

Abstract
The formation of mate recognition memory in mice is associated with neural changes at the reciprocal dendrodendritic synapses between glutamatergic mitral cell (MC) projection neurons and GABAergic granule cell (GC) interneurons in the accessory olfactory bulb (AOB). Although noradrenaline (NA) plays a critical role in the formation of the memory, the mechanism by which it exerts this effect remains unclear. Here we used extracellular field potential and whole-cell patch-clamp recordings to assess the actions of bath-applied NA (10 µM) on the glutamatergic transmission and its plasticity at the MC-to-GC synapse in the AOB. Stimulation (400 stimuli) of MC axons at 10 Hz but not at 100 Hz effectively induced N-methyl-d-aspartate (NMDA) receptor-dependent long-term potentiation (LTP), which exhibited reversibility. NA paired with subthreshold 10-Hz stimulation (200 stimuli) facilitated the induction of NMDA receptor-dependent LTP via the activation of α2-adrenergic receptors (ARs). We next examined how NA, acting at α2-ARs, facilitates LTP induction. In terms of acute actions, NA suppressed GC excitatory postsynaptic current (EPSC) responses to single pulse stimulation of MC axons by reducing glutamate release from MCs via G-protein coupled inhibition of calcium channels. Consequently, NA reduced recurrent inhibition of MCs, resulting in the enhancement of evoked EPSCs and spike fidelity in GCs during the 10-Hz stimulation used to induce LTP. These results suggest that NA, acting at α2-ARs, facilitates the induction of NMDA receptor-dependent LTP at the MC-to-GC synapse by shifting its threshold through disinhibition of MCs.
The mammalian main olfactory bulb (MOB) at the first stage of olfactory processing receives a wealth of centrifugal noradrenergic projections from the locus coeruleus (LC) that is instrumental for a broad range of olfactory behaviors (Macrides et al. 1981; Shipley et al. 1985; McLean et al. 1989). Enhanced release of noradrenaline (NA) within the MOB plays an important role in long-term synaptic plasticity and odor learning in a variety of contexts. These include olfactory conditioning in neonatal rats (Sullivan et al. 1989, 1992, 2000; Harley et al. 2006; Zhang et al. 2010; Shakhawat et al. 2012), the learning of newborn lamb odors after parturition in sheep (Pissonnier et al. 1985), odor discrimination after memory formation in mice (Doucette et al. 2007; Shea et al. 2008; Moreno et al. 2012), a specific long-term suppression of mitral cell (MC) responses to paired odors in mice (Shea et al. 2008), a long-term reduction of paired-pulse inhibition in neonatal rats (Wilson and Leon 1988), long-term enhancement of synchronized γ frequency oscillations in rat MOB slices (Gire and Schoppa 2008; Pandipati et al. 2010), long-term potentiation (LTP) of synaptic strength in rat MOB slices (Yuan 2009; Zhang et al. 2010), and a long-term suppression of presynaptic input to MCs in mice (Eckmeier and Shea 2014).
LC activation enhances MC responses to weak olfactory nerve input (Jiang et al. 1996). This may be due to a transient reduction of the recurrent inhibition of MCs by LC activation (Okutani et al. 1998). Disinhibition of MCs may contribute to long-term synaptic plasticity in the MOB (Pandipati et al. 2010). Therefore, a plausible candidate mechanism that could initiate long-term changes to the MOB network would be the synergy of sensitization and disinhibition of MCs (Brennan and Keverne 1997; Gire and Schoppa 2008; Shea et al. 2008; Pandipati et al. 2010; Eckmeier and Shea 2014).
Similarly, enhanced release of NA within the accessory olfactory bulb (AOB) is vital for the expression of mate recognition memory in mice, in which a female forms a memory to the urinary chemosignals of her male partner during a critical postmating window (Keverne and de la Riva 1982; Rosser and Keverne 1985). This mate recognition memory is essential for preventing the activation of the neuroendocrine mechanisms leading to pregnancy loss (Keverne and Rosser 1986). It has been hypothesized that the mate recognition memory functions as a gate to suppress or modulate the mate's chemosignals at the level of the AOB (Brennan et al. 1990; Kaba and Nakanishi 1995; Brennan and Zufall 2006; Griffiths and Brennan 2015). Synaptic connections in the external plexiform layer (EPL) of the AOB are dominated by the reciprocal dendrodendritic synapses between MCs, a single class of projection neurons, and granule cell (GC) interneurons. Glutamate released from MC dendrites activates the GC dendrites, which in turn mediate the GABAergic dendrodendritic inhibition (DDI) of the MC dendrites (Jia et al. 1999; Taniguchi and Kaba 2001). This feedback inhibition at the reciprocal synapses regulates MC output (Jia et al. 1999; Taniguchi and Kaba 2001; Castro et al. 2007; Taniguchi et al. 2013).
The mate recognition memory involves the formation of an association of the mate's chemosignals and the mating signal in the AOB. The most likely source of this mating signal is the noradrenergic projection from the LC (Shipley et al. 1985; McLean et al. 1989). Artificial vagino-cervical stimulation (Rosser and Keverne 1985) or mating (Brennan et al. 1995) promotes NA release in the AOB. A blockade of α-adrenergic receptors (ARs), but not β-ARs, in the AOB immediately after mating prevented the formation of the mate recognition memory (Kaba and Keverne 1988), as did the destruction of the noradrenergic innervation of the AOB prior to mating (Rosser and Keverne 1985). In addition, memory formation is associated with neurochemical and morphological changes at the MC–GC reciprocal synapses (Brennan et al. 1995; Matsuoka et al. 1997, 2004).
Despite advances such as these, the cellular and synaptic mechanisms underlying the noradrenergic modulation of the mate recognition memory are unknown. Here we report that NA, acting at α2-ARs, facilitates the induction of an N-methyl-d-aspartate (NMDA) receptor-dependent LTP at the MC-to-GC synapse by enhancing responses and spike fidelity in GCs during 10-Hz inputs through disinhibition of MCs.
Results
10-Hz stimulation induced NMDA receptor-dependent LTP in the AOB
Prior electron-microscopic analyses revealed an increase in the length of the postsynaptic densities of the MC-to-GC glutamatergic side of the reciprocal synapses in the AOB, 24 h after the association of mating and exposure to male chemosignals (Matsuoka et al. 1997, 2004). Because such morphological changes may represent a morphological correlate of synaptic plasticity, we focused on the MC-to-GC excitatory synapse as a primary site for the modifiability of synaptic strength via LTP and other processes. The MC output neurons of the AOB project mainly to the amygdala via the lateral olfactory tract (LOT). Therefore, stimulation of the LOT antidromically activates MCs and engages the MC-to-GC synapse. In the present experiments, field potentials were recorded from the EPL containing the reciprocal dendrodendritic synapses. The stimulation of the LOT in the mouse slice preparation evoked two sequential deflections, as originally reported in rats by Jia et al. (1999) (Fig. 1A,B). Immediately after the stimulus artifact, there was a brief negativity (N1), which represents antidromic activation of the MCs. This was followed by a slower negative deflection (N2), which represents a field excitatory postsynaptic potential (fEPSP) evoked in GC peripheral dendrites in the EPL, as the corresponding potential has been closely correlated with the intracellularly recorded EPSP (Jia et al. 1999). The synaptic response was little affected by the NMDA receptor antagonist DL-2-amino-5-phosphonopentanoic acid (AP5), but was completely blocked by the non-NMDA receptor antagonist 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX). The initial response, however, was resistant to both AP5 and CNQX, but was eliminated by the sodium channel blocker tetrodotoxin (TTX) (Fig. 1B). These results confirm those obtained by Fang et al. (2008).
Figure 1.

Properties of LTP in the mouse AOB, induced by 10-Hz stimulation. (A) Schematic diagram of major synaptic interconnections and positions of the recording (Rec) and stimulating (Stim) electrodes in the AOB. (EPL) external plexiform layer, (GL) glomerular layer, (Glu) glutamate, (GRL) granule cell layer, (IPL/LOT) internal plexiform layer/lateral olfactory tract, (NP) noradrenergic projection, (VNL) vomeronasal nerve layer. (B) Pharmacological properties of LOT-evoked negative field potentials, N1 and N2. Only TTX (1 µM) blocked N1, whereas N2 was blocked by CNQX (30 µM) but not by AP5 (50 µM). Calibration: 5 msec, 0.5 mV. (C,D) Dependence on stimulus frequency and NMDA receptors. Four trains of high-frequency stimulation (100-Hz, 100-pulse train spaced 3 min apart) did not induce LTP. Five or 10 trains of 10-Hz stimulation (10-Hz, 20-pulse train spaced 3 min apart) induced a transient potentiation that decayed to baseline, whereas 20 trains induced stable LTP that lasted at least 3 h. This LTP was blocked by AP5. (Inset) Superimposed traces taken at the indicated times. (E) Depotentiation. Low-frequency stimulation (1 Hz, 15 min) depotentiated the LTP. Downward arrows represent the times at which 100- or 10-Hz stimulation started. The upward arrow represents the time at which 1-Hz stimulation started.
