

Abstract
Changes in splicing are known to affect the function and regulation of genes. We analyzed splicing events that take place during the postnatal development of the prefrontal cortex in humans, chimpanzees, and rhesus macaques based on data obtained from 168 individuals. Our study revealed that among the 38,822 quantified alternative exons, 15% are differentially spliced among species, and more than 6% splice differently at different ages. Mutations in splicing acceptor and/or donor sites might explain more than 14% of all splicing differences among species and up to 64% of high-amplitude differences. A reconstructed trans-regulatory network containing 21 RNA-binding proteins explains a further 4% of splicing variations within species. While most age-dependent splicing patterns are conserved among the three species, developmental changes in intron retention are substantially more pronounced in humans.
Keywords: transcriptomics, RNA-seq, alternative splicing, brain development
INTRODUCTION
Alternative splicing (AS) allows a single gene to produce several transcripts and, consequently, proteins, by way of the differential usage of splicing sites during the removal of introns from the pre-mRNA by the spliceosome (Maniatis 1991). Up to 95% of primate protein-coding genes may undergo AS (Takeda et al. 2010). In addition to its role in the expansion of the protein repertoire, AS that affects mRNA stability and/or export to cytoplasm is involved in the regulation of gene expression (Reed and Hurt 2002; Mockenhaupt and Makeyev 2015). In particular, intron retention may regulate gene expression during cell differentiation through the nonsense-mediated decay (NMD) mechanism (Chang et al. 2007). AS is regulated by splicing factors (SF), proteins that bind RNA in a sequence-dependent manner and affect, positively or negatively, the assembly of spliceosomes (Black 2003). AS plays a significant role in cell differentiation, organ development, and diseases (Konieczny et al. 2014; Danan-Gotthold et al. 2015; Li et al. 2015; Xiong et al. 2015). Recent studies have shown that the interspecies variability of AS events exceeds intertissue variability and is mainly explained by changes in cis-regulatory elements (Barbosa-Morais et al. 2012; Merkin et al. 2012). Vice versa, tissue-specific AS events are more conserved, implying conservation of trans-acting regulatory circuits during evolution.
Humans differ strikingly from their close evolutionary relatives, such as chimpanzees, in terms of social behavior and cognitive abilities (Sherwood et al. 2008). Investigations of molecular mechanisms of these differences could shed light on the functioning and evolution of the human brain and provide new clues for the treatment of mental illnesses. For example, it has recently been shown that the rapid evolution of age-related gene expression regulation in the prefrontal cortex (PFC) in the human lineage is linked with an extended period of brain plasticity in humans compared to other primates (Liu et al. 2012). Previously, we have reported substantial changes in AS in human PFC during brain development and aging (Mazin et al. 2013). Here, we extend this analysis using data from 168 human, chimpanzee, and macaque PFC samples obtained at different ages: from the late fetal stages to advanced age.
RESULTS
Primary data analysis and determination of a consensus gene set
To investigate human-specific splicing changes taking place during the development of the prefrontal cortex (PFC), we analyzed PFC transcriptome data from 40 humans, 39 chimpanzees, and 40 macaques produced using the Illumina platform (data set 1, DS1) (He et al. 2014; Liu et al. 2016). The ages of these individuals covered the entirety of postnatal development and much of adulthood in each species (humans: 0–61 yr; chimpanzees: 0–43 yr; macaques: 0–21 yr). In the case of macaques, samples also included late fetal developmental stages (Fig. 1A; Supplemental Table S1). For each sample there was an average of 10 million single-end 100-nt-long sequence reads, 85% of which could be mapped unambiguously to the respective genome.
FIGURE 1.

Splicing variability in the human, chimpanzee, and macaque brain. (A) Age distribution of individuals used in the study. Each symbol represents one individual (circle—female, triangle—male). The rectangles indicate individual samples pooled in DS3. The colors show species: humans—red, chimpanzees—blue, macaques—green. (B,C) Multidimensional scaling of samples based on the Pearson correlation of the PSI values. (D) The numbers and the types of gene segments showing: (i) significant splicing differences between species (H–C, H–M, C–M), (ii) species-specific splicing differences (H-spec, C-spec, M-spec), and (iii) age-dependent splicing changes within species (H-age, C-age, M-age). (E) Comparison of splicing variation between species detected based on DS1 and DS3. The colors show species’ comparisons. Only segments with significant splicing differences in DS1 are shown. (F) Assessment of splicing differences detected based on RNA-seq data using RT-PCR. (G) Example of large splicing difference between species in the SNHG11 gene. Shown are the average coverage plots for each species: (humans—red, chimpanzees—blue, macaques—green), human gene annotation (black), and PSI distributions for selected gene segments (boxplots). (H) Neighbor joining tree based on splicing divergence (one minus Person correlation coefficient). The numbers of divergent splicing events are shown in the legend.
In addition to DS1, we analyzed splicing variation in the primate PFC development using two smaller RNA-seq data sets. One data set (DS2) includes published human and macaque data from 13 and 15 individuals, respectively (Mazin et al. 2013), as well as new data from 15 chimpanzees, containing a total of 1.3 billion sequenced reads (Supplemental Table S2). The other data set (DS3) consists of 12 human, four chimpanzee, and four macaque samples, each pooled from five individuals of similar ages (Li et al. 2013; Mazin et al. 2013). DS3 covers two brain regions, PFC and cerebellar cortex (CBC), with a total of 157 million reads (Supplemental Table S3).
The quality and completeness of human, chimpanzee, and macaque genome annotations differ substantially. To circumvent this problem, we performed a de novo annotation for each of these species based on our RNA-seq data, and used it to construct a consensus gene set. Specifically, we mapped all DS1 reads to the respective genomes using TopHat (Ghosh and Chan 2016) without prior information about exon coordinates, and then used UCSC pairwise genome alignments (Rosenbloom et al. 2015) to define orthologous splicing sites (that is, sites that could be aligned among all three species). Only exon–exon junctions generated by two orthologous splicing sites (orthologous junctions) were used in further analyses. We required expression of the corresponding junction reads in at least one species (Supplemental File S4). We defined segments as genomic fragments with sufficient read coverage located between two adjacent splicing sites regardless of their type. Based on these data, we built a graph with splicing sites as nodes and junctions or segments as edges. Genes were defined as linked components of this graph. This resulted in a total of 26,260 genes containing 278,225 segments and 221,541 junctions. Segments that are completely covered by annotated coding DNA sequence (CDS, Ensembl77) were defined as protein-coding (Supplemental Files S1–S3).
