

SUMMARY
Altering AMPA receptor (AMPAR) content at synapses is a key mechanism underlying the regulation of synaptic strength during learning and memory. Previous work demonstrated that SynDIG1 (synapse differentiation-induced gene 1) encodes a transmembrane AMPAR-associated protein that regulates excitatory synapse strength and number. Here we show that the related protein SynDIG4 (also known as Prrt1) modifies AMPAR gating properties in a subunit-dependent manner. Young SynDIG4 knockout (KO) mice have weaker excitatory synapses, as evaluated by immunocytochemistry and electrophysiology. Adult SynDIG4 KO mice show complete loss of tetanus-induced long-term potentiation (LTP), while mEPSC amplitude is reduced by only 25%. Furthermore, SynDIG4 KO mice exhibit deficits in two independent cognitive assays. Given that SynDIG4 colocalizes with the AMPAR subunit GluA1 at non-synaptic sites, we propose that SynDIG4 maintains a pool of extrasynaptic AMPARs necessary for synapse development and function underlying higher-order cognitive plasticity.
In Brief
Matt et al. show that mice lacking the AMPAR-associated protein SynDIG4/Prrt1 display deficits in synaptic plasticity and cognition. SynDIG4 modifies AMPAR biophysical properties in heterologous cells, but synaptic AMPAR kinetics are unchanged, suggesting that SynDIG4 establishes a pool of extrasynaptic AMPARs necessary for higher-order cognitive plasticity.
INTRODUCTION
AMPA receptors (AMPARs) are responsible for fast excitatory synaptic transmission in the brain. AMPARs are implicated in plasticity mechanisms such as long-term potentiation (LTP) (Huganir and Nicoll, 2013) and synaptic scaling (Lee et al., 2014; Turrigiano, 2012). A diverse family of AMPAR auxiliary factors regulates AMPAR trafficking and gating (Jackson and Nicoll, 2011). Stargazin, the prototypical transmembrane AMPAR regulating protein (TARP), promotes surface expression and alters biophysical properties of AMPARs in heterologous cells such that they more closely resemble those of endogenous receptors (Chen et al., 2000). In addition to TARPs, several other unrelated protein families have been identified as auxiliary AMPAR factors with distinct and overlapping functions (Chen et al., 2000; Chen et al., 2014; Díaz, 2010; Jackson and Nicoll, 2011; Schwenk et al., 2009, 2014; Shanks et al., 2012; von Engelhardt et al., 2010).
SynDIG1 (synapse differentiation-induced gene 1) encodes a type II transmembrane protein that interacts with AMPARs in brain and heterologous cells and regulates synaptic strength (Kalashnikova et al., 2010). SynDIG1 mutant mice exhibit deficits in excitatory synapse maturation (Chenaux et al., 2016). In contrast to other AMPAR accessory proteins, SynDIG1 does not regulate AMPAR dynamics (Lovero et al., 2013), suggesting that SynDIG1 is an atypical AMPAR auxiliary factor.
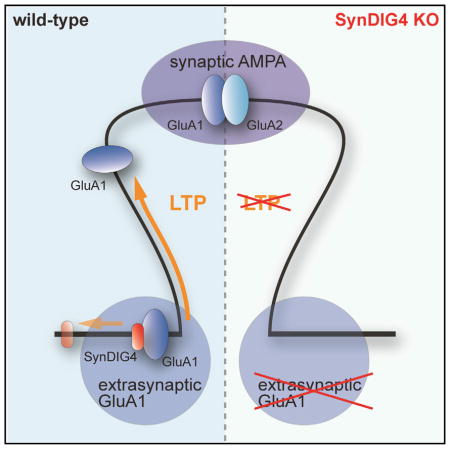
SynDIG defines a family of genes that encodes brain-specific transmembrane proteins (Kalashnikova et al., 2010). SynDIG4, also known as Prrt1 (proline-rich transmembrane protein 1), was identified in several independent proteomic studies as a candidate AMPAR-associated protein (Chen et al., 2014; Schwenk et al., 2012, 2014; Shanks et al., 2012; von Engelhardt et al., 2010), as well as a component of the postsynaptic density (PSD) (Jordan et al., 2004). Surprisingly, SynDIG4 is not enriched at synapses; the major fraction of SynDIG4 colocalizes with the AMPAR subunit GluA1 at extrasynaptic sites in rat cortical neurons (Kirk et al., 2016). Here we show that SynDIG4 plays a critical role in excitatory synapse function with a combination of electrophysiology, immunocytochemistry, biochemistry, and behavior. We propose that SynDIG4 maintains a pool of extrasynaptic AMPARs necessary for synapse development and function underlying higher-order cognitive plasticity.
