

Abstract
Since their inception, tetracycline (Tet)-inducible systems have become the method of choice for transgenic research. The Tet-Off systems have a number of advantages, including robust target induction using a relatively benign effector molecule. However, use of the Tet-On system has been fraught with difficulties, including high background expression in the absence of effector molecules and inconsistent gene induction. Recently, secondgeneration Tet-On transactivators (TAs) have been described. In HeLa cells, they are far more efficient than the original reverse TA protein, and they exhibit lower background activity in the absence of effectors. Here we examine the most promising TA in transgenic Drosophila and characterize its in vivo properties. We report that low levels of doxycycline, when added to normal fly food, efficiently and rapidly induce target transgenes in adults, larvae, and embryos. This TA is superior to all other Tet-On proteins, and its performance is comparable to that of the widely used Tet-Off TA. In addition, combining the improved Tet-On TA with the Gal4-UAS (upstream-activating sequence) system produces robust, spatially restricted, temporally controlled transgene induction. Because this Tet-On TA is significantly more efficient than previous ones used in Drosophila, it is also possible to modulate gene induction by controlling the dosage of the antibiotic in the food.
Reverse genetic techniques are essential tools for researchers in the postgenome project era. The ability to express gene products in a temporally restricted manner has been an essential experimental strategy in determining gene function in tissue culture and in vivo, where mutant or knockout studies are often not practical. Currently, the best-characterized and most versatile inducible approaches use the tetracycline (Tet)-mediated expression systems (see ref. 1 for review). These rely on the specific, high-affinity binding of the Escherichia coli Tet repressor protein (TetR), or its derivatives, to the Tet operator (TetO) sequences. When TetR is fused to the herpes simplex virus VP16 activation domain, the hybrid TetR/VP16 protein becomes a powerful Tet-responsive transactivator (tTA). In the absence of Tet or its analogs, the tTA protein binds TetO and initiates transcription from artificial promoters containing these sequences. In the presence of the antibiotic, the TA binds the drug, resulting in a conformational change that disrupts DNA binding and inactivates transcription. The Tet-Off system has been invaluable in addressing many biological problems where a tightly regulated genetic switch is desirable.
A second Tet-regulated system was developed to address biological problems where prolonged exposure to antibiotics is undesirable (such as during development) or to express potentially toxic target genes during very discrete periods of time. A random mutagenesis of the TetR gene produced a protein that bound to TetO sequences only in the presence of the Tet analog doxycycline (dox) (2). Fusion of this mutant to VP16 produced a reverse TA (rtTA), a protein that required drug interaction to bind TetO sequences and activate transcription. This Tet-On TA works well in most tissue culture applications, but is less effective in transgenic animals, where there are only a handful of papers reporting its use (3–6). Although the system works for certain applications in mice, anecdotal reports indicate that it is uneven in its performance. In many of these cases, induction kinetics is slow and the fold induction is modest. It is likely that the reported successes partially result from the determination of particular combinations of transgenes that produce significant induction. This empirical screening process, which is expensive and time consuming, involves analyzing many TA and target transgenic lines, and optimizing their combinatorial effectiveness (1). In Drosophila, the production and analysis of transgenic flies is routine and rapid, but combinatorial analysis is still difficult, albeit not impossible. In mice, this type of analysis becomes prohibitive.
Previously, the best performance of Tet-On in flies (a 10-fold increase in β-galactosidase activity) required 2 days of dox feeding (at 1 mg/ml) and 3 additional days of accumulation (5). This level of induction, although significant, was slow and modest, despite the fact that the rtTA gene was expressed under the control of the strong actin5C promoter. In this case, 13 TA lines and six target lines had to be screened to find combinations that produced this modest level of induction. In addition, many of the combinations of transgenes exhibited high levels of leaky expression (expression in the absence of the effector). Thus it is not surprising that other Drosophila labs have reported that the Tet-On system does not work in their experimental settings, because a requisite number of combinations may not have been screened (7).
Partially in response to the limitations of the first-generation rtTA, a second screen for more effective Tet-On TAs was undertaken. This screen produced a second-generation Tet-On TA, rtTAs-M2, with increased induction capabilities and tighter regulation (8). This TA consists of a second set of mutations that reverse the binding properties of the protein. In addition, several other changes were made to increase expression of the TA protein. When assayed in HeLa cells, the combination of the M2 mutations and the changes to increase its expression result in better induction at lower drug concentrations and less leakiness.
