and third-row transition metals such as RuVI and OsVI,238−242 manganese remains the only known first-row transition metal that can form a trans-dioxo species. The isolated trans-dioxoMn(V) porphyrins were less reactive toward typical substrates but could be activated via decreasing the pH. This pH-dependent reactivity supports the previously proposed acid–base equilibria between the trans-dioxoMn(V), oxo–hydroxo Mn(V), and oxo-aqua Mn(V) species and their increasing reactivity for oxidation reactions.227,243 DFT calculations indicated that this trend correlated with corresponding energy gaps between unreactive singlet ground state and higher-energy triplet and quintet states.244
2.5. Compound II and Its Protonation State–ferryl or Hydroxoiron(IV)
The one-electron-reduction of oxoiron(IV) porphyrin cation radicals, cpd I, affords a ferryl porphyrin (cpd II), which also has an iron(IV) center (S = 1), similar to that of cpd I but lacking the porphyrin cation radical. The protonation state of cpd II has generated considerable research interest because of its importance in determining cpd I reactivity (see section 3.1).245 HRP-II, Mb-II, and CcP compound ES were among the earliest cpd II or cpd II-like species to be spectroscopically characterized.115,122,173,246−248 The first synthetic ferryl porphyrin model compound was described by Balch et al.249 X-ray absorption measurements of these intermediates showed a Fe–O bond lengths in the range of 1.64–1.70 Å, indicating a Fe=O unit.248,250−252 The Fe=O structure is also consistent with the resonance Raman studies in which Fe(IV)–O stretching vibrations were detected in the range from 740 to 870 cm–1.236 These results, however, contradicted data obtained with X-ray crystallography, in which much larger (>1.80 Å) Fe–O bond distances were determined.48,253,254 This discrepancy arose mainly from the radiolytic reduction caused by the hydrated electrons generated by the X-ray irradiation during data collection. For instance, Poulos and co-workers found that the Fe–O distance of CcP-ES changed linearly with the radiation dosage. The Fe–O distance measured at high-X-ray doses was 0.17 Å longer than that measured at low doses (1.9 Å vs 1.73 Å).255 Structures obtained by nonionizing neutron cryo-crystallography also support a Fe(IV)=O structure for CcP-ES with a Fe–O distance around 1.6 Å.126 Since HRP-II, Mb-II, and CcP-ES exist in Fe(IV)=O state with pH as low as 4.5, their conjugate acid (Fe(IV)–OH) would have to be highly acidic (Table 2).245 Recently, a more accurate measurement of the Mb-II pKa was performed by Green et al. An upper limit of 2.7 for pKa(Mb-II) was determined by probing Mb-II over a wide pH range (3.9–9.5) with a combination of EXAFS, Mössbauer, and resonance Raman spectroscopies.256 No evidence of a protonated Mb-II was obtained.
Table 2. Structural and Spectroscopic Parameters of Representative Compound II of Heme Proteinsa.
| entry | d(FeIV–O)b (Å) | δ (mm/s) | ΔEQ (mm/s) | pKa(FeIVO–H) | ref |
|---|---|---|---|---|---|
| HRP-II | 1.70 ± 0.02 (6.0) | 0.03 (6.9) | 1.61 (6.9) | ≤3.5 | (245) |
| Mb-II | 1.66 (3.9) | 0.07 (3.9) | 1.58 (3.9) | ≤2.7 | (256) |
| CcP-ES | 1.67 (6.0) | 0.05 (7.0) | 1.55 (7.0) | ≤4 | (245) |
| Catalase-II | 1.78 (5.3) | 0.02 (5) | 2.28 (5) | 13.1 | (262) |
| CPO-II | 1.82 (6.5) | 0.10 (6.5)c | 2.06 (6.5)c | ≥8.2 | (250)(259), |
| CYP119-II | – | 0.12 (8.0) | 1.94 (8.0) | 12.2 | (260) |
| CYP158-II | 1.84 (9.0) | 0.10 (9.0) | 2.05 (9.0) | 12.0 | (260) |
pHs are shown in parentheses.
Fe–O bond distances obtained from EXAFS studies.
Parameters for the major component. A minor component is present with δ = 0.11 mm/s and ΔEQ = 1.59 mm/s.
The cpd II of chloroperoxidase (CPO) stands in sharp contrast to HRP-II, Mb-II, and CcP-ES. The EXAFS measurements by Green, Dawson, and Gray revealed a Fe–O length of 1.82 Å at pH 5.5 and pH 6.7.250 The visible spectrum of CPO-II remained unchanged from pH 3–7, suggesting a pKa(CPO-II) ≥ 8.2 (assuming CPO-II is 95% protonated at pH = 6.9). The unusual basicity of CPO-II is explained by the strong electron donation by the axial thiolate, which leads to a more basic ferryl oxygen. This “push” effect of a cysteine axial ligand suggests that cpd II of other heme-thiolate proteins might also be basic.257−259 In 2013, Green and co-workers studied the protonation event of a P450 cpd II (CYP158-II) using rapid-mixing pH-jump experiments coupled with a variety of spectroscopic techniques. Both deprotonated and protonated forms of CYP158-II were well-characterized, and a remarkably basic pKa of 12 was determined.260 The ferryl protonation led to an elongation of Fe–O bond from 1.68 to 1.84 Å and a shortening of Fe–S bond from 2.36 to 2.27 Å along with an increase in quadrupole splitting and isomer shift, consistent with the generation of a Fe(IV)–OH species. The pKa(cpd II) of another P450, CYP119, was also determined in the same study. Unlike CYP158, wild-type CYP119 did not accumulate cpd II. Mutational studies have shown that Y352 is important for generation of CYP158-II, as it can serve as an electron source to reduce cpd I to cpd II. In CYP119, a leucine residue (L316) is in the position corresponding to Y352 in CYP158. The redox inactive leucine might prevent efficient cpd I reduction to generate cpd II. Indeed, an L316Y mutation was found to substantially increase the yield of CYP119-II. Despite having a different active site environment, CYP119 displayed a very similar pKa(cpd II) to that of CYP158. Very recently, the protonation of cpd II of a heme-thiolate peroxygenase, AaeAPO, has been investigated by Groves and co-workers.261 A pKa of 10.0 was obtained from pH-jump double-mixing stopped–flow experiments.
Very recently, Green et al. reported a pKa of 13.1 for cpd II of Helicobacter pylori catalase (HPC).262 The high pKa value of HPC-II suggests that the tyrosinate axial ligand of catalase is electron-donating enough to maintain a highly basic ferryl, even though it forms a hydrogen bond with a conserved arginine residue. Another case of a basic cpd II without a thiolate axial ligand was recently reported by Raven and Moody. They showed that cpd II of ascorbate peroxidase (APX-II) exists as FeIV–OH.263 A neutron diffraction analysis of APX-II prepared via soaking APX crystals in m-CPBA solution revealed a long Fe–O bond distance of 1.84 Å, apparently confirming an earlier X-ray determination for that protein by the same authors.264 The cause of protonation in APX-II and the protonation state of compound II of other peroxidases remains to be determined.
The basicity of metal–oxo moieties in model compounds has been much less studied. Recently, Boaz and Groves investigated the protonation of cpd II of several water-soluble sulfonated iron porphyrins.265 A two-proton equilibrium with an apparent pKaobs = 5.5 was determined for the protonation of [O=FeIV(TMPS)(OH2)] (56, Figure 27a). The resulting bis-aqua complex is an iron(III) porphyrin cation radical species 55 that is at the same net oxidation state. The absence of the accumulation of monoprotonated species 57 indicated that the first ferryl protonation with pKa1 (∼4) must produce a much more basic intermediate, pKa2 ∼ 7. The increase in basicity is apparently due to a change in iron oxidation state from a protonated ferryl to a hydroxoferric porphyrin cation radical through an electromeric equilibrium after the first protonation. The effect of this two-proton electromeric equilibrium was shown to have a significant effect on the C–H scission reactivity of the corresponding compound I analog, [O=FeIV(TMPS•+)(OH2)]. In terms of the Bordwell equation, such a two-proton equilibrium leads to an increase in the overall driving force of 5–6 kcal/mol because two O–H bonds are being formed at the transition state (defined as D(OH2) in Figure 27a), with one proton coming from the substrate and the other from the medium.
Figure 27.

(a) Basicities of model hydroxo- and oxoiron(IV) porphyrin complexes. The second protonation provides an additional 5 kcal/mol driving force for HAT. (b) Acidities of several MnIV–OH complexes.
Several oxoMn(IV) porphyrins have been synthesized and characterized in the presence of strong base.221,222 Fujii et al. successfully prepared and characterized a series of MnIV=O and MnIV–OH complexes with salen ligands via direct deprotonation of corresponding MnIII–OH2 salen cation radical complexes.266,267 But the pKa’s of these Mn–OH species have yet to be reported. Since manganese is less electronegative than iron, Mn(IV)–OH should be less acidic than Fe(IV)–OH. There are several acidity data reported for MnIV–OH complexes with corrolazine or nonheme ligands (Figure 27b).268 For instance, a lower limit around 15 was estimated for a MnIV–OH corrolazine complex 58 (Figure 27b).269 A similar MnIV–OH pKa (∼15) was also estimated for a nonheme MnIV–OH species 59.270 Busch et al. synthesized a dihydroxomanganese(IV) complex 60 based on a cross-bridged cyclam ligand. pKa of 6.86 and 10 were determined for the first and the second deprotonation, respectively.271
2.6. Preventing Protein Oxidative Damage by Cpd I
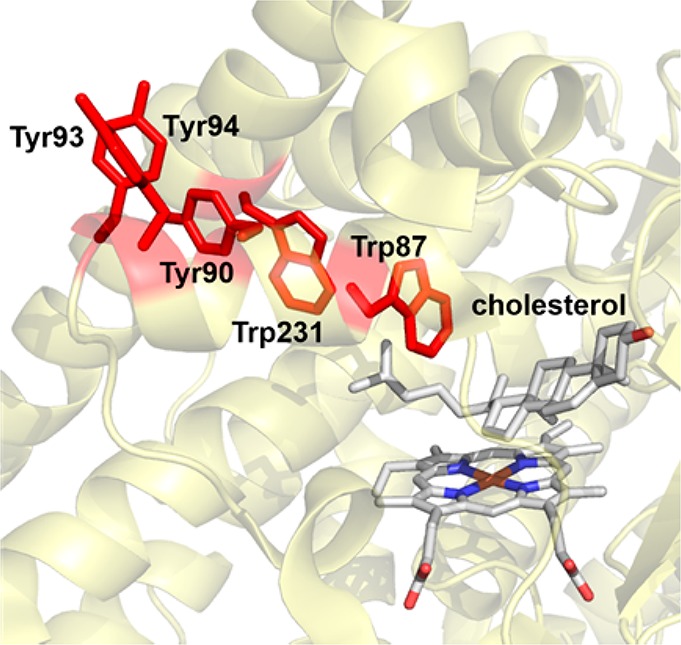
Compounds I of P450s and UPOs have remarkable activity and can activate very inert C–H bonds in substrates. It is not a coincidence that they also exhibit the highest compound II basicities among heme proteins. As discussed in detail in section 3.1.1, the thermodynamic driving force for C–H activation by cpd I depends on the reduction potential of cpd I and the basicity of cpd II. According to the Bordwell equation,272−274 an increase of one pK unit in cpd II basicity would enhance the O–H bond strength of cpd II by 1.37 kcal/mol. Therefore, the high cpd II basicity of P450s and UPOs would allow them to break strong C–H bonds with relatively low reduction potentials, which, at least in part, prevents the oxidative damage of protein superstructure by cpd I. The presence of chains of near-by tyrosine and tryptophan residues could provide further protection of enzymes during these oxidative transformations.275,276 These redox-active residues have been proposed to transport oxidizing equivalents of cpd I to protein surfaces. This protection machinery could be important for P450 catalysis, in which compounds I that are not consumed due to inefficient substrate oxidation could be effectively quenched. For instance, a five-residue Tyr/Trp chain has been identified in CYP11A1, which includes residues Trp87, Trp231, Tyr90, Tyr93, and Tyr94 (Scheme 1). Trp87 is critical for triggering the protection machinery. Once Trp87 gets oxidized by cpd I to form a cation radical, the oxidizing equivalent on Trp87 will rapidly transit through Trp231 and Tyr90 to surface-exposed Tyr93 and Tyr94. Kinetic simulations of CYP11A1 have indicated a ∼3 μs survival time for cpd I before its oxidizing equivalents are transferred to the protein surface.275 In this regard, C–H activation by cpd I could be viewed as an inner sphere process that competes with a slower, long-range outer-sphere electron transfer.
Scheme 1. Chain of Redox Active Trp/Tyr Residues in CYP11A1 (PDB: 3N9Y).
3. C–H Functionalization Reactions Mediated by Oxoiron(IV) Porphyrin Cation Radicals (Compound I)
The majority of reactions mediated by cpd I are initiated via hydrogen atom transfer (HAT). These reactions not only include aliphatic hydroxylation as introduced above but also encompass a broad range of oxidative transformations such as desaturation, decarboxylation, C–C bond scission, and a variety of rearrangement reactions. Like C–H hydroxylation, these alternative reaction pathways involve an initial hydrogen atom transfer (HAT) via cpd I and the generation of an intermediate substrate radical. A common mechanistic feature shared by these reactions is the redirection of substrate to other reaction pathways leading to nonoxygenated products.277−279 This strategy represents a general approach to develop novel catalytic systems for developing novel C–H functionalization reactions. In this section, we will first discuss fundamental aspects of HAT and oxygen rebound steps in cpd I-mediated reactions, as well as various factors that affect these two steps. Further, we will introduce several representative examples of nonconventional C–H activation transformations catalyzed by P450s and metalloporphyrins. In particular, we will delineate how substrate radical intermediates are controlled to achieve different types of reactions.
3.1. Factors that Affect Oxoiron(IV) Porphyrin Cation Radical Reactivity for HAT
3.1.1. Thermodynamic Properties of Cpd I
For a given C–H bond, the driving force of HAT reactions will be determined by the free energy of the O–H bond formed in cpd II [DO−H(cpd II)]. Using thermochemical cycles to estimate bond dissociation free energies from measurable pKa values and oxidation potentials has a long history,280 which was systematically developed by Bordwell and extended to metal–oxo species by Mayer.272−274 In accordance with the Bordwell equation (eqs 1 and 2, Scheme 2).272−274 The bond dissociation free energies of an X–H bond can be derived from the X–H pKa and the X– oxidation potential using a Hess cycle. Thus, an increase in cpd II ferryl basicity [higher pKa for Fe(IV)O–H] would lead to a stronger O–H bond formed in cpd II and a more thermodynamic driving force (1.37 kcal/mol per pKa unit) for the C–H abstraction by cpd I. For instance, the more electron-donating cysteine (thiolate) axial ligand in P450s can increase the basicity of compound II by over 7 pK units compared to the histidine-ligated myoglobin and horseradish peroxidase.19,260,281 Considering the pKa term in the Bordwell equation, this change in basicity corresponds to at least a 9 kcal/mol increase in O–H BDE of compound II.260,261
Scheme 2. Determination of D(O–H) from the One-Electron Reduction Potential of Compound I and the pKa of Compound II.
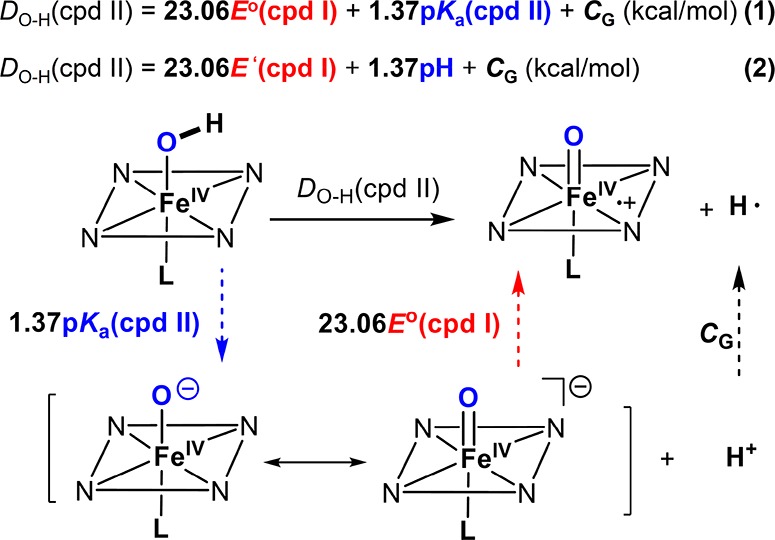
CG is a constant depending on the solvent and electrode (57.6 kcal/mol for aqueous solution and the normal hydrogen electrode). When E′(cpd I) at a given pH is determined, eq 2 can be used to calculate D(O−H). D(O–H) obtained via eq 1 and eq 2 is the bond dissociation free energy (BDFE). If the entropic difference between protonated and unprotonated cpd II is negligible, the same equations can be used to calculate the bond dissociation enthalpies (BDE) of O–H bonds using constant CH (55.8 kcal/mol for aqueous solution and normal hydrogen electrode).274
The reduction potential of cpd I to cpd II also plays an important role in determining cpd I reactivity. E′(cpd I/cpd II) of horseradish peroxidase (HRP) has been determined to be ∼0.95 V versus NHE at pH 6.0 (Table 3).282 Intriguingly, HRP showed almost identical reduction potentials for cpd I and cpd II. It is much harder to determine E′(cpd I/cpd II) for P450s and CPO because of the short lifetimes of these intermediates. Instead, the two-electron reduction potential E′(cpd I/ferric) has usually been measured, which, in turn, can be used to estimate E′(cpd I/cpd II) with certain assumptions. For instance, assuming a similar redox potential for CPO-I and CPO-II, Green, Dawson, and Gray estimated the redox potential of CPO-I to be 1.3 ± 0.1 V at pH 6, which is 0.35 V higher than that of HRP, corresponding to an ∼8 kcal/mol increase in DOH(cpd II).250 This enhancement together with the contribution of a basic CPO-II led to an overall over 11 kcal/mol increase in DOH(CPO-II) compared to DOH(HRP-II).
Table 3. pKa and BDEOH of Cpd II and Reduction Potentials of Cpd I.
Very recently, Wang and Groves were able to determine the thermodynamic properties of the heme-thiolate peroxygenase AaeAPO-I.283 This approach utilized the rapid, reversible oxygen atom transfer between cpd I and halide ions to determine the two-electron cpd I reduction potential. The measured E′(cpd I/ferric) was in the range of 1.12–1.32 V versus NHE from pH 7.0 to 3.0. From plots of the rate for C–H abstraction versus substrate C–H BDE, the DOH(CPO-II) was estimated to be 100 kcal/mol. This value leads to an E′(cpd I/cpd II) of 1.4 V and E′(cpd II/ferric) of 0.8 V at pH 7.0 (Figure 28). This estimate was further supported by the reaction of nitroxyl radicals with AaeAPO-I. 3-Carboxyproxyl (Eo ∼ 0.9 V) reacted rapidly with APO-I to form APO-II, which decayed much more slowly under these conditions. This pronounced reactivity difference shows that APO-II is a much milder oxidant. By contrast, the two analogous reduction potentials of HRP-I are both around 0.9 V. Thus, APO has increased the reduction potential of APO-I to perform the difficult C–H scission at the expense of APO-II, which needs only to interact with the incipient substrate radical. With nitroxyl radical as a selective reductant for APO-I, APO-II could be produced in high yield and the pKa has been determined to be 10.0 via rapid-mixing, pH-jump spectrophotometry. With these values, the DOH(APO-II) was calculated to be around 100 kcal/mol, consistent with the estimate above determined from kinetic measurements.
Figure 28.
Selective reduction of APO-I to APO-II by a nitroxyl radical with Eo = 0.9 V. Redox potentials are obtained from ref (283)
3.1.2. Effect of Axial Ligands and Porphyrin Peripheral Substituents on Cpd I Reactivity
It is clear from the above discussion that axial ligands can have a huge impact on cpd I reactivity. Such effects have been observed in a number of model compound studies. For example, a 650-fold rate enhancement was reported for cyclohexene hydroxylation by changing the axial ligand of FeIII(TPFPP)L from CF3SO3– to Cl–.284 Subsequent DFT calculations showed that this ligand replacement was predicted to cause a 6 kcal/mol increase in FeIVO–H BDE, and that the main attribute of this BDE enhancement is the increase in cpd II basicity.285 The axial ligand effect on C–H hydroxylation was also investigated for FeIV(O)(TMP•+) with para-substituted pyridine N-oxide (p-Y-PyO) axial ligands.285 In this system, the hydroxylation reactivity increased with the increasing electron-donating ability of para-substituent Y. A good correlation between calculated activation energies and FeIVO–H acidity was obtained, which, again, suggested that axial ligand could affect cpd I reactivity via tuning cpd II basicity.
While the increase in cpd II ferryl basicity via introducing electron-donating axial ligands has now been observed, the effect of axial ligands on cpd I reduction potential [Eo(cpd I)] is less clear. A number of theoretical calculations have shown that Eo(cpd I) decreased with increasing electron-donation from axial ligand and that cpd I with negatively charged ligands such as F– and Cl– exhibited lower Eo(cpd I) than those with neutral ligands such as imidazole.286−289 However, Fujii et al. measured the reduction potential of a series FeIV(O)(TMP•+)L and found an opposite trend (Figure 29). With L = ClO4– as a reference, a 0.08–0.16 V positive shift of Eo(cpd I) was determined for anionic axial ligands such as Cl–, F–, CH3CO2–, and benzoate, whereas a negative shift (−0.08 to −0.12 V) was found for neutral imidazole-type ligands.290 The cause of this discrepancy between theory and experiments is not clear at this writing and further investigations are needed.
Figure 29.

Redox potentials (vs SCEa) of cpd I models in CH2Cl2 at −60 °C. aUnder these conditions, the redox potential (E1/2) of ferrocene was 0.460 V at −60 °C and 0.528 V at 20 °C. bValues in parentheses were obtained from differential pulse voltammetry. cIn CH3CN at −35 °C. dIn CH2Cl2/CH3CN (1:1). Adapted from ref (290). Copyright 2011 American Chemical Society.
Synthetic porphyrin ligands almost always contain substitutions at the pyrrole β-carbon and/or the meso-porphyrin positions. These substitutions, as discussed in previous sections, are important for the reactivity of synthetic metalloporphyrins. They can prevent undesired side reactions that diminish the reactivity of metalloporphyrin catalysts during the oxidative reactions, such as the formation of μ-oxo dimers and the destruction of porphyrin ligands. As discussed in section 2.4.2, the electronic properties of meso- and pyrrole-β substituents substantially affect the energy of porphyrin a1u and a2u π–orbitals, thus changing the singly occupied orbitals of porphyrin radicals of compounds I. Although the electronic structure of the porphyrin radical (a1u or a2u) apparently does not affect cpd I reactivity very much,27 the introduction of electron-withdrawing groups at both positions were shown to greatly increase the reduction potential of cpd I (Figure 29)290,291 and thus increase the thermodynamic driving force of HAT.209,290 Indeed, the second order-rate constants for aliphatic hydroxylation mediated by an electron-deficient cpd I FeIV(O)(TPFPP•+) were 2–10 times larger than those obtained with FeIV(O)(TMP•+).211
3.1.3. Correlation of Thermodynamic Driving Force with Rate Constants of HAT
The thermodynamic driving force of HAT via cpd I is the net free-energy change resulting from the C–H bond that is broken and the O–H bond that is formed at the ferryl oxygen. In many C–H activation reactions mediated by iron–oxo intermediates, an empirical Brønsted-Evans–Polanyi (BEP) correlation has been observed,292 in which the free-energy barriers (ΔG‡), and thus the rates of hydrogen atom transfer (HAT) are negatively correlated with bond dissociation energies (BDE) of the scissile C–H bonds (eq 3, Scheme 3a).273,293 We use BDE here instead of bond dissociation free energies for simplicity because comparative data is more readily available and the differences are small. The slope α (Brønsted coefficient, 0 < α < 1) is interpreted to reflect the position of hydrogen along the reaction coordinate in the linear transition state [Fe–O---H---C]‡, with α = 0 referring to a reactant-like (“early”) transition state with a strong C–H bond character and α = 1 for a product-like (“late”) transition state with a strong O–H bond feature.294 While being broadly applied to a variety of HAT processes, the BEP relationship is less successful at correlating reactants with disparate molecular features. For instance, phenol O–H bonds have similar BDEs to benzylic C–H bonds but show much faster rates for HAT to iron–oxo intermediates.261 Such limitations of the BEP equation have led Mayer et al. to apply the Marcus cross correlation to analyze HAT, in analogy to Marcus electron transfer theory.295,296 Thus, for a HAT reaction, AH + B· → BH + A· (eq 4, Scheme 3b), the reaction rate for HAT (kAH/B) is estimated from the equilibrium constant Keq of the reaction and the two self-exchange rate constants (kAH/A and kBH/B) for the individual reactants A·/AH and B·/BH (eq 5 and 6). Since the rates of the self-exchange reactions can be experimentally determined in favorable cases (e.g., via NMR line broadening), Mayer’s method offers a powerful approach to predict kinetic properties of HAT from intrinsic properties of individual reactants.273 With this method, it was found that the self-exchange rate constants of phenols were ∼108 larger than those of alkanes, which matched the observed difference in their HAT rate constants.295 The origin of the faster phenol rates is likely due to the fact that a phenol is a good hydrogen bond donor while an aliphatic C–H proton is not.
Scheme 3. (a) Brønsted–Evans–Polanyi (BEP) Correlation for HAT Reactions and (b) Cross Correlation of HAT Reactions.

BDEs (or BDFEs) are purely thermodynamic properties and should not necessarily mirror the structural and entropy effects that affect transition-state energies of HAT events. As recently summarized by Bietti et al. for aliphatic C–H activation by alkoxyl radicals, structural effects (such as polar effects, stereoelectronic effects, and strain-release) and medium effects can have very large impacts on the reactivity of C–H bonds.297 Thus, the thermodynamically weakest C–H bond is not necessarily the most reactive one. This phenomenon has also been observed in C–H activation reactions of heme proteins and metalloporphyrins. A good example is the deactivation of α C–H bonds near electron-withdrawing groups such as ketones, esters, and nitriles in metalloporphyrin-catalyzed C–H hydroxylations and halogenations,298 although these bonds have lower BDEs than other reactants. This is a typical polar effect. Another factor that can affect rates of HAT is the entropic contribution to the free-energy barriers. Entropy effects would be most evident for C–H activation of substrates with weak C–H BDEs. These reactions will have early transition states with the C–H bonds elongated to a minimal extent. Thus, activation enthalpies (ΔH‡) are insensitive to C–H BDEs in this scenario, and overall free-energy barriers will be determined by the entropy term TΔS‡. Tanko et al. have measured rate constants of C–H abstraction by tBuO·, for which the O–H bond formed during HAT has a BDE of 104 kcal/mol.299 A good BEP correlation was found for C–H BDEs greater than 92 kcal/mol, whereas for C–H BDEs below 92 kcal/mol, a flat log kH versus BDE plot was observed. Further analysis showed similar ΔH‡ values (∼2 kcal/mol) for the flat region, and the entropy contribution outweighs that of enthalpy for most hydrogen abstractions by tBuO·.
We have found a similar nonlinear BEP correlation for C–H hydroxylation by the fungal heme-thiolate peroxygenase AaeAPO compound I (AaeAPO-I) (Figure 30).200 A Brønsted coefficient of about 0.4 was determined for log kH versus BDE for substrates with C–H bonds stronger than 90 kcal/mol, indicating a nearly symmetrical [Fe–O---H---C]‡ transition state in this region. But for BDE < 90 kcal/mol, the rate constant was not sensitive to C–H BDEs. Since the observed rate constants in the flat region were shown to be slower than both the active site substrate exchange and the diffusion limit, HAT by AaeAPO-I for C–H BDEs less than 90 kcal/mol is entropy-controlled and is more dependent on factors such as orientation, accessibility, and trajectory toward the transition state. These results are consistent with a FeIVO–H BDE in AaeAPO-II of ∼100 kcal/mol. The entropy effect was also found to affect the chemoselectivity of oxygenation reactions catalyzed by metalloporphyrins. Fujii et al. determined the activation parameters for epoxidation and allylic hydroxylation for cyclohexene oxidation mediated by FeIV(O)(TMP•+)Cl. By varying temperatures between 193 and 233 K, they determined the enthalpy and entropy of activation (ΔH‡ and ΔS‡) to be 8.0 ± 0.4 kcal/mol, −24.0 ± 1.6 cal mol–1 K–1 and 4.6 ± 0.6 kcal/mol and −40.3 ± 2.1 cal mol–1 K–1 for epoxidation and allylic hydroxylation, respectively.300 On the basis of these values, the contribution of the entropy term −TΔS‡ would exceed that of enthalpy term in the free energy of activation (ΔG‡) for allylic oxidation at temperatures above 114 K, while the entropy term provides the major contribution to ΔG‡ for epoxidation at temperatures above 334 K.
Figure 30.
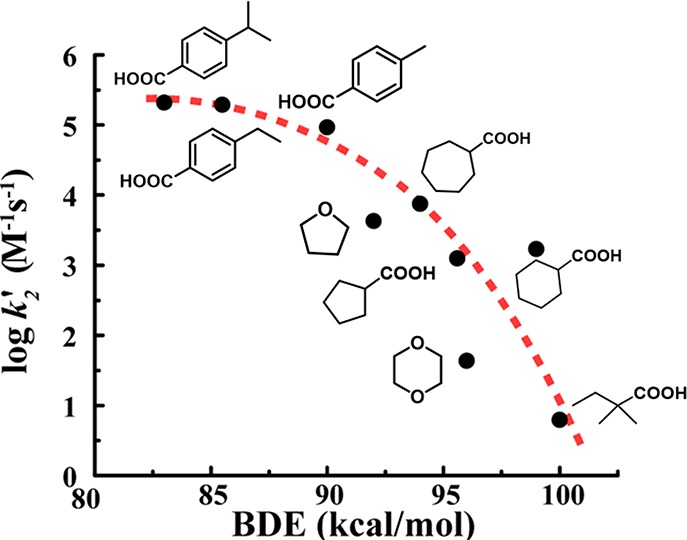
Nonlinear BEP correlation for C–H hydroxylation by AaeAPO-I. Adapted from ref (200). Copyright 2012 American Chemical Society.
3.1.4. Two-State and Multistate Reactivity
Compared to simple organic radicals such as tBuO·, a distinct feature of paramagnetic transition metal complexes is the existence of multiple spin states that can be close in energy. Thus, a given reaction may occur along different spin surfaces, and a higher-energy spin state could have the lowest-energy transition state. Such a situation can lead to spin crossover along the reaction coordinate from reactants to products. This concept was first proposed by Shaik, Schröder, and Schwarz and termed “two-state reactivity” (TSR) or “multistate reactivity” (MSR) (Figure 31a).301−303 HAT by compound I was one of the first systems to be studied for TSR in transition metal catalysis (Figure 31b). DFT calculations of cpd I of iron porphine revealed that the quartet and doublet spin states, wherein the porphyrin cation radical is either ferromagnetically or antiferromagnetically coupled to the S = 1 ferryl, were very close in energy and had comparable activation barriers for C–H activation, suggesting important contributions of both spin states for HAT.304 QM/MM studies also showed similar mechanistic features for HAT by P450 compound I.305,306
Figure 31.
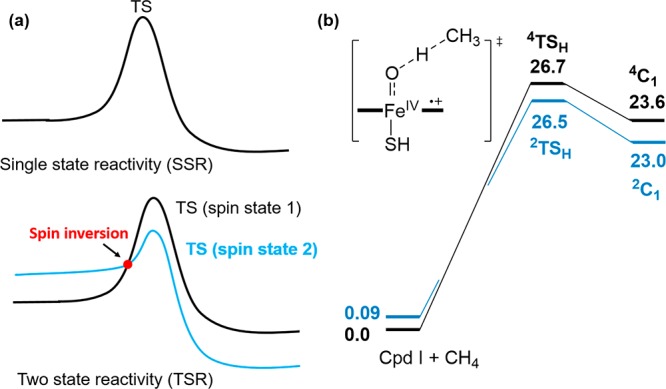
(a) Schematic illustration of two-state reactivity (TSR). (b) Two-state reactivity for C–H activation via an iron porphine catalyst (energies in kcal/mol, obtained from ref (305)).
C–H activation mediated by oxoMn(V) porphyrins represents another example of potential TSR in metalloporphyrin-mediated reactions (Figure 32).226,227 DFT calculations showed that the singlet ground state of oxoMn(V),243 which is known from the experiment,227,233,243 had a very high energy barrier for C–H activation.244,307,308 The triplet spin state, which is calculated to be only 3.4 kcal/mol higher than the singlet ground state, had the lowest-energy transition state (5.3 kcal/mol relative to the singlet ground state). The energy of the quintet state is much higher (11.1 kcal/mol) than the singlet state and is likely not accessible, despite the low activation energy of this spin state (2.2 kcal/mol). Therefore, during C–H activation, the unreactive singlet oxoMn(V) would undergo spin-crossing to the triplet energy surface.
Figure 32.
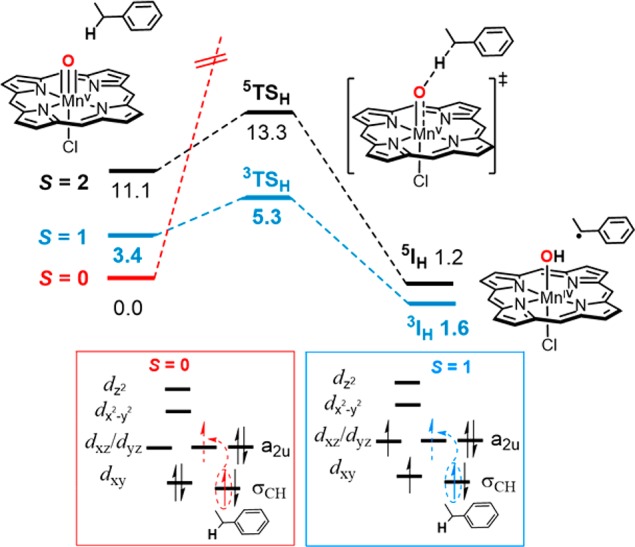
Energy profile for C–H activation mediated by an oxoMn(V) porphyrin complex.
Shaik recently introduced the concept of exchange-enhanced reactivity (EER) as a general principle to evaluate the preferred spin state for a given reaction state especially catalyzed by 3d metals.309,310 The main idea is that transition states with an increased number of unpaired spin-identical electrons at metal center would be stabilized by enhanced electron-exchange. Such an exchange can be assessed by multiplying the exchange interaction of two unpaired electrons (Kdd) with the number of such interactions (n). The gained exchange-energy (ΔKdd) versus the orbital promotion energy (ΔEorb) needed for promoting an electron into a higher-lying orbital would determine the preferred spin state. This concept can be utilized to analyze the preference for the triplet state in oxoMn(V)-mediated C–H activation in Figure 32.311 The transition state on the S = 0 surface is an open shell singlet with no spin-exchange interactions, while for the triplet surface, three exchange interactions are present. The overall stabilization effect in the triplet state can be estimated by 3Kdd – ΔEorb(dxy → dxz/yz), in which ΔEorb(dxy → dxz/yz) is the energy needed for orbital excitation from dxy to dxz/yz. Although these energies have not been determined for manganese porphyrins, the Kdd of 11.1 kcal/mol and ΔEorb(dxy → dxz/yz) of 26.4 kcal/mol have been estimated for an analogous oxoMn(V) corrolazine species, indicating a net stabilization of ∼6.9 kcal/mol. This stabilization energy is comparable to the calculated energy barrier difference between singlet and triplet pathways for this species.311
The kinetic isotope effect (KIE) for hydrogen/deuterium HAT has been proposed to be a good mechanistic probe to probe the reactive spin state of a reaction. Theoretical calculations have shown that different spin states can display very different KIEs.312−314 For instance, in the demethylation reaction of para-substituted N,N-dimethylaniline (p-Y-DMA), a positive linear correlation was found between experimental KIE values (KIEexp) and the Hammett substituent parameter of para-substituent Y.315−317 The calculation showed that only the low-spin doublet state showed this trend, while the calculated KIEs of high-spin quartet state (KIEHS) were much larger than the experimental values and were not sensitive to the para-substituent.318 This result suggested that the low spin state is the primary state for cpd I reactivity of N-demethylation. A further analysis of KIEexp using a blend of KIELS and KIEHS showed that for Y = H and Cl, the reaction occurred exclusively on the LS surface, while for Y = CN and NO2, the reaction occurred through both states with 70/30% and 50/50%, respectively.
The nature of the axial ligand can also impact the relative energies of different spin states and the spin-state crossing. For instance, the HAT activity of oxoMnV porphyrins with axial ligand X (X = H2O, OH–, and O2–) has shown to increase in the following order: H2O > OH– > O2–.227,233 DFT calculations indicated that this trend correlated with corresponding energy gaps between unreactive singlet ground state and higher-energy triplet and quintet states.244 In a related study of less reactive oxoMnV corrolazine (Cz) complexes, de Visser and Goldberg showed that fluoride and cyanide axial ligands increased the rate of HAT by 103–105-fold compared to that with no axial ligand.319 Computational studies showed 12 and 17 kcal/mol increases in the O–H BDE of Mn(IV)–OH for fluoride and cyanide axial ligands, respectively. The origin of the rate-enhancement exerted by axial ligands is a matter of debate. A computational study by Shaik indicated that fluoride and cyanide also stabilized the triplet transition state by 14–16 kcal/mol, due to a reduction in the σxy → π*xz/yz excitation energy and an increase in exchange coupling of unpaired d electrons in the triplet transition state.311 However, DFT and CASSCF calculations by the Goldberg and de Visser groups on MnV(O)(Cz)-mediated sulfoxidation of thioanisole indicated small spin–orbit coupling constants, suggesting a singlet-to-triplet spin-state crossing before the rate-limiting step is unlikely.320 In support of this statement, only the singlet spin state can computationally reproduce a V-shaped pattern observed in a Hammett analysis with para-substituted thioanisoles, in which a negative slope was obtained for electro-donating para-substituents but a positive slope was observed for electron-withdrawing substituents.320
In addition to increasing the reduction potential of cpd I, the porphyrin peripheral substituents can also affect cpd I reactivity via regulating the spin state crossover. Groves et al. have studied the influence of two cationic meso-substituents 2-N-methylpyridyl (2-TMPyP) and 4-N-methylpyridyl (4-TMPyP) on oxygen transfer reactivity of oxoMn(V) porphyrins.227 Surprisingly, the less electron-deficient [MnV(O)(4-TMPyP)], with the N-methyl pyridium ions farther from the porphyrin ring, was 10–103 times more reactive than MnV(O)(2-TMPyP). Computational studies showed that the 4-pyridyl isomer [MnV(O)(4-TMPyP)] had a smaller low-spin-high-spin promotion energy and was therefore kinetically favored in a two-state energy profile involving spin state crossing from the starting singlet oxoMn(V) species.243 Furthermore, the meso-substituents were also shown to affect the acid–base equilibria involving the axial water ligand. As discussed in section 2.4.3, MnV(O)(4-TMPyP) and MnV(O)(2-TMPyP) have three different prototropic forms in water depending on the protonation state of the axial ligand, oxo-aqua, oxo–hydroxo, and trans-dioxo.232 The reactivity of these three states increased in the following order: trans-dioxo < oxo–hydroxo < oxo-aqua. The less electron-deficient MnV(O)(4-TMPyP) exhibited higher basicity for the axial aqua and hydroxo ligands (Figure 33). Thus, at the same pH, the kinetically more reactive protonated species are more accessible for MnV(O)(4-TMPyP), leading to the observed higher reactivity.227
Figure 33.
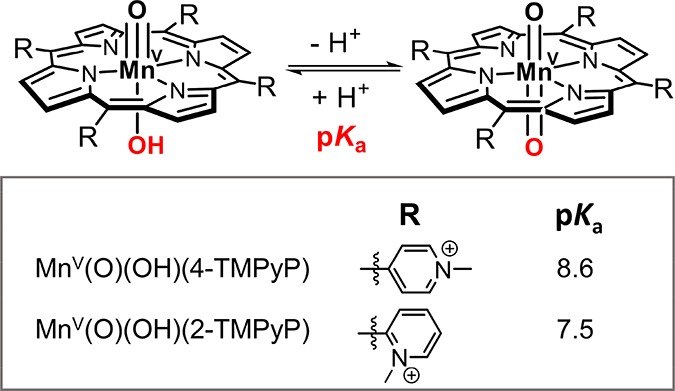
pKa of the hydroxo ligand of [MnV(O)(OH)(4-TMPyP)] and [MnV(O)(OH)(2-TMPyP)].
3.1.5. Quantum Tunneling in Hydrogen Atom Transfer
Hydrogen is a small particle and as such can show quantum tunneling effects.321 As a result, hydrogen can “tunnel” through the transition state barrier and thus increase the rate of HAT (Figure 34a). This energy reduction (ΔΔE‡tunneling) associated with proton tunneling is commonly between 1–4 kcal/mol.322 The kinetic hydrogen isotope effect is a useful tool to probe the tunneling phenomenon in HAT. There are three criteria proposed first by Kreevoy and co-workers with respect to organic radicals that are widely used for assessing a tunneling process: (1) a KIE significantly larger than 6.4 at 20 °C; (2) an activation energy difference between deuterium transfer and hydrogen transfer (Ea(D) – Ea(H)) larger than >5.0 kJ/mol (1.2 kcal/mol); and (3) a ratio of pre-exponential factors AH/AD < 0.7.323,324
Figure 34.