RESULTS
SynDIG4 Modifies AMPAR Gating Kinetics in a Subunit-Dependent Manner
To test direct effects of SynDIG4 on AMPAR properties, heterologous expression in Xenopus oocytes and outside-out patch-clamp electrophysiological recordings were used to compare it with TARPγ8, which is highly expressed in hippocampus (Tomita et al., 2003). SynDIG4 slows deactivation kinetics of both GluA1 homomers and GluA1/2 heteromers, albeit less than TARPγ8; however, a synergistic effect was observed with both SynDIG4 and TARPγ8 (Figure 1A). SynDIG4 reduces desensitization of GluA1 homomers with or without TARPγ8 but has no significant effect on desensitization of heteromeric GluA1/2 (Figure 1B). SynDIG4 does not alter recovery from desensitization of GluA1 homomers or GluA1/2 heteromers (Table S1).
Figure 1. SynDIG4 Modifies AMPAR Gating Kinetics in a Subunit-Dependent Manner.

(A and B) Representative normalized current responses of AMPAR recorded upon 1 ms (A) and 500 ms (B) application of 10 mM glutamate (G, indicated above the current trace) to giant outside-out patches excised from Xenopus laevis oocytes expressing homomeric GluA1 (top) and heteromeric GluA1/2 (bottom) alone (black) or in combination with SynDIG4 (red), TARPγ8 (green), or both (blue). Graphs summarize weighted time constants for deactivation (τW deact) and desensitization (τW des), as well as steady-state to peak current (ss/peak). Data shown are mean ± SEM; n = 10–20 patches.
Significance (one-way ANOVA): */$ p < 0.05; **/$$ p < 0.01; ***/$$$ p < 0.001; ns, not significant. See also Table S1.
Expression Profile of Prrt1/SynDIG4 Mutant Reporter Mice
These results motivated investigation of a SynDIG4 null reporter line [Prrt1tm1(KOMP)Vlcg] obtained from the Knock Out Mouse Project (KOMP) consortium (Valenzuela et al., 2001), in which the SynDIG4 protein-coding region is replaced with a β-galactosidase (β-gal) cassette (Figure 2A). The resulting reporter protein, driven by the SynDIG4 promoter, is retained within the soma. To verify loss of SynDIG4 protein, brain lysates were collected from homozygous mutant mouse (referred to here as SynDIG4−/−) and compared with wild-type (WT) littermates at postnatal day (P) 14. No detectable levels of SynDIG4 in postnuclear (S1), membrane (P2), synaptosomal (Syn), or PSD-enriched fractions were detected in SynDIG4−/− mice (Figure 2B). PSD-95 was enriched and synaptophysin was undetectable in PSD fractions, while β-actin indicated equivalent loading between fractions.
Figure 2. Loss of SynDIG4 Leads to Altered GluA1 Distribution and Weaker Synapses.

(A) Schematic showing the replacement of the coding region of the SynDIG4 locus with a lacZ reporter and loxP-flanked neomycin selection cassette.
(B) Immunoblots (10 μg of protein loaded per lane) stained for SynDIG4, PSD-95, synaptophysin, and β-actin show postnuclear (S1), membrane (P2), synaptosomal (Syn), and PSD biochemical fractions from postnatal day (P14) WT (+/+) and SynDIG4 homozygous mutant (−/−) mouse brain tissue.