In parallel, we identified and removed a cryptic splicing site in the TetR gene. When TetR is expressed in eukaryotic cells, alternative splicing in the middle of the TetR coding region results in nonproductive mRNA. Presumably, this usage of cryptic splicing sites also occurs when TetR is fused in the tTA and rtTA genes. We also adjusted the codon usage of the TetR moiety to reflect mammalian frequencies, and the rtTA expression cassette was flanked with the Drosophila boundary elements SCS and SCS′ (9, 10). These changes, referred to as altering the rtTA gene, produced the rtTA-alt TA. In this study, the second-generation mutations from the rtTAs-M2 allele are incorporated into our altered rtTA gene, producing the rtTA-M2-alt TA. We use the actin5C promoter to ubiquitously express the rtTA-M2-alt TA in transgenic Drosophila and demonstrate its utility in adult flies, embryos, and larvae. This TA is compared directly to all of the currently available Tet-inducible systems in Drosophila. Finally, we have combined the Tet-On system with the Gal4-upstream-activating sequence (UAS) system to achieve both spatial and temporal control of target transgene expression. This study demonstrates that the rtTAs-M2 mutations, when combined into the rtTA-M2-alt TA, function efficiently in transgenic animals.
Methods
Construction of the rtTA-alt and rtTA-M2-alt Genes and Transgenic Flies.
The original bacterial Tn10 TetR coding sequence was optimized for expression in mammalian cells. The signal sequences that defined a cryptic intron located between amino acids 8 and 144 were altered to block splicing. The overall G + C content of the gene was increased from ≈40% in the wild-type TetR gene to ≈50% in the optimized sequence. Although we increased the overall G + C content, the occurrence of CpG dinucleotides in the optimized sequence was maintained at a low frequency consistent with mammalian coding sequences.
The codon usage of TetR was altered to improve expression in mammalian cells by using the codon frequencies of porcine albumin as a guide and through repeated codon bias analysis with macvector software. The final gene was assembled in pUC8 from four 100- to 160-bp double- stranded oligonucleotides to produce the plasmid pSyntetR. This plasmid was then used as the basis for producing the rtTA-alt genes. Site-directed mutagenesis of pSyntetR was used to introduce the mutations that change the binding properties of the TetR protein. This mutated TetR gene was then fused to the VP16 activation domain, producing a rtTA gene (rtTA-alt). This gene was placed in an expression cassette, downstream of the actin5C promoter, but upstream of a poly(A) addition site. A similar strategy was used to assemble the rtTA-M2-alt gene, except that different oligonucleotides were used for the site-directed mutagenesis and introduction of the M2 mutations.
Expression cassettes consisting of either the rtTA-alt or rtTA-M2-alt genes under the control of the actin5C promoter were subcloned into a miniwhite P-element transformation vector (pBlocker5). The plasmid pJY2005, which contains the rtTA-alt gene, was assembled so that the boundary elements SCS and SCS′ flank the promoter, TA gene, and a poly(A) addition sequence (Fig. 1). The plasmid pJY2006, which contains the rtTA-M2-alt gene, was similarly constructed.
Figure 1.
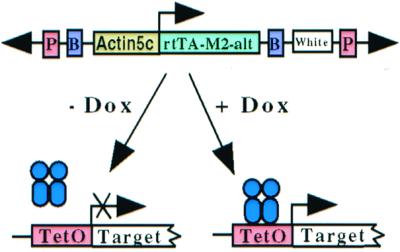
Our Drosophila Tet-On system. Two separate P-element constructs, one using the actin5C promotor to ubiquitously express the rtTA-M2-alt protein, the other carrying a target gene of interest under the control of the Tet response elements (TetO), are combined in a single animal. In the presence of the effector molecule, dox, transcription of the target transgene is activated. In the absence of dox, target transgene remains silent. The miniwhite gene is used to select for transgenic lines. We use the boundary elements SCS and SCS′ (labeled B) to insulate the rtTA-M2-alt expression cassette from position effects.
A target transgene, consisting of seven tandem TetO sequences, the cytomegalovirus minimal promoter, the luciferase cDNA, a chicken β-globin intron, and a poly(A) signal was subcloned into a standard miniwhite P-element transformation vector (pJY2007). To facilitate usage of this system, we also constructed universal acceptor vectors for insertion of desired cDNAs. The uninsulated, universal vector (pJY2004) contains seven tandem TetO sequences, the cytomegalovirus minimal promoter, the unique restriction sites 5′-XhoI and HpaI-3′ for cDNA insertion, the chicken β-globin intron, and a poly(A) signal inside of a standard P-element transformation vector that was derived from pCaSpeR4. These plasmids and detailed information on the other ones used in this paper are available on request. Transgenic flies were made and raised according to standard procedures (11, 12). (For more information on TetO-rpr flies contact B.B.)
Dox Treatment.
