

Abstract
N6-methyladenine (m6A) is found on many eukaryotic RNAs including mRNAs. m6A modification has been implicated in mRNA stability and turnover, localization, or translation efficiency. A heterodimeric enzyme complex composed of METTL3 and METTL14 generates m6A on mRNAs. METTL3/14 is found in the nucleus where it is localized to nuclear speckles and the splicing regulator WTAP is required for this distinct nuclear localization pattern. Although recent crystal structures revealed how the catalytic MT-A70 domains of METTL3 and METTL14 interact with each other, a more global architecture including WTAP and RNA interactions has not been reported so far. Here, we used recombinant proteins and mapped binding surfaces within the METTL3/14-WTAP complex. Furthermore, we identify nuclear localization signals and identify phosphorylation sites on the endogenous proteins. Using an in vitro methylation assay, we confirm that monomeric METTL3 is soluble and inactive while the catalytic center of METTL14 is degenerated and thus also inactive. In addition, we show that the C-terminal RGG repeats of METTL14 are required for METTL3/14 activity by contributing to RNA substrate binding. Our biochemical work identifies characteristic features of METTL3/14-WTAP and reveals novel insight into the overall architecture of this important enzyme complex.
Keywords: METTL14, METTL3, RNA modification, m6A, methyltransferase
INTRODUCTION
Newly synthesized RNA molecules contain unmodified bases attached to a ribose. However, many RNAs receive specific modifications after transcription, which can be found at the 2′ OH of the ribose or at specific positions of the bases. More than 100 such modifications are known to date and many are found in the well-studied tRNA and ribosomal RNAs (rRNAs) (Motorin and Helm 2011). Most prominent in these RNAs are 2′ O-methylations and conversion of uridines to pseudouridines (Matera et al. 2007). Modifications on these RNAs but also other RNAs including mRNAs have been known for decades (Rottman et al. 1974, 1994; Lane et al. 1995). Their specific functions on mRNAs, however, are still largely unclear. The most studied and thus best understood base modification in eukaryotic mRNAs is N6-methyladenine (m6A) (Meyer and Jaffrey 2017). The availability of an antibody specifically recognizing this modification allowed for mapping of m6A on a genome-wide scale in different organisms including yeast and mammals, and it was found that such modifications affect mRNA stability (Dominissini et al. 2012; Meyer et al. 2012; Wang et al. 2014). Depending on the identity of the mRNA and the specific location of the modified adenosine on the mRNA, additional functional consequences have been proposed. For example, m6A residues are enriched in the 3′ untranslated region (UTR) of mRNAs predominantly around the stop codon. Furthermore, the m6A sites seem to correlate with the presence of predicted microRNA (miRNA) binding sites, suggesting a functional link (Meyer et al. 2012). They are also enriched in large internal exon sequences and are found in 5′ UTRs (Yue et al. 2015). m6A modifications in the 5′ UTR are involved in translational control and seem to affect cap-independent translation (Meyer et al. 2015; Zhou et al. 2015). Very recently, it has been found that m6A modification of the first nucleotide adjacent to the 7-methylguanosine cap protects the mRNA from decapping and leads to mRNA stabilization (Mauer et al. 2017). Interestingly, m6A modifications are induced on distinct mRNAs upon UV light-induced DNA damage. These mRNAs serve as beacons for DNA damage response factors in U2OS cells (Xiang et al. 2017). Furthermore, m6A modification is required for clearance of maternal mRNAs in zebrafish, leading to efficient maternal-to-zygotic transition (Zhao et al. 2017).
Thousands of m6A methylation sites have been reported but how are specific sites recognized? Very early biochemical studies (Rottman et al. 1974, 1994) revealed and the above-mentioned profiling studies confirmed that m6A modifications occur within a distinct motif composed of RRACH, where R is a purine and H is A, U, or C (Fu et al. 2014; Maity and Das 2016). An enzyme complex composed of a heterodimer of METTL3 and METTL14 recognizes these sites and transfers a methyl group to the adenosine (Liu et al. 2014). Therefore, these enzymes are termed m6A “writers.” Since m6A is shown to be dynamic and involved in many regulatory processes, m6A “erasers” remove the modification from mRNAs. So far, the obesity-associated gene FTO and the demethylase ALKBH5 have been characterized as m6A erasers in different processes (Jia et al. 2011; Zheng et al. 2013). m6A “reader” proteins recognize the methyl group and facilitate downstream mRNA effects. Readers contain a YTH domain and are therefore collectively called YTH proteins. They can be found in the nucleus (YTHDC1) or the cytoplasm (YTHDF1-3) of eukaryotic cells and are engaged in different pathways (Wang et al. 2014, 2015; Zhou et al. 2015; Xiao et al. 2016; Shi et al. 2017). For example, the above-mentioned maternal-to-zygotic transition involves YTHDF2, which targets maternal mRNAs for deadenylation and decay (Zhao et al. 2017).
METTL3 and METTL14 belong to a large and conserved family of methyltransferases. They all contain a MT-A70 domain (also referred to as methyltransferase domain, MTD) that catalyzes the methyl transfer to adenosine. Recent crystal structures of truncated variants of the METTL3/14 heterodimer revealed that both proteins are tightly bound to each other. METTL3 contributes the catalytic residues, while METTL14's catalytic center is degenerated and is therefore not involved in the chemical reaction. Nevertheless, it contributes to the architecture of the catalytic center and serves as RNA binding scaffold (Śledź and Jinek 2016; Wang et al. 2016a,b).
The METTL3/14 heterodimer is predominantly localized to the nucleus where it interacts with the Wilms’ tumor 1-associated protein (WTAP) (Liu et al. 2014; Ping et al. 2014). WTAP has been functionally linked to alternative splicing (Horiuchi et al. 2013) and targets METTL3/14 into nuclear speckles (Ping et al. 2014). In addition, the human ortholog of Drosophila virilizer (vir) termed KIAA1429, a protein involved in alternative splicing as well, has been found to interact with WTAP and knockdown affects m6A methylation (Ortega et al. 2003; Schwartz et al. 2014). Finally, RBM15 interacts with RNA substrates near m6A sites, and it has been suggested that RBM15 recruits METTL3/14 to the RNA for modification (Patil et al. 2016).
Here, we used biochemical experiments to further delineate the overall architecture of the METTL3–METTL14–WTAP complex. We identify interaction surfaces, which the three proteins use to contact each other. In addition, we discover nuclear localization signals that are used for nuclear transport. Using a monoclonal antibody against METTL3, we isolate endogenous proteins and characterize phosphorylation sites. Using an in vitro methylation assay, we demonstrate that monomeric METTL3, although stable in solution, is catalytically inactive. Interestingly, deleting the C-terminal RGG repeats of METTL14 abolished catalytic activity of the METTL3/14 dimer. Using binding and cross-linking experiments, we show that loss of catalytic activity is caused by strongly reduced RNA binding, thus defining the RGG repeats of METTL14 as a second RNA contact site besides the catalytic domain.
RESULTS
Mapping of METTL3–METTL14–WTAP interaction surfaces
METTL3 and METTL14 form a heterodimeric complex, which interacts with WTAP. The precise interaction domains of these proteins, however, are not fully characterized. To map interactions in vitro, we coexpressed and purified METTL3 and METTL14 as well as a number of truncations from insect cells (Fig. 1A–C). The complex was purified via a GST-tag on METTL3 (Fig. 1A) or METTL3 (76–580), and METTL14 and its truncations were copurified (Fig. 1B,C). Coexpression of truncated METTL3/14 proteins revealed that the predominant interaction of both proteins is mediated by the methyltransferase domain (MTD) (Fig. 1C,D). These findings are consistent with recent crystal structures of the isolated MTD domains of METTL3 and METTL14 in complex with each other, which have been reported while our work was in progress (Śledź and Jinek 2016; Wang et al. 2016a,b).