(a) Quantum tunneling (wavy arrow) leads to an effective energy barrier (ΔE‡eff) 1–4 kcal/mol below TS. (b) Reactivity pattern for [FeIV(O)(TMC)(L)]z+ in HAT from alkanes (red line) and oxygen-transfer to phosphines (back line). Figure is adapted from ref (328). Copyright 2015 American Chemical Society.
Tunneling effects have been found in HAT processes mediated by a variety of metalloenzymes and related metal–oxygen model compounds.325−328 Many of these systems are nonheme iron-containing enzymes and/or their model compounds.325−329 In contrast, there are few examples of tunneling effects in cytochrome P450s and metalloporphyrins. Although KIEs (10–32) larger than usually adopted theoretical limit (∼7) for nontunneling reactions have been reported for several P450-catalyzed reactions,330−334 the extent of hydrogen tunneling in these reactions is not conclusive because of the difficulties in determining the activation energies and pre-exponential factors of these systems. In P450 model compounds, an early example of a possible tunneling event was reported by Sorokin and Khenkin.335 They determined a KIE value of ∼22 (20 °C) for cyclohexane oxidation catalyzed by Fe(TMP)Cl/NaOCl in benzene. They further determined a [Ea(D) – Ea(H)] of 4.5 ± 0.5 kcal/mol and A(H)/A(D) = 0.01, indicating the presence of a tunneling effect. Similar effects have also been observed by Meunier et al. in a study of intramolecular KIEs of adamantane-1,3-d2 hydroxylation catalyzed by a variety of metalloporphyrins.333 For an Fe(TMP)Cl/NaOCl system, they observed a KIE of 9 (10 °C), [Ea(D) – Ea(H)] of 1.83 kcal/mol, and A(H)/A(D) of 0.35. Recently, large KIE values indicative of tunneling effects were also reported by Newcomb et al. for benzyl alcohol oxidation mediated by FeIV(O)(TMP•+)(ClO4–)336 and by Fujii et al. for C–H(D) oxidations of xanthene and tetralin promoted by FeIV(O)(TMP•+)(L) (L = NO3–, Cl–, imidazole).337 It is worth noting that the KIE and tunneling effects are very dependent on the position of transition state along the reaction coordinate, which will be dramatically affected by the properties of ligands, metal centers, solvents, and substrates.326,327,338 For instance, while most studies of tunneling effect have been conducted on iron porphyrins with electron-donating tetramesityl porphyrin ligand, manganese porphyrins and electron-withdrawing iron porphyrins like Fe(TDCPP)Cl exhibited much smaller KIEs (3–7) and catalyzed the reactions through classical HAT pathways.333,335−337
H-tunneling could potentially contribute to the reactivity difference between hydrogen atom transfer (HAT) and oxygen atom transfer (OAT), as OAT reactions will not be affected by H-tunneling effect. For example, in a series of nonheme iron(IV)–oxo tetramethylcyclam (TMC) complexes with different axial ligands L ([FeIV(O)(TMC)(L)]z+ (z = 1, 2)), the reactivity for HAT increased with the electron-donating ability of L, while oxygen atom transfer (OAT) showed an opposite trend (Figure 34b).339−341 This reactivity difference was attributed to a combined effect of spin inversion probability and quantum tunneling.328,342 In addition to decreasing the triplet/quintet energy gap and facilitating spin crossover, the most electron-donating thiolate axial ligand (compound 63, Figure 34b) also decreased the energy barrier by 3.4 kcal/mol via a quantum tunneling effect, in comparison to the 1.4 kcal/mol obtained with the CH3CN axial ligand. Such a quantum tunneling effect was absent in oxygen atom transfer reactions and is the main attribute of the reactivity difference between HAT and OAT. The reactivity difference between HAT and OAT reactions were also explored for cyclohexene oxidation mediated by FeIV(O)(TMP•+)L. It was found that ratio of epoxidation/allylic oxidation products decreased dramatically with decreasing temperatures.343 This trend is further studied by Fukuzumi and Nam et al.344 They found that while epoxidation was preferred at higher temperatures (213–233 K), the allylic hydroxylation took over at lower temperatures (193–213 K). This switchover was accompanied by a drastic increase in KIEs for allylic hydroxylation (1.4 at 233 K vs 13 at 193 K). The large difference in activation energy [Ea(D) – Ea(H) = 5.0 kcal/mol] and a small ratio of exponential factors (AH/AD ∼ 4 × 10–5) suggest a significant contribution from tunneling for the allylic hydroxylation pathway.
The enhancement of HAT reactivity by H-tunneling is also manifested in an unusually reactive hydroxoiron(III) porphyrazine complex 65 (Figure 35) reported very recently by Gao and Groves.345 This hydroxoferric species abstracts benzylic C–H bonds with rate constants of 2.22 × 103 M–1 s–1 for xanthene and 81 M–1 s–1 for dihydroanthracene. These rate constants are 5–6 orders of magnitude faster than other typical FeIII–OH complexes and more reactive than many ferryl complexes (Figure 35a).34565 displayed an unusually high reduction potential of 0.84 V versus NHE and a very small activation enthalpy of 6.1 ± 0.3 kcal/mol for C–H scission. A large tunneling component was inferred from the observed deuterium isotope parameters (Figure 35b),327,346,347 KIE of 20.2, Ea(D) – Ea(H) = 2.5 ± 0.4 kcal/mol, and A(H)/A(D) = 0.28 ± 0.14 for xanthene oxidation, contributing to the unusual activity of 65.
Figure 35.

(a) Correlation of C–H HAT rate constants with O–H BDE for a variety of metal–oxo and metal–hydroxo oxidants at 298 K. The black dashed line passes through permanganate, t-butylperoxyl, and t-butoxyl radicals for reference. The red ■ is (PyPz)FeIII–OH. Reported hydroxometal(III) complexes (green ▲), Fe(IV)=O complexes (blue ▲) and nitroxyl radicals HBTO and PINO (magenta ◆) are also shown for comparison. (b) A highly reactive hydroxo-iron(III) porphyrazine complex 65 for HAT. Adapted from ref (345). Copyright 2017 American Chemical Society.
3.2. Intermediacy of Substrate Radicals–Oxygen Rebound
In P450-catalyzed hydroxylations, hydrogen atom transfer (HAT) has been shown to generate substrate radicals with short lifetimes. The results can be best understood as leading to an active site caged substrate radical [Fe(IV)–OH ·R] that subsequently rebounds to the FeIV–OH intermediate to afford hydroxylation products. The intermediacy of substrate radicals in such processes was first implicated in model reactions related to Fenton’s reagent, a mixture of Fe2+ and H2O2. The use of cyclohexanol (66) and trans,trans-3,5-dideuterocyclohexanol (67) as diagnostic substrates clearly indicated a radical epimerization process after HAT (Scheme 4a).188 Later, similar radical epimerization or rearrangement processes were found in the hydroxylation of exo,exo,exo,exo-2,3,5,6-tetradeuteronorbornane (68) and selectively deuterated cyclohexene (69) by phenobarbital-induced P450LM2 (CYP2B4) as well as reactions mediated by synthetic iron porphyrins (Scheme 4b).189,348 These results clearly indicated a stepwise, nonconcerted mechanism for C–H hydroxylation mediated by cpd I.
Scheme 4. (a) C–H Hydroxylation of Cyclohexanol and Dideuterocyclohexanol Revealed a Step-Wise Radical Mechanism for C–H Hydroxylation Mediated by Fe2+/H2O2 and (b) Radical Epimerization and Rearrangement Were Also Observed for Hydroxylation Catalyzed by P450s and Model Compounds.
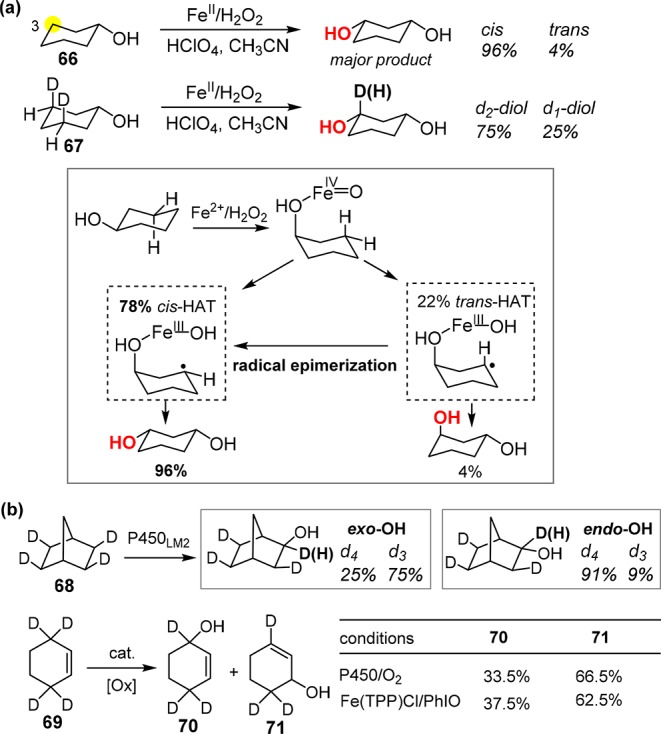
Additional insights regarding this stepwise radical C–H cleavage event were obtained using mechanistically diagnostic substrates and radical clock probes, which form radicals that undergo characteristic stereochemical/structural changes at known rates.349 A highly informative diagnostic clock is the simple hydrocarbon norcarane, which will afford different rearranged products for pathways involving radical and cationic intermediates (Scheme 5).350 Thus, the 2-norcaranyl radical (72 in Scheme 5) will ring-open to a kinetically favored homoallylic radical (73), whereas formation of the thermodynamically more stable cyclohepten-4-yl cation (75) is the favored rearrangement pathway for the 2-norcaranyl cation (74). Other small-ring systems, such as bicyclo[2.1.0]pentane and a variety of substituted cyclopropanes are also common radical clocks for the study of HAT in P450s and model systems.16 Radical clocks provide a facile way to measure the average lifetime of radical intermediates from the ratio of unrearranged products (U) to rearranged products (R) (Scheme 5). It is now known that P450s and synthetic iron porphyrins generally have very short radical lifetimes, tens to hundreds of picoseconds, with corresponding rebound rate constants between 1010 and 1011 s–1.16 A similar time frame is also measured for hydroxylation catalyzed by several nonheme systems such as soluble methane monooxygenase (sMMO) and toluene 4-monooxygenase (T4moH).351−356 Recently, with norcarane radical clock, the rebound rate of AaeUPO was determined to be 1.06 × 1011 s–1 (radical lifetime: 9.4 ps), which is much faster than the values of other functional P450s.199 Notably, substrate radical lifetimes in the picosecond time regime provide time for many molecular vibrations and rotations in an enzyme active site or solution solvent cage. However, there would not be time for such a short-lived radical to diffuse away from the active site.
Scheme 5. Radical and Cation Rearrangement of Norcarane and Radical Lifetimes of Representative P450s and Nonheme Oxygenases As Determined by the Measured Ratios of Rearranged (R) and Unrearranged (U) Products.
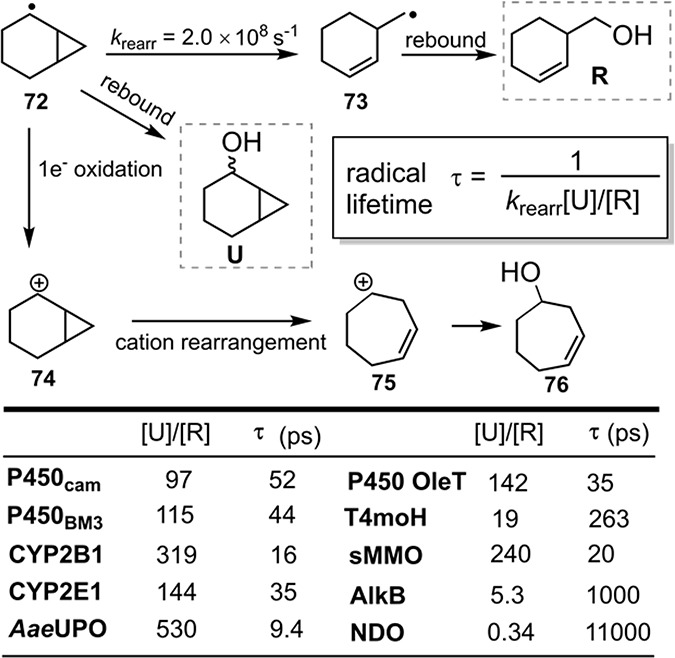
Direct study of the radical rebound step is difficult because of the fast recombination rate between substrate radicals and FeIV–OH species (1010–1011 s–1) and the difficulty of preparing discrete FeIV–OH intermediates. Very recently, Green and Goldberg et al. have successfully prepared a stable monomeric FeIV–OH complex with a sterically hindered tris(2,4,6-triphenyl)phenyl corrole (ttppc) ligand.357 This complex (denoted as [FeIV(ttppc)(OH)]) exhibits a Fe–O bond distance of 1.857 Å, a resonance Raman vibration ν(Fe–O) of 555 cm–1, a small Mössbauer isomer shift (0.13 mm/s) and a large quadrupole splitting (2.21 mm/s). These features are characteristic of a FeIV–OH species. Treating [FeIV(ttppc)(OH)] with stable radicals like the trityl radical (Ph3C·) led to the formation of the hydroxylation product (Ph3COH) and FeIII(ttppc) (Scheme 6). Kinetic measurements of this radical recombination step with a series of para-substituted trityl radicals afforded second-order rate constants (k), ranging from 12.6 to 357 M–1 s–1. A Hammett analysis with σ+para parameter showed a small slope (ρ = −0.55). A Marcus cross-relation between (RT/F) ln(k) and redox potentials of substituted trityl radicals (Eox[(p-X-C6H4)3C+/•]) also yielded a small slope (−0.15). These results clearly indicate that, despite the low oxidation potential of trityl radicals, their recombination with [FeIV(ttppc)(OH)] follows a concerted pathway with small charge separation, as opposed to a stepwise mechanism in which the carbon-centered radical is first oxidized to cations and then recombines with hydroxide.
Scheme 6. Radical Recombination between Trityl Radicals and [FeIV(ttppc)(OH)]. Adapted from ref (357). Copyright 2017 American Chemical Society.
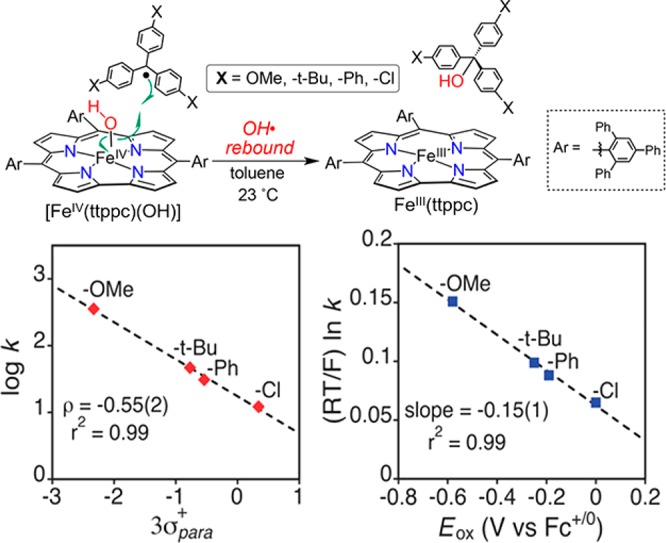
After HAT, substrate radicals R· in the resulting [FeIV–OH ·R] cage are not static. Factors such as tightness of the cage, ease of solvent reorganization, the rate of radical rebound, and the nature of the substrate radical can all dramatically affect the behavior of R·.13 An instructive manifestation of the interplay between these factors is the C–H hydroxylations catalyzed by nonheme diiron hydroxylase AlkB, in which the use of three clock substrates with very different rearrangement rates (kr) showed similar R/U ratios and led to drastically different apparent radical lifetimes, all in the low nanosecond time regime (1–200 ns) (Scheme 7).358,359 The cause of this enigmatic behavior is a competition between the relatively slow radical rebound and diffusive radical cage escape to generate a solvent-separated radical pair (RPss). If the rate of radical cage escape (ke) is comparable to that of radical rebound kR and larger than the radical rearrangement rates krearr, then R/U ratios will be insensitive to the differences in krearr but only reflect the competition between cage escape and radical rebound ke/kR. Significantly, measured radical lifetimes for AlkB hydroxylases were found to be in the nanosecond time regime, much longer than those obtained for heme proteins such as P450 and APO. An embodiment of the cage escape effect in AlkB system is a decrease in radical lifetimes by increasing the substrate concentration. Higher substrate concentration would lead to more substrate molecules present in the long and hydrophobic substrate channel in AlkB,360 which could displace and reorganize the water in the active site and create a crowded environment that reduces the radical cage escape.
Scheme 7. Oxidation of Radical Clocks with Different Rearrangement Rate Constants.

The similar ratios of rearranged/unrearranged products (R/U) result from the competition between cage escape and rebound of substrate radicals. Adapted with permission from ref (358). Copyright 2008 Wiley-VCH.
In addition to the environment of the radical cage, the nature of the metal center can also dramatically affect the radical lifetime. For instance, manganese porphyrin-catalyzed hydroxylation exhibited long radical lifetime (ns), whereas no rearranged product was observed for ruthenium porphyrin-catalyzed reactions.220,361 The former might relate to a lower reduction potential of MnIV–OH compared to FeIV–OH, and the latter might be caused by an unusually high electrophilic nature of RuV=O.362 Like HAT, radical rebound can also occur along different spin surfaces and are subject to spin crossover events (Figure 36). DFT calculations for methane hydroxylation by a simplified heme cpd I showed that the rebound barrier on the quartet transition state is much higher than that on the doublet spin state due to the difference in frontier orbital interactions.304 In the quartet state, the unpaired electron of the radical goes into a higher-energy 3dz2 orbital, whereas in the doublet state, the electron transfers into a lower-energy singly occupied dxz (or dyz) orbital.
Figure 36.
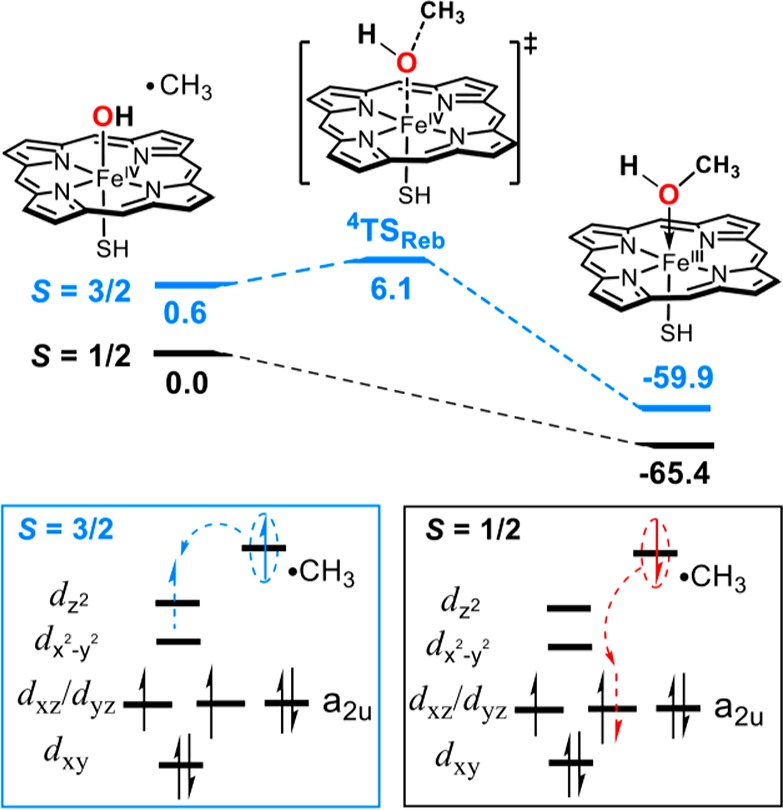
Oxygen rebound of different spin states (energies in kcal/mol, obtained from ref (305)).
Groves et al. have found a significant impact of the donor properties of the axial ligands on rates of radical rebound. Manganese porphyrins with anionic electron-donating ligands such as OH– and F– were found to afford mainly radical rearranged products in hydroxylation of norcarane, whereas radical unrearranged products prevailed in reactions catalyzed by Mn porphyrins with neutral axial ligands such as pyridine and CH3CN (Scheme 8).298,363 This effect is reminiscent of axial ligand effect on HAT. Electron-donating ligands would diminish the reduction potential of MnIV–OH and destabilize frontier orbitals (3dz2 and dxz/dyz), leading to a higher rebound barrier. The deceleration of rebound would facilitate the escaping of substrate radicals from the radical cage. Indeed, a large amount of radical dimerization products were observed in xanthene hydroxylation by manganese porphyrins in the presence of F– with yield dependent on the concentration of F–.
Scheme 8. Effect of Axial Ligands on Radical Rebound to MnIV–OH Porphyrins.

The rebound step in the hydroxylation of deuterated ethylbenzene catalyzed by iron and manganese porphyrins has been explored recently.363 The results showed that more electron-deficient porphyrins displayed less stereochemical inversion at the benzylic hydroxylation site (Figure 37). The extent of stereochemical inversion was found to be sensitive to the metal center, with only 8–20% retention of configuration for Cl–MnV(O)(Por), while Cl–FeIV(O)(Por•+) showed 80–90% retention. Significantly, F–FeIV(O)(Por•+) afforded only 60–70% retention, showing that the donor properties of the ligand can also affect iron porphyrin stereochemistry. These results are also consistent with the general idea that oxygen rebound is faster for stronger oxidants and more able to compete with radical cage escape. The radical cage escape event was recently explored in nonheme systems by Nam and Shaik.364 It was determined that such processes occurred in a number of nonheme metal–oxo systems such as MnIVO, FeIVO, CrIVO, FeVO, and RuIVO. DFT calculations revealed small dissociation energies and relatively large rebound barriers for these systems.
Figure 37.
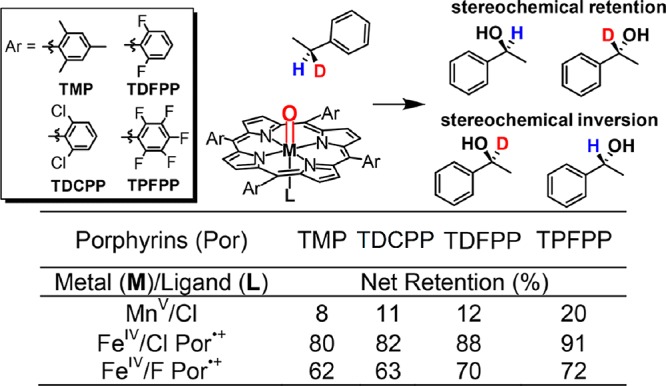
Stereochemistry of the rebound process for iron and manganese porphyrins. Adapted from ref (363). Copyright 2017 American Chemical Society.
3.3. Redirecting Substrate Radicals to Reaction Pathways Other than Oxygen Rebound
3.3.1. Decarboxylation Mediated by P450 OleTJE
P450 OleTJE, found in Jeotgalicoccus sp., converts Cn fatty acids to Cn–1 terminal alkenes through a desaturative decarboxylation (eq 9, Figure 38).365 This enzyme has attracted considerable interest for its potential applications in biofuel production and fine chemical synthesis.366−369 P450 OleTJE belongs to the P450 peroxygenase family CYP152. Other notable members in this family are P450SPα (CYP152B1) and P450BSβ (CYP152A1),135,140,370 which have 36% and 40% amino acid identity to P450 OleTJE, respectively. All three enzymes utilize an active-site arginine and the carboxylate group provided by the substrate to effect the heterolytic cleavage of H2O2 to generate compound I. Despite the similarity in H2O2 activation and active site structures to P450 OleTJE, P450SPα and P450BSβ catalyze hydroxylations of fatty acid substrates rather than decarboxylation (Figure 38).135,136,277,370 P450SPα hydroxylates only at the Cα position (eq 10), whereas P450BSβ hydroxylates both the Cα and Cβ positions of fatty acid substrates (eq 11).
Figure 38.
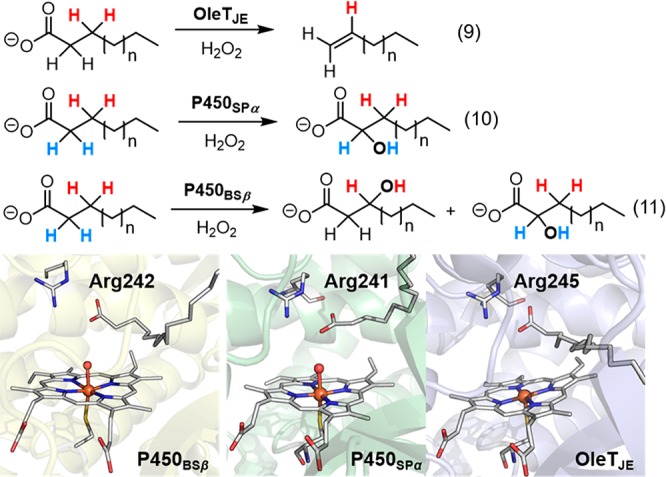
Native reactions catalyzed by P450 OleTJE (PDB: 4L40), P450SPα (PDB: 3AWM), and P450BSβ (PDB: 1IZO). All three enzymes have a conserved arginine residue that can form a salt bridge with the carboxylate group of fatty acid substrates.
The decarboxylation reaction catalyzed by P450 OleTJE is initiated by a hydrogen atom transfer (HAT) from the Cβ position, as indicated by the large primary kinetic isotope effect (KIE ≥ 8.1) for fully deuterated fatty acid substrates.371 This finding indicates that the oxygen rebound that normally occurs after HAT in P450 catalysis is inhibited in P450 OleTJE. The substrate radical is thus redirected to decarboxylation presumably via a single electron transfer to cpd II and a subsequent C–C cleavage with concomitant loss of CO2 (Figure 39). It is worth noting that a similar Cβ-H scission step has also been identified as the key step for the decarboxylative desaturation catalyzed by UndA, a nonheme iron oxidase that can synthesize medium-chain 1-alkenes from fatty acids.372,373 Very recently, Makris and co-workers observed OleT compound I and compound II using stopped-flow spectroscopic techniques by employing deuterated substrates to slow the initial HAT step.374 Intriguingly, OleT compound II was remarkably stable, having a lifetime of hundreds of milliseconds after its preparation from compound I via substrate C–H abstraction. The unusual stability of OleT-II supports the proposal that this enzyme imposes an inhibition of oxygen rebound, which would normally occur with rate constants in the range of 1010–1011 s–1 for heme-thiolate proteins. What causes this remarkable slow-down of the radical rebound process is still not entirely clear. But the fact that the amount of hydroxylated side products varied substantially with fatty acid chain length suggests that the binding and the positioning of substrates play a crucial role in affecting the reaction pathway.369,375 Evidence in favor of rebound inhibition by substrate–protein interactions has recently been provided by the observation that the radical clock probe norcarane is a substrate for OleTJE.376 Further, the radical lifetime for the norcaranyl radical intermediate was typical for other P450 proteins, in the picosecond time regime. Apparently, the radical intermediate of a small hydrocarbon substrate cannot be restrained by the active site architecture and rebound proceeds normally.
Figure 39.

Proposed mechanism for the oxidative decarboxylation catalyzed by OleTJE.
Li et al. have recently examined several key active site residues of OleTJE and found that Arg254, His85, and Ile170 are all required for the decarboxylation activity (Figure 40).377 The roles of these residues are thought to be the formation of a salt-bridge with the substrate carboxyl group (Arg245), acting as a proton donor (His85), and regulating substrate positioning in the active site (Ile170). As discussed in section 2.3.1, the active site structure of the CYP152 family of enzymes is similar to that of AaeUPO in that both proteins have an arginine in the distal pocket that forms a salt bridge with a nearby carboxylate that facilitates hydrogen peroxide binding and O–O bond scission. In AaeUPO, the carboxyl group is provided by a glutamate residue, while in CYP152 the carboxyl group is from the fatty acid substrate. Makris et al. recently found that a mutation of Pro246 to Asp in OleT circumvents the requirement for fatty acid substrates for H2O2 activation.367 The resulting OleT P246D variant is not active toward native fatty acids but showed high activity for a broad range of hydrocarbon substrates.
Figure 40.
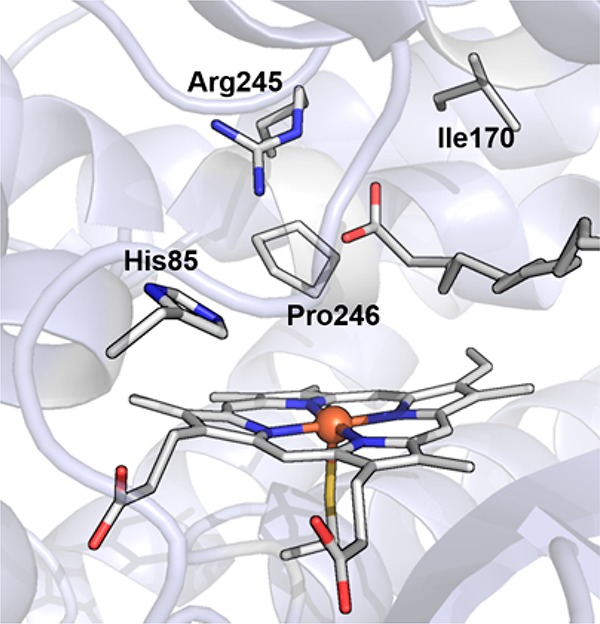
Key active site residues of OleTJE (PDB: 4L40).
3.3.2. P450-Catalyzed Desaturation Reactions
In addition to obtaining olefins via decarboxylative elimination, P450 has also shown capabilities for forming C=C bonds via dehydrogenation. An early example is the formation of Δ4-valproic acid (Δ4-VPA) during the metabolism of the antiepileptic drug valproic acid (VPA) (Figure 41a).378 Other representative substrates that undergo P450-catalyzed desaturation include warfarin, ethyl carbamate, and retinol.279,379 The fact that purified P450 catalyzed both desaturation and hydroxylation in most cases and the alcohol and desaturation products did not interconvert indicates a bifurcation of the usual rebound mechanism. Furthermore, almost identical values for the primary kinetic isotope effects were determined for hydroxylation and desaturation products of VPA.380 On the basis of these results, it is proposed that the substrate radical resulting from the initial hydrogen-atom transfer (HAT) by cpd I will undergo a second HAT or proton-coupled electron transfer process (PCET) by compound II at C–H bonds vicinal to the carbon-centered radical, affording the desaturation product and H2O (Figure 41a). This mechanism is reminiscent of the desaturation pathway of fatty acid desaturases, in which a bis-μ-oxo diiron(IV) cluster mediates two sequential HAT to form C=C bonds in long-chain fatty acyl derivatives (Figure 41b).381,382 A recent mechanistic study of the histidine-rich nonheme diiron enzyme AlkB using diagnostic substrates norcarane and deuterated norcarane revealed H atom abstraction occurred at both C2 and C3 (Figure 41b).359 While the C2 radical leads to the formation of 2-norcaranol and the radical-rearranged homoallylic alcohol product, desaturation and hydroxylation products were obtained from the C3 radical with similarly large KIEs (∼20). From these results, a bifurcation of the reaction pathways leading from the Fe–OH/R· intermediate appears to be a common feature for both heme- and nonheme-catalyzed desaturation reactions.
Figure 41.

(a) P450-catalyzed hydroxylation and desaturation of valproic acid (VPA) and the proposed mechanism. (b) Proposed mechanism of fatty acid desaturases and the hydroxylation and desaturation of norcarane catalyzed by nonheme diiron hydroxylase AlkB.
The factors that control the hydroxylation/desaturation pathways have been studied via computational methods.383,384 In general, the desaturation pathway was found to be less exothermic than the hydroxylation pathway. The energy barriers of two reaction channels varied depending on the spin state and electronic state of the FeIVOH intermediate, the properties of the substrate radicals, and the active site environment. For instance, comparable energy barriers were calculated for desaturation and oxygen rebound on low-spin doublet surface, whereas the desaturation step is more unfavorable along quartet surfaces.383,384 Shaik also suggested that FeIV–OH species can also exist as a FeIII–OH porphyrin cation radical electromer, whose energy barrier for the desaturation pathway is 8–20 kcal/mol higher than that of oxygen rebound.384 The desaturation pathway also requires the approach of FeIV–OH to C–H bonds vicinal to the substrate radical. If this step is restricted by the substrate/enzyme interaction then the desaturation pathway will be inhibited. In the case of camphor, gas-phase calculations showed similar barriers for desaturation and oxygen rebound on both doublet and quartet surfaces, however, the desaturation pathway was over 15 kcal/mol higher than oxygen rebound in the enzyme active site. An energy scan along the reaction coordinate revealed that the tight binding of camphor in the active site disfavored the reactive conformation that leads to the desaturation product.384
3.3.3. Oxidative Rearrangement Catalyzed by PntM (CYP161C2)
CYP161C2 catalyzes the conversion of pentalenolactone F (77) to pentalenolactone (78), a sesquiterpenoid antibiotic synthesized in Streptomyces arenae (Figure 42a).385 This transformation features an oxidative cationic rearrangement, which is very unusual in P450-mediated transformations but similar to the cation rearrangement pathways typically encountered in terpene cycloases. The reaction involves an initial hydrogen atom abstraction of Hsi at C1 followed by an electron-transfer oxidation of the substrate radical by compound II. The carbocation intermediate can then undergo 1,2-migration of a methyl group with a subsequent loss of a proton at C3 (Figure 42a). Cane et al. have studied the mechanism of PntM using X-ray crystallography, kinetic measurements, site-directed mutagenesis, and product analyses.386 The inhibition of the rebound step in PntM could arise from the active site residues that stabilize the carbon cation intermediate. Examination of substrate-bound crystal structure of PntM revealed three active-site residues (F232, M77, and M81) that are in close proximity to C1, C2, and C3 and are unique among all known P450s (Figure 42b). However, mutation of these sites still afforded the pentalenolactone 78 as the only product, albeit with lower activity. Another possible cause of the inhibition of the rebound step is steric hindrance at the carbon-centered radical. After hydrogen abstraction at C1–Hsi, the subsequent rebound step can be retarded by methyl and vinylidene groups at the si face of C1, whereas the outer-sphere 1e– transfer would not be affected by the steric bulkiness at C1. The hydroxylation of 1-deoxypentalenic acid (79, Figure 42), which is structurally analogous to pentalenolactone F, by CYP105D7 provides additional support for this hypothesis. CYP105D7 abstracts the hydrogen at the re side of C1 followed by a normal oxygen rebound process (Figure 42b).387 Compared to the si face, C1 re face has much smaller steric hindrance with only a hydrogen atom and a methyl group at neighboring carbons (Figure 42c).
Figure 42.

(a) Proposed mechanism for the oxidative rearrangement of pentalenolactone F catalyzed by PntM. (b) Active site structures of PntM with bound pentalenolactone F (PDB: 5L1O). (c) Steric environment of C1 si and re sides of pentalenolactone F and hydroxylation of 1-deoxypentalenic acid by CYP105D7.
3.3.4. Mn-Catalyzed C–H Halogenations
In the early studies of Mn porphyrin-catalyzed hydroxylations, chlorination side products were observed. The chlorine source in those side reactions was determined to be the chloride ligand on the Mn porphyrin catalysts.220 Radical clock studies indicated an unusually long radical lifetime for the chlorination side product (∼25 ns). A lifetime this long suggests that the substrate radical intermediate may be freely diffusing. A detailed mechanistic study later revealed that radical lifetimes in Mn porphyrin-catalyzed hydroxylation can be controlled by axial ligands, as noted above, in which the rebound can be slowed down by anionic electron-donating axial ligands.298,388 Another important finding was that a Mn(IV)-X type of intermediate (X = halogens), such as the well-characterized trans-difluoromanganese(IV) porphyrins, can trap independently generated alkyl radicals to yield halogenation products in a process analogous to the oxygen rebound (Figure 43a).389 These two mechanistic findings enabled the development of a series of aliphatic C–H halogenation reactions catalyzed by manganese porphyrin or manganese salen complexes (Figure 43, panels b and c).298,390−394 The catalyst-dependent regioselectivity and large primary kinetic isotope effect (KIE > 5) unambiguously indicated the involvement of oxoMn(V) intermediate in the C–H activation step. The radical nature of the halogen transfer step was implied by a large amount of radical rearranged products observed with the probe substrate norcarane. Furthermore, moderate enantioselectivity (20%–70%) was observed when a chiral manganese salen catalyst was used (Scheme 9a). Kagan analysis has shown a linear relationship between enantiomer excess (ee) of products and ee of manganese salen chiral catalysts.394 These results indicate that halogen transfer proceeded through a catalyst-bound halogen intermediate, namely Mn(IV)-X.
Figure 43.

(a) Formation of fluorination products via trapping of in situ generated alkyl radicals by MnIV(TMP)F2. (b) Concept of manganese-catalyzed C–H halogenation reactions via heteroatom rebound. (c) Mn-catalyzed C–H halogenation and pseudohalogenation reactions.
Scheme 9. (a) Enantioselective C–H Halogenation-Catalyzed by a Chiral Manganese Salen Catalyst and (b) Radical lifetimes of Mn-Catalyzed C–H Halogenation Reactions.
What determines the bifurcation of substrate radicals to halogen rebound instead of oxygen rebound in this case? Clearly the nature of metal center is important, as switching from manganese porphyrins to iron porphyrins afforded mainly oxygenation products.363 In addition, hydroxylation products prevailed if more electron-withdrawing manganese porphyrins such as manganese 5,10,15,20-tetrapentafluorophenylporphyrin (TPFPP) were used. These results are consistent with a deceleration of oxygen rebound, as both the iron center and electron-withdrawing porphyrins were known to cause a faster oxygen recombination. This analysis is consistent with radical clock analysis, as all C–H halogenations exhibit unusually long radical lifetimes (2–30 ns) (Scheme 9b). Although the identity of the Mn(IV)-X intermediate is still not fully elucidated, especially the nature of the axial ligand trans to the halogen. DFT calculations showed that halogen rebound of alkyl radicals to Mn(IV)-X has low energy barriers, comparable to or even lower than the oxygen rebound.389,391 The facile halogen transfer is also indicated by the successful application of C–H fluorination for 18F labeling.390 Unlike normal catalytic conditions, [18F]fluoride is present only in minuscule amounts (pmol–nmol) and other reagents (substrate, oxidant, and catalysts) are all in vast excess. Thus, Mn(IV)-18F intermediates must be competing effectively with a large amount of Mn(IV)–OH intermediates to trap alkyl radicals via fluorine transfer. Therefore, the observed high 18F conversion into target molecules indicates a preferential radical rebound to Mn(IV)–F.
These Mn-catalyzed halogenations are reminiscent of C–H chlorinations catalyzed by α-ketoglutarate (αKG)-dependent nonheme iron halogenases, in which a chloroferryl intermediate (Cl–FeIV=O) has been detected spectroscopically. This chloroferyl intermediate abstracts a C–H bond from substrates, and substrate radicals recombine with the chlorine rather than the hydroxyl group cis to the chloride in the rebound intermediate, Cl–FeIII–OH (Figure 44a).395−401 It has been shown by Bollinger and Krebs that substrate orientation is crucial for the preference for chlorine rebound.395,396,402,403 Computational and structural studies revealed that the oxo group repositions itself from an axial position to the equatorial plane defined by the chloride and α-KG (Figure 44, panels a and b).395,396,399 This geometry facilitates π pathways for both the C–H abstraction and the subsequent radical recombination step. Solomon et al. analyzed the frontier molecular orbitals of Cl–FeIII–OH. Although the Fe–OH bond is intrinsically weaker, the dπ*(Fe–OH) was found to be 4.5 kcal/mol higher in energy than dπ*(Fe–Cl), leading to a lower energy barrier for chlorine rebound (Figure 44c).404 Further, their results showed that the chlorine rebound to a substrate radical is best described as an asynchronous electron transfer from the substrate radical to iron followed by late ligand transfer to the substrate in the same single transition state, instead of a direct ligand radical transfer. This description should apply as well for the fluorine transfer step in manganese porphyrin-catalyzed fluorinations, as the generation of fluorine radicals is unlikely due to the enormously high one-electron oxidation potential of fluoride.405
Figure 44.
(a) Mechanism of C–H chlorination catalyzed by αKG nonheme halogenases. (b) Active site structure of a nonheme halogenase, WelO5 (PDB: 5IQT), with a bound 12-epi-Fisherindole U substrate. (c) Orbital diagram of dπ*(Fe–OH) and dπ*(Fe–Cl).
3.4. Oxygen-Atom Transfer Reactions Catalyzed by Compound I
In addition to activating aliphatic C–H bonds, P450s and their model compounds could also catalyze a myriad of oxygen-atom transfer reactions. These reactions, including alkene epoxidation, aromatic hydroxylation, and N- and S-oxidation, have been extensively reviewed.25,26,92,306,379 Herein we will only briefly discuss their mechanistic features. O-, S-, and N-dealkylations, although not belonging to OAT reactions, will also be discussed in this section for comparison with corresponding OAT reactions.
Alkene epoxidations catalyzed by P450s are widely present in natural product biosynthesis and drug metabolism.406 Also, the infamous epoxidation of benzpyrene by CYP1A1 is an important initial step in the etiology of human cancer.407 The mechanism of this oxygen transfer event appears to begin with an electrophilic ferryl addition to a C=C bond to form an alkyl radical intermediate, which undergoes a subsequent radical rebound step to forge the epoxide ring (Figure 45a). DFT calculations have indicated that the addition and rebound steps occur nonsynchronously in a single transition state or in a stepwise fashion with a rate-determining C–O bond forming step followed by a low-energy-barrier ring closure step.306,408,409 In several alkene oxidation reactions, a cationic intermediate, presumably generated via single electron transfer from alkyl radical to the ferryl center, was inferred from the formation of aldehydes, ketone, or heme N-alkylated side products.17,92,146,410,411 The ketone or aldehyde products were generated via a 1,2-hydride shift, while heme N-alkylation products were likely to be formed via direct electrophilic attack of the carbocation from a pyrrole nitrogen of the porphyrin ligand.412 Very recently, Hammer, Arnold, and co-workers have harnessed the side activity of P450s for aldehyde formation to develop the first olefin anti-Markovnikov oxygenase (aMOx) via directed evolution (Figure 45b).413 aMOx contains 12 mutations from its parent P450LA1, a P450 from the rhodobacterium Labrenzia aggregata, and exhibits high anti-Markovnikov oxidation activity (up to 4500 TTN) and aldehyde/epoxide selectivity (up to 16:1). Furthermore, aMOx can convert α-methylstyrene to (S)-2-phenylpropionaldehyde with 91:9 enantiomeric ratio (er), representing the first catalytic asymmetric aldehyde synthesis via direct anti-Markovnikov oxidation. This result combined with a deuterium-labeling experiment indicate a mechanism involving an initial oxo-transfer followed by a highly ordered 1,2-hydride migration. It is still not clear how the 12 mutations in aMOx benefit the anti-Markovnikov activity. Some of these mutations are in the distal pocket and might help lock the substrate conformation for 1,2-hydride transfer. Intriguingly, one mutation (M391L) is in the highly conserved proximal cysteine loop, which indicates that the proximal heme ligand might also play an important role for anti-Markovnikov activity, as changes in Cys loop are known to affect the electron donation ability of proximal cysteine thiolate.181
Figure 45.

