

Summary
Mammalian mtDNA encodes only 13 proteins, all essential components of respiratory complexes, synthesized by mitochondrial ribosomes. Mitoribosomes contain greatly truncated RNAs transcribed from mtDNA, including a structural tRNA in place of 5S RNA as a scaffold for binding 82 nucleus-encoded proteins, mitoribosomal proteins (MRPs). Cryoelectron microscopy (cryo-EM) studies have determined the structure of the mitoribosome, but its mechanism of assembly is unknown. Our SILAC pulse-labeling experiments determine the rates of mitochondrial import of MRPs and their assembly into intact mitoribosomes, providing a basis for distinguishing MRPs that bind at early and late stages in mitoribosome assembly to generate a working model for mitoribosome assembly. Mitoribosome assembly is a slow process initiated at the mtDNA nucleoid driven by excess synthesis of individual MRPs. MRPs that are tightly associated in the structure frequently join the complex in a coordinated manner. Clinically significant MRP mutations reported to date affect proteins that bind early on during assembly.
Graphical abstract
Recent cryo-EM studies provided detailed structures of the protein-rich mammalian mitoribosome, but the process of their assembly is poorly understood. Bogenhagen et al. use SILAC pulse labeling to determine the order of addition of mitoribosomal proteins (MRPs) to provide a structure-based kinetic model for assembly of mammalian mitoribosomes.
Introduction
Normal cell physiology relies on a close interaction between the nuclear and mitochondrial genomes to support oxidative metabolism. Biogenesis of the mitochondrial respiratory chain depends on coordinated expression of 13 proteins encoded in mtDNA with nearly 70 proteins that are nuclear gene products. The synthesis of this small number of mtDNA-encoded proteins occurs on mitochondrial ribosomes that are entirely distinct from cytoplasmic ribosomes. The mtDNA genome contributes only the RNA components of mitoribosomes. The rRNA genes are expressed by a single subunit mtRNA polymerase, POLRMT, along with mRNAs and tRNAs in long polycistronic precursors. In higher eukaryotes, although not in yeast, the rRNAs are co-localized with genes for tRNAF and tRNAV in a relatively long-lived precursor termed RNA 4 (Gelfand and Attardi, 1981). The biogenesis of mitoribosomes depends on coordinated synthesis of 82 mitochondrial ribosomal proteins (MRPs) encoded in nuclear DNA, which must be translated on cytoplasmic ribosomes and individually imported into mitochondria. It is apparent that building a mitoribosome is a difficult logistical task comparable to the complexity of assembling the entire respiratory chain.
The structures of mitoribosomes have been elucidated at near atomic resolution by two groups employing recent advances in cryoelectron microscopy (cryo-EM) methods (Amunts et al., 2015; Greber et al., 2015). Some of the unique features of the mitoribosomes are as follows: (1) As inferred from the human mtDNA sequence (Anderson et al., 1981), the rRNAs of large and small subunits of the mammalian ribosome were reduced in size during evolution to ∼60% of the size of the corresponding E. coli rRNAs, with numerous deletions that shorten or eliminate secondary structure elements. (2) The 5S rRNA found in the central protuberance of the large subunit of nearly all standard pro-karyotic and eukaryotic cytoplasmic ribosomes is absent from mammalian mitoribosomes, where it has been replaced by tRNA (generally tRNAV or tRNAF, which are adjacent to the 12S rRNA gene in mtDNA). (3) Several protein subunits conserved in other ribosomes are absent from mitoribosomes, often reflecting corresponding deletions of their rRNA binding sites. (4) Despite truncation of the rRNA component, the overall mass is essentially conserved, as mitoribosomes contain a significant number of novel mitochondrial-specific polypeptides whose locations were only elucidated when the cryo-EM structures were resolved. (5) The tight organization of mitochondrial rRNA genes completely lacks intergenic spacer regions found in other rRNA gene regions, requiring precise incisions by RNases P and ELAC2 for rRNA maturation. (6) S. cerevisiae has followed a different evolutionary path to adopt unlinked rRNAs even larger than those found in E. coli, along with a distinct pattern of altered ribosomal proteins (Amunts et al., 2014; Desai et al., 2017). Thus, this organism is not a suitable model for studies of mammalian mitoribosome structure or function, although it bears its own intrinsic interest.
Despite the exciting advances in understanding the structure of mitoribosomes, we have few insights into the assembly of these macromolecular machines. In contrast, decades of research exploring the mechanism of bacterial ribosome assembly have provided robust models for assembly of both the large and small subunits (Shajani et al., 2011). This wealth of information is not readily applicable to mitoribosomes due to extensive structural changes during evolution as noted above. Coordinating assembly of a mitoribosome is clearly a daunting task, since it requires import of as many structural proteins as the entire respiratory chain. We know that the assembly of a simpler structure, NADH dehydrogenase, requires nearly 24 hr for the 44 component proteins to join the complex (Guerrero-Castillo et al., 2017; Ugalde et al., 2004). The kinetics of mitoribo-some assembly may be limited by the time required to import the 82 individual MRPs into mitochondria. It is evident that novel approaches are required to study mitoribosome assembly.
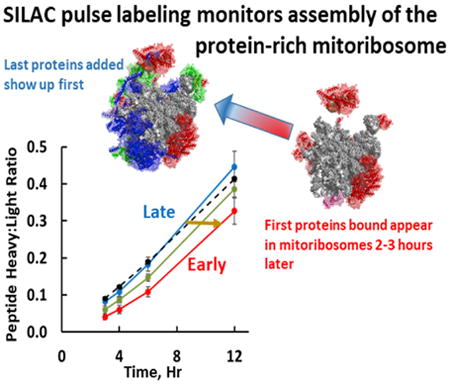
In this paper, we report proteomic experiments to study the kinetics of accumulation of nascent MRPs in mitochondria and their general order of addition during mitoribosome assembly. These experiments employed pulse labeling with stable isotopes (SILAC) to track the appearance of newly synthesized MRPs in isolated mitochondria and in mature mitoribosomes. SILAC permits detection of the newly synthesized polypeptides that incorporated 13C-labeled lysine and arginine using mass spectrometry. We previously reported that a subset of newly synthesized MRPs is selectively associated with mtRNA at the mtDNA nucleoid, suggesting that these MRPs represent some of the first proteins to bind nascent rRNA (Figure 1; Bogenhagen et al., 2014). The current study tests the hypothesis that SILAC labeling can be used to dissect later steps in mitoribosome assembly. We found that MRPs are synthesized and imported into mitochondria at rates greater than required for mitoribosome assembly and are either built into mitoribosomes or, if they cannot be assembled, degraded. Some MRPs appear in mature ribosomes much more rapidly than others. We reasoned that the first MRPs to appear in mature mitoribosomes would be the last polypeptides added to complete assembly (Figure 1). In contrast, the first MRPs bound to nascent rRNA only appear in mature mitoribosomes after a delay. The data revealed several examples of pairs or small groups of MRPs that share extensive protein-protein interactions and join the mitoribosome with similar kinetics in a coordinated manner. Therefore, we analyzed the interactions between MRPs in the mitoribosome structure. The combination of SILAC pulse-labeling results and structural analysis provides a working model for the kinetic mechanism of mitoribosome assembly interpretable in light of the cryo-EM structure of the mitoribosome.
Figure 1. SILAC Labeling Resolves Steps in Mitoribosome Assembly.