(C–F) Primary dissociated hippocampal cultures (14 DIV) were used for immunocytochemistry. Representative images of WT and SynDIG4−/− neurons stained with GluA1 and vGlut1 (C). Graphs depict quantification of synaptic (colocalized with vGlut1) and extrasynaptic (no colocalization with vGlut1) GluA1 puncta density (D), area (E), and integrated density (ID) (F). Data are averaged from two independent experiments; n = 24–25 cells per genotype per experiment, and three dendrites per cell were selected for measurement. Scale bar, 5 μm.
(G–K) Hippocampal pyramidal neurons in acute slices from 12- to 15-day-old mice were used to record AMPAR mEPSC at −70 mV. Traces from representative recordings (G). Averaged events from one representative cell per genotype are presented to scale (left) and normalized to peak (right) (H). Graphs represent average mEPSC amplitude (I), frequency (J), and decay time (K) in SynDIG4−/− and WT mice.
Significance (Student’s t test): *p < 0.05, ***p < 0.001. See also Figures S1 and S2 and Tables S1, S2, and S3.
To investigate potential changes in subcellular composition of mutant brain tissue, candidate synaptic proteins were analyzed (Table S2). Except for an increase in GluA2 in the synaptosomal fraction of SynDIG4−/− samples (p = 0.045), there were no significant changes in glutamate receptor subunits (GluA1, GluA2, GluN1, and GluN2B), and the distribution of synaptic scaffolds PSD-93 or PSD-95 was not altered in SynDIG4−/− (Figures S1A–S1D).
To investigate SynDIG4 expression in vivo, mutant mouse brain sections were stained for β-gal activity. Sagittal sections from P7 SynDIG4−/− mice show β-gal reporter activity throughout hippocampus, with weak expression in olfactory bulb and neocortex (Figure S1E). Coronal sections of P14, P28, and P62 brains show β-gal expression remains high throughout hippocampus and increased in olfactory bulb and neocortex (Figure S1F).
Reduced Extrasynaptic AMPARs and Weaker Synapses in SynDIG4−/− Mice
To investigate effects of SynDIG4 deficiency, a combination of electrophysiology and immunocytochemistry was employed. Dissociated hippocampal neurons from WT and SynDIG4−/− littermates were fixed and stained for GluA1 at synapses (defined as overlap with vGlut1) and at extrasynaptic sites (defined as no overlap with vGlut1) at 14 days in vitro (DIV) (Figure 2C). We observed decreased GluA1 density at extrasynaptic sites and a corresponding increased density of GluA1 at synapses in SynDIG4−/− neurons compared with WT (Figure 2D; Table S1). GluA1 puncta size and intensity were reduced at both synaptic and extrasynaptic sites in SynDIG4−/− neurons compared with WT (Figures 2E and 2F; Table S1). We did not observe significant changes in synaptic GluA2 puncta; however, extrasynaptic GluA2 puncta density was significantly reduced (Figures S2A and S2B).
Extrasynaptic AMPARs are localized to different locations: mobile pools at the cell surface and intracellular compartments, including recycling endosomes and transport vesicles that might not be fully captured in our analysis of extrasynaptic puncta. Therefore, we evaluated the level and area of total GluA1 and GluA2 signal that does not overlap with vGlut1; similar results were obtained with this analysis (Table S3). SynDIG4−/− neurons did not show significant differences in dendrite complexity compared with WT (Figures S2C and S2D). Staining with primary antibodies individually and both secondary antibodies indicated no cross-reactivity or bleed-through that might contribute to the immunofluorescence signal observed (Figures S2E and S2F).
To assess whether the change in AMPAR distribution reflected functional alterations, acute slices from 2- to 3-week-old WT and SynDIG4−/− mice were used in whole-cell patch-clamp experiments. Miniature excitatory postsynaptic currents (mEPSCs) were recorded in hippocampal CA1 pyramidal cells (Figures 2G and 2H). The mEPSC amplitude in SynDIG4−/− acute slices was significantly reduced and the mEPSC frequency was increased compared to WT (Figures 2I and 2J; Table S1). However, there was no significant difference in decay kinetics between WT and SynDIG4−/− neurons (Figure 2K; Table S1).