FIGURE 1.

Binding studies of METTL3, METTL14, and WTAP. (A) Coomassie gel of purified recombinant full-length GST-METTL3 and copurified METTL14. The coexpression was conducted in insect cells (SF21). The complex was purified via the GST-tag on METTL3. (B) Schematic representation of the different truncation constructs of F/H-METTL3 and myc-METTL14 used to narrow down the binding sites of these proteins. (C) Coomassie gel of recombinantly expressed and purified complexes composed of the indicated truncated METTL14 and METTL3 (76–580) constructs. Complexes were purified via GST-METTL3 (76–580) and GST was subsequently cleaved off during elution. (D) Schematic view of the interaction surface between METTL3 and METTL14 revealed by coexpression studies. (E) Scheme of the different WTAP N- and C-terminal truncation constructs. (F) Western blots of coimmunoprecipitations of full-length F/H-METTL3 with N-terminal truncations of myc-WTAP. The upper panel was incubated with α-HA antibody, the lower one with α-myc antibody. The arrows show the bands for the different constructs. (G) Western blots of coimmunoprecipitations as in F but with C-terminally truncated constructs of myc-WTAP. (H) Western blot of coimmunoprecipitated full-length myc-WTAP and LH-deletion mutants of F/H-METTL3 (ΔLH-METTL3). (I) Western blots of coimmunoprecipitations of the N-terminal part of F/H-METTL3 (leader helix, short LH) with C-terminally truncated constructs of myc-WTAP. The upper panel shows the HA-antibody treated blot, the lower blot shows the α-myc-western blot. (J) Schematic cartoon of the regions of interaction between WTAP and METTL3 based on the results of coimmunoprecipitation experiments.
We next mapped interactions between METTL3 and WTAP. Structure predictions suggest that WTAP contains an extended N-terminal coiled coil region followed by a rather unstructured C-terminal part (Fig. 1E; Supplemental Fig. 1). We subsequently shortened WTAP from the N terminus as well as the C terminus and transfected the myc-tagged variants into HEK 293T cells together with full-length FLAG/HA (F/H)-tagged METTL3 (Fig. 1F,G). Immunoprecipitations of F/H-METTL3 revealed that all WTAP constructs lacking the first 110 amino acids do not bind to METTL3 (NT2, NT3) (Fig. 1F). When shortening WTAP from the C terminus, a fragment containing the N-terminal 150 amino acids (CT3) still binds, suggesting the METTL3 binding surface is within the first 150 amino acids (Fig. 1G). Of note, shortening of the coiled coil region from the N or the C terminus may differently affect local folding thus restricting a more precise mapping in our experiments.
Finally, we analyzed the WTAP interaction surface on METTL3. In silico structural analysis suggests a helical structure at the very N terminus of METTL3 referred to as the leader helix (LH) (Fig. 1D; Supplemental Fig. 1). We hypothesized that this leader helix might be the interaction surface for WTAP, because it contains hydrophobic residues with optimal spacing for coiled coil interactions. Myc-WTAP or -GFP as control, were cotransfected into HEK 293T cells together with either full-length F/H-METTL3 or F/H-METTL3 lacking the leader helix (F/H-METTL3ΔLH). Anti-myc immunoprecipitation revealed that binding of WTAP to F/H-METTL3ΔLH is strongly reduced while F/H-METTL3 can be readily detected (Fig. 1H). To further solidify these findings, we fused the leader helix to GFP and cotransfected this construct with the myc-tagged C-terminal truncations (CT1–4) (Fig. 1E) of WTAP into HEK 293T cells (Fig. 1I). Anti-myc immunoprecipitations show that the leader helix alone interacts with the CT1 and CT2 constructs (Fig. 1I), which is consistent with the data obtained with the full-length METTL3 (Fig. 1G). Our results demonstrate that the leader helix alone is necessary and also sufficient for the interaction of the two proteins. Taken together, using recombinant proteins and coimmunoprecipitations, we define the major interaction surfaces within the METTL3–METTL14–WTAP complex (Fig. 1D,J).
Nuclear localization signals on WTAP and METTL3 mediate nuclear import of WTAP and the METTL3/14 heterodimer
WTAP and METTL3/14 localize to the nucleus of human cells, where they are enriched in nuclear speckles (Horiuchi et al. 2013; Ping et al. 2014). It has been demonstrated that WTAP regulates the localization of METTL3/14 into speckles (Ping et al. 2014). How these proteins reach the nucleus, however, is currently unknown. To address this question, we predicted potential nuclear localization signals (NLS) on WTAP, METTL3, and METTL14 (Fig. 2). WTAP possesses a potential NLS at its N terminus. To directly validate whether this is indeed a functional NLS, key residues were mutated and myc-tagged wild-type (wt) and mutant WTAP were transfected into HeLa cells (Fig. 2A). While wt WTAP localizes to the nucleus, the NLS-deleted variant appears to be solely in the cytoplasm, indicating that we have identified the NLS on WTAP. For METTL3, we predicted a potential NLS in a helical domain located between the N-terminal leader helix and the MTD. F/H-METTL3 localizes predominantly to the nucleus (Fig. 2B). A mutant variant (METTL3-NLS-mut), however, is retained in the cytoplasm, suggesting that the predicted NLS is indeed used for nuclear import. To rule out that the cytoplasmic localization of METTL3-NLS-mut is caused by impaired nuclear retention by binding to WTAP, we performed coimmunoprecipitations (Fig. 2C). F/H-METTL3 and METTL3-NLS-mut efficiently interacted with cotransfected myc-WTAP, indicating that cytoplasmic localization is caused by deletion of the NLS rather than disturbed WTAP interaction (Fig. 2C).
FIGURE 2.

Localization studies of METTL3, METTL14, and WTAP. (A) Immunofluorescence staining of HeLa cells transfected with wild-type (WT) myc-WTAP (upper panels) and myc-WTAP-NLS mutant (mut) (lower panels). The schematic cartoon under the immunofluorescences shows the very N-terminal location and the sequence of the NLS in the protein. (B) Immunofluorescence of HeLa cells which were transfected with WT (upper panels) and NLS-mutated F/H-METTL3 (lower panels). The computed NLS is predicted in the potential RBD between the leader helix and the MT-A70 domain as shown in the schematic view beneath the stainings. (C) Western blots of coimmunoprecipitations between F/H-METTL3-NLS mutant and myc-WTAP. GFP containing either a myc- or a F/H-tag serves as control. (D) HeLa cells were transfected with myc-METTL14 WT (upper panels) and the predicted F/H-NLS mutant (lower panels). The cartoon shows the position and sequence of the potential NLS in the protein. (E) For the upper panel, HEK293T cells were transfected with myc-METTL14 and F/H-METTL3. The lower panels show immunofluorescence stainings of myc-METTL14 and the predicted F/H-METTL3-NLS mutant. (F) Western blot of the coimmunoprecipitation of different F/H-METTL3 and myc-METTL14 constructs. GFP was used as control.