(a) Proposed mechanism for alkene epoxidation catalyzed by P450s. (b) Olefin anti-Markovnikov oxidation catalyzed by an engineered cytochrome P450 (aMOx). The homology model of wild-type P450LA1 is shown with mutation sites highlighted by red sphere. Adapted with permission from ref (413). Copyright 2017 American Association for the Advancement of Science.
Aromatic hydroxylation is a paradigmatic cpd I-mediated reaction that has attracted decades of research interest, because of its importance in drug metabolism and the potential applications in organic synthesis. The mechanism involves an initial addition of cpd I to the arene π-system to form a tetrahedral σ-complex (Figure 46).414−419 While many mechanistic studies support a direct electrophilic attack of cpd I to arenes (step a, Figure 46),417−421 a recent study by Fujii et al. using a Marcus-type analysis showed a significant contribution of an electron transfer process in the rate-limiting step. The results were interpreted to indicate a mechanism involving a fast electron transfer equilibrium between cpd I and the arene, which is coupled to a subsequent C–O bond formation (steps b and c, Figure 46).422 The tetrahedral σ-complex could adopt two forms: a cationic species with a ferric center and a radical form with a ferryl center. DFT calculations suggest that two electromers are admixed in the σ-complex.417−420 A common mechanistic feature of P450-mediated aromatic hydroxylation is the migration of the ipso–hydrogen to the adjacent carbon (“NIH shift”) and the partial retention of this hydrogen in the final product.30,423,424 There are several proposed mechanisms for the hydrogen migration and the formation of the hydroxylation product. The “NIH shift” could occur directly at the cationic σ-complex and led to the formation of a ketone product, which undergoes keto–enol tautomerization to form the product with partial retention of the migrated hydrogen. This mechanism is generally supported by DFT calculations,25 showing that the hydrogen migration can proceed through a 1,2-hydride shift (step d, Figure 46),419 or through a proton-shuttle mechanism mediated by the pyrrole nitrogen (step e).417,418 In an alternative mechanism, an arene oxide intermediate is formed from the σ-complex (step f), which then converts into the hydroxylation product via “NIH shift” and tautomerization.30,423,424 There are also examples in which the complete loss of the ipso–hydrogen was observed, indicating the formation of aromatic ring via direct deprotonation of the ipso–hydrogen without migration or that the keto–enol rearrangement is mediated by the active site architecture.425,426
Figure 46.

Proposed mechanisms for aromatic hydroxylation.
Molecules containing O-, S-, N-alkyl groups undergo dealkylation, which are commonly seen in xenobiotic and drug metabolism.427,428 Dealkylation reactions are triggered by the formation of α-hydroxylation intermediates, which decompose to afford aldehyde and dealkylated products.25 There are two mechanisms proposed for the generation of α-hydroxylation intermediates: H atom-transfer (HAT) and single-electron transfer (SET) followed by proton transfer (PT) (Figure 47a). The O-dealkylation reactions generally follow an HAT mechanism as evidenced by the large primary kinetic isotope effect.429−431 The mechanism for N-dealkylation is a topic of long debate. In support of an HAT mechanism, the use of radical clock substrates such as N-cyclopropyl-N-methylaniline afforded the ring-intact products.432−434 Additionally, a large kinetic isotope effect (KIE > 6) was observed for dealkylation of N-alkylamides.435,436 However, the KIE values of dealkylation of N-alkylamines are much lower (generally <3).437,438 Furthermore, the reaction rates of dealkylation of substituted N-alkylalniline correlate well with their redox potentials and Hammett electronic parameters with negative slopes.25 All these results seem to favor an SET–first mechanism. DFT calculations have provided critical insight for the understanding of N-delakylation mechanism and support the HAT–first mechanism. It has been shown that the SET step is not energetically favorable. Moreover, the low KIE and the linear correlation with substrate oxidation potentials could be reproduced with calculations based on the HAT mechanism.318,439 Further analysis showed that the nitrogen lone pair can interact with the radical character developed at the carbon center during the C–H bond cleavage. This interaction leads to partial positive charge accumulation at the amine moiety in the transition state, causing the experimentally observed substituent effect.
Figure 47.
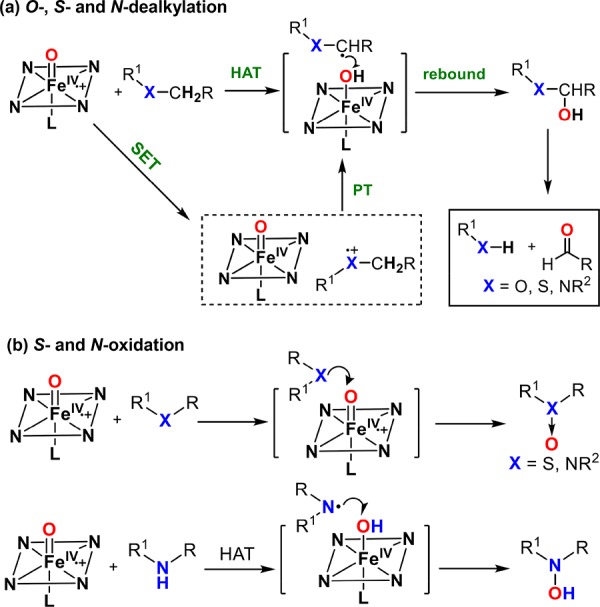
Proposed mechanisms for (a) O-, S-, and N-dealkylation. (b) S- and N-oxidation.
In addition to dealkylation, N-alkyl and S-alkyl groups could undergo direct heteroatom oxidation to afford N-oxide and sulfoxide products (Figure 47b). The mechanisms for both reactions have been shown to be a concerted direct oxygen atom transfer.313,440−443 In theory, the heteroatom oxidation and dealkylation could compete with each other. Indeed, both products were observed in some examples.17,444,445 Intriguingly, it seems that N-alkyl groups favor dealkylation while S-alkyl groups prefer sulfoxidation. A recent computational study reproduced this trend and found that the activation energies of dealkylation pathway was largely affected by the radical stabilization ability of the neighboring heteroatom.443 The nitrogen inversion barrier of tertiary amines was recently found to affect the N-oxidation. Olsen et al. studied a series of tertiary amines and found that those compounds with N-oxidation reactivity had lower N-inversion barriers.446
Computational studies of sulfoxidation and N-oxidation have shown that the energy of the quartet transition state was much higher than that of the doublet transition state. Thus, the reaction is predicted to occur along the low-spin pathway.313,440−443,446,447 This is in sharp contrast to aliphatic hydroxylation, epoxidation, and aromatic hydroxylation reactions,305,306 in which close-lying transition states were found for both doublet and quartet spin surfaces.408,409,421,448−451 The main distinction of S- and N-oxidation compared to other oxidation reactions is that they were indicated to proceed through a synchronous 2e– transfer process. Therefore, in the high-spin quartet transition state, one electron from sulfur or nitrogen has to migrate to a high-energy σz2* orbital,441 giving rise to the high energy barriers of quartet transition states. Primary and secondary amines can also be oxidized via N-hydroxylation. This reaction has been much less explored. DFT calculations support two possible mechanisms with comparable activation energies, a direct oxygen atom transfer (OAT)/isomerization pathway and a hydrogen atom transfer (HAT)/radical rebound pathway.452−455
The axial ligand effect has also been explored for oxygen atom transfer reactions (OATs). Initial studies by Gross et al. showed that electron-donating ligands (such as F– and Cl–) substantially increased the epoxidation reactivity of model cpd I complexes, whereas cpd I species with weakly coordinating ligands, such as CF3SO3– and ClO4– displayed lower reactivity.456−458 A decrease in FeIV=O bond strength for electron-donating ligands was indicated by their lower stretching frequencies (νFe=O = 801 cm–1 as determined by resonance Ramon spectroscopy) compared to those with weak-ligating ligands (νFe=O = 834–835 cm–1).457,458 Recently, phenolate and electron-neutral imidazole axial ligands were found to accelerate epoxidations with second-order rate constants 10 times faster than that of chloride ligand.459 The effect of para-substituted pyridine N-oxides ligands (p-Y-PyO) were also explored for alkene epoxidation and aromatic hydroxylation, and a rate enhancement was observed for both reactions by increasing the electron-donating ability of para-substituent Y.285 DFT calculations were employed to study the impact of axial ligands on a number of OAT reactions, including alkene epoxidation,409,449 aromatic hydroxylation,448,451 and sulfoxidation.440 In all of these reactions, calculated activation energies were not dependent on the reduction potential of cpd I but did exhibit a good negative linear correlation with BDEOH of cpd II. This correlation might suggest that the C–O or S–O bond formation step in OATs shares similarities with the O–H bond formation in HAT reactions. Alternatively, it might imply the importance of FeIV=O weakening in cpd I for OAT, since calculations showed that, in addition to increasing the cpd II basicity, the electron donation from axial ligand also weakened the FeIV=O bond in cpd I.285
3.5. O–O Bond Formation Mediated by Oxometalloporphyrins
3.5.1. Oxygen Evolution from Oxometal Complexes
A key step in photosynthesis is the oxidation of water to dioxygen. This four-electron, four-proton process generates the electrons finally used to convert CO2 to carbohydrates.460 In photosystem II, the water oxidation occurs via multiple proton-coupled electron transfers (PCETs) at the Mn4Ca oxygen-evolving complex (OEC).461 The development of molecular catalysts that can catalyze this process under mild conditions and low energy cost is important for solar energy conversion and storage.461 Metalloporphyrins, with their ability to form high-valent metal–oxo intermediates, are promising catalysts for water oxidation and oxygen evolution.33,462,463 The major pathways considered for metal–oxo-mediated water oxidation include nucleophilic attack of water/hydroxide by oxoMn(V) and the coupling of two metal–oxo (or μ-oxo) units (Scheme 10). The former is formally the microscopic reverse of the O–O bond cleavage for cpd I generation, while the coupling between two μ-oxo units in the latter pathway will afford a (μ-η2:η2-peroxo)dimetal complex similar to the oxygen adduct of hemocyanine.464−466 In a third mechanism, the O–O bond is formed via coupling of a metal-bound oxide with a metal-ligated μ-oxo bridge.467−470 This mechanism is mainly proposed for O2 evolution of photosystem II OEC and related polynuclear metal–oxygen complexes.
Scheme 10. Proposed Mechanisms for Water Oxidation Mediated by Metal–Oxo Intermediates.
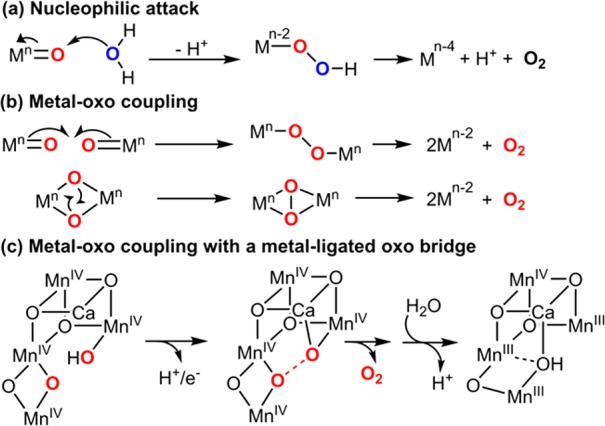
An early example of a water–oxidation catalyst based on metalloporphyrins is a dimeric manganese porphyrin complex 81 synthesized by Naruta et al. (Figure 48a).471 This species can catalyze the anodic water oxidation in aqueous acetonitrile solution containing nBu4NOH with a potential range of 1.2–2.0 V (vs Ag/Ag+) and Faradaic efficiency of 5–17%. An dimeric oxoMnV intermediate 52 was later prepared by treating the Mn(III) dimer complex with mCPBA as a potential reactive intermediate for water oxidation (Figure 48a).231 This species is very stable under basic conditions but decomposes to Mn(III) and dioxygen upon neutralization with acid. Åkermark, Sun, and co-workers reported a monomeric manganese porphyrinoid system for water oxidation (Figure 48b).472 A manganese corrole complex 83 was shown to efficiently mediate the oxygen evolution at a low potential ∼0.8 V versus Ag/AgNO3. An oxoMnV intermediate 84 was prepared from 83. The addition of hydroxide 84 led to the formation of O2 presumably via the nucleophilic attack of OH–. A Mn(IV)-peroxo intermediate 86 was detected during this process, which was likely formed from the further oxidation of the Mn(III)-hydroperoxo intermediate 85. Nam also reported the formation of a MnIV-peroxo intermediate upon adding base to a oxoMn(V) corrole complex, MnV(O)(TFMPC) (TFMPC = 5,10,15-tris(3,5-trifluoromethylphenyl)corrolato trianion).473 This step is reversible as the addition of acid to peroxo-MnIV(TFMPC) could regenerate the oxoMn(V) intermediate.
Figure 48.

(a) Water oxidation mediated by a dimeric manganese porphyrin complex. Adapted from ref (231). Copyright 2004 Wiley-VCH. (b) Water oxidation catalyzed by a manganese corrole complex 83. Adapted from ref (472). Copyright 2009 American Chemical Society.
In addition to manganese, cobalt is another base metal that has been extensively studied for water oxidation. Nocera et al. reported the first cobalt porphyrinoid system for water oxidation (87, Scheme 11a) using a cobalt “hangman” β-octafluoro corrole complex 87. When immobilized in Nafion films, 87 can catalyze water oxidation at 1.4 V versus Ag/AgCl at pH 7 with turnover frequency (TOF) of 0.81 s–1.474 Sakai et al. reported several Co(III) porphyrins, including Co(TPP), Co(TMPyP), and Co(TPPS), for water oxidation via photon-initiation using RuII(bpy)32+/Na2S2O8 (Scheme 11b). The maximum TOFs were in the range of 0.16–0.35 s–1 at pH 11.475 A second-order dependence of the initial rate on catalyst concentration was observed, suggesting a bimolecular oxyl–oxyl coupling pathway for O–O bond formation. The degradation of the catalyst has been observed presumably due to the highly oxidizing sulfate radical anions and single dioxygen. More recently, the same group developed a fluorinated Co(III) porphyrin catalyst CoFPS for water oxidation with improved stability and efficiency (TOF = 1.1 s–1).476 In contrast to their previous studies, a first-order catalyst concentration dependence was observed for CoFPS, suggesting a nucleophilic attack pathway for the water oxidation reaction catalyzed by CoFPS.
Scheme 11. (a) Cobalt Hangman Corrole Complex for Water Oxidation and (b) Several Cobalt Porphyrins Utilized for Water Oxidation.

Reprinted from ref (474). Copyright 2011 American Chemical Society.
Groves and Wang developed a series of electron-deficient cationic cobalt porphyrins for highly efficient electrocatalytic water oxidation.477 For CoIII(TDMImP) (88, Figure 49), at a potential of 1.3 V versus Ag/AgCl at pH 7, the O2 formation rate was determined to be 170 nmol cm–2 min–1 (kobs = 1.4 × 103 s–1) with a near 90% Faradaic efficiency. The authors conducted detailed mechanistic studies of these cobalt-based water–oxidation systems. They identified that a CoIV–O porphyrin cation radical species P•+CoIV–O (92, Figure 49) is the active intermediate for water oxidation. A linear dependence between catalytic current and catalyst concentration suggested that the rate-limiting O–O bond formation occurred at a single cobalt site. Intriguingly, the buffer anion was identified to play multiple important roles in the catalytic cycle. First, it acts as a proton acceptor in the conversion of P•+CoIII–OH to P•+CoIV–O, which is found to be a single PCET process with a slope of −54 mV pKa–1 for a plot of onset potential of water oxidation versus buffer pKa. Furthermore, it acts as a base to deprotonate water in the rate-limiting O–O formation step, as indicated by a linear correlation between (icat/iwater)2 versus buffer base concentration and a KIE of 2.8 measured in H2O versus in D2O. Finally, an inhibition of water oxidation was observed at high buffer concentrations, presumably resulting from the formation of an inactive bis-coordinated Co(III) intermediate 91 with buffer anions. The unchanged intensity and UV–vis spectroscopic features of 88 after bulk electrolysis, the absence of an inhibition effect of EDTA, and the clean electrode surface with no sign of heterogeneous cobalt phase clearly exclude the possibility of forming heterogeneous metal oxide films at electrode surface, which is known to be a potential issue for homogeneous water–oxidation catalysts.478 More recently, Cao et al. reported a nickel-based water–oxidation catalyst with a cationic porphyrin ligand.479 A TOF of 0.67 s–1 was observed at a potential of 1.32 V (vs NHE). The reactive intermediate of this system is proposed to be a Ni(III)–oxyl radical species. As in the case of analogous cobalt cationic porphyrins, buffer base was shown to participate in the rate-determining O–O bond formation step. An inhibition effect of buffer base at high concentrations was also observed.
Figure 49.

Proposed mechanism for water oxidation catalyzed by Co-porphyrins (B = buffer anions). Adapted with permission from ref (477). Copyright 2013 National Academy of Sciences.
3.5.2. Chlorite Dismutase
The conversion of chlorite to chloride and dioxygen is another reaction that might involve the O–O bond formation mediated by cpd I. This process is important for the microbial perchlorate respiration, as it removes the toxic end-product chlorite.480 In nature, the decomposition of chlorite is catalyzed by chlorite dismutase (Cld), a heme-containing protein with a histidine proximal ligand.481 The active site structure of Cld is shown in Figure 50, which is featured by an arginine positioned above the heme and the lack of other hydrogen-bonding residues that are essential for O–O bond heterolytic cleavage.482 The proposed mechanism for functional Cld involves an initial formation of a ferric–chlorito intermediate (FeIII–OClO), analogous to compounds 0 in P450s and peroxidases (Figure 50c).483 A subsequent O–Cl bond heterolytic cleavage will generate cpd I and hypochlorite, which will undergo nucleophilic attack at cpd I to generate Cl– and O2.484 Alternatively, the homolysis of Cl–O bond of FeIII–OClO will afford compound II (cpd II) and chlorine monoxide radical (ClO·. Cl– and O2 are formed via a rebound process between ClO· and cpd II. The latter mechanism is analogous to the mechanism proposed for NO oxidation catalyzed by nitric oxide dioxygenase (see section 4.2). The distal arginine can adopt either “in” and “out” conformations (Figure 50a and 50b),482 which is proposed to play an important role in positioning the transiently formed hypochlorite to ensure its efficient capturing by cpd I (or cpd II).485−487 There are two phylogenetically distinct class of Clds (clades 1 and 2). Clade 1 Clds (including NdCld and DaCld shown in Figure 50) are pentameric and hexameric, whereas clade 2 Clds are dimeric. Very recently, Obinger and co-workers have carried out detail structural and kinetic studies of a dimeric clade 2 Cld from Cyanothece sp. PCC7425 (CCld).488 The spectral features of various intermediates of dimeric CCld appeared to be much better resolved than those of clade 1 Clds, which allowed for the presteady-state kinetic studies of CCld. Their results showed that mixing CCld with chlorite under stopped-flow conditions led to the formation of cpd II within milliseconds at pH > 5.0. Furthermore, no O2 formation was observed by treating CCld cpd I with excess hypochlorite (HOCl/–OCl). These results suggest a homolysis pathway for chlorite degradation by CCld.
Figure 50.

(a) Active site of chlorite dismutase (Cld) from Candidatus Nitrospira defluvii (NdCld) with an imidazole ligand (PDB: 3NN1). Catalytically important arginine (Arg173) exhibits an “out” conformation in this structure. (b) Active site of Cld from Dechloromonas aromatica (DaCld) with a nitrite ligand (PDB: 3Q09). Catalytically important arginine (Arg183) exhibits an “in” conformation in this structure. (c) The proposed mechanism for chlorite dismutase.
The chlorite decomposition reactions have also been explored with metalloporphyrins. In a Mn porphyrin-catalyzed alkane oxidation with chlorite as oxidant, Collman et al. found a catalytic side reaction that produced Cl– and O2 from chlorite.489 The disproportionation of ClO2– into Cl– and ClO3– was also observed especially in the absence of hydrocarbon substrates. More recently, Abu–Omar and co-workers studied the reaction between chlorite and several water-soluble iron porphyrins (Figure 51a).490 They also identified two degradation pathways of ClO2–, in which one afforded ClO3– and Cl– and the other yielded O2 and Cl–. The more electron-withdrawing fluorinated Fe(TF4MAP) catalyzed both reactions, while less electron-deficient Fe(TMAP) and Fe(TPPS) only catalyzed the disproportionation of ClO2– to ClO3– and Cl–. On the basis of isotope labeling studies, they proposed Cl– and O2 were formed via a cpd I/rebound pathway similar to that proposed for Cld (cycle A, Figure 51), whereas that ClO3–/Cl– pathway proceeds through a Fe(II) porphyrin/cpd II catalytic cycle (cycle B, Figure 51).
Figure 51.

(a) Iron porphyrins to mimic Cld reactivity. (b) The proposed mechanism of ClO2– decomposition to form Cl– and O2 and that of ClO2– disproportionation to ClO3– and Cl– catalyzed by iron porphyrins. Adapted from ref (490). Copyright 2009 American Chemical Society.
Recently, electron-deficient cationic manganese porphyrins have been shown to catalyze the generation of chlorine dioxide from chlorite by Groves and Abu–Omar groups, respectively.491−493 The reaction proceeds rapidly under mild conditions. For Mn(TDMImP), TOFs were determined to be 0.47 s–1 at pH 6.8 and 1.0 s–1 at pH 4.7. The catalytic efficiency can be dramatically increased by changing Mn(TDMImP) to an electron-deficient manganese porphyrazine catalyst MnTM23PyPz (TOF = 32.0 s–1 at pH 4.5) (Figure 52a).492 A detailed mechanistic study by Groves et al. with MnIII(TDMImP) catalyst revealed an initial generation of oxoMn(V) and ClO–, which then diffuses to oxidize a second Mn(III) site to oxoMn(V) (Figure 52b).492 This is in sharp contrast to Cld and iron porphyrins, in which an in-cage recombination between cpd I and ClO– would afford O2 and Cl–. Each oxoMn(V) species could produce two ClO2 molecules via successive out-sphere electron transfer to ClO2–. The oxidation of ClO2– to ClO2 by oxoMnV and oxoMnIV showed comparable rates at pH 6.8 (kapp = 3.13–6.30 × 103 M–1 s–1). The oxidation of ClO2– by oxoMnIV is also reversible [kapp = (4.59 ± 2.37) × 102 M–1 s–1]. Furthermore, the evolved ClO2 can be further oxidized to ClO3– by oxoMnV and oxoMnIV in a subsequent phase of the reaction. Similar mechanistic features but at much slower rates have been described by Abu–Omar et al. for a Mn(TF4TMAP)/NaClO2 system.494
Figure 52.

(a) Mn porphyrin and porphyrazine catalysts for generation of ClO2 from chlorite and their turnover frequencies. (b) Mechanism of chlorite dismutation catalyzed by Mn(TDMImP) and the apparent second-order rate constants for the elementary steps. Hypochlorite is written in protonated form as the measurements were carried out at a pH range of 4.7–6.8. Adapted from ref (492). Copyright 2011 American Chemical Society.
3.6. Single Electron Transfer (SET) Reactions of Cpd I
In addition to participating in hydrogen atom transfer (HAT) and oxygen atom transfer (OAT) reactions, compound I can also display single electron transfer (SET) reactivity, which is commonly seen in heme-containing peroxidases. Unlike most P450s, peroxidase compound I is generated directly from the resting ferric heme and H2O2 or organic hydroperoxides via the “peroxide shunt” mechanism. Peroxidase cpd I can carry out two discrete single-electron transfers to two substrate molecules and regenerate the ferric heme state (Scheme 12). This three-step mechanism was first proposed by Dunford et al. for the oxidation of ferrocyanide by H2O2 and HRP.246 The resulting substrate radicals can undergo further transformations.495 Thus, substrates of heme peroxidases are generally good one-electron donors, such as electron-rich aromatics, ascorbate, ferrocytochrome c, etc.496 Even strongly oxidizing Mn3+ can be produced from Mn2+ in this way by manganese peroxidase.
Scheme 12. Heme Peroxidase Generates Cpd I with H2O2 and Catalyzes Two Discrete Single Electron Transfer (SET) Reactions to Substrates.
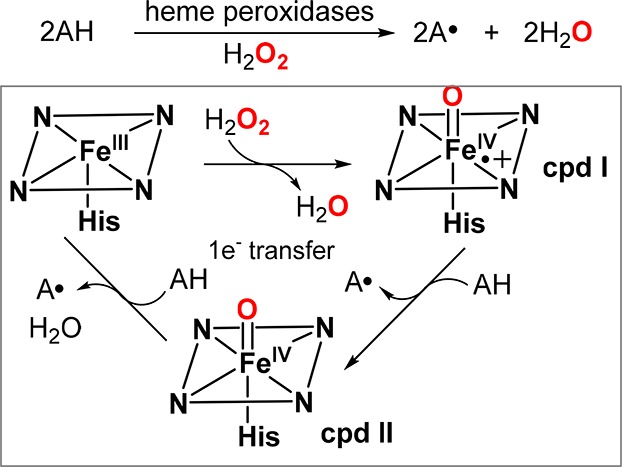
Heme peroxidases are not the only enzymes that utilize H2O2 to form compound I. Unspecific peroxygenases (UPO) and chloroperoxidase (CPO) employ a similar “peroxide shunt” pathway to generate compound I to catalyze HAT and OAT reactions.497 As discussed in section 3.1, a major cause of this reactivity difference is the much larger O–H BDEs of UPO-II and CPO-II (98–100 kcal/mol) compared to that of HRP-II, originating from the elevated reduction potentials of UPO-I and CPO-I and the decreased acidities of UPO-II and CPO-II. Furthermore, the 1e– reactions in peroxidases are facilitated by the proper positioning of substrates.498 The local protein environment of peroxidases hinders the approach of substrates to the FeIV=O moiety. Therefore, many substrates bind in the heme periphery and participate in a hydrogen-bonding network with heme propionate and active site residues (Figure 53).499 This network can serve as a relay to mediate the proton and electron transfer.500,501 Alternatively, substrates can be oxidized at a remote site through a chain of redox-active amino acids via long-range electron transfer.502−505
Figure 53.
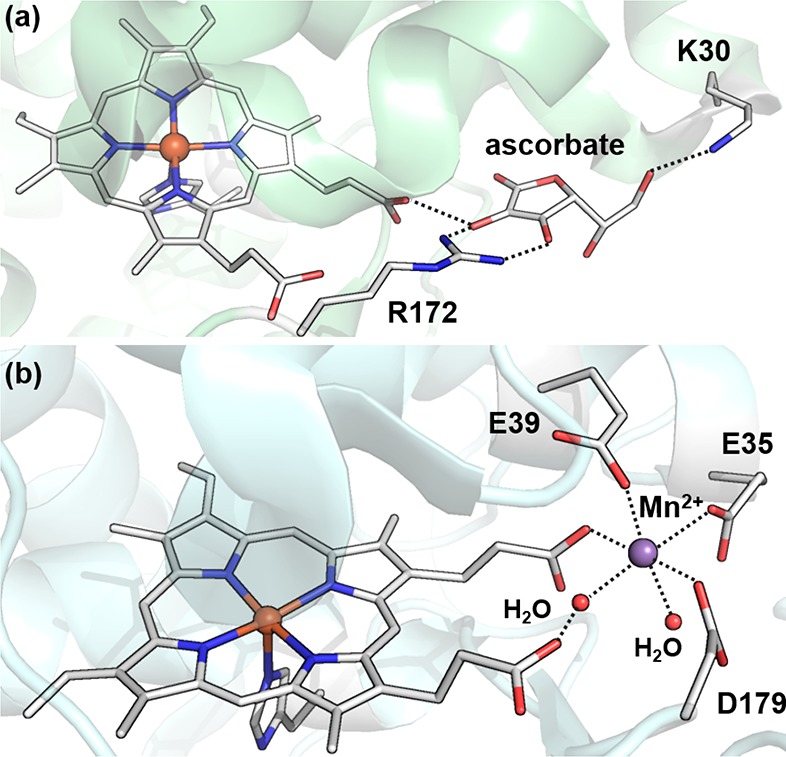
Examples of substrate binding to the heme periphery of peroxidases and forming hydrogen bonds with heme propionates and active site residues. (a) Ascorbate peroxidase (APX, PDB: 1OAF). (b) Manganese peroxidase (MnP, PDB: 1YYD).
In some peroxidases, the porphyrin cation radical form of compound I is transient and will rapidly transfer one electron to a nearby redox-active residue like tyrosine or tryptophan. One example is the compound ES of cytochrome c peroxidase (CcP), which is an iron(IV)-oxo intermediate with an uncoupled Trp191 cation radical near the proximal histidine.184 Although the Trp191 radical in CcP just serves as a storage for one oxidation equivalent, in some heme proteins, such radicals can directly mediate reactions. For instance, in prostaglandin H synthase (PGHS), the compound I formed via the normal peroxide activation pathway will oxidize a tyrosine (Tyr385) about 10 Å away to give a tyrosyl radical, which abstracts a hydrogen atom from arachidonic acid (Figure 54).506,507 The resulting substrate radical will further react with two O2 molecules to give product PGG2 and finally prostaglandin H2 (PGH2). This reaction also represents an example of substrate activation for O2 reactions in heme proteins.2
Figure 54.
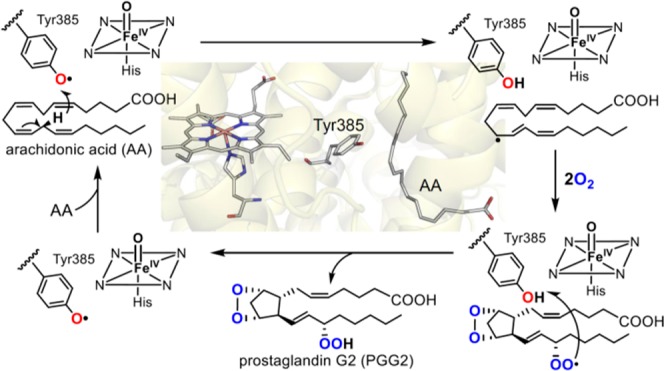
Conversion of arachidonic acid (AA) into PGG2 by PGHS.
Another example of using protein radicals for substrate activation is the synthesis of heme b by coproheme decarboxylase (HemQ).508 HemQ catalyzes a H2O2-dependent decarboxylation of two propionates in coproheme (94, Scheme 13). Recent mechanistic studies by DuBois et al. showed that Tyr145, which hydrogen bonds with one of the cleaved propionates, is first converted to a tyrosyl radical by coproheme compound I (95, Scheme 13).509,510 The Tyr145 radical then abstracts a hydrogen from the heme propionate group presumably at the Cβ position (96). The resulting carbon-centered radical transfers one electron through porphyrin to Fe(IV) and reduces it to iron(III) with loss of CO2 (97 → 98). The decomposition of the second propionate is thought to follow a similar mechanism, but the identity of the mediator of Cβ-H cleavage is not clear.
Scheme 13. HemQ-Catalyzed Synthesis of Heme b from Coproheme III.

Adapted from ref (509). Copyright 2017 American Chemical Society.
4. Reactions Mediated by Heme–Oxygen Intermediates Other than Cpd I
In addition to cpd I, other heme–oxygen intermediates generated during O2 activation also exhibit rich redox chemistries that are harnessed by heme proteins for a variety of oxidative transformations, giving rise to the diverse biological functions of heme proteins. In this section, we analyze several representative reactions catalyzed by heme proteins and metalloporphyrins to delineate their mechanistic features and to discuss how the fundamental properties of heme–oxygen intermediates and their active site environments impact reaction outcomes.
4.1. Homolytic O–O Bond Cleavage of Peroxides by Heme Proteins
In addition to generating compound I via O–O bond heterolysis, organic peroxides can also react with ferric hemes via single-electron-transfer processes, which will oxidize the ferric heme to ferryl with concurrent homolytic cleavage of O–O bond. These transformations play important roles in the synthesis of important bioactive molecules, especially oxylipins.511 Natural sources of peroxides include fatty acid hydroperoxides formed via O2 trapping of substrate allylic radicals.512 Subsequent O–O bond homolysis leads to oxy radicals that can undergo a plethora of rearrangements, bond cleavages, and other transformations.
One example is the synthesis of prostacyclin from prostaglandin H2 (PGH2) via prostacyclin synthase (Scheme 14a).507,513,514 This transformation starts with the interaction between endoperoxide of PGH2 and ferric heme center of prostacyclin synthase (104, Scheme 14a), leading to the homolytic O–O bond cleavage of endoperoxide to afford the C9-alkoxyl radical and ferryl heme intermediate (105). The C9-alkoxyl radical then add to the C5–C6 alkene to forge the new five-membered heterocycle and C5 radical (106), which is later oxidized to carbocation by the ferryl heme to regenerate the C5–C6 alkene moiety via loss of a proton (107). The homolytic cleavage of endoperoxide of PGH2 is also used in the synthesis of thromboxane A2 (TXA2) mediated by thromboxane A synthase (Scheme 14b).515−517 In this reaction, the ferric heme center interacts with O9 and afforded C11-alkoxyl radical (109 and 110, Scheme 14b), which is later rearranged to an allylic radical at C12 (111). Subsequent electron transfer and ring cyclization would afford TXA2.
Scheme 14. (a) Synthesis of Prostacyclin from PGH2 Mediated by Prostacyclin Synthase and (b) Synthesis of Thromboxane A2 (TXA2) Catalyzed by Thromboxane A Synthase.

In another instructive example, 13S-hydroperoxyoctadecatrienoic acid [13(S)-HPOT] can be activated by the CYP74 family of enzymes, affording three different products: 12,13(S)-allene oxide, etherolenic acid, and an analogous hemiacetal (Figure 55a).518,519 The first step in all of these three transformations is the homolysis of an iron-bound O–O bond to afford an epoxyallylic radical and a cysteine-ligated FeIV–OH intermediate similar to compound II in P450 monooxygenases. The reactions then diverge depending on how the incipient substrate radical interacts with the FeIV–OH intermediate. In allene oxide synthase (AOS), the C11 epoxyallylic radical is oxidized by electron transfer to FeIV–OH to generate a substrate-derived carbocation intermediate, which eliminates a proton to form the new C=C bond. In hydroperoxide lyase (HPL), by contrast, the radical undergoes rearrangement to form a radical at C13, which then recombines with FeIV–OH via an oxygen-rebound mechanism. In the divinyl ether synthase (DES) pathway, the product is thought to be formed via a hydrogen atom abstraction from C14 by FeIV–OH or via an electron-transfer process followed by cation rearrangement and proton elimination. Sequence analysis and mutagenesis studies revealed that, in all three enzymes, an asparagine positioned directly above heme is important for mediating the O–O bond homolysis.520 The crystal structure of Arabidopsis thaliana AOS (AtAOS) complexed with 12R,13S-vernolic acid, a mimic of the epoxyallylic intermediate, showed that C11 is in proximity to F137 (Figure 55b), whereas in HPL and DES, a similar position is occupied by a leucine. The aromatic ring of F137 is thought to stabilize the C11 carbocation and thus facilitate the single electron transfer in the AOS pathway. In support of this rationale, a F137L mutation induced HPL activity for AOS.
Figure 55.

(a) Reactions catalyzed by the CYP74 family of enzymes. The mechanisms of all three pathways involve a common epoxyallylic radical intermediate. (b) Active site structure of Arabidopsis thaliana AOS complexed with vernolic acid (PDB: 2RCL). (c) Key N321 and I328 residues of AtAOS (PDB: 3CLI). The cysteine loop region is highlighted in red. (d) Key threonine residue of P450BM3 (PDB: 1BVY). The cysteine loop region is highlighted in yellow.
It is intriguing to compare the structural difference between CYP74 and P450 hydroxylases, since CYP74 has a typical P450 protein fold but does not perform O–O bond heterolysis. In addition to the critical Asn residue (N321) located above the heme, there are two more structural features that distinguish CYP74 from P450 hydroxlyases.520 As can be seen from the crystal structure of AtAOS, the highly conserved threonine in the proton relay channel of P450 hydroxylases, which is crucial for O–O bond heterolysis, is replaced by an isoleucine (I328) (Figure 55, panels c and d). Further, there is a nine-residue insert in the heme-binding loop of AOS, which seems to contribute to the long Fe–S bond (2.4 Å) of AOS. A long Fe–S distance has been shown to decrease the electron-donating ability of cysteine thiolate, which, in turn, would lead to a higher Fe reduction potential and a more facile SET reactivity of compound II.181
In addition to CYP74, there is another enzyme that can also catalyze the synthesis of allene oxides via homolytic cleavage of fatty acid hydroperoxides. This enzyme is structurally similar to catalase and thus termed catalase-related allene oxide synthase (cAOS).521 cAOS converts 8R-hydroperoxyarachidonic acid (8R-HPETE) into corresponding 8,9-epoxy allene oxide in the biosynthesis of preclavulone A. The crystal structure comparison between cAOS and catalase showed very similar active site structures, including the proximal heme-ligating tyrosine and the critical distal histidine and asparagine that are important for the O–O bond activation in catalase (Figure 56). A key difference is identified at a residue adjacent to the distal histidine, which is normally a valine in catalases but is changed to a threonine (T66) in cAOS.522 The histidine in cAOS hydrogen bonds with both T66 and asparagine. This hydrogen bonding network could function to dampen H2O2 activation in cAOS by inhibiting the hydrogen bonding between histidine and the hydroperoxo moiety of FeIII–OOH (cpd 0). Indeed, a T66V mutation greatly increased the catalase activity of cAOS, albeit with short lifetime due to the heme degradation. cAOS is thought to proceed through a similar epoxy allylic radical intermediate. As in the case of CYP74, products other than allene oxide were also observed in reactions catalyzed by cAOS and its relatives.523−528
Figure 56.

(a) Reactions catalyzed by cAOS and the active site structure of cAOS (PDB: 1U5U). (b) Active site structure of human catalase (PDB: 1QQW) and the mechanism of O–O bond activation in catalase-compound 0.
4.2. Reactions Initiated by Ferric-Superoxo Intermediates
In most heme proteins, ferric-superoxo complexes either serve as stable complexes, such as in oxymyoglobin and gas sensing proteins such as heme-nitric oxide/oxygen (H-NOX) binding proteins,529−531 or rapidly convert to high-valent iron–oxo intermediates via reduction and O–O bond scission. Tryptophan 2,3-dioxygenase (TDO) and indoleamine 2,3-dioxygenase (IDO), however, utilize ferric-superoxo intermediates to initiate the oxidation of substrates.532,533 TDO and IDO are histidine-ligated heme-containing dioxygenases involved in tryptophan oxidation.534−536 TDO employs dioxygen to cleave the 2,3-bond of l-tryptophan to form N-formyl kynurenine, whereas IDO catalyzes the same reaction but has a much broader substrate scope, including a variety of indoleamine derivatives such as d-tryptophan, melatonin, and serotonin. IDO has recently gained substantial research interest because of its immunomodulatory potential for cancer treatment.537,538
The initially proposed mechanism for TDO and IDO-catalyzed tryptophan oxygenation involved a histidine-catalyzed proton abstraction of the indole N–H, followed by electrophilic attack at C3 of indoleamine by the distal oxygen of ferric-superoxo intermediate (Figure 57a).539 But this mechanism is not consistent with later findings showing that N-methyl-tryptophan was an active substrate and that human IDO lacked the proposed proton-abstracting histidine.540−542 DFT calculations indicated high energy barriers for the formation of either a dioxetane intermediate or a Criegee-type rearrangement.543 Further studies by Yeh et al. using resonance Raman and UV–vis spectroscopies revealed the presence of a ferryl species during catalytic turnover of IDO.544−546 Similar ferryl species can also be generated by treating IDO and TDO using H2O2.541,547−549 On the basis of these results, a new mechanism has been proposed (Figure 57b), in which the ferric-superoxo complexes directly attacks the indoleamine ring via either electrophilic addition or radical addition to afford an indoline-2,3-epoxide intermediate and a ferryl compound II. Subsequent reactions between the epoxide and Fe(IV)=O are currently speculative but do lead to the formation of kynurenine type product and regenerate the ferrous heme. While the ferryl intermediate of IDO accumulates under steady-state conditions, TDO cpd II has yet to be observed during turnover. Rather, the [Fe(II)–O2, l-Trp] ternary complex accumulates under catalytic conditions. Raven et al. have recently suggested that this difference is caused by a change of the rate-limiting step from compound II decay in IDO to oxygen addition to formation of the 2,3-epoxide intermediate in TDO.550
Figure 57.

(a) Previously proposed base-abstraction mechanism followed by either Criegee rearrangement or dioxetane formation. Adapted from ref (539). Copyright 2011 American Chemical Society. (b) A recently proposed mechanism involves the formation of an FeIV=O intermediate and a 2,3-indoline epoxide intermediate. Adapted from ref (550). Copyright 2016 American Chemical Society.
Another enzyme that utilizes an ferric-superoxo intermediate for oxygenation is nitric oxide dioxygenase (NOD).551 As shown in Figure 58a, NO first reacts with an ferric-superoxo intermediate to form a proposed ferric-peroxynitrite intermediate. This intermediate then undergoes homolytic cleavage to generate NO2 and compound II.552 The recombination between NO2 and compound II yields nitrate and ferric heme. This recombination process is reminiscent of the oxygen rebound of carbon-centered radicals in P450s. The study of peroxynitrite decomposition mediated by met-myoglobin offered critical insight into the radical rebound mechanism, especially the radical cage escape effect.553,554 Unlike carbon-centered radicals, the diffusion of NO2 can be easily detected via fluorogenic probes such as fluorescein. Kinetic simulations revealed that 10% of the NO2 escaped from the radical cage [MbFeIV=O ·NO2] formed after peroxynitrite decomposition. Cytochrome P450 TxtE from Streptomyces scabies also employs a ferric-superoxo intermediate to generate NO2 from NO, which is then used to nitrate l-tryptophan to form 4-nitro-l-tryptophan.555,556 The proposed mechanism involves the formation of a ferric-peroxynitrite intermediate by the trapping of NO by the ferric-superoxo intermediate (Figure 58b). The NO2 generated after peroxynitrite decomposition can then add to the aromatic ring, which is further oxidized by compound II to afford the nitration product. An alternative mechanism involves heterolytic cleavage to generate nitronium ion (NO2+) for direct electrophilic nitration.555
Figure 58.