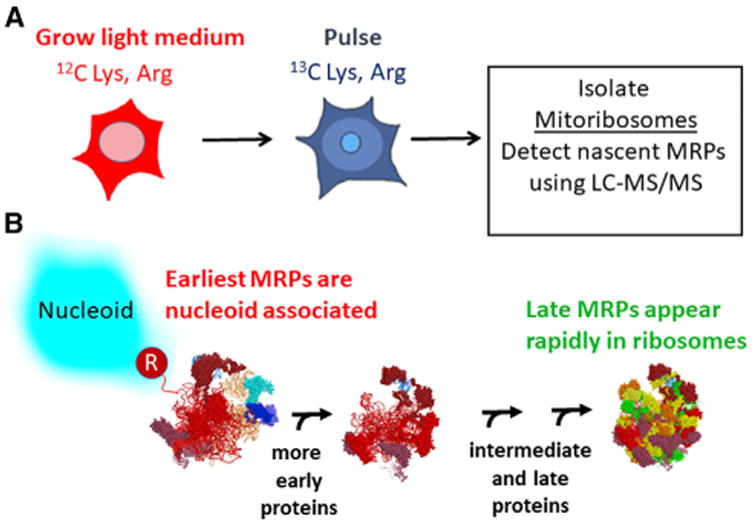
(A) The SILAC pulse-labeling approach.
(B) Newly synthesized rRNA is shown as a hypothetical folded structure emanating from mtRNA polymerase (R) with 12S rRNA (orange) still linked to tRNAV and 16S rRNA (red) as in the RNA 4 precursor. The actual conformation of the nascent RNA is unknown. MRP polypeptides begin to bind nascent rRNA at the nucleoid (Bogenhagen et al., 2014) and assembly intermediates progress to complete mitoribosomes after some delay. In contrast, latebinding proteins are less likely to be found in nucleoid preparations and appear more rapidly in fully assembled, mature mitoribosomes at rates distinguishable by SILAC pulse labeling.
Results
Kinetic Analysis of Mitoribosome Assembly
We conducted a series of SILAC experiments in which HeLa cells were pulse labeled with 13C6 lysine and arginine (K6R6) for various time intervals (Experimental Procedures) to track the kinetic appearance of newly synthesized MRPs containing a high content of 13C6 in completed mitoribosomes, indicated by a high ratio of “heavy” to “light” peptides, or a high H:L ratio. This ratio is analogous to a specific activity measurement in a radioactive labeling study. A broad survey of protein turnover rates included data suggesting that MRPs labeled during long-term SILAC are generally stable through the time course of our experiments (Schwanhäusser et al., 2011), so that the content of unlabeled MRPs, the denominator in the H:L ratio calculation, is not changed significantly over this interval. In growing cells, we would expect that labeling for a period of time equal to one cell generation time would result in 50% “heavy” MRPs. Similarly, simple exponential growth kinetics (using N(t) = No eIn2t/Tg) predict that a 4-hr labeling should result in 12% “heavy” MRPs if the generation time (Tg) is 24 hr. A more complete description of the predicted labeling kinetics is provided in Supplemental Experimental Procedures. The SILAC approach tests the hypothesis that mitoribosome assembly is a rapid process, in which case all MRPs in fully assembled mitoribosomes would be labeled to a comparable extent. Mitoribosome purification followed the classical method developed by the O'Brien lab (Matthews et al., 1982) using two successive sucrose gradient separations under high-salt conditions as adapted in recent cryo-EM studies of the mitoribosome (Amunts et al., 2015; Greber et al., 2015). Gradient fractions containing intact 55S, 39S, and 28S mitoribosomes were identified by immunoblotting with antibodies to individual subunits (usually uL10m or uL13m and uS15m or uS17m; Figure S1). Proteins were recovered and submitted for digestion and mass spectrometry. To examine early kinetics of assembly, we used shorter labeling times of 3, 4, 6, or 12 hr. The results of triplicate experiments (Table S1) show that individual MRPs exhibit distinct kinetics of accumulation in mature mitoribosomes, with H:L ratios varying by over 2-fold at early labeling intervals. Figures 2A and 2B show the H:L ratios of individual MRPs after 3- and 4-hr labeling intervals for large subunit (LSU) and small subunit (SSU) proteins, respectively, indicating that proteins behaved consistently at a variety of pulse times. Higher values for the ribosome H:L ratio after short labeling intervals reflect more rapid appearance of newly synthesized protein in mature mitoribosomes (blue in Figure 2), while others appear more slowly (red) or at intermediate rates (green). Figures 2C and 2D illustrate the average kinetic patterns observed for the rapidly appearing class C MRPs (blue) in contrast to more slowly incorporated class A MRPs (red) for the LSU and SSU, respectively. The class C MRPs appear in completed mitoribosomes at a rate comparable to that predicted by a mathematical model (Supplemental Methods) shown by the dashed line in Figure 2 that assumes preexisting mitoribosomes are relatively stable and new MRPs are synthesized to maintain a steady cellular content of mitoribosomes in growing cells.
Figure 2. MRPs Appear in Mitoribosomes at Different Rates.
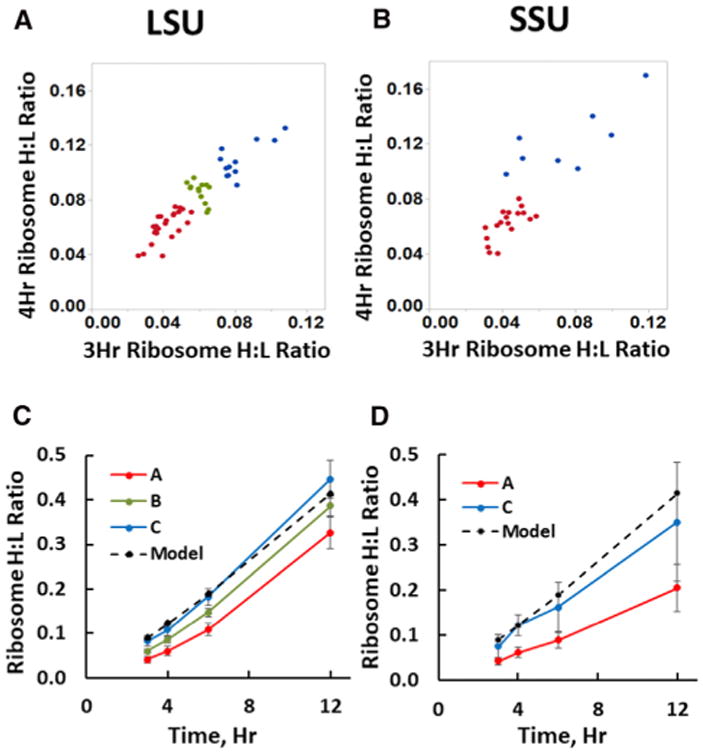
(A and B) Scatterplots of H:L ratios observed for individual LSU (A) and SSU (B) MRPs after 3 and 4 hours of SILAC labeling. Points representing MRPs that have consistently low H:L ratios (Table S1) are designated class A (red), while those that appear more rapidly in mitoribosomes are class C (blue). For the larger set of LSU polypeptides, an intermediate class B is also designated (green).
(C and D) Average H:L ratios (± SD) for the early, intermediate and late classes of MRPs for the LSU (C) and SSU (D), as identified in Table S1. The dashed line shows the H:L ratios expected for the rate of mitoribosome synthesis required to sustain a constant cellular complement of mitoribosomes in growing cells (see Supplemental Experimental Procedures). Some MRPs were not included in this analysis because of insufficient numbers of peptides or, for bL33m, atypically rapid accumulation (discussed in the text). See also Figures S1 and S2.
We consider three alternative hypotheses to account for the varied rates of accumulation of MRPs in mitoribosomes. First, the logic in Figure 1 suggests that the class A MRPs may accumulate more slowly if they are incorporated into assembly intermediates that require more time to appear in intact mitoribo-somes. An alternative hypothesis is that some nascent MRPs may be incorporated more rapidly by exchanging into preexisting mitoribosomes. A third consideration is that some nascent MRPs imported into mitochondria may be effectively diluted by larger pools of preexisting unlabeled MRPs, which could delay their appearance in intact mitoribosomes.