SynDIG4−/− Mice Show Impaired Schaffer-Collateral LTP
To test synaptic transmission, we recorded excitatory postsynaptic potentials (EPSPs) as extracellular field potentials (fEPSPs) in 8- to 12-week-old mice. By applying paired pulses, we did not observe alterations in presynaptic facilitation in SynDIG4−/− mice (Figure S3A). No differences were detected in signal strength in relation to stimulus intensity (Figure S3B). To test synaptic plasticity, we induced LTP of Schaffer-collateral synapses using a single 100 Hz/1 s tetanus, a paradigm that successfully elicits significant LTP in WT mice. Surprisingly, SynDIG4−/− synapses were not potentiated following tetanic stimulation; rather, there was a slight depression in transmission strength (Figure 3A; Table S1). To test whether LTP induction in SynDIG4−/− mice was generally impaired, we recorded LTP induced with 10 theta-burst stimulations, a different stimulus paradigm also known to elicit robust LTP in Schaffer-collateral synapses. TBS produced robust LTP in 8- to 12-week-old WT mice, as well as SynDIG4−/− mice (Figure 3B; Table S1). These results were recapitulated in 2- to 4-week-old animals (Figures S3C and S3D).
Figure 3. LTP Induction by a Single Tetanus, but Not by Theta-Burst or Pairing Stimulation, Is Impaired in SynDIG4−/− Mice.

(A and B) Schaffer-collateral fEPSPs were recorded from acute forebrain slices of 8- to 12-week-old mice. A 100 Hz/1 s tetanus elicited LTP in WT mice while leading to a depression of the fEPSP slope in SynDIG4−/− mice (A). Theta-burst stimulus (TBS) led to robust LTP in SynDIG4−/− mice that was not significantly different from LTP in WT (B).
(C) Hippocampal pyramidal neurons in acute slices from 12- to 15-day-old mice were used to record evoked EPSC in whole-cell patch-clamp configuration following Schaffer-collateral stimulation. Cells were held at −70 mV to record AMPAR-mediated currents. LTP elicited by pairing presynaptic stimulation with postsynaptic depolarization to 0 mV was not significantly different in cells from SynDIG4−/− mice compared to WT.
Insets show sample traces before (black) and 30 min after (gray) tetanization. Significance is calculated by one-way ANOVA with Bonferroni’s post-test between baseline and tetanized for each genotype and between tetanized of both genotypes. See also Figure S3 and Table S1.
To investigate further the difference in LTP between WT and SynDIG4−/−, we used the pairing-induced LTP paradigm in which the presynaptic high-frequency stimulus is applied while the postsynaptic cell is depolarized using a patch electrode. Pairing removes the Mg2+ block from the NMDA-type receptor (NMDAR), forgoing the necessity of AMPAR-dependent postsynaptic depolarization. We found no difference in pairing-induced LTP between 2-week-old WT and SynDIG4−/− neurons (Figure 3C; Table S1).
SynDIG4 KO Mice Display Deficits on Two Independent Cognitive Tasks
We investigated the impact of SynDIG4 deletion on cognitive function in vivo using two established learning and memory tasks: Morris water maze and novel object recognition. A battery of general health parameters did not indicate any significant differences that might contribute to behavioral analysis (Table S4).
In the Morris water maze assay, mice were trained over a period of 5 days to swim to a hidden, submerged platform using visual cues, according to methods previously described (Brielmaier et al., 2012). The average latency to find the platform decreased over the training period for WT animals, while the average latency for SynDIG4−/− mice only decreased slightly and was significantly different when compared to WT animals (Figure 4A; Table S1). After the last training trial, the platform was removed and mice underwent a probe trial to measure time spent exploring the target quadrant that previously contained the hidden platform. WT mice spent significantly more time in the target quadrant than in the other three quadrants, in contrast to SynDIG4−/− mice (Figure 4B; Table S1), confirming that the deficit in spatial learning was caused by failure to use distal environmental cues to acquire the spatial location of the hidden platform.