(a) Proposed mechanism for NO oxidation catalyzed by NOD. (b) Proposed mechanism for tryptophan nitration catalyzed by P450 TxtE.
4.3. O2 Reduction Catalyzed by Heme/Cu Terminal Oxidases and Related Model Compounds
Heme/Cu terminal oxidases such as cytochrome c oxidases (CcO) catalyze the 4e–/4H+ reduction of O2 as a final step in respiration with concomitant membrane proton-translocation leading to ATP synthesis.557 The active site structure of CcO is unusual compared to many heme proteins as it contains a binuclear heme a3/CuB center (BNC) with a Fe–Cu separation of around 5 Å (Figure 59a). CuB is coordinated to three histidine residues, one of which is covalently cross-linked to a tyrosine residue.558−560 During O2 reduction by CcO, electrons from cyt c are delivered one at a time to BNC via a dicopper complex (CuA) and a low-spin heme a. The mechanism of CcO action has been extensively studied. The catalytic cycle involves four consecutive proton and electron transfer steps and multiple intermediates are involved. Figure 59b showed a simplified mechanism of CcO-catalyzed oxygen reduction.561 The Fe(III)–Cu(II) resting state of BNC (compound O) is converted to an E state and then to a Fe(II)–Cu(I) reduced state (R) via two one-electron reductions and two proton transfer steps. The ferrous heme a3 in R will bind oxygen rapidly (∼1.4 × 108 M–1 s–1), generating a low-spin end-on Fe–O2 intermediate (A) similar to other oxygen-binding heme proteins. O–O bond cleavage in A, which can be viewed as an O–O hemolysis in a bridged Fe(III)–O–O–Cu(II) intermediate, yields a four-electron oxidized intermediate PM, containing a ferric heme a, a Fe(IV)=O, Cu(II)–OH, and a tyrosyl radical. Four subsequent 1e– reductions and proton transfers will lead sequentially through the F, O and E states, poising the protein for another oxygen-binding event. It is worth noting that in Figure 59b, we only showed intermediates formed after both electron- and proton-transfer steps. Furthermore, depending on the status of copper ligation and redox potentials of copper center, the cross-linked tyrosine could exist as tyrosyl radical with Cu(II) center being reduced to Cu(I).561,562 Each of these reduction/protonation steps (scalar protons) are also coupled to one vectorial proton pumped across the membrane, thus maintaining the proton gradient required for ATPase phosphorylation.561
Figure 59.

(a) Cofactor arrangement in a cytochrome c oxidase (PDB: 2YEV) and the structure of binuclear heme a3/CuB center (BNC). (b) General catalytic cycle of CcO and important intermediates involved (R, A, PM, F, O, and E). This mechanism is produced according to the catalytic cycles shown in refs (561) and (562).
While many aspects of the CcO mechanism have been clarified, the details of the O–O cleavage to generate compound PM and the role of the CuB His-Tyr unit in this step remains elusive. As noted above, a putative ferric peroxo intermediate (FeIII–O–O–CuII) is thought to be generated via 1e– reduction of ferric-superoxo intermediate by Cu(I) center (Figure 60a).558 A subsequent proton-coupled electron transfer (PCET) from tyrosine to FeIII–O–O–CuII could promote the O–O cleavage and generate the ferryl center and tyrosyl radical. Yoshikawa et al. have examined the binding of diatomic molecules (CO, NO, and CN–) to the reduced bovine CcO via X-ray structural analysis.563 Compared to CO- and NO-bound structures, CN– binding leads to the displacement and dissociation of a copper-bound histidine and the coordination of cyanide nitrogen to the copper. Furthermore, a water molecule is incorporated to form a hydrogen bond with both CN– and covalently ligated tyrosine (Tyr244) (Figure 60b). These structural features were thought to be highly relevant to the ferric-superoxo intermediate of CcO, where O2 is negatively polarized similarly to CN–. The hydrogen-bonding network in the active site could provide a channel for PCET from Tyr244 to the peroxo moiety. Another intriguing feature of this CN-bound CcO structure is the significant bending of the Fe–C–N angle (96°). This bent structure is in line with previous resonance Raman characterizations and might be caused by the weak π-accepting ability of cyanide, which makes it easier to adopt a bent geometry compared to the isoelectronic Fe-CO complexes.564−567
Figure 60.

(a) The proposed ferric-peroxo-cupric intermediate for the generation of PM state. (b) A CN-bound CcO might share similar structural features to the putative ferric-peroxo-cupric intermediate (PDB: 3AG4).
Model compounds have offered critical insights to the O–O heterolytic cleavage in CcO.23,568 The study of CcO model compounds has led to the development of electrocatalytic systems for the four-electron reduction of O2 to water, which would have potential applications in fuel cells.20,33,569−571 These systems have been reviewed in detail very recently.33 Common strategies for preparing CcO models include treating ferrous porphyrins with covalently appended copper(I) with O2 or adding Cu(I) complexes to preformed, discrete ferric peroxo porphyrins. In early attempts, μ-oxo heme–FeIII–O–CuII species were obtained, presumably from the disproportionation of the corresponding complexes.23 The oxygen of FeIII-μ-oxo-CuII species are basic and can undergo reversible protonation (Scheme 15). A pKa of 8 has been reported for [(F8TPP)FeIII-(OH)-CuII(TMPA)]2+ (F8TPP = tetrakis(2,6-difluorophenyl)porphyrinate, TMPA = tris(2-pyridylmethyl)amine).572
Scheme 15. Formation of FeIII-μ-oxo-CuII Complexes by Treating Fe(II) Porphyrin/CuI Complex with O2.

The first well-characterized heme-peroxo-copper adduct was reported by Collman with a complex “capped” porphyrin scaffold with an appended triazacyclononane (TACN) ligand for copper binding (114, Figure 61a).573 This species has a high spin (S = 2) ground state with a ν(O–O) of 758 cm–1. More recently, a variety of heme-peroxo-copper adducts were characterized and shown to display a rich coordination chemistry. The peroxo moiety can adopt μ-η2:η1, μ-η1:η1, or μ-η2:η2 bonding modes depending on the precise copper ligand environment and heme axial ligand (Figure 61, panels b and c).20,568,574 Karlin has described a μ-η2:η1 heme-peroxo-copper compound obtained by treating a ferric peroxo complex [(F8TPP)FeIII(O22–)]− with a tetracoordinated CuII complex CuII(TMPA) (115, Figure 61c).575,576 The compound showed a high spin (S = 2) electronic state with a ν(O–O) of 804 cm–1. Naruta has successfully obtained the crystal structure of a μ-η2:η1 heme-peroxo-copper adduct, [(TMP)FeIII-(O2)-(5MeTPA)CuII)BPh4 (116, Figure 61c).577 The μ-η2:η1 peroxo core showed two Fe–O bond distances of 2.031(4) and 1.890(6), respectively. Like [(F8TPP)FeIII-(O2)-CuII(TMPA)]+, [(TMP)FeIII-(O2)-CuII(5MeTPA)]BPh4 is EPR silent with an S = 2 spin state resulting from a strong antiferromagnetic coupling between high spin FeIII (S = 5/2) and CuII (S = 1/2).
Figure 61.

(a) A heme-peroxo-copper model compound based on “capped” porphyrin with an appended TACN ligand for copper binding. (b) Different peroxo bonding mode for heme-peroxo-copper complexes. (c) Several representative heme-peroxo-copper model compounds.
The peroxo bonding mode can be regulated by copper coordination and axial ligand of iron porphyrins. For example, addition of a tridentate Cu(II) complex Cu(AN) (AN = 3,3′-iminobis(N,N-dimethylpropylamine)) to [(F8TPP)FeIII(O22–)]− afforded a μ-η2:η2 peroxo coordination (117, Figure 61c), whereas adding imidazole-type axial ligand to [[(F8TPP)FeIII-(O2)-CuII(AN)] shifted the peroxo coordination to the μ-η1:η1 mode (118, Figure 61c).578−581 The end on μ-η1:η1 heme-peroxo-copper complex is diamagnetic with a low spin S = 1/2 Fe(III) antiferromagnetically coupled to Cu(II) (S = 1/2).581,582 Very recently, Karlin et al. reported a [FeIII-(O22–)-CuII(MeTHF)3]+ (MeTHF = 2-methyltetrahydrofuran) complex (119, Figure 61c). The weak coordination of the MeTHF ether oxygen allows for facile ligand exchange, providing ready access to a variety of heme-peroxo-CuII species.583 In addition to the heme-peroxo-CuII intermediate, isoelectronic FeIII-superoxide-CuI complexes were synthesized using picket fence porphyrins.584,585 These FeIII-superoxide-CuI complexes are relatively stable at room temperature, suggesting that CuI could also play a nonredox role in stabilizing iron dioxygen adducts.
The reactivity of CcO models toward phenols was also explored. Collman et al. found that an FeIII-superoxide-CuI complex could react with phenol both intra- and intermolecularly to afford a ferryl species, a tyrosyl radical, and Cu(II)–OH,586,587 a process that resembles the transition from CcO A state to PM state (Figure 62). Solomon and Karlin have tested the reactivity of both low- and high-spin heme-peroxo-copper complexes by treating them with phenols (Figure 63).578 While high-spin [(F8TPP)FeIII-(O22–)-CuII(AN)]+ was not reactive toward phenols, the low-spin [(DCHIm)(F8TPP)FeIII-(O22–)-CuII(AN)]+ (DCHIm = dicyclohexyl imidazole) reacted with phenols readily, even at −80 °C, to afford a ferryl intermediate, Cu(II)–OH, and a tyrosyl radical.578
Figure 62.

Reactions between FeIII-superoxide-CuI complexes and phenols.
Figure 63.
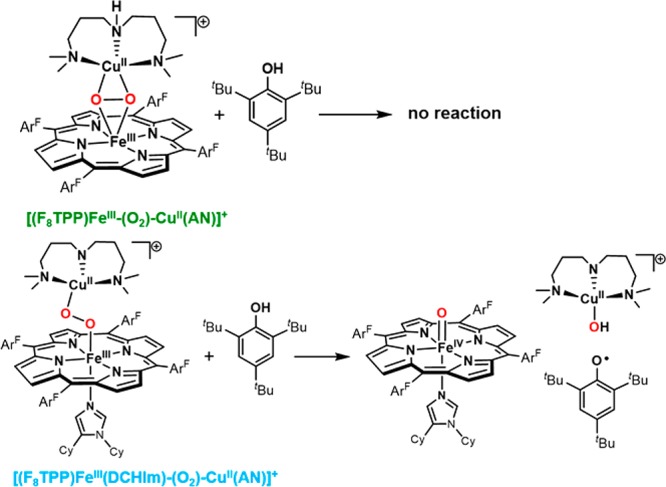
Comparison of reactions with phenols between low-spin and high-spin FeIII-peroxo-CuII complexes.
The impact of copper complex structure and the nature of the proton source on the O–O bond activation step was recently investigated.588 Two FeIII-peroxo-CuII complexes with tridentate and tetradentate copper complexes, respectively (LS-3DCHIm and LS-4DCHIm), were prepared and examined for their reactivity toward different acids or electron donor decamethylferrocene (Fc*) (Figure 64). While LS-4DCHIm alone was not reactive toward Fc*, the addition of 4-nitrophenol led to the formation of an [LS-4DHCIm···4-NO2-phenol] association complex, which underwent facile O–O bond cleavage via proton-coupled electron transfer from 4-nitrophenol and Fc* to peroxo moiety. Intriguingly, under similar conditions, LS-3DCHIm was not reactive. Further examination showed that LS-3DCHIm could not form the association complex with phenol presumably due to a more compressed and sterically inaccessible peroxo core. Strong acids like triflic acid led to the direct decomposition of both LS-3DCHIm and LS-4DHCIm and the release of hydrogen peroxide, regardless of the steric environment of peroxo group.
Figure 64.

(a) Phenol association will promote the O–O bond cleavage of LS-4DCHIm in the presence of electron donor Fc*. (b) LS-3DCHIm could not form the phenol association complex and is not reactive toward Fc*. DFT calculation showed a more compressed structure of LS-3DCHIm. Adapted from ref (588). Copyright 2017 American Chemical Society.
In addition to small molecule models of CcO, redesigned metalloproteins that mimic the reactivity of CcO represents another powerful approach to study its mechanism.589 Lu et al. found that the introduction of two mutations (L29H and F43H) would create a copper binding site in the heme pocket of myoglobin (Figure 65a).590 The resulting copper-bound myoglobin (CuBMb) showed heme oxygenase reactivity that degrades heme to verdoheme. In 2012, Lu and Wang generated a CuBMb with a cross-linked Tyr-His ligand via genetic incorporation of the unnatural amino acid imiTyr at the F33 position (Figure 65). The imiTyr-CuBMb exhibited enhanced oxidase reactivity (2/min).591 In the same year, Lu et al. found that the introduction of a tyrosine mutation to CuBMb at F33 also improved O2 reduction activity (24/min). Intriguingly, the observed oxidase activity is not copper-dependent. A subsequent study showed that a ferric–hydroperoxo species was formed for F33Y–CuBMb upon cryoreduction at 77K, whereas a peroxo–ferric state was formed for wild-type Mb.592 The X-ray crystal structure of F33Y–CuBMb revealed an extensive H-bond network with two water molecules (W1 and W2), in which W1 is H-bonded to H43 and Y33 and W2 is stabilized by H-bonds to W1 and H29 (Figure 65b).593 This hydrogen-bonding network could polarize the O–O bond and facilitate its reduction and protonation. Very recently, Lu and co-workers studied the effect of nonheme metals on oxidase activity of the repurposed Mb.594 While Zn-Mb formed a stable ferric-superoxo intermediate, both Cu-Mb and Fe-Mb showed oxidase reactivity with Cu-Mb almost three times more reactive than Fe-Mb. The higher reactivity of CuI is attributed to the higher CuII/CuI potential and an enhanced weakening of O–O bond through dπ-pπ back bonding, as revealed by the longer O–O bond length (1.391 Å) in Cu-Mb than that in Fe-Mb (1.351 Å).
Figure 65.