High-frequency stimulation that normally produces robust LTP in other areas was not effective in inducing reliable LTP (118% ± 3%, n = 8) (Fig. 1C). In contrast, 10-Hz stimulation, which is within the physiological range of natural neuronal activity (Luo et al. 2003; Leszkowicz et al. 2012), effectively induced LTP. Five or 10 trains of 10-Hz stimulation induced only short-term potentiation of the synaptic response that decayed back to its control value (101% ± 4% for five trains, n = 7; 102% ± 3% for 10 trains, n = 7), whereas 20 trains induced LTP that remained potentiated for at least 3 h (144% ± 3%, P < 0.001, n = 8, paired t-test) (Fig. 1D). Moreover, in the presence of AP5 (50 µM) only short-term potentiation was elicited (105% ± 6%, n = 7) (Fig. 1D), indicating that this form of LTP depends on NMDA receptor activation.
A feature that is shared by LTP and learning is that, for the most part, both are reversible. Low-frequency stimulation can depress synapses that have recently undergone LTP in other brain areas (Stäubli and Chun 1996). We tested the reversibility of LTP induced in N2 by using a similar protocol. Low-frequency stimulation (1 Hz, 15 min) produced a significant amount of depotentiation when given 30 min after the completion of 10-Hz stimulation lasting 1 h (17% ± 3%, expressed as the magnitude of depotentiation, P = 0.03, n = 9, paired t-test) (Fig. 1E).
NA facilitated LTP induction via α2-ARs
We hypothesized that NA released into the AOB at mating facilitates the induction of LTP of synaptic strength at the MC-to-GC synapses that are activated by the mate's chemosignals. Subsequent exposure to unfamiliar males would result in these males’ pregnancy-blocking signals being transmitted, whereas the pregnancy-blocking signals from the mate would be selectively inhibited by an increase in GABAergic inhibitory feedback from the GCs via the potentiated MC-to-GC synapses. We tested this possibility by pairing 10-Hz stimulation with bath-applied NA (10 µM, a concentration widely used in studies with similar slice preparations).
Indeed, we observed robust LTP when we paired NA with subthreshold stimuli (200 pulses at 10 Hz) that by themselves are insufficient for inducing LTP and only produced short-term potentiation (161%, n = 8, P < 0.001, paired t-test) (Fig. 2A). These results indicate that NA can facilitate LTP induction at the MC-to-GC synapse. NA-facilitated LTP was also blocked by the NMDA receptor antagonist AP5 (99% ± 4%, n = 8) (Fig. 2A). We further determined which of the AR subtypes was responsible for the noradrenergic enhancement of LTP (Fig. 2B–D). NA-facilitated LTP was blocked by the α-AR antagonist phentolamine (n = 6), but not by the β-AR antagonist propranolol (n = 6) (Fig. 2B). In addition, NA-facilitated LTP was blocked by the α2-AR antagonist idazoxan (n = 7), but not by the α1-AR antagonist prazosin (n = 8) (Fig. 2C), and it was mimicked by the α2-AR agonist clonidine (n = 8) (Fig. 2D). Figure 2E summarizes the effects of these drugs on LTP induction. These results clearly demonstrate that NA facilitates LTP induction via α-ARs of the α2-type.
Figure 2.

The pairing of α2-AR activation and 10-Hz stimulation induced LTP. (A) Records showing NA-facilitated LTP. Ten trains of 10-Hz stimulation elicited the short-term potentiation of transmission that decayed back to baseline (Control). However, pairing this stimulation with a perfusion of NA (10 µM) resulted in the generation of robust LTP. AP5 (50 µM) blocked NA-facilitated LTP as well. Note that NA reduced the baseline fEPSP slope during test stimulation and enhanced it immediately after the drug washed out. (B) Phentolamine (Phe), but not propranolol (Pro), blocked the NA effect. (C) Blockade of the NA effect was also obtained by idazoxan (Ida) but not by prazosin (Pra). (D) The NA effect was mimicked by clonidine (Clo). The bars indicate drug application. (E) The effects of the AR agonists and antagonists. (***) P < 0.001 versus the no-drug control (Cont), ANOVA (F(6,43) = 10.99, P < 0.001) followed by Dunnett's test.
NA suppressed glutamate release from MCs in response to single pulse stimulation
How does NA, acting at α2-ARs, facilitate LTP induction at the MC-to-GC synapse? To address this question, we used whole-cell patch-clamp techniques. We examined the effect of NA on evoked excitatory postsynaptic currents (eEPSCs) recorded from GCs. EPSCs were elicited by single pulse stimulation of the LOT. NA reduced the peak amplitude of eEPSCs by 41% ± 1% (n = 8, P < 0.001, paired t-test) without affecting the decay of the synaptic currents, and this effect was mimicked by the α2-AR agonist clonidine, which reduced the peak amplitude of eEPSCs by 48% ± 2% (n = 7, P < 0.001, paired t-test) (Fig. 3A,B). These results indicate that NA suppresses glutamatergic transmission from MCs to GCs via presynaptic α2-ARs.
Figure 3.
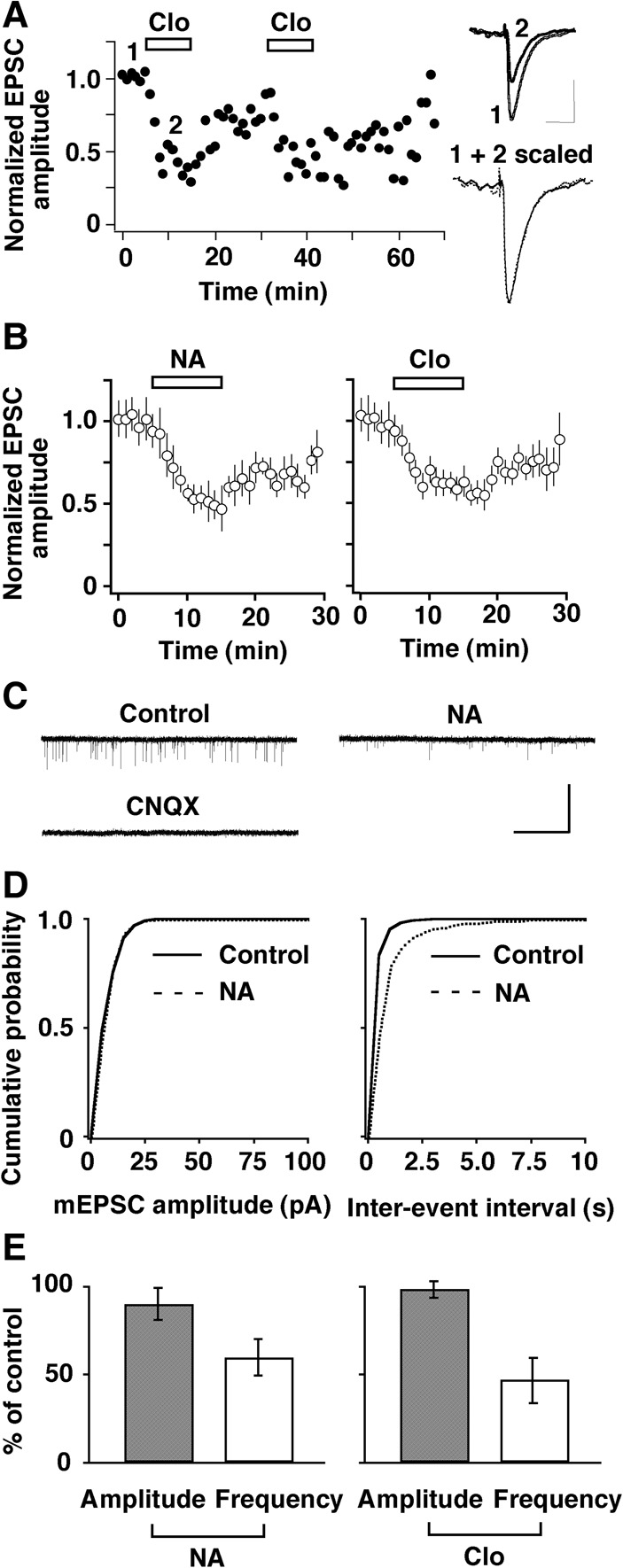
Activation of α2-AR suppressed the excitatory transmission from MCs to GCs via a presynaptic mechanism. (A) A representative example of the reproducible and reversible reduction in the amplitude (normalized to predrug values) of eEPSCs recorded from GCs by the α2-AR agonist clonidine (Clo, 5 µM). (Inset) Superimposed traces taken at the indicated times. Calibration: 50 msec, 40 pA. The EPSC in the presence of Clo is scaled to the size of the control response, and both traces are superimposed to show a comparable decay. (B) Summarized results showing the time course of the reduction in eEPSC amplitude by NA (10 µM, left) and Clo (right). Bars indicate drug application. (C) Traces showing typical mEPSCs recorded from a GC before and during the application of NA (10 µM) and blockade of the currents by CNQX (30 µM). NA suppressed the frequency but not the size of events. Calibration: 20 sec, 10 pA. (D) Plots of the probability ratios of events from the cell shown in (C) against their amplitudes and inter-event intervals. NA shifted the frequency curve to the right, but the amplitude curve was unchanged. (E) Pooled data illustrating relative changes in mean amplitude and mean frequency of mEPSCs caused by NA (left) and clonidine (Clo, 5 µM, right).