The hypothesis that the kinetics in Figure 2 could be explained by rapid exchange of MRPs into preexisting mitoribosomes is addressed by considering the behavior of one protein, bL33m, which accumulates at a rate far faster than any other MRP (Table S1), and which was omitted from the analysis for this reason. Figures S2A and S2B show that the mitoribosomal H:L ratio of bL33m was 0.21 after only 3 hr of labeling, increasing to 1.11 in a 12-hr labeling, which requires replacement of over half the copies of bL33m within 12 hr. Other rapidly appearing class C MRPs accumulated at a rate not appreciably greater than predicted by the mathematical model shown with the dashed lines in Figures 2C and 2D. No other MRP showed similar labeling kinetics. The exceptional assembly kinetics of bL33m suggest that exchange of newly synthesized MRPs into preexisting mitoribosomes is not a major factor for all other MRPs.
The second alternative hypothesis to explain differential assembly kinetics in Figure 2 is that the isotopically labeled MRPs that assemble more slowly may be diluted by larger pools of preexisting free, unlabeled polypeptides. We addressed this in two ways. First, we note that neither the rapidly appearing uS15m nor the slowly appearing uL10m featured a large pool of free protein in Figure S1. We surveyed six other class A MRPs for the presence or absence of large pools of free precursor proteins using immunoblotting of sucrose gradient fractions (Figure S2C), revealing several with small pools of free protein (uS17m, bL19m, and mL44) and others with larger pools (mL45, mS23, and mS27). This showed no correlation between assembly class and the relative size of precursor pools. In addition, we performed a separate analysis of the relative synthesis rates, assembly rates, and stability of MRPs below. The results of these analyses are consistent with the hypothesis that the class A MRPs bind early on during ribosome assembly while the class C MRPs appear first in intact mitoribosomes, because they represent the last MRPs to bind to complete the assembly of stable 39S and 28S subunits.
Nascent Early-Binding MRPs Complete Their Assembly into Mature Mitoribosomes during a Chase Interval
We performed additional pulse-chase labeling experiments to test whether the early binding proteins would ultimately appear in intact mitoribosomes along with the late-binding proteins. HeLa cells pulse labeled with K6R6 for 4 hr were incubated for an additional 10-hr “chase” with normal 12C-labeled lysine and arginine. Figures 3A and 3B show comparisons of the labeling intensity for individual 39S and 28S MRPs after the pulse and pulse-chase incubations. The early-binding (class A) MRPs identified in Table S1 and Figure 2 (red in Figure 3) tend to reside in the lower regions of these plots, since their low H:L ratios after the pulse increase during the chase, as expected if labeled copies of these MRPs initially associated within incomplete assembly intermediates during the pulse. Distinct behavior of early and late MRPs can also be expressed by the ratio of H:L values with and without the chase, the P4C10/P4 ratio (Figures 3C and 3D). Clearly, the putative early-binding proteins increase significantly in H:L ratio during the chase as the intermediates in which they bind initially complete assembly. In contrast, the late-binding MRPs (blue) tend to reside in the upper region of the plots in Figures 3A and 3B, and the average H:L ratio for this group actually decreases slightly during the chase (Figures 3C and 3D), revealing a statistically significant difference from the early-binding proteins (p < 0.0001). The late-binding proteins were incorporated quickly into mitoribosomes during the pulse. The continuous synthesis and assembly of unlabeled protein during the chase led to a slight decline in their H:L ratios.
Figure 3. Mitoribosome Assembly Intermediates Containing Early-Binding Proteins Complete Assembly during Chase Incubation.
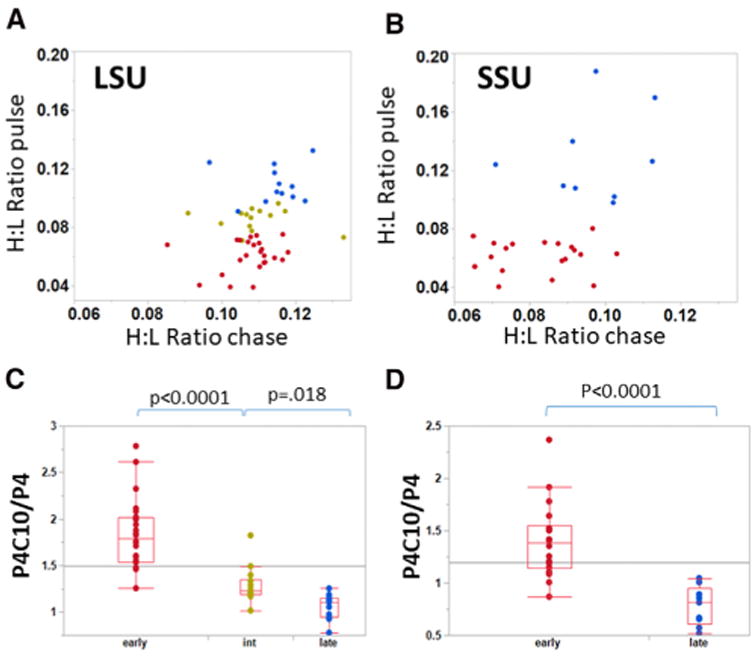
(A and B) Scatterplots of the H:L ratios of individual LSU (A) and SSU (B) polypeptides, after the 4-hr pulse (ordinate) or after an additional 10-hr chase incubation (abscissa). Early- and late-binding proteins identified in Figure 2 are in red and blue, respectively, with intermediate LSU proteins in gold-green. (C and D) The early, intermediate, and late classes of assembled MRPs in the LSU (C) and SSU (D) show statistically significant differences in P4C10/P4, the ratio of H:L values after and before the chase. Here and in Figure 4E, the horizontal lines indicate the mean value for all data points. See also Table S1.
MRPs Are Synthesized and Imported in Excess and Can Be Unstable if Not Assembled
Our results support the model that the distinct kinetics of incorporation of different MRPs into mitoribosomes principally reflects their relative assembly order. To further test the possibility that differences in the ability of individual nascent MRPs to exchange with the pool of free polypeptides or their general stability might influence the apparent assembly kinetics, we also monitored the rates of synthesis and mitochondrial import and the stability of MRPs by determining the H:L ratio of the total mitochondrial pool of each protein in pulse-chase SILAC experiments. In parallel with our mitoribosome pulse-chase experiment, samples of total mitochondrial protein obtained after 4-hr pulse labeling with and without a 10-hr chase (P4C10 and P4, respectively) were analyzed by liquid chromatography-tandem mass spectrometry (LC-MS/MS) to determine H:L ratios of the combined population of free and mitoribosome-bound copies of each MRP. Table S2 shows the results obtained for MRPs and for a set of standard proteins identified in Table S2 and summarized in the histogram in Figure 4A. The standard proteins declined from an initial average H:L value of 0.172 to 0.127 during the chase. This decrease is explained by the continued growth of the cells in 12C medium during the chase, which we expect to dilute labeled proteins and to reduce the H:L value by 25%. Clearly, the rates of synthesis and importation of these standard proteins are commensurate with the cell growth rate, and there is negligible wastage of newly made protein. In contrast, the MRPs are synthesized and imported at high rates, with H:L ratios averaging ∼0.3. Many copies of these nascent MRPs are degraded in the 10-hr chase as the H:L ratio declines to statistically the same level as the standard proteins (Figure 4A). The magnitude of this decline cannot be explained by dilution with other newly synthesized protein but must reflect degradation. The final H:L ratio of the total mitochondrial pool of MRPs is very close to that in intact mitoribosomes (Figure 4B). Thus, the degradation process mostly operates on free copies of MRPs in this time frame.
Figure 4. MRPs Are Synthesized and Imported in Excess and Can Be Unstable if Not Assembled.