A 10× dox (Sigma) concentrate in PBS (pH ≈7.2) was added directly to our standard fly food. Fly food was liquefied in a microwave and allowed to cool to 50°C. The 10× dox concentrate was added directly to the food and mixed, and the food was allowed to resolidify. Because dox oxidizes rapidly in the light, food is stored in the dark for a maximum of 24 h. Flies treated with dox for longer than 24 h were transferred to fresh dox-containing food daily. Larvae treated with dox-supplemented food were kept in the dark with periodic light flashes to minimize oxidation of dox. Larvae were not transferred to fresh dox-containing food. For experiments on embryos, mothers were fed a yeast paste (containing dox, or not) spread on standard agar egg-laying plates. The agar also contained dox, or not.
For injection experiments, ≈10 nl of dox was injected directly into adult flies by using a Tritech Research (Los Angeles) injection setup with pulled glass needles. We adjusted the pH of dox at 10 mg/ml to ≈7 by using 5 M NaOH. Needles were inserted directly into the thorax (at the pterapleura) of anesthetized flies, which were allowed to recover for at least 30 min before time points were taken.
Western Blot Analysis.
The Western blots shown in Fig. 2 were performed with a mAb raised against firefly luciferase protein. Purified luciferase protein was injected into mice, and mAbs were raised by using standard techniques. Equal amounts of protein were separated on standard protein gels, transferred to nitrocellulose, blocked with 4% nonfat dry milk in TBST (Tris-buffered saline with Tween 20), and probed with concentrated luciferase antibody (diluted 1:750 in TBST). The blots were washed and probed with a goat anti-mouse horseradish peroxidase-conjugated secondary antibody before being visualized with standard chemiluminescent techniques. The Western blot in Fig. 3 was performed by using a rabbit polyclonal antibody against the TetR protein (W.H., unpublished work). Twenty to 30 flies were flash-frozen and subsequently homogenized in 8 M urea. Protein concentrations were determined by using the Bio-Rad Protein Assay according to the manufacturer's instructions. Twenty micrograms of protein from each sample was separated by using SDS/PAGE and transferred, and Western blots were performed as described (13). The Western blot in Fig. 3 was repeated and also probed with a polyclonal ADF-1 antibody (14) to confirm that all lanes were loaded and transferred equally (data not shown).
Figure 2.

Analysis of the Tet-On TA (rtTA-M2-alt) in adult flies. (A) Dose curve for target gene induction. Double transgenic flies were maintained on dox-free food (0) or food supplemented with 1, 10, 100, or 1,000 μg/ml of dox for 24 h. Whole fly extracts were analyzed on Western blots using a luciferase mAb. Extracts also were made from control flies carrying two copies of the luciferase target transgene (−), without a TA transgene, and not fed antibiotic. (B) Induction kinetics of flies fed dox at 10 μg/ml. Double transgenic flies were frozen immediately (0), 6, 12, 24, 36, or 48 h after being placed on dox-containing food (10 μg/ml), extracts were prepared, and Western blot analysis was done with the luciferase antibody. Extracts also were made from control flies carrying two copies of the target transgene without a TA (−), and not fed antibiotic. (C) Induction kinetics of flies fed dox at 1 mg/ml. Double transgenic flies were frozen immediately (0), 1, 3, 6, 12, 24, or 48 h after being placed on dox-containing food, extracts were prepared, and Western blot analysis was done with the luciferase antibody. Extracts also were made from control flies carrying two copies of the target transgene without a TA (−), and not fed antibiotic. (D) Induction kinetics of flies injected with dox. Double transgenic flies were injected with ≈1 nl of a 10 mg/ml dox solution, and extracts were prepared 30 min, 1, 3, 6, or 12 h after injection. Extracts also were made from flies injected with PBS (Mock), or single transgenic flies missing the TA (−) and uninjected. Levels of induction: (E) Effect of TA gene dosage on target gene expression levels. Flies carrying one or two copies of the rtTA-M2-alt and two copies of a luciferase target transgene were frozen at 0, 12, or 24 h after being placed on dox-containing food (1 mg/ml). Extracts also were made from control flies containing two copies of the luciferase target transgene without a TA source (−) and not fed antibiotic. Western blot analysis was done with the luciferase antibody. (G) Quantitative analysis using in vitro luciferase assays. Twelve hours after the start of drug feeding, extracts from flies containing one (rtTA-M2-alt/CyO) or two (rtTA-M-alt/rtTA-M2-alt) copies of the TA were assayed for luciferase activity. (F) Estimate of the absolute amount of luciferase protein that is induced. Western blot analysis of extracts using a luciferase-specific mAb. Twenty micrograms of total protein from double transgenic flies fed antibiotic (+Dox), or not (−Dox), was compared with extracts from wild-type flies supplemented with known quantities (1, 2.5, or 5 ng) of purified luciferase protein.
Figure 3.