Figure 4. SynDIG4−/− Mice Are Deficient in Two Cognitive Learning and Memory Tasks.
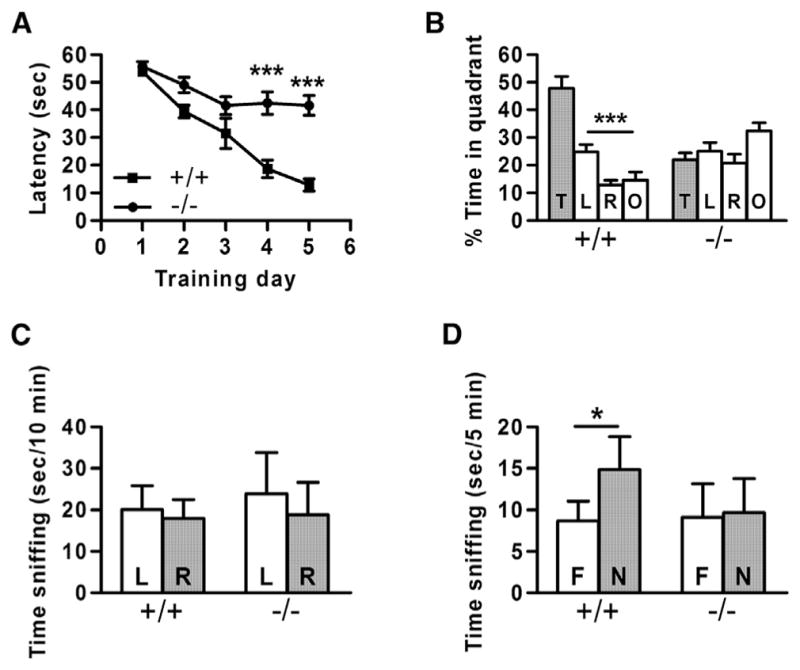
(A) Latency to find a hidden platform did not decrease significantly in SynDIG4−/− mice over a 5-day training period. Asterisks indicate significant differences in latency between WT (+/+) and SynDIG4 homozygous mutant (−/−) mice on days 4 and 5.
(B) During the probe trial, WT mice spent significantly more time in the target quadrant than in other quadrants, whereas SynDIG4−/− mice did not. Quadrants: T, target; L, left; R, right; O, opposite.
(C) Neither WT nor SynDIG4−/− mice exhibited left-right bias in the habituation session of the novel object recognition task. L, left object; R, right object.
(D) WT mice spent significantly more time investigating the novel object (N) than the familiar object (F), while SynDIG4−/− animals showed no preference, indicating a deficit in object recognition.
For all experiments, WT, n = 9; SynDIG4−/−, n = 11. Significance: *p < 0.05, ***p < 0.001.
See also Figure S4 and Tables S1 and S4.
We further tested whether SynDIG4−/− mice had deficits in a second cognitive task with different sensory and motor demands: the novel object recognition task (Brielmaier et al., 2012; Cohen et al., 2013; Vogel-Ciernia and Wood, 2014). Given a choice between two neutral objects of equal salience but with differing shapes and textures, one familiar and one novel, rodents will usually spend more time investigating the novel object. As previously described (Yang et al., 2015), mice were first habituated to the open-field testing environment, and a familiarization session with two identical objects indicated no left-right biases in either genotype (Figure 4C; Table S4). After exposure to the two identical objects, the chamber was cleaned and animals were allowed to explore one of the now-familiar objects and one novel object. WT mice displayed normal novel object recognition, spending significantly more time investigating the novel object than the familiar object. SynDIG4−/− mice failed to display novel object recognition, spending approximately equal time with the novel and the familiar objects (Figure 4D; Table S4).