(a) Crystal structure of oxy-Mb (PDB: 1A6M). (b) Crystal structure and the hydrogen bonding network in the active site of oxy–F33Y–CuBMb (PDB: 5HAV).
5. Conclusions and Outlook
The central goal of this review was to survey the range of redox reactions mediated by heme proteins. Parallel developments with model metalloporphyrins and computational approaches are also reviewed and have served both to elucidate the nature of reactive enzymatic intermediates and to discover new chemistry, some of which is unknown in the repertoire of heme protein reactions. Another goal was to provide context for the recent advances with key discoveries from the formative years of the field. This coverage took inspiration from a course we developed at Princeton University called Metals in Biology: From Stardust to DNA. It is interesting to consider that heme and heme proteins apparently evolved long before oxygen was present in the environment in significant quantities. From this perspective, it is clear that oxygen binding and oxygen activation are relatively recent innovations in biology. Accordingly, it is likely that access to hydrogen peroxide allowed heme proteins to be repurposed by exploring the rich chemistry made possible by the ferryl group as well as ferric and iron(IV) hydroxides. In this context, heme and nonheme iron proteins are essentially similar, tied together by the sequential reduction of oxygen to superoxide, hydrogen peroxide, and ultimately water. As we have seen in the examples discussed above, the porphyrin ring can be oxidized at ∼1 V versus NHE to a porphyrin cation radical. Accordingly, there is a direct mechanistic analogy between heme iron and nonheme diiron proteins such as sMMO and AlkB, wherein the second oxidizing equivalent resides on the second iron rather than the porphyrin ligand. There are also nonheme, monoiron systems such as TauD and naphthalene dioxygenase that exploit Fe(IV)/Fe(II) and Fe(III)/Fe(V) couples, respectively, in their substrate oxidations. Studies of heme protein and metalloporphyrin reaction mechanisms and the insights gained from that work have in recent years led to significant developments in drug design. Clopidogrel (Plavix) and similar drugs are effective at lowering the risk of heart attack and stroke due to the nature of their oxidative metabolism by liver cytochrome P450s. Likewise, inhibitors of P450 aromatase proteins, such as anastrazole, are emerging as key agents for the treatment of certain human breast cancers. New methods of C–H functionalization, such as C–H fluorination with fluoride ion-manganese systems, and their applications have offered novel and practical routes to more efficacious drugs and to the development of potential PET imaging agents. More generally, the recent upsurge of radical chemistry in organic synthesis has provided an explosion of methods for generating substrate radicals under mild conditions.595−597 One can well imagine that a merger of these radical-generating processes (such as photoredox catalysis and electrocatalysis) with metalloenzymes and their model systems could lead to the development of entirely new synthetic transformations that are inaccessible today. These developments are further strengthened by recent advancements in biocatalysis for non-natural transformations, in which existing enzymes or proteins can be adapted and repurposed to catalyze abiological chemistry via directed evolution.598,599 We envision that an orchestration between metalloenzymes/metal–oxygen systems and these emerging fields in catalysis have the potential to push the frontiers of biocatalysis and organic synthesis as well as the conceptual underpinnings of the field that continue to provide fresh air.
Acknowledgments
We are deeply indebted to our co-workers at Princeton University and at the University of Michigan in the early years, for their imaginative and energetic contributions to the work from our laboratories described herein. We are also grateful for the continuous support of this research by federal granting agencies. Research directed toward C–H oxidative functionalization was supported by the U.S. National Science Foundation Awards CHE-1148597 and CHE-1464578 and by the Center for Catalytic Hydrocarbon Functionalization, an Energy Frontier Research Center, U.S. Department of Energy, Office of Science, Basic Energy Sciences, under Award DESC0001298. Cytochrome P450 and APO enzymology has been supported by the National Institutes of Health (2R37 GM036298). X.H. thanks Merck, Inc., the Howard Hughes Foundation, and NIH (Award F32GM125231) for fellowship support.
Glossary
Abbreviations
- AOS
allene oxide synthase
- APX
ascorbate peroxidase
- CcO
cytochrome c oxidase
- Cld
chlorite dismutase
- Cpd 0
compound 0
- Cpd I
compound I
- Cpd II
compound II
- DES
divinyl ether synthase
- HAT
hydrogen atom transfer
- Hb
hemoglobin
- HRP
horseradish peroxidase
- Mb
myoglobin
- OAT
oxygen atom transfer
- TPFPBr8
2,3,7,8,12,13,17,18-octabromo-5,10,15,20-tetrakis(pentafluorophenyl)porphyrin
- TpivPP
5,10,15,20-tetra(α,α,α,α-O-pivalamidophenyl)porphyrin
- TPP
5,10,15,20-tetraphenylporphyrin
- TMP
5,10,15,20-tetramesitylporphyrin
- TPFPP
5,10,15,20-tetrakis(pentafluorophenyl)porphyrin
- TDMPP
meso-tetrakis(2,6-dimethylphenyl)porphyrin
- TTMPP
meso-tetrakis(3,4,5-trimethoxypbenyl)porphyrin
- 4-TMPyP
meso-tetrakis(4-N-methylpyridyl)porphyrin
- 2-TMPyP
meso-tetrakis(2-N-methylpyridyl)porphyrin
- TF4TMAP
meso-tetrakis(2,3,5,6-tetrafluoro-N,N,N-trimethyl-4-aniliniumyl)porphinato dianion
- TFMPC
5,10,15-tris(3,5-trifluoromethylphenyl)corrolato trianion
- PHM
peptidylglycine α-hydroxylating monooxygenase
- TDO
tryptophan 2,3-dioxygenase
- IDO
indoleamine 2,3-dioxygenase
Biographies
Xiongyi Huang received his B.S. degree in chemistry in 2010 from University of Science and Technology of China (USTC), where he performed undergraduate research in computational chemistry under the guidance of Prof. Yao Fu and Prof. Jing Shi. He received his Ph.D. in 2016 at Princeton University with Prof. John T. Groves. His Ph.D. work mainly focused on developing new synthetic strategies for the construction of aliphatic C–halogen, C–F, and C–N bonds using biomimetic manganese catalysts. While at Princeton, Xiongyi was a recipient of the HHMI International Student Research Fellowship (2013–2015) and the 2016 Chinese Government Award for Outstanding Students Abroad. He is currently an NIH Ruth L. Kirschstein postdoctoral fellow in the laboratory of Prof. Frances H. Arnold at Caltech, where he works on developing new enzymatic reactions via directed evolution.
John T. Groves is the Hugh Stott Taylor Chair of Chemistry at Princeton University. He received an S.B. in Chemistry at MIT and a Ph.D. in Organic Chemistry at Columbia University, where he worked with Ronald Breslow. Groves’ early academic career was at the University of Michigan where he was director of the Michigan Center for Catalysis and Surface Science and received the Phi Lambda Upsilon Award for Teaching and Leadership. Moving to Princeton University as Professor of Chemistry, he became Department Chair in 1988. He has received the Arthur C. Cope Scholar Award, the Alfred Bader Award in Bioorganic and Bioinorganic Chemistry, the Ira Remsen Award and the Award in Inorganic Chemistry, all from the American Chemical Society. He was co-Chair of the ACS-NSF Committee and Workshop on the Molecular Basis of Life Processes. He is a fellow of the American Academy of Arts and Sciences, the American Association for the Advancement of Science, the Japan Society for the Promotion of Science, and the Royal Society of Chemistry. Groves is a member of the U.S. National Academy of Sciences. International awards include the Grand Prix, Maison de la Chimie (Paris), the Frontiers in Biological Chemistry Award from the Max Planck Institute (Mülheim), and the Hans Fischer Award in Porphyrin Chemistry. Groves’ research has focused on the roles of metal–oxo species in chemical and biological oxidations.
The authors declare no competing financial interest.
References
- Kovaleva E. G.; Lipscomb J. D. Versatility of Biological Non-Heme Fe(II) Centers in Oxygen Activation Reactions. Nat. Chem. Biol. 2008, 4, 186–193. 10.1038/nchembio.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pau M. Y. M.; Lipscomb J. D.; Solomon E. I. Substrate Activation for O2 Reactions by Oxidized Metal Centers in Biology. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 18355–18362. 10.1073/pnas.0704191104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groves J. T. The Bioinorganic Chemistry of Iron in Oxygenases and Supramolecular Assemblies. Proc. Natl. Acad. Sci. U. S. A. 2003, 100, 3569–3574. 10.1073/pnas.0830019100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groves J. T. High-Valent Iron in Chemical and Biological Oxidations. J. Inorg. Biochem. 2006, 100, 434–447. 10.1016/j.jinorgbio.2006.01.012. [DOI] [PubMed] [Google Scholar]
- Sheng Y.; Abreu I. A.; Cabelli D. E.; Maroney M. J.; Miller A.-F.; Teixeira M.; Valentine J. S. Superoxide Dismutases and Superoxide Reductases. Chem. Rev. 2014, 114, 3854–3918. 10.1021/cr4005296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decker A.; Solomon E. I. Dioxygen Activation by Copper, Heme and Non-Heme Iron Enzymes: Comparison of Electronic Structures and Reactivities. Curr. Opin. Chem. Biol. 2005, 9, 152–163. 10.1016/j.cbpa.2005.02.012. [DOI] [PubMed] [Google Scholar]
- Poulos T. L. Heme Enzyme Structure and Function. Chem. Rev. 2014, 114, 3919–3962. 10.1021/cr400415k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mbughuni M. M.; Chakrabarti M.; Hayden J. A.; Bominaar E. L.; Hendrich M. P.; Münck E.; Lipscomb J. D. Trapping and Spectroscopic Characterization of an FeIII-Superoxo Intermediate from a Nonheme Mononuclear Iron-Containing Enzyme. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 16788–16793. 10.1073/pnas.1010015107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh S. Mononuclear Copper Active-Oxygen Complexes. Curr. Opin. Chem. Biol. 2006, 10, 115–122. 10.1016/j.cbpa.2006.02.012. [DOI] [PubMed] [Google Scholar]
- Mirica L. M.; Ottenwaelder X.; Stack T. D. P. Structure and Spectroscopy of Copper–Dioxygen Complexes. Chem. Rev. 2004, 104, 1013–1046. 10.1021/cr020632z. [DOI] [PubMed] [Google Scholar]
- Elwell C. E.; Gagnon N. L.; Neisen B. D.; Dhar D.; Spaeth A. D.; Yee G. M.; Tolman W. B. Copper–Oxygen Complexes Revisited: Structures, Spectroscopy, and Reactivity. Chem. Rev. 2017, 117, 2059–2107. 10.1021/acs.chemrev.6b00636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costas M.; Mehn M. P.; Jensen M. P.; Que L. Dioxygen Activation at Mononuclear Nonheme Iron Active Sites: Enzymes, Models, and Intermediates. Chem. Rev. 2004, 104, 939–986. 10.1021/cr020628n. [DOI] [PubMed] [Google Scholar]
- Huang X.; Groves J. T. Beyond Ferryl-Mediated Hydroxylation: 40 Years of the Rebound Mechanism and C–H Activation. JBIC, J. Biol. Inorg. Chem. 2017, 22, 185–207. 10.1007/s00775-016-1414-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Eldik R. Fascinating Inorganic/Bioinorganic Reaction Mechanisms. Coord. Chem. Rev. 2007, 251, 1649–1662. 10.1016/j.ccr.2007.02.004. [DOI] [Google Scholar]
- Oszajca M.; Franke A.; Brindell M.; Stochel G.; van Eldik R. Redox Cycling in the Activation of Peroxides by Iron Porphyrin and Manganese Complexes. ‘Catching’ Catalytic Active Intermediates. Coord. Chem. Rev. 2016, 306, 483–509. 10.1016/j.ccr.2015.01.013. [DOI] [Google Scholar]
- Ortiz de Montellano P. R. Hydrocarbon Hydroxylation by Cytochrome P450 Enzymes. Chem. Rev. 2010, 110, 932–948. 10.1021/cr9002193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortiz de Montellano P. R.; De Voss J. J. Oxidizing Species in the Mechanism of Cytochrome P450. Nat. Prod. Rep. 2002, 19, 477–493. 10.1039/b101297p. [DOI] [PubMed] [Google Scholar]
- Krest C. M.; Onderko E. L.; Yosca T. H.; Calixto J. C.; Karp R. F.; Livada J.; Rittle J.; Green M. T. Reactive Intermediates in Cytochrome P450 Catalysis. J. Biol. Chem. 2013, 288, 17074–17081. 10.1074/jbc.R113.473108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yosca T. H.; Ledray A. P.; Ngo J.; Green M. T. A New Look at the Role of Thiolate Ligation in Cytochrome P450. JBIC, J. Biol. Inorg. Chem. 2017, 22, 209–220. 10.1007/s00775-016-1430-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collman J. P.; Boulatov R.; Sunderland C. J.; Fu L. Functional Analogues of Cytochrome c Oxidase, Myoglobin, and Hemoglobin. Chem. Rev. 2004, 104, 561–588. 10.1021/cr0206059. [DOI] [PubMed] [Google Scholar]
- Denisov I. G.; Makris T. M.; Sligar S. G.; Schlichting I. Structure and Chemistry of Cytochrome P450. Chem. Rev. 2005, 105, 2253–2278. 10.1021/cr0307143. [DOI] [PubMed] [Google Scholar]
- Jones R. D.; Summerville D. A.; Basolo F. Synthetic Oxygen Carriers Related to Biological-Systems. Chem. Rev. 1979, 79, 139–179. 10.1021/cr60318a002. [DOI] [Google Scholar]
- Kim E.; Chufán E. E.; Kamaraj K.; Karlin K. D. Synthetic Models for Heme–Copper Oxidases. Chem. Rev. 2004, 104, 1077–1134. 10.1021/cr0206162. [DOI] [PubMed] [Google Scholar]
- Momenteau M.; Reed C. A. Synthetic Heme-Dioxygen Complexes. Chem. Rev. 1994, 94, 659–698. 10.1021/cr00027a006. [DOI] [Google Scholar]
- Shaik S.; Cohen S.; Wang Y.; Chen H.; Kumar D.; Thiel W. P450 Enzymes: Their Structure, Reactivity, and Selectivity-Modeled by QM/MM Calculations. Chem. Rev. 2010, 110, 949–1017. 10.1021/cr900121s. [DOI] [PubMed] [Google Scholar]
- Costas M. Selective C-H oxidation catalyzed by metalloporphyrins. Coord. Chem. Rev. 2011, 255, 2912–2932. 10.1016/j.ccr.2011.06.026. [DOI] [Google Scholar]
- Fujii H. Electronic Structure and Reactivity of High-Valent Oxo Iron Porphyrins. Coord. Chem. Rev. 2002, 226, 51–60. 10.1016/S0010-8545(01)00441-6. [DOI] [Google Scholar]
- Fukuzumi S.; Kojima T.; Lee Y.-M.; Nam W. High-Valent Metal-Oxo Complexes Generated in Catalytic Oxidation Reactions Using Water as an Oxygen Source. Coord. Chem. Rev. 2017, 333, 44–56. 10.1016/j.ccr.2016.09.018. [DOI] [Google Scholar]
- Sahu S.; Goldberg D. P. Activation of Dioxygen by Iron and Manganese Complexes: A Heme and Nonheme Perspective. J. Am. Chem. Soc. 2016, 138, 11410–11428. 10.1021/jacs.6b05251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sono M.; Roach M. P.; Coulter E. D.; Dawson J. H. Heme-Containing Oxygenases. Chem. Rev. 1996, 96, 2841–2888. 10.1021/cr9500500. [DOI] [PubMed] [Google Scholar]
- Watanabe Y.; Nakajima H.; Ueno T. Reactivities of Oxo and Peroxo Intermediates Studied by Hemoprotein Mutants. Acc. Chem. Res. 2007, 40, 554–562. 10.1021/ar600046a. [DOI] [PubMed] [Google Scholar]
- Nam W. High-Valent Iron(IV)–Oxo Complexes of Heme and Non-Heme Ligands in Oxygenation Reactions. Acc. Chem. Res. 2007, 40, 522–531. 10.1021/ar700027f. [DOI] [PubMed] [Google Scholar]
- Zhang W.; Lai W.; Cao R. Energy-Related Small Molecule Activation Reactions: Oxygen Reduction and Hydrogen and Oxygen Evolution Reactions Catalyzed by Porphyrin- and Corrole-Based Systems. Chem. Rev. 2017, 117, 3717–3797. 10.1021/acs.chemrev.6b00299. [DOI] [PubMed] [Google Scholar]
- Sorokin A. B. Phthalocyanine Metal Complexes in Catalysis. Chem. Rev. 2013, 113, 8152–8191. 10.1021/cr4000072. [DOI] [PubMed] [Google Scholar]
- Baglia R. A.; Zaragoza J. P. T.; Goldberg D. P.. Biomimetic Reactivity of Oxygen-Derived Manganese and Iron Porphyrinoid Complexes. Chem. Rev. 2017, asap, 11713320. 10.1021/acs.chemrev.7b00180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marti M. A.; Crespo A.; Capece L.; Boechi L.; Bikiel D. E.; Scherlis D. A.; Estrin D. A. Dioxygen Affinity in Heme Proteins Investigated by Computer Simulation. J. Inorg. Biochem. 2006, 100, 761–770. 10.1016/j.jinorgbio.2005.12.009. [DOI] [PubMed] [Google Scholar]
- Scott E. E.; Gibson Q. H.; Olson J. S. Mapping the Pathways for O2 Entry into and Exit from Myoglobin. J. Biol. Chem. 2001, 276, 5177–5188. 10.1074/jbc.M008282200. [DOI] [PubMed] [Google Scholar]
- Tsai A.-L.; Berka V.; Martin E.; Olson J. S. A “Sliding Scale Rule” for Selectivity among NO, CO, and O2 by Heme Protein Sensors. Biochemistry 2012, 51, 172–186. 10.1021/bi2015629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai A. L.; Martin E.; Berka V.; Olson J. S. How Do Heme-Protein Sensors Exclude Oxygen? Lessons Learned from Cytochrome c ′, Nostoc puntiforme Heme Nitric Oxide/Oxygen-Binding Domain, and Soluble Guanylyl Cyclase. Antioxid. Redox Signaling 2012, 17, 1246–1263. 10.1089/ars.2012.4564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson J. A.; Ishimura Y.; Griffin B. W. Pseudomonas putida Cytochrome P450: Characterization of an Oxygenated Form of the Hemoprotein. Arch. Biochem. Biophys. 1972, 149, 197–208. 10.1016/0003-9861(72)90315-3. [DOI] [PubMed] [Google Scholar]
- Zangar R. C.; Davydov D. R.; Verma S. Mechanisms That Regulate Production of Reactive Oxygen Species by Cytochrome P450. Toxicol. Appl. Pharmacol. 2004, 199, 316–331. 10.1016/j.taap.2004.01.018. [DOI] [PubMed] [Google Scholar]
- Grinstaff M.; Hill M.; Labinger J.; Gray H. Mechanism of Catalytic Oxygenation of Alkanes by Halogenated Iron Porphyrins. Science 1994, 264, 1311–1313. 10.1126/science.8191283. [DOI] [PubMed] [Google Scholar]
- Pauling L.; Coryell C. D. The Magnetic Properties and Structure of Hemoglobin, Oxyhemoglobin and Carbonmonoxyhemoglobin. Proc. Natl. Acad. Sci. U. S. A. 1936, 22, 210–216. 10.1073/pnas.22.4.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauling L. Nature of the Iron-Oxygen Bond in Oxyhaemoglobin. Nature 1964, 203, 182–183. 10.1038/203182b0. [DOI] [PubMed] [Google Scholar]
- Collman J. P.; Gagne R. R.; Reed C. A.; Robinson W. T.; Rodley G. A. Structure of an Iron(II) Dioxygen Complex; A Model for Oxygen Carrying Hemeproteins. Proc. Natl. Acad. Sci. U. S. A. 1974, 71, 1326–1329. 10.1073/pnas.71.4.1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips S. E. V. Structure and Refinement of Oxymyoglobin at 1.6 Å Resolution. J. Mol. Biol. 1980, 142, 531–554. 10.1016/0022-2836(80)90262-4. [DOI] [PubMed] [Google Scholar]
- Shaanan B. The Iron-Oxygen Bond in Human Oxyhaemoglobin. Nature 1982, 296, 683–684. 10.1038/296683a0. [DOI] [PubMed] [Google Scholar]
- Berglund G. I.; Carlsson G. H.; Smith A. T.; Szoke H.; Henriksen A.; Hajdu J. The Catalytic Pathway of Horseradish Peroxidase at High Resolution. Nature 2002, 417, 463–468. 10.1038/417463a. [DOI] [PubMed] [Google Scholar]
- Schlichting I.; Berendzen J.; Chu K.; Stock A. M.; Maves S. A.; Benson D. E.; Sweet R. M.; Ringe D.; Petsko G. A.; Sligar S. G. The Catalytic Pathway of Cytochrome P450cam at Atomic Resolution. Science 2000, 287, 1615–1622. 10.1126/science.287.5458.1615. [DOI] [PubMed] [Google Scholar]
- Nagano S.; Poulos T. L. Crystallographic Study on the Dioxygen Complex of Wild-Type and Mutant Cytochrome P450cam. Implications for the Dioxygen Activation Mechanism. J. Biol. Chem. 2005, 280, 31659–31663. 10.1074/jbc.M505261200. [DOI] [PubMed] [Google Scholar]
- Nagano S.; Cupp-Vickery J. R.; Poulos T. L. Crystal Structures of the Ferrous Dioxygen Complex of Wild-Type Cytochrome P450eryF and Its Mutants, A245S and A245T: Investigation of the Proton Transfer System in p450eryF. J. Biol. Chem. 2005, 280, 22102–22107. 10.1074/jbc.M501732200. [DOI] [PubMed] [Google Scholar]
- Vojtěchovský J.; Chu K.; Berendzen J.; Sweet R. M.; Schlichting I. Crystal Structures of Myoglobin-Ligand Complexes at Near-Atomic Resolution. Biophys. J. 1999, 77, 2153–2174. 10.1016/S0006-3495(99)77056-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H.; Ikeda-Saito M.; Shaik S. Nature of the Fe–O2 Bonding in Oxy-Myoglobin: Effect of the Protein. J. Am. Chem. Soc. 2008, 130, 14778–14790. 10.1021/ja805434m. [DOI] [PubMed] [Google Scholar]
- Bren K. L.; Eisenberg R.; Gray H. B. Discovery of the Magnetic Behavior of Hemoglobin: a Beginning of Bioinorganic Chemistry. Proc. Natl. Acad. Sci. U. S. A. 2015, 112, 13123–13127. 10.1073/pnas.1515704112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss J. J. Nature of the Iron-Oxygen Bond in Oxyhaemoglobin. Nature 1964, 202, 83–84. 10.1038/202083b0. [DOI] [PubMed] [Google Scholar]
- Goddard W. A.; Olafson B. D. Ozone Model for Bonding of an O2 to Heme in Oxyhemoglobin. Proc. Natl. Acad. Sci. U. S. A. 1975, 72, 2335–2339. 10.1073/pnas.72.6.2335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClure D. S. Electronic Structure of Transition-Metal Complex Ions. Radiat. Res., Suppl. 1960, 2, 218–242. 10.2307/3583598. [DOI] [Google Scholar]
- Lang G.; Marshali W. Mössbauer Effect in Some Haemoglobin Compounds. Proc. Phys. Soc., London 1966, 87, 3–34. 10.1088/0370-1328/87/1/302. [DOI] [PubMed] [Google Scholar]
- Sharrock M.; Debrunner P. G.; Schulz C.; Lipscomb J. D.; Marshall V.; Gunsalus I. C. Cytochrome P450cam and Its Complexes, MöSsbauer Parameters of the Heme Iron. Biochim. Biophys. Acta, Protein Struct. 1976, 420, 8–26. 10.1016/0005-2795(76)90340-8. [DOI] [PubMed] [Google Scholar]
- Das T. K.; Couture M.; Ouellet Y.; Guertin M.; Rousseau D. L. Simultaneous Observation of the O-O and Fe-O2 Stretching Modes in Oxyhemoglobins. Proc. Natl. Acad. Sci. U. S. A. 2001, 98, 479–484. 10.1073/pnas.98.2.479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiro T. G.; Strekas T. C. Resonance Raman Spectra of Heme Proteins. Effects of Oxidation and Spin State. J. Am. Chem. Soc. 1974, 96, 338–345. 10.1021/ja00809a004. [DOI] [PubMed] [Google Scholar]
- Yamamoto T.; Palmer G.; Gill D.; Salmeen I. T.; Rimai L. The Valence and Spin State of Iron in Oxyhemoglobin as Inferred from Resonance Raman Spectroscopy. J. Biol. Chem. 1973, 248, 5211–5213. [PubMed] [Google Scholar]
- Wilson S. A.; Green E.; Mathews I. I.; Benfatto M.; Hodgson K. O.; Hedman B.; Sarangi R. X-Ray Absorption Spectroscopic Investigation of the Electronic Structure Differences in Solution and Crystalline Oxyhemoglobin. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 16333–16338. 10.1073/pnas.1315734110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson S. A.; Kroll T.; Decreau R. A.; Hocking R. K.; Lundberg M.; Hedman B.; Hodgson K. O.; Solomon E. I. Iron L-Edge X-ray Absorption Spectroscopy of Oxy-Picket Fence Porphyrin: Experimental Insight into Fe–O2 Bonding. J. Am. Chem. Soc. 2013, 135, 1124–1136. 10.1021/ja3103583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang C. K.; Traylor T. G. Proximal Base Influence on the Binding of Oxygen and Carbon Monoxide to Heme. J. Am. Chem. Soc. 1973, 95, 8477–8479. 10.1021/ja00806a062. [DOI] [PubMed] [Google Scholar]
- Collman J. P. Synthetic Models for the Oxygen-Binding Hemoproteins. Acc. Chem. Res. 1977, 10, 265–272. 10.1021/ar50115a006. [DOI] [Google Scholar]
- Li J.; Noll B. C.; Oliver A. G.; Schulz C. E.; Scheidt W. R. Correlated Ligand Dynamics in Oxyiron Picket Fence Porphyrins: Structural and Mössbauer Investigations. J. Am. Chem. Soc. 2013, 135, 15627–15641. 10.1021/ja408431z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson J. S.; Gallagher A. T.; Mason J. A.; Harris T. D. A Five-Coordinate Heme Dioxygen Adduct Isolated within a Metal–Organic Framework. J. Am. Chem. Soc. 2014, 136, 16489–16492. 10.1021/ja5103103. [DOI] [PubMed] [Google Scholar]
- Basolo F.; Hoffman B. M.; Ibers J. A. Synthetic Oxygen Carriers of Biological Interest. Acc. Chem. Res. 1975, 8, 384–392. 10.1021/ar50095a004. [DOI] [Google Scholar]
- Collman J. P.; Brauman J. I.; Doxsee K. M.; Halbert T. R.; Hayes S. E.; Suslick K. S. Oxygen Binding to Cobalt Porphyrins. J. Am. Chem. Soc. 1978, 100, 2761–2766. 10.1021/ja00477a031. [DOI] [Google Scholar]
- Kitagawa T.; Ondrias M. R.; Rousseau D. L.; Ikedasaito M.; Yonetani T. Evidence for Hydrogen-Bonding of Bound Dioxygen to the Distal Histidine of Oxycobalt Myoglobin and Hemoglobin. Nature 1982, 298, 869–871. 10.1038/298869a0. [DOI] [PubMed] [Google Scholar]
- Wang M. Y. R.; Hoffman B. M.; Shire S. J.; Gurd F. R. N. Oxygen Binding to Myoglobins and Their Cobalt Analogs. J. Am. Chem. Soc. 1979, 101, 7394–7397. 10.1021/ja00518a044. [DOI] [Google Scholar]
- Brucker E. A.; Olson J. S.; Phillips G. N.; Dou Y.; IkedaSaito M. High Resolution Crystal Structures of the Deoxy, Oxy, and Aquomet Forms of Cobalt Myoglobin. J. Biol. Chem. 1996, 271, 25419–25422. 10.1074/jbc.271.41.25419. [DOI] [PubMed] [Google Scholar]
- Li J.; Noll B. C.; Oliver A. G.; Scheidt W. R. Structural Insights into Ligand Dynamics: Correlated Oxygen and Picket Motion in Oxycobalt Picket Fence Porphyrins. J. Am. Chem. Soc. 2012, 134, 10595–10606. 10.1021/ja303475a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallagher A. T.; Kelty M. L.; Park J. G.; Anderson J. S.; Mason J. A.; Walsh J. P. S.; Collins S. L.; Harris T. D. Dioxygen Binding at a Four-Coordinate Cobaltous Porphyrin Site in a Metal-Organic Framework: Structural, EPR, and O2 Adsorption Analysis. Inorg. Chem. Front. 2016, 3, 536–540. 10.1039/C5QI00275C. [DOI] [PubMed] [Google Scholar]
- Friesen B. A.; Bhattarai A.; Mazur U.; Hipps K. W. Single Molecule Imaging of Oxygenation of Cobalt Octaethylporphyrin at the Solution/Solid Interface: Thermodynamics from Microscopy. J. Am. Chem. Soc. 2012, 134, 14897–14904. 10.1021/ja304431b. [DOI] [PubMed] [Google Scholar]
- Weschler C. J.; Hoffman B. M.; Basolo F. Synthetic Oxygen Carrier. Dioxygen Adduct of a Manganese Porphyrin. J. Am. Chem. Soc. 1975, 97, 5278–5280. 10.1021/ja00851a043. [DOI] [PubMed] [Google Scholar]
- Gonzalez B.; Kouba J.; Yee S.; Reed C. A.; Kirner J. F.; Scheidt W. R. Manganese(II) Porphyrins - Synthesis, Structures, and Preference for 5-Coordination. J. Am. Chem. Soc. 1975, 97, 3247–3249. 10.1021/ja00844a070. [DOI] [Google Scholar]
- Reed C. A.; Kouba J. K.; Grimes C. J.; Cheung S. K. Manganese(II) and Chromium(II) Porphyrin Complexes - Synthesis and Characterization. Inorg. Chem. 1978, 17, 2666–2670. 10.1021/ic50187a057. [DOI] [Google Scholar]
- Hoffman B. M.; Weschler C. J.; Basolo F. The Dioxygen Adduct of Meso-Tetraphenylporphyrinmanganese(II), a Synthetic Oxygen Carrier. J. Am. Chem. Soc. 1976, 98, 5473–5482. 10.1021/ja00434a012. [DOI] [PubMed] [Google Scholar]
- Hoffman B. M.; Szymanski T.; Brown T. G.; Basolo F. Dioxygen Adducts of Several Manganese(II) Porphyrins - Electron-Paramagnetic Resonance Studies. J. Am. Chem. Soc. 1978, 100, 7253–7259. 10.1021/ja00491a022. [DOI] [Google Scholar]
- Watanabe T.; Ama T.; Nakamoto K. Matrix-Isolation Infrared-Spectra of (Octaethylporphyrinato)Manganese(II) and (Phthalocyaninato)Manganese(II) and Their Dioxygen Adducts. Inorg. Chem. 1983, 22, 2470–2472. 10.1021/ic00159a027. [DOI] [Google Scholar]
- Collman J. P.; Brauman J. I.; Fitzgerald J. P.; Sparapany J. W.; Ibers J. A. Reversible Binding of Dinitrogen and Dioxygen by a Ruthenium Picnic-Basket Porphyrin. J. Am. Chem. Soc. 1988, 110, 3486–3495. 10.1021/ja00219a024. [DOI] [Google Scholar]
- Paulson D. R.; Addison A. W.; Dolphin D.; James B. R. Preparation of Ruthenium(II) and Ruthenium(III) Myoglobin and the Reaction of Dioxygen, and Carbon Monoxide, with Ruthenium(II) Myoglobin. J. Biol. Chem. 1979, 254, 7002–7006. [PubMed] [Google Scholar]
- Farrell N.; Dolphin D. H.; James B. R. Reversible Binding of Dioxygen to Ruthenium(II) Porphyrins. J. Am. Chem. Soc. 1978, 100, 324–326. 10.1021/ja00469a076. [DOI] [Google Scholar]
- Cheung S. K.; Grimes C. J.; Wong J.; Reed C. A. Chromium(II) Porphyrins and an Irreversible Dioxygen Complex. J. Am. Chem. Soc. 1976, 98, 5028–5030. 10.1021/ja00432a061. [DOI] [Google Scholar]
- Blumenfeld L. A.; Davydov R. M.; Fel N. S.; Magonov S. N.; Vilu R. O. Studies on Conformational-Changes of Metalloproteins Induced by Electrons in Water-Ethylene Glycol Solutions at Low-Temperatures - Cytochrome c. FEBS Lett. 1974, 45, 256–258. 10.1016/0014-5793(74)80856-2. [DOI] [PubMed] [Google Scholar]
- Blumenfeld L. A.; Davydov R. M.; Magonov S. N.; Vilu R. O. Studies on Conformational-Changes of Metalloproteins Induced by Electrons in Water-Ethylene-Glycol Solutions at Low-Temperatures - Hemoglobin. FEBS Lett. 1974, 49, 246–248. 10.1016/0014-5793(74)80522-3. [DOI] [PubMed] [Google Scholar]
- Symons M. C. R.; Petersen R. L. Electron Capture at the Iron-Oxygen Centre in Single Crystals of Oxymyoglobin Studied by Electron Spin Resonance Spectroscopy. Biochim. Biophys. Acta, Protein Struct. 1978, 535, 241–246. 10.1016/0005-2795(78)90090-9. [DOI] [PubMed] [Google Scholar]
- Denisov I. G.; Makris T. M.; Sligar S. G. In Cytochrome P450, Part C; Johnson E. F., Waterman M. R., Eds.; Elsevier Academic Press: San Diego, 2002; Vol. 357. [Google Scholar]
- Davydov R.; Hoffman B. M. Active Intermediates in Heme Monooxygenase Reactions as Revealed by Cryoreduction/Annealing, EPR/ENDOR Studies. Arch. Biochem. Biophys. 2011, 507, 36–43. 10.1016/j.abb.2010.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denisov I. G.; Sligar S. G. In Cytochrome P450: Structure, Mechanism, and Biochemistry; Ortiz de Montellano P. R., Ed.; Springer International Publishing: Cham, 2015. [Google Scholar]
- Davydov R.; Chemerisov S.; Werst D. E.; Rajh T.; Matsui T.; Ikeda-Saito M.; Hoffman B. M. Proton Transfer at Helium Temperatures During Dioxygen Activation by Heme Monooxygenases. J. Am. Chem. Soc. 2004, 126, 15960–15961. 10.1021/ja044646t. [DOI] [PubMed] [Google Scholar]
- Davydov R.; Makris T. M.; Kofman V.; Werst D. E.; Sligar S. G.; Hoffman B. M. Hydroxylation of Camphor By-Reduced Oxy-Cytochrome P450cam: Mechanistic Implications of EPR and ENDOR Studies of Catalytic Intermediates in Native and Mutant Enzymes. J. Am. Chem. Soc. 2001, 123, 1403–1415. 10.1021/ja003583l. [DOI] [PubMed] [Google Scholar]
- Denisov I. G.; Makris T. M.; Sligar S. G. Formation and Decay of Hydroperoxo-Ferric Heme Complex in Horseradish Peroxidase Studied by Cryoradiolysis. J. Biol. Chem. 2002, 277, 42706–42710. 10.1074/jbc.M207949200. [DOI] [PubMed] [Google Scholar]
- Mak P. J.; Thammawichai W.; Wiedenhoeft D.; Kincaid J. R. Resonance Raman Spectroscopy Reveals pH-Dependent Active Site Structural Changes of Lactoperoxidase Compound 0 and Its Ferryl Heme O–O Bond Cleavage Products. J. Am. Chem. Soc. 2015, 137, 349–361. 10.1021/ja5107833. [DOI] [PubMed] [Google Scholar]
- Kuhnel K.; Derat E.; Terner J.; Shaik S.; Schlichting I. Structure and Quantum Chemical Characterization of Chloroperoxidase Compound 0, a Common Reaction Intermediate of Diverse Heme Enzymes. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 99–104. 10.1073/pnas.0606285103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unno M.; Chen H.; Kusama S.; Shaik S.; Ikeda-Saito M. Structural Characterization of the Fleeting Ferric Peroxo Species in Myoglobin: Experiment and Theory. J. Am. Chem. Soc. 2007, 129, 13394–13395. 10.1021/ja076108x. [DOI] [PubMed] [Google Scholar]
- Hersleth H. P.; Hsiao Y. W.; Ryde U.; Gorbitz C. H.; Andersson K. K. The Crystal Structure of Peroxymyoglobin Generated Through Cryoradiolytic Reduction of Myoglobin Compound Iii During Data Collection. Biochem. J. 2008, 412, 257–264. 10.1042/BJ20070921. [DOI] [PubMed] [Google Scholar]
- Burstyn J. N.; Roe J. A.; Miksztal A. R.; Shaevitz B. A.; Lang G.; Valentine J. S. Magnetic and Spectroscopic Characterization of an Iron Porphyrin Peroxide Complex - Peroxoferrioctaethylporphyrin(1-). J. Am. Chem. Soc. 1988, 110, 1382–1388. 10.1021/ja00213a009. [DOI] [Google Scholar]
- McCandlish E.; Miksztal A. R.; Nappa M.; Sprenger A. Q.; Valentine J. S.; Stong J. D.; Spiro T. G. Reactions of Superoxide with Iron Porphyrins in Aprotic Solvents. a High Spin Ferric Porphyrin Peroxo Complex. J. Am. Chem. Soc. 1980, 102, 4268–4271. 10.1021/ja00532a053. [DOI] [Google Scholar]
- Welborn C. H.; Dolphin D.; James B. R. One-Electron Electrochemical Reduction of a Ferrous Porphyrin Dioxygen Complex. J. Am. Chem. Soc. 1981, 103, 2869–2871. 10.1021/ja00400a067. [DOI] [Google Scholar]
- Tajima K. A Possible Model of a Hemoprotein Hydrogen-Peroxide Complex. Inorg. Chim. Acta 1989, 163, 115–122. 10.1016/S0020-1693(00)87150-9. [DOI] [Google Scholar]
- Tajima K.; Shigematsu M.; Jinno J.; Ishizu K.; Ohyanishiguchi H. Generation of Fe(III)OEP-Hydrogen Peroxide Complex (OEP = Octaethylporphyrinato) by Reduction of Fe(II)OEP-O2 with Ascorbic Acid Sodium Salt. J. Chem. Soc., Chem. Commun. 1990, 144–145. 10.1039/C39900000144. [DOI] [Google Scholar]
- Tajima K.; Oka S.; Edo T.; Miyake S.; Mano H.; Mukai K.; Sakurai H.; Ishizu K. Optical-Absorption and Epr Studies on a 6-Coordinate Iron(III)-Tetramesitylporphyrin-Hydrogen Peroxide Complex Having a Nitrogenous Axial Ligand. J. Chem. Soc., Chem. Commun. 1995, 0, 1507–1508. 10.1039/C39950001507. [DOI] [Google Scholar]
- Liu J. G.; Ohta T.; Yamaguchi S.; Ogura T.; Sakamoto S.; Maeda Y.; Naruta Y. Spectroscopic Characterization of a Hydroperoxo-Heme Intermediate: Conversion of a Side-On Peroxo to an End-On Hydroperoxo Complex. Angew. Chem., Int. Ed. 2009, 48, 9262–9267. 10.1002/anie.200904572. [DOI] [PubMed] [Google Scholar]
- de Visser S. P.; Valentine J. S.; Nam W. A Biomimetic Ferric Hydroperoxo Porphyrin Intermediate. Angew. Chem., Int. Ed. 2010, 49, 2099–2101. 10.1002/anie.200906736. [DOI] [PubMed] [Google Scholar]
- Garcia-Serres R.; Davydov R. M.; Matsui T.; Ikeda-Saito M.; Hoffman B. M.; Huynh B. H. Distinct Reaction Pathways Followed upon Reduction of Oxy-Heme Oxygenase and Oxy-Myoglobin as Characterized by Mössbauer Spectroscopy. J. Am. Chem. Soc. 2007, 129, 1402–1412. 10.1021/ja067209i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J. G.; Shimizu Y.; Ohta T.; Naruta Y. Formation of an End-On Ferric Peroxo Intermediate upon One-Electron Reduction of a Ferric Superoxo Heme. J. Am. Chem. Soc. 2010, 132, 3672–3673. 10.1021/ja1001955. [DOI] [PubMed] [Google Scholar]
- Nagaraju P.; Ohta T.; Liu J.-G.; Ogura T.; Naruta Y. The Secondary Coordination Sphere Controlled Reactivity of a Ferric-Superoxo Heme: Unexpected Conversion to a Ferric Hydroperoxo Intermediate by Reaction with a High-Spin Ferrous Heme. Chem. Commun. 2016, 52, 7213–7216. 10.1039/C6CC02162J. [DOI] [PubMed] [Google Scholar]
- Sengupta K.; Chatterjee S.; Dey A. Catalytic H2O2 Disproportionation and Electrocatalytic O2 Reduction by a Functional Mimic of Heme Catalase: Direct Observation of Compound 0 and Compound I in Situ. ACS Catal. 2016, 6, 1382–1388. 10.1021/acscatal.5b02668. [DOI] [Google Scholar]
- Oliveira R.; Zouari W.; Herrero C.; Banse F.; Schöllhorn B.; Fave C.; Anxolabéhère-Mallart E. Characterization and Subsequent Reactivity of an Fe-Peroxo Porphyrin Generated by Electrochemical Reductive Activation of O2. Inorg. Chem. 2016, 55, 12204–12210. 10.1021/acs.inorgchem.6b01804. [DOI] [PubMed] [Google Scholar]
- Costentin C.; Dridi H.; Savéant J.-M. Molecular Catalysis of O2 Reduction by Iron Porphyrins in Water: Heterogeneous versus Homogeneous Pathways. J. Am. Chem. Soc. 2015, 137, 13535–13544. 10.1021/jacs.5b06834. [DOI] [PubMed] [Google Scholar]
- Sengupta K.; Chatterjee S.; Samanta S.; Dey A. Direct Observation of Intermediates Formed During Steady-State Electrocatalytic O2 Reduction by Iron Porphyrins. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 8431–8436. 10.1073/pnas.1300808110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keilin D.; Mann T. On the Haematin Compound of Peroxidase. Proc. R. Soc. London, Ser. B 1937, 122, 119–133. 10.1098/rspb.1937.0015. [DOI] [Google Scholar]
- Theorell H. Crystalline Peroxidase. Enzymologia 1942, 10, 250–252. [Google Scholar]
- Stern K. G. On the Mechanism of Enzyme Action: A study of the Decomposition of Monoethyl Hydrogen Peroxide by Catalse and of an Intermediate Enzyme Substrate Compound. J. Biol. Chem. 1936, 114, 473–494. [Google Scholar]
- Chance B.; Rosted C. O.; Sillén L. G.; Linnasalmi A.; Laukkanen P. An Intermediate Compound in the Catalase-Hydrogen Peroxide Reaction. Acta Chem. Scand. 1947, 1, 236–267. 10.3891/acta.chem.scand.01-0236. [DOI] [Google Scholar]
- Nicholls P. Classical Catalase: Ancient and Modern. Arch. Biochem. Biophys. 2012, 525, 95–101. 10.1016/j.abb.2012.01.015. [DOI] [PubMed] [Google Scholar]
- Jones P.; Dunford H. B. On the Mechanism of Compound I Formation from Peroxidases and Catalases. J. Theor. Biol. 1977, 69, 457–470. 10.1016/0022-5193(77)90152-7. [DOI] [PubMed] [Google Scholar]
- Baek H. K.; Van Wart H. E. Elementary Steps in the Formation of Horseradish Peroxidase Compound I: Direct Observation of Compound 0, a New Intermediate with a Hyperporphyrin Spectrum. Biochemistry 1989, 28, 5714–5719. 10.1021/bi00440a003. [DOI] [PubMed] [Google Scholar]
- Dunford H. B.; Stillman J. S. On the Function and Mechanism of Action of Peroxidases. Coord. Chem. Rev. 1976, 19, 187–251. 10.1016/S0010-8545(00)80316-1. [DOI] [Google Scholar]
- Jones P.; Dunford H. B. The Mechanism of Compound I Formation Revisited. J. Inorg. Biochem. 2005, 99, 2292–2298. 10.1016/j.jinorgbio.2005.08.009. [DOI] [PubMed] [Google Scholar]
- Poulos T. L.; Kraut J. The Stereochemistry of Peroxidase Catalysis. J. Biol. Chem. 1980, 255, 8199–8205. [PubMed] [Google Scholar]
- Poulos T. L.; Freer S. T.; Alden R. A.; Edwards S. L.; Skoglund U.; Takio K.; Eriksson B.; Xuong N.; Yonetani T.; Kraut J. The Crystal Structure of Cytochrome c Peroxidase. J. Biol. Chem. 1980, 255, 575–580. [PubMed] [Google Scholar]
- Casadei C. M.; Gumiero A.; Metcalfe C. L.; Murphy E. J.; Basran J.; Concilio M. G.; Teixeira S. C. M.; Schrader T. E.; Fielding A. J.; Ostermann A.; et al. Neutron Cryo-Crystallography Captures the Protonation State of Ferryl Heme in a Peroxidase. Science 2014, 345, 193–197. 10.1126/science.1254398. [DOI] [PubMed] [Google Scholar]
- Groves J. T.; Boaz N. C. Fishing for Peroxidase Protons. Science 2014, 345, 142–143. 10.1126/science.1256754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson J. Probing Structure-Function Relations in Heme-Containing Oxygenases and Peroxidases. Science 1988, 240, 433–439. 10.1126/science.3358128. [DOI] [PubMed] [Google Scholar]
- Usharani D.; Zazza C.; Lai W.; Chourasia M.; Waskell L.; Shaik S. A Single-Site Mutation (F429H) Converts the Enzyme CYP 2B4 into a Heme Oxygenase: A QM/MM Study. J. Am. Chem. Soc. 2012, 134, 4053–4056. 10.1021/ja211905e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson J. H.; Sono M. Cytochrome P450 and Chloroperoxidase: Thiolate-Ligated Heme Enzymes. Spectroscopic Determination of Their Active-Site Structures and Mechanistic Implications of Thiolate Ligation. Chem. Rev. 1987, 87, 1255–1276. 10.1021/cr00081a015. [DOI] [Google Scholar]
- Fita I.; Rossmann M. G. The Active Center of Catalase. J. Mol. Biol. 1985, 185, 21–37. 10.1016/0022-2836(85)90180-9. [DOI] [PubMed] [Google Scholar]
- Sundaramoorthy M.; Terner J.; Poulos T. L. The Crystal Structure of Chloroperoxidase: a Heme Peroxidase–Cytochrome P450 Functional Hybrid. Structure 1995, 3, 1367–1378. 10.1016/S0969-2126(01)00274-X. [DOI] [PubMed] [Google Scholar]
- Yi X.; Conesa A.; Punt P. J.; Hager L. P. Examining the Role of Glutamic Acid 183 in Chloroperoxidase Catalysis. J. Biol. Chem. 2003, 278, 13855–13859. 10.1074/jbc.M210906200. [DOI] [PubMed] [Google Scholar]
- Piontek K.; Strittmatter E.; Ullrich R.; Grobe G.; Pecyna M. J.; Kluge M.; Scheibner K.; Hofrichter M.; Plattner D. A. Structural Basis of Substrate Conversion in a New Aromatic Peroxygenase: Cytochrome P450 Functionality with Benefits. J. Biol. Chem. 2013, 288, 34767–34776. 10.1074/jbc.M113.514521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee D. S.; Yamada A.; Sugimoto H.; Matsunaga I.; Ogura H.; Ichihara K.; Adachi S.; Park S. Y.; Shiro Y. Substrate Recognition and Molecular Mechanism of Fatty Acid Hydroxylation by Cytochrome P450 from Bacillus Subtilis. Crystallographic, Spectroscopic, and Mutational Studies. J. Biol. Chem. 2003, 278, 9761–9767. 10.1074/jbc.M211575200. [DOI] [PubMed] [Google Scholar]