Comparison of the various TAs. (A) Western blot analysis of TA levels. Equivalent amounts of total extract protein from flies carrying one copy of the various TAs were probed with a TetR-specific antibody. All TAs were expressed from the actin5C promoter. Wild-type flies (−) without any TA and flies containing an unaltered Tet-Off (tTA), unaltered Tet-On (rtTA), altered Tet-On (rtTA-alt), and altered M2 (rtTA-M2-alt) TAs were compared. (B) Comparison of TA activity using in vitro luciferase assays. Double transgenic Tet-On flies were fed dox-containing food (1 mg/ml) for 24 h, then assayed for luciferase activity. Double transgenic Tet-Off flies were maintained on regular food before luciferase measurements. Flies containing the unaltered, original Tet-On TA (rtTA) produced no detectable luciferase induction, and this value was set to one. All other values are relative to this amount of luciferase.
In Vitro Luciferase Assay.
Twenty adult flies were flash-frozen in liquid nitrogen, ground into a powder, and refrozen in the presence of buffer. The samples were thawed and homogenized, and protein concentrations were determined by using the Bio-Rad Dc protein assay. Luciferase assays were performed according to the manufacturer's instructions (Promega, Luciferase Assay Kit). SEM was calculated by using standard equations.
Embryonic Immunocytochemistry.
Embryonic immunocytochemistry was performed as described (14, 15). Concentrated luciferase hybridoma supernatant was diluted 1:50 and preadsorbed against 0- to 18-h wild-type embryos. Embryos were dechorinated, fixed, and incubated with preadsorbed antibody (diluted to a final concentration of 1:250) overnight at 4°C. The secondary antibody was an horseradish peroxidase-conjugated goat anti-mouse diluted to a final concentration of 1:500 (Jackson ImmunoResearch). Ni-diaminobenzidine-peroxidase was used to visualize immune complexes as described (14).
5-Bromo-4-Chloro-3-Indolyl β-d-Galactoside (X-Gal) Staining of Larvae.
Wandering third-instar larvae were collected and washed in PBS before being dissected in 4% paraformaldehyde (PBS). Discs, brains, and various other larval parts were transferred to X-gal staining solution for ≈10 min. Larval parts were washed repeatedly in PBS before being serially dehydrated in EtOH. Larval brains and discs were cleared and mounted in Hemo-De before being imaged.
Results
The Tet-On System in Adult Drosophila.
Fig. 1 is a schematic cartoon of the Tet-On system. The actin5C promoter is used to ubiquitously express the rtTA-M2-alt protein. These transgenic flies are crossed to flies carrying a target transgene whose expression is controlled by seven tandem TetO sequences and a minimal promoter. In this study, we use either the TetO-luciferase or TetO-lacZ reporters as the target transgenes. Boundary (insulator) elements are used to insulate the TA construct from the effects of transcriptional elements flanking the site of transgene insertion.
Dosage.
Dox feeding induces reporter expression in adult flies. Transgenic flies homozygous for both the rtTA-M2-alt and TetO-luciferase transgenes were placed on standard fly food supplemented with varying amounts of dox. After 24 h, equal amounts of total fly protein were analyzed on Western blots by using a luciferase-specific mAb. There was a direct relationship between the dosage of dox fed to the flies and the level of luciferase induction (Fig. 2A). Target gene expression was barely detectable in flies that did not have dox in their diet (Fig. 2A, lane 0). Significant induction was observed with antibiotic concentrations as low as 1 μg/ml, and there was a graded response up to 1,000 μg/ml, demonstrating the practicality of modulating levels of induction by varying the drug concentration in the food. Luciferase expression was undetectable in flies that carried two copies of the target transgene without a TA source (Fig. 2A, lane designated −).
Kinetics of Induction.
The kinetics of target gene induction depends on the concentration of dox in the food. Flies were maintained on dox-containing food (10 μg/ml, Fig. 2B, or 1 mg/ml, Fig. 2C) for varying amounts of time, and equal amounts of total fly protein were analyzed for luciferase expression by using Western blots. When flies were fed dox at 10 μg/ml, luciferase induction was detected within 12 h. The amount of luciferase protein continued to increase until some time point between 36 and 48 h, when expression reached a plateau (data not shown). When flies were fed dox at 1 mg/ml, significant luciferase induction was detectable in 3 h, and this induction peaked sometime between 24 and 48 h. There was no further increase beyond the levels achieved at 48 h (data not shown). Flies maintained on food supplemented with dox at concentrations higher than 1 mg/ml for 48 h appeared slightly sluggish (data not shown) and were not analyzed further.