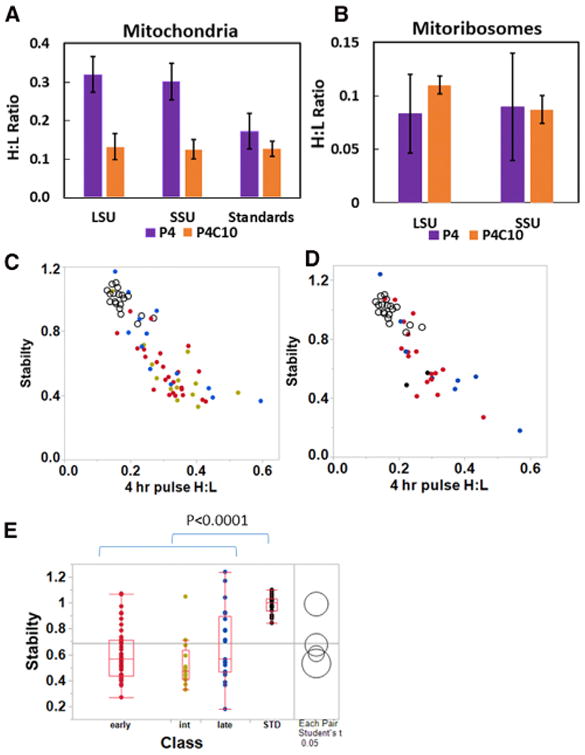
(A) The H:L ratio immediately after 4 hr of pulse labeling is greater for MRPs than for a standard set of mitochondrial proteins, indicating a higher rate of synthesis and mitochondrial import, but the H:L ratio of MRPs declines during a 10-hr chase to a value not significantly different from the stable standard proteins. Mean values are shown ± SD.
(B) The H:L values observed for MRPs in assembled mitoribosomes before and after the chase are shown for comparison. The large SE after the pulse reflects variable assembly kinetics of individual proteins and is significantly smaller after the chase. Note the change in scale of the ordinate compared to (A).
(C and D) The stability of LSU (C) and SSU (D) MRPs in early (red), intermediate (green), and late (blue) assembly classes is plotted against the H:L ratio after the pulse. Standard proteins are shown with open circles in both plots.
(E) The stabilities (P4C10/expected) of early, intermediate, and late assembly classes of MRPs are significantly lower than those of the standard proteins, but not significantly different from one another.
See also Tables S1 and S2.
We next sought to determine whether the putative early and late MRPs exhibited differences in stability. This is an important issue, since it may be argued that the rapid appearance in intact mitoribosomes of the MRPs we designate as late might be due to their exchange with polypeptides within preexisting mitoribosomes. To express the behavior of proteins concisely, we defined a stability parameter as the ratio of the observed H:L value after the 10-hr chase to that expected due to the dilution of labeled protein during the chase. A very long-lived protein would be expected to have a stability parameter of 1.0. In keeping with this, the 19 standard proteins had an average stability (± SD) of 0.99 ± 0.07. Figures 4C and 4D show that most of the early and late LSU and SSU MRPs, respectively, are much less stable than the standard proteins shown as open circles. Statistical analysis using pairwise t tests showed that all three classes of early, intermediate, and late MRPs were significantly less stable than the standard proteins (p < 0.0001), but there was no significant difference in stability among these three classes of MRPs (Figure 4E). Similar results were obtained in other experiments with 3-hr pulse labeling followed by an 8-hr chase (data not shown). While further experiments will be required to explore the degradation rates of nascent MRPs in greater detail, we found no evidence that protein stability or exchange into preexisting mitoribosomes plays a major role in the different assembly kinetics of early and late MRPs, except for the counter-example of bL33m noted above. This does not rule out the possibility that there may be some slower exchange of other proteins.
Developing a Kinetic Model for Mitoribosome Assembly
The foregoing results distinguish broad classes of early- and late-binding MRPs. The positions these proteins occupy in the final model of the mitoribosome are important to understand the process of mitoribosome assembly. The mammalian mitoribosome has a much higher protein/RNA content and smaller rRNA scaffolds than the bacterial ribosome, so that protein-protein interactions are likely to be more important in mitoribosome assembly. Table S3 shows the rosters of early-, intermediate-, and late-binding MRPs in the SSU and LSU. The assembly order is not solely determined by the extent of a protein's interaction with rRNA (Figure S3). We noted many instances in which proteins with similar assembly kinetics shared extensive binding interfaces, raising the possibility that adjacent proteins might assemble in a coordinated manner. The binding affinity of a protein-protein interaction in a crystal structure is generally related to the buried surface area (BSA) (Kastritis and Bonvin, 2012), except in cases where conformational changes take place upon binding, which is likely at least for some proteins in the context of ribosome assembly. We conducted a comprehensive analysis of the size of protein-protein interaction surfaces for the model of the human mitoribosome (PDB: 3J9M) determined using the PDBePISA site (www.ebi.ac.uk/pdbe/pisa/). BSA calculations have been used previously in the analysis of mitoribosome cryo-EM structures (Brown et al., 2014). BSA values larger than 600–850 Å2 are considered to indicate a biologically significant interface (Krissinel, 2010). We estimated that BSA >1,000 Å2 would likely result in strong interactions that would influence the assembly pathway. The exact value set as a threshold for strong interactions is somewhat arbitrary, but we selected this value because most homodimers have a BSA >1,000 Å2 (Bahadur et al., 2003). The BSAs for pairs of MRPs are shown in Figure S4, where proteins are ordered as in Table S3. It is apparent that small sets of proteins with similar assembly kinetics can often be grouped into modules that share extensive interactions. Indeed, this principle guided the subdivision of proteins within groups shown in Table S3 and Figure S4. Our goal was not to determine a unique sequence of binding events, since it is well accepted even for the simpler bacterial ribosome that assembly is best described as a general landscape that may proceed through a variety of detailed pathways (Talkington et al., 2005). Instead, the model indicates where early- and late-binding proteins reside in the mature mitoribosome structure with a full realization that large-scale structural remodeling is likely to occur at each stage of assembly. This model provides a frame of reference for further studies to refine the assembly process. The model is limited by the fact that mL52, bL36m, bS18m, and mS37 did not produce a sufficient number of peptides for our mass spectrometry (MS) analysis and that uL1m and bL12m are not present in the structural model we used, 3J9M.
SSU Assembly
Models for bacterial ribosome assembly cannot be applied directly to the mitoribosome, since a number of critical early-binding or primary proteins of the bacterial ribosome such as bS20, bS4, and bS8 were lost during evolution. Table S3 identifies the set of early-binding 28S proteins, most of which reside in two relatively large groups at the head and lower body/foot of the elongated 12S RNA core (Figure 5A). Figure 5 illustrates early- and late-binding MRPs as red and blue, respectively. Individual MRPs within binding clusters are illustrated in Figure S5. In one cluster, bS16m, mS22, and mS40 interact in the lower body in a complex that makes RNA contacts almost exclusively with the 5′ domain of 12S rRNA (Figures 5, S3, and S5B). The fact that this set of proteins interacts with the first 12S rRNA sequences available as transcription proceeds is consistent with the hypothesis that the organization of this domain may seed the entire assembly process shown in Figure 5C. Interestingly, one member of this set, mS22, does not contact 12S rRNA directly but joins at an early stage through its interactions with bS16m and mS40. This module extends to the foot with mS34 and mS27 interacting with the 3′ domain of 12S rRNA. Binding of this subgroup is followed closely by that of uS5m, likely due to the tight interaction of this protein with uS22m and mS40. uS5m is differentially colored in Figure 5 to illustrate how it bridges the two major groups of early proteins. The second set of early SSU proteins binding to the major 3′ domain of 12S rRNA in the head region includes three closely interacting proteins with extensive RNA contacts (uS7m, uS9m, and mS29) as well as three other proteins (mS31, mS35, and mS39) that substantially lack RNA interactions (Figure S3) and appear to be recruited by the other group members. A smaller set of early proteins in Table S3 includes mS23, which shares a binding surface of 1,797 Å2 with uS2m as well as a substantial interaction with bS1m (Figure S5D). This module interacts strongly with early proteins uS9m and uS5m. Remaining early proteins uS11m, uS12m, and uS17m bind as individual proteins in the center (Figure 5A). Since these three proteins do not contact other early-binding proteins, their addition appears to depend upon their substantial interactions with 12S rRNA (Figure S3). Late-binding proteins also showed some tendency to cluster, with one group (uS14m, uS10m, uS3m, and mS33) binding in the head region in association with the early uS7m-mS29 group. The significant contrast in the assembly kinetics displayed by the early-binding uS7-mS29 group (Figure S5C) and the late-binding uS3m group (Figure S5E) suggests that the former recruits the latter. A second set of late-binding proteins, uS15m, mS25, and mS26, joins the body near the early bS16m-mS22 complex. The superficial location and late-binding kinetics of uS15m, mS25, and mS26 qualify these as tertiary binding proteins dependent on the presence of early proteins with which they interact, including members of the bS16m-mS22 set as well as uS9m and uS17m. It is notable that the early proteins all have a substantial presence in the nucleoid fraction (Bogenhagen et al., 2014) and bind to the outer surface of the SSU, away from the interface with the LSU. In contrast, the late-binding proteins (blue in Figure 5B) tend to localize closer to the interface with the LSU.