- Belcher J.; McLean K. J.; Matthews S.; Woodward L. S.; Fisher K.; Rigby S. E. J.; Nelson D. R.; Potts D.; Baynham M. T.; Parker D. A.; et al. Structure and Biochemical Properties of the Alkene Producing Cytochrome P450 OleTJE (CYP152L1) from the Jeotgalicoccus sp. 8456 Bacterium. J. Biol. Chem. 2014, 289, 6535–6550. 10.1074/jbc.M113.527325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B.; Li C.; Dubey K. D.; Shaik S. Quantum Mechanical/Molecular Mechanical Calculated Reactivity Networks Reveal How Cytochrome P450cam and Its T252A Mutant Select Their Oxidation Pathways. J. Am. Chem. Soc. 2015, 137, 7379–7390. 10.1021/jacs.5b02800. [DOI] [PubMed] [Google Scholar]
- Cho K.-B.; Derat E.; Shaik S. Compound I of Nitric Oxide Synthase: The Active Site Protonation State. J. Am. Chem. Soc. 2007, 129, 3182–3188. 10.1021/ja066662r. [DOI] [PubMed] [Google Scholar]
- Ramanan R.; Dubey K. D.; Wang B.; Mandal D.; Shaik S. Emergence of Function in P450-Proteins: A Combined Quantum Mechanical/Molecular Mechanical and Molecular Dynamics Study of the Reactive Species in the H2O2-Dependent Cytochrome P450SPα and Its Regio- and Enantioselective Hydroxylation of Fatty Acids. J. Am. Chem. Soc. 2016, 138, 6786–6797. 10.1021/jacs.6b01716. [DOI] [PubMed] [Google Scholar]
- Shoji O.; Watanabe Y. Peroxygenase Reactions Catalyzed by Cytochromes P450. JBIC, J. Biol. Inorg. Chem. 2014, 19, 529–539. 10.1007/s00775-014-1106-9. [DOI] [PubMed] [Google Scholar]
- Shoji O.; Fujishiro T.; Nakajima H.; Kim M.; Nagano S.; Shiro Y.; Watanabe Y. Hydrogen Peroxide Dependent Monooxygenations by Tricking the Substrate Recognition of Cytochrome P450BSβ. Angew. Chem., Int. Ed. 2007, 46, 3656–3659. 10.1002/anie.200700068. [DOI] [PubMed] [Google Scholar]
- Kumar D.; Altun A.; Shaik S.; Thiel W. Water as Biocatalyst in Cytochrome P450. Faraday Discuss. 2011, 148, 373–383. 10.1039/C004950F. [DOI] [PubMed] [Google Scholar]
- Li Q.-S.; Ogawa J.; Schmid R. D.; Shimizu S. Engineering Cytochrome P450 BM3 for Oxidation of Polycyclic Aromatic Hydrocarbons. Appl. Environ. Microbiol. 2001, 67, 5735–5739. 10.1128/AEM.67.12.5735-5739.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirino P. C.; Arnold F. H. A Self-Sufficient Peroxide-Driven Hydroxylation Biocatalyst. Angew. Chem., Int. Ed. 2003, 42, 3299–3301. 10.1002/anie.200351434. [DOI] [PubMed] [Google Scholar]
- Li Q. S.; Ogawa J.; Shimizu S. Critical Role of the Residue Size at Position 87 in H2O2-Dependent Substrate Hydroxylation Activity and H2O2 Inactivation of Cytochrome P450BM3. Biochem. Biophys. Res. Commun. 2001, 280, 1258–1261. 10.1006/bbrc.2001.4261. [DOI] [PubMed] [Google Scholar]
- Shoji O.; Fujishiro T.; Nishio K.; Kano Y.; Kimoto H.; Chien S.-C.; Onoda H.; Muramatsu A.; Tanaka S.; Hori A.; et al. A Substrate-Binding-State Mimic of H2O2-Dependent Cytochrome P450 Produced by One-Point Mutagenesis and Peroxygenation of Non-Native Substrates. Catal. Sci. Technol. 2016, 6, 5806–5811. 10.1039/C6CY00630B. [DOI] [Google Scholar]
- Traylor T. G.; Lee W. A.; Stynes D. V. Model Compound Studies Related to Peroxidases. Mechanisms of Reactions of Hemins with Peracids. J. Am. Chem. Soc. 1984, 106, 755–764. 10.1021/ja00315a050. [DOI] [Google Scholar]
- Lee W. A.; Bruice T. C. Homolytic and Heterolytic Oxygen-Oxygen Bond Scissions Accompanying Oxygen Transfer to Iron(III) Porphyrins by Percarboxylic Acids and Hydroperoxides. a Mechanistic Criterion for Peroxidase and Cytochrome P-450. J. Am. Chem. Soc. 1985, 107, 513–514. 10.1021/ja00288a046. [DOI] [Google Scholar]
- Groves J. T.; Watanabe Y. Oxygen Activation by Metalloporphyrins Related to Peroxidase and Cytochrome P450. Direct Observation of the Oxygen-Oxygen Bond Cleavage Step. J. Am. Chem. Soc. 1986, 108, 7834–7836. 10.1021/ja00284a058. [DOI] [PubMed] [Google Scholar]
- Traylor T. G.; Xu F. A Biomimetic Model for Catalase: the Mechanisms of Reaction of Hydrogen Peroxide and Hydroperoxides with Iron(III) Porphyrins. J. Am. Chem. Soc. 1987, 109, 6201–6202. 10.1021/ja00254a059. [DOI] [Google Scholar]
- Groves J. T.; Watanabe Y. Reactive Iron Porphyrin Derivatives Related to the Catalytic Cycles of Cytochrome P450 and Peroxidase. Studies of the Mechanism of Oxygen Activation. J. Am. Chem. Soc. 1988, 110, 8443–8452. 10.1021/ja00233a021. [DOI] [Google Scholar]
- Yamaguchi K.; Watanabe Y.; Morishima I. Direct Observation of the Push Effect on the Oxygen-Oxygen Bond Cleavage of Acylperoxoiron(III) Porphyrin Complexes. J. Am. Chem. Soc. 1993, 115, 4058–4065. 10.1021/ja00063a026. [DOI] [Google Scholar]
- Hessenauer-Ilicheva N.; Franke A.; Meyer D.; Woggon W.-D.; van Eldik R. Low-Temperature Rapid-Scan Detection of Reactive Intermediates in Epoxidation Reactions Catalyzed by a New Enzyme Mimic of Cytochrome P450. J. Am. Chem. Soc. 2007, 129, 12473–12479. 10.1021/ja073266f. [DOI] [PubMed] [Google Scholar]
- Seo M. S.; Kamachi T.; Kouno T.; Murata K.; Park M. J.; Yoshizawa K.; Nam W. Experimental and Theoretical Evidence for Nonheme Iron(III) Alkylperoxo Species as Sluggish Oxidants in Oxygenation Reactions. Angew. Chem., Int. Ed. 2007, 46, 2291–2294. 10.1002/anie.200604219. [DOI] [PubMed] [Google Scholar]
- Soper J. D.; Kryatov S. V.; Rybak-Akimova E. V.; Nocera D. G. Proton-Directed Redox Control of O–O Bond Activation by Heme Hydroperoxidase Models. J. Am. Chem. Soc. 2007, 129, 5069–5075. 10.1021/ja0683032. [DOI] [PubMed] [Google Scholar]
- Franke A.; Fertinger C.; van Eldik R. Which Oxidant Is Really Responsible for P450 Model Oxygenation Reactions? A Kinetic Approach. Angew. Chem., Int. Ed. 2008, 47, 5238–5242. 10.1002/anie.200800907. [DOI] [PubMed] [Google Scholar]
- Han A.-R.; Jin Jeong Y.; Kang Y.; Yoon Lee J.; Sook Seo M.; Nam W. Direct Evidence for an Iron(IV)-Oxo Porphyrin π-Cation Radical as an Active Oxidant in Catalytic Oxygenation Reactions. Chem. Commun. 2008, 1076–1078. 10.1039/b716558g. [DOI] [PubMed] [Google Scholar]
- Fertinger C.; Hessenauer-Ilicheva N.; Franke A.; van Eldik R. Direct Comparison of the Reactivity of Model Complexes for Compounds 0, I, and II in Oxygenation, Hydrogen-Abstraction, and Hydride-Transfer Processes. Chem. - Eur. J. 2009, 15, 13435–13440. 10.1002/chem.200901804. [DOI] [PubMed] [Google Scholar]
- Nam W.; Choi H. J.; Han H. J.; Cho S. H.; Lee H. J.; Han S.-Y. Use of 2-Methyl-1-Phenylpropan-2-yl Hydroperoxide (Mpph) as a Mechanistic Probe for the Heterolytic Versus Homolytic O-O Bond Cleavage of Tert-Alkyl Hydroperoxide by Iron(III) Porphyrin Complex. Chem. Commun. 1999, 387–388. 10.1039/a809876j. [DOI] [Google Scholar]
- Almarsson O.; Bruice T. C. A Homolytic Mechanism of O-O Bond Scission Prevails in the Reactions of Alkyl Hydroperoxides with an Octacationic Tetraphenylporphinato-Iron(III) Complex in Aqueous Solution. J. Am. Chem. Soc. 1995, 117, 4533–4544. 10.1021/ja00121a012. [DOI] [Google Scholar]
- Nam W.; Goh Y. M.; Lee Y. J.; Lim M. H.; Kim C. Biomimetic Alkane Hydroxylations by an Iron(III) Porphyrin Complex with H2O2 and by a High-Valent Iron(IV) Oxo Porphyrin Cation Radical Complex. Inorg. Chem. 1999, 38, 3238–3240. 10.1021/ic980670u. [DOI] [Google Scholar]
- Nam W.; Lim M. H.; Oh S. Y.; Lee J. H.; Lee H. J.; Woo S. K.; Kim C.; Shin W. Remarkable Anionic Axial Ligand Effects of Iron(III) Porphyrin Complexes on the Catalytic Oxygenations of Hydrocarbons by H2O2 and the Formation of Oxoiron(IV) Porphyrin Intermediates by M-Chloroperoxybenzoic Acid. Angew. Chem., Int. Ed. 2000, 39, 3646–3649. . [DOI] [PubMed] [Google Scholar]
- Yang S. J.; Nam W. Water-Soluble Iron Porphyrin Complex-Catalyzed Epoxidation of Olefins with Hydrogen Peroxide and tert-Butyl Hydroperoxide in Aqueous Solution. Inorg. Chem. 1998, 37, 606–607. 10.1021/ic9710519. [DOI] [Google Scholar]
- Traylor T. G.; Kim C.; Richards J. L.; Xu F.; Perrin C. L. Reactions of Iron(III) Porphyrins with Oxidants. Structure-Reactivity Studies. J. Am. Chem. Soc. 1995, 117, 3468–3474. 10.1021/ja00117a015. [DOI] [Google Scholar]
- Traylor T. G.; Xu F. Mechanisms of Reactions of Iron(III) Porphyrins with Hydrogen Peroxide and Hydroperoxides: Solvent and Solvent Isotope Effects. J. Am. Chem. Soc. 1990, 112, 178–186. 10.1021/ja00157a029. [DOI] [Google Scholar]
- Brausam A.; Eigler S.; Jux N.; van Eldik R. Mechanistic Investigations of the Reaction of an Iron(III) Octa-Anionic Porphyrin Complex with Hydrogen Peroxide and the Catalyzed Oxidation of Diammonium-2,2′-azinobis(3-ethylbenzothiazoline-6-sulfonate). Inorg. Chem. 2009, 48, 7667–7678. 10.1021/ic9005955. [DOI] [PubMed] [Google Scholar]
- Franke A.; Fertinger C.; van Eldik R. Axial Ligand and Spin-State Influence on the Formation and Reactivity of Hydroperoxo–Iron(III) Porphyrin Complexes. Chem. - Eur. J. 2012, 18, 6935–6949. 10.1002/chem.201103036. [DOI] [PubMed] [Google Scholar]
- Groves J. T.; Watanabe Y.; McMurry T. J. Oxygen Activation by Metalloporphyrins. Formation and Decomposition of an Acylperoxymanganese(III) Complex. J. Am. Chem. Soc. 1983, 105, 4489–4490. 10.1021/ja00351a068. [DOI] [Google Scholar]
- Schappacher M.; Weiss R. Formation of Manganese(IV)-Oxo-Porphyrin Derivatives by Decomposition of Peroxycarbonate Complexes. Inorg. Chem. 1987, 26, 1189–1190. 10.1021/ic00255a001. [DOI] [Google Scholar]
- Yuan L. C.; Bruice T. C. Influence of Nitrogen Base Ligation and Hydrogen Bonding on the Rate Constants for Oxygen Transfer from Percarboxylic Acids and Alkyl Hydroperoxides to (meso-Tetraphenylporphinato)Manganese(III) Chloride. J. Am. Chem. Soc. 1986, 108, 1643–1650. 10.1021/ja00267a039. [DOI] [Google Scholar]
- Jin N.; Lahaye D. E.; Groves J. T. A ″Push-Pull″ Mechanism for Heterolytic O-O Bond Cleavage in Hydroperoxo Manganese Porphyrins. Inorg. Chem. 2010, 49, 11516–11524. 10.1021/ic1015274. [DOI] [PubMed] [Google Scholar]
- Schulz C. E.; Devaney P. W.; Winkler H.; Debrunner P. G.; Doan N.; Chiang R.; Rutter R.; Hager L. P. Horseradish Peroxidase Compound I: Evidence for Spin Coupling Between the Heme Iron and a ‘Free’ Radical. FEBS Lett. 1979, 103, 102–105. 10.1016/0014-5793(79)81259-4. [DOI] [PubMed] [Google Scholar]
- Schulz C. E.; Rutter R.; Sage J. T.; Debrunner P. G.; Hager L. P. Mössbauer and Electron Paramagnetic Resonance Studies of Horseradish Peroxidase and Its Catalytic Intermediates. Biochemistry 1984, 23, 4743–4754. 10.1021/bi00315a033. [DOI] [PubMed] [Google Scholar]
- Khindaria A.; Aust S. D. EPR Detection and Characterization of Lignin Peroxidase Porphyrin π-Cation Radical. Biochemistry 1996, 35, 13107–13111. 10.1021/bi961297+. [DOI] [PubMed] [Google Scholar]
- Patterson W. R.; Poulos T. L.; Goodin D. B. Identification of a Porphyrin.pi. Cation Radical in Ascorbate Peroxidase Compound I. Biochemistry 1995, 34, 4342–4345. 10.1021/bi00013a024. [DOI] [PubMed] [Google Scholar]
- Benecky M. J.; Frew J. E.; Scowen N.; Jones P.; Hoffman B. M. Epr and Endor Detection of Compound I from Micrococcus lysodeikticus Catalase. Biochemistry 1993, 32, 11929–11933. 10.1021/bi00095a024. [DOI] [PubMed] [Google Scholar]
- Green M. T. The Structure and Spin Coupling of Catalase Compound I: A Study of Noncovalent Effects. J. Am. Chem. Soc. 2001, 123, 9218–9219. 10.1021/ja010105h. [DOI] [PubMed] [Google Scholar]
- Green M. T. Imidazole-Ligated Compound I Intermediates: The Effects of Hydrogen Bonding. J. Am. Chem. Soc. 2000, 122, 9495–9499. 10.1021/ja994377k. [DOI] [Google Scholar]
- Rutter R.; Hager L. P.; Dhonau H.; Hendrich M.; Valentine M.; Debrunner P. Chloroperoxidase Compound I: Electron Paramagnetic Resonance and Mössbauer Studies. Biochemistry 1984, 23, 6809–6816. 10.1021/bi00321a082. [DOI] [PubMed] [Google Scholar]
- Yosca T. H.; Green M. T. Preparation of Compound I in P450cam: The Prototypical P450. Isr. J. Chem. 2016, 56, 834–840. 10.1002/ijch.201600013. [DOI] [Google Scholar]
- Krest C. M.; Silakov A.; Rittle J.; Yosca T. H.; Onderko E. L.; Calixto J. C.; Green M. T. Significantly Shorter Fe-S Bond in Cytochrome p450-I Is Consistent with Greater Reactivity Relative to Chloroperoxidase. Nat. Chem. 2015, 7, 696–702. 10.1038/nchem.2306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rittle J.; Green M. T. Cytochrome P450 Compound I: Capture, Characterization, and C-H Bond Activation Kinetics. Science 2010, 330, 933–937. 10.1126/science.1193478. [DOI] [PubMed] [Google Scholar]
- Coulson A. F.; Yonetani T. Oxidation of Cytochrome c Peroxidase with Hydrogen Peroxide: Identification of the ″Endogenous Donor″. Biochem. Biophys. Res. Commun. 1972, 49, 391–398. 10.1016/0006-291X(72)90423-8. [DOI] [PubMed] [Google Scholar]
- Sivaraja M.; Goodin D.; Smith M.; Hoffman B. Identification by Endor of trp191 as the Free-Radical Site in Cytochrome c Peroxidase Compound Es. Science 1989, 245, 738–740. 10.1126/science.2549632. [DOI] [PubMed] [Google Scholar]
- Dolphin D.; Forman A.; Borg D. C.; Fajer J.; Felton R. H. Compounds I of Catalase and Horse Radish Peroxidase: π-Cation Radicals. Proc. Natl. Acad. Sci. U. S. A. 1971, 68, 614–618. 10.1073/pnas.68.3.614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palcic M. M.; Rutter R.; Araiso T.; Hager L. P.; Dunford H. B. Spectrum of Chloroperoxidase Compound I. Biochem. Biophys. Res. Commun. 1980, 94, 1123–1127. 10.1016/0006-291X(80)90535-5. [DOI] [PubMed] [Google Scholar]
- Chance B.; Powers L.; Ching Y.; Poulos T.; Schonbaum G. R.; Yamazaki I.; Paul K. G. X-Ray Absorption Studies of Intermediates in Peroxidase Activity. Arch. Biochem. Biophys. 1984, 235, 596–611. 10.1016/0003-9861(84)90234-0. [DOI] [PubMed] [Google Scholar]
- Groves J. T.; Vanderpuy M. Stereospecific Aliphatic Hydroxylation by Iron-Hydrogen Peroxide - Evidence for a Stepwise Process. J. Am. Chem. Soc. 1976, 98, 5290–5297. 10.1021/ja00433a039. [DOI] [Google Scholar]
- Groves J. T.; McClusky G. A.; White R. E.; Coon M. J. Aliphatic Hydroxylation by Highly Purified Liver Microsomal Cytochrome P450. Evidence for a Carbon Radical Intermediate. Biochem. Biophys. Res. Commun. 1978, 81, 154–160. 10.1016/0006-291X(78)91643-1. [DOI] [PubMed] [Google Scholar]
- Groves J. T.; McClusky G. A. Aliphatic Hydroxylation via Oxygen Rebound. Oxygen Transfer Catalyzed by Iron. J. Am. Chem. Soc. 1976, 98, 859–861. 10.1021/ja00419a049. [DOI] [Google Scholar]
- Egawa T.; Shimada H.; Ishimura Y. Evidence for Compound I Formation in the Reaction of Cytochrome-P450cam with m-Chloroperbenzoic Acid. Biochem. Biophys. Res. Commun. 1994, 201, 1464–1469. 10.1006/bbrc.1994.1868. [DOI] [PubMed] [Google Scholar]
- Spolitak T.; Dawson J. H.; Ballou D. P. Reaction of Ferric Cytochrome p450cam with Peracids: Kinetic Characterization of Intermediates on the Reaction Pathway. J. Biol. Chem. 2005, 280, 20300–20309. 10.1074/jbc.M501761200. [DOI] [PubMed] [Google Scholar]
- Kellner D. G.; Hung S.-C.; Weiss K. E.; Sligar S. G. Kinetic Characterization of Compound I Formation in the Thermostable Cytochrome P450 CYP119. J. Biol. Chem. 2002, 277, 9641–9644. 10.1074/jbc.C100745200. [DOI] [PubMed] [Google Scholar]
- van Rantwijk F.; Sheldon R. A. Selective Oxygen Transfer Catalysed by Heme Peroxidases: Synthetic and Mechanistic Aspects. Curr. Opin. Biotechnol. 2000, 11, 554–564. 10.1016/S0958-1669(00)00143-9. [DOI] [PubMed] [Google Scholar]
- Chandrasena R. E. P.; Vatsis K. P.; Coon M. J.; Hollenberg P. F.; Newcomb M. Hydroxylation by the Hydroperoxy-Iron Species in Cytochrome P450 Enzymes. J. Am. Chem. Soc. 2004, 126, 115–126. 10.1021/ja038237t. [DOI] [PubMed] [Google Scholar]
- Newcomb M.; Aebisher D.; Shen R. N.; Chandrasena R. E. P.; Hollenberg P. F.; Coon M. J. Kinetic Isotope Effects Implicate Two Electrophilic Oxidants in Cytochrome P450-Catalyzed Hydroxylations. J. Am. Chem. Soc. 2003, 125, 6064–6065. 10.1021/ja0343858. [DOI] [PubMed] [Google Scholar]
- Wang Q.; Sheng X.; Horner J. H.; Newcomb M. Quantitative Production of Compound I from a Cytochrome P450 Enzyme at Low Temperatures. Kinetics, Activation Parameters, and Kinetic Isotope Effects for Oxidation of Benzyl Alcohol. J. Am. Chem. Soc. 2009, 131, 10629–10636. 10.1021/ja9031105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofrichter M.; Ullrich R. Oxidations Catalyzed by Fungal Peroxygenases. Curr. Opin. Chem. Biol. 2014, 19, 116–125. 10.1016/j.cbpa.2014.01.015. [DOI] [PubMed] [Google Scholar]
- Peter S.; Kinne M.; Wang X.; Ullrich R.; Kayser G.; Groves J. T.; Hofrichter M. Selective Hydroxylation of Alkanes by an Extracellular Fungal Peroxygenase. FEBS J. 2011, 278, 3667–3675. 10.1111/j.1742-4658.2011.08285.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X. S.; Peter S.; Kinne M.; Hofrichter M.; Groves J. T. Detection and Kinetic Characterization of a Highly Reactive Heme-Thiolate Peroxygenase Compound I. J. Am. Chem. Soc. 2012, 134, 12897–12900. 10.1021/ja3049223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onderko E. L.; Silakov A.; Yosca T. H.; Green M. T. Characterization of a Selenocysteine-Ligated P450 Compound I Reveals Direct Link between Electron Donation and Reactivity. Nat. Chem. 2017, 9, 623–628. 10.1038/nchem.2781. [DOI] [PubMed] [Google Scholar]
- Groves J. T.; Haushalter R. C.; Nakamura M.; Nemo T. E.; Evans B. J. High-Valent Iron-Porphyrin Complexes Related to Peroxidase and Cytochrome P450. J. Am. Chem. Soc. 1981, 103, 2884–2886. 10.1021/ja00400a075. [DOI] [Google Scholar]
- Pennerhahn J. E.; Mcmurry T. J.; Renner M.; Latosgrazynsky L.; Eble K. S.; Davis I. M.; Balch A. L.; Groves J. T.; Dawson J. H.; Hodgson K. O. X-Ray Absorption Spectroscopic Studies of High Valent Iron Porphyrins - Horseradish-Peroxidase Compound-I and Compound-II and Synthetic Models. J. Biol. Chem. 1983, 258, 2761–2764. [PubMed] [Google Scholar]
- Boso B.; Lang G.; Mcmurry T. J.; Groves J. T. Mossbauer-Effect Study of Tight Spin Coupling in Oxidized Chloro-5,10,15,20-Tetra(Mesityl)Porphyrinatoiron(III). J. Chem. Phys. 1983, 79, 1122–1126. 10.1063/1.445913. [DOI] [Google Scholar]
- Groves J. T.; Nemo T. E. Aliphatic Hydroxylation Catalyzed by Iron Porphyrin Complexes. J. Am. Chem. Soc. 1983, 105, 6243–6248. 10.1021/ja00358a009. [DOI] [Google Scholar]
- Hohenberger J.; Ray K.; Meyer K. The Biology and Chemistry of High-Valent Iron–Oxo and Iron–Nitrido Complexes. Nat. Commun. 2012, 3, 720. 10.1038/ncomms1718. [DOI] [PubMed] [Google Scholar]
- Jung C. The Mystery of Cytochrome P450 Compound I: a Mini-Review Dedicated to Klaus Ruckpaul. Biochim. Biophys. Acta, Proteins Proteomics 2011, 1814, 46–57. 10.1016/j.bbapap.2010.06.007. [DOI] [PubMed] [Google Scholar]
- Dolphin D.; Traylor T. G.; Xie L. Y. Polyhaloporphyrins: Unusual Ligands for Metals and Metal-Catalyzed Oxidations. Acc. Chem. Res. 1997, 30, 251–259. 10.1021/ar960126u. [DOI] [Google Scholar]
- Fujii H. Effects of the Electron-Withdrawing Power of Substituents on the Electronic Structure and Reactivity in Oxoiron(IV) Porphyrin π-Cation Radical Complexes. J. Am. Chem. Soc. 1993, 115, 4641–4648. 10.1021/ja00064a027. [DOI] [Google Scholar]
- Fujii H.; Yoshimura T.; Kamada H. ESR Studies of A1u and A2u Oxoiron(IV) Porphyrin π-Cation Radical Complexes. Spin Coupling between Ferryl Iron and A1u/A2u Orbitals. Inorg. Chem. 1996, 35, 2373–2377. 10.1021/ic9513752. [DOI] [PubMed] [Google Scholar]
- Pan Z. Z.; Zhang R.; Newcomb M. Kinetic Studies of Reactions of Iron(IV)-Oxo Porphyrin Radical Cations with Organic Reductants. J. Inorg. Biochem. 2006, 100, 524–532. 10.1016/j.jinorgbio.2005.12.022. [DOI] [PubMed] [Google Scholar]
- Bell S. R.; Groves J. T. A Highly Reactive P450 Model Compound I. J. Am. Chem. Soc. 2009, 131, 9640–9641. 10.1021/ja903394s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng X.; Horner J. H.; Newcomb M. Spectra and Kinetic Studies of the Compound I Derivative of Cytochrome P450 119. J. Am. Chem. Soc. 2008, 130, 13310–13320. 10.1021/ja802652b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Afanasiev P.; Sorokin A. B. μ-Nitrido Diiron Macrocyclic Platform: Particular Structure for Particular Catalysis. Acc. Chem. Res. 2016, 49, 583–593. 10.1021/acs.accounts.5b00458. [DOI] [PubMed] [Google Scholar]
- Afanasiev P.; Kudrik E. V.; Millet J.-M. M.; Bouchu D.; Sorokin A. B. High-Valent Diiron Species Generated from N-Bridged Diiron Phthalocyanine and H2O2. Dalton Trans. 2011, 40, 701–710. 10.1039/C0DT00958J. [DOI] [PubMed] [Google Scholar]
- Kudrik E. V.; Afanasiev P.; Alvarez L. X.; Dubourdeaux P.; Clémancey M.; Latour J.-M.; Blondin G.; Bouchu D.; Albrieux F.; Nefedov S. E.; et al. An N-Bridged High-Valent Diiron–Oxo Species on a Porphyrin Platform That Can Oxidize Methane. Nat. Chem. 2012, 4, 1024–1029. 10.1038/nchem.1471. [DOI] [PubMed] [Google Scholar]
- Sorokin A. B.; Kudrik E. V.; Bouchu D. Bio-Inspired Oxidation of Methane in Water Catalyzed by N-Bridged Diiron Phthalocyanine Complex. Chem. Commun. 2008, 2562–2564. 10.1039/b804405h. [DOI] [PubMed] [Google Scholar]
- Isci U.; Faponle A. S.; Afanasiev P.; Albrieux F.; Briois V.; Ahsen V.; Dumoulin F.; Sorokin A. B.; de Visser S. P. Site-Selective Formation of an Iron(IV)-Oxo Species at the More Electron-Rich Iron Atom of Heteroleptic μ-Nitrido Diiron Phthalocyanines. Chem. Sci. 2015, 6, 5063–5075. 10.1039/C5SC01811K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quesne M. G.; Senthilnathan D.; Singh D.; Kumar D.; Maldivi P.; Sorokin A. B.; de Visser S. P. Origin of the Enhanced Reactivity of μ-Nitrido-Bridged Diiron(IV)-Oxo Porphyrinoid Complexes over Cytochrome P450 Compound I. ACS Catal. 2016, 6, 2230–2243. 10.1021/acscatal.5b02720. [DOI] [Google Scholar]
- Groves J. T.; Kruper W. J.; Haushalter R. C. Hydrocarbon Oxidations with Oxometalloporphinates. Isolation and Reactions of a (Porphinato)manganese(V) Complex. J. Am. Chem. Soc. 1980, 102, 6375–6377. 10.1021/ja00540a050. [DOI] [Google Scholar]
- Czernuszewicz R. S.; Su Y. O.; Stern M. K.; Macor K. A.; Kim D.; Groves J. T.; Spiro T. G. Oxomanganese(IV) Porphyrins Identified by Resonance Raman and Infrared-Spectroscopy - Weak Bonds and the Stability of the Half-Filled t2g Subshell. J. Am. Chem. Soc. 1988, 110, 4158–4165. 10.1021/ja00221a010. [DOI] [Google Scholar]
- Groves J. T.; Stern M. K. Synthesis, Characterization, and Reactivity of Oxomanganese(IV) Porphyrin Complexes. J. Am. Chem. Soc. 1988, 110, 8628–8638. 10.1021/ja00234a009. [DOI] [Google Scholar]
- Bernadou J.; Fabiano A.-S.; Robert A.; Meunier B. ″Redox Tautomerism″ in High-Valent Metal-Oxo-Aquo Complexes. Origin of the Oxygen Atom in Epoxidation Reactions Catalyzed by Water-Soluble Metalloporphyrins. J. Am. Chem. Soc. 1994, 116, 9375–9376. 10.1021/ja00099a083. [DOI] [Google Scholar]
- Groves J. T.; Marla S. S. Peroxynitrite-Induced DNA Strand Scission Mediated by a Manganese Porphyrin. J. Am. Chem. Soc. 1995, 117, 9578–9579. 10.1021/ja00142a032. [DOI] [Google Scholar]
- Pitie M.; Bernadou J.; Meunier B. Oxidation at Carbon-1′ of DNA Deoxyriboses by the Mn-TMPyP/KHSO5 System Results from a Cytochrome P-450-type Hydroxylation Reaction. J. Am. Chem. Soc. 1995, 117, 2935–2936. 10.1021/ja00115a032. [DOI] [Google Scholar]
- Groves J. T.; Lee J. B.; Marla S. S. Detection and Characterization of an Oxomanganese(V) Porphyrin Complex by Rapid-Mixing Stopped-Flow Spectrophotometry. J. Am. Chem. Soc. 1997, 119, 6269–6273. 10.1021/ja962605u. [DOI] [Google Scholar]
- Jin N.; Groves J. T. Unusual Kinetic Stability of a Ground-State Singlet Oxomanganese(V) Porphyrin. Evidence for a Spin State Crossing Effect. J. Am. Chem. Soc. 1999, 121, 2923–2924. 10.1021/ja984429q. [DOI] [Google Scholar]
- Nick R. J.; Ray G. B.; Fish K. M.; Spiro T. G.; Groves J. T. Evidence for a Weak Mn:O Bond and a Non-Porphyrin Radical in Manganese-Substituted Horseradish Peroxidase Compound I. J. Am. Chem. Soc. 1991, 113, 1838–1840. 10.1021/ja00005a062. [DOI] [Google Scholar]
- Nam W.; Kim I.; Lim M. H.; Choi H. J.; Lee J. S.; Jang H. G. Isolation of an Oxomanganese(V) Porphyrin Intermediate in the Reaction of a Manganese(III) Porphyrin Complex and H2O2 in Aqueous Solution. Chem. - Eur. J. 2002, 8, 2067–2071. . [DOI] [PubMed] [Google Scholar]
- Zhang R.; Horner J. H.; Newcomb M. Laser Flash Photolysis Generation and Kinetic Studies of Porphyrin–Manganese–Oxo Intermediates. Rate Constants for Oxidations Effected by Porphyrin–MnV–Oxo Species and Apparent Disproportionation Equilibrium Constants for Porphyrin–MnIV–Oxo Species. J. Am. Chem. Soc. 2005, 127, 6573–6582. 10.1021/ja045042s. [DOI] [PubMed] [Google Scholar]
- Shimazaki Y.; Nagano T.; Takesue H.; Ye B.-H.; Tani F.; Naruta Y. Characterization of a Dinuclear MnV=O Complex and Its Efficient Evolution of O2 in the Presence of Water. Angew. Chem., Int. Ed. 2004, 43, 98–100. 10.1002/anie.200352564. [DOI] [PubMed] [Google Scholar]
- Jin N.; Bourassa J. L.; Tizio S. C.; Groves J. T. Rapid, Reversible Oxygen Atom Transfer Between an Oxomanganese(V) Porphyrin and Bromide: a Haloperoxidase Mimic with Enzymatic Rates. Angew. Chem., Int. Ed. 2000, 39, 3849–3851. . [DOI] [PubMed] [Google Scholar]
- Jin N.; Ibrahim M.; Spiro T. G.; Groves J. T. Trans-Dioxo Manganese(V) Porphyrins. J. Am. Chem. Soc. 2007, 129, 12416–12417. 10.1021/ja0761737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gross Z. The Groves–Spiro Dioxomanganese(V) Story. Angew. Chem., Int. Ed. 2008, 47, 2737–2739. 10.1002/anie.200705750. [DOI] [PubMed] [Google Scholar]
- Song W. J.; Seo M. S.; DeBeer George S.; Ohta T.; Song R.; Kang M. J.; Tosha T.; Kitagawa T.; Solomon E. I.; Nam W. Synthesis, Characterization, and Reactivities of Manganese(V)-Oxo Porphyrin Complexes. J. Am. Chem. Soc. 2007, 129, 1268–1277. 10.1021/ja066460v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green M. T. Application of Badger’s Rule to Heme and Non-Heme Iron–Oxygen Bonds: An Examination of Ferryl Protonation States. J. Am. Chem. Soc. 2006, 128, 1902–1906. 10.1021/ja054074s. [DOI] [PubMed] [Google Scholar]
- Badger R. M. The Relation Between the Internuclear Distances and Force Constants of Molecules and Its Application to Polyatomic Molecules. J. Chem. Phys. 1935, 3, 710–714. 10.1063/1.1749581. [DOI] [Google Scholar]
- Ho C.; Leung W.-H.; Che C.-M. Kinetics of C-H Bond and Alkene Oxidation by Trans-Dioxoruthenium(VI) Porphyrins. J. Chem. Soc., Dalton Trans. 1991, 2933–2939. 10.1039/DT9910002933. [DOI] [Google Scholar]
- Leung W. H.; Che C. M. High-Valent Ruthenium(IV) and -(VI) Oxo Complexes of Octaethylporphyrin. Synthesis, Spectroscopy, and Reactivities. J. Am. Chem. Soc. 1989, 111, 8812–8818. 10.1021/ja00206a007. [DOI] [Google Scholar]
- Groves J. T.; Quinn R. Models of Oxidized Heme Proteins. Preparation and Characterization of a Trans-Dioxoruthenium(VI) Porphyrin Complex. Inorg. Chem. 1984, 23, 3844–3846. 10.1021/ic00192a004. [DOI] [Google Scholar]
- Che C. M.; Poon C. K.; Chung W. C.; Gray H. B. Synthesis and Characterization of Osmium Porphyrins. Inorg. Chem. 1985, 24, 1277–1278. 10.1021/ic00202a037. [DOI] [Google Scholar]
- Che C. M.; Chung W. C.; Lai T. F. Synthesis, Reactivity, and X-Ray Structural Characterization of Trans-Dioxoosmium(VI) Porphyrin Complexes. Inorg. Chem. 1988, 27, 2801–2804. 10.1021/ic00289a012. [DOI] [Google Scholar]
- De Angelis F.; Jin N.; Car R.; Groves J. T. Electronic Structure and Reactivity of Isomeric Oxo-Mn(V) Porphyrins: Effects of Spin-State Crossing and pKa Modulation. Inorg. Chem. 2006, 45, 4268–4276. 10.1021/ic060306s. [DOI] [PubMed] [Google Scholar]
- Balcells D.; Raynaud C.; Crabtree R. H.; Eisenstein O. A Rational Basis for the Axial Ligand Effect in C-H Oxidation by MnO(porphyrin)(X)(+) (X = H2O, OH–, O2–) from a DFT Study. Inorg. Chem. 2008, 47, 10090–10099. 10.1021/ic8013706. [DOI] [PubMed] [Google Scholar]
- Behan R. K.; Green M. T. On the Status of Ferryl Protonation. J. Inorg. Biochem. 2006, 100, 448–459. 10.1016/j.jinorgbio.2005.12.019. [DOI] [PubMed] [Google Scholar]
- Dunford H. B.; Hasinoff B. B. Kinetics of the Oxidation of Ferrocyanide by Horseradish Peroxidase Compounds I and II. Biochemistry 1970, 9, 4930–4939. 10.1021/bi00827a015. [DOI] [PubMed] [Google Scholar]
- Sitter A. J.; Reczek C. M.; Terner J. Observation of the Fe(IV)=O Stretching Vibration of Ferryl Myoglobin by Resonance Raman Spectroscopy. Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol. 1985, 828, 229–235. 10.1016/0167-4838(85)90301-2. [DOI] [PubMed] [Google Scholar]
- Chance M.; Powers L.; Poulos T.; Chance B. Cytochrome c Peroxidase Compound ES Is Identical with Horseradish Peroxidase Compound I in Iron-Ligand Distances. Biochemistry 1986, 25, 1266–1270. 10.1021/bi00354a011. [DOI] [PubMed] [Google Scholar]
- Chin D.-H.; La Mar G. N.; Balch A. L. Mechanism of Autoxidation of Iron(II) Porphyrins. Detection of a Peroxo-Bridged Iron(III) Porphyrin Dimer and the Mechanism of Its Thermal Decomposition to the Oxo-Bridged Iron(III) Porphyrin Dimer. J. Am. Chem. Soc. 1980, 102, 4344–4350. 10.1021/ja00533a009. [DOI] [Google Scholar]
- Green M. T.; Dawson J. H.; Gray H. B. Oxoiron(IV) in Chloroperoxidase Compound II Is Basic: Implications for P450 Chemistry. Science 2004, 304, 1653–1656. 10.1126/science.1096897. [DOI] [PubMed] [Google Scholar]
- Penner-Hahn J. E.; Smith Eble K.; McMurry T. J.; Renner M.; Balch A. L.; Groves J. T.; Dawson J. H.; Hodgson K. O. Structural Characterization of Horseradish Peroxidase Using Exafs Spectroscopy. Evidence for Fe=O Ligation in Compounds I and II. J. Am. Chem. Soc. 1986, 108, 7819–7825. 10.1021/ja00284a054. [DOI] [PubMed] [Google Scholar]
- Chance M.; Powers L.; Kumar C.; Chance B. X-Ray Absorption Studies of Myoglobin Peroxide Reveal Functional Differences Between Globins and Heme Enzymes. Biochemistry 1986, 25, 1259–1265. 10.1021/bi00354a010. [DOI] [PubMed] [Google Scholar]
- Bonagura C. A.; Bhaskar B.; Shimizu H.; Li H.; Sundaramoorthy M.; McRee D. E.; Goodin D. B.; Poulos T. L. High-Resolution Crystal Structures and Spectroscopy of Native and Compound I Cytochrome c Peroxidase. Biochemistry 2003, 42, 5600–5608. 10.1021/bi034058c. [DOI] [PubMed] [Google Scholar]
- Hersleth H.-P.; Dalhus B.; Görbitz C.; Andersson K. An Iron Hydroxide Moiety in the 1.35 Å Resolution Structure of Hydrogen Peroxide Derived Myoglobin Compound II at pH 5.2. JBIC, J. Biol. Inorg. Chem. 2002, 7, 299–304. 10.1007/s007750100296. [DOI] [PubMed] [Google Scholar]
- Meharenna Y. T.; Doukov T.; Li H.; Soltis S. M.; Poulos T. L. Crystallographic and Single-Crystal Spectral Analysis of the Peroxidase Ferryl Intermediate. Biochemistry 2010, 49, 2984–2986. 10.1021/bi100238r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yosca T. H.; Behan R. K.; Krest C. M.; Onderko E. L.; Langston M. C.; Green M. T. Setting an Upper Limit on the Myoglobin Iron(IV)Hydroxide pKa: Insight into Axial Ligand Tuning in Heme Protein Catalysis. J. Am. Chem. Soc. 2014, 136, 9124–9131. 10.1021/ja503588n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Behan R. K.; Hoffart L. M.; Stone K. L.; Krebs C.; Green M. T. Evidence for Basic Ferryls in Cytochromes P450. J. Am. Chem. Soc. 2006, 128, 11471–11474. 10.1021/ja062428p. [DOI] [PubMed] [Google Scholar]
- Stone K. L.; Behan R. K.; Green M. T. Resonance Raman Spectroscopy of Chloroperoxidase Compound Ii Provides Direct Evidence for the Existence of an Iron(IV)-Hydroxide. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 12307–12310. 10.1073/pnas.0603159103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone K. L.; Hoffart L. M.; Behan R. K.; Krebs C.; Green M. T. Evidence for Two Ferryl Species in Chloroperoxidase Compound II. J. Am. Chem. Soc. 2006, 128, 6147–6153. 10.1021/ja057876w. [DOI] [PubMed] [Google Scholar]
- Yosca T. H.; Rittle J.; Krest C. M.; Onderko E. L.; Silakov A.; Calixto J. C.; Behan R. K.; Green M. T. Iron(IV)hydroxide pKa and the Role of Thiolate Ligation in C-H Bond Activation by Cytochrome P450. Science 2013, 342, 825–829. 10.1126/science.1244373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X. S.; Ullrich R.; Hofrichter M.; Groves J. T. Heme-Thiolate Ferryl of Aromatic Peroxygenase Is Basic and Reactive. Proc. Natl. Acad. Sci. U. S. A. 2015, 112, 3686–3691. 10.1073/pnas.1503340112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yosca T. H.; Langston M. C.; Krest C. M.; Onderko E. L.; Grove T. L.; Livada J.; Green M. T. Spectroscopic Investigations of Catalase Compound II: Characterization of an Iron(IV) Hydroxide Intermediate in a Non-thiolate-Ligated Heme Enzyme. J. Am. Chem. Soc. 2016, 138, 16016–16023. 10.1021/jacs.6b09693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon H.; Basran J.; Casadei C. M.; Fielding A. J.; Schrader T. E.; Ostermann A.; Devos J. M.; Aller P.; Blakeley M. P.; Moody P. C. E.; et al. Direct Visualization of a Fe(IV)–OH Intermediate in a Heme Enzyme. Nat. Commun. 2016, 7, 13445. 10.1038/ncomms13445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gumiero A.; Metcalfe C. L.; Pearson A. R.; Raven E. L.; Moody P. C. E. Nature of the Ferryl Heme in Compounds I and II. J. Biol. Chem. 2011, 286, 1260–1268. 10.1074/jbc.M110.183483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boaz N. C.; Bell S. R.; Groves J. T. Ferryl Protonation in Oxoiron(IV) Porphyrins and Its Role in Oxygen Transfer. J. Am. Chem. Soc. 2015, 137, 2875–2885. 10.1021/ja508759t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurahashi T.; Kikuchi A.; Tosha T.; Shiro Y.; Kitagawa T.; Fujii H. Transient Intermediates from Mn(salen) with Sterically Hindered Mesityl Groups: Interconversion between MnIV-Phenolate and MnIII-Phenoxyl Radicals as an Origin for Unique Reactivity. Inorg. Chem. 2008, 47, 1674–1686. 10.1021/ic702061y. [DOI] [PubMed] [Google Scholar]
- Kurahashi T.; Kikuchi A.; Shiro Y.; Hada M.; Fujii H. Unique Properties and Reactivity of High-Valent Manganese–Oxo versus Manganese–Hydroxo in the Salen Platform. Inorg. Chem. 2010, 49, 6664–6672. 10.1021/ic100673b. [DOI] [PubMed] [Google Scholar]
- Chen Z.; Yin G. The Reactivity of the Active Metal Oxo and Hydroxo Intermediates and Their Implications in Oxidations. Chem. Soc. Rev. 2015, 44, 1083–1100. 10.1039/C4CS00244J. [DOI] [PubMed] [Google Scholar]
- Lansky D. E.; Goldberg D. P. Hydrogen Atom Abstraction by a High-Valent Manganese(V)–Oxo Corrolazine. Inorg. Chem. 2006, 45, 5119–5125. 10.1021/ic060491+. [DOI] [PubMed] [Google Scholar]
- Parsell T. H.; Yang M.-Y.; Borovik A. S. C–H Bond Cleavage with Reductants: Re-Investigating the Reactivity of Monomeric MnIII/IV–Oxo Complexes and the Role of Oxo Ligand Basicity. J. Am. Chem. Soc. 2009, 131, 2762–2763. 10.1021/ja8100825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin G.; Danby A. M.; Kitko D.; Carter J. D.; Scheper W. M.; Busch D. H. Understanding the Selectivity of a Moderate Oxidation Catalyst: Hydrogen Abstraction by a Fully Characterized, Activated Catalyst, the Robust Dihydroxo Manganese(IV) Complex of a Bridged Cyclam. J. Am. Chem. Soc. 2007, 129, 1512–1513. 10.1021/ja0673229. [DOI] [PubMed] [Google Scholar]
- Bordwell F. G. Equilibrium Acidities in Dimethyl Sulfoxide Solution. Acc. Chem. Res. 1988, 21, 456–463. 10.1021/ar00156a004. [DOI] [Google Scholar]
- Mayer J. M. Understanding Hydrogen Atom Transfer: From Bond Strengths to Marcus Theory. Acc. Chem. Res. 2011, 44, 36–46. 10.1021/ar100093z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warren J. J.; Tronic T. A.; Mayer J. M. Thermochemistry of Proton-Coupled Electron Transfer Reagents and its Implications. Chem. Rev. 2010, 110, 6961–7001. 10.1021/cr100085k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray H. B.; Winkler J. R. Hole Hopping Through Tyrosine/Tryptophan Chains Protects Proteins from Oxidative Damage. Proc. Natl. Acad. Sci. U. S. A. 2015, 112, 10920–10925. 10.1073/pnas.1512704112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkler J. R.; Gray H. B. Electron Flow Through Biological Molecules: Does Hole Hopping Protect Proteins from Oxidative Damage?. Q. Rev. Biophys. 2015, 48, 411–420. 10.1017/S0033583515000062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guengerich F. P.; Munro A. W. Unusual Cytochrome P450 Enzymes and Reactions. J. Biol. Chem. 2013, 288, 17065–17073. 10.1074/jbc.R113.462275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guengerich F. P.; Waterman M. R.; Egli M. Recent Structural Insights into Cytochrome P450 Function. Trends Pharmacol. Sci. 2016, 37, 625–640. 10.1016/j.tips.2016.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramlinger V. M.; Nagy L. D.; Fujiwara R.; Johnson K. M.; Phan T. T. N.; Xiao Y.; Enright J. M.; Toomey M. B.; Corbo J. C.; Guengerich F. P. Human Cytochrome P450 27C1 Catalyzes 3,4-Desaturation of Retinoids. FEBS Lett. 2016, 590, 1304–1312. 10.1002/1873-3468.12167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breslow R.; Balasubramanian K. The pKa of Triphenylcyclopropene. Electrochemical Determination of an Inaccessible Equilibrium Constant. J. Am. Chem. Soc. 1969, 91, 5182–5183. 10.1021/ja01046a055. [DOI] [Google Scholar]
- Groves J. T. Enzymatic C-H Bond Activation: Using Push to Get Pull. Nat. Chem. 2014, 6, 89–91. 10.1038/nchem.1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi Y.; Yamazaki I. The Oxidation-Reduction Potentials of Compound I/Compound II and Compound II/Ferric Couples of Horseradish Peroxidases A2 and C. J. Biol. Chem. 1979, 254, 9101–9106. [PubMed] [Google Scholar]
- Wang X. S.; Peter S.; Ullrich R.; Hofrichter M.; Groves J. T. Driving Force for Oxygen-Atom Transfer by Heme-Thiolate Enzymes. Angew. Chem., Int. Ed. 2013, 52, 9238–9241. 10.1002/anie.201302137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song W. J.; Ryu Y. O.; Song R.; Nam W. Oxoiron(IV) Porphyrin π-Cation Radical Complexes with a Chameleon Behavior in Cytochrome P450 Model Reactions. JBIC, J. Biol. Inorg. Chem. 2005, 10, 294–304. 10.1007/s00775-005-0641-9. [DOI] [PubMed] [Google Scholar]
- Kang Y.; Chen H.; Jeong Y. J.; Lai W.; Bae E. H.; Shaik S.; Nam W. Enhanced Reactivities of Iron(IV)-Oxo Porphyrin π-Cation Radicals in Oxygenation Reactions by Electron-Donating Axial Ligands. Chem. - Eur. J. 2009, 15, 10039–10046. 10.1002/chem.200901238. [DOI] [PubMed] [Google Scholar]
- Castro L.; Bühl M. Calculations of One-Electron Redox Potentials of Oxoiron(IV) Porphyrin Complexes. J. Chem. Theory Comput. 2014, 10, 243–251. 10.1021/ct400975w. [DOI] [PubMed] [Google Scholar]
- Rydberg P.; Sigfridsson E.; Ryde U. On the Role of the Axial Ligand in Heme Proteins: a Theoretical Study. JBIC, J. Biol. Inorg. Chem. 2004, 9, 203–223. 10.1007/s00775-003-0515-y. [DOI] [PubMed] [Google Scholar]
- Dey A.; Jiang Y.; Ortiz de Montellano P.; Hodgson K. O.; Hedman B.; Solomon E. I. S K-edge XAS and DFT Calculations on Cytochrome P450: Covalent and Ionic Contributions to the Cysteine-Fe Bond and Their Contribution to Reactivity. J. Am. Chem. Soc. 2009, 131, 7869–7878. 10.1021/ja901868q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Visser S. P. Trends in Substrate Hydroxylation Reactions by Heme and Nonheme Iron(IV)-Oxo Oxidants Give Correlations between Intrinsic Properties of the Oxidant with Barrier Height. J. Am. Chem. Soc. 2010, 132, 1087–1097. 10.1021/ja908340j. [DOI] [PubMed] [Google Scholar]
- Takahashi A.; Kurahashi T.; Fujii H. Redox Potentials of Oxoiron(IV) Porphyrin π-Cation Radical Complexes: Participation of Electron Transfer Process in Oxygenation Reactions. Inorg. Chem. 2011, 50, 6922–6928. 10.1021/ic102564e. [DOI] [PubMed] [Google Scholar]
- Sugimoto H.; Tung H. C.; Sawyer D. T. The Formation, Characterization, and Reactivity of the Oxene Adduct of [Tetrakis(2,6-Dichlorophenyl)Porphinato]Iron(III) Perchlorate in Acetonitrile. Model for the Reactive Intermediate of Cytochrome P450. J. Am. Chem. Soc. 1988, 110, 2465–2470. 10.1021/ja00216a020. [DOI] [Google Scholar]