If the kinetics depends on the concentration of dox in the food, direct injection of the antibiotic into adult flies should greatly accelerate the process, because it bypasses the slow and uncontrolled steps of feeding and metabolism. To test this, we injected dox (10 mg/ml) into the thoracic cavity of flies. Groups of flies were frozen at various times after injection, and equal amounts of protein were analyzed with Western blots (Fig. 2D). Robust luciferase induction occured within 30 min, and there was no further increase at later time points. These data indicate that the kinetics of drug delivery to the fly limits the kinetics of target gene induction and can be optimized by directly injecting the antibiotic into the animal. There was no detectable increase in luciferase expression in flies mock-injected with PBS.
Leakiness.
There was detectable, but low-level, leakiness in the absence of drug. Flies that contained only the target transgene (Fig. 2 A–C, lanes marked −) had undetectable levels of luciferase expression. When the TA transgene was crossed into these flies, but the flies were not fed any antibiotic, a low level of luciferase expression could be detected (Fig. 2 A–C, lanes designated 0). The difference in expression between these flies (Fig. 2 A–C, − versus 0) was defined as the amount of leakiness. The background activity detected was likely to be caused by the low affinity of rtTA-M2 protein for TetO sequences in the absence of effector, but was significantly lower than levels observed with the original rtTA protein. However, when compared with the maximal induced levels, the amount of leaky expression in the rtTA-M2 system was insignificant.
Amount of Induction.
What limits the level of induction?
It is likely that the concentration of the TA/drug complex limits the amount of target gene induction. We compared the effect of TA gene dosage on luciferase induction. Flies heterozygous or homozygous for the TA gene were fed antibiotic (1 mg/ml), and equal amounts of protein were analyzed with Western blots. After both 12 and 24 h of feeding, homozygous flies produced more luciferase than flies with a single copy (Fig. 2E). Quantitative analysis confirmed the Western blot analysis (Fig. 2G). Homozygous flies fed dox for 12 h produced roughly twice as much luciferase activity as heterozygous flies. Together, the Western blots and luciferase assays show that increasing the copy number, and presumably the amount of the TA protein, increases target gene induction.
Absolute level of target protein that is induced.
We estimate that the dox-induced luciferase protein represents about 1 in 20,000 total protein molecules. Flies homozygous for both TA and target transgenes were fed dox (1 mg/ml) for 48 h (Fig. 2F, +Dox). Twenty micrograms of total fly protein was compared with protein from untreated flies (Fig. 2F, −Dox) and protein from wild-type flies that were supplemented with defined amounts (1–5 ng) of purified luciferase protein. Western blot analysis showed that about 1–2 ng of luciferase protein was induced per 20 μg of total protein. When simplifying assumptions are made, this amount of induced protein corresponds to ≈1 in 20,000 total molecules.
Comparison of Various TAs.
TA expression levels.
Altering the codon usage, removing a cryptic splicing site, and insulating from position effects increases TA expression. We refer to these three modifications collectively as altering the TA. Three versions of the rtTA and the tTA were expressed under the control of the actin5C promoter and compared for protein expression. Equal amounts of whole fly protein from the different transgenic flies were analyzed by using Western blots and a polyclonal antibody raised against the TetR protein (Fig. 3A). When either the original Tet-On (Fig. 3A, rtTA-alt = lane 2) or the new M2 (Fig. 3A, rtTA-M2-alt = lane 1) genes were altered, TA protein could be detected. TA protein was undetectable in wild-type flies (Fig. 3A, lane 5), or flies with either the unaltered Tet-Off (Fig. 3A, tTA = lane 4) or the original, unaltered Tet-On (Fig. 3A, rtTA = lane 3) TA (5). Thus, the net effect of altering the TA was to increase expression.
Induction capabilities.
rtTA-M2-alt lines function comparably to the Tet-Off TA lines. Double transgenic flies carrying the various Tet-On TAs and target were fed dox (1 mg/ml) for 24 h. Transgenic Tet-Off flies (with targets) were maintained on regular food. Whole fly extracts were prepared, and equal amounts of protein were assayed for luciferase activity (Fig. 3B). Consistent with a previous report, the rtTA failed to induce significant amounts of luciferase when flies were fed for only 24 h at 1 mg/ml dox (5). This value was arbitrarily set to a value of one, and all other levels were normalized to this number. Altering the TA resulted in an increase in TA expression and an approximate 8-fold induction of luciferase activity (rtTA versus rtTA-alt). This result demonstrates that increasing the expression levels of the TA leads to greater luciferase induction. The new M2 allele, when altered, results in a 68-fold induction of luciferase activity. When compared with the rtTA-alt, which is expressed at comparable levels, it results in 8.5-fold greater induction. This improvement results from the set of mutations that make the M2 protein a better TA. The Tet-Off TA produces a 119-fold induction, or about twice that of rtTA-M2-alt. However, the tTA is expressed at significantly lower levels, indicating that the tTA protein is far more active than the rtTA-M2-alt protein.
The rtTA-M2-alt Functions in Both Embryos and Larvae.
Embryos.