We also predicted a putative NLS for METTL14, which is found within the N-terminal domain (Fig. 2D). Mutation of this NLS did not alter nuclear localization of myc-METTL14, suggesting that the analyzed sequence element does not serve as NLS (Fig. 2D). How does METTL14 reach the nucleus if it does not contain a functional NLS? Since METTL3 and METTL14 form a heterodimer and this appears to be the functional complex, we reasoned that this complex may form in the cytoplasm and is transported as a heterodimer. To directly test this hypothesis, we cotransfected myc-METTL14 together with METTL3-NLS-mut, which is retained in the cytoplasm (Fig. 2E). Indeed, under these conditions, METTL14 is retained in the cytoplasm as well, suggesting that a functional NLS on METTL3 is needed to transport METTL14 into the nucleus.
Our localization data suggest that METTL14 reaches the nucleus by binding to METTL3. To further validate this model, we analyzed interactions of the NLS mutant and wt METTL proteins (Fig. 2F). Myc- and F/H-tagged proteins were cotransfected and F/H-proteins were immunoprecipitated using anti-FLAG antibodies. The isolated proteins were visualized by western blotting against the HA- or myc-tag. These experiments revealed that both the METTL3-NLS-mut and the METTL14-NLS-mut efficiently interact with the respective wt binding partners, suggesting that METTL14 retention could indeed be due to the formation of a METTL3-NLS-mut/METTL14 heterodimer that is retained in the cytoplasm.
In summary, we have identified the NLS of WTAP and METTL3. Furthermore, we suggest a model in which METTL14 does not contain its own NLS but is transported in complex with METTL3.
The METTL3/14 complex is phosphorylated at several sites
The formation and function of macromolecular protein complexes can be mediated or supported by post-translational modifications such as phosphorylation. We therefore analyzed whether METTL3/14 carry phosphorylations that are important for assembly and/or function. Since overexpression of proteins can lead to unspecific results due to unphysiological levels, we generated a rat monoclonal antibody against METTL3 allowing for the analysis of endogenous proteins (Fig. 3). Peptides spanning the METTL3 amino acids 17–31 (SLRERLQRRRKQDSG) were injected into rats, hybridomas were generated and validated by ELISA, western blot, and immunoprecipitation. We established the monoclonal antibody clone METTL3 29C8 (IgG2a/k), which efficiently immunoprecipitates ectopic (Fig. 3A, upper panel) as well as endogenous METTL3 (lower panel). In western blot analyses, only overexpressed or enriched METTL3 is well detected (data not shown). The 29C8 antibody is suitable for complex purification since METTL3 and -14 are prominent in immunoprecipitates separated on coomassie stained gels (Fig. 3B).
FIGURE 3.

Establishment of a monoclonal antibody against METTL3. (A) The upper western blot shows IPs of F/H-METTL3 using an α-FLAG antibody for precipitation on the one hand and the established α-METTL3 antibody 29C8 on the other. The blot was incubated with an α-HA antibody. The lower panel shows a western blot of an endogenous METTL3-IP using the METTL3 antibody. (B) Coomassie gel of endogenous METTL3 purified using the anti-METTL3 antibody 29C8. METTL14 is coimmunoprecipitated. The control IP does not show any bands.
For phospho-analysis, endogenous METTL3 was immunoprecipitated from nuclear HeLa cell lysates. Since the 29C8 antibody efficiently coimmunoprecipitated endogenous METTL14, both proteins can be analyzed simultaneously. The two protein bands were excised from the gel and analyzed by mass spectrometry (Fig. 4A). We were able to map several phosphorylation sites on METTL3. Phosphorylations are found at the N terminus clustering around the leader helix (S2, S43, S48, S50), the central region close to the NLS (S219) and adjacent to the MTD (S243, T348, S350) (Fig. 4B). Alignment of METTL3 from different species revealed that S2, S43, S48, S50, S219, and S243 are conserved in vertebrates but not in Drosophila. S350 is conserved in mammals but not in Zebrafish and Drosophila and T348 is not conserved, suggesting a human-specific function or a potentially nonrelevant phosphorylation (Fig. 4C). For METTL14, we found only one phosphorylation site, located in a C-terminal motif adjacent to the MTD (Fig. 4B), which is conserved in all organisms analyzed (Fig. 4C). Taken together, we comprehensively mapped phosphorylation sites on endogenous METTL3/14 from HeLa cells and find modifications at regions that might be functionally important.
FIGURE 4.

Identification and characterization of phosphorylation sites in METTL3/14 complex. (A) Coimmunoprecipitation of METTL3 and METTL14. To analyze phosphorylation sites, endogenous METTL3 was purified from nuclear HeLa S3 lysate by α-METTL3 29C8 antibody. Endogenous METTL14 was coimmunoprecipitated. (B) Mass spectrometric measurements of human METTL3/14 complex. Schematic representation of phosphorylation sites in METTL3 and METTL14 proteins. The structural domains (leader helix [LH]; nuclear localization sequence [NLS]; N-terminal α-helical motif [NHM]; C-terminal motif [CTM]) are shown in gray and phosphorylation sites are represented by red bars. (C) Conservation of the phosphorylation sites detected in METTL3 and METTL14. Phosphorylation sites of METTL3 and METTL14 measured in our analysis are shown in red. (D) Nuclear localization of METTL3 S219 phosphorylation variants. F/H-METTL3 constructs were detected by α-FLAG antibody staining (green). Myc-METTL14 was visualized by α-myc antibody (red). Nuclei were stained with DAPI (blue). (E) Impact of the LH-surrounding phosphorylation sites on WTAP interaction. HEK293T cells were transfected with F/H-METTL3 phosphorylation mutants together with myc-WTAP. Complexes were purified by α-FLAG-IPs. F/H-METTL3 constructs and copurified myc-WTAP were subsequently visualized by western blotting.
METTL3 phosphorylation does not affect subcellular localization, WTAP interaction, or catalytic activity
The phospho-sites on METTL3 are either at the N terminus, where WTAP interactions occur, close to the NLS or the catalytic MTD (Fig. 4B). Therefore, we generated phospho-mimicking glutamate (E) or phospho-lacking alanine (A) mutants of all the identified phosphorylated residues and analyzed localization, WTAP binding, or catalytic activity. F/H-METTL3 wt, S219A and S219E were cotransfected with myc-tagged METTL14 into HEK293T cells and localization was examined by immunofluorescence experiments (Fig. 4D). Both mutants were indistinguishable from wt and therefore, we concluded that phosphorylation of S219 does not affect localization. Although the phosphorylated residue is located close to the NLS, phosphorylation has no effect on subcellular localization. Next, we analyzed phosphorylation sites, located at the N terminus of METTL3, where we mapped the interaction with WTAP (Fig. 4E; see Supplemental Fig. 2 for specificity control). F/H-METTL3 S2A/E, S43A/E, S48A/E, and S50A/E were cotransfected together with myc-tagged WTAP and anti-FLAG immunoprecipitations were performed. All mutants efficiently coimmunoprecipitated WTAP, suggesting that phosphorylation does not affect WTAP interactions (Fig. 4E).