- Evans M. G.; Polanyi M. Inertia and Driving Force of Chemical Reactions. Trans. Faraday Soc. 1938, 34, 11–24. 10.1039/tf9383400011. [DOI] [Google Scholar]
- Panov G. I.; Parfenov M. V.; Parmon V. N. The Brønsted–Evans–Polanyi Correlations in Oxidation Catalysis. Catal. Rev.: Sci. Eng. 2015, 57, 436–477. 10.1080/01614940.2015.1074487. [DOI] [Google Scholar]
- Mayr H.; Ofial A. R. The Reactivity–Selectivity Principle: An Imperishable Myth in Organic Chemistry. Angew. Chem., Int. Ed. 2006, 45, 1844–1854. 10.1002/anie.200503273. [DOI] [PubMed] [Google Scholar]
- Roth J. P.; Yoder J. C.; Won T.-J.; Mayer J. M. Application of the Marcus Cross Relation to Hydrogen Atom Transfer Reactions. Science 2001, 294, 2524–2526. 10.1126/science.1066130. [DOI] [PubMed] [Google Scholar]
- Warren J. J.; Mayer J. M. In Proton-Coupled Electron Transfer: A Carrefour of Chemical Reactivity Traditions; The Royal Society of Chemistry, 2012. [Google Scholar]
- Salamone M.; Bietti M. Tuning Reactivity and Selectivity in Hydrogen Atom Transfer from Aliphatic C–H Bonds to Alkoxyl Radicals: Role of Structural and Medium Effects. Acc. Chem. Res. 2015, 48, 2895–2903. 10.1021/acs.accounts.5b00348. [DOI] [PubMed] [Google Scholar]
- Liu W.; Groves J. T. Manganese Catalyzed C-H Halogenation. Acc. Chem. Res. 2015, 48, 1727–1735. 10.1021/acs.accounts.5b00062. [DOI] [PubMed] [Google Scholar]
- Finn M.; Friedline R.; Suleman N. K.; Wohl C. J.; Tanko J. M. Chemistry of the t-Butoxyl Radical: Evidence that Most Hydrogen Abstractions from Carbon are Entropy-Controlled. J. Am. Chem. Soc. 2004, 126, 7578–7584. 10.1021/ja0493493. [DOI] [PubMed] [Google Scholar]
- Takahashi A.; Kurahashi T.; Fujii H. Activation Parameters for Cyclohexene Oxygenation by an Oxoiron(IV) Porphyrin π-Cation Radical Complex: Entropy Control of an Allylic Hydroxylation Reaction. Inorg. Chem. 2007, 46, 6227–6229. 10.1021/ic7009379. [DOI] [PubMed] [Google Scholar]
- Schröder D.; Shaik S.; Schwarz H. Two-State Reactivity as a New Concept in Organometallic Chemistry. Acc. Chem. Res. 2000, 33, 139–145. 10.1021/ar990028j. [DOI] [PubMed] [Google Scholar]
- Shaik S.; Danovich D.; Fiedler A.; Schröder D.; Schwarz H. Two-State Reactivity in Organometallic Gas-Phase Ion Chemistry. Helv. Chim. Acta 1995, 78, 1393–1407. 10.1002/hlca.19950780602. [DOI] [Google Scholar]
- Usharani D.; Wang B.; Sharon D. A.; Shaik S. In Spin States in Biochemistry and Inorganic Chemistry; John Wiley & Sons, Ltd: Chichester, UK, 2015. [Google Scholar]
- Ogliaro F.; Harris N.; Cohen S.; Filatov M.; de Visser S. P.; Shaik S. A Model “Rebound” Mechanism of Hydroxylation by Cytochrome P450: Stepwise and Effectively Concerted Pathways, and Their Reactivity Patterns. J. Am. Chem. Soc. 2000, 122, 8977–8989. 10.1021/ja991878x. [DOI] [Google Scholar]
- Shaik S.; de Visser S. P.; Ogliaro F.; Schwarz H.; Schroder D. Two-State Reactivity Mechanisms of Hydroxylation and Epoxidation by Cytochrome P450 Revealed by Theory. Curr. Opin. Chem. Biol. 2002, 6, 556–567. 10.1016/S1367-5931(02)00363-0. [DOI] [PubMed] [Google Scholar]
- Shaik S.; Kumar D.; de Visser S. P.; Altun A.; Thiel W. Theoretical Perspective on the Structure and Mechanism of Cytochrome p450 Enzymes. Chem. Rev. 2005, 105, 2279–2328. 10.1021/cr030722j. [DOI] [PubMed] [Google Scholar]
- Balcells D.; Raynaud C.; Crabtree R. H.; Eisenstein O. C-H Oxidation by Hydroxo Manganese(V) Porphyrins: a DFT Study. Chem. Commun. 2009, 1772–1774. 10.1039/b821029b. [DOI] [PubMed] [Google Scholar]
- Balcells D.; Raynaud C.; Crabtree R. H.; Eisenstein O. The Rebound Mechanism in Catalytic C-H Oxidation by MnO(TPP)Cl from DFT Studies: Electronic Nature of the Active Species. Chem. Commun. 2008, 744–746. 10.1039/B715939K. [DOI] [PubMed] [Google Scholar]
- Usharani D.; Janardanan D.; Li C. S.; Shaik S. A Theory for Bioinorganic Chemical Reactivity of Oxometal Complexes and Analogous Oxidants: The Exchange and Orbital-Selection Rules. Acc. Chem. Res. 2013, 46, 471–482. 10.1021/ar300204y. [DOI] [PubMed] [Google Scholar]
- Shaik S.; Chen H.; Janardanan D. Exchange-Enhanced Reactivity in Bond Activation by Metal-Oxo Enzymes and Synthetic Reagents. Nat. Chem. 2011, 3, 19–27. 10.1038/nchem.943. [DOI] [PubMed] [Google Scholar]
- Janardanan D.; Usharani D.; Shaik S. The Origins of Dramatic Axial Ligand Effects: Closed-Shell MnVO Complexes Use Exchange-Enhanced Open-Shell States to Mediate Efficient H Abstraction Reactions. Angew. Chem., Int. Ed. 2012, 51, 4421–4425. 10.1002/anie.201200689. [DOI] [PubMed] [Google Scholar]
- Li C. S.; Wu W.; Kumar D.; Shaik S. Kinetic Isotope Effect Is a Sensitive Probe of Spin State Reactivity in C-H Hydroxylation of N,N-Dimethylaniline by Cytochrome P450. J. Am. Chem. Soc. 2006, 128, 394–395. 10.1021/ja055987p. [DOI] [PubMed] [Google Scholar]
- Li C.; Wu W.; Cho K.-B.; Shaik S. Oxidation of Tertiary Amines by Cytochrome P450—Kinetic Isotope Effect as a Spin-State Reactivity Probe. Chem. - Eur. J. 2009, 15, 8492–8503. 10.1002/chem.200802215. [DOI] [PubMed] [Google Scholar]
- Klinker E. J.; Shaik S.; Hirao H.; Que L. A Two-State Reactivity Model Explains Unusual Kinetic Isotope Effect Patterns in C–H Bond Cleavage by Nonheme Oxoiron(IV) Complexes. Angew. Chem., Int. Ed. 2009, 48, 1291–1295. 10.1002/anie.200804029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manchester J. I.; Dinnocenzo J. P.; Higgins; Jones J. P. A New Mechanistic Probe for Cytochrome P450: An Application of Isotope Effect Profiles. J. Am. Chem. Soc. 1997, 119, 5069–5070. 10.1021/ja9705250. [DOI] [Google Scholar]
- Dinnocenzo J. P.; Karki S. B.; Jones J. P. On Isotope Effects for the Cytochrome P450 Oxidation of Substituted N,N-Dimethylanilines. J. Am. Chem. Soc. 1993, 115, 7111–7116. 10.1021/ja00069a007. [DOI] [Google Scholar]
- Karki S. B.; Dinnocenzo J. P.; Jones J. P.; Korzekwa K. R. Mechanism of Oxidative Amine Dealkylation of Substituted N,N-Dimethylanilines by Cytochrome P450: Application of Isotope Effect Profiles. J. Am. Chem. Soc. 1995, 117, 3657–3664. 10.1021/ja00118a001. [DOI] [Google Scholar]
- Wang Y.; Kumar D.; Yang C.; Han K.; Shaik S. Theoretical Study of N-Demethylation of Substituted N,N-Dimethylanilines by Cytochrome P450: The Mechanistic Significance of Kinetic Isotope Effect Profiles. J. Phys. Chem. B 2007, 111, 7700–7710. 10.1021/jp072347v. [DOI] [PubMed] [Google Scholar]
- Prokop K. A.; de Visser S. P.; Goldberg D. P. Unprecedented Rate Enhancements of Hydrogen-Atom Transfer to a Manganese(V)–Oxo Corrolazine Complex. Angew. Chem., Int. Ed. 2010, 49, 5091–5095. 10.1002/anie.201001172. [DOI] [PubMed] [Google Scholar]
- Yang T.; Quesne M. G.; Neu H. M.; Cantú Reinhard F. G.; Goldberg D. P.; de Visser S. P. Singlet versus Triplet Reactivity in an Mn(V)–Oxo Species: Testing Theoretical Predictions Against Experimental Evidence. J. Am. Chem. Soc. 2016, 138, 12375–12386. 10.1021/jacs.6b05027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohen A.; Klinman J. P. Enzyme Catalysis: Beyond Classical Paradigms. Acc. Chem. Res. 1998, 31, 397–404. 10.1021/ar9701225. [DOI] [Google Scholar]
- Truhlar D. G.; Gao J.; Alhambra C.; Garcia-Viloca M.; Corchado J.; Sánchez M. L.; Villà J. The Incorporation of Quantum Effects in Enzyme Kinetics Modeling. Acc. Chem. Res. 2002, 35, 341–349. 10.1021/ar0100226. [DOI] [PubMed] [Google Scholar]
- Kim Y.; Kreevoy M. M. The Experimental Manifestations of Corner-Cutting Tunneling. J. Am. Chem. Soc. 1992, 114, 7116–7123. 10.1021/ja00044a024. [DOI] [Google Scholar]
- Kwart H. Temperature Dependence of the Primary Kinetic Hydrogen Isotope Effect as a Mechanistic Criterion. Acc. Chem. Res. 1982, 15, 401–408. 10.1021/ar00084a004. [DOI] [Google Scholar]
- Kwon Y. H.; Mai B. K.; Lee Y. M.; Dhuri S. N.; Mandal D.; Cho K. B.; Kim Y.; Shaik S.; Nam W. Determination of Spin Inversion Probability, H-Tunneling Correction, and Regioselectivity in the Two-State Reactivity of Nonheme Iron(IV)-Oxo Complexes. J. Phys. Chem. Lett. 2015, 6, 1472–1476. 10.1021/acs.jpclett.5b00527. [DOI] [PubMed] [Google Scholar]
- Klinman J. P. The Role of Tunneling in Enzyme Catalysis of C–H Activation. Biochim. Biophys. Acta, Bioenerg. 2006, 1757, 981–987. 10.1016/j.bbabio.2005.12.004. [DOI] [PubMed] [Google Scholar]
- Layfield J. P.; Hammes-Schiffer S. Hydrogen Tunneling in Enzymes and Biomimetic Models. Chem. Rev. 2014, 114, 3466–3494. 10.1021/cr400400p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandal D.; Ramanan R.; Usharani D.; Janardanan D.; Wang B.; Shaik S. How Does Tunneling Contribute to Counterintuitive H-Abstraction Reactivity of Nonheme Fe(IV)O Oxidants with Alkanes?. J. Am. Chem. Soc. 2015, 137, 722–733. 10.1021/ja509465w. [DOI] [PubMed] [Google Scholar]
- Mandal D.; Shaik S. Interplay of Tunneling, Two-State Reactivity, and Bell–Evans–Polanyi Effects in C–H Activation by Nonheme Fe(IV)O Oxidants. J. Am. Chem. Soc. 2016, 138, 2094–2097. 10.1021/jacs.5b12077. [DOI] [PubMed] [Google Scholar]
- Zhang Y.; Lin H. Quantum Tunneling in Testosterone 6β-Hydroxylation by Cytochrome P450: Reaction Dynamics Calculations Employing Multiconfiguration Molecular–Mechanical Potential Energy Surfaces. J. Phys. Chem. A 2009, 113, 11501–11508. 10.1021/jp901850c. [DOI] [PubMed] [Google Scholar]
- Krauser J. A.; Guengerich F. P. Cytochrome P450 3A4-catalyzed Testosterone 6β-Hydroxylation Stereochemistry, Kinetic Deuterium Isotope Effects, and Rate-limiting Steps. J. Biol. Chem. 2005, 280, 19496–19506. 10.1074/jbc.M501854200. [DOI] [PubMed] [Google Scholar]
- Nelson S. D.; Trager W. F. The Use of Deuterium Isotope Effects to Probe the Active Site Properties, Mechanism of Cytochrome P450-Catalyzed Reactions, and Mechanisms of Metabolically Dependent Toxicity. Drug Metab. Dispos. 2003, 31, 1481–1497. 10.1124/dmd.31.12.1481. [DOI] [PubMed] [Google Scholar]
- Sorokin A.; Robert A.; Meunier B. Intramolecular Kinetic Isotope Effects in Alkane Hydroxylations Catalyzed by Manganese and Iron Porphyrin Complexes. J. Am. Chem. Soc. 1993, 115, 7293–7299. 10.1021/ja00069a031. [DOI] [Google Scholar]
- White R. E.; Miller J. P.; Favreau L. V.; Bhattacharyya A. Stereochemical Dynamics of Aliphatic Hydroxylation by Cytochrome P450. J. Am. Chem. Soc. 1986, 108, 6024–6031. 10.1021/ja00279a059. [DOI] [PubMed] [Google Scholar]
- Sorokin A. B.; Khenkin A. M. The Contribution of Tunnelling to High Values of Kinetic Isotope Effect in Aliphatic Hydroxylation by a Cytochrome P450 Model. J. Chem. Soc., Chem. Commun. 1990, 45–46. 10.1039/c39900000045. [DOI] [Google Scholar]
- Pan Z.; Horner J. H.; Newcomb M. Tunneling in C–H Oxidation Reactions by an Oxoiron(IV) Porphyrin Radical Cation: Direct Measurements of Very Large H/D Kinetic Isotope Effects. J. Am. Chem. Soc. 2008, 130, 7776–7777. 10.1021/ja802484n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong Z.; Kinemuchi H.; Kurahashi T.; Fujii H. Factors Affecting Hydrogen-Tunneling Contribution in Hydroxylation Reactions Promoted by Oxoiron(IV) Porphyrin π-Cation Radical Complexes. Inorg. Chem. 2014, 53, 10632–10641. 10.1021/ic501737j. [DOI] [PubMed] [Google Scholar]
- Meisner J.; Kästner J. Atom Tunneling in Chemistry. Angew. Chem., Int. Ed. 2016, 55, 5400–5413. 10.1002/anie.201511028. [DOI] [PubMed] [Google Scholar]
- Bukowski M. R.; Koehntop K. D.; Stubna A.; Bominaar E. L.; Halfen J. A.; Münck E.; Nam W.; Que L. A Thiolate-Ligated Nonheme Oxoiron(IV) Complex Relevant to Cytochrome P450. Science 2005, 310, 1000–1002. 10.1126/science.1119092. [DOI] [PubMed] [Google Scholar]
- Rohde J.-U.; Que L. Axial Coordination of Carboxylate Activates the Non-heme FeIV=O Unit. Angew. Chem., Int. Ed. 2005, 44, 2255–2258. 10.1002/anie.200462631. [DOI] [PubMed] [Google Scholar]
- Sastri C. V.; Lee J.; Oh K.; Lee Y. J.; Lee J.; Jackson T. A.; Ray K.; Hirao H.; Shin W.; Halfen J. A.; et al. Axial Ligand Tuning of a Nonheme Iron(IV)–Oxo Unit for Hydrogen Atom Abstraction. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 19181–19186. 10.1073/pnas.0709471104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirao H.; Que L.; Nam W.; Shaik S. A Two-State Reactivity Rationale for Counterintuitive Axial Ligand Effects on the C-H Activation Reactivity of Nonheme Fe(IV)=O Oxidants. Chem. - Eur. J. 2008, 14, 1740–1756. 10.1002/chem.200701739. [DOI] [PubMed] [Google Scholar]
- Groves J. T.; Gross Z. In Bioinorganic Chemistry: An Inorganic Perspective of Life; Kessissoglou D. P., Ed.; Springer: Netherlands, 1995. [Google Scholar]
- Gupta R.; Li X. X.; Cho K. B.; Guo M.; Lee Y. M.; Wang Y.; Fukuzumi S.; Nam W. Tunneling Effect That Changes the Reaction Pathway from Epoxidation to Hydroxylation in the Oxidation of Cyclohexene by a Compound I Model of Cytochrome P450. J. Phys. Chem. Lett. 2017, 8, 1557–1561. 10.1021/acs.jpclett.7b00461. [DOI] [PubMed] [Google Scholar]
- Gao H.; Groves J. T. Fast Hydrogen Atom Abstraction by a Hydroxo Iron(III) Porphyrazine. J. Am. Chem. Soc. 2017, 139, 3938–3941. 10.1021/jacs.6b13091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewandowska-Andralojc A.; Grills D. C.; Zhang J.; Bullock R. M.; Miyazawa A.; Kawanishi Y.; Fujita E. Kinetic and Mechanistic Studies of Carbon-to-Metal Hydrogen Atom Transfer Involving Os-Centered Radicals: Evidence for Tunneling. J. Am. Chem. Soc. 2014, 136, 3572–3578. 10.1021/ja4123076. [DOI] [PubMed] [Google Scholar]
- Slaughter L. M.; Wolczanski P. T.; Klinckman T. R.; Cundari T. R. Inter- and Intramolecular Experimental and Calculated Equilibrium Isotope Effects for (Silox)2(tBu3SiND)TiR + RH (Silox = tBu3SiO): Inferred Kinetic Isotope Effects for Rh/D Addition to Transient (Silox)2Ti = NSitBu3. J. Am. Chem. Soc. 2000, 122, 7953–7975. 10.1021/ja000112q. [DOI] [Google Scholar]
- Groves J. T.; Subramanian D. V. Hydroxylation by Cytochrome P450 and Metalloporphyrin Models. Evidence for Allylic Rearrangement. J. Am. Chem. Soc. 1984, 106, 2177–2181. 10.1021/ja00319a044. [DOI] [Google Scholar]
- Griller D.; Ingold K. U. Free-Radical Clocks. Acc. Chem. Res. 1980, 13, 317–323. 10.1021/ar50153a004. [DOI] [Google Scholar]
- Rozhkova-Novosad E. A.; Chae J. C.; Zylstra G. J.; Bertrand E. M.; Alexander-Ozinskas M.; Deng D. Y.; Moe L. A.; van Beilen J. B.; Danahy M.; Groves J. T.; et al. Profiling Mechanisms of Alkane Hydroxylase Activity in vivo Using the Diagnostic Substrate Norcarane. Chem. Biol. 2007, 14, 165–172. 10.1016/j.chembiol.2006.12.007. [DOI] [PubMed] [Google Scholar]
- Baik M. H.; Newcomb M.; Friesner R. A.; Lippard S. J. Mechanistic Studies on the Hydroxylation of Methane by Methane Monooxygenase. Chem. Rev. 2003, 103, 2385–2419. 10.1021/cr950244f. [DOI] [PubMed] [Google Scholar]
- Brazeau B. J.; Austin R. N.; Tarr C.; Groves J. T.; Lipscomb J. D. Intermediate Q from Soluble Methane Monooxygenase Hydroxylates the Mechanistic Substrate Probe Norcarane: Evidence for a Stepwise Reaction. J. Am. Chem. Soc. 2001, 123, 11831–11837. 10.1021/ja016376+. [DOI] [PubMed] [Google Scholar]
- Jin Y.; Lipscomb J. D. Probing the Mechanism of C-H Activation: Oxidation of Methylcubane by Soluble Methane Monooxygenase from Methylosinus Trichosporium OB3b. Biochemistry 1999, 38, 6178–6186. 10.1021/bi990068v. [DOI] [PubMed] [Google Scholar]
- Jin Y.; Lipscomb J. D. Mechanistic Insights into C-H Activation from Radical Clock Chemistry: Oxidation of Substituted Methylcyclopropanes Catalyzed by Soluble Methane Monooxygenase from Methylosinus Trichosporium OB3b. Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol. 2000, 1543, 47–59. 10.1016/S0167-4838(00)00199-0. [DOI] [PubMed] [Google Scholar]
- Austin R. N.; Buzzi K.; Kim E.; Zylstra G. J.; Groves J. T. Xylene Monooxygenase, a Membrane-Spanning Non-Heme Diiron Enzyme That Hydroxylates Hydrocarbons via a Substrate Radical Intermediate. J. Biol. Inorg. Chem. 2003, 8, 733–740. [DOI] [PubMed] [Google Scholar]
- Moe L. A.; Hu Z. B.; Deng D. Y.; Austin R. N.; Groves J. T.; Fox B. G. Remarkable Aliphatic Hydroxylation by the Diiron Enzyme Toluene 4-Monooxygenase in Reactions with Radical or Cation Diagnostic Probes Norcarane, 1,1-Dimethylcyclopropane, and 1,1-Diethylcyclopropane. Biochemistry 2004, 43, 15688–15701. 10.1021/bi040033h. [DOI] [PubMed] [Google Scholar]
- Zaragoza J. P. T.; Yosca T. H.; Siegler M. A.; Moënne-Loccoz P.; Green M. T.; Goldberg D. P. Direct Observation of Oxygen Rebound with an Iron-Hydroxide Complex. J. Am. Chem. Soc. 2017, 139, 13640–13643. 10.1021/jacs.7b07979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Austin R. N.; Luddy K.; Erickson K.; Pender-Cudlip M.; Bertrand E.; Deng D.; Buzdygon R. S.; van Beilen J. B.; Groves J. T. Cage Escape Competes with Geminate Recombination during Alkane Hydroxylation by the Diiron Oxygenase AlkB. Angew. Chem., Int. Ed. 2008, 47, 5232–5234. 10.1002/anie.200801184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper H. L.; Mishra G.; Huang X.; Pender-Cudlip M.; Austin R. N.; Shanklin J.; Groves J. T. Parallel and Competitive Pathways for Substrate Desaturation, Hydroxylation, and Radical Rearrangement by the Non-Heme Diiron Hydroxylase AlkB. J. Am. Chem. Soc. 2012, 134, 20365–20375. 10.1021/ja3059149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertrand E.; Sakai R.; Rozhkova-Novosad E.; Moe L.; Fox B. G.; Groves J. T.; Austin R. N. Reaction Mechanisms of Non-Heme Diiron Hydroxylases Characterized in Whole Cells. J. Inorg. Biochem. 2005, 99, 1998–2006. 10.1016/j.jinorgbio.2005.06.020. [DOI] [PubMed] [Google Scholar]
- Cooper H. L. R.; Groves J. T. Molecular Probes of the Mechanism of Cytochrome P450. Oxygen Traps a Substrate Radical Intermediate. Arch. Biochem. Biophys. 2011, 507, 111–118. 10.1016/j.abb.2010.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogliaro F.; de Visser S. P.; Groves J. T.; Shaik S. Chameleon States: High-Valent Metal–Oxo Species of Cytochrome P450 and Its Ruthenium Analogue. Angew. Chem., Int. Ed. 2001, 40, 2874–2878. . [DOI] [PubMed] [Google Scholar]
- Liu W.; Cheng M.-J.; Nielsen R. J.; Goddard W. A.; Groves J. T. Probing the C–O Bond-Formation Step in Metalloporphyrin-Catalyzed C–H Oxygenation Reactions. ACS Catal. 2017, 7, 4182–4188. 10.1021/acscatal.7b00655. [DOI] [Google Scholar]
- Cho K. B.; Hirao H.; Shaik S.; Nam W. To Rebound or Dissociate? This Is the Mechanistic Question in C-H Hydroxylation by Heme and Nonheme Metal-Oxo Complexes. Chem. Soc. Rev. 2016, 45, 1197–1210. 10.1039/C5CS00566C. [DOI] [PubMed] [Google Scholar]
- Rude M. A.; Baron T. S.; Brubaker S.; Alibhai M.; Del Cardayre S. B.; Schirmer A. Terminal Olefin (1-Alkene) Biosynthesis by a Novel P450 Fatty Acid Decarboxylase from Jeotgalicoccus Species. Appl. Environ. Microbiol. 2011, 77, 1718–1727. 10.1128/AEM.02580-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J.-b.; Lonsdale R.; Reetz M. T. Exploring Substrate Scope and Stereoselectivity of P450 Peroxygenase OleTJE in Olefin-Forming Oxidative Decarboxylation. Chem. Commun. 2016, 52, 8131–8133. 10.1039/C6CC04345C. [DOI] [PubMed] [Google Scholar]
- Hsieh C. H.; Makris T. M. Expanding the Substrate Scope and Reactivity of Cytochrome P450 OleT. Biochem. Biophys. Res. Commun. 2016, 476, 462–466. 10.1016/j.bbrc.2016.05.145. [DOI] [PubMed] [Google Scholar]
- Wise C. E.; Grant J. L.; Amaya J. A.; Ratigan S. C.; Hsieh C. H.; Manley O. M.; Makris T. M. Divergent Mechanisms of Iron-Containing Enzymes for Hydrocarbon Biosynthesis. JBIC, J. Biol. Inorg. Chem. 2017, 22, 221–235. 10.1007/s00775-016-1425-0. [DOI] [PubMed] [Google Scholar]
- Dennig A.; Kuhn M.; Tassoti S.; Thiessenhusen A.; Gilch S.; Bulter T.; Haas T.; Hall M.; Faber K. Oxidative Decarboxylation of Short-Chain Fatty Acids to 1-Alkenes. Angew. Chem., Int. Ed. 2015, 54, 8819–8822. 10.1002/anie.201502925. [DOI] [PubMed] [Google Scholar]
- Fujishiro T.; Shoji O.; Nagano S.; Sugimoto H.; Shiro Y.; Watanabe Y. Crystal Structure of H2O2-Dependent Cytochrome P450SPα With Its Bound Fatty Acid Substrate: Insight into the Regioselective Hydroxylation of Fatty Acids at the Alpha Position. J. Biol. Chem. 2011, 286, 29941–29950. 10.1074/jbc.M111.245225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant J. L.; Hsieh C. H.; Makris T. M. Decarboxylation of Fatty Acids to Terminal Alkenes by Cytochrome P450 Compound I. J. Am. Chem. Soc. 2015, 137, 4940–4943. 10.1021/jacs.5b01965. [DOI] [PubMed] [Google Scholar]
- Kourist R. A New Class of Enzymes Discovered: A Non-Heme Oxidase Produces Medium-Chain 1-Alkenes. Angew. Chem., Int. Ed. 2015, 54, 4156–4158. 10.1002/anie.201500466. [DOI] [PubMed] [Google Scholar]
- Rui Z.; Li X.; Zhu X.; Liu J.; Domigan B.; Barr I.; Cate J. H. D.; Zhang W. Microbial Biosynthesis of Medium-Chain 1-Alkenes by a Nonheme Iron Oxidase. Proc. Natl. Acad. Sci. U. S. A. 2014, 111, 18237–18242. 10.1073/pnas.1419701112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grant J. L.; Mitchell M. E.; Makris T. M. Catalytic Strategy for Carbon-Carbon Bond Scission by the Cytochrome P450 OleT. Proc. Natl. Acad. Sci. U. S. A. 2016, 113, 10049–10054. 10.1073/pnas.1606294113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zachos I.; Gaβmeyer S. K.; Sieber V.; Hollmann F.; Kourist R.; Bauer D. Photobiocatalytic Decarboxylation for Olefin Synthesis. Chem. Commun. 2015, 51, 1918–1921. 10.1039/C4CC07276F. [DOI] [PubMed] [Google Scholar]
- Hsieh C. H.; Huang X.; Amaya J. A.; Rutland C. D.; Keys C. L.; Groves J. T.; Austin R. N.; Makris T. M. The Enigmatic P450 Decarboxylase OleT Is Capable of, but Evolved To Frustrate, Oxygen Rebound Chemistry. Biochemistry 2017, 56, 3347–3357. 10.1021/acs.biochem.7b00338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang B.; Xu H.; Liu Y.; Qi F.; Zhang W.; Chen H.; Wang C.; Wang Y.; Yang W.; Li S. Mutagenesis and Redox Partners Analysis of the P450 Fatty Acid Decarboxylase OleTJE. Sci. Rep. 2017, 7, 44258. 10.1038/srep44258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rettie A.; Rettenmeier A.; Howald W.; Baillie T. Cytochrome P450-Catalyzed Formation of Delta 4-VPA, a Toxic Metabolite of Valproic Acid. Science 1987, 235, 890–893. 10.1126/science.3101178. [DOI] [PubMed] [Google Scholar]
- Guengerich F. P. Common and Uncommon Cytochrome P450 Reactions Related to Metabolism and Chemical Toxicity. Chem. Res. Toxicol. 2001, 14, 611–650. 10.1021/tx0002583. [DOI] [PubMed] [Google Scholar]
- Rettie A. E.; Boberg M.; Rettenmeier A. W.; Baillie T. A. Cytochrome P450-Catalyzed Desaturation of Valproic Acid in vitro. Species Differences, Induction Effects, and Mechanistic Studies. J. Biol. Chem. 1988, 263, 13733–13738. [PubMed] [Google Scholar]
- Buist P. H. Fatty Acid Desaturases: Selecting the Dehydrogenation Channel. Nat. Prod. Rep. 2004, 21, 249–262. 10.1039/b302094k. [DOI] [PubMed] [Google Scholar]
- Behrouzian B.; Buist P. H. Fatty Acid Desaturation: Variations on an Oxidative Theme. Curr. Opin. Chem. Biol. 2002, 6, 577–582. 10.1016/S1367-5931(02)00365-4. [DOI] [PubMed] [Google Scholar]
- Ji L.; Faponle A. S.; Quesne M. G.; Sainna M. A.; Zhang J.; Franke A.; Kumar D.; van Eldik R.; Liu W.; de Visser S. P. Drug Metabolism by Cytochrome P450 Enzymes: What Distinguishes the Pathways Leading to Substrate Hydroxylation Over Desaturation?. Chem. - Eur. J. 2015, 21, 9083–9092. 10.1002/chem.201500329. [DOI] [PubMed] [Google Scholar]
- Lai W.; Chen H.; Cohen S.; Shaik S. Will P450cam Hydroxylate or Desaturate Alkanes? QM and QM/MM Studies. J. Phys. Chem. Lett. 2011, 2, 2229–2235. 10.1021/jz2007534. [DOI] [Google Scholar]
- Zhu D.; Seo M.-J.; Ikeda H.; Cane D. E. Genome Mining in Streptomyces. Discovery of an Unprecedented P450-Catalyzed Oxidative Rearrangement That Is the Final Step in the Biosynthesis of Pentalenolactone. J. Am. Chem. Soc. 2011, 133, 2128–2131. 10.1021/ja111279h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan L.; Jogl G.; Cane D. E. The Cytochrome P450-Catalyzed Oxidative Rearrangement in the Final Step of Pentalenolactone Biosynthesis: Substrate Structure Determines Mechanism. J. Am. Chem. Soc. 2016, 138, 12678–12689. 10.1021/jacs.6b08610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cane D. E.; Oliver J. S.; Harrison P. H. M.; Abell C.; Hubbard B. R.; Kane C. T.; Lattman R. Biosynthesis of Pentalenene and Pentalenolactone. J. Am. Chem. Soc. 1990, 112, 4513–4524. 10.1021/ja00167a059. [DOI] [Google Scholar]
- Liu W.; Groves J. T. Manganese Porphyrins Catalyze Selective C-H Bond Halogenations. J. Am. Chem. Soc. 2010, 132, 12847–12849. 10.1021/ja105548x. [DOI] [PubMed] [Google Scholar]
- Liu W.; Huang X. Y.; Cheng M. J.; Nielsen R. J.; Goddard W. A.; Groves J. T. Oxidative Aliphatic C-H Fluorination with Fluoride Ion Catalyzed by a Manganese Porphyrin. Science 2012, 337, 1322–1325. 10.1126/science.1222327. [DOI] [PubMed] [Google Scholar]
- Huang X.; Liu W.; Ren H.; Neelamegam R.; Hooker J. M.; Groves J. T. Late Stage Benzylic C-H Fluorination with F-18 Fluoride for PET Imaging. J. Am. Chem. Soc. 2014, 136, 6842–6845. 10.1021/ja5039819. [DOI] [PubMed] [Google Scholar]
- Huang X. Y.; Bergsten T. M.; Groves J. T. Manganese-Catalyzed Late-Stage Aliphatic C-H Azidation. J. Am. Chem. Soc. 2015, 137, 5300–5303. 10.1021/jacs.5b01983. [DOI] [PubMed] [Google Scholar]
- Liu W.; Huang X.; Groves J. T. Oxidative Aliphatic C-H Fluorination with Manganese Catalysts and Fluoride Ion. Nat. Protoc. 2013, 8, 2348–2354. 10.1038/nprot.2013.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W.; Groves J. T. Manganese-Catalyzed Oxidative Benzylic C-H Fluorination by Fluoride Ions. Angew. Chem., Int. Ed. 2013, 52, 6024–6027. 10.1002/anie.201301097. [DOI] [PubMed] [Google Scholar]
- Huang X.; Zhuang T.; Kates P. A.; Gao H.; Chen X.; Groves J. T. Alkyl Isocyanates via Manganese-Catalyzed C-H Activation for the Preparation of Substituted Ureas. J. Am. Chem. Soc. 2017, 139, 15407–15413. 10.1021/jacs.7b07658. [DOI] [PubMed] [Google Scholar]
- Martinie R. J.; Livada J.; Chang W. C.; Green M. T.; Krebs C.; Bollinger J. M.; Silakov A. Experimental Correlation of Substrate Position with Reaction Outcome in the Aliphatic Halogenase, SyrB2. J. Am. Chem. Soc. 2015, 137, 6912–6919. 10.1021/jacs.5b03370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong S. D.; Srnec M.; Matthews M. L.; Liu L. V.; Kwak Y.; Park K.; Bell C. B. III; Alp E. E.; Zhao J.; Yoda Y.; et al. Elucidation of the Fe(IV)=O Intermediate in the Catalytic Cycle of the Halogenase SyrB2. Nature 2013, 499, 320–323. 10.1038/nature12304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blasiak L. C.; Vaillancourt F. H.; Walsh C. T.; Drennan C. L. Crystal Structure of the Non-Haem Iron Halogenase SyrB2 in Syringomycin Biosynthesis. Nature 2006, 440, 368–371. 10.1038/nature04544. [DOI] [PubMed] [Google Scholar]
- Vaillancourt F. H.; Yin J.; Walsh C. T. SyrB2 in Syringomycin E Biosynthesis Is a Nonherne FeII Alpha-Ketoglutarate- and O2-Dependent Halogenase. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 10111–10116. 10.1073/pnas.0504412102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell A. J.; Zhu Q.; Maggiolo A. O.; Ananth N. R.; Hillwig M. L.; Liu X.; Boal A. K. Structural Basis for Halogenation by Iron- and 2-Oxo-Glutarate-Dependent Enzyme WelO5. Nat. Chem. Biol. 2016, 12, 636–640. 10.1038/nchembio.2112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillwig M. L.; Zhu Q.; Ittiamornkul K.; Liu X. Discovery of a Promiscuous Non-Heme Iron Halogenase in Ambiguine Alkaloid Biogenesis: Implication for an Evolvable Enzyme Family for Late-Stage Halogenation of Aliphatic Carbons in Small Molecules. Angew. Chem., Int. Ed. 2016, 55, 5780–5784. 10.1002/anie.201601447. [DOI] [PubMed] [Google Scholar]
- Wong C.; Fujimori D. G.; Walsh C. T.; Drennan C. L. Structural Analysis of an Open Active Site Conformation of Nonheme Iron Halogenase CytC3. J. Am. Chem. Soc. 2009, 131, 4872–4879. 10.1021/ja8097355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulik H. J.; Blasiak L. C.; Marzari N.; Drennan C. L. First-Principles Study of Non-heme Fe(II) Halogenase SyrB2 Reactivity. J. Am. Chem. Soc. 2009, 131, 14426–14433. 10.1021/ja905206k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews M. L.; Neumann C. S.; Miles L. A.; Grove T. L.; Booker S. J.; Krebs C.; Walsh C. T.; Bollinger J. M. Substrate Positioning Controls the Partition Between Halogenation and Hydroxylation in the Aliphatic Halogenase, SyrB2. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 17723–17728. 10.1073/pnas.0909649106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srnec M.; Solomon E. I. Frontier Molecular Orbital Contributions to Chlorination versus Hydroxylation Selectivity in the Non-Heme Iron Halogenase SyrB2. J. Am. Chem. Soc. 2017, 139, 2396–2407. 10.1021/jacs.6b11995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furuya T.; Kamlet A. S.; Ritter T. Catalysis for Fluorination and Trifluoromethylation. Nature 2011, 473, 470–477. 10.1038/nature10108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thibodeaux C. J.; Chang W.-C.; Liu H.-W. Enzymatic Chemistry of Cyclopropane, Epoxide, and Aziridine Biosynthesis. Chem. Rev. 2012, 112, 1681–1709. 10.1021/cr200073d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexandrov K.; Cascorbi I.; Rojas M.; Bouvier G.; Kriek E.; Bartsch H. CYP1A1 and GSTM1 Genotypes Affect Benzo[a]Pyrene Dna Adducts in Smokers’ Lung: Comparison with Aromatic/Hydrophobic Adduct Formation. Carcinogenesis 2002, 23, 1969–1977. 10.1093/carcin/23.12.1969. [DOI] [PubMed] [Google Scholar]
- de Visser S. P.; Ogliaro F.; Harris N.; Shaik S. Multi-State Epoxidation of Ethene by Cytochrome P450: a Quantum Chemical Study. J. Am. Chem. Soc. 2001, 123, 3037–3047. 10.1021/ja003544+. [DOI] [PubMed] [Google Scholar]
- Kumar D.; Karamzadeh B.; Sastry G. N.; de Visser S. P. What Factors Influence the Rate Constant of Substrate Epoxidation by Compound I of Cytochrome P450 and Analogous Iron(IV)-Oxo Oxidants?. J. Am. Chem. Soc. 2010, 132, 7656–7667. 10.1021/ja9106176. [DOI] [PubMed] [Google Scholar]
- Gross Z.; Nimri S.; Barzilay C. M.; Simkhovich L. Reaction Profile of the Last Step in Cytochrome P450 Catalysis Revealed by Studies of Model Complexes. JBIC, J. Biol. Inorg. Chem. 1997, 2, 492–506. 10.1007/s007750050161. [DOI] [Google Scholar]
- Groves J. T.; Gross Z.; Stern M. K. Preparation and Reactivity of Oxoiron(IV) Porphyrins. Inorg. Chem. 1994, 33, 5065–5072. 10.1021/ic00100a035. [DOI] [Google Scholar]
- de Visser S. P.; Kumar D.; Shaik S. How Do Aldehyde Side Products Occur During Alkene Epoxidation by Cytochrome P450? Theory Reveals a State-Specific Multi-State Scenario Where the High-Spin Component Leads to All Side Products. J. Inorg. Biochem. 2004, 98, 1183–1193. 10.1016/j.jinorgbio.2004.01.015. [DOI] [PubMed] [Google Scholar]
- Hammer S. C.; Kubik G.; Watkins E.; Huang S.; Minges H.; Arnold F. H. Anti-Markovnikov Alkene Oxidation by Metal-Oxo–Mediated Enzyme Catalysis. Science 2017, 358, 215–218. 10.1126/science.aao1482. [DOI] [PubMed] [Google Scholar]
- Jerina D. M.; Daly J. W.; Witkop B.; Zaltzman-Nirenberg P.; Udenfriend S. 1,2-Naphthalene Oxide as an Intermediate in the Microsomal Hydroxylation of Naphthalene. V. Biochemistry 1970, 9, 147–156. 10.1021/bi00803a019. [DOI] [PubMed] [Google Scholar]
- Korzekwa K. R.; Swinney D. C.; Trager W. F. Isotopically Labeled Chlorobenzenes as Probes for the Mechanism of Cytochrome P450 Catalyzed Aromatic Hydroxylation. Biochemistry 1989, 28, 9019–9027. 10.1021/bi00449a010. [DOI] [PubMed] [Google Scholar]
- Rietjens I. M. C. M.; Soffers A. E. M. F.; Veeger C.; Vervoort J. Regioselectivity of Cytochrome P450 Catalyzed Hydroxylation of Fluorobenzenes Predicted by Calculated Frontier Orbital Substrate Characteristics. Biochemistry 1993, 32, 4801–4812. 10.1021/bi00069a015. [DOI] [PubMed] [Google Scholar]
- Bathelt C. M.; Ridder L.; Mulholland A. J.; Harvey J. N. Mechanism and Structure-Reactivity Relationships for Aromatic Hydroxylation by Cytochrome P450. Org. Biomol. Chem. 2004, 2, 2998–3005. 10.1039/B410729B. [DOI] [PubMed] [Google Scholar]
- de Visser S. P.; Shaik S. A Proton-Shuttle Mechanism Mediated by the Porphyrin in Benzene Hydroxylation by Cytochrome P450 Enzymes. J. Am. Chem. Soc. 2003, 125, 7413–7424. 10.1021/ja034142f. [DOI] [PubMed] [Google Scholar]
- Bathelt C. M.; Ridder L.; Mulholland A. J.; Harvey J. N. Aromatic Hydroxylation by Cytochrome P450: Model Calculations of Mechanism and Substituent Effects. J. Am. Chem. Soc. 2003, 125, 15004–15005. 10.1021/ja035590q. [DOI] [PubMed] [Google Scholar]
- Shaik S.; Milko P.; Schyman P.; Usharani D.; Chen H. Trends in Aromatic Oxidation Reactions Catalyzed by Cytochrome P450 Enzymes: A Valence Bond Modeling. J. Chem. Theory Comput. 2011, 7, 327–339. 10.1021/ct100554g. [DOI] [PubMed] [Google Scholar]
- Cantú Reinhard F. G.; Sainna M. A.; Upadhyay P.; Balan G. A.; Kumar D.; Fornarini S.; Crestoni M. E.; de Visser S. P. A Systematic Account on Aromatic Hydroxylation by a Cytochrome P450 Model Compound I: A Low-Pressure Mass Spectrometry and Computational Study. Chem. - Eur. J. 2016, 22, 18608–18619. 10.1002/chem.201604361. [DOI] [PubMed] [Google Scholar]
- Asaka M.; Fujii H. Participation of Electron Transfer Process in Rate-Limiting Step of Aromatic Hydroxylation Reactions by Compound I Models of Heme Enzymes. J. Am. Chem. Soc. 2016, 138, 8048–8051. 10.1021/jacs.6b03223. [DOI] [PubMed] [Google Scholar]
- Jerina D. M.; Daly J. W. Arene Oxides: A New Aspect of Drug Metabolism. Science 1974, 185, 573–582. 10.1126/science.185.4151.573. [DOI] [PubMed] [Google Scholar]
- Ortiz de Montellano P. R. In Cytochrome P450: Structure, Mechanism, and Biochemistry; Ortiz de Montellano P. R., Ed.; Springer International Publishing: Cham, 2015. [Google Scholar]
- Preston B. D.; Miller J. A.; Miller E. C. Non-Arene Oxide Aromatic Ring Hydroxylation of 2,2′,5,5′-Tetrachlorobiphenyl as the Major Metabolic Pathway Catalyzed by Phenobarbital-Induced Rat Liver Microsomes. J. Biol. Chem. 1983, 258, 8304–8311. [PubMed] [Google Scholar]
- Tomaszewski J. E.; Jerina D. M.; Daly J. W. Deuterium Isotope Effects During Formation of Phenols by Hepatic Monoxygenases. Evidence for an Alternative to the Arene Oxide Pathway. Biochemistry 1975, 14, 2024–2031. 10.1021/bi00680a033. [DOI] [PubMed] [Google Scholar]
- Zanger U. M.; Schwab M. Cytochrome P450 Enzymes in Drug Metabolism: Regulation of Gene Expression, Enzyme Activities, and Impact of Genetic Variation. Pharmacol. Ther. 2013, 138, 103–141. 10.1016/j.pharmthera.2012.12.007. [DOI] [PubMed] [Google Scholar]
- Guengerich F. P. Enzymatic Oxidation of Xenobiotic Chemicals. Crit. Rev. Biochem. Mol. Biol. 1990, 25, 97–153. 10.3109/10409239009090607. [DOI] [PubMed] [Google Scholar]
- Fukuto J. M.; Kumagai Y.; Cho A. K. Determination of the Mechanism of Demethylenation of (Methylenedioxy)Phenyl Compounds by Cytochrome P450 Using Deuterium Isotope Effects. J. Med. Chem. 1991, 34, 2871–2876. 10.1021/jm00113a028. [DOI] [PubMed] [Google Scholar]
- Ichinose R.; Kurihara N. Deuterium and Tritium Isotope Effects on the Oxidative Demethylation Rate of Methoxychior in Rat Liver Microsomes. J. Pestic. Sci. 1986, 11, 231–236. 10.1584/jpestics.11.231. [DOI] [Google Scholar]
- Miwa G. T.; Walsh J. S.; Lu A. Y. Kinetic Isotope Effects on Cytochrome P-450-Catalyzed Oxidation Reactions. J. Biol. Chem. 1984, 259, 3000–3004. [PubMed] [Google Scholar]
- Roberts K. M.; Jones J. P. Anilinic N-Oxides Support Cytochrome P450-Mediated N-Dealkylation through Hydrogen-Atom Transfer. Chem. - Eur. J. 2010, 16, 8096–8107. 10.1002/chem.201000185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaffer C. L.; Morton M. D.; Hanzlik R. P. N-Dealkylation of an N-Cyclopropylamine by Horseradish Peroxidase. Fate of the Cyclopropyl Group. J. Am. Chem. Soc. 2001, 123, 8502–8508. 10.1021/ja0111479. [DOI] [PubMed] [Google Scholar]
- Shaffer C. L.; Morton M. D.; Hanzlik R. P. Enzymatic N-Dealkylation of an N-Cyclopropylamine: An Unusual Fate for the Cyclopropyl Group. J. Am. Chem. Soc. 2001, 123, 349–350. 10.1021/ja003048l. [DOI] [PubMed] [Google Scholar]
- Constantino L.; Rosa E.; iley J. The Microsomal Demethylation of N,N-Dimethylbenzamides: Substituent and Kinetic Deuterium Isotope Effects. Biochem. Pharmacol. 1992, 44, 651–658. 10.1016/0006-2952(92)90399-4. [DOI] [PubMed] [Google Scholar]
- Hall L. R.; Hanzlik R. P. Kinetic Deuterium Isotope Effects on the N-Demethylation of Tertiary Amides by Cytochrome P-450. J. Biol. Chem. 1990, 265, 12349–12355. [PubMed] [Google Scholar]
- Guengerich F. P.; Yun C. H.; Macdonald T. L. Evidence for a 1-Electron Oxidation Mechanism in N-Dealkylation of N,N-Dialkylanilines by Cytochrome P450 2B1. Kinetic Hydrogen Isotope Effects, Linear Free Energy Relationships, Comparisons with Horseradish Peroxidase, and Studies with Oxygen Surrogates. J. Biol. Chem. 1996, 271, 27321–27329. 10.1074/jbc.271.44.27321. [DOI] [PubMed] [Google Scholar]
- Miwa G. T.; Walsh J. S.; Kedderis G. L.; Hollenberg P. F. The Use of Intramolecular Isotope Effects to Distinguish Between Deprotonation and Hydrogen Atom Abstraction Mechanisms in Cytochrome P-450- and Peroxidase-Catalyzed N-Demethylation Reactions. J. Biol. Chem. 1983, 258, 14445–14449. [PubMed] [Google Scholar]
- Li C.; Wu W.; Kumar D.; Shaik S. Kinetic Isotope Effect is a Sensitive Probe of Spin State Reactivity in C–H Hydroxylation of N,N-Dimethylaniline by Cytochrome P450. J. Am. Chem. Soc. 2006, 128, 394–395. 10.1021/ja055987p. [DOI] [PubMed] [Google Scholar]
- Kumar D.; Sastry G. N.; de Visser S. P. Effect of the Axial Ligand on Substrate Sulfoxidation Mediated by Iron(IV)–Oxo Porphyrin Cation Radical Oxidants. Chem. - Eur. J. 2011, 17, 6196–6205. 10.1002/chem.201003187. [DOI] [PubMed] [Google Scholar]
- Shaik S.; Wang Y.; Chen H.; Song J.; Meir R. Valence Bond Modelling and Density Functional Theory Calculations of Reactivity and Mechanism of Cytochrome P450 Enzymes: Thioether Sulfoxidation. Faraday Discuss. 2010, 145, 49–70. 10.1039/B906094D. [DOI] [Google Scholar]
- Li C.; Zhang L.; Zhang C.; Hirao H.; Wu W.; Shaik S. Which Oxidant Is Really Responsible for Sulfur Oxidation by Cytochrome P450?. Angew. Chem., Int. Ed. 2007, 46, 8168–8170. 10.1002/anie.200702867. [DOI] [PubMed] [Google Scholar]
- Rydberg P.; Ryde U.; Olsen L. Sulfoxide, Sulfur, and Nitrogen Oxidation and Dealkylation by Cytochrome P450. J. Chem. Theory Comput. 2008, 4, 1369–1377. 10.1021/ct800101v. [DOI] [PubMed] [Google Scholar]
- Afzelius L.; Hasselgren Arnby C.; Broo A.; Carlsson L.; Isaksson C.; Jurva U.; Kjellander B.; Kolmodin K.; Nilsson K.; Raubacher F.; et al. State-of-the-art Tools for Computational Site of Metabolism Predictions: Comparative Analysis, Mechanistical Insights, and Future Applications. Drug Metab. Rev. 2007, 39, 61–86. 10.1080/03602530600969374. [DOI] [PubMed] [Google Scholar]
- Seto Y.; Guengerich F. P. Partitioning Between N-Dealkylation and N-Oxygenation in the Oxidation of N,N-Dialkylarylamines Catalyzed by Cytochrome P450 2B1. J. Biol. Chem. 1993, 268, 9986–9997. [PubMed] [Google Scholar]
- Rydberg P.; Jørgensen M. S.; Jacobsen T. A.; Jacobsen A.-M.; Madsen K. G.; Olsen L. Nitrogen Inversion Barriers Affect the N-Oxidation of Tertiary Alkylamines by Cytochromes P450. Angew. Chem., Int. Ed. 2013, 52, 993–997. 10.1002/anie.201206207. [DOI] [PubMed] [Google Scholar]
- Li C.; Zhang L.; Zhang C.; Hirao H.; Wu W.; Shaik S. Which Oxidant is Really Responsible for Sulfur Oxidation by Cytochrome P450?. Angew. Chem., Int. Ed. 2008, 47, 8148–8148. 10.1002/anie.200890215. [DOI] [PubMed] [Google Scholar]