It is important to clearly establish that the NA site of action is just at the presynaptic side of this reciprocal synaptic dyad. We thus analyzed the frequency and amplitude of miniature EPSCs (mEPSCs) recorded from GCs (Fig. 3C–E). NA did not change the amplitude of events, but the frequency of the events dropped markedly so that the cumulative frequency curve shifted to the right (Fig. 3D). On average, the mean frequency decreased during NA application to 60% ± 11% of the control frequency (n = 9, P = 0.024, paired t-test), whereas the mean amplitude was not significantly affected (90% ± 10% of control, P = 0.88, paired t-test) (Fig. 3E, left). The synaptic effects of NA on mEPSCs were mimicked by the α2-AR agonist clonidine (n = 7, 47% ± 13% of control frequency, P = 0.013, paired t-test; 99% ± 3% of control amplitude, P = 0.99, paired t-test) (Fig. 3E, right), confirming NA's presynaptic mode of action.
Mechanism of the noradrenergic suppression of glutamate release from MCs
Presynaptic receptors of central neurons are believed to reduce the transmitter release most commonly by inhibiting voltage-gated Ca2+ channels (Miller 1998). Here we first examined the effects of NA and clonidine on Ba2+ currents in MCs. A depolarizing voltage step from a holding potential of −70 mV to various test potentials evoked a sustained inward current that Cd2+ (200 µM) completely blocked (Fig. 4A,B). This inward current activated near −40 mV, peaked at −10 mV, and reversed near +45 mV. These characteristics suggest that the current was from high-voltage-gated Ca2+ channels. NA suppressed the high-threshold Ba2+ currents in a reversible manner in 14 of 17 MCs tested.
Figure 4.

NA inhibited high-voltage-activated Ca2+ channels in MCs via a G-protein. (A) Individual current records before, during, and after bath application of NA (10 µM). Ba2+ was used as the charge carrier. Calibration: 50 msec, 0.5 nA. (B) Current-voltage relationships taken before and during NA (10 µM, left) or clonidine (Clo, 5 µM, right) and after Cd2+(200 µM). (C) Individual current records before and during the application of Clo (5 µM) in slices treated with vehicle or PTX. Calibration: 50 msec, 0.5 nA. (D) Pooled data showing that the Gi/o-protein inhibitor PTX reverses the effect of Clo. (**) P < 0.01 versus the predrug control.
The current-voltage plot shows a near-uniform current suppression at all voltages, suggesting that the activation parameters are not shifted along the voltage axis (Fig. 4B, left). In the MCs that responded, the mean suppression of the peak was 30% ± 3% (n = 14, P = 0.009, paired t-test). The effect of NA was mimicked by the α2-AR agonist clonidine (Fig. 4B, right); the mean suppression was 29% ± 4% (n = 13, P = 0.015, paired t-test). Consistent with the findings of Dong et al. (2009), our results showed that NA suppressed the high-threshold Ca2+ currents via α2-AR activation.
Presynaptic α2-adrenergic inhibition is most frequently mediated by proteins of the Gi/Go class (Saunders and Limbird 1999). We next examined whether this type of G protein mediates the suppressive action of NA in MCs in slices treated with vehicle or pertussis toxin (PTX), which inactivates the Gi/o family of G proteins (Fig. 4C). Clonidine suppressed Ca2+ currents in MCs treated with vehicle; the mean suppression was 40% ± 3% (n = 12, P < 0.01, Tukey–Kramer's test) (Fig. 4D). In contrast, clonidine failed to suppress Ca2+ currents in MCs treated with PTX; the mean suppression was 8% ± 0.4% (n = 18, P > 0.05, Tukey–Kramer's test) (Fig. 4D), suggesting Gi/o protein-mediated inhibition of Ca2+ currents by α2-AR activation.
Presynaptic receptors may reduce transmitter release not only by inhibiting Ca2+ channels, but also by facilitating voltage-gated K+ channels (Miller 1998), which regulate high-fidelity synaptic transmission (Nakamura and Takahashi 2007; Yang et al. 2014). We thus tested the effects of clonidine on outward K+ currents in MCs. The results showed that clonidine failed to affect outward currents evoked by ramp depolarizations from −100 to +70 mV (Supplemental Fig. S1). Finally, the recordings of miniature inhibitory postsynaptic currents (mIPSCs) from MCs and voltage-gated Ba2+ currents from GCs showed that α2-AR activation had no effects on GABAergic transmission from GCs to MCs (Supplemental Figs. S2, S3). Taken together, our results indicate that α2-AR activation suppresses presynaptic glutamate release from MCs by a Gi/o-protein-mediated inhibition of Ca2+ channels without affecting GABAergic transmission from GCs to MCs.
NA-enhanced GC responses to 10-Hz stimulation
The NA-induced reduction in MC-to-GC transmission raises the possibility that NA, acting at α2-AR, decreases inhibitory feedback from GCs, disinhibiting MCs. We examined the effect of the α2-AR agonist clonidine on reciprocal transmission between MCs and GCs by stimulating an MC and recording the evoked IPSCs from the same cell. Conventional whole-cell recordings with Cs+-internal solution in the pipette were performed in the presence of TTX (1 µM) to block Na+ channels and thus prevent any contribution of axonal transmission. A depolarizing voltage step reported to evoke a relatively slow inward Ca2+ current (Isaacson and Strowbridge 1998; Schoppa et al. 1998; Taniguchi and Kaba 2001; Castro et al. 2007) was followed by IPSCs (Fig. 5A). Following the addition of clonidine (5 µM), there was a significant reduction in reciprocal DDI from 357 (median) to 85 pA·sec (n = 7, P = 0.0156, Wilcoxon signed-rank test), which was reversible (Fig. 5B–E). These results showed that α2-AR activation suppressed the reciprocal synaptic currents triggered by endogenous glutamate release from MCs, thereby disinhibiting MCs.
Figure 5.

NA disinhibited MCs, enhancing GC responses to 10-Hz stimulation. (A) IPSCs recorded from an MC bathed in ACSF containing no Mg2+. (B) IPSCs recorded from the same cell bathed in ACSF containing clonidine (Clo, 5 µM), and no Mg2+. (C) IPSCs observed after the removal of Clo. (D) Normalized magnitudes of the DDI evoked by voltage steps. The magnitudes of the DDI were normalized to the mean magnitude recorded in ACSF containing TTX and no Mg2+. All traces and data points were obtained from the same cell. To illustrate the synaptic component of the evoked responses more clearly, the Ca2+ current generated during the voltage step is removed from the traces. Bars above the scatter graph indicate the periods of extracellular application of TTX, Mg2+-free ACSF and Clo. (E) Summarized results showing that Clo produced a significant decrease in the DDI. Data are given as box plots. The line inside the boxes marks the median, and the box boundaries indicate the 25th and 75th percentiles. Error bars the range from minimum to maximum. (*) P < 0.05 versus the control. (F,G) Normalized peak amplitudes of the consecutive GC eEPSCs elicited by 10-Hz stimulation of the LOT before and during bath application of NA (10 µM; F) or Clo (5 µM; G). (*) P < 0.05 versus the first response. (H) Local field potential records showing changes in the amplitude of the consecutive N2 evoked by 10-Hz stimulation of the LOT in the presence of ACSF (Control), NA (10 µM) or Clo (5 µM). (I) NA attenuated the reduction in the amplitude of the consecutive N2 evoked by 10-Hz stimulation of the LOT, and this effect was mimicked by Clo. (*) P < 0.05, (**) P < 0.01 for NA versus the control; (#) P < 0.05, (##) P < 0.01 for Clo versus the control. (J) Decay time courses of N2 amplitudes. The data shown in (I) were normalized to the first N2 amplitudes. Under control conditions, the decay time course of N2 amplitudes was fitted by a single exponential with a time constant of 272 ± 21 ms. The decay time constants of N2 amplitudes were significantly larger in the presence of NA or Clo than in the presence of ACSF (Control). (*) P < 0.05 for NA versus the control; (##) P < 0.01 for Clo versus the control.