METTL3 interacts with METTL14 via its MTD. Since several phospho-sites were located close to this domain, we investigated METTL14 interaction (Fig. 5A; Supplemental Fig. 2). F/H-tagged A or E mutants of the residues S219, S243, T348, or S350 were cotransfected together with myc-METTL14 and coimmunoprecipitations were performed. Again, all mutations had no effect on the interaction with METTL14 indicating that the analyzed phospho-sites are dispensable for METTL14 interaction. Finally, we reasoned that the phospho-sites close to the MTD might influence catalytic activity. Thus, we performed in vitro methylation assays based on the conditions reported before (Wang et al. 2016a). An RNA substrate (4× ggacu [Wang et al. 2016a]) was incubated with the recombinant METTL3/14 complex and the transfer of a methyl group from radiolabeled SAM to the RNA substrate was monitored (Fig. 5B). Indeed, efficient RNA methylation was observed under these conditions. A METTL3 variant containing a mutated catalytic center (DPPW to APPA) was inactive (Fig. 5B). A METTL14 variant with mutations in the putative catalytic center (EPPL to APPA), however, is indistinguishable from wt, confirming the structural model, in which the catalytic center of the METTL3/14 complex is solely formed by METTL3. These experiments served as controls for our studies. Next, we tested S348 and S350 A and E mutants. Recombinant wt and mutated METTL3 in complex with METTL14 was generated and in vitro methylation assays were performed as described above (Fig. 5C). None of the mutants affected methylation activity. Taken together, phosphorylation at the detected sites appears to be dispensable for the basic METTL3 functions.
FIGURE 5.

Phosphorylation sites of METTL3 and METTL14 are not essential for methylation activity. (A) Phosphorylation sites in the central region of METTL3 do not influence the interaction with myc-METTL14. HEK293T cells were transfected with F/H-METTL3 phosphorylation mutants together with myc-METTL14. Complexes were purified by α-FLAG-IPs. F/H-METTL3 constructs and copurified myc-METTL14 were visualized by western blotting. (B) Measurement of methyltransferase activity of METTL3/14 wild-type and mutated variants. Point mutations in the catalytic center of both proteins were introduced as indicated in the schematic representation. Data are shown as mean ± SD from six independent replicates. As control, 1 µg of recombinant protein was separated on a 10% SDS gel and stained with Coomassie blue. (C) Methyltransferase activity of WT METTL3/14 complex and variants in the phosphorylation residues T348 and S350. Data are shown as mean ± SD from five independent replicates. As control, 1 µg of recombinant protein was separated on a 10% SDS gel and stained with Coomassie blue. (D) Structural environment of phosphorylated residue S399 in METTL14 (green) relating to R471 in METTL3 (blue). (Left) pS399 position within the METTL3/14 interaction surface. (Right) Potential contact of pS399 to the R471 residue. (E) Coimmunoprecipitation of F/H-METTL3 with S399A and S399E mutants of myc-METTL14. HEK293T cells were transfected with F/H-METTL3 and myc-METTL14 constructs. F/H-METTL3 was purified by α-FLAG-IP. All myc-METTL14 constructs were copurified and visualized by western blotting. (F) In vitro methyltransferase activity of WT METTL3/14 and mutants in the phosphorylated S399 residue of METTL14. Phosphorylation residues S399 of METTL14 was modified as shown in the schematic representation. Data are shown as mean ± SD from six independent replicates. For control, 1 µg of recombinant protein was separated on a 10% SDS gel and stained with Coomassie blue. Of note, primary antibodies against the HA- and the myc-tags were added simultaneously to the same western blot membrane. Since F/H-METTL3 and myc-METTL14 migrate identically on the gel, western blots appear very similar (A,E).
Mutation of METTL14 S399 does not affect METTL3 binding and methylation activity
We next assessed, whether phosphorylation of METTL14 affects the function of the enzyme complex. METTL14 S399 is located close to the MTD. Interestingly, one of the recent structural studies suggested that phosphorylation of METTL14 S399 could be important for the interaction with METTL3 since it could form a salt bridge with R471 of METTL3 (Fig. 5D; Wang et al. 2016b). We therefore analyzed, whether S399 phosphorylation affects METTL3–METTL14 interactions (Fig. 5E; Supplemental Fig. 2). F/H-METTL3 was cotransfected with myc-METTL14 or myc-METTL14-S399A or -S399E mutants and coimmunoprecipitations were performed as described above. Both phospho-mutants bound efficiently to METTL3. In contrast to the suggested model in which S399 phosphorylation contributes to METTL3–METTL14 interactions (Wang et al. 2016a), our data show that phosphorylation is dispensable for this interaction.
Although METTL14 S399 phosphorylation is not important for binding to METTL3, it could nevertheless affect methylation activity. We next investigated whether METTL14 S399 phosphorylation may alter catalytic activity. Consistently with our binding and localization data, both the METTL14 S399A and S399E mutants in complex with wt METTL3 show similar methylation activity as observed for the wt proteins (Fig. 5F), indicating that methylation activity is independent of S399 phosphorylation.
Unlike suggested before (Wang et al. 2016a), we found that phosphorylation of METTL14 at S399 is not required for efficient complex formation and methylation activity.
METTL14 contacts the RNA substrate via its RGG repeats
Besides its methyltransferase domain, METTL14 contains Arginine-Glycine repeats (RGG) at the C terminus. The function of these repeats is unknown. We therefore analyzed whether the RGG repeats are important for METTL3/14 activity. METTL3/14 or METTL3/14ΔRGG were coexpressed in insect cells and purified via GST-METTL3. In addition, a TEV cleavage site was inserted in front of the RGG repeats allowing for removal after purification (Fig. 6A, right panel). Both the METTL3/14ΔRGG complex and the TEV cleaved complex lost methylation activity while wt complexes remained active (Fig. 6A, left panel). These data indicate that the RGG repeats of METTL14 contribute to catalytic activity of the METTL3/14 complex. Of note, METTL3 alone can be stably expressed and forms monomers (data not shown). However, although it contains the critical catalytic residues, it remains inactive without binding to METTL14 (Fig. 6A). These observations are consistent with a proposed role for monomeric METTL3 in translational control (Lin et al. 2016).
FIGURE 6.

RGG deletion affects catalytic activity of METTL3/14 and substrate binding (A) Analysis of methyltransferase activity of METTL3 and METTL14 lacking the RGG repeats. Truncations were introduced to the METTL14 sequence (METTL14 ΔRGG) directly or mediated by TEV protease cleavage (METTL14 TEV-RGG) as indicated in the schematic representation. Data are shown as mean ± SD from five independent replicates. As control, 1 µg of recombinant protein was separated on a SDS gel and stained with Coomassie blue. (B) RNA binding of full-length METTL3/14 and METTL3/14 ΔRGG. The proteins were diluted serially in a 50 µL reaction mixture. (C) RNA-binding studies of F/H-METTL14 using cross-linking and immunoprecipitation (CLIP). F/H-METTL3 with F/H-METTL14-WT or F/H-METTL14-ΔRGG were transfected into HEK293T cells grown in 4SU-containg medium and UV cross-linked (356 nm). After α-FLAG-IP and RNase T1 digestion, the bound RNA was radioactively labeled, the protein-RNA complex was separated by SDS-PAGE and blotted onto a nitrocellulose membrane. Cross-linked RNA was visualized by autoradiography.