We observed moderately higher exploratory activity in SynDIG4−/− mice compared to WT in the open field (Figures S4A and S4B). On the elevated plus-maze, more open arm time and entries were seen in SynDIG4−/− mice compared to WT; however, total number of entries was elevated (Figures S4C and S4D), indicating higher general exploratory activity rather than reduced anxiety-related behavior. Similarly, the number of light ↔ dark transitions was higher in SynDIG4−/− mice than in WT (Figures S4E and S4F). Higher exploratory locomotion in SynDIG4−/− mice in these three assays suggests that deficits in cognitive function are unlikely to be due to a motor disability.
DISCUSSION
Although SynDIG4 shares sequence similarity with SynDIG1, it has distinct expression in rat brain and is enriched with GluA1-containing AMPARs at extrasynaptic sites (Kirk et al., 2016). Based on the results presented here, we propose that SynDIG4 establishes a pool of extrasynaptic AMPARs necessary for synapse development and function underlying higher-order cognitive plasticity.
SynDIGs belong to a larger superfamily, named dispanin, based on sequence similarity (Sällman Almén et al., 2012). However, SynDIG4/Prrt1 shares only 35% sequence similarity with SynDIG1. In contrast to other AMPAR-associated transmembrane proteins, SynDIG1 does not promote AMPAR surface expression or alter channel gating when coexpressed with GluA1/2 subunits in HEK cells (Lovero et al., 2013), suggesting that SynDIG1 is an atypical AMPAR auxiliary factor. We tested whether coexpression of SynDIG1 altered biophysical properties of AMPARs in oocytes, and we did not find significant changes (data not shown), consistent with results in HEK cells (Lovero et al., 2013). However, there was a significant and high reduction in total current amplitude measured by whole-cell two-electrode voltage-clamp (TEVC) recordings (data not shown), so there may be minor changes associated with SynDIG1 coexpression that we could not detect in oocytes.
In contrast, SynDIG4 influences AMPAR gating properties in a subunit-dependent manner. We did not measure whether SynDIG4 influences AMPAR surface expression in oocytes directly. However, average current amplitude measured by TEVC recordings in the presence of the desensitization blocker cyclothiazide indicated ~35% reduction for GluA1 coexpressed with SynDIG4 (p = 0.02) and ~20% reduction for GluA1/2 coexpressed with SynDIG4 that did not reach significance (p = 0.06). These results are consistent with the outside-out patch recordings for SynDIG4. There was no correlation between current amplitude and the decay time in patch-clamp current recordings. That is, comparing currents with the same amplitude with or without SynDIG4 (as well as with or without TARPγ8) exhibited a significant slower deactivation in the presence SynDIG4. Therefore, we have not explored this observation further, although SynDIG4 might negatively affect surface delivery of AMPARs. These data support a direct and specific interaction of SynDIG4 with GluA1-containing AMPARs. SynDIG4 increased deactivation of GluA1 or GluA1/2 in oocytes, yet no significant change in decay time of mEPSC events in SynDIG4−/− neurons was observed, suggesting that SynDIG4 does not modulate GluA1 at synapses. In addition, we observed synergistic effects on AMPAR biophysical properties upon coexpression of both SynDIG4 and TARPγ8, indicating that these accessory proteins can interact simultaneously with AMPARs. TARPγ8 is a critical Ca2+/calmodulin-dependent protein kinase IIa (CaMKIIa) substrate for hippocampal LTP, learning, and memory (Park et al., 2016). Future studies are needed to address the relationship, if any, between SynDIG4 and TARPγ8-dependent LTP.