Significant induction of luciferase was detected in embryos from mothers that were fed dox-containing food. Embryos (6–18 h) were collected from mothers homozygous for both the rtTA-M2-alt and TetO-luciferase transgenes. Robust, ubiquitous luciferase staining was detected in embryos from mothers maintained on food supplemented with dox (100 μg/ml) for 48 h before embryo collection (Fig. 4A Upper). Luciferase was undetectable in embryos from mothers who were maintained on dox-free food (Fig. 4A Lower). Early embryos (0–2 h) from double transgenic animals, wild-type embryos, or embryos lacking the TA transgene did not produce any detectable staining (data not shown). The lack of detectable staining in early embryos indicates that the staining in older embryos is likely to be a result of zygotic target transgene expression.
Figure 4.

Analysis of our Tet-On TA in embryos and larvae. (A) Mothers carrying two copies of both the actin5C-rtTA-M2-alt transgene and the TetO-luciferase transgene were fed yeast paste supplemented with dox (100 μg/ml) for 48 h (+), or dox-free yeast paste (−). Embryos of 6–18 h were dechorinated, fixed, and incubated with an antiluciferase mAb overnight at 4°C. Antibody complexes were detected with an horseradish peroxidase-conjugated secondary antibody and Ni- diaminobenzidine staining. (B–D) Wandering third-instar larvae carrying one copy of both the actin5C-rtTA-M2-alt and the TetO-LacZ transgenes were dissected, fixed, and stained with X-gal. Brains (B), wing discs (C), and leg discs (D) from larvae fed dox-containing (+) food (10 μg/ml) or regular food (−) were compared. No staining was detected in wild-type larvae or larvae carrying only the TetO-LacZ transgene (data not shown).
Larvae.
Strong induction of a TetO-LacZ transgene (5) was detected in larvae raised on food supplemented with dox. Larvae heterozygous for both the rtTA-M2-alt and TetO-LacZ transgenes were raised on food supplemented with dox (10 μg/ml) or on regular food. Wandering third-instar larvae were dissected, fixed, and stained for β-galactosidase activity. Strong, dox-dependent X-gal staining was detected in dissected larval brains, wing discs, and leg discs (Fig. 4 B–D, +). Discs from mothers that were fed drug-free food (Fig. 4 B–D, −) showed no staining. Single transgenic larvae, carrying either the TA or target transgenes only, or wild-type larvae, did not produce any detectable X-gal staining in their discs (data not shown). Previous studies using an actin5C-driven rtTA transgene required high dox concentrations (up to 250 μg/ml) to produce target gene induction (5). In our hands, larvae fed dox at these high concentrations suffered clear effects on developmental morphology and behavior consistent with previous findings (5). We saw no adverse effects on larvae when they were fed dox at 10 μg/ml.
Discussion
We describe the application of a second-generation Tet-On TA (rtTA-M2-alt) to transgenic animals and compare its performance to previously available Tet-regulated systems in vivo. Rapid and strong induction of target transgenes occurs throughout the animal. This expression is dox-dependent, sensitive to the dosage of drug in the food, and requires the presence of both TA and target transgenes. These properties, in adult flies, embryos, and larvae, validate the use of the system for transgenic applications. The TA protein produced from our rtTA-M2-alt gene is slightly different from the one previously reported (8). Our protein has a cryptic splicing site removed in the TetR moiety, and the VP16 activation domain is larger. It is unclear which version of the M2 TA is more efficient. However, it is very clear that the M2 mutations result in a better TA, with faster induction kinetics, lower drug requirements, and greater induction.
In adult flies, induction of a luciferase target can be achieved when standard Drosophila food is supplemented with dox at concentrations as low as 1 μg/ml, although maximum induction requires concentrations of 100–1,000 μg/ml. The kinetics of induction is concentration-dependent, and significant induction can occur within 3 h. Induction continues to increase until a plateau is reached sometime between 24 and 48 h. When the drug is directly injected into adult flies, maximal induction is detected within 30 min, and these flies are healthy and viable (data not shown). These results represent a significant improvement over the performance of the original Tet-On system in flies (5), where significant accumulation of β-galactosidase required at least 48 h of feeding followed by 3 days of “recovery.” Also, in the previous study, dox was administered in a Tris-sucrose solution. Altering feeding conditions can have a dramatic effect on many adult behaviors. By placing dox in standard fly food, researchers will be able to avoid the complications of changing feeding conditions.