Splicing variation among species
To assess splicing variation among species, we used 38,822 segments from 7427 genes covered by at least 10 reads in at least 20 samples of each species in DS1. Consistent with previous reports (Barbosa-Morais et al. 2012; Merkin et al. 2012), differences between species had the strongest influence on splicing, explaining 38% of the total splicing variation in our data. A further 7.3% of splicing variation was explained by age. Other factors, such as sex or RNA integrity number (RIN), each explained less than 1% of the total splicing variation (Fig. 1B,C; Supplemental Fig. S1).
To identify differentially spliced gene segments, we used the SAJR algorithm (Mazin et al. 2013) to calculate the PSI (percent spliced in) for each segment in each sample. SAJR uses generalized linear models with quasi-binomial distribution to model segment inclusion and exclusion read counts (see Materials and Methods). Of the 38,822 segments, 5788 (15%) showed significant splicing differences between at least one pair of species (likelihood ratio test, BH corrected P < 0.05, and dPSI [PSI difference] > 10%) (Fig. 1D; Supplemental File S4). The majority of these splicing differences (an average of 90%) were consistent across the data sets (Fig. 1E). The results were further validated by semiquantitative RT-PCR: Nine of 10 significant splicing differences between species pairs detected by RNA-seq were reproduced by RT-PCR. Furthermore, PSI values, as well as PSI differences between species, correlated strongly and positively between the RNA-seq and RT-PCR measurements (r = 0.92 and 0.94, respectively) (Fig. 1F; Supplemental Table S4). Among these differences, two human-specific splicing events and one chimpanzee-specific event were localized in one gene, snoRNA-host gene SNHG11 (Fig. 1G).
Of 5788 gene segments showing significant splicing differences between species, 1675 (29%) represented protein-coding segments. Among the remaining noncoding segments, the majority, 2628 (64%), were retained introns (Fig. 1D). The assignment of observed splicing changes to the human and chimpanzee evolutionary lineages using macaque as an outgroup resulted in 461 human-specific and 645 chimpanzee-specific changes. The direction of the splicing change was, however, asymmetric for different types of gene segments. In both lineages, constitutive protein-coding segments tended to become alternatively spliced (isoform gain), while only a few alternatively spliced segments became constitutive (isoform loss). Similarly, many intronic sequences were included into transcripts as retained introns in a lineage-specific manner, while only a few retained introns were lost (Fig. 1H). Thus, in both evolutionary lineages, the overall trend was toward an increase in the proportion of alternatively spliced gene segments, with a gain/loss ratio of 6.3-fold for the human lineage and 18.6-fold for the chimpanzee lineage.
Age-related splicing changes in prefrontal cortex development
Age is the second largest factor explaining splicing variation in our data and by far the largest factor explaining splicing variation within each species (Supplemental Fig. S2). Indeed, 2477 (6.4%) of 38,822 segments showed significant age-dependent splicing differences in at least one species (likelihood ratio test, BH corrected P < 0.05) (Fig. 2A; Supplemental File S5). Almost all (89%–92%) of these segments exhibited a positive Pearson correlation of PSI between the data sets, and for 72%–83% of them, the correlation was above 0.5 (Fig. 2B). A functional analysis showed that the set of genes with age-related coding segments are enriched in such biological processes as “cell adhesion,” “neuron differentiation,” and “synaptic transmission,” while genes with age-related retained introns are involved in the “regulation of GTPase activity” and the “regulation of ion transport” (Supplemental Table S5).
FIGURE 2.

Age-dependent splicing changes in the human, chimpanzee, and macaque brain. (A) The numbers of gene segments showing age-dependent splicing in each species. Here and further the colors indicate the following: humans—red, chimpanzees—blue, and macaques—green. (B) The distributions of the Pearson correlation coefficients based on age-dependent splicing changes measured using DS1 and DS2. (C) The distribution of age-scale coefficients. The dashed lines show location of the distribution modes shown in the legend. (D) The distributions of the Pearson correlation coefficients based on age-dependent splicing changes detected in both compared species. (E,F) Six age-dependent splicing patterns identified for protein-coding segments and retained introns. Each panel represents one pattern; the dots show the mean z-score transformed PSI. The curves are drawn using cubic splines with four degrees of freedom. Numbers of segments in each cluster are shown in title. (G) Mean normalized expression profile for 24 human-specific genes showing age-related intron retention.
To assess the role of changes of cell type composition in splicing variation we used the Cybersort deconvolution algorithm (Newman et al. 2015) based on the expression of genes specific to different neuronal cell types (Darmanis et al. 2015). This analysis revealed a transition from fetal quiescent neurons to adult neurons at approximately 3 mo postnatal (Supplemental Fig. S7). From this age on, there were no substantial changes in the total neuronal proportion estimated by the algorithm. Furthermore, a study based on the anatomical data reported little change in the overall neuronal proportion with age, up to 60 yr, in cognitively healthy individuals (Hwang et al. 2016). These results suggest that the detected age-related splicing changes are not influenced substantially by changes in cell type composition.
To compare age-dependent splicing changes between the species, we first addressed differences in the human, chimpanzee, and macaque lifespan by searching for the best age-scaling coefficient for each age-dependent segment for each pair of species (see Materials and Methods). For each comparison, the distribution of age-scaling coefficients had a single mode: 1.6 for scaling the chimpanzee age to the human age and 2.8 for scaling the macaque age to the human age (Fig. 2C).
Based on the scaled lifespan, the majority of age-dependent splicing changes (an average of 81%) exhibited high correlation between the species that is almost equal to the agreement between two data sets of the same species, indicating general conservation of age-dependent splicing changes among primates (Pearson correlation r > 0.5) (Fig. 2D). To assess this further, we sorted age-dependent splicing changes according to their PSI patterns into six clusters using an unsupervised hierarchical clustering technique (Fig. 2E,F). While the result confirmed the general conservation of age-dependent splicing patterns among humans, chimpanzees, and macaques, two unexpected observations emerged. First, in contrast to protein-coding segments showing a balanced ratio of the age-dependent increase and decrease in PSI (60% vs. 40%, respectively), 81% of retained introns showed a PSI decrease during development (Fig. 2E,F). Second, in contrast to protein-coding segments showing similar numbers of age-related segments in all three species, retained introns were twice as abundant in humans as in other species (Fig. 2A).