Figure 5. Assembly Scheme for the SSU.
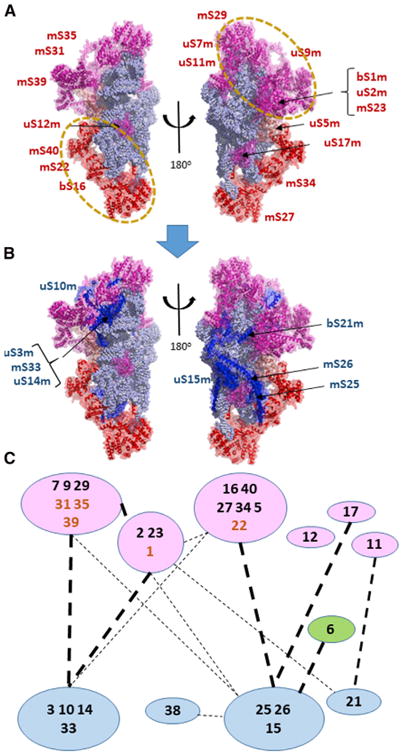
, The early- and late-binding MRPs are illustrated in (A) and (B), respectively, from two points of view, one rotated 180° with respect to the other. The 12S rRNA is shown with light blue spheres representing individual residues. Proteins are shown as cartoon structures within transparent surfaces.
(A) Early MRPs are shown in magenta and, for the group containing mS27, in red. uS5m is shown in a salmon color to indicate that it bridges the upper and lower groups, which are outlined by dashed circles and illustrated in greater detail in Figure S5.
(B) Late-binding proteins are shown in blue along with the early proteins as in (A).
(C) The assembly scheme for the SSU showing protein-protein interactions between individual or grouped polypeptides, with their longer standard names truncated to numbers for simplicity. Heavy dashed lines indicate interaction surface areas greater than 1,000 Å2 (see Figure S4), while lighter dashed lines indicate interactions between 1,000 Å2 and 350 Å2. mS22, mS31, mS35, mS39, and bS1m are shown in boldface brown type, since they are early-binding proteins lacking extensive RNA contacts. Since they depend on other proteins for assembly, they may be considered secondary binding proteins associated with the indicated early clusters. uS6m and mS38 are shown as independent proteins with variable association with the mS26 group but did not yield sufficient proteomic data for definitive kinetic assignment.
LSU Assembly
The earliest proteins to bind 16S rRNA include 24 proteins, some of which have very extensive RNA contacts (Figure S3), including bL20 and two pairs that extend deeply into the 16S rRNA, uL3m-bL19m and uL4m-uL15m. The working model for LSU assembly (Figures 6 and 7) features subgroups of proteins with strong interactions (Figures S4B, S6, and S7). However, there are few strong interactions between these subgroups. The similar assembly kinetics displayed by these three early-binding modules is consistent with the possibility that the proteins within each subgroup bind the structure in a coordinated manner. Some early-binding groups surprisingly include proteins that have no direct contact with RNA, such as mL39 and mL50, like early-binding SSU proteins mS22, mS31, mS35, and mS39. mL50 is recruited as an early-binding protein by its substantial interactions with the uL4m-uL15m heterodimer and mL49. Similarly, a group with broad-based mutual interactions, uL3m, bL19m, uL14m, bL17m, uL22m, and bL32m, appears to anchor binding of mL39 and, later, mL45, which also has minimal direct RNA contact. The incorporation of mL45 at an early stage may serve to tether the 39S subunit at the inner membrane during subsequent steps in assembly (Englmeier et al., 2017; Greber et al., 2014).
Figure 6. Model for Assembly of the LSU.
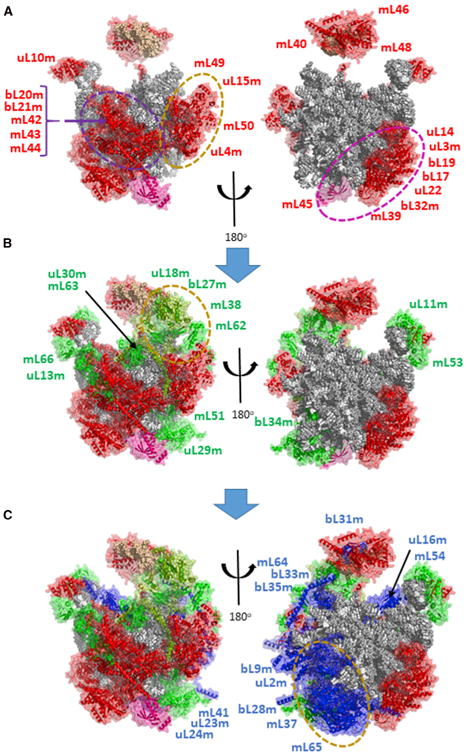
16S rRNA and tRNAV are shown with spheres representing individual residues in gray or tan, respectively. Proteins are shown as cartoon structures within transparent surfaces. Major clusters of coordinately assembled interacting proteins are surrounded by dashed ovals and illustrated in Figures S6 and S7.
(A) Early MRPs are shown in red, but with mL45 shown in magenta to provide a marker
(B) Intermediate proteins are shown in green, but uL18m and mL63 are colored pea green, since they exhibit earlier binding kinetics than other intermediate proteins.
(C) Late-binding proteins are shown in blue.
Figure 7. Scheme for Assembly of the LSU.
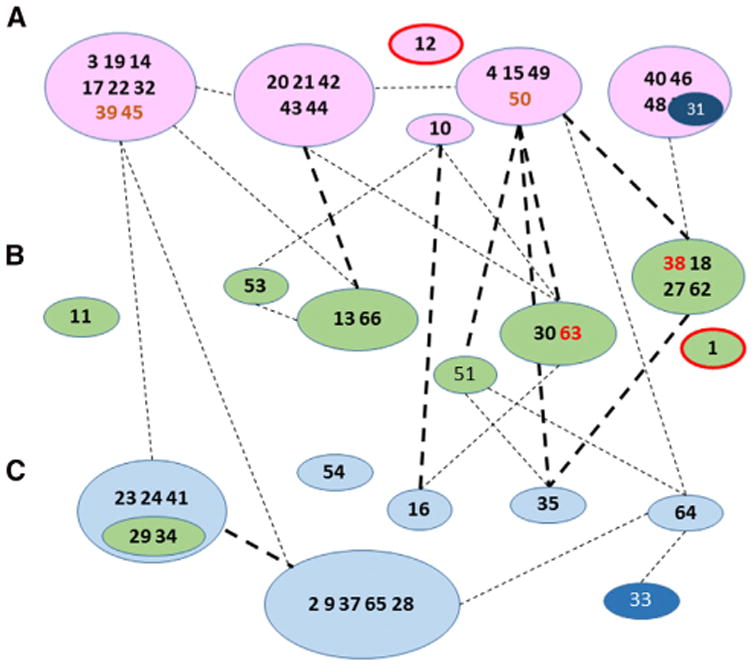
, Assembly scheme showing protein-protein interactions between individual or grouped polypeptides with line designations based on Figure S4 as in Figure 5C and with the longer standard names of individual proteins truncated to numbers for simplicity. uL12m and uL1m are included in the early and intermediate groups based on the kinetic proteomic results but are outlined in red, since they were not identified in the cryo-EM structure. bL31m is shown as an atypical member of an early group that appears to join the structure at a later stage (see text for details). mL39, mL45, and mL50 are shown in boldface brown type, since they are early-binding proteins but do not have extensive RNA contacts. Since they depend on other proteins for assembly, they may be considered secondary binding proteins. mL38 and mL63 are shown in boldface red type, since their assembly kinetics resemble those of early-binding proteins, but they have very close associations with intermediate binding proteins. uL29m and bL34m are shown within a green ellipse associated with a late group, since they showed somewhat earlier assembly kinetics than other group members.