The amount of induction depends on the dosage of antibiotic and the duration of feeding. Maximal induction is easily achieved with dox concentrations as low as 10 μg/ml when flies are fed for 48 h. At 1 mg/ml, which is the highest concentration that we recommend feeding adult flies, the maximum amount of induction (≈68-fold) is still sensitive to the gene dosage of the TA. Therefore, transgenic lines that have higher basal level expression of the TA should result in greater induction of the target gene. The amount of induction we measure by using our Tet-On TA results from a combination of two factors. Altering the TA leads to better expression, which contributes about an 8-fold improvement in induction. The M2 mutations result in a better reverse TA protein, which contributes approximately another 8-fold. Together, our Tet-On TA performed 68-fold better than the rtTA protein, and this amount of induction was within 2-fold of what is achievable with the Tet-Off TA. Therefore, in transgenic animals, greater induction and faster kinetics can be achieved by finding transgenic lines with more TA expression or delivering the antibiotic to the tissue of choice in as direct a manner as possible.
We also have demonstrated that the Tet-On system can be used to induce target transgenes throughout embryos and larvae. If mothers are fed dox (100 μg/ml), their embryos show strong induction. Massive induction of a LacZ transgene is detected in larvae reared on food supplemented with dox at 10 μg/ml in all tissues tested. The previously published rtTA-based system had marginal utility in larvae. Larvae had to be fed dox at 250 μg/ml to induce expression from the same LacZ transgene that we are inducing effectively at 10 μg/ml (5). There are clear morphological and behavioral effects on larvae when they are fed dox at the higher concentration. Larval studies using the rtTA-M2-alt system will now be able to proceed without fear of toxicity.
The combination of the Tet-On and Gal4-UAS systems provides a powerful reverse genetic tool (see Results in the supporting information and Figs. 5 and 6, which are published on the PNAS web site, www.pnas.org). We demonstrate that a sevenless-Gal4 driver is able to bring expression of rtTA-M2-alt to high enough levels to achieve a 40-fold induction of a luciferase target transgene upon dox feeding. Several other eye- and brain-specific drivers showed similar results (not shown). However, it should be noted that the fold induction will vary greatly depending on the Gal4 driver and target transgenes used. In adults there is only a 1.3-fold difference in expression levels between flies with and without a Gal4 driver. These results suggest that almost all of the TA protein is inactive in the absence of drug. Despite the low leakiness the system exhibits high induction ratios. Because of the instability of a luciferin-luciferase complex, luciferase accumulation is unlikely to contribute to an overestimation of the amount of induction. It is likely that the spatially restricted system will have similar kinetics to the actin-based system because the amount of induction at 24 h is similar in both systems. However, the detailed pharmacokinetics of drug delivery may vary in different tissues and needs to be empirically determined.
Tissue-specific, concentration-dependent expression is further demonstrated when the system is used to cause defects in wing structures upon expression of rpr. The improved Tet-On system allows temporal control of gene induction and concentration-dependent modulation of the amount of induction. These properties should facilitate analyses of late phenotypes, because many enhancers express in earlier patterns, and these may interfere with analyses of later phenotypes. This principle is illustrated with the Salm enhancer, because traditional Salm-Gal4/UAS expression of rpr is lethal, but control of drug dosage and rpr expression allows development to proceed normally, revealing late wing and eye phenotypes. Spatially restricted induction also should complement currently available induction systems to define tissue and temporal requirements for gene activity in any biological process.
It is our sense that part of the reason for our success is the use of boundary elements to minimize position effects of random insertions (9, 10, 16–19). In this report, we insulate only the TA expression cassette and do not insulate the target. When target transgenes are insulated and compared with uninsulated ones, we find that insulators generally increase expression levels, but do not change the fold induction (M.J.S., unpublished data). Most insulated target transgenes had the same baseline and induced expression levels. Similarly, four combinations of insulated TA lines performed equally when combined with the same target line. This equivalence decreased the number of transgenes, and combinations of transgenes, that we examined, because most were nearly identical (M.J.S., unpublished data).
We and others have shown that the second-generation Tet-On TA works well in transfected HeLa cells (8), primary neurons (Z. Lin, personal communication), mice (19), and transgenic flies. These experimental systems differ in many details that affect transcription. However, all of the important measures of the performance of an inducible system (induction ratio, kinetics of induction, leakiness, and possible toxic or pleiotropic effects of dox) are similar in the various cells, regardless of species, developmental stage, or tissue type. This type of uniformity of performance bodes well for future applications in other transgenic and cellular contexts. The improvements in the Tet-On system have allowed us to successfully combine the Gal4-UAS and Tet-On systems, resulting in spatially restricted, temporally regulated gene induction upon drug feeding. The combined Gal4 Tet-On system represents a major improvement to inducible technology in Drosophila with implications for other transgenic model systems. Finally, this study provides a benchmark to which future improvements to the Tet-On system can be compared.