Intron retention and gene expression
Most human introns contain stop codons in all three frames, and thus their retention should lead to mRNA degradation through the NMD pathway (Braunschweig et al. 2014). Additionally, retention of the last intron was shown to play a role in the regulation of the expression of neuronal genes in mouse-brain development via mRNA degradation by the nucleolar exosome (Yap et al. 2012). Due to the limited sequencing depth of the study, our analysis was restricted to 21,625 retained introns from 5978 genes that passed the coverage cutoff. Our analysis showed that the PSI of age-related retained introns, but not of protein-coding segments, correlates negatively with the expression of its host gene significantly more often than expected by chance (Wilcoxon test, P < 0.0001) (Fig. 3A). Notably, the proportion of the last introns is significantly higher among retained introns showing age-dependent behavior in humans, but not in the other two species (Fisher exact test, P = 0.027) (Fig. 3B).
FIGURE 3.

Splicing regulation and function. (A) The distribution of the Pearson correlation coefficients based on the PSI of age-dependent retained introns and the expression level of the respective genes (solid lines) or randomly selected genes (dashed lines). The legend shows P-values of the Wilcoxon test comparing the observed and control distributions. (B) The proportions of the last (top) and the second-to-last (bottom) non-age-dependent introns or age-dependent introns in one of the species among all retained introns. The error bars show 95% confidence intervals. Only genes with at least five introns were included. (C) The relationship between PSI variation between species and splicing propensity differences at the alternative donor sites. (D) The proportions of splicing differences between species that could be explained by the sequence differences within the core splicing sites at a different PSI change amplitudes. (E,F) The distributions of PSI values at the alternative donor sites within the PARP2 and ULK3 genes. Reference and alternative alleles (red) are shown under the consensus donor site sequence. In the ULK3 sequence, SNP overlaps both ancestral and alternative donor sites. (G) The distributions of the average primate phastCons scores for protein-coding cassette exons and adjacent introns.
The prevalence of human-specific retained introns, their unusually uniform age-dependent PSI pattern, and the inverse relationship between the retained introns’ PSI and gene expression, suggest the contribution of retained introns to gene expression changes unique to the development of the human brain. Indeed, among 1013 genes containing age-dependent retained introns, 24 were genes with human-specific age-related regulation identified in Liu et al. (2012), which is significantly more than expected by chance (Fisher exact test, P < 0.0005, odds ratio > 2.3). Expression of these genes increased during development in humans, but not in the other two species (Fig. 2G). Furthermore, this pattern is opposite to the pattern observed for the inclusion ratio of retained introns. Thus, retained introns appear to play a role in the regulation of gene expression via NMD in a human-specific manner.
Splicing of microexons
Microexons (exons not longer than 27 nt) were shown to be specifically included in the neuronal tissue (Irimia et al. 2014). Similarly to previous reports, we found that the length of alternative microexons were more frequently a multiple of three than the length of longer exons (Fisher exact test, P < 0.0001, odds ratio > 3.6) and their host genes were significantly associated with neuronal development (Supplemental Table S6). In contrast, no such tendencies were observed for short constant exons. Interestingly, microexons were significantly overrepresented among age-dependent segments (Fisher exact test P < 0.0001, odds ratio > 4.6) and species-specific segments (Fisher exact test P < 0.0001, odds ratio > 2).
Mutations in splice sites
Most intraspecies splicing divergence is thought to be due to sequence changes in cis-regulatory elements (Barbosa-Morais et al. 2012; Merkin et al. 2012). To assess the influence of such changes on splicing differences between humans, chimpanzees, and macaques, we built positional weight matrices (PWMs) for acceptor and donor sites and used them to calculate the splicing propensity for each segment. The analysis showed that 61% of significant splicing differences between species were accompanied by sequence changes affecting predicted splicing propensity. In the majority of cases (83% for cases with PSI changes above 0.5, Fisher exact test, P < 0.0001, odds ratio > 22) predicted splicing efficiency changes coincided with PSI change directions (Fig. 3C). The effect of sequence changes in splice sites on segment PSI was significant for all types of splicing events, but particularly pronounced for alternative donor sites (Supplemental Fig. S3). In total, mutations in the core splicing sites might explain approximately 20% of all interspecies differences and up to 80% for the high amplitude differences (Fig. 3D).
The gene PARP2 involved in the base excision DNA repair pathway represents an interesting example of splicing cis-regulation potentially driven by a mutation that is not fixed in the human population. Human SNP rs2297616, which is present in 21% of the population, switches the use of an alternative donor site in the second exon of PARP2 (Coulombe-Huntington et al. 2009). We observed a clear three-modal distribution of PSI for the respective segment in humans with modes at 0, ∼0.5 and 1, while we never observed its inclusion in chimpanzees and macaques (Fig. 3E). A search for other coding segments with similar patterns of PSI distribution revealed one new case: SNP rs12898397 appeared to be linked with a human-specific alternative shortening of exon 14 of the ULK3 serine/threonine protein kinase, which regulates Sonic hedgehog signaling and autophagy (Fig. 3F; Maloverjan et al. 2010).
Splicing trans-regulation and splicing factor target identification
We found that cassette exons showing age-dependent splicing changes in our data, as well as their adjacent introns, are more conserved at the sequence level than the constitutive exons or non-age-dependent cassette exons (Fig. 3G). This could reflect the presence of a stabilizing selection operating to preserve the binding sequence of trans-acting splicing regulators (splicing factors or SFs). To investigate this, we compared the binding affinity of 219 SF with known binding motifs (Ray et al. 2013) in the vicinity of age-dependent and non-age-dependent cassette exons (see Materials and Methods). We indeed found a significantly greater affinity of 23 motifs at locations proximal to age-dependent cassette exons (Wilcoxon test, BH corrected, P < 0.05). Many of these motifs showed positional preferences to upstream or downstream positions relative to the splice site (Table 1).
TABLE 1.