A number of early-binding proteins associate closely with tRNAV in the central protuberance of the 39S subunit, raising the prospect that they may serve to retain the tRNA in the structure at an early stage. There are two possible sources for the tRNA in the structure. Either it is recruited from a free pool of tRNAs or the copy of tRNAV or tRNAF that is co-transcribed with 12S and 16S rRNA is built into the mitoribosome. The latter possibility seems more likely, since only tRNAs adjacent to 12S rRNA are incorporated into the mitoribosome. However, this would require a large-scale relocation of the nascent tRNAV to a position far from its location in the primary transcript, since it is not found near the 5′ end of 16S rRNA in the LSU. How the tRNA is retained in the structure during assembly is not known. In the final mitoribosome structure, the tRNA is nestled between two groups of proteins (Figures 6 and S7B) that have roughly similar incorporation kinetics. One group of proteins with strong mutual interactions, mL40, mL46, and mL48, binds one facet of the tRNA, while a second group containing mL38, uL18m, and bL27m binds the other. The incorporation kinetics for these proteins suggest that the mL40-46-48 group binds at a slightly earlier stage along with mL38, while bL27m, uL18m, and mL62 bind slightly later (Figure S7B). The assembly of the central protuberance presents a structural problem not observed elsewhere in the mitoribosome. The three proteins on one side of the tRNA binding pocket, mL40, mL46, and mL48, all share extensive binding surfaces with bL31m, but bL31m behaved as a late-binding protein in our SILAC data (Figures S7A and S7B). bL31m is rather firmly enclosed by these three proteins in the final cryo-EM structure. Future experiments may shed light on the question of whether the mL40-46-48 trimer is sufficiently flexible to incorporate bL31m at a later stage in assembly.
Overall, 21 of the 24 early-binding LSU proteins are members of four large clusters that also share some weaker binding interactions. All of these clusters contain proteins with high enrichment of nascent protein in the nucleoid fraction (Bogenhagen et al., 2014). Together, the early proteins form a nearly complete band decorating a swath of outer surface of the LSU but largely avoiding the intersubunit face (Figure 6). Interestingly, a recent cryo-EM analysis of late-stage 39S subunit assembly intermediates has also found that the morphology of the intersubunit interface is well organized and well resolved only at a late stage in assembly (Brown et al., 2017).
Intermediate-binding LSU proteins reside in a more scattered distribution on the 39S subunit, often sharing interactions with early-binding proteins (Figure 6B). uL13m-mL66, with a BSA of 2,312 Å2, bind the L10 stalk through interactions with RNA as well as early-binding proteins. These two proteins and uL11m tend to surround uL10m. A major set of intermediate-late proteins (mL41, uL23m, uL24m, uL29, and bL34m) joins the base of the LSU to contribute to the formation of the peptide exit tunnel (Figure S7C). These proteins make few contacts to early proteins or to other intermediate proteins. They likely bind to an RNA surface that becomes available only after rearrangement of the 16S RNA structure following binding of early proteins.
At a later assembly stage, uL2m, mL37, and mL65 join the structure to add a prominent mass adjacent to uL23m:uL29m approaching the interface of the LSU with the SSU (Figures 6C and S7D), where uL2m forms one of the intersubunit bridges (to bS6m). mL37 joins bL28m in extended contacts wrapping along intermediate-binding protein uL29. mL37 and mL65 share a substantial protein-protein interface and serve to compensate for a large evolutionary RNA deletion in domain III of the LSU rRNA (Brown et al., 2014). This pair continues the theme that some proteins that are tightly associated with one another and join the structure at approximately the same stage may bind in a coordinate manner. uL2m is of particular interest, since it shares a relatively large interaction surface with 16S rRNA (Figure S3), but it was not observed at nucleoids. We do not know what prevents binding of uL2m during early stages of mitoribosome assembly, but we suggest that its binding requires RNA conformational changes induced by the binding of primary MRPs.
Discussion
The mitoribosome differs from the bacterial ribosome in its high ratio of protein to RNA as described in elegant recent cryo-EM studies (Amunts et al., 2015; Greber et al., 2015). The assembly of the mammalian mitoribosome is a poorly studied, complex process requiring independent synthesis and mitochondrial import of 82 structural proteins. We report results of an extensive series of SILAC pulse-labeling experiments revealing great variability in the kinetics of appearance of MRPs in intact mitoribosomes. We considered several factors that might contribute to this variability, including the exchange of nascent MRPs into previously assembled mitoribosomes. We found that this mechanism can explain the rapid appearance of one MRP, bL33m, in mitoribosomes, but no others. We also considered the possibility that some MRPs may exhibit delayed assembly because newly imported nascent copies may be diluted by large pools of previously synthesized, unlabeled proteins. We concluded that this was a minor contributing factor, since we found that some of the proteins that assemble slowly do not have large pools of free protein. Thus, we consider that SILAC can distinguish between MRPs that participate at early and late stages in mitoribosome assembly. Interestingly, several MRPs implicated to date in serious mitochondrial disorders participate at early stages in mitoribosome assembly, including uL3, mL44, uS7m, mS22, and uS16m (Baertling et al., 2015; Carroll et al., 2013; Distelmaier et al., 2015; Emdadul Haque et al., 2008; Menezes et al., 2015; Miller et al., 2004; Saada et al., 2007; Smits et al., 2011). Lack of an early-binding protein may be more likely to disrupt mitoribosome structure and subsequent steps in assembly.
Our results (Figure 2) suggested that complete assembly requires ∼2–3 hr, and additional pulse-chase labeling experiments confirmed that early-binding pulse labeled MRPs can be effectively “chased” into completed mitoribosomes (Figure 3). Expanding our pulse-chase SILAC approach to study the entire mitochondrial pool of MRPs (free plus assembled copies) revealed that most MRPs are synthesized and imported in considerable excess over the amount required to support assembly. This influx of nascent MRPs provides a supply of parts for mitoribosome assembly, but the accessibility of the proper proteins exactly when required may still limit the overall pace of assembly. We found that unassembled copies of MRPs are degraded relatively rapidly. This turnover may be essential to degrade MRPs imported into mitochondria not actively involved in mitoribosome assembly to avoid excessive accumulation. These results have important implications for mitochondrial proteostasis and quality control that may be explored in future experiments (Rugarli and Langer, 2012).