Since α2-AR activation disinhibited MCs, we reasoned that NA, acting at α2-ARs, might enhance GC responses to 10-Hz stimulation, contrary to single shock stimulation of the LOT. We therefore examined the effects of NA on seven consecutive GC eEPSC responses elicited by 10-Hz stimulation of the LOT. In contrast to the control, NA significantly increased the peak amplitudes of the second to sixth eEPSCs compared to those of the first eEPSCs (n = 7, P < 0.05, Tukey–Kramer's test) (Fig. 5F). This effect of NA was mimicked by clonidine (n = 7, P < 0.05, Tukey–Kramer's test) (Fig. 5G). We further examined whether the NA-induced enhancement of the eEPSC amplitude results in an increase in the amplitude of fEPSP N2 evoked by 10-Hz stimulation of the LOT (Fig. 5H–J). The amplitudes of the second to seventh N2 were significantly larger and decayed significantly more slowly in the presence of NA (amplitude, n = 7, P < 0.05 or P < 0.01, Tukey–Kramer's test; τ = 568 ± 76 msec, n = 7, P = 0.0117, Tukey–Kramer's test) or clonidine (amplitude, n = 7, P < 0.05 or P < 0.01, Tukey–Kramer's test; τ = 579 ± 102 msec, n = 7, P = 0.0093, Tukey–Kramer's test) than in the presence of artificial cerebrospinal fluid (ACSF: control, τ = 272 ± 21 msec).
NMDA receptor-dependent LTP underlies several forms of memory formation and has been extensively documented in hippocampal pyramidal neurons, where dependence on the conjunction of pre- and postsynaptic activity is one of the cardinal features (Bliss and Collingridge 1993). We tested the effect of NA on a postsynaptic GC maintained in the current-clamp mode during 10-Hz stimulation used for inducing LTP (Fig. 6A,B). The application of NA significantly reduced the size of the mean plateau depolarization and concomitantly increased spike fidelity in response to presynaptic stimulation at a frequency of 10 Hz (n = 9, P = 0.031 for plateau depolarization, P = 0.003 for spike fidelity, paired t-test) (Fig. 6B). The effects of NA on the depolarizing plateau and spike fidelity were mimicked by the α2-AR agonist clonidine (n = 7, P = 0.019 for plateau depolarization, P = 0.010 for spike fidelity, paired t-test) (Fig. 6B), as reported at the calyx of Held synapse in the rat medial nucleus of the trapezoid body (Leão and Von Gersdorff 2002).
Figure 6.
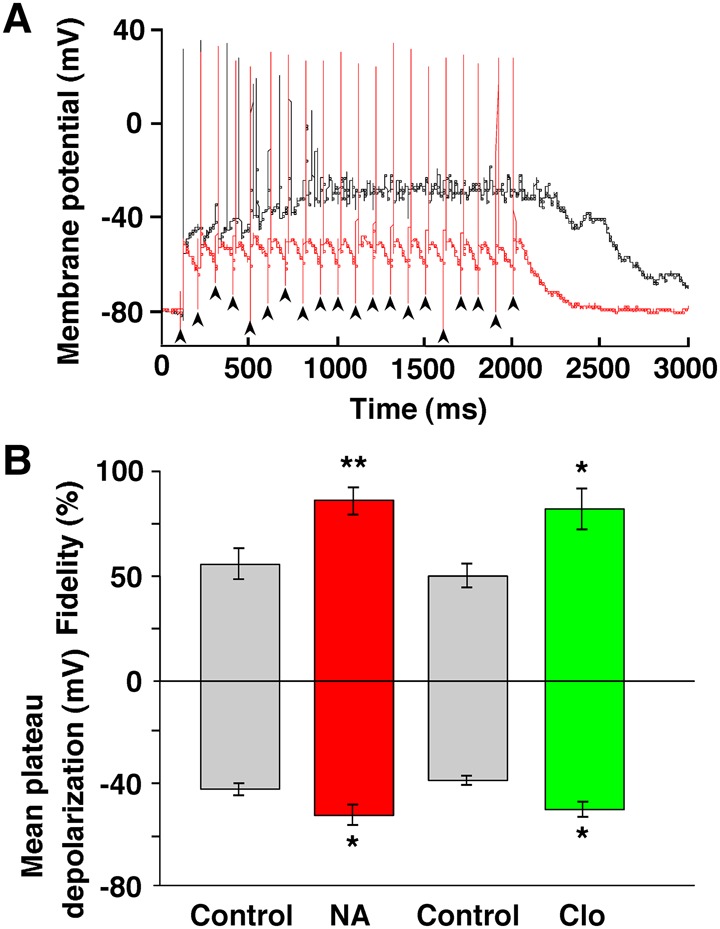
NA reduced postsynaptic plateau depolarization and conversely increased the fidelity of postsynaptic spiking. (A) A representative example of postsynaptic spiking during a 10-Hz, 20-pulse train of LOT stimulation in a GC maintained in current-clamp, before (black line) and after (red line) the bath application of NA (10 µM). Note that the GC reliably fired within 20 msec after each presynaptic stimulus in a 10-Hz train in the presence of NA. Arrowheads indicate the timing of stimuli. (B) Summarized results showing that NA produced a significant decrease in mean plateau depolarization and a significant increase in spike fidelity, and that clonidine (Clo, 5 µM) mimicked NA's effect. Spike fidelity in the ordinate was defined as (20−n)/20 × 100%, where n represents the number of transmission failures and aberrant APs during 20 stimuli. (*) P < 0.05, (**) P < 0.01 versus the control.
Discussion
The major findings of the present study on mouse AOB slices were as follows. (1) NMDA receptor-dependent LTP can be effectively elicited at the MC-to-GC synapse by physiologically relevant stimulation at a frequency of 10 Hz. (2) NA, acting at α2-ARs but not α1- or β-ARs, facilitates the NMDA receptor-dependent LTP. (3) In terms of acute effects, NA, acting at α2-ARs, suppresses GC eEPSC responses to single pulse stimulation of MC axons by reducing glutamate release from MCs via G-protein coupled inhibition of calcium channels. (4) As a consequence, NA reduces recurrent DDI of MCs, which in turn enhances eEPSCs and spike fidelity in GCs during 10-Hz stimulation. These findings thus reveal a novel mechanism of gating in which NA facilitates LTP induction at the MC-to-GC synapse by the enhancement of eEPSCs and spike fidelity in GCs through disinhibition of MCs.
Our present experiments yielded two surprising conclusions. First, in contrast to the abundant evidence regarding the NA action of facilitating or inducing LTP via β-ARs (Berridge and Waterhouse 2003; Marzo et al. 2009; Tully and Bolshakov 2010; O'Dell et al. 2015), there is limited evidence that this occurs via α2-ARs. The crucial role of α2-ARs in long-term synaptic plasticity might be attributable to the unique properties of reciprocal dendrodendritic synapses between MCs and GCs in the AOB. This is also true for reciprocal synapses in the MOB, in which α2-AR activation induces long-term enhancement of synchronized γ oscillations in extracellular local field potential signals (Pandipati et al. 2010).
Second, the acute effects of NA on eEPSCs elicited by electrical stimulation of MC axons differed according to whether single pulse or 10-Hz stimulation was delivered. NA suppressed GC responses to single pulse stimulation, whereas NA conversely enhanced GC responses to 10-Hz stimulation through disinhibition of MCs. With respect to the NA site of action, NA can affect inhibitory GABAergic transmission from GCs to MCs not only due to direct actions at the GC-to-MC synapse but also indirectly by affecting MC-to-GC excitatory glutamatergic transmission. In the present experiments, the NA-induced reduction in eEPSCs recorded from GCs was compatible with the finding that NA induced a reduction in the frequency of mEPSCs recorded from GCs without affecting their amplitude, implying that NA reduces glutamate release from MCs. Both effects of NA were mimicked by the α2-AR agonist clonidine. In addition, clonidine had no effect on mIPSCs recorded from MCs. Collectively, these findings clearly show that the actions of NA at α2-ARs are confined to MC-to-GC transmission but not GC-to-MC transmission at reciprocal dendrodendritic synapses in the AOB. In contrast to our findings in the AOB, Pandipati et al. (2010) reported that NA reduces GC-to-MC transmission in the MOB.
Link between the NA enhancement of eEPSCs and spike fidelity in GCs and LTP that may underlie olfactory learning
How does NA shift the threshold for LTP induction from 400 to 200 stimuli at 10 Hz, and thereby achieve activation of NMDA receptors to generate LTP? Acute disinhibition of MCs due to the NA-induced reduction in MC-to-GC transmission could be linked to the induction of LTP, based on several findings. First, both acute disinhibition of MCs and LTP induction were mimicked by α2-AR activation. Second, acute disinhibition of MCs from GABAergic GCs is a key step leading to olfactory learning, because local infusions of the GABAA receptor antagonist bicuculline into the AOB of female mice created a global memory in the absence of mating (Kaba et al. 1989). Such a mechanism is consistent with the finding that artificial vagino-cervical stimulation of anesthetized female mice caused a disinhibition of MC firing in the AOB (Otsuka et al. 2001). Finally, local infusions of the α-AR antagonist phentolamine, but not the β-AR antagonist timolol, into the AOB immediately after mating prevented mate recognition memory formation (Kaba and Keverne 1988).