RGG repeats are frequently found in RNA binding proteins and it has been reported that these repeats can be involved in RNA binding (Thandapani et al. 2013). To directly test RGG-mediated RNA binding, we performed filter-binding assays (Fig. 6B). Purified METTL3/14 and METTL3/14ΔRGG were incubated with radiolabeled substrate RNA and the binding reactions were subsequently transferred to nitrocellulose membranes that do not bind RNA. Thus, only protein-bound RNA is retained on the filter. Strikingly, we found that removal of the RGG repeats strongly reduces binding of METTL14 to the RNA substrate. To further strengthen these observations, we performed CLIP experiments (Fig. 6C; see Supplemental Fig. 3 for loading control). F/H-METTL3 together with F/H-METTL14 wt or -ΔRGG was transfected into HEK293T cells, which were grown in 4-thio-uridine (4SU) containing medium. Cells were treated with UV light to cross link endogenous proteins to 4SU-labeled RNA. F/H-METTL3 and F/H-METTL14 were then immunoprecipitated, the cross-linked RNA radioactively labeled, and the RNA protein complexes analyzed by SDS-PAGE (Hafner et al. 2010). In agreement with our filter-binding assays, F/H-METTL14 was efficiently cross-linked to RNA, while the ΔRGG mutant showed much weaker signals (Fig. 6C; Supplemental Fig. 3). Taken together, our experiments identify the RGG repeats of METTL14 as a second RNA contact region besides the catalytic MTD within the METTL3/14 enzyme complex.
DISCUSSION
More than 100 different RNA modifications have been found on cellular RNAs. Some of these modifications have not only been found on noncoding RNAs such as rRNAs or tRNAs but also on mRNAs (Roundtree et al. 2017). Due to the availability of a potent antibody, m6A mRNA modification has been broadly studied (Helm and Motorin 2017). Biochemical and structural work has demonstrated that m6A is generated by the METTL3/14 enzyme complex, which associates with WTAP and KIAA1429 (virilizer) in the nucleus. To better understand the biology of this complex, we have further characterized its molecular architecture. Using truncated versions and coimmunoprecipitation studies, we found that the two MTDs of METTL3 and 14 tightly bind to each other and form the catalytic center for adenine methylation. However, catalytic activity is solely provided by METTL3. Our data confirm the recent structural studies of the dimeric METTL3/14 MTD that were reported while our work was in progress (Śledź and Jinek 2016; Wang et al. 2016a,b). One of the reported structures identified a phosphorylated serine (S399) on METTL14 that is placed adjacent to the METTL3/14 interaction surface (Wang et al. 2016a). We performed mass spectrometry and identified S399 phosphorylation also in our protein preparations. When mutating S399 to alanine or glutamate, neither methylation activity nor binding to METTL3 was affected. Therefore, we suggest that the phosphorylated residue S399 is not required for METTL3/14 activity. Our mass spectrometry analyses revealed a number of additional phosphorylations all located on METTL3 (Fig. 7). However, none of them are required for protein interaction and localization (data not shown). It is conceivable that these phosphorylations play a role under specific physiological conditions (e.g., cross talk with signaling pathways) but are most likely not relevant for in vitro activity.
FIGURE 7.
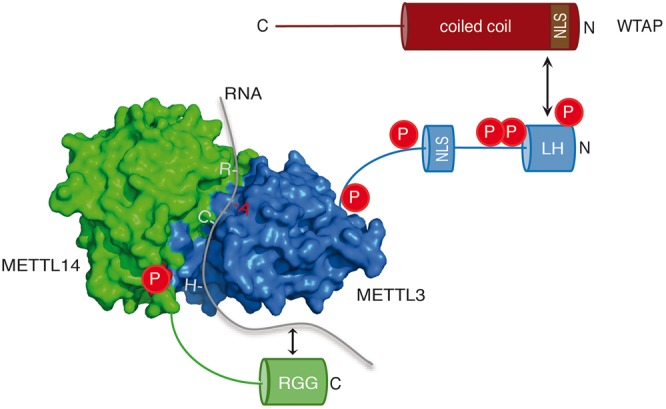
Schematic representation of the interaction surfaces of METTL3 (blue), METTL14 (green), and WTAP (red). Proposed working model includes phosphorylation sites (red) of METTL3/14 as well as the NLS motifs of METTL3 (blue) and WTAP (brown). METTL3 catalyzes methylation of the adenosine base (red) within the RRACH motif. METTL14 coordinates and stabilizes the RNA binding.
Our functional mapping studies further revealed that the N-terminal leader helix (LH) of METTL3 interacts with the N-terminal part of WTAP. Structural prediction of the leader helix suggests a high probability for a coiled coil and therefore the leader helix most likely engages in interactions with the N-terminal part of the coiled coil of WTAP (Fig. 7). WTAP is found in the nucleus and is required for METTL3/14 localization to speckles (Ping et al. 2014). Thus, METTL3ΔLH should not be found in speckles. Although our experiments suggest such a model, due to over expression effects, we were not able to clearly distinguish between speckles and the nucleoplasm. This needs to be analyzed in more detail.
We further identified NLS both on METTL3 and WTAP. Mutation of these sequences leads to cytoplasmic retention, suggesting that WTAP and METTL3 are transported to the nucleus independently and find each other in the nucleus, again supporting the model that WTAP targets METTL3 to specific sites such as nuclear speckles. Interestingly, METTL14 does not contain a functional NLS and is cotransported to the nucleus together with METTL3. The dimeric complex forms already in the cytoplasm and is imported as one functional unit. METTL3 has been shown to function without METTL14 in the cytoplasm and promotes translation of specific mRNAs independently of its catalytic activity (Lin et al. 2016). It will be interesting to see which signals prevent METTL3 from binding to its partner METTL14. This is particularly intriguing in the light of the tight binding of the two proteins to each other and the formation of an active enzymatic center. It is tempting to speculate that specific cytoplasmic protein-binding partners or post-translational modifications are involved in the formation of different METTL3 complexes.
Finally, we studied the C-terminal RGG repeats of METTL14. Deletion of these repeats leads to reduced methylation activity of the complex, which is due to strongly reduced substrate RNA binding (Fig. 7). RGG repeats are often found in nucleic acid binding proteins and it has indeed been shown for some of them that they are directly involved in RNA binding (Thandapani et al. 2013). They do not bind specific RNA sequences but rather recognize secondary structures such as G-quartets. Therefore, it appears unlikely that additional sequence motifs surrounding the methylated RRACH motif are recognized and bound by METTL14. This would be consistent with the sequence of our substrate RNA, which is a quartet of the RRACH motif without additional sequences. It is more likely, however, that the RRACH motif may form a distinct secondary structure or conformation and such structures require the RGG motif of METTL14. It should also be noted that RBM15 has been suggested to support interactions between the METTL3/14 complex and substrate RNA (Patil et al. 2016). It is therefore conceivable that RBM15 recruits the complex to distinct sites in a sequence-specific manner while the RGG repeats of METTL14 facilitate efficient RNA binding sequence-unspecifically. Future experiments and structural analysis of the full-length METTL3/14 dimer in complex with its substrate RNA will help elucidate the function of the RGG repeats of METTL14.
MATERIALS AND METHODS
Cell culture and transfections
HEK 293T, HeLa, and adherent HeLa S3 cells were cultivated under standard conditions (37°C and 5% CO2 atmosphere) by using Dulbecco's modified Eagle's medium (DMEM, Sigma-Aldrich) supplemented with 10% FBS (Gibco) and 1% penicillin-streptomycin (Sigma-Aldrich). Adapted HeLa S3 cells were grown in Joklik's medium (Sigma-Aldrich) supplemented with l-glutamine, nonessential amino acids, sodium hydrogen carbonate, 10% FBS and 1% penicillin–streptomycin in a rotating cell culture system at 37°C. Sf21 cells were cultured in SF-900 II SFM serum-free medium (Gibco) at 27°C, shaking at 80 r.p.m.