Supplementary Material
Acknowledgments
We thank Jim DeZazzo for ADF-1 antibody, reagents, advice, and comments on early versions of the manuscript. We also thank Grisha Enikolopov, Maurice Kernan, Paul Schedl, and Sid Strickland for valuable discussions and Hong Zhou for technical assistance. We acknowledge Walter Gehring, Andreas Keller, and Martin Heisenberg for help in the rpr-mediated disruption experiment and analysis of the eye phenotype. This work was supported by grants from the National Institutes of Health (R01 NS35575), the McKnight Foundation, and start-up funds from the Cold Spring Harbor Laboratory (to J.C.P.Y.).
Abbreviations
- Tet
tetracycline
- TA
transactivator
- TetR
Tet repressor protein
- TetO
Tet operator
- tTA
Tet-responsive TA
- rtTA
reverse TA
- dox
doxycycline
- UAS
upstream-activating sequence
- X-gal
5-bromo-4-chloro-3-indolyl β-d-galactoside
References
- 1.Baron U, Bujard H. Methods Enzymol. 2000;327:401–421. doi: 10.1016/s0076-6879(00)27292-3. [DOI] [PubMed] [Google Scholar]
- 2.Gossen M, Freundlieb S, Bender G, Muller G, Hillen W, Bujard H. Science. 1995;268:1766–1769. doi: 10.1126/science.7792603. [DOI] [PubMed] [Google Scholar]
- 3.Kistner A, Gossen M, Zimmermann F, Jerecic J, Ullmer C, Lubbert H, Bujard H. Proc Natl Acad Sci USA. 1996;93:10933–10938. doi: 10.1073/pnas.93.20.10933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mansuy I M, Winder D G, Moallem T M, Osman M, Mayford M, Hawkins R D, Kandel E R. Neuron. 1998;21:257–265. doi: 10.1016/s0896-6273(00)80533-4. [DOI] [PubMed] [Google Scholar]
- 5.Bieschke E T, Wheeler J C, Tower J. Mol Gen Genet. 1998;258:571–579. doi: 10.1007/s004380050770. [DOI] [PubMed] [Google Scholar]
- 6.Malleret G, Haditsch U, Genoux D, Jones M W, Bliss T V, Vanhoose A M, Weitlauf C, Kandel E R, Winder D G, Mansuy I M. Cell. 2001;104:675–686. doi: 10.1016/s0092-8674(01)00264-1. [DOI] [PubMed] [Google Scholar]
- 7.Bello B, Resendez-Perez D, Gehring W J. Development (Cambridge, UK) 1998;125:2193–2202. doi: 10.1242/dev.125.12.2193. [DOI] [PubMed] [Google Scholar]
- 8.Urlinger S, Baron U, Thellmann M, Hasan M T, Bujard H, Hillen W. Proc Natl Acad Sci USA. 2000;97:7963–7968. doi: 10.1073/pnas.130192197. . (First Published June 20, 2000; 10.1073/pnas.130192197) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kellum R, Schedl P. Cell. 1991;64:941–950. doi: 10.1016/0092-8674(91)90318-s. [DOI] [PubMed] [Google Scholar]
- 10.Kellum R, Schedl P. Mol Cell Biol. 1992;12:2424–2431. doi: 10.1128/mcb.12.5.2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rubin G M, Spradling A C. Science. 1982;218:348–353. doi: 10.1126/science.6289436. [DOI] [PubMed] [Google Scholar]
- 12.Spradling A C, Rubin G M. Science. 1982;218:341–347. doi: 10.1126/science.6289435. [DOI] [PubMed] [Google Scholar]
- 13.Belvin M P, Zhou H, Yin J C. Neuron. 1999;22:777–787. doi: 10.1016/s0896-6273(00)80736-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.DeZazzo J, Sandstrom D, de Belle S, Velinzon K, Smith P, Grady L, DelVecchio M, Ramaswami M, Tully T. Neuron. 2000;27:145–158. doi: 10.1016/s0896-6273(00)00016-7. [DOI] [PubMed] [Google Scholar]
- 15.Patel N H, Martin-Blanco E, Coleman K G, Poole S J, Ellis M C, Kornberg T B, Goodman C S. Cell. 1989;58:955–968. doi: 10.1016/0092-8674(89)90947-1. [DOI] [PubMed] [Google Scholar]
- 16.Chung J H, Whiteley M, Felsenfeld G. Cell. 1993;74:505–514. doi: 10.1016/0092-8674(93)80052-g. [DOI] [PubMed] [Google Scholar]
- 17.Udvardy A, Schedl P. Mol Cell Biol. 1993;13:7522–7530. doi: 10.1128/mcb.13.12.7522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vazquez J, Schedl P. EMBO J. 1994;13:5984–5993. doi: 10.1002/j.1460-2075.1994.tb06944.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hasan M T, Schonig K, Berger S, Graewe W, Bujard H. Genesis. 2001;29:116–122. doi: 10.1002/gene.1014. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}