A plausible mechanism for the NA-paired LTP would be the NA enhancement of eEPSCs and spike fidelity in GCs through disinhibition of MCs during 10-Hz stimulation. The NA enhancement of eEPSCs and spike fidelity in GCs may contribute to the induction of LTP by regulating synaptic efficacy or cellular excitability during 10-Hz stimulation, allowing for more effective activation of NMDA receptors to trigger LTP. For LTP induction, both pre- and postsynaptic neurons need to be active at the same time, because the postsynaptic neuron must be depolarized when glutamate is released from the presynaptic bouton to fully relieve the Mg+ block of NMDA receptors (Lüscher and Malenka 2012). Much stronger activation of NMDA receptors leading to much larger increases in postsynaptic calcium is required to trigger LTP (Malenka 1994). In the present experiments, the NA enhancement of GC eEPSCs during 10-Hz stimulation might have rendered GCs more responsive to NMDA receptor activation.
Precisely timed spiking of postsynaptic neurons following synaptic inputs has been shown to be crucial for LTP induction (Gustafsson et al. 1987; Bi and Poo 1998). Such manipulations lead to both a synaptic potential and an action potential that back-propagates into the dendrites, which presumably provides the additional depolarization required to facilitate calcium influx through NMDA receptors and voltage-gated calcium channels (Madison et al. 1991; Bliss and Collingridge 1993). The present finding that α2-AR activation increased the temporal precision between presynaptic MCs and postsynaptic GCs is consistent with the idea that the regulation of high-fidelity synaptic transmission by α2-ARs converts the normally subthreshold 10-Hz stimulation into a stimulation to elicit LTP by allowing postsynaptic calcium entry through a more effective activation of NMDA receptors and voltage-gated calcium channels. Indeed, NA has been shown to increase the temporal precision between excitatory afferent input and postsynaptic responses, enhancing synchrony between sensory input and spike output (Kössl and Vater 1989; Bouret and Sara 2002; Lecas 2004; Moxon et al. 2007).
In GCs of the rat MOB, action potentials evoke robust dendritic calcium transients (Egger et al. 2003). In contrast to observations in CA1 pyramidal neurons (Spruston et al. 1995) and neocortical layer 2/3 pyramidal neurons (Svoboda et al. 1999), these amplitudes do not decrease, but they do increase with distance from the soma and attain a plateau level within the EPL, where the reciprocal spines are located (Egger et al. 2003). The decay of dendritic calcium transients is slow (τ = 780 msec). The dendritic calcium transients require Na+-dependent action potentials and are strongly dependent on the membrane potential, decreasing with depolarization and increasing with hyperpolarization from rest, which is consistent with the properties of T-type calcium channels (Egger et al. 2003). T-type calcium channels are known to be expressed in GCs of the AOB (Yunker et al. 2003). Taken together, the action of NA on the spike fidelity could enhance the dendritic calcium entry through NMDA receptors and possibly T-type calcium channels, boosting 10-Hz synaptic inputs and consequently shifting the threshold for LTP induction from 400 to 200 stimuli.
In addition to regulating synaptic efficacy during 10-Hz stimulation, NA could regulate synaptic plasticity by modifying cellular excitability during 10-Hz stimulation. Indeed, Gao et al. (2017) used targeted whole-cell recordings of identified neurons activated by the stud male in ex vivo brain slices from female mice and found a striking reduction in intrinsic excitability in MCs, suggesting a cellular basis for encoding sensory memories in the AOB.
A state of reduced external interference is required for the consolidation of memories (Barnes and Wilson 2014). As shown in Figure 2A, NA enhanced the induction of LTP after the drug was washed out. This characteristic, also observed for the NA-induced long-term changes in γ oscillations in MOB slices, was similar to that of area CA3 of the rat hippocampal slices, where LTP and sharp ripple activity thought to underlie memory replay processes were induced after NA washout (Ul Haq et al. 2016).
Function of NA and its receptor subtypes in AOB sensory processing
Concerning the role of the AR subtypes in information processing in the AOB, studies on mouse AOB slices have shown that stimulation of α1-ARs, but not α2-ARs, increases the release of GABA from GCs and subsequently inhibits MC activity (Araneda and Firestein 2006; Smith et al. 2009). In contrast, our study revealed that α2-AR activation decreased the inhibitory feedback from GCs, thereby disinhibiting MCs. This finding is in line with the previous report that α2-AR activation decreased Ca2+ currents via N-type/R-type Ca2+ channels in MCs in AOB slice preparations (Dong et al. 2009), which would be expected to have a disinhibitory effect on MC activity. NA has multiple effects on bulbar cellular and synaptic parameters in the MOB, and these effects are strongly concentration dependent (Linster et al. 2011). In the present study, NA reduced the baseline fEPSP slope (Fig. 2A). Judging from the effects of an AR agonist and antagonists on this slope (Fig. 2B–D), NA appears to modulate the tightly coupled dendrodendritic interactions between MCs and GCs through the activation of different AR subtypes. Recent electrophysiological recordings from AOB neurons in ex vivo preparations indicated that NA prominently inhibited MC responses to chemosensory cues, but NA's effects were heterogeneous (Doyle and Meeks 2017).
With respect to the long-term effects of NA on the MC-to-GC synapse, the present results show that the expression of NA-facilitated LTP is dependent on α2-ARs, but not α1- or β-ARs. Interestingly, NA infusion into the AOB in awake mice induces long-term increases in the power of oscillatory local field potentials (Leszkowicz et al. 2012), similar to those observed following memory formation (Binns and Brennan 2005). Likewise, in vitro studies on rat MOB slices have shown that pairing either bath-applied NA or an α2-AR agonist with θ-patterned stimulation of olfactory nerve afferents induces a long-term enhancement of γ oscillations (Gire and Schoppa 2008; Pandipati et al. 2010). Collectively, these findings suggest that the NA-facilitated LTP is linked to NA-induced enhancement of oscillatory local field potentials in the AOB and they hold promise for understanding the relationship between memory formation and synaptic plasticity.
Functional implications
Single-unit recordings in freely behaving mice showed that putative MCs in the AOB respond to conspecific chemosignals with relatively low firing frequencies (spanning the β range) compared to MCs in the MOB (Luo et al. 2003). It is particularly noteworthy that the long-term stimulation at 10 Hz that is required to induce LTP is compatible with the prolonged exposure to the mate's chemosignals that is required for the formation of mate recognition memories (Rosser and Keverne 1985). Therefore, the 10-Hz stimulation protocol, which is within the physiological range of natural neuronal activity, could potentially provide a more physiologically relevant model of the synaptic changes during learning.
A simple hypothesis may explain how mate recognition is achieved by the neural circuitry of the AOB (Fig. 7). It is conceivable that the tightly coupled feedback inhibition acting on MCs could act as a gate to regulate the transmission of pregnancy-blocking signals. During learning, the association of the mate's chemosignals with an increased release of NA in the AOB at the time of mating would reduce the recurrent inhibition of MCs responding to the male's chemosignals, resulting in the enhancement of eEPSCs and spike fidelity in GCs. In addition to the enhancement of eEPSCs in GCs, the precisely timed action potentials generated in GCs would back-propagate into the dendrites, which presumably would provide the additional depolarization required to facilitate calcium influx through NMDA receptors and possibly voltage-gated calcium channels, leading to NMDA receptor-dependent LTP at the MC-to-GC side of the reciprocal synapses that are activated by the mate's chemosignals. The LTP would lead to a sequence of changes in the morphology of the reciprocal synapses (Matsuoka et al. 1997, 2004) and an increased release of the inhibitory neurotransmitter GABA in the AOB (Brennan et al. 1995).
Figure 7.

A hypothesized schematic model of synaptic mechanisms underlying mate recognition and selective gating of the pregnancy-blocking signals in the mouse AOB. See Discussion for details. (α2) α2-adrenergic receptor, (AP) action potential, (EPSC) excitatory postsynaptic current, (G) G protein, (GC) granule cell, (Glu) glutamate, (MC) mitral cell, (NA) noradrenaline, (NMDAR) NMDA receptor, (VGCC) voltage-gated calcium channel. The signaling pathway that inhibits Glu release is marked by an encircled “–.”
After learning, the subpopulation of MCs that respond to the mate's chemosignals would be subject to enhanced feedback inhibition from GCs via the potentiated synapses. The enhanced feedback inhibition of the subpopulation of MCs would selectively suppress the transmission of the familiar mate's pregnancy-blocking signals at the level of the AOB, preventing it from activating the neuroendocrine response leading to pregnancy loss. Exposure to chemosignals from unfamiliar males, during the vulnerable preimplantation period, would activate a different subpopulation of MCs without potentiated synapses, and would result in their pregnancy-blocking signals being finally transmitted to the hypothalamus.