The list of CisBP-DB motifs enriched near age-dependent cassette exons
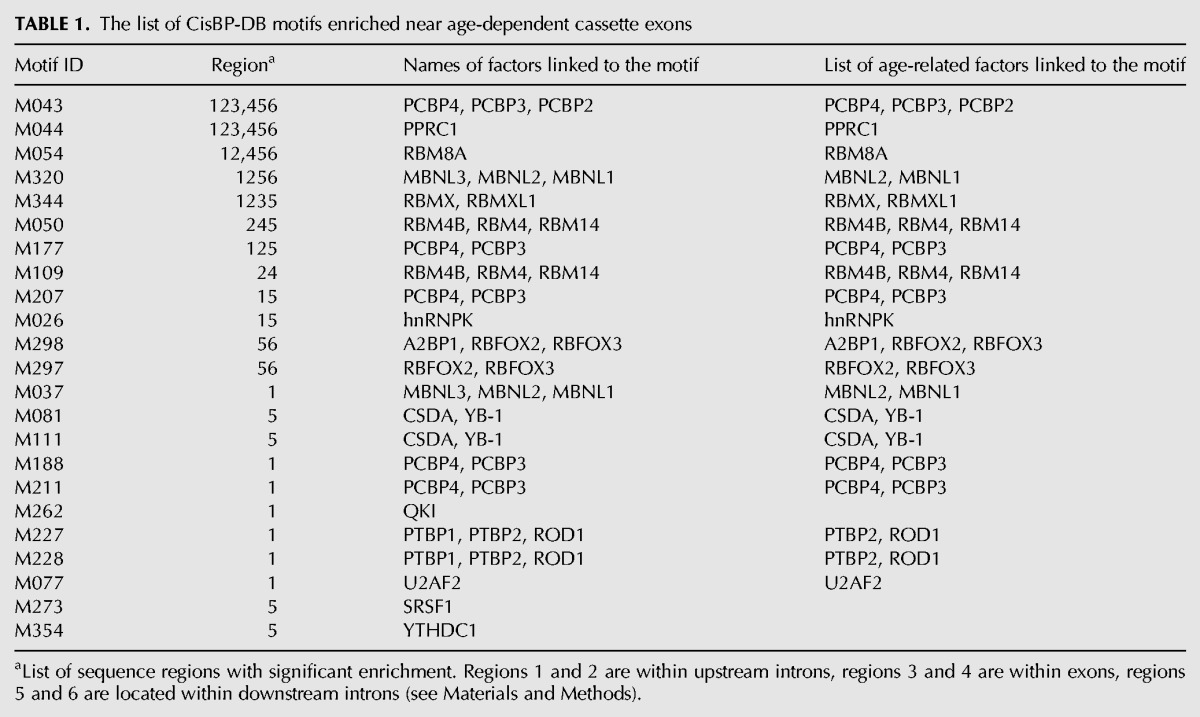
The 23 motifs are bound by 26 SFs expressed at detectable levels in our data. Of them, 21 showed significant age-dependent expression changes in at least one species (ANOVA, BH corrected P < 0.05), which is significantly more than expected by chance (Fisher test, P < 0.025, odds ratio > 3, Table 2).
TABLE 2.
SFs with motifs enriched near age-dependent cassette exons
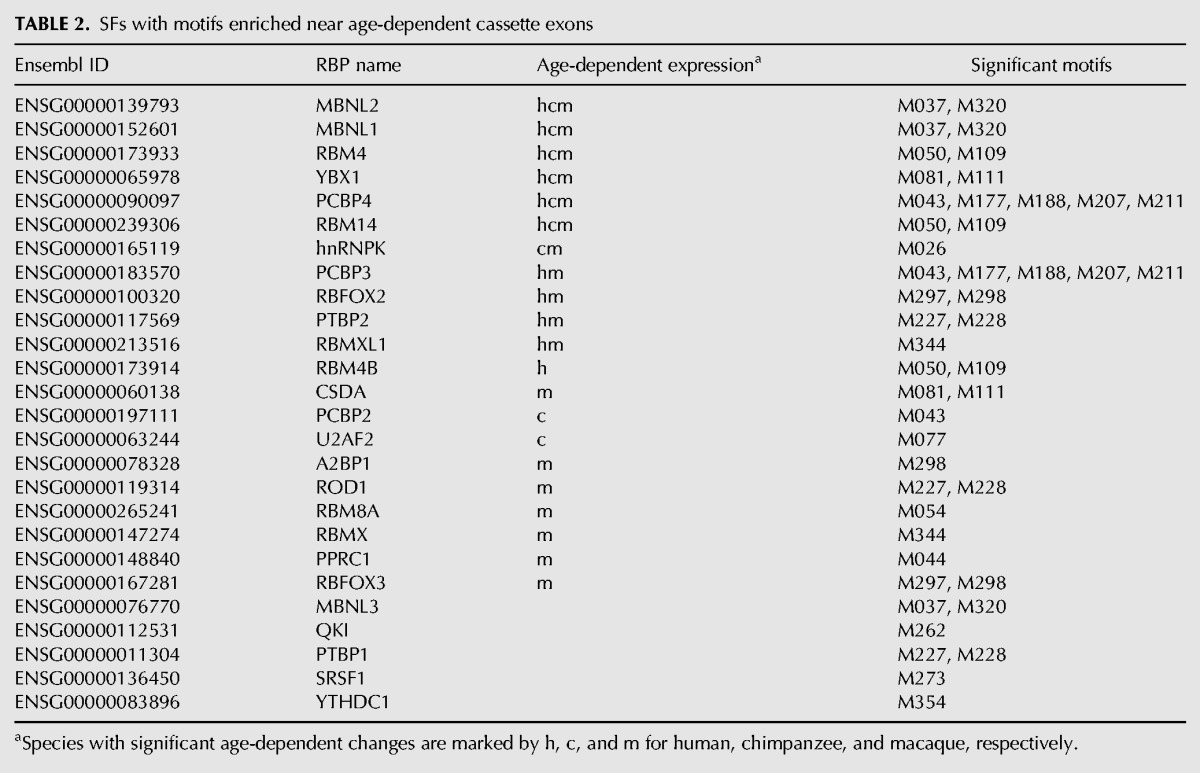
Six SFs change expression significantly in all three species. Of them, four are known to play roles in brain development and/or disease: MBNL2 and MBNL1 are linked with brain pathology in patients with myotonic dystrophy (Charizanis et al. 2012), RBM4 regulates the splicing of the tau protein, which is involved in Alzheimer's disease (Kar et al. 2006), YBX1 is linked with autism (Klenova et al. 2004). SFs that bind to enriched motifs and significantly change expression with age in a subset of species include RBFOX2 (involved in brain development [Gehman et al. 2012]) and RBM8A (known to play a role in intellectual disabilities, autism, schizophrenia, and microcephaly through the NMD-related regulation of gene expression [Zou et al. 2015]).