For immunoprecipitations of overexpressed proteins, HEK 293T cells were cultured on dishes with a diameter of 15 cm. Then cells were calcium phosphate-transfected with 4–10 µg of plasmid DNA per plate. Cells were harvested 48 h after transfection. For immunofluorescence, HEK 293T and HeLa cells were transfected with 125–500 ng of plasmid DNA at a confluency of 100% on 24-wells using Lipofectamine 2000 (Thermo Fisher Scientific). Twenty four hours after transfection, 50% of transfected HeLa cells were transferred on cover slips. For baculoviruses amplification, Sf21 cells were seeded on 6-wells at a cell count of 0.5 × 106 cell/mL and transfected with FuGENE (Promega).
Immunoprecipitation, Coomassie staining, and western blot analysis
For immunoprecipitation experiments, the antibody of interest was coupled to protein G Sepharose beads (GE Healthcare). Five micrograms of purified antibody was added to 30 µL of beads. Prior to coupling, the beads were washed twice with 1× PBS. The antibody was coupled to the beads overnight at 4°C. Cells lysis was induced by lysis buffer (25 mM Tris/HCl, pH 7.5, 150 mM KCl, 0.1% [v/v] NP-40, 2 mM EDTA, 1 mM NaF, 0.5 mM DTT, and 1 mM AEBSF; used in Fig. 1E,F,G, and Fig. 2E,F) or NP-40 buffer (20 mM Tris/HCl, pH 8, 137 mM NaCl, 1% [v/v] NP-40, 10% [v/v] glycerol, 2 mM EDTA, 0.5 mM DTT, and 1 mM AEBSF; used in Fig. 4). The reaction was carried out for 30 min on ice. Lysates were cleared by centrifugation at 17,000g for 15 min at 4°C and added to the beads. The setups were then incubated for 3 h at 4°C while rotating. The beads were washed four times with Co-IP washing buffer (50 mM Tris HCl, pH 7.4, 300 mM KCl, 0.1% [v/v] NP-40, 1.5 mM MgCl2, 0.5 mM AEBSF, 1 mM DTT). Elution of the proteins was conducted by adding 5× Laemmli sample buffer to the beads and boiling the IP setup for 5 min at 95°C. The supernatant was loaded onto a 10% SDS-polyacrylamide gel. Coomassie staining was performed by incubating the gel for 1 to 2 h in 10% (v/v) acetic acid, 30% (v/v) ethanol, and 0.25% (w/v) Coomassie R250. Gels were afterwards rinsed three times with Milli Q-grade H2O and destained with 10% (v/v) acetic acid and 30% (v/v) ethanol. For western blotting, proteins were transferred onto a Nitrocellulose membrane (GE Healthcare) using Towbin blotting buffer (192 mM glycine, 25 mM Tris/HCl pH 8.6, 20% [v/v] methanol). Membranes were blocked in 1× TBST (150 mM NaCl, 10 mM Tris/HCl, pH 8, 0.1% [v/v] Tween 20) containing 5% (w/v) skim milk for 1 h at 4°C. After incubation with the primary antibodies (α-HA [Covance] 1:1000; α-myc [Sigma] 1:500; α-METTL3 29C8), membranes were washed three times with 1× TBST. Of note, α-HA and α-myc antibodies were applied simultaneously to the same membrane. After incubating with the secondary antibody (α-mouse and α-rabbit [Licor] 1:10000, which were also applied simultaneously), the membrane was again washed three times with 1× TBST. The documentation was conducted using the Odyssey scanner system (LI-COR Biosciences).
Immunofluorescence staining
HEK 293T cells were seeded on coverslips with Gelatin solution Type B (Sigma). Fixation and permeabilization were performed 24 h later at −20°C with ice-cold acetone (Alshammari et al. 2016). After blocking with 6% (w/v) BSA in 1× TBS containing 0.3% (v/v) Triton X-100, cells were incubated with 3% (w/v) BSA in PBS containing 0.1% (v/v) Tween-20 and primary antibody (α-FLAG 1:1000; α-myc [Sigma] 1:500) over night at 4°C. Incubation with secondary antibody (α-mouse Alexa 488 and α-rabbit Alexa 555 [Invitrogen] 1:400) diluted in 3% (v/v) BSA in PBS containing 0.1% (v/v) Tween-20 was carried out for 1 h at room temperature. The immunofluorescence experiments of HeLa cells were conducted as described previously (Schraivogel et al. 2015). The first and secondary antibodies were incubated in blocking buffer (0.05% [v/v] Triton X-100, 50 g/L BSA [Cohn-Fraction V] in PBS). After incubation with the antibodies, HEK293T cells were washed five times 5 min in PBS, HeLa cells once with blocking buffer and four times with PBS. Subsequently the cells were mounted using Prolong Gold containing DAPI (Thermo Fisher Scientific–Life Technologies). Confocal microscopy was done on a TCSSP8 (Leica Microsystems) equipped with acousto-optical beam splitter, 405 nm laser (for DAPI), argon laser (488 nm for Alexa 488), and DPSS laser 561 nm.
In silico prediction of nuclear localization signals
Nuclear localization sequences (NLS) were predicted using the Eukaryotic Linear Motif resource tool (http://elm.eu.org/search/). The default settings of this tool were used for the prediction of NLS within METTL3, METTL14, and WTAP.
Preparation of nuclear extract from HeLa S3
Nuclear fractionation was performed according to the protocol of Kataoka and Dreyfuss (2008). HeLa S3 cells were used at the growth stage of 4–6 × 106 cell/mL. Briefly, before cell disruption with a dounce homogenizer, cells were swollen for 10 min in buffer A (10 mM HEPES-KOH [pH 7.9], 10 mM KCL, 1.5 mM MgCl2, 0.5 mM AEBSF, 1 mM DTT, 5 mM NaF, and PhosSTOP phospatase inhibitor [Roche]). Then, nuclei were precipitated by centrifugation at 1900g for 10 min and resuspended in buffer C (20 mM HEPES-KOH [pH 7.9], 600 mM KCL, 1.5 mM MgCl2, 0.2 mM EDTA, 25% [v/v] glycerol 0.5 mM AEBSF, 1 mM DTT, 5 mM NaF, and PhosSTOP). The nuclei were extracted by several strokes with a loose pestle followed by incubation 30 min at 4°C while shaking. The nuclei extract was cleared by centrifugation at 39,000g for 30 min at 4°C and supernatant was dialyzed against lysis buffer (25 mM Tris/HCl, pH 7.5, 150 mM KCl, 0.1% [v/v] NP-40, 2 mM EDTA, 5 mM NaF, 0.5 mM DTT, and 1 mM AEBSF) two times for 30 min at 4°C. The precipitate was removed by centrifugation at 33,000g at 4°C before the nuclear extract was used for immunoprecipitation.
Protein purification
Full-length METTL3 and METTL14 as well as mutants and truncations were coexpressed from pFB-HT-A in SF21 cells using the BAC-to-BAC baculovirus expression system. Seventy-two hours after infection the cells were harvested and lysed by sonication in PBS buffer (pH 7) supplemented with 1.5 M NaCl and 2 mM DTT. The complex was bound to a Glutathione Sepharose column (GE Healthcare) using the N-terminal GST-tag contained in all METTL3 constructs. After elution with 10 mM glutathione the GST-tag was cleaved off by addition of tobacco etch virus (TEV) protease for which a specific cleavage site was engineered between METTL3 and the tag. The cleavage reaction was incubated overnight and the solution was subsequently concentrated to 1 mL total volume and run over a Superdex S200 10/30 gel filtration column (GE Healthcare) in a buffer containing 50 mM HEPES pH 7.5 200mM NaCl and 2 mM DTT. METTL3 and METTL14 cofractionated in a well-defined peak, which was collected and concentrated by ultrafiltration.