Although SynDIG4 primarily colocalizes with GluA1-containing AMPARs at extrasynaptic sites, biochemical fractionation indicates a portion of SynDIG4 is present in the PSD-enriched fraction (Kirk et al., 2016). One possibility is that SynDIG4 traffics with GluA1-containing AMPARs between extrasynaptic and synaptic sites, perhaps due to an activity-dependent post-translational modification. For example, a CCFW motif in the juxta-transmembrane-associated region is conserved in all SynDIG proteins. The two cysteine residues in SynDIG1 are palmitoylated in an activity-dependent manner to regulate stability, localization, and function (Kaur et al., 2016) and preliminary experiments indicate that SynDIG4 is also palmitoylated (data not shown). It will be informative to investigate the relationship, if any, between SynDIG4 palmitoylation and its role in synapse function. However, the SynDIG family does not contain a recognizable intracellular domain such as a PDZ binding motif, and it is unclear how SynDIG proteins are localized at synapses. Therefore, an alternative model is that SynDIG4 might physically restrain GluA1-containing AMPARs at extrasynaptic sites to maintain an extrasynaptic pool of AMPARs. Upon stimulation, SynDIG4 might release GluA1-containing AMPARs from extrasynaptic sites and allow other auxiliary factors such as TARPγ8 to transport them to synapses. Given that SynDIG4 increased deactivation of GluA1 and GluA1/2 in oocytes yet no significant change in decay time of mEPSC events in SynDIG4−/− neurons was observed, we interpret these results as evidence that SynDIG4 does not act primarily on synaptic AMPARs. However, SynDIG4 is not the only protein in which heterologous cell expression did not match the phenotype observed in vivo. For example, CKAMP44, which is a synaptic protein interacting with synaptic AMPARs, also slows AMPAR deactivation and is additive to TARPγ8; however, there was no effect on mEPSC decay time upon overexpression or in CKAMP44 knockout (KO) mice (von Engelhardt et al., 2010). The reason for the discrepancy is still not known. While we interpret our result as evidence for a primary role of SynDIG4 on extrasynaptic AMPARs, which is consistent with its localization, our findings do not rule out a direct role of SynDIG4 on synaptic AMPARs.
A particularly intriguing aspect of this study is that synaptic plasticity is disrupted in SynDIG4−/− mice when tetanic stimulation is used to induce plasticity, while TBS is normal. A previous study showed that TBS-induced LTP can be induced in GluA1 null mice, whereas tetanic-induced LTP cannot (Romberg et al., 2009). Given that SynDIG4 preferentially colocalizes with GluA1-containing AMPARs at non-synaptic sites (Kirk et al., 2016), and loss of SynDIG4 significantly reduces non-synaptic GluA1 and GluA2 shown here, these data are consistent with a model by which SynDIG4 selectively regulates an extrasynaptic pool of AMPARs during tetanic-induced LTP. Evidence suggests that LTP requires a reserve pool of extrasynaptic glutamate receptors independent of receptor subunit type (Granger et al., 2013). Furthermore, GluA1/2 heteromers constitute 95% of the extrasynaptic AMPAR pool (Lu et al., 2009), consistent with the observed reduced density of extrasynaptic GluA1/2 in SynDIG4−/− neurons. The importance of SynDIG4 in cognitive plasticity is underscored by the deficit in two independent mouse learning and memory behaviors.
The ~25% reduction in mEPSC amplitude in SynDIG4−/− neurons reflects a reduction in postsynaptic AMPAR responses, which could contribute to a decrease in tetanus-induced LTP by decreasing NMDAR channel activity, but it cannot explain the complete lack of LTP that we observe. More than 50% inhibition of AMPARs by 20 μM GYKI52466 or 0.25 μM NBQX does not diminish induction of LTP by tetanic stimulation in CA1 fEPSP recordings (Kapus et al., 2000). Furthermore, knockdown of PORCN, which fosters AMPAR secretory trafficking, reduces AMPAR mEPSC amplitude by ~25%, as does SynDIG4 KO. However, it only reduces LTP induced by 100 Hz/1 s stimulation proportional to the degree it reduces basal postsynaptic AMPAR responses, while the magnitude of LTP relative to basal synaptic transmission is the same in WT and PORCN knockdown neurons (Erlenhardt et al., 2016). Similarly, in knockin mice in which the last 4 residues of TARPγ8 were deleted to impair PSD-95 binding, postsynaptic AMPAR activity was reduced by ~40%, but the degree of tetanus-induce LTP relative to basal synaptic transmission was comparable between WT and knockin mice (Sumioka et al., 2011). Thus, it seems unlikely that a 25% reduction in postsynaptic AMPAR activity would by itself abolish tetanus-induced LTP in SynDIG4−/− mice. Abrogation of LTP induced by tetanic stimulation, but not by TBS or pairing, is consistent with the possibility that SynDIG4 is necessary for certain forms of synaptic plasticity through its role to establish an extrasynaptic pool of AMPARs that might also be needed to maintain normal basal synaptic function involving AMPAR trafficking.