We used affinities of the found motifs and expression values of corresponding SFs to model the age-related regulation of cassette exons. Our results show a weak but significant correlation between observed and modeled PSI values in cross-validation: The median correlation coefficient for 100 replicates is 0.11 in the actual data, which is significantly different from zero (Wilcoxon test, P < 0.0001), but in randomized or shuffled data sets (Supplemental Fig. S4) it is not. To further verify the role of identified trans-factors in the regulation of alternative splicing, we used data generated in SF knockdown experiments in K562 cells (Hong et al. 2016). Out of six SFs associated with enriched motifs and showing age-related expression in all three species (Table 2), only one (MBNL1) had such data. A reanalysis of these data yielded 83 cassette exons significantly affected by the MBNL1 knockdown (likelihood ratio test, BH corrected P < 0.05, dPSI > 10%). Notably, these exons were significantly enriched among exons showing significant splicing changes during human brain development (Fisher exact test, P < 0.0001, odds ratio > 11) (Supplemental Fig. S5C). Further, the direction of developmental change agreed with the results of the knockdown experiment (Supplemental Fig. S5D). Furthermore, high affinity MBNL1 binding sites were significantly enriched in intronic regions located 50–100 nt downstream from the exons affected by MBNL1 knockdown compared to unaffected exons with a comparable expression level (Fisher exact test, P < 0.0008) (Supplemental Fig. S5E).
DISCUSSION
Our analysis demonstrated that, in agreement with previous studies, age contributes substantially to splicing variability in humans and nonhuman primates, affecting the inclusion of more than 6.4% (2477) of detected exons located within 21% (1581) of expressed genes. Our analyses of splicing differences between species have further shown that the majority of high-amplitude changes could be explained by the single-nucleotide substitutions within core splicing sites.
For all types of splicing events there is an increase in the number of alternatively spliced events in the human and the chimpanzee lineages. This can be explained by the fact that most gene segments are spliced constitutively and their splicing site sequences are close to the consensus. Thus, any mutation in the splicing sites of constitutive exons might result in the emergence of splicing alternatives.
In contrast to high interspecies splicing variability, the age-dependent regulation of the splicing of protein-coding segments is highly conserved in the primate brain. The only notable exception to this conservation is an approximate twofold excess of age-dependent intron retention events in humans compared to chimpanzees and macaques. Our results show that intron retention might result in the suppression of gene expression at early stages of the postnatal development of the human brain. The same mechanism might be present in other primates, but may occur at prenatal developmental stages that are not covered by our samples. This notion is in agreement with age-scaling results, showing that the splicing profile in chimpanzee and macaque newborns correspond to more advanced postnatal developmental stages in humans.
We identified 21 SFs as potential regulators of age-dependent splicing changes during primate PFC development and maturation, and constructed a simple mechanistic model explaining approximately 4% of the age-dependent splicing variation. While the role of some of these proteins, such as MBNL1, MBNL2, RBM4, YB-1, and RBFOX2, in the regulation of AS in neuronal tissues has been shown, our list contains many proteins that had not previously been implicated in the regulation of brain development, such as RNA-binding proteins PCBP4 and RBM14.
One of the limitations of our work is a relatively low sequencing depth that restricts our analysis to approximately 7.5 thousand genes, approximately half of the genes expressed in the brain. We also did not consider species-specific exons whose sequences cannot be unambiguously aligned across species.
Our study also does not discriminate between hereditary and environmentally induced splicing differences between species. Still, while the environmental effects might cause some of the interspecies differences, their influence is unlikely to be strong. Although we do not know of any studies that have examined environmental effects on human or primate brain splicing, a study showed very limited effects of human and nonhuman primate diet on brain transcriptome in mice (Somel et al. 2008). Second, even specifically trained apes adopted at most hundreds of words, and their communicative abilities were comparable to a 2-yr-old human child (Terrace et al. 1979; Savage-Rumbaugh and Society for Research in Child Development 1993), indicating the existence of a strong genetic component underlying cognitive differences between human and nonhuman primates. Additionally, all primates used in this study lived in captivity, which is more similar to the modern human lifestyle than to living in the wild in terms of diet, physical activity levels, lifespan, and causes of death.
Splicing changes observed in our work might also be partially caused by changes in NMD efficiency. Yet, since the majority (67%) of age-related protein-coding segments have a length that is divisible by three and lacks inframe stop codons, they should not be affected by NMD. To the contrary, the majority (87%) of retained introns should induce NMD or nucleolar exosome degradation (Yap et al. 2012) if included. Consistently, human genes containing age-related retained introns exhibit strong enrichment (Fisher exact test, odds ratio = 1.7, P < 0.001) in experimentally defined NMD targets (Colombo et al. 2017). In contrast, there is no such enrichment for human genes that contain age-related protein-coding segments (Fisher exact test, odds ratio = 0.95, P = 1). To explain observed interspecies differences in intron retention, however, changes in NMD efficiency with age would need to differ between humans and nonhuman primates. Yet, we observed no significant interspecies differences in the expression of six major NMD factors (Supplemental Fig. S8).
Overall, our results show that splicing in the human PFC changes substantially with age and differs from the splicing in the PFC of chimpanzees and macaques. While the functional significance of these observations remains to be investigated, it is clear that the transcriptome complexity of the human brain cannot be understood based singly on analogies with model organisms; rather, this requires studies of the human brain itself at different stages of development and aging.
MATERIALS AND METHODS
Sample collection and preparation
Chimpanzee samples (Supplemental Table S2) were obtained from the Alamogordo Primate Facility in New Mexico, USA, the Anthropological Institute and Museum of the University of Zürich-Irchel in Switzerland, and the Biomedical Primate Research Centre in the Netherlands. All primates used in this study suffered sudden deaths for reasons other than their participation in this study and without any relation to the tissue used. PFC dissections were made from the frontal part of the superior frontal gyrus and all samples contained an approximately 2:1 gray matter to white matter volume ratio.
Total RNA was isolated using TRIzol reagent (Invitrogen). Sequencing libraries were prepared with a TruSeq RNA Sample Preparation Kit (Illumina) in accordance with the manufacturer's instructions. Briefly, poly(T) oligo-attached magnetic beads were used to isolate long polyadenylated RNA from 1 μg of total RNA. RIN was assessed using the Agilent 2100 Bioanalyzer System. After fragmentation, first-strand cDNA was reverse transcribed with random hexamer-primers, followed by second-strand cDNA synthesis, end repair, adenylation of 3′ ends and ligation of the adapters. The fragments were then enriched by PCR, and sequenced on the Illumina Hi-seq 2000 system, using the 100-bp single-end sequencing protocol. All samples were randomized prior to library preparation and RNA sequencing.
The chimpanzee sequences from DS2 generated for this study, as well as sequences from two human samples that were not analyzed previously, were deposited into SRA under the accession numbers SRP092096 and SRP108819.