In vitro methylation assay
For m6A methylation assay a RNA probe of four repetitive RRACH sequences 5′-GGACUGGACUGGACUGGACU-3′ (Wang et al. 2016b) was used. The reaction mixture of 40 µL contained 20 mM Tris/HCl (pH 7.5), 0.01% (v/v) Triton-X, 1 mM DTT, 50 µM ZnCl2, 0.2 U/mL RiboLock (Thermo Fisher Scientific), 1% (v/v) glycerol, 50 nM purified protein (Li et al. 2016) and 2.25 × 10−3 µCi [3H]-SAM. The methylation reactions were incubated at 30°C for 1 h. The methylation reaction was stopped at 65°C for at least 5 min and purified afterwards using MicroSpin G-25 columns (GE Healthcare Life Sciences) according to the manufacturer's protocol. The levels of the incorporated 3H-methyl group were measured in counts per minute (c.p.m.) using a multipurpose scintillation counter (Beckmann Coulter LS6500). The purified RNA was dissolved in 10 mL scintillations cocktail before (Zinsser Analytic). The average c.p.m. was determined from five to six independent measurements.
32P labeling of RNA
The RNA of four repetitive RRACH sequences 5′-GGACUGGACUGGACUGGACU-3′ (Wang et al. 2016b) used also in the m6A methylation assay was labeled with radioactive phosphate. One micromole of RNA oligonucleotides were incubated with 20 µCi of [γ-32P]ATP (Hartmann Analytic) and 0.5 U/µL T4 Polynucleotide Kinase (PNK) in 1× PNK buffer A (Thermo Scientific) at 37°C for 30–60 min. The labeling reaction was stopped by EDTA, pH 8.0 and purified afterwards using MicroSpin G-25 columns (GE Healthcare Life Sciences) according to the manufacturer's protocol.
In vitro filter binding assay
Serial dilutions of WT METTL3/METTL14 complex and METTL3/METTL14 ΔRGG mutant were incubated with 2 Bq/cm2 of 32P-labeled RNA in 20 mM Tris/HCl pH 7.5, 0.01% (v/v) Triton-X, 1 mM DTT, 50 µL ZnCl2, 0.2 U/mL RiboLock (Thermo Fisher), 1% (v/v) glycerol. The binding reactions were carried out at 25°C for 30 min and were then transferred by vacuum to a nitrocellulose membrane. The amount of the protein-bound RNA was determined by liquid scintillation counting. The nitrocellulose membrane was incubated in 10 mL scintillation cocktail (Zinsser Analytic) and counts per minutes (c.p.m.) were measured with a multipurpose scintillation counter (Beckmann Coulter LS6500). The measurements were performed in four independent replicates.
Cross-linking and immunoprecipitation (CLIP)
The PAR-CLIP protocol was conducted as described before (Baltz et al. 2012) with minor changes. In short, transfected HEK293T cells were treated with 100 µM 4-SU 16 h prior to harvesting and UV cross-linked at 365 nm, washed with cold PBS and harvested. The UV negative cells were harvested directly. The pellets were resuspended in NP-40 lysis buffer (50 mM HEPES-KOH, pH 7.5, 150 mM KCl, 2 mM EDTA-NaOH, pH 8, 1 mM NaF, 0.5% [v/v] NP-40 substitute). The cleared lysate was RNase T1 digested for 7 min at 22°C. α-FLAG M2 beads (Sigma) were used for IP (2 h at 4°C, rotating). After IP, the beads were washed with IP wash buffer (50 mM HEPES-KOH, pH 7.5, 300 mM KCl, 0.05% [v/v] NP-40 substitute) and again RNase T1 digested as before. After a High Salt wash step (50 mM HEPES-KOH, pH 7.5, 500 mM KCl, 0.05% [v/v] NP-40 substitute), the beads were dephosphorylated (50 mM Tris-HCl, pH 7.9, 100 mM NaCl, 10 mM MgCl2) with Calf Intestinal Alkaline Phosphatase (NEB) for 1 h at 37°C. Beads were washed with phosphatase buffer (50 mM Tris-HCl, pH 7.9, 20 mM EGTA-NaOH, pH 7.5, 0.5% [v/v] NP-40 substitute) and phosphorylated with [γ-32P]ATP, using PNK (Thermo Fisher) at 37°C for 1 h. For the last washing step, PNK buffer (50 mM Tris-HCl, pH 7.5, 50 mM NaCl, 10 mM MgCl2) was used. The beads were then resuspended in 5× Laemmli sample buffer. The supernatant was loaded onto a SDS gel, which was blotted on a Hybond ECL membrane (GE Healthcare). The signals were detected with the Phospho-imager (PMI, Bio-Rad).
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
We thank S. Ammon and C. Friederich for technical support, E. Hochmuth for mass spectrometric assistance, and R. Hett for antibody purification. Our work is supported by the Deutsche Forschungsgemeinschaft (SPP 1784/1), the Bavarian Systems-Biology Network (BioSysNet), and the European Research Council (ERC grant “moreRNA”).
Footnotes
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.064063.117.
REFERENCES
- Alshammari MA, Alshammari TK, Laezza F. 2016. Improved methods for fluorescence microscopy detection of macromolecules at the axon initial segment. Front Cell Neurosci 10: 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baltz AG, Munschauer M, Schwanhausser B, Vasile A, Murakawa Y, Schueler M, Youngs N, Penfold-Brown D, Drew K, Milek M, et al. 2012. The mRNA-bound proteome and its global occupancy profile on protein-coding transcripts. Mol Cell 46: 674–690. [DOI] [PubMed] [Google Scholar]
- Dominissini D, Moshitch-Moshkovitz S, Schwartz S, Salmon-Divon M, Ungar L, Osenberg S, Cesarkas K, Jacob-Hirsch J, Amariglio N, Kupiec M, et al. 2012. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature 485: 201–206. [DOI] [PubMed] [Google Scholar]
- Fu Y, Dominissini D, Rechavi G, He C. 2014. Gene expression regulation mediated through reversible m6A RNA methylation. Nat Rev Genet 15: 293–306. [DOI] [PubMed] [Google Scholar]
- Hafner M, Landthaler M, Burger L, Khorshid M, Hausser J, Berninger P, Rothballer A, Ascano M Jr, Jungkamp AC, Munschauer M, et al. 2010. Transcriptome-wide identification of RNA-binding protein and microRNA target sites by PAR-CLIP. Cell 141: 129–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helm M, Motorin Y. 2017. Detecting RNA modifications in the epitranscriptome: predict and validate. Nat Rev Genet 18: 275–291. [DOI] [PubMed] [Google Scholar]
- Horiuchi K, Kawamura T, Iwanari H, Ohashi R, Naito M, Kodama T, Hamakubo T. 2013. Identification of Wilms’ tumor 1-associating protein complex and its role in alternative splicing and the cell cycle. J Biol Chem 288: 33292–33302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia G, Fu Y, Zhao X, Dai Q, Zheng G, Yang Y, Yi C, Lindahl T, Pan T, Yang YG, et al. 2011. N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat Chem Biol 7: 885–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kataoka N, Dreyfuss G. 2008. Preparation of efficient splicing extracts from whole cells, nuclei, and cytoplasmic fractions. Methods Mol Biol 488: 357–365. [DOI] [PubMed] [Google Scholar]
- Lane BG, Ofengand J, Gray MW. 1995. Pseudouridine and O2′-methylated nucleosides. Significance of their selective occurrence in rRNA domains that function in ribosome-catalyzed synthesis of the peptide bonds in proteins. Biochimie 77: 7–15. [DOI] [PubMed] [Google Scholar]