The proline-rich N terminus of SynDIG4/Prrt1 is shared with Prrt2 (67% sequence similarity,) and Prrt2 has been identified as a candidate AMPAR-associated protein (Chen et al., 2014; Schwenk et al., 2012, 2014; Shanks et al., 2012; von Engelhardt et al., 2010). However, a study demonstrated a presynaptic role for Prrt2 in regulated exocytosis of neurotransmitter via interaction with synaptotagmin (Valente et al., 2016). Presynaptic release failure is a valid concern in the interpretation of tetanic stimulation-induced LTP for SynDIG4−/− mice discussed earlier. However, paired-pulse facilitation is not affected. We recognize that this result does not rule out all possible presynaptic deficiencies, but it does not indicate possible defects either. Furthermore, the input-output relationship is unaltered (if anything, SynDIG4−/− displays a tendency for increased fEPSP slope). If release probability was altered, we would expect differences either at high or at low stimulus intensities, depending on the deficit (Ca2+ sensitivity of the release machinery versus vesicle loading or density). Moreover, mEPSC frequency in SynDIG4−/− is increased. If release probability was decreased (to account for reduced LTP) we would expect a reduction in mEPSC frequency. The increase in mEPSC frequency could also reflect a postsynaptic effect by activating silent synapses in SynDIG4−/− to which extrasynaptic GluA1-containing AMPARs are recruited (indicated by increased synaptic GluA1 in SynDIG4−/−). The reduction in mEPSC amplitude may reflect the reduction of synaptic GluA1 puncta area and intensity. Altogether, it is unlikely that presynaptic effects are the reason for the reduced LTP; however, we cannot rule out this possibility at present.
Given that both SynDIG1 and SynDIG4 are expressed in hippocampus, it is not unexpected that there are some similarities in phenotypes. For example, we observed that loss of SynDIG4 leads to increased synapse number similar to that observed in SynDIG1 mutant mice (Chenaux et al., 2016). The increased synapse density observed in SynDIG4−/− or SynDIG1 mutant mice could be a consequence of reduced synaptic strength. Homeostatic mechanisms lead to an increase in synapse number in the absence of potentiation to maintain total input strength (Bourne and Harris, 2011; Turrigiano, 2008). However, there are key differences between phenotypes in SynDIG4−/− and SynDIG1 mutant mice that illustrate the unique role of SynDIG4 in synapse function. First, the magnitude of reduction in mEPSC amplitude is greater in SynDIG4−/− compared with SynDIG1 mutant mice. Second, LTP induced by tetanic stimulation is abolished in both young and adult SynDIG4−/− mice, while mEPSC amplitudes were reduced by 25% and basal fEPSP transmission was unchanged, which suggests an additional effect of SynDIG4 on synaptic plasticity beyond its role in synapse development. In contrast, such LTP is reduced only in young SynDIG1 mutant mice, likely as a consequence of reduced synaptic transmission (Chenaux et al., 2016).
Altogether, our studies use a combination of approaches, including biochemistry, immunocytochemistry, electrophysiology, and behavior, to provide strong evidence that SynDIG4 establishes a pool of extrasynaptic AMPARs necessary for excitatory synapse development and function underlying higher-order cognitive plasticity.
EXPERIMENTAL PROCEDURES
Full experimental procedures are available in the Supplemental Information.
Animals
Frogs
Xenopus laevis frogs (females, age 1–3 years old) were used as the source for oocytes for heterologous expression and outside-out patch-clamp electrophysiological recordings. Maintenance of frogs and extraction of oocytes were performed in accordance with the NIH guidelines for the Care and Use of Laboratory Animals and Israeli law for animal experimentation and were approved by the Institutional Animal Care and Use Committee (IACUC) of the Hebrew University of Jerusalem.
Mice
The mutant allele [