Validation of splicing changes using PCR
First-strand cDNA was synthesized using SuperScript II reverse transcriptase (Invitrogen), using the same pooled adult PFC RNA samples that were used for the RNA-seq DS3 as templates. To check the inclusion ratio change of each selected segment, a primer pair was designed at the two flanks of the segment for each species. PCR reactions were performed at the condition of 95°C for 5 min, 30 cycles of 95°C for 30 sec, 60°C for 30 sec and 72°C for 30 sec, followed by 1 cycle of 72°C for 10 min with rTaq DNA polymerase (Toyobo). Then 5 μL of each PCR product were used for electrophoresis in a 2.5% (w/v) agarose gel. The sizes of the fragments was estimated using visual estimation based on a DL 2000 DNA marker (Takara). After ethidium bromide (EB) staining, six of a total of seven segments tested by PCR showed two bands with expected product lengths. All primer sequences are listed in (Supplemental Table S4). The results of RT-PCR were quantified manually using the ImageJ program. The intensity of the band that corresponds to the longer isoform was divided by the sum of intensities of both bands to obtain PSI.
Read mapping
Reads from DS1 were mapped twice. First, all reads from a given species were merged and mapped together by TopHat (v2.0.13) with the following parameters: “--read-mismatches 4 --read-edit-dist 4 --read-realign-edit-dist 0 --max-intron-length 3000000 --max-segment-intron 3000000” to respective genomes (hg38, panTro4, and rheMac3). Mapped reads were used to reconstruct the gene annotation (see below). Then, all samples from all three species were mapped by TopHat sample by sample. During this second round, TopHat was supplied with information about exon–exon junction coordinates from the reconstructed annotation and run with the following parameters: “--read-mismatches 4 --read-edit-dist 4 --read-realign-edit-dist 0 --max-intron-length 3000000 --max-segment-intron 3000000 --no-novel-juncs”. The results of the second mapping were used in further analysis.
Gene annotation construction
The genome assembly and gene annotation quality vary between the studied species. To avoid any possible bias, we reannotated all three genomes de novo using our data (DS1). Our procedure ensures that in all three species the annotation contains only orthologous genes that, in turn, have completely the same exon–intron structure in all species. The procedure consists of the following steps:
All exon–exon junctions found by TopHat during the first mapping round were extracted.
Exon–exon junctions were aligned between the three species using pairwise alignments from UCSC and the liftOver tool. All junctions that could not be unambiguously aligned between all three species were removed. The remaining junctions were considered to be orthologous junctions.
We defined segments as regions between two adjacent orthologous splicing sites (boundaries of orthologous junctions) of any type with an average read coverage above 1 and gaps in the read coverage not longer than 5 nt. Two orthologous splicing sites were considered to be connected by a segment if the segment was found in at least one species.
The splicing graph was constructed with orthologous splicing sites as nodes and orthologous junctions and segments as edges. All linked components in the resulting splicing graph were considered to be genes.
Differential splicing analysis
The differential splicing analysis was performed by the SAJR method, as described in Mazin et al. (2013). Briefly, for each segment, the number of inclusion reads (reads that overlap the segment by at least 1 nt) and the number of exclusion reads (reads mapping to exon–exon junctions that span the segment) were calculated in each sample. Only segments that had at least ten inclusion plus exclusion reads in at least twenty samples in DS1 were used in the analysis. The inclusion and exclusion reads were considered as results of binomial trials and modeled by the pseudobinomial distribution using the generalized linear model (the glm function in R). Since most of age-related changes take place in the first few years of life (Mazin et al. 2013) we used a fourth root transformed age scale. A square root age transformation was used to account for possible nonlinear changes at fourth root transformed age scale. For the interspecies comparison we used the following model:
Segments with a BH-corrected P-value for the “species” term (pseudo-log-likelihood test) below 0.05 and an interspecies PSI difference above 0.1 were considered as significant. An analysis of age-related splicing changes was performed in each species independently using the following model:
Segments with at least one term having the BH-corrected P-value below 0.05 and with a PSI amplitude above 0.1 were considered to be age-related. The PSI amplitude was calculated for each segment by sorting samples by PSI of the segments and taking the difference between the second highest PSI and the second lowest PSI.
Species-specific splicing analysis
A segment was considered to have specific splicing in species A if: (i) the difference between A and the other two species B and C was significant; (ii) the difference between B and C was not significant; (iii) mean PSI in A (psiA) is greater or less than mean PSI in both B and C; and (iv) min {abs (psiA–psiB), (psiA–psiC)}/max {abs (psiA–psiB), abs (psiA–psiC)} ≥ 0.7.
Age-scaling
To find the optimal age-scaling coefficients between a pair of species, we considered segments that were age-related in both species. First, we approximated the dependence of PSI on age (in the 0.25 power scale) using a cubic spline with four degrees of freedom in each species. We centered both approximation to zero and then looked for age-scaling coefficient k in the form of age (species 1) = k × age(species 2) that minimizes the average distance between two approximations using the optimize function from the R statistical package. For details, see Supplemental File S6.
Interspecies and inter-data sets AS correlation
To calculate the correlation coefficient between two age-related PSI patterns with unmatched age-points (different data sets for same species or different species) we approximated the dependence of PSI on age (in the 0.25 power scale) using a cubic spline with four degrees of freedom for both patterns in common age-span, and calculated the correlation coefficient between the approximations.
GO-enrichment analysis
We linked orthologous primate genes identified in this work to human genes from Ensembl (version 77) based on shared exon–exon junctions in humans. Genes were linked if they shared at least one junction. We annotated genes with all GO terms of all Ensembl genes linked to the orthologous genes. We considered only GO categories with at least three genes. We used the goseq package (Young et al. 2010) to perform functional enrichment analysis, for age-related segments we used the Wallenius method with host gene expression (in log scale) as the bias data. For GO-analysis of microexons we used the hypergeometric test. Only overrepresentation was tested. Terms with BH-corrected P-values below 0.2 were considered significant.
Splicing propensity
We used all splicing sites of our human annotation to build position weight matrices (PWM) for donor and acceptor sites (nucleotide positions from –3 to +6 and from –24 to +1, respectively). Then, we used these PWMs to calculate the total weights of all orthologous splicing sites in all three species. We used these weights to estimate the splicing propensity (SP) of each segment in each species. For cassette exons, SP was calculated as the sum of weights of donor and acceptor sites; for retained introns, SP was calculated as minus sum of weights of donor and acceptor sites; for alternative donor and acceptor sites SP, as the difference between the weights of the outer and inner sites.