With respect to the selective gating of biologically relevant odors, in vivo or ex vivo studies have demonstrated that olfactory memories result from a long-lasting suppression of MC responses to the learned odors in neonatal rats (Wilson and Leon 1988), adult male mice (Shea et al. 2008) and newly mated female mice (Gao et al. 2017). Taken together, the prior and present findings indicate that the NA modulation of long-term plasticity may serve as a common mechanism for coding biologically relevant odor information in both the MOB and AOB regardless of age or sex.
Materials and Methods
Slice preparation
Acute slices were prepared from Balb/c mice (postnatal days 22–42; Japan SLC) following a protocol approved by the Kochi Medical School Animal Care and Use Committee and conformed to international guidelines on the ethical use of animals. All efforts were made to minimize the number of animals used. Mice were decapitated and the AOB, together with the MOB, was rapidly dissected out from the skull and placed in an ice-cold ACSF solution containing (in mM) the following: 119 NaCl, 2.5 KCl, 1.25 NaH2PO4, 1.0 MgSO4, 2.5 CaCl2, 26 NaHCO3, and 11 D-glucose, bubbled with 95% O2/5% CO2, pH 7.3–7.4. Coronal slices (350 µm thick containing the AOB) were prepared, incubated in oxygenated ACSF at 32°C ± 1°C for at least 1 h, and then transferred to a recording chamber mounted on an upright microscope (Axioskop 2FS, Zeiss) equipped with infrared differential interference contrast optics (filter, 850 nm) video microscopy and a CCD camera (C2400; Hamamatsu Photonics). For field potential or whole-cell patch-clamp recordings were made at room temperature (22°C–26°C). Oxygenated ACSF was continuously perfused through the chamber at 1.0–1.5 mL/min.
Field potential recordings
Dendritic fEPSP responses were evoked with a bipolar platinum-iridium electrode (30 µm diameter, 50- to 100-µm tip separation) positioned on the LOT. A glass microelectrode (∼2 MΩ) was placed in the EPL to record fEPSPs, which provide a reliable and stable measure of excitatory synaptic transmission. Responses were amplified using an amplifier (MEZ-8301, Nihon Kohden). The electrical stimulus consists of a single square waveform of 50-µsec duration given at intensities of 0.12–0.18 mA generated by a stimulator (SEN-7203, Nihon Kohden) equipped with a stimulus isolation unit (SS-203J, Nihon Kohden). The initial slope of the fEPSP, which provides a measure of the strength of excitatory synaptic transmission, was measured. Baseline stimulation parameters were selected to evoke a response of 50%–60% of the maximum slope. Baseline fEPSPs were then elicited every 30 sec for the entire length of the experiment.
Whole-cell patch-clamp recordings
Whole-cell patch-clamp recordings were made from individual MCs or GCs in voltage- or current-clamp mode with an EPC9 amplifier (HEKA Electronik). The MCs and GCs were identified by their morphology and location in the slices using a 40× water-immersion objective. Patch electrodes (3–6 MΩ) were pulled from borosilicate glass and filled with a solution containing (in mM) the following: 120 KCl, 5 NaCl, 1 MgCl2, 0.2 EGTA, 10 HEPES, 2 Mg-ATP, and 0.1 Na-GTP, pH 7.2 with KOH, osmolarity adjusted to 290–300 mOsm. The membrane potential was clamped at −70 mV unless otherwise stated.
For the recording of eEPSCs from GCs, QX-314 (5 mM) was added to the pipette solution to block sodium spikes, and the LOT was stimulated every 30 sec or at 10 Hz with an intensity (0.2–0.4 mA, 0.1 msec) that was adjusted to 1.5-fold the threshold. For the recording of mEPSCs and mIPSCs, TTX (1 µM) was added to the external solution to block evoked synaptic transmission. In addition, mEPSCs were isolated by blocking mIPSCs with picrotoxin (100 µM), whereas blocking the mEPSCs with CNQX (30 µM) and AP5 (50 µM) isolated the mIPSCs.
In the experiments in which Ba2+ was used as the charge carrier, the external solution contained (in mM) the following: 140 NaCl, 3 KCl, 10 HEPES, 2 MgCl2, 5 BaCl2, 11 D-glucose, 0.001 TTX, and 0.1 picrotoxin; the internal solution contained (in mM) the following: 100 CsCl, 30 TEA-Cl, 5 NaCl, 1 MgCl2, 10 EGTA, 10 HEPES, 4 Mg-ATP, and 0.2 Na-GTP, pH 7.2–7.3 with CsOH. Ba2+ currents were elicited by 100 msec voltage steps from a holding potential of –70 mV to various test potentials at 20-sec intervals.
For the experiments involving PTX treatment, slices were incubated for 4–6 h at 37°C with ACSF containing PTX (5 µg/mL). For the experiments examining Ca2+ currents, the external solution was ACSF with TTX (1 µM) and picrotoxin (0.1 mM); the CsCl-TEA internal solution was used. For the recording of voltage-activated K+ currents, the external solution contained TTX (1 µM). The K+ currents were evoked by 400-msec ramp depolarizations from –100 to +70 mV.
For the experiments examining postsynaptic plateau depolarization and spike fidelity, GCs were held at a potential that was somewhat hyperpolarized (–80 mV) in order to prevent spiking before LOT stimulation, which was applied with an intensity just above the threshold for evoking an action potential in GCs. To quantify the mean plateau depolarization during a 10-Hz train of 20 stimuli, we chose to take the value at the midpoint of the stimulation period. The fidelity of synaptic transmission was evaluated from the number (n) of transmission failures and aberrant APs during 20 stimuli and expressed as (20–n)/20 × 100%. Postsynaptic responses generated at 50–100 msec post-stimulus onset were not counted in the APs.
For the experiments examining DDI evoked in MCs, patch pipettes (5–7 MΩ) were filled with an internal solution (Cs+-internal) containing (in mM) the following: 140 CsCl, 5 NaCl, 1 MgCl2, 1 EGTA, 10 HEPES, 2 Na2ATP, and 0.2 Na3GTP (pH 7.4). TTX (1 µM) was dissolved in ACSF containing no Mg2+. To evoke DDI, a depolarizing voltage step from a holding potential of –70 to 0 mV (10 msec) was applied to an MC.
Data acquisition and analysis
Evoked field potentials were recorded, digitized at 10 kHz, and analyzed using the PowerLab/4sp system (ADInstruments). One-minute records (two traces) were averaged. We measured the maximal initial slope of the fEPSP in the falling phase to monitor the strength of synaptic transmission. Unless otherwise stated, the magnitude of potentiation or depotentiation was calculated by comparing the average of the fEPSP slope measurements made for the last 10 min of the recording period with the average of the 10 min preceding 1-, 10-, or 100-Hz stimulation or drug application. Potentiation and depotentiation were elicited with the same intensity and pulse duration as the test stimuli.
These experiments were classified as successful LTP when the average of the fEPSP slope measurements made for the last 10 min of the recording period showed an increase of at least 20% above baseline values (Malenka 1991). Membrane currents were recorded, filtered at 1–2 kHz, digitized at 1–5 kHz, and stored for off-line analysis. The peak amplitude of eEPSCs elicited by single pulse stimulation was measured from the average of two responses. The mEPSCs were detected using the Mini Analysis Program v5.6.3 (Synaptosoft). The area and amplitude thresholds were set above the noise level and kept constant in each experiment. Events that did not show a typical synaptic waveform were rejected manually. The Ba2+ or Ca2+ current amplitude was measured as the maximum value of the 10-msec period after the onset of the voltage step. We subtracted linear components of capacitative and leak currents using the standard P/4 protocol. Series resistance compensations of 70%–80% were used. We quantified the DDI by calculating the current integral recorded via voltage-clamp recordings over a 3-sec period beginning 50–230 msec after the end of the voltage step.
Statistical analysis
Data were tested for normality (Shapiro–Wilk test) and equal variance (F-test) first. Standard parametric tests were only used when data passed the normality and equal variance tests (P > 0.05). Nonparametric tests were used otherwise. Cumulative histograms were compared using the Kolmogorov–Smirnov test. Significance was accepted at P < 0.05. Data are expressed as the mean ± standard error of the mean (SEM) unless otherwise stated.
Drugs
Drugs were obtained from the following sources: CNQX, DL-AP5, TTX, (±)-noradrenaline(+)-hydrogentartrate, (±)-propranolol hydrochloride, prazosin hydrochloride, idazoxan hydrochloride, clonidine hydrochloride, picrotoxin, QX-314 were from Sigma; phentolamine mesylate was from Research Biochemicals International; and PTX was from Wako Chemicals USA. Ascorbic acid (100 µM) was added to the NA solution to prevent oxidation.