- Li F, Kennedy S, Hajian T, Gibson E, Seitova A, Xu C, Arrowsmith CH, Vedadi M. 2016. A radioactivity-based assay for screening human m6A-RNA methyltransferase, METTL3-METTL14 complex, and demethylase ALKBH5. J Biomol Screen 21: 290–297. [DOI] [PubMed] [Google Scholar]
- Lin S, Choe J, Du P, Triboulet R, Gregory RI. 2016. The m6A methyltransferase METTL3 promotes translation in human cancer cells. Mol Cell 62: 335–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Yue Y, Han D, Wang X, Fu Y, Zhang L, Jia G, Yu M, Lu Z, Deng X, et al. 2014. A METTL3-METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat Chem Biol 10: 93–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maity A, Das B. 2016. N6-methyladenosine modification in mRNA: machinery, function and implications for health and diseases. FEBS J 283: 1607–1630. [DOI] [PubMed] [Google Scholar]
- Matera AG, Terns RM, Terns MP. 2007. Non-coding RNAs: lessons from the small nuclear and small nucleolar RNAs. Nat Rev Mol Cell Biol 8: 209–220. [DOI] [PubMed] [Google Scholar]
- Mauer J, Luo X, Blanjoie A, Jiao X, Grozhik AV, Patil DP, Linder B, Pickering BF, Vasseur JJ, Chen Q, et al. 2017. Reversible methylation of m6Am in the 5′ cap controls mRNA stability. Nature 541: 371–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer KD, Jaffrey SR. 2017. Rethinking m6A readers, writers, and erasers. Annu Rev Cell Dev Biol 33: 319–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer KD, Saletore Y, Zumbo P, Elemento O, Mason CE, Jaffrey SR. 2012. Comprehensive analysis of mRNA methylation reveals enrichment in 3′ UTRs and near stop codons. Cell 149: 1635–1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer KD, Patil DP, Zhou J, Zinoviev A, Skabkin MA, Elemento O, Pestova TV, Qian SB, Jaffrey SR. 2015. 5′ UTR m6A promotes cap-independent translation. Cell 163: 999–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motorin Y, Helm M. 2011. RNA nucleotide methylation. Wiley Interdiscip Rev RNA 2: 611–631. [DOI] [PubMed] [Google Scholar]
- Ortega A, Niksic M, Bachi A, Wilm M, Sánchez L, Hastie N, Valcárcel J. 2003. Biochemical function of female-lethal (2)D/Wilms’ tumor suppressor-1-associated proteins in alternative pre-mRNA splicing. J Biol Chem 278: 3040–3047. [DOI] [PubMed] [Google Scholar]
- Patil DP, Chen CK, Pickering BF, Chow A, Jackson C, Guttman M, Jaffrey SR. 2016. m6A RNA methylation promotes XIST-mediated transcriptional repression. Nature 537: 369–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ping XL, Sun BF, Wang L, Xiao W, Yang X, Wang WJ, Adhikari S, Shi Y, Lv Y, Chen YS, et al. 2014. Mammalian WTAP is a regulatory subunit of the RNA N6-methyladenosine methyltransferase. Cell Res 24: 177–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rottman F, Shatkin AJ, Perry RP. 1974. Sequences containing methylated nucleotides at the 5′ termini of messenger RNAs: possible implications for processing. Cell 3: 197–199. [DOI] [PubMed] [Google Scholar]
- Rottman FM, Bokar JA, Narayan P, Shambaugh ME, Ludwiczak R. 1994. N6-adenosine methylation in mRNA: substrate specificity and enzyme complexity. Biochimie 76: 1109–1114. [DOI] [PubMed] [Google Scholar]
- Roundtree IA, Evans ME, Pan T, He C. 2017. Dynamic RNA modifications in gene expression regulation. Cell 169: 1187–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schraivogel D, Schindler SG, Danner J, Kremmer E, Pfaff J, Hannus S, Depping R, Meister G. 2015. Importin-β facilitates nuclear import of human GW proteins and balances cytoplasmic gene silencing protein levels. Nucleic Acids Res 43: 7447–7461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz S, Mumbach MR, Jovanovic M, Wang T, Maciag K, Bushkin GG, Mertins P, Ter-Ovanesyan D, Habib N, Cacchiarelli D, et al. 2014. Perturbation of m6A writers reveals two distinct classes of mRNA methylation at internal and 5′ sites. Cell Rep 8: 284–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi H, Wang X, Lu Z, Zhao BS, Ma H, Hsu PJ, Liu C, He C. 2017. YTHDF3 facilitates translation and decay of N6-methyladenosine-modified RNA. Cell Res 27: 315–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Śledź P, Jinek M. 2016. Structural insights into the molecular mechanism of the m6A writer complex. Elife 5: e18434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thandapani P, O'Connor TR, Bailey TL, Richard S. 2013. Defining the RGG/RG motif. Mol Cell 50: 613–623. [DOI] [PubMed] [Google Scholar]
- Wang X, Lu Z, Gomez A, Hon GC, Yue Y, Han D, Fu Y, Parisien M, Dai Q, Jia G, et al. 2014. N6-methyladenosine-dependent regulation of messenger RNA stability. Nature 505: 117–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Zhao BS, Roundtree IA, Lu Z, Han D, Ma H, Weng X, Chen K, Shi H, He C. 2015. N6-methyladenosine modulates messenger RNA translation efficiency. Cell 161: 1388–1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P, Doxtader KA, Nam Y. 2016a. Structural basis for cooperative function of Mettl3 and Mettl14 methyltransferases. Mol Cell 63: 306–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Feng J, Xue Y, Guan Z, Zhang D, Liu Z, Gong Z, Wang Q, Huang J, Tang C, et al. 2016b. Structural basis of N6-adenosine methylation by the METTL3-METTL14 complex. Nature 534: 575–578. [DOI] [PubMed] [Google Scholar]
- Xiang Y, Laurent B, Hsu CH, Nachtergaele S, Lu Z, Sheng W, Xu C, Chen H, Ouyang J, Wang S, et al. 2017. RNA m6A methylation regulates the ultraviolet-induced DNA damage response. Nature 543: 573–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao W, Adhikari S, Dahal U, Chen YS, Hao YJ, Sun BF, Sun HY, Li A, Ping XL, Lai WY, et al. 2016. Nuclear m6A reader YTHDC1 regulates mRNA splicing. Mol Cell 61: 507–519. [DOI] [PubMed] [Google Scholar]
- Yue Y, Liu J, He C. 2015. RNA N6-methyladenosine methylation in post-transcriptional gene expression regulation. Genes Dev 29: 1343–1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao BS, Wang X, Beadell AV, Lu Z, Shi H, Kuuspalu A, Ho RK, He C. 2017. m6A-dependent maternal mRNA clearance facilitates zebrafish maternal-to-zygotic transition. Nature 542: 475–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng G, Dahl JA, Niu Y, Fedorcsak P, Huang CM, Li CJ, Vågbø CB, Shi Y, Wang WL, Song SH, et al. 2013. ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol Cell 49: 18–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Wan J, Gao X, Zhang X, Jaffrey SR, Qian SB. 2015. Dynamic m6A mRNA methylation directs translational control of heat shock response. Nature 526: 591–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}