Splicing factor analysis
For each segment and each motif from CISBP-DB (Ray et al. 2013), we calculated the average affinity (as described in Lee and Bussemaker 2010) for six 50 nt sequences: [–100, –51], [–50, –1], and [1, 49] relative to the 5′ exon end, and [–49, –1], [1, 50], and [51, 100] relative to the 3′ exon end. Then we compared these affinities between age-related and non-age-related segments using the Mann–Whitney test. A motif was considered to be significantly associated with age-related cassette exons in one of six sequences if the BH-corrected P-value was below 0.05.
Splicing SNPs
We searched for exons with splicing affected by SNPs. For such cases we expected that one allele corresponds to almost complete inclusion of the exon, while the other allele results in exon exclusion. So, we expect that the PSI of such exons should be close to 0, 0.5, or 1. Following this expectation, we looked for exons that satisfy the following criteria: There is at least one sample with PSI below 0.1, there is at least one sample with PSI above 0.9, and there is at least one sample with PSI within the [0.25, 0.75] interval. In addition, for each sample we found the closest value out of 0, 0.5 or 1 and required the average squared distance of real PSI from these values to be below 0.01. Only two segments (from PARP2 and ULK3) satisfied these criteria and both of them contained SNPs in the core splicing sites with the frequency of alternative alleles above 0.2 and with strong (more than 2 bits) effects on the SP that is significantly more than expected by chance (Fisher test, P < 0.001).
Modelling of AS regulation
To model the regulation of cassette exons by SF, we used the following linear model:
where m runs through all significant motifs and sf runs through all age-related SF associated with at least one significant motif. Prior to modeling, all PSI were z-score transformed. To avoid overfitting, we fit this model using l1 regularization. The constraint was set to 0.01 based on the best performance in cross-validation (see Supplemental Fig. S6). To perform cross validation, all human age-related segments were randomly divided into training (70% of segments) and test (30% of segments) sets. To validate the results, we repeated the same procedure using shuffled data (PSI values were randomly shuffled relative to predictor matrices) and using randomly generated PSI (normal distribution with mean 0 and variance 1).
MBNL1 knockdown analysis
Raw data from control and knockdown experiments were downloaded from ENCODE (library IDs are ENCLB547AMZ, ENCLB023TDY, ENCLB023BAH, ENCLB042UYK). Reads were mapped and AS was quantified the same way as for DS3. MBNL1 target exons were identified using the GLM, likelihood ratio test followed by BH-correction.
Cell-type analysis
A list of 140 marker genes with cell-type specific expression and their expression levels in each cell population was taken from (Darmanis et al. 2015). We deconvoluted the cell type composition in the DS1 data based on expression levels of these marker genes in DS1 using the Cybersort tool (Newman et al. 2015). The results were compared to 500 permutations of marker gene assignments. The other parameters were default.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
Supplementary Material
ACKNOWLEDGMENTS
We thank Anna Tkachev and Ingrid Burke for their helpful comments on the manuscript. This study was supported by the National Natural Science Foundation of China (grant 31420103920), the Strategic Priority Research Program of the Chinese Academy of Sciences (grant XDB13010200), the National Natural Science Foundation of China (grant 91331203), the China National One Thousand Foreign Experts Plan (grant WQ20123100078), the Bureau of International Cooperation, Chinese Academy of Sciences (grant GJHZ201313), and the Russian Science Foundation (grant 16-14-00220) to P.K. M.S.G. was supported by the Russian Science Foundation under grant 14-50-00150.
Footnotes
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.064931.117.
REFERENCES
- Barbosa-Morais NL, Irimia M, Pan Q, Xiong HY, Gueroussov S, Lee LJ, Slobodeniuc V, Kutter C, Watt S, Colak R, et al. 2012. The evolutionary landscape of alternative splicing in vertebrate species. Science 338: 1587–1593. [DOI] [PubMed] [Google Scholar]
- Black DL. 2003. Mechanisms of alternative pre-messenger RNA splicing. Annu Rev Biochem 72: 291–336. [DOI] [PubMed] [Google Scholar]
- Braunschweig U, Barbosa-Morais NL, Pan Q, Nachman EN, Alipanahi B, Gonatopoulos-Pournatzis T, Frey B, Irimia M, Blencowe BJ. 2014. Widespread intron retention in mammals functionally tunes transcriptomes. Genome Res 24: 1774–1786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang YF, Imam JS, Wilkinson MF. 2007. The nonsense-mediated decay RNA surveillance pathway. Annu Rev Biochem 76: 51–74. [DOI] [PubMed] [Google Scholar]
- Charizanis K, Lee KY, Batra R, Goodwin M, Zhang C, Yuan Y, Shiue L, Cline M, Scotti MM, Xia G, et al. 2012. Muscleblind-like 2-mediated alternative splicing in the developing brain and dysregulation in myotonic dystrophy. Neuron 75: 437–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colombo M, Karousis ED, Bourquin J, Bruggmann R, Mühlemann O. 2017. Transcriptome-wide identification of NMD-targeted human mRNAs reveals extensive redundancy between SMG6- and SMG7-mediated degradation pathways. RNA 23: 189–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coulombe-Huntington J, Lam KCL, Dias C, Majewski J. 2009. Fine-scale variation and genetic determinants of alternative splicing across individuals. PLoS Genet 5: e1000766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danan-Gotthold M, Golan-Gerstl R, Eisenberg E, Meir K, Karni R, Levanon EY. 2015. Identification of recurrent regulated alternative splicing events across human solid tumors. Nucleic Acids Res 43: 5130–5144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darmanis S, Sloan SA, Zhang Y, Enge M, Caneda C, Shuer LM, Hayden Gephart MG, Barres BA, Quake SR. 2015. A survey of human brain transcriptome diversity at the single cell level. Proc Natl Acad Sci 112: 7285–7290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gehman LT, Meera P, Stoilov P, Shiue L, O'Brien JE, Meisler MH, Ares M Jr., Otis TS, Black DL. 2012. The splicing regulator Rbfox2 is required for both cerebellar development and mature motor function. Genes Dev 26: 445–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosh S, Chan CK. 2016. Analysis of RNA-seq data using TopHat and Cufflinks. Methods Mol Biol 1374: 339–361. [DOI] [PubMed] [Google Scholar]