An additional factor that may contribute to the apparently long duration required for mitoribosome assembly is the unusual extent to which this must be coordinated with mtRNA processing events. The mitochondrial rRNAs are tandemly transcribed to generate a precursor termed RNA 4 (Gelfand and Attardi, 1981) containing 12S-tRNAV-16S RNA and possibly tRNAF that is cleaved by RNase P and ELAC2 to separate the mature RNAs. These incisions, led by RNase P, are not necessarily rapid steps, as evidenced by the facile detection of the RNA 4 precursor. mtRNA processing is clearly essential for mitoribosome assembly, as illustrated by the deleterious effects on mitoribosome assembly resulting from genetic inactivation of the RNase P nuclease, MRPP3 (Rackham et al., 2016). It will be important to determine how potential impairment of RNase P activity may contribute to the clinical impact of mutations in RNase P subunit genes (Alsmadi et al., 2009; Zschocke, 2012). Since RNA processing and protein-RNA binding occur in the same compartment and the RNA scaffold lacks extragenic spacer regions it is important that early-binding MRPs should not block cleavage by RNase P or RNase Z, or access of RNA modification enzymes. Our previous finding that SSU MRPs are more abundant in nucleoid preparations than LSU proteins (Bogenhagen et al., 2014) implies that SSU assembly may progress further than LSU assembly before either intermediate is liberated from the nucleoid and transferred to a nearby RNA granule (Jourdain et al., 2016), where mitoribosome assembly is thought to continue. Super-resolution structured illumination microscopy (SIM) and stochastic optical reconstruction microscopy (STORM) imaging shows that most RNA granules in cultured HeLa cells are in intimate contact with nucleoids to facilitate this transfer (D.F.B., unpublished data).
SILAC labeling suggested a temporal hierarchy for mitoribosome assembly events. Examination of the roster of early-binding MRPs identified numerous examples of sets of proteins that shared both similar binding kinetics and extensive buried surface areas (BSAs) in the final structure. Detailed analysis of protein-protein and protein-RNA interactions identified twelve instances where three or more very closely associated proteins join the complex at the same stage in assembly, along with two other closely linked pairs. These coordinately assembled modules represent 60 of the 82 MRPs, indicating that protein-protein interactions have a far greater relative importance in assembly of the mitoribosome than in the bacterial ribosome. In both systems, assembly is rather probabilistic in nature as it characterizes a general landscape of protein binding that most likely does not follow a strictly ordered sequence of events. Coordinated assembly appears to be necessary to recruit eight proteins that bind at early stages in mitoribosome assembly but lack substantial contact with rRNAs, including mS22, mS31, mS35, mS39, bS1m, mL39, mL50, and mL45. Mitoribosome assembly would be facilitated by association of MRPs in pre-assembled sub-complexes, as has been documented for assembly of mammalian respiratory complex I (Guerrero-Castillo et al., 2017). While this is not absolutely required for coordinated assembly, the model generated by our SILAC labeling approach provides a guide for future experiments to search for such intermediates and to dissect the assembly sequence in greater detail.
Experimental Procedures
Cell Culture and Mitoribosome Preparation
HeLa cell culture, SILAC pulse labeling, and mitochondrial isolation were conducted as described previously (Bogenhagen et al., 2014; Lee and Bogenhagen, 2016). Each preparation typically started with ∼1.5 to 2 × 108 adherent cells. Mitochondrial lysis and mitoribosome preparation followed Matthews and O'Brien (Matthews et al., 1982) as adapted by Brown et al. (2014) to permit mitochondrial isolation and mitoribosome purification within 24–26 hr without freezing. Detailed procedures are in Supplemental Experimental Procedures.
Peptide Identification and Quantification by LC-MS/MS
Trypsin digestion and LC-MS/MS analysis of peptides was performed using standard methods as described in Supplemental Experimental Procedures.
Statistical Methods of Data Analysis
Peptide data produced by SCIEX Protein Pilot software in text file format was imported into Excel and searched to select only peptides derived from human MRPs. In some experiments, results were filtered for all mitochondrial proteins in the Mitocarta 2.0 collection (Calvo et al., 2016). Peptides were sorted to eliminate those with low heavy or light peptide signals, confidence scores <90%, and those that might represent another peptide. Peptides with in appropriately high H:L ratios in the highest 2.5% of the distribution were manually removed as outliers. Peptide information was imported into SAS JMP 13 software to generate average H:L ratios and SE calculations using robust fitting (Huber, 1973). Tests of statistical significance used one-way ANOVA with pair-wise Student's t test. Structural renderings were generated using Pymol.
Supplementary Material
Highlights.
Pulse-chase SILAC provides a coarse-grained model for mitoribosome assembly
Assembly of the protein-rich mitoribosome requires 2–3 hr
Most mitoribosomal proteins are synthesized in excess and degraded if not assembled
Mitoribosomal proteins implicated in human disease bind early during assembly
Acknowledgments
D.F.B. would like to acknowledge the accomplishments of his Stony Brook University colleague Dr. Melvin Simpson in making the first observations of translation in mitochondria and reaching his 96th birthday. This work was supported by grants to D.F.B. from NIH (1R01GM112790) and the United Mitochondrial Disease Foundation.
Footnotes
Supplemental Information: Supplemental Information includes Supplemental Experimental Procedures, seven figures, three tables, and two datasets and can be found with this article online at https://doi.org/10.1016/j.celrep.2018.01.066.
Author Contributions: D.F.B. performed all cell labeling, fractionation, and preparation of samples for proteomic analysis with technical assistance from A.G.O.-F. J.D.H. performed protein digestion and LC-MS/MS analysis. J.D.H. and D.F.B. analyzed proteomic data. M.G.-D. and D.F.B. conducted the structural biology analyses and wrote the paper.
Declaration of Interests: The authors declare no competing interests.
References
- Alsmadi O, Muiya P, Khalak H, Al-Saud H, Meyer BF, Al-Mohanna F, Alshahid M, Dzimiri N. Haplotypes encompassing the KIAA0391 and PSMA6 gene cluster confer a genetic link for myocardial infarction and coronary artery disease. Ann Hum Genet. 2009;73:475–483. doi: 10.1111/j.1469-1809.2009.00534.x. [DOI] [PubMed] [Google Scholar]
- Amunts A, Brown A, Bai XC, Llácer JL, Hussain T, Emsley P, Long F, Murshudov G, Scheres SHW, Ramakrishnan V. Structure of the yeast mitochondrial large ribosomal subunit. Science. 2014;343:1485–1489. doi: 10.1126/science.1249410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amunts A, Brown A, Toots J, Scheres SHW, Ramakrishnan V. Ribosome. The structure of the human mitochondrial ribosome Science. 2015;348:95–98. doi: 10.1126/science.aaa1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson S, Bankier AT, Barrell BG, de Bruijn MHL, Coulson AR, Drouin J, Eperon IC, Nierlich DP, Roe BA, Sanger F, et al. Sequence and organization of the human mitochondrial genome. Nature. 1981;290:457–465. doi: 10.1038/290457a0. [DOI] [PubMed] [Google Scholar]
- Baertling F, Haack TB, Rodenburg RJ, Schaper J, Seibt A, Strom TM, Meitinger T, Mayatepek E, Hadzik B, Selcan G, et al. MRPS22 mutation causes fatal neonatal lactic acidosis with brain and heart abnormalities. Neurogenetics. 2015;16:237–240. doi: 10.1007/s10048-015-0440-6. [DOI] [PubMed] [Google Scholar]