Supplementary Material
Acknowledgments
This work was supported in part by a Grant-in-Aid for Scientific Research on Priority Areas from MEXT (grant number 17021031). G.-Z.H. and J.-J.Z. were supported by JSPS Postdoctoral Fellowships for Foreign Researchers. Y.-B.Z. was supported by an AIEJ Short-term Student Exchange Program Scholarship.
Footnotes
[Supplemental material is available for this article.]
Article is online at http://www.learnmem.org/cgi/doi/10.1101/lm.046391.117.
Freely available online through the Learning & Memory Open Access option.
References
- Araneda RC, Firestein S. 2006. Adrenergic enhancement of inhibitory transmission in the accessory olfactory bulb. J Neurosci 26: 3292–3298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes DC, Wilson DA. 2014. Sleep and olfactory cortical plasticity. Front Behav Neurosci 8: 134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berridge CW, Waterhouse BD. 2003. The locus coeruleus-noradrenergic system: modulation of behavioral state and state-dependent cognitive processes. Brain Res Brain Res Rev 42: 33–84. [DOI] [PubMed] [Google Scholar]
- Bi GQ, Poo MM. 1998. Synaptic modifications in cultured hippocampal neurons: dependence on spike timing, synaptic strength, and postsynaptic cell type. J Neurosci 18: 10464–10472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Binns KE, Brennan PA. 2005. Changes in electrophysiological activity in the accessory olfactory bulb and medial amygdala associated with mate recognition in mice. Eur J Neurosci 21: 2529–2537. [DOI] [PubMed] [Google Scholar]
- Bliss TV, Collingridge GL. 1993. A synaptic model of memory: long-term potentiation in the hippocampus. Nature 361: 31–39. [DOI] [PubMed] [Google Scholar]
- Bouret S, Sara SJ. 2002. Locus coeruleus activation modulates firing rate and temporal organization of odour-induced single-cell responses in rat piriform cortex. Eur J Neurosci 16: 2371–2382. [DOI] [PubMed] [Google Scholar]
- Brennan PA, Keverne EB. 1997. Neural mechanisms of mammalian olfactory learning. Prog Neurobiol 51: 457–481. [DOI] [PubMed] [Google Scholar]
- Brennan PA, Zufall F. 2006. Pheromonal communication in vertebrates. Nature 444: 308–315. [DOI] [PubMed] [Google Scholar]
- Brennan P, Kaba H, Keverne EB. 1990. Olfactory recognition: a simple memory system. Science 250: 1223–1226. [DOI] [PubMed] [Google Scholar]
- Brennan PA, Kendrick KM, Keverne EB. 1995. Neurotransmitter release in the accessory olfactory bulb during and after the formation of an olfactory memory in mice. Neuroscience 69: 1075–1086. [DOI] [PubMed] [Google Scholar]
- Castro JB, Hovis KR, Urban NN. 2007. Recurrent dendrodendritic inhibition of accessory olfactory bulb mitral cells requires activation of group I metabotropic glutamate receptors. J Neurosci 27: 5664–5671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong C, Godwin DW, Brennan PA, Hegde AN. 2009. Protein kinase Cα mediates a novel form of plasticity in the accessory olfactory bulb. Neuroscience 163: 811–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doucette W, Milder J, Restrepo D. 2007. Adrenergic modulation of olfactory bulb circuitry affects odor discrimination. Learn Mem 14: 539–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle WI, Meeks JP. 2017. Heterogeneous effects of norepinephrine on spontaneous and stimulus-driven activity in the male accessory olfactory bulb. J Neurophysiol 117: 1342–1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckmeier D, Shea SD. 2014. Noradrenergic plasticity of olfactory sensory neuron inputs to the main olfactory bulb. J Neurosci 34: 15234–15243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egger V, Svoboda K, Mainen ZF. 2003. Mechanisms of lateral inhibition in the olfactory bulb: efficiency and modulation of spike-evoked calcium influx into granule cells. J Neurosci 23: 7551–7588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang LY, Quan RD, Kaba H. 2008. Oxytocin facilitates the induction of long-term potentiation in the accessory olfactory bulb. Neurosci Lett 438: 133–137. [DOI] [PubMed] [Google Scholar]
- Gao Y, Budlong C, Durlacher E, Davison IG. 2017. Neural mechanisms of social learning in the female mouse. Elife 6 pii: e25421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gire DH, Schoppa NE. 2008. Long-term enhancement of synchronized oscillations by adrenergic receptor activation in the olfactory bulb. J Neurophysiol 99: 2021–2025. [DOI] [PubMed] [Google Scholar]
- Griffiths PR, Brennan PA. 2015. Roles for learning in mammalian chemosensory responses. Horm Behav 68: 91–102. [DOI] [PubMed] [Google Scholar]
- Gustafsson B, Wigström H, Abraham WC, Huang YY. 1987. Long-term potentiation in the hippocampus using depolarizing current pulses as the conditioning stimulus to single volley synaptic potentials. J Neurosci 7: 774–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harley CW, Darby-King A, McCann J, McLean JH. 2006. β1-adrenoceptor or α1-adrenoceptor activation initiates early odor preference learning in rat pups: support for the mitral cell/cAMP model of odor preference learning. Learn Mem 13: 8–13. [DOI] [PubMed] [Google Scholar]
- Isaacson JS, Strowbridge BW. 1998. Olfactory reciprocal synapses: dendritic signaling in the CNS. Neuron 20: 749–761. [DOI] [PubMed] [Google Scholar]
- Jia C, Chen WR, Shepherd GM. 1999. Synaptic organization and neurotransmitters in the rat accessory olfactory bulb. J Neurophysiol 81: 345–355. [DOI] [PubMed] [Google Scholar]
- Jiang M, Griff ER, Ennis M, Zimmer LA, Shipley MT. 1996. Activation of locus coeruleus enhances the responses of olfactory bulb mitral cells to weak olfactory nerve input. J Neurosci 16: 6319–6329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaba H, Keverne EB. 1988. The effect of microinfusions of drugs into the accessory olfactory bulb on the olfactory block to pregnancy. Neuroscience 25: 1007–1011. [DOI] [PubMed] [Google Scholar]
- Kaba H, Nakanishi S. 1995. Synaptic mechanisms of olfactory recognition memory. Rev Neurosci 6: 125–141. [DOI] [PubMed] [Google Scholar]
- Kaba H, Rosser A, Keverne B. 1989. Neural basis of olfactory memory in the context of pregnancy block. Neuroscience 32: 657–662. [DOI] [PubMed] [Google Scholar]
- Keverne EB, de la Riva C. 1982. Pheromones in mice: reciprocal interaction between the nose and brain. Nature 296: 148–150. [DOI] [PubMed] [Google Scholar]
- Keverne EB, Rosser AE. 1986. The evolutionary significance of the olfactory block to pregnancy. In Chemical Signals in Vertebrates 4, (ed. Duvall D, Müller-Schwarze D, Silverstein RM) pp. 433–439. Plenum Press, New York. [Google Scholar]
- Kössl M, Vater M. 1989. Noradrenaline enhances temporal auditory contrast and neuronal timing precision in the cochlear nucleus of the mustached bat. J Neurosci 9: 4169–4178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leão RM, Von Gersdorff H. 2002. Noradrenaline increases high-frequency firing at the calyx of Held synapse during development by inhibiting glutamate release. J Neurophysiol 87: 2297–2306. [DOI] [PubMed] [Google Scholar]
- Lecas JC. 2004. Locus coeruleus activation shortens synaptic drive while decreasing spike latency and jitter in sensorimotor cortex. Implications for neuronal integration. Eur J Neurosci 19: 2519–2530. [DOI] [PubMed] [Google Scholar]
- Leszkowicz E, Khan S, Ng S, Ved N, Swallow DL, Brennan PA. 2012. Noradrenaline-induced enhancement of oscillatory local field potentials in the mouse accessory olfactory bulb does not depend on disinhibition of mitral cells. Eur J Neurosci 35: 1433–1445. [DOI] [PubMed] [Google Scholar]
- Linster C, Nai Q, Ennis M. 2011. Nonlinear effects of noradrenergic modulation of olfactory bulb function in adult rodents. J Neurophysiol 105: 1432–1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo M, Fee MS, Katz LC. 2003. Encoding pheromonal signals in the accessory olfactory bulb of behaving mice. Science 299: 1196–1201. [DOI] [PubMed] [Google Scholar]
- Lüscher C, Malenka RC. 2012. NMDA receptor-dependent long-term potentiation and long-term depression (LTP/LTD). Cold Spring Harb Perspect Biol 4 pii: a005710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macrides F, Davis BJ, Youngs WM, Nadi NS, Margolis FL. 1981. Cholinergic and catecholaminergic afferents to the olfactory bulb in the hamster: a neuroanatomical, biochemical, and histochemical investigation. J Comp Neurol 203: 495–514. [DOI] [PubMed] [Google Scholar]