- He Z, Bammann H, Han D, Xie G, Khaitovich P. 2014. Conserved expression of lincRNA during human and macaque prefrontal cortex development and maturation. RNA 20: 1103–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong EL, Sloan CA, Chan ET, Davidson JM, Malladi VS, Strattan JS, Hitz BC, Gabdank I, Narayanan AK, Ho M, et al. 2016. Principles of metadata organization at the ENCODE data coordination center. Database 2016: baw001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang T, Park CK, Leung AK, Gao Y, Hyde TM, Kleinman JE, Rajpurohit A, Tao R, Shin JH, Weinberger DR. 2016. Dynamic regulation of RNA editing in human brain development and disease. Nat Neurosci 19: 1093–1099. [DOI] [PubMed] [Google Scholar]
- Irimia M, Weatheritt RJ, Ellis JD, Parikshak NN, Gonatopoulos-Pournatzis T, Babor M, Quesnel-Vallieres M, Tapial J, Raj B, O'Hanlon D, et al. 2014. A highly conserved program of neuronal microexons is misregulated in autistic brains. Cell 159: 1511–1523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kar A, Havlioglu N, Tarn WY, Wu JY. 2006. RBM4 interacts with an intronic element and stimulates tau exon 10 inclusion. J Biol Chem 281: 24479–24488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klenova E, Scott AC, Roberts J, Shamsuddin S, Lovejoy EA, Bergmann S, Bubb VJ, Royer HD, Quinn JP. 2004. YB-1 and CTCF differentially regulate the 5-HTT polymorphic intron 2 enhancer which predisposes to a variety of neurological disorders. J Neurosci 24: 5966–5973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konieczny P, Stepniak-Konieczna E, Sobczak K. 2014. MBNL proteins and their target RNAs, interaction and splicing regulation. Nucleic Acids Res 42: 10873–10887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee E, Bussemaker HJ. 2010. Identifying the genetic determinants of transcription factor activity. Mol Syst Biol 6: 412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Bammann H, Li M, Liang H, Yan Z, Phoebe Chen YP, Zhao M, Khaitovich P. 2013. Evolutionary and ontogenetic changes in RNA editing in human, chimpanzee, and macaque brains. RNA 19: 1693–1702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li YI, Sanchez-Pulido L, Haerty W, Ponting CP. 2015. RBFOX and PTBP1 proteins regulate the alternative splicing of micro-exons in human brain transcripts. Genome Res 25: 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Somel M, Tang L, Yan Z, Jiang X, Guo S, Yuan Y, He L, Oleksiak A, Zhang Y, et al. 2012. Extension of cortical synaptic development distinguishes humans from chimpanzees and macaques. Genome Res 22: 611–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Han D, Somel M, Jiang X, Hu H, Guijarro P, Zhang N, Mitchell A, Halene T, Ely JJ, et al. 2016. Disruption of an evolutionarily novel synaptic expression pattern in Autism. PLoS Biol 14: e1002558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maloverjan A, Piirsoo M, Michelson P, Kogerman P, Osterlund T. 2010. Identification of a novel serine/threonine kinase ULK3 as a positive regulator of Hedgehog pathway. Exp Cell Res 316: 627–637. [DOI] [PubMed] [Google Scholar]
- Maniatis T. 1991. Mechanisms of alternative pre-mRNA splicing. Science 251: 33–34. [DOI] [PubMed] [Google Scholar]
- Mazin P, Xiong J, Liu X, Yan Z, Zhang X, Li M, He L, Somel M, Yuan Y, Phoebe Chen YP, et al. 2013. Widespread splicing changes in human brain development and aging. Mol Syst Biol 9: 633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merkin J, Russell C, Chen P, Burge CB. 2012. Evolutionary dynamics of gene and isoform regulation in mammalian tissues. Science 338: 1593–1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mockenhaupt S, Makeyev EV. 2015. Non-coding functions of alternative pre-mRNA splicing in development. Semin Cell Dev Biol 47–48: 32–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman AM, Liu CL, Green MR, Gentles AJ, Feng W, Xu Y, Hoang CD, Diehn M, Alizadeh AA. 2015. Robust enumeration of cell subsets from tissue expression profiles. Nat Methods 12: 453–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray D, Kazan H, Cook KB, Weirauch MT, Najafabadi HS, Li X, Gueroussov S, Albu M, Zheng H, Yang A, et al. 2013. A compendium of RNA-binding motifs for decoding gene regulation. Nature 499: 172–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reed R, Hurt E. 2002. A conserved mRNA export machinery coupled to pre-mRNA splicing. Cell 108: 523–531. [DOI] [PubMed] [Google Scholar]
- Rosenbloom KR, Armstrong J, Barber GP, Casper J, Clawson H, Diekhans M, Dreszer TR, Fujita PA, Guruvadoo L, Haeussler M, et al. 2015. The UCSC Genome Browser database: 2015 update. Nucleic Acids Res 43: D670–D681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savage-Rumbaugh ES, Society for Research in Child Development. 1993. Language comprehension in ape and child. University of Chicago Press, Chicago, IL. [PubMed] [Google Scholar]
- Sherwood CC, Subiaul F, Zawidzki TW. 2008. A natural history of the human mind: tracing evolutionary changes in brain and cognition. J Anat 212: 426–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somel M, Creely H, Franz H, Mueller U, Lachmann M, Khaitovich P, Pääbo S. 2008. Human and chimpanzee gene expression differences replicated in mice fed different diets. PloS ONE 3: e1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda J, Suzuki Y, Sakate R, Sato Y, Gojobori T, Imanishi T, Sugano S. 2010. H-DBAS: human-transcriptome database for alternative splicing: update 2010. Nucleic Acids Res 38: D86–D90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terrace HS, Petitto LA, Sanders RJ, Bever TG. 1979. Can an ape create a sentence? Science 206: 891–902. [DOI] [PubMed] [Google Scholar]
- Xiong HY, Alipanahi B, Lee LJ, Bretschneider H, Merico D, Yuen RK, Hua Y, Gueroussov S, Najafabadi HS, Hughes TR, et al. 2015. RNA splicing. The human splicing code reveals new insights into the genetic determinants of disease. Science 347: 1254806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yap K, Lim ZQ, Khandelia P, Friedman B, Makeyev EV. 2012. Coordinated regulation of neuronal mRNA steady-state levels through developmentally controlled intron retention. Genes Dev 26: 1209–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young MD, Wakefield MJ, Smyth GK, Oshlack A. 2010. Gene ontology analysis for RNA-seq: accounting for selection bias. Genome Biol 11: R14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou D, McSweeney C, Sebastian A, Reynolds DJ, Dong F, Zhou Y, Deng D, Wang Y, Liu L, Zhu J, et al. 2015. A critical role of RBM8a in proliferation and differentiation of embryonic neural progenitors. Neural Dev 10: 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.