- Bahadur RP, Chakrabarti P, Rodier F, Janin J. Dissecting sub-unit interfaces in homodimeric proteins. Proteins. 2003;53:708–719. doi: 10.1002/prot.10461. [DOI] [PubMed] [Google Scholar]
- Bogenhagen DF, Martin DW, Koller A. Initial steps in RNA processing and ribosome assembly occur at mitochondrial DNA nucleoids. Cell Metab. 2014;19:618–629. doi: 10.1016/j.cmet.2014.03.013. [DOI] [PubMed] [Google Scholar]
- Brown A, Amunts A, Bai XC, Sugimoto Y, Edwards PC, Murshudov G, Scheres SHW, Ramakrishnan V. Structure of the large ribosomal subunit from human mitochondria. Science. 2014;346:718–722. doi: 10.1126/science.1258026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown A, Rathore S, Kimanius D, Aibara S, Bai XC, Rorbach J, Amunts A, Ramakrishnan V. Structures of the human mitochondrial ribosome in native states of assembly. Nat Struct Mol Biol. 2017;24:866–869. doi: 10.1038/nsmb.3464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calvo SE, Clauser KR, Mootha VK. MitoCarta2.0: an updated inventory of mammalian mitochondrial proteins. Nucleic Acids Res. 2016;44(D1):D1251–D1257. doi: 10.1093/nar/gkv1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll CJ, Isohanni P, Pöyhönen R, Euro L, Richter U, Brilhante V, Götz A, Lahtinen T, Paetau A, Pihko H, et al. Whole-exome sequencing identifies a mutation in the mitochondrial ribosome protein MRPL44 to underlie mitochondrial infantile cardiomyopathy. J Med Genet. 2013;50:151–159. doi: 10.1136/jmedgenet-2012-101375. [DOI] [PubMed] [Google Scholar]
- Desai N, Brown A, Amunts A, Ramakrishnan V. The structure of the yeast mitochondrial ribosome. Science. 2017;355:528–531. doi: 10.1126/science.aal2415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Distelmaier F, Haack TB, Catarino CB, Gallenmüller C, Rodenburg RJ, Strom TM, Baertling F, Meitinger T, Mayatepek E, Prokisch H, Klopstock T. MRPL44 mutations cause a slowly progressive multisystem disease with childhood-onset hypertrophic cardiomyopathy. Neurogenetics. 2015;16:319–323. doi: 10.1007/s10048-015-0444-2. [DOI] [PubMed] [Google Scholar]
- Emdadul Haque M, Grasso D, Miller C, Spremulli LL, Saada A. The effect of mutated mitochondrial ribosomal proteins S16 and S22 on the assembly of the small and large ribosomal subunits in human mitochondria. Mitochondrion. 2008;8:254–261. doi: 10.1016/j.mito.2008.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Englmeier R, Pfeffer S, Förster F. Structure of the human mitochondrial ribosome studied in situ by cryoelectron tomography. Structure. 2017;25:1574–1581. doi: 10.1016/j.str.2017.07.011. e2. [DOI] [PubMed] [Google Scholar]
- Gelfand R, Attardi G. Synthesis and turnover of mitochondrial ribonucleic acid in HeLa cells: the mature ribosomal and messenger ribonucleic acid species are metabolically unstable. Mol Cell Biol. 1981;1:497–511. doi: 10.1128/mcb.1.6.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greber BJ, Boehringer D, Leitner A, Bieri P, Voigts-Hoffmann F, Erzberger JP, Leibundgut M, Aebersold R, Ban N. Architecture of the large subunit of the mammalian mitochondrial ribosome. Nature. 2014;505:515–519. doi: 10.1038/nature12890. [DOI] [PubMed] [Google Scholar]
- Greber BJ, Bieri P, Leibundgut M, Leitner A, Aebersold R, Boehringer D, Ban N. Ribosome. The complete structure of the 55S mammalian mitochondrial ribosome Science. 2015;348:303–308. doi: 10.1126/science.aaa3872. [DOI] [PubMed] [Google Scholar]
- Guerrero-Castillo S, Baertling F, Kownatzki D, Wessels HJ, Arnold S, Brandt U, Nijtmans L. The assembly pathway of mitochondrial respiratory chain complex I Cell Metab. 2017;25:128–139. doi: 10.1016/j.cmet.2016.09.002. [DOI] [PubMed] [Google Scholar]
- Huber PJ. Robust regression: asymptotics, conjectures and Monte Carlo. Ann Stat. 1973;1:799–821. [Google Scholar]
- Jourdain AA, Boehm E, Maundrell K, Martinou JC. Mitochondrial RNA granules: Compartmentalizing mitochondrial gene expression. J Cell Biol. 2016;212:611–614. doi: 10.1083/jcb.201507125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kastritis PL, Bonvin AM. On the binding affinity of macromolecular interactions: daring to ask why proteins interact. J R Soc Interface. 2012;10:20120835. doi: 10.1098/rsif.2012.0835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krissinel E. Crystal contacts as nature's docking solutions. J Comput Chem. 2010;31:133–143. doi: 10.1002/jcc.21303. [DOI] [PubMed] [Google Scholar]
- Lee KW, Bogenhagen D. Scalable isolation of mammalian mitochondria for nucleic acid and nucleoid analysis In Mitochondrial DNA, M McKenzie, ed. (Springer); 2016. pp. 67–79. [DOI] [PubMed] [Google Scholar]
- Matthews DE, Hessler RA, Denslow ND, Edwards JS, O'Brien TW. Protein composition of the bovine mitochondrial ribosome. J Biol Chem. 1982;257:8788–8794. [PubMed] [Google Scholar]
- Menezes MJ, Guo Y, Zhang J, Riley LG, Cooper ST, Thorburn DR, Li J, Dong D, Li Z, Glessner J, et al. Mutation in mitochondrial ribosomal protein S7 (MRPS7) causes congenital sensorineural deafness, progressive hepatic and renal failure and lactic acidemia. Hum Mol Genet. 2015;24:2297–2307. doi: 10.1093/hmg/ddu747. [DOI] [PubMed] [Google Scholar]
- Miller C, Saada A, Shaul N, Shabtai N, Ben-Shalom E, Shaag A, Hersh-kovitz E, Elpeleg O. Defective mitochondrial translation caused by a ribosomal protein (MRPS16) mutation. Ann Neurol. 2004;56:734–738. doi: 10.1002/ana.20282. [DOI] [PubMed] [Google Scholar]
- Rackham O, Busch JD, Matic S, Siira SJ, Kuznetsova I, Atanassov I, Ermer JA, Shearwood AM, Richman TR, Stewart JB, et al. Hierarchical RNA processing is required for mitochondrial ribosome assembly. Cell Rep. 2016;16:1874–1890. doi: 10.1016/j.celrep.2016.07.031. [DOI] [PubMed] [Google Scholar]
- Rugarli EI, Langer T. Mitochondrial quality control: A matter of life and death for neurons. EMBO J. 2012;31:1336–1349. doi: 10.1038/emboj.2012.38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saada A, Shaag A, Arnon S, Dolfin T, Miller C, Fuchs-Telem D, Lombes A, Elpeleg O. Antenatal mitochondrial disease caused by mitochondrial ribosomal protein (MRPS22) mutation. J Med Genet. 2007;44:784–786. doi: 10.1136/jmg.2007.053116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwanhäusser B, Busse D, Li N, Dittmar G, Schuchhardt J, Wolf J, Chen W, Selbach M. Global quantification of mammalian gene expression control. Nature. 2011;473:337–342. doi: 10.1038/nature10098. [DOI] [PubMed] [Google Scholar]
- Shajani Z, Sykes MT, Williamson JR. Assembly of bacterial ribosomes. Annu Rev Biochem. 2011;80:501–526. doi: 10.1146/annurev-biochem-062608-160432. [DOI] [PubMed] [Google Scholar]
- Smits P, Saada A, Wortmann SB, Heister AJ, Brink M, Pfundt R, Miller C, Haas D, Hantschmann R, Rodenburg RJT, et al. Mutation in mitochondrial ribosomal protein MRPS22 leads to Cornelia de Lange-like phenotype, brain abnormalities and hypertrophic cardiomyopathy. Eur J Hum Genet. 2011;19:394–399. doi: 10.1038/ejhg.2010.214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talkington MWT, Siuzdak G, Williamson JR. An assembly landscape for the 30S ribosomal subunit. Nature. 2005;438:628–632. doi: 10.1038/nature04261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ugalde C, Vogel R, Huijbens R, Van Den Heuvel B, Smeitink J, Nijtmans L. Human mitochondrial complex I assembles through the combination of evolutionary conserved modules: a framework to interpret complex I deficiencies. Hum Mol Genet. 2004;13:2461–2472. doi: 10.1093/hmg/ddh262. [DOI] [PubMed] [Google Scholar]
- Zschocke J. HSD10 disease: clinical consequences of mutations in the HSD17B10 gene. J Inherit Metab Dis. 2012;35:81–89. doi: 10.1007/s10545-011-9415-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.