- de Visser S. P.; Tahsini L.; Nam W. How Does the Axial Ligand of Cytochrome P450 Biomimetics Influence the Regioselectivity of Aliphatic versus Aromatic Hydroxylation?. Chem. - Eur. J. 2009, 15, 5577–5587. 10.1002/chem.200802234. [DOI] [PubMed] [Google Scholar]
- Kumar D.; Latifi R.; Kumar S.; Rybak-Akimova E. V.; Sainna M. A.; de Visser S. P. Rationalization of the Barrier Height for p-Z-styrene Epoxidation by Iron(IV)-Oxo Porphyrin Cation Radicals with Variable Axial Ligands. Inorg. Chem. 2013, 52, 7968–7979. 10.1021/ic4005104. [DOI] [PubMed] [Google Scholar]
- Sainna M. A.; Kumar S.; Kumar D.; Fornarini S.; Crestoni M. E.; de Visser S. P. A Comprehensive Test Set of Epoxidation Rate Constants for Iron(IV)-Oxo Porphyrin Cation Radical Complexes. Chem. Sci. 2015, 6, 1516–1529. 10.1039/C4SC02717E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar D.; Sastry G. N.; de Visser S. P. Axial Ligand Effect On The Rate Constant of Aromatic Hydroxylation By Iron(IV)–Oxo Complexes Mimicking Cytochrome P450 Enzymes. J. Phys. Chem. B 2012, 116, 718–730. 10.1021/jp2113522. [DOI] [PubMed] [Google Scholar]
- Ji L.; Schüürmann G. Model and Mechanism: N-Hydroxylation of Primary Aromatic Amines by Cytochrome P450. Angew. Chem., Int. Ed. 2013, 52, 744–748. 10.1002/anie.201204116. [DOI] [PubMed] [Google Scholar]
- Ripa L.; Mee C.; Sjö P.; Shamovsky I. Theoretical Studies of the Mechanism of N-Hydroxylation of Primary Aromatic Amines by Cytochrome P450 1A2: Radicaloid or Anionic?. Chem. Res. Toxicol. 2014, 27, 265–278. 10.1021/tx400376u. [DOI] [PubMed] [Google Scholar]
- Seger S. T.; Rydberg P.; Olsen L. Mechanism of the N-Hydroxylation of Primary and Secondary Amines by Cytochrome P450. Chem. Res. Toxicol. 2015, 28, 597–603. 10.1021/tx500371a. [DOI] [PubMed] [Google Scholar]
- Lonsdale R.; Fort R. M.; Rydberg P.; Harvey J. N.; Mulholland A. J. Quantum Mechanics/Molecular Mechanics Modeling of Drug Metabolism: Mexiletine N-Hydroxylation by Cytochrome P450 1A2. Chem. Res. Toxicol. 2016, 29, 963–971. 10.1021/acs.chemrestox.5b00514. [DOI] [PubMed] [Google Scholar]
- Gross Z.; Nimri S. A Pronounced Axial Ligand Effect on the Reactivity of Oxoiron(IV) Porphyrin Cation Radicals. Inorg. Chem. 1994, 33, 1731–1732. 10.1021/ic00087a001. [DOI] [Google Scholar]
- Czarnecki K.; Nimri S.; Gross Z.; Proniewicz L. M.; Kincaid J. R. Direct Resonance Raman Evidence for a Trans Influence on the Ferryl Fragment in Models of Compound I Intermediates of Heme Enzymes. J. Am. Chem. Soc. 1996, 118, 2929–2935. 10.1021/ja952177c. [DOI] [Google Scholar]
- Gross Z. The Effect of Axial Ligands on the Reactivity and Stability of the Oxoferryl Moiety in Model Complexes of Compound I of Heme-Dependent Enzymes. JBIC, J. Biol. Inorg. Chem. 1996, 1, 368–371. 10.1007/s007750050067. [DOI] [Google Scholar]
- Takahashi A.; Kurahashi T.; Fujii H. Effect of Imidazole and Phenolate Axial Ligands on the Electronic Structure and Reactivity of Oxoiron(IV) Porphyrin π-Cation Radical Complexes: Drastic Increase in Oxo-Transfer and Hydrogen Abstraction Reactivities. Inorg. Chem. 2009, 48, 2614–2625. 10.1021/ic802123m. [DOI] [PubMed] [Google Scholar]
- Barber J. Photosynthetic Energy Conversion: Natural and Artificial. Chem. Soc. Rev. 2009, 38, 185–196. 10.1039/B802262N. [DOI] [PubMed] [Google Scholar]
- McEvoy J. P.; Brudvig G. W. Water-Splitting Chemistry of Photosystem II. Chem. Rev. 2006, 106, 4455–4483. 10.1021/cr0204294. [DOI] [PubMed] [Google Scholar]
- Blakemore J. D.; Crabtree R. H.; Brudvig G. W. Molecular Catalysts for Water Oxidation. Chem. Rev. 2015, 115, 12974–13005. 10.1021/acs.chemrev.5b00122. [DOI] [PubMed] [Google Scholar]
- Karkas M. D.; Verho O.; Johnston E. V.; Akermark B. Artificial Photosynthesis: Molecular Systems for Catalytic Water Oxidation. Chem. Rev. 2014, 114, 11863–12001. 10.1021/cr400572f. [DOI] [PubMed] [Google Scholar]
- Solomon E. I.; Heppner D. E.; Johnston E. M.; Ginsbach J. W.; Cirera J.; Qayyum M.; Kieber-Emmons M. T.; Kjaergaard C. H.; Hadt R. G.; Tian L. Copper Active Sites in Biology. Chem. Rev. 2014, 114, 3659–3853. 10.1021/cr400327t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolman W. B. Making and Breaking the Dioxygen O-O Bond: New Insights from Studies of Synthetic Copper Complexes. Acc. Chem. Res. 1997, 30, 227–237. 10.1021/ar960052m. [DOI] [Google Scholar]
- Halfen J. A.; Mahapatra S.; Wilkinson E. C.; Kaderli S.; Young V. G.; Que L.; Zuberbuhler A. D.; Tolman W. B. Reversible Cleavage and Formation of the Dioxygen O-O Band Within a Dicopper Complex. Science 1996, 271, 1397–1400. 10.1126/science.271.5254.1397. [DOI] [PubMed] [Google Scholar]
- Cox N.; Retegan M.; Neese F.; Pantazis D. A.; Boussac A.; Lubitz W. Electronic Structure of the Oxygen-Evolving Complex in Photosystem II Prior to O-O Bond Formation. Science 2014, 345, 804–808. 10.1126/science.1254910. [DOI] [PubMed] [Google Scholar]
- Siegbahn P. E. M. Structures and Energetics for O2 Formation in Photosystem II. Acc. Chem. Res. 2009, 42, 1871–1880. 10.1021/ar900117k. [DOI] [PubMed] [Google Scholar]
- Askerka M.; Brudvig G. W.; Batista V. S. The O2-Evolving Complex of Photosystem II: Recent Insights from Quantum Mechanics/Molecular Mechanics (QM/MM), Extended X-ray Absorption Fine Structure (EXAFS), and Femtosecond X-ray Crystallography Data. Acc. Chem. Res. 2017, 50, 41–48. 10.1021/acs.accounts.6b00405. [DOI] [PubMed] [Google Scholar]
- Suga M.; Akita F.; Sugahara M.; Kubo M.; Nakajima Y.; Nakane T.; Yamashita K.; Umena Y.; Nakabayashi M.; Yamane T.; et al. Light-Induced Structural Changes and the Site of O=O Bond Formation in PSII Caught by XFEL. Nature 2017, 543, 131–135. 10.1038/nature21400. [DOI] [PubMed] [Google Scholar]
- Naruta Y.; Sasayama M.-a.; Sasaki T. Oxygen Evolution by Oxidation of Water with Manganese Porphyrin Dimers. Angew. Chem., Int. Ed. Engl. 1994, 33, 1839–1841. 10.1002/anie.199418391. [DOI] [Google Scholar]
- Gao Y.; Åkermark T.; Liu J.; Sun L.; Åkermark B. Nucleophilic Attack of Hydroxide on a MnV Oxo Complex: A Model of the O–O Bond Formation in the Oxygen Evolving Complex of Photosystem II. J. Am. Chem. Soc. 2009, 131, 8726–8727. 10.1021/ja901139r. [DOI] [PubMed] [Google Scholar]
- Kim S. H.; Park H.; Seo M. S.; Kubo M.; Ogura T.; Klajn J.; Gryko D. T.; Valentine J. S.; Nam W. Reversible O–O Bond Cleavage and Formation between Mn(IV)-Peroxo and Mn(V)-Oxo Corroles. J. Am. Chem. Soc. 2010, 132, 14030–14032. 10.1021/ja1066465. [DOI] [PubMed] [Google Scholar]
- Dogutan D. K.; McGuire R.; Nocera D. G. Electocatalytic Water Oxidation by Cobalt(III) Hangman β-Octafluoro Corroles. J. Am. Chem. Soc. 2011, 133, 9178–9180. 10.1021/ja202138m. [DOI] [PubMed] [Google Scholar]
- Nakazono T.; Parent A. R.; Sakai K. Cobalt Porphyrins as Homogeneous Catalysts for Water Oxidation. Chem. Commun. 2013, 49, 6325–6327. 10.1039/c3cc43031f. [DOI] [PubMed] [Google Scholar]
- Nakazono T.; Parent A. R.; Sakai K. Improving Singlet Oxygen Resistance during Photochemical Water Oxidation by Cobalt Porphyrin Catalysts. Chem. - Eur. J. 2015, 21, 6723–6726. 10.1002/chem.201500716. [DOI] [PubMed] [Google Scholar]
- Wang D.; Groves J. T. Efficient Water Oxidation Catalyzed by Homogeneous Cationic Cobalt Porphyrins with Critical Roles for the Buffer Base. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 15579–15584. 10.1073/pnas.1315383110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artero V.; Fontecave M. Solar Fuels Generation and Molecular Systems: Is It Homogeneous or Heterogeneous Catalysis?. Chem. Soc. Rev. 2013, 42, 2338–2356. 10.1039/C2CS35334B. [DOI] [PubMed] [Google Scholar]
- Han Y.; Wu Y.; Lai W.; Cao R. Electrocatalytic Water Oxidation by a Water-Soluble Nickel Porphyrin Complex at Neutral pH with Low Overpotential. Inorg. Chem. 2015, 54, 5604–5613. 10.1021/acs.inorgchem.5b00924. [DOI] [PubMed] [Google Scholar]
- Coates J. D.; Achenbach L. A. Microbial Perchlorate Reduction: Rocket-Fuelled Metabolism. Nat. Rev. Microbiol. 2004, 2, 569–580. 10.1038/nrmicro926. [DOI] [PubMed] [Google Scholar]
- DuBois J. L.; Ojha S. In Sustaining Life on Planet Earth: Metalloenzymes Mastering Dioxygen and Other Chewy Gases; Kroneck P. M. H., Sosa Torres M. E., Eds.; Springer International Publishing: Cham, 2015. [PubMed] [Google Scholar]
- Schaffner I.; Hofbauer S.; Krutzler M.; Pirker K. F.; Furtmüller P. G.; Obinger C. Mechanism of Chlorite Degradation to Chloride and Dioxygen by the Enzyme Chlorite Dismutase. Arch. Biochem. Biophys. 2015, 574, 18–26. 10.1016/j.abb.2015.02.031. [DOI] [PubMed] [Google Scholar]
- Lee A. Q.; Streit B. R.; Zdilla M. J.; Abu-Omar M. M.; DuBois J. L. Mechanism of and Exquisite Selectivity for O–O Bond Formation by the Heme-Dependent Chlorite Dismutase. Proc. Natl. Acad. Sci. U. S. A. 2008, 105, 15654–15659. 10.1073/pnas.0804279105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayfield J. A.; Blanc B.; Rodgers K. R.; Lukat-Rodgers G. S.; DuBois J. L. Peroxidase-Type Reactions Suggest a Heterolytic/Nucleophilic O–O Joining Mechanism in the Heme-Dependent Chlorite Dismutase. Biochemistry 2013, 52, 6982–6994. 10.1021/bi4005599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celis A. I.; Geeraerts Z.; Ngmenterebo D.; Machovina M. M.; Kurker R. C.; Rajakumar K.; Ivancich A.; Rodgers K. R.; Lukat-Rodgers G. S.; DuBois J. L. A Dimeric Chlorite Dismutase Exhibits O2-Generating Activity and Acts as a Chlorite Antioxidant in Klebsiella pneumoniae MGH 78578. Biochemistry 2015, 54, 434–446. 10.1021/bi501184c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofbauer S.; Gruber C.; Pirker K. F.; Sündermann A.; Schaffner I.; Jakopitsch C.; Oostenbrink C.; Furtmüller P. G.; Obinger C. Transiently Produced Hypochlorite Is Responsible for the Irreversible Inhibition of Chlorite Dismutase. Biochemistry 2014, 53, 3145–3157. 10.1021/bi500401k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sündermann A.; Reif M. M.; Hofbauer S.; Obinger C.; Oostenbrink C. Investigation of Ion Binding in Chlorite Dismutases by Means of Molecular Dynamics Simulations. Biochemistry 2014, 53, 4869–4879. 10.1021/bi500467h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaffner I.; Mlynek G.; Flego N.; Pühringer D.; Libiseller-Egger J.; Coates L.; Hofbauer S.; Bellei M.; Furtmüller P. G.; Battistuzzi G.; et al. Molecular Mechanism of Enzymatic Chlorite Detoxification: Insights from Structural and Kinetic Studies. ACS Catal. 2017, 7, 7962–7976. 10.1021/acscatal.7b01749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slaughter L. M.; Collman J. P.; Eberspacher T. A.; Brauman J. I. Radical Autoxidation and Autogenous O2 Evolution in Manganese–Porphyrin Catalyzed Alkane Oxidations with Chlorite. Inorg. Chem. 2004, 43, 5198–5204. 10.1021/ic049922j. [DOI] [PubMed] [Google Scholar]
- Zdilla M. J.; Lee A. Q.; Abu-Omar M. M. Concerted Dismutation of Chlorite Ion: Water-Soluble Iron-Porphyrins As First Generation Model Complexes for Chlorite Dismutase. Inorg. Chem. 2009, 48, 2260–2268. 10.1021/ic801681n. [DOI] [PubMed] [Google Scholar]
- Umile T. P.; Groves J. T. Catalytic Generation of Chlorine Dioxide from Chlorite Using a Water-Soluble Manganese Porphyrin. Angew. Chem., Int. Ed. 2011, 50, 695–698. 10.1002/anie.201004482. [DOI] [PubMed] [Google Scholar]
- Umile T. P.; Wang D.; Groves J. T. Dissection of the Mechanism of Manganese Porphyrin-Catalyzed Chlorine Dioxide Generation. Inorg. Chem. 2011, 50, 10353–10362. 10.1021/ic201430v. [DOI] [PubMed] [Google Scholar]
- Hicks S. D.; Petersen J. L.; Bougher C. J.; Abu-Omar M. M. Chlorite Dismutation to Chlorine Dioxide Catalyzed by a Water-Soluble Manganese Porphyrin. Angew. Chem., Int. Ed. 2011, 50, 699–702. 10.1002/anie.201005128. [DOI] [PubMed] [Google Scholar]
- Hicks S. D.; Xiong S.; Bougher C. J.; Medvedev G. A.; Caruthers J.; Abu-Omar M. M. Mechanistic Study of a Manganese Porphyrin Catalyst for On-Demand Production of Chlorine Dioxide in Water. J. Porphyrins Phthalocyanines 2015, 19, 492–499. 10.1142/S1088424615500376. [DOI] [Google Scholar]
- de Montellano P. R. O. In Biocatalysis Based on Heme Peroxidases: Peroxidases as Potential Industrial Biocatalysts; Torres E., Ayala M., Eds.; Springer: Berlin, 2010. [Google Scholar]
- Raven E. L. In Encyclopedia of Biophysics; Roberts G. C. K., Ed.; Springer: Berlin, 2013. [Google Scholar]
- Hofrichter M.; Kellner H.; Pecyna M. J.; Ullrich R. In Monooxygenase, Peroxidase and Peroxygenase Properties and Mechanisms of Cytochrome P450; Hrycay E. G., Bandiera S. M., Eds.; Springer International Publishing: Cham, 2015. [Google Scholar]
- Kwon H.; Moody P. C. E.; Raven E. L. In Heme Peroxidases; The Royal Society of Chemistry: Cambridge, UK, 2016. [Google Scholar]
- Macdonald I. K.; Badyal S. K.; Ghamsari L.; Moody P. C. E.; Raven E. L. Interaction of Ascorbate Peroxidase with Substrates: A Mechanistic and Structural Analysis. Biochemistry 2006, 45, 7808–7817. 10.1021/bi0606849. [DOI] [PubMed] [Google Scholar]
- Efimov I.; Badyal S. K.; Metcalfe C. L.; Macdonald I.; Gumiero A.; Raven E. L.; Moody P. C. E. Proton Delivery to Ferryl Heme in a Heme Peroxidase: Enzymatic Use of the Grotthuss Mechanism. J. Am. Chem. Soc. 2011, 133, 15376–15383. 10.1021/ja2007017. [DOI] [PubMed] [Google Scholar]
- Guallar V. Heme Electron Transfer in Peroxidases: The Propionate e-Pathway. J. Phys. Chem. B 2008, 112, 13460–13464. 10.1021/jp806435d. [DOI] [PubMed] [Google Scholar]
- Ruiz-Duenas F. J.; Pogni R.; Morales M.; Giansanti S.; Mate M. J.; Romero A.; Martinez M. J.; Basosi R.; Martinez A. T. Protein Radicals in Fungal Versatile Peroxidase: Catalytic Tryptophan Radical in Both Compound I and Compound II and Studies on W164Y, W164H, and W164S Variants. J. Biol. Chem. 2009, 284, 7986–7994. 10.1074/jbc.M808069200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray H. B.; Winkler J. R. Long-Range Electron Transfer. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 3534–3539. 10.1073/pnas.0408029102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sáez-Jiménez V.; Baratto M. C.; Pogni R.; Rencoret J.; Gutiérrez A.; Santos J. I.; Martínez A. T.; Ruiz-Dueñas F. J. Demonstration of Lignin-To-Peroxidase Direct Electron Transfer: a Transient-State Kinetics, Directed Mutagenesis, EPR, and NMR Study. J. Biol. Chem. 2015, 290, 23201–23213. 10.1074/jbc.M115.665919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey P. J.; Palmer J. M. Oxidation of Phenolic Compounds by Ligninase. J. Biotechnol. 1990, 13, 169–179. 10.1016/0168-1656(90)90102-H. [DOI] [Google Scholar]
- Tsai A.-L.; Kulmacz R. J. Prostaglandin H Synthase: Resolved and Unresolved Mechanistic Issues. Arch. Biochem. Biophys. 2010, 493, 103–124. 10.1016/j.abb.2009.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouzer C. A.; Marnett L. J. Mechanism of Free Radical Oxygenation of Polyunsaturated Fatty Acids by Cyclooxygenases. Chem. Rev. 2003, 103, 2239–2304. 10.1021/cr000068x. [DOI] [PubMed] [Google Scholar]
- Dailey H. A.; Gerdes S.; Dailey T. A.; Burch J. S.; Phillips J. D. Noncanonical Coproporphyrin-Dependent Bacterial Heme Biosynthesis Pathway That Does Not Use Protoporphyrin. Proc. Natl. Acad. Sci. U. S. A. 2015, 112, 2210–2215. 10.1073/pnas.1416285112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Celis A. I.; Gauss G. H.; Streit B. R.; Shisler K.; Moraski G. C.; Rodgers K. R.; Lukat-Rodgers G. S.; Peters J. W.; DuBois J. L. Structure-Based Mechanism for Oxidative Decarboxylation Reactions Mediated by Amino Acids and Heme Propionates in Coproheme Decarboxylase (HemQ). J. Am. Chem. Soc. 2017, 139, 1900–1911. 10.1021/jacs.6b11324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Streit B. R.; Celis A. I.; Shisler K.; Rodgers K. R.; Lukat-Rodgers G. S.; DuBois J. L. Reactions of Ferrous Coproheme Decarboxylase (HemQ) with O2 and H2O2 Yield Ferric Heme b. Biochemistry 2017, 56, 189–201. 10.1021/acs.biochem.6b00958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreou A.; Brodhun F.; Feussner I. Biosynthesis of Oxylipins in Non-Mammals. Prog. Lipid Res. 2009, 48, 148–170. 10.1016/j.plipres.2009.02.002. [DOI] [PubMed] [Google Scholar]
- Brash A. R. Mechanistic Aspects of CYP74 Allene Oxide Synthases and Related Cytochrome P450 Enzymes. Phytochemistry 2009, 70, 1522–1531. 10.1016/j.phytochem.2009.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahn U.; Galano J.-M.; Durand T. Beyond Prostaglandins—Chemistry and Biology of Cyclic Oxygenated Metabolites Formed by Free-Radical Pathways from Polyunsaturated Fatty Acids. Angew. Chem., Int. Ed. 2008, 47, 5894–5955. 10.1002/anie.200705122. [DOI] [PubMed] [Google Scholar]
- Das S.; Chandrasekhar S.; Yadav J. S.; Grée R. Recent Developments in the Synthesis of Prostaglandins and Analogues. Chem. Rev. 2007, 107, 3286–3337. 10.1021/cr068365a. [DOI] [PubMed] [Google Scholar]
- Ullrich V.; Brugger R. Prostacyclin and Thromboxane Synthase: New Aspects of Hemethiolate Catalysis. Angew. Chem., Int. Ed. Engl. 1994, 33, 1911–1919. 10.1002/anie.199419111. [DOI] [Google Scholar]
- Smith W. L.; Urade Y.; Jakobsson P.-J. Enzymes of the Cyclooxygenase Pathways of Prostanoid Biosynthesis. Chem. Rev. 2011, 111, 5821–5865. 10.1021/cr2002992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Audran G.; Brémond P.; Marque S. R. A.; Siri D.; Santelli M. Energetics of the Biosynthesis of Prostanes from Arachidonate. Tetrahedron 2015, 71, 6920–6927. 10.1016/j.tet.2015.07.015. [DOI] [Google Scholar]
- Grechkin A. N. Hydroperoxide Lyase and Divinyl Ether Synthase. Prostaglandins Other Lipid Mediators 2002, 68–69, 457–470. 10.1016/S0090-6980(02)00048-5. [DOI] [PubMed] [Google Scholar]
- Hughes R. K.; De Domenico S.; Santino A. Plant Cytochrome CYP74 Family: Biochemical Features, Endocellular Localisation, Activation Mechanism in Plant Defence and Improvements for Industrial Applications. ChemBioChem 2009, 10, 1122–1133. 10.1002/cbic.200800633. [DOI] [PubMed] [Google Scholar]
- Lee D.-S.; Nioche P.; Hamberg M.; Raman C. S. Structural Insights into the Evolutionary Paths of Oxylipin Biosynthetic Enzymes. Nature 2008, 455, 363–368. 10.1038/nature07307. [DOI] [PubMed] [Google Scholar]
- Oldham M. L.; Brash A. R.; Newcomer M. E. The Structure of Coral Allene Oxide Synthase Reveals a Catalase Adapted for Metabolism of a Fatty Acid Hydroperoxide. Proc. Natl. Acad. Sci. U. S. A. 2005, 102, 297–302. 10.1073/pnas.0406352102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mashhadi Z.; Newcomer M. E.; Brash A. R. The Thr–His Connection on the Distal Heme of Catalase-Related Hemoproteins: A Hallmark of Reaction with Fatty Acid Hydroperoxides. ChemBioChem 2016, 17, 2000–2006. 10.1002/cbic.201600345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brash A. R.; Hughes M. A.; Hawkins D. J.; Boeglin W. E.; Song W. C.; Meijer L. Allene Oxide and Aldehyde Biosynthesis in Starfish Oocytes. J. Biol. Chem. 1991, 266, 22926–22931. [PubMed] [Google Scholar]
- Gao B.; Boeglin W. E.; Zheng Y.; Schneider C.; Brash A. R. Evidence for an Ionic Intermediate in the Transformation of Fatty Acid Hydroperoxide by a Catalase-related Allene Oxide Synthase from the Cyanobacterium Acaryochloris marina. J. Biol. Chem. 2009, 284, 22087–22098. 10.1074/jbc.M109.013151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teder T.; Lohelaid H.; Boeglin W. E.; Calcutt W. M.; Brash A. R.; Samel N. A Catalase-Related Hemoprotein in Coral Is Specialized for Synthesis of Short-Chain Aldehydes: Discovery of P450-Type Hydroperoxide Lyase Activity in a Catalase. J. Biol. Chem. 2015, 290, 19823–19832. 10.1074/jbc.M115.660282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brash A. R.; Niraula N. P.; Boeglin W. E.; Mashhadi Z. An Ancient Relative of Cyclooxygenase in Cyanobacteria Is a Linoleate 10S-Dioxygenase That Works in Tandem with a Catalase-related Protein with Specific 10S-Hydroperoxide Lyase Activity. J. Biol. Chem. 2014, 289, 13101–13111. 10.1074/jbc.M114.555904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung R.; Lee S.; Bennett J. W. Fungal Volatile Organic Compounds and Their Role in Ecosystems. Appl. Microbiol. Biotechnol. 2015, 99, 3395–3405. 10.1007/s00253-015-6494-4. [DOI] [PubMed] [Google Scholar]
- Schneider C.; Niisuke K.; Boeglin W. E.; Voehler M.; Stec D. F.; Porter N. A.; Brash A. R. Enzymatic Synthesis of a Bicyclobutane Fatty Acid by a Hemoprotein–Lipoxygenase Fusion Protein from the Cyanobacterium Anabaena PCC 7120. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 18941–18945. 10.1073/pnas.0707148104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plate L.; Marletta M. A. Nitric Oxide-Sensing H-NOX Proteins Govern Bacterial Communal Behavior. Trends Biochem. Sci. 2013, 38, 566–575. 10.1016/j.tibs.2013.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cary S. P. L.; Winger J. A.; Derbyshire E. R.; Marletta M. A. Nitric Oxide Signaling: No Longer Simply on or Off. Trends Biochem. Sci. 2006, 31, 231–239. 10.1016/j.tibs.2006.02.003. [DOI] [PubMed] [Google Scholar]
- Shimizu T.; Huang D.; Yan F.; Stranava M.; Bartosova M.; Fojtíková V.; Martínková M. Gaseous O2, NO, and CO in Signal Transduction: Structure and Function Relationships of Heme-Based Gas Sensors and Heme-Redox Sensors. Chem. Rev. 2015, 115, 6491–6533. 10.1021/acs.chemrev.5b00018. [DOI] [PubMed] [Google Scholar]
- Millett E. S.; Efimov I.; Basran J.; Handa S.; Mowat C. G.; Raven E. L. Heme-Containing Dioxygenases Involved in Tryptophan Oxidation. Curr. Opin. Chem. Biol. 2012, 16, 60–66. 10.1016/j.cbpa.2012.01.014. [DOI] [PubMed] [Google Scholar]
- Geng J.; Liu A. Heme-Dependent Dioxygenases in Tryptophan Oxidation. Arch. Biochem. Biophys. 2014, 544, 18–26. 10.1016/j.abb.2013.11.009. [DOI] [PubMed] [Google Scholar]
- Hayaishi O.; Rothberg S.; Mehler A. H.; Saito Y. Studies on Oxygenases; Enzymatic Formation of Kynurenine from Tryptophan. J. Biol. Chem. 1957, 229, 889–896. [PubMed] [Google Scholar]
- Yamamoto S.; Hayaishi O. Tryptophan Pyrrolase of Rabbit Intestine. D- and L-Tryptophan-Cleaving Enzyme or Enzymes. J. Biol. Chem. 1967, 242, 5260–5266. [PubMed] [Google Scholar]
- Hirata F.; Hayaishi O.; Tokuyama T.; Senoh S. In Vitro and in vivo Formation of Two New Metabolites of Melatonin. J. Biol. Chem. 1974, 249, 1311–1313. [PubMed] [Google Scholar]
- Mellor A. L.; Munn D. H. Ido Expression by Dendritic Cells: Tolerance and Tryptophan Catabolism. Nat. Rev. Immunol. 2004, 4, 762–774. 10.1038/nri1457. [DOI] [PubMed] [Google Scholar]
- Prendergast G. C. Immune Escape as a Fundamental Trait of Cancer: Focus on Ido. Oncogene 2008, 27, 3889–3900. 10.1038/onc.2008.35. [DOI] [PubMed] [Google Scholar]
- Efimov I.; Basran J.; Thackray S. J.; Handa S.; Mowat C. G.; Raven E. L. Structure and Reaction Mechanism in the Heme Dioxygenases. Biochemistry 2011, 50, 2717–2724. 10.1021/bi101732n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chauhan N.; Thackray S. J.; Rafice S. A.; Eaton G.; Lee M.; Efimov I.; Basran J.; Jenkins P. R.; Mowat C. G.; Chapman S. K.; et al. Reassessment of the Reaction Mechanism in the Heme Dioxygenases. J. Am. Chem. Soc. 2009, 131, 4186–4187. 10.1021/ja808326g. [DOI] [PubMed] [Google Scholar]
- Geng J.; Dornevil K.; Liu A. Chemical Rescue of the Distal Histidine Mutants of Tryptophan 2,3-Dioxygenase. J. Am. Chem. Soc. 2012, 134, 12209–12218. 10.1021/ja304164b. [DOI] [PubMed] [Google Scholar]
- Lu C.; Lin Y.; Yeh S.-R. Inhibitory Substrate Binding Site of Human Indoleamine 2,3-Dioxygenase. J. Am. Chem. Soc. 2009, 131, 12866–12867. 10.1021/ja9029768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung L. W.; Li X.; Sugimoto H.; Shiro Y.; Morokuma K. Density Functional Theory Study on a Missing Piece in Understanding of Heme Chemistry: The Reaction Mechanism for Indoleamine 2,3-Dioxygenase and Tryptophan 2,3-Dioxygenase. J. Am. Chem. Soc. 2008, 130, 12299–12309. 10.1021/ja803107w. [DOI] [PubMed] [Google Scholar]
- Lewis-Ballester A.; Batabyal D.; Egawa T.; Lu C.; Lin Y.; Marti M. A.; Capece L.; Estrin D. A.; Yeh S.-R. Evidence for a Ferryl Intermediate in a Heme-Based Dioxygenase. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 17371–17376. 10.1073/pnas.0906655106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booth E. S.; Basran J.; Lee M.; Handa S.; Raven E. L. Substrate Oxidation by Indoleamine 2,3-Dioxygenase: EVIDENCE FOR A COMMON REACTION MECHANISM. J. Biol. Chem. 2015, 290, 30924–30930. 10.1074/jbc.M115.695684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sachiko Y.; Keiko Y.; Yumi H.; Masaki H.; Hiroshi S.; Yoshitsugu S.; Appelman E. H.; Takashi O.. Identification of the Fe–O2 and the Fe=O Heme Species for Indoleamine 2,3-Dioxygenase during Catalytic Turnover. Chem. Lett. 2010, 39, 36–37 10.1246/cl.2010.36. [DOI] [Google Scholar]
- Freewan M.; Rees M. D.; Plaza T. S.; Glaros E.; Lim Y. J.; Wang X. S.; Yeung A. W.; Witting P. K.; Terentis A. C.; Thomas S. R. Human Indoleamine 2,3-Dioxygenase Is a Catalyst of Physiological Heme Peroxidase Reactions: Implications for the Inhibition of Dioxygenase Activity by Hydrogen Peroxide. J. Biol. Chem. 2013, 288, 1548–1567. 10.1074/jbc.M112.410993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu C.; Yeh S.-R. Ferryl Derivatives of Human Indoleamine 2,3-Dioxygenase. J. Biol. Chem. 2011, 286, 21220–21230. 10.1074/jbc.M111.221507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu R.; Gupta R.; Geng J.; Dornevil K.; Wang S.; Zhang Y.; Hendrich M. P.; Liu A. Enzyme Reactivation by Hydrogen Peroxide in Heme-based Tryptophan Dioxygenase. J. Biol. Chem. 2011, 286, 26541–26554. 10.1074/jbc.M111.253237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basran J.; Booth E. S.; Lee M.; Handa S.; Raven E. L. Analysis of Reaction Intermediates in Tryptophan 2,3-Dioxygenase: A Comparison with Indoleamine 2,3-Dioxygenase. Biochemistry 2016, 55, 6743–6750. 10.1021/acs.biochem.6b01005. [DOI] [PubMed] [Google Scholar]
- Gardner P. R.; Gardner A. M.; Martin L. A.; Salzman A. L. Nitric Oxide Dioxygenase: an Enzymic Function for Flavohemoglobin. Proc. Natl. Acad. Sci. U. S. A. 1998, 95, 10378–10383. 10.1073/pnas.95.18.10378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner P. R. Nitric Oxide Dioxygenase Function and Mechanism of Flavohemoglobin, Hemoglobin, Myoglobin and Their Associated Reductases. J. Inorg. Biochem. 2005, 99, 247–266. 10.1016/j.jinorgbio.2004.10.003. [DOI] [PubMed] [Google Scholar]
- Su J.; Groves J. T. Mechanisms of Peroxynitrite Interactions with Heme Proteins. Inorg. Chem. 2010, 49, 6317–6329. 10.1021/ic902157z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su J.; Groves J. T. Direct Detection of the Oxygen Rebound Intermediates, Ferryl Mb and NO2, in the Reaction of metMyoglobin with Peroxynitrite. J. Am. Chem. Soc. 2009, 131, 12979–12988. 10.1021/ja902473r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barry S. M.; Kers J. A.; Johnson E. G.; Song L.; Aston P. R.; Patel B.; Krasnoff S. B.; Crane B. R.; Gibson D. M.; Loria R.; et al. Cytochrome P450–Catalyzed L-Tryptophan Nitration in Thaxtomin Phytotoxin Biosynthesis. Nat. Chem. Biol. 2012, 8, 814–816. 10.1038/nchembio.1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dodani S. C.; Kiss G.; Cahn J. K. B.; Su Y.; Pande V. S.; Arnold F. H. Discovery of a Regioselectivity Switch in Nitrating P450s Guided by Molecular Dynamics Simulations and Markov Models. Nat. Chem. 2016, 8, 419–425. 10.1038/nchem.2474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshikawa S.; Shimada A. Reaction Mechanism of Cytochrome c Oxidase. Chem. Rev. 2015, 115, 1936–1989. 10.1021/cr500266a. [DOI] [PubMed] [Google Scholar]
- Yoshikawa S.; Shinzawa-Itoh K.; Nakashima R.; Yaono R.; Yamashita E.; Inoue N.; Yao M.; Fei M. J.; Libeu C. P.; Mizushima T.; et al. Redox-Coupled Crystal Structural Changes in Bovine Heart Cytochrome c Oxidase. Science 1998, 280, 1723–1729. 10.1126/science.280.5370.1723. [DOI] [PubMed] [Google Scholar]
- Ostermeier C.; Harrenga A.; Ermler U.; Michel H. Structure at 2.7 Å Resolution of the Paracoccus Denitrificans Two-Subunit Cytochrome C Oxidase Complexed with an Antibody Fv Fragment. Proc. Natl. Acad. Sci. U. S. A. 1997, 94, 10547–10553. 10.1073/pnas.94.20.10547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyons J. A.; Aragao D.; Slattery O.; Pisliakov A. V.; Soulimane T.; Caffrey M. Structural Insights into Electron Transfer in caa3-Type Cytochrome Oxidase. Nature 2012, 487, 514–518. 10.1038/nature11182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blomberg M. R. A.; Siegbahn P. E. M. How Cytochrome c Oxidase Can Pump Four Protons per Oxygen Molecule at High Electrochemical Gradient. Biochim. Biophys. Acta, Bioenerg. 2015, 1847, 364–376. 10.1016/j.bbabio.2014.12.005. [DOI] [PubMed] [Google Scholar]
- Sharma V.; Wikström M. The Role of the K-Channel and the Active-Site Tyrosine in the Catalytic Mechanism of Cytochrome c Oxidase. Biochim. Biophys. Acta, Bioenerg. 2016, 1857, 1111–1115. 10.1016/j.bbabio.2016.02.008. [DOI] [PubMed] [Google Scholar]
- Muramoto K.; Ohta K.; Shinzawa-Itoh K.; Kanda K.; Taniguchi M.; Nabekura H.; Yamashita E.; Tsukihara T.; Yoshikawa S. Bovine Cytochrome c Oxidase Structures Enable O2 Reduction with Minimization of Reactive Oxygens and Provide a Proton-Pumping Gate. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 7740–7745. 10.1073/pnas.0910410107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirota S.; Ogura T.; Shinzawa-Itoh K.; Yoshikawa S.; Kitagawa T. Observation of Multiple CN-Isotope-Sensitive Raman Bands for CN- Adducts of Hemoglobin, Myoglobin, and Cytochrome c Oxidase: Evidence for Vibrational Coupling between the Fe–C–N Bending and Porphyrin In-Plane Modes. J. Phys. Chem. 1996, 100, 15274–15279. 10.1021/jp953190m. [DOI] [Google Scholar]
- Pinakoulaki E.; Vamvouka M.; Varotsis C. Resonance Raman Detection of the Fe2+–C–N Modes in Heme–Copper Oxidases: A Probe of the Active Site. Inorg. Chem. 2004, 43, 4907–4910. 10.1021/ic035216r. [DOI] [PubMed] [Google Scholar]
- Pinakoulaki E.; Vamvouka M.; Varotsis C. The Active Site Structure of Heme a33+-C⋮N-CuB2+ of Cytochrome aa3 Oxidase as Revealed from Resonance Raman Scattering. J. Phys. Chem. B 2003, 107, 9865–9868. 10.1021/jp034326g. [DOI] [Google Scholar]
- Kim Y.; Babcock G. T.; Surerus K. K.; Fee J. A.; Dyer R. B.; Woodruff W. H.; Oertling W. A. Cyanide Binding and Active Site Structure in Heme-Copper Oxidases: Normal Coordinate Analysis of Iron-Cyanide Vibrations of a32+ CN– Complexes of Cytochromes ba3 and aa3. Biospectroscopy 1998, 4, 1–15. . [DOI] [PubMed] [Google Scholar]
- Hematian S.; Garcia-Bosch I.; Karlin K. D. Synthetic Heme/Copper Assemblies: Toward an Understanding of Cytochrome c Oxidase Interactions with Dioxygen and Nitrogen Oxides. Acc. Chem. Res. 2015, 48, 2462–2474. 10.1021/acs.accounts.5b00265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mukherjee S.; Mukherjee A.; Bhagi-Damodaran A.; Mukherjee M.; Lu Y.; Dey A. A Biosynthetic Model of Cytochrome c Oxidase as an Electrocatalyst for Oxygen Reduction. Nat. Commun. 2015, 6, 8467. 10.1038/ncomms9467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halime Z.; Kotani H.; Li Y.; Fukuzumi S.; Karlin K. D. Homogeneous Catalytic O2 Reduction to Water by a Cytochrome c Oxidase Model with Trapping of Intermediates and Mechanistic Insights. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 13990–13994. 10.1073/pnas.1104698108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collman J. P.; Devaraj N. K.; Decréau R. A.; Yang Y.; Yan Y.-L.; Ebina W.; Eberspacher T. A.; Chidsey C. E. D. A Cytochrome c Oxidase Model Catalyzes Oxygen to Water Reduction Under Rate-Limiting Electron Flux. Science 2007, 315, 1565–1568. 10.1126/science.1135844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox S.; Nanthakumar A.; Wikström M.; Karlin K. D.; Blackburn N. J. XAS Structural Comparisons of Reversibly Interconvertible Oxo- and Hydroxo-Bridged Heme-Copper Oxidase Model Compounds. J. Am. Chem. Soc. 1996, 118, 24–34. 10.1021/ja951686b. [DOI] [Google Scholar]
- Collman J. P.; Herrmann P. C.; Boitrel B.; Zhang X.; Eberspacher T. A.; Fu L.; Wang J.; Rousseau D. L.; Williams E. R. Synthetic Analog for the Oxygen Binding Site in Cytochrome c Oxidase. J. Am. Chem. Soc. 1994, 116, 9783–9784. 10.1021/ja00100a067. [DOI] [Google Scholar]
- Chufán E. E.; Puiu S. C.; Karlin K. D. Heme–Copper/Dioxygen Adduct Formation, Properties, and Reactivity. Acc. Chem. Res. 2007, 40, 563–572. 10.1021/ar700031t. [DOI] [PubMed] [Google Scholar]
- Chufán E. E.; Karlin K. D. An Iron–Peroxo Porphyrin Complex: New Synthesis and Reactivity Toward a Cu(II) Complex Giving a Heme–Peroxo–Copper Adduct. J. Am. Chem. Soc. 2003, 125, 16160–16161. 10.1021/ja036632d. [DOI] [PubMed] [Google Scholar]
- del Río D.; Sarangi R.; Chufán E. E.; Karlin K. D.; Hedman B.; Hodgson K. O.; Solomon E. I. Geometric and Electronic Structure of the Heme–Peroxo–Copper Complex [(F8TPP)FeIII–(O2)–CuII(TMPA)](ClO4). J. Am. Chem. Soc. 2005, 127, 11969–11978. 10.1021/ja043374r. [DOI] [PubMed] [Google Scholar]
- Chishiro T.; Shimazaki Y.; Tani F.; Tachi Y.; Naruta Y.; Karasawa S.; Hayami S.; Maeda Y. Isolation and Crystal Structure of a Peroxo-Bridged Heme–Copper Complex. Angew. Chem. 2003, 115, 2894–2897. 10.1002/ange.200351415. [DOI] [PubMed] [Google Scholar]
- Halime Z.; Kieber-Emmons M. T.; Qayyum M. F.; Mondal B.; Gandhi T.; Puiu S. C.; Chufán E. E.; Sarjeant A. A. N.; Hodgson K. O.; Hedman B.; et al. Heme–Copper–Dioxygen Complexes: Toward Understanding Ligand-Environmental Effects on the Coordination Geometry, Electronic Structure, and Reactivity. Inorg. Chem. 2010, 49, 3629–3645. 10.1021/ic9020993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chufán E. E.; Mondal B.; Gandhi T.; Kim E.; Rubie N. D.; Moënne-Loccoz P.; Karlin K. D. Reactivity Studies on FeIII–(O2)–CuII Compounds: Influence of the Ligand Architecture and Copper Ligand Denticity. Inorg. Chem. 2007, 46, 6382–6394. 10.1021/ic700363k. [DOI] [PubMed] [Google Scholar]
- Kim E.; Helton M. E.; Wasser I. M.; Karlin K. D.; Lu S.; Huang H.-w.; Moënne-Loccoz P.; Incarvito C. D.; Rheingold A. L.; Honecker M.; et al. Superoxo, μ-Peroxo, and μ-Oxo Complexes from Heme/O2 and Heme-Cu/O2 Reactivity: Copper Ligand Influences in Cytochrome c Oxidase Models. Proc. Natl. Acad. Sci. U. S. A. 2003, 100, 3623–3628. 10.1073/pnas.0737180100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kieber-Emmons M. T.; Li Y.; Halime Z.; Karlin K. D.; Solomon E. I. Electronic Structure of a Low-Spin Heme/Cu Peroxide Complex: Spin-State and Spin-Topology Contributions to Reactivity. Inorg. Chem. 2011, 50, 11777–11786. 10.1021/ic2018727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kieber-Emmons M. T.; Qayyum M. F.; Li Y.; Halime Z.; Hodgson K. O.; Hedman B.; Karlin K. D.; Solomon E. I. Spectroscopic Elucidation of a New Heme/Copper Dioxygen Structure Type: Implications for O–O Bond Rupture in Cytochrome c Oxidase. Angew. Chem., Int. Ed. 2012, 51, 168–172. 10.1002/anie.201104080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Bosch I.; Adam S. M.; Schaefer A. W.; Sharma S. K.; Peterson R. L.; Solomon E. I.; Karlin K. D. A “Naked” FeIII-(O2)-CuII Species Allows for Structural and Spectroscopic Tuning of Low-Spin Heme-Peroxo-Cu Complexes. J. Am. Chem. Soc. 2015, 137, 1032–1035. 10.1021/ja5115198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collman J. P.; Sunderland C. J.; Berg K. E.; Vance M. A.; Solomon E. I. Spectroscopic Evidence for a Heme–Superoxide/Cu(I) Intermediate in a Functional Model of Cytochrome c Oxidase. J. Am. Chem. Soc. 2003, 125, 6648–6649. 10.1021/ja034382v. [DOI] [PubMed] [Google Scholar]
- Liu J.-G.; Naruta Y.; Tani F. A Functional Model of the Cytochrome c Oxidase Active Site: Unique Conversion of a Heme−μ-peroxo–CuII Intermediate into Heme– superoxo/CuI. Angew. Chem., Int. Ed. 2005, 44, 1836–1840. 10.1002/anie.200462582. [DOI] [PubMed] [Google Scholar]
- Collman J. P.; Decreau R. A.; Sunderland C. J. Single-Turnover Intermolecular Reaction Between a Fe-Superoxide-Cu Cytochrome C Oxidase Model and Exogeneous Tyr244 Mimics. Chem. Commun. 2006, 3894–3896. 10.1039/B607277A. [DOI] [PubMed] [Google Scholar]
- Collman J. P.; Decréau R. A.; Yan Y.; Yoon J.; Solomon E. I. Intramolecular Single-Turnover Reaction in a Cytochrome c Oxidase Model Bearing a Tyr244 Mimic. J. Am. Chem. Soc. 2007, 129, 5794–5795. 10.1021/ja0690969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adam S. M.; Garcia-Bosch I.; Schaefer A. W.; Sharma S. K.; Siegler M. A.; Solomon E. I.; Karlin K. D. Critical Aspects of Heme–Peroxo–Cu Complex Structure and Nature of Proton Source Dictate Metal–Operoxo Breakage versus Reductive O–O Cleavage Chemistry. J. Am. Chem. Soc. 2017, 139, 472–481. 10.1021/jacs.6b11322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nastri F.; Chino M.; Maglio O.; Bhagi-Damodaran A.; Lu Y.; Lombardi A. Design and Engineering of Artificial Oxygen-Activating Metalloenzymes. Chem. Soc. Rev. 2016, 45, 5020–5054. 10.1039/C5CS00923E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigman J. A.; Kim H. K.; Zhao X.; Carey J. R.; Lu Y. The Role of Copper and Protons in Heme-Copper Oxidases: Kinetic Study of an Engineered Heme-Copper Center in Myoglobin. Proc. Natl. Acad. Sci. U. S. A. 2003, 100, 3629–3634. 10.1073/pnas.0737308100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X.; Yu Y.; Hu C.; Zhang W.; Lu Y.; Wang J. Significant Increase of Oxidase Activity through the Genetic Incorporation of a Tyrosine–Histidine Cross-Link in a Myoglobin Model of Heme–Copper Oxidase. Angew. Chem., Int. Ed. 2012, 51, 4312–4316. 10.1002/anie.201108756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miner K. D.; Mukherjee A.; Gao Y.-G.; Null E. L.; Petrik I. D.; Zhao X.; Yeung N.; Robinson H.; Lu Y. A Designed Functional Metalloenzyme that Reduces O2 to H2O with Over One Thousand Turnovers. Angew. Chem., Int. Ed. 2012, 51, 5589–5592. 10.1002/anie.201201981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrik I. D.; Davydov R.; Ross M.; Zhao X.; Hoffman B.; Lu Y. Spectroscopic and Crystallographic Evidence for the Role of a Water-Containing H-Bond Network in Oxidase Activity of an Engineered Myoglobin. J. Am. Chem. Soc. 2016, 138, 1134–1137. 10.1021/jacs.5b12004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhagi-Damodaran A.; Michael M. A.; Zhu Q.; Reed J.; Sandoval B. A.; Mirts E. N.; Chakraborty S.; Moënne-Loccoz P.; Zhang Y.; Lu Y. Why Copper is Preferred over Iron for Oxygen Activation and Reduction in Haem-Copper Oxidases. Nat. Chem. 2016, 9, 257–263. 10.1038/nchem.2643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan M.; Lo J. C.; Edwards J. T.; Baran P. S. Radicals: Reactive Intermediates with Translational Potential. J. Am. Chem. Soc. 2016, 138, 12692–12714. 10.1021/jacs.6b08856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prier C. K.; Rankic D. A.; MacMillan D. W. C. Visible Light Photoredox Catalysis with Transition Metal Complexes: Applications in Organic Synthesis. Chem. Rev. 2013, 113, 5322–5363. 10.1021/cr300503r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Studer A.; Curran D. P. Catalysis of Radical Reactions: A Radical Chemistry Perspective. Angew. Chem., Int. Ed. 2016, 55, 58–102. 10.1002/anie.201505090. [DOI] [PubMed] [Google Scholar]
- Renata H.; Wang Z. J.; Arnold F. H. Expanding the Enzyme Universe: Accessing Non-Natural Reactions by Mechanism-Guided Directed Evolution. Angew. Chem., Int. Ed. 2015, 54, 3351–3367. 10.1002/anie.201409470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prier C. K.; Arnold F. H. Chemomimetic Biocatalysis: Exploiting the Synthetic Potential of Cofactor-Dependent Enzymes To Create New Catalysts. J. Am. Chem. Soc. 2015, 137, 13992–14006. 10.1021/jacs.5b09348. [DOI] [PubMed] [Google Scholar]