- Madison DV, Malenka RC, Nicoll RA. 1991. Mechanisms underlying long-term potentiation of synaptic transmission. Annu Rev Neurosci 14: 379–397. [DOI] [PubMed] [Google Scholar]
- Malenka RC. 1991. Postsynaptic factors control the duration of synaptic enhancement in area CA1 of the hippocampus. Neuron 6: 53–60. [DOI] [PubMed] [Google Scholar]
- Malenka RC. 1994. Synaptic plasticity in the hippocampus: LTP and LTD. Cell 78: 535–538. [DOI] [PubMed] [Google Scholar]
- Marzo A, Bai J, Otani S. 2009. Neuroplasticity regulation by noradrenaline in mammalian brain. Curr Neuropharmacol 7: 286–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuoka M, Kaba H, Mori Y, Ichikawa M. 1997. Synaptic plasticity in olfactory memory formation in female mice. Neuroreport 8: 2501–2504. [DOI] [PubMed] [Google Scholar]
- Matsuoka M, Kaba H, Moriya K, Yoshida-Matsuoka J, Costanzo RM, Norita M, Ichikawa M. 2004. Remodeling of reciprocal synapses associated with persistence of long-term memory. Eur J Neurosci 19: 1668–1672. [DOI] [PubMed] [Google Scholar]
- McLean JH, Shipley MT, Nickell WT, Aston-Jones G, Reyher CK. 1989. Chemoanatomical organization of the noradrenergic input from locus coeruleus to the olfactory bulb of the adult rat. J Comp Neurol 285: 339–349. [DOI] [PubMed] [Google Scholar]
- Miller RJ. 1998. Presynaptic receptors. Annu Rev Pharmacol Toxicol 38: 201–227. [DOI] [PubMed] [Google Scholar]
- Moreno MM, Bath K, Kuczewski N, Sacquet J, Didier A, Mandairon N. 2012. Action of the noradrenergic system on adult-born cells is required for olfactory learning in mice. J Neurosci 32: 3748–3758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moxon KA, Devilbiss DM, Chapin JK, Waterhouse BD. 2007. Influence of norepinephrine on somatosensory neuronal responses in the rat thalamus: a combined modeling and in vivo multi-channel, multi-neuron recording study. Brain Res 1147: 105–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura Y, Takahashi T. 2007. Developmental changes in potassium currents at the rat calyx of Held presynaptic terminal. J Physiol 581(Pt 3): 1101–1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Dell TJ, Connor SA, Guglietta R, Nguyen PV. 2015. β-Adrenergic receptor signaling and modulation of long-term potentiation in the mammalian hippocampus. Learn Mem 22: 461–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okutani F, Kaba H, Takahashi S, Seto K. 1998. The biphasic effects of locus coeruleus noradrenergic activation on dendrodendritic inhibition in the rat olfactory bulb. Brain Res 783: 272–279. [DOI] [PubMed] [Google Scholar]
- Otsuka T, Ishii K, Osako Y, Okutani F, Taniguchi M, Oka T, Kaba H. 2001. Modulation of dendrodendritic interactions and mitral cell excitability in the mouse accessory olfactory bulb by vaginocervical stimulation. Eur J Neurosci 13: 1833–1838. [DOI] [PubMed] [Google Scholar]
- Pandipati S, Gire DH, Schoppa NE. 2010. Adrenergic receptor-mediated disinhibition of mitral cells triggers long-term enhancement of synchronized oscillations in the olfactory bulb. J Neurophysiol 104: 665–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pissonnier D, Thiery JC, Fabre-Nys C, Poindron P, Keverne EB. 1985. The importance of olfactory bulb noradrenalin for maternal recognition in sheep. Physiol Behav 35: 361–363. [DOI] [PubMed] [Google Scholar]
- Rosser AE, Keverne EB. 1985. The importance of central noradrenergic neurones in the formation of an olfactory memory in the prevention of pregnancy block. Neuroscience 15: 1141–1147. [DOI] [PubMed] [Google Scholar]
- Saunders C, Limbird LE. 1999. Localization and trafficking of α2-adrenergic receptor subtypes in cells and tissues. Pharmacol Ther 84: 193–205. [DOI] [PubMed] [Google Scholar]
- Schoppa NE, Kinzie JM, Sahara Y, Segerson TP, Westbrook GL. 1998. Dendrodendritic inhibition in the olfactory bulb is driven by NMDA receptors. J Neurosci 18: 6790–6802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shakhawat AM, Harley CW, Yuan Q. 2012. Olfactory bulb α2-adrenoceptor activation promotes rat pup odor-preference learning via a cAMP-independent mechanism. Learn Mem 19: 499–502. [DOI] [PubMed] [Google Scholar]
- Shea SD, Katz LC, Mooney R. 2008. Noradrenergic induction of odor-specific neural habituation and olfactory memories. J Neurosci 28: 10711–10719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shipley MT, Halloran FJ, de la Torre J. 1985. Surprisingly rich projection from locus coeruleus to the olfactory bulb in the rat. Brain Res 329: 294–299. [DOI] [PubMed] [Google Scholar]
- Smith RS, Weitz CJ, Araneda RC. 2009. Excitatory actions of noradrenaline and metabotropic glutamate receptor activation in granule cells of the accessory olfactory bulb. J Neurophysiol 102: 1103–1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spruston N, Schiller Y, Stuart G, Sakmann B. 1995. Activity-dependent action potential invasion and calcium influx into hippocampal CA1 dendrites. Science 268: 297–300. [DOI] [PubMed] [Google Scholar]
- Stäubli U, Chun D. 1996. Factors regulating the reversibility of long-term potentiation. J Neurosci 16: 853–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan RM, Wilson DA, Leon M. 1989. Norepinephrine and learning-induced plasticity in infant rat olfactory system. J Neurosci 9: 3998–4006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan RM, Zyzak DR, Skierkowski P, Wilson DA. 1992. The role of olfactory bulb norepinephrine in early olfactory learning. Brain Res Dev Brain Res 70: 279–282. [DOI] [PubMed] [Google Scholar]
- Sullivan RM, Stackenwalt G, Nasr F, Lemon C, Wilson DA. 2000. Association of an odor with activation of olfactory bulb noradrenergic β-receptors or locus coeruleus stimulation is sufficient to produce learned approach responses to that odor in neonatal rats. Behav Neurosci 114: 957–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svoboda K, Helmchen F, Denk W, Tank DW. 1999. Spread of dendritic excitation in layer 2/3 pyramidal neurons in rat barrel cortex in vivo. Nat Neurosci 2: 65–73. [DOI] [PubMed] [Google Scholar]
- Taniguchi M, Kaba H. 2001. Properties of reciprocal synapses in the mouse accessory olfactory bulb. Neuroscience 108: 365–370. [DOI] [PubMed] [Google Scholar]
- Taniguchi M, Yokoi M, Shinohara Y, Okutani F, Murata Y, Nakanishi S, Kaba H. 2013. Regulation of synaptic currents by mGluR2 at reciprocal synapses in the mouse accessory olfactory bulb. Eur J Neurosci 37: 351–358. [DOI] [PubMed] [Google Scholar]
- Tully K, Bolshakov VY. 2010. Emotional enhancement of memory: how norepinephrine enables synaptic plasticity. Mol Brain 3: 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ul Haq R, Anderson M, Liotta A, Shafiq M, Sherkheli MA, Heinemann U. 2016. Pretreatment with β-adrenergic receptor agonists facilitates induction of LTP and sharp wave ripple complexes in rodent hippocampus. Hippocampus 26: 1486–1492. [DOI] [PubMed] [Google Scholar]
- Wilson DA, Leon M (1988). Noradrenergic modulation of olfactory bulb excitability in the postnatal rat. Brain Res 470: 69–75. [DOI] [PubMed] [Google Scholar]
- Yang YM, Wang W, Fedchyshyn MJ, Zhou Z, Ding J, Wang LY. 2014. Enhancing the fidelity of neurotransmission by activity-dependent facilitation of presynaptic potassium currents. Nat Commun 5: 4564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan Q. 2009. θ bursts in the olfactory nerve paired with β-adrenoceptor activation induce calcium elevation in mitral cells: a mechanism for odor preference learning in the neonate rat. Learn Mem 16: 676–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yunker AM, Sharp AH, Sundarraj S, Ranganathan V, Copeland TD, McEnery MW. 2003. Immunological characterization of T-type voltage-dependent calcium channel CaV3.1 (α 1G) and CaV3.3 (α 1I) isoforms reveal differences in their localization, expression, and neural development. Neuroscience 117: 321–335. [DOI] [PubMed] [Google Scholar]
- Zhang JJ, Okutani F, Huang GZ, Taniguchi M, Murata Y, Kaba H. 2010. Common properties between synaptic plasticity in the main olfactory bulb and olfactory learning in young rats. Neuroscience 170: 259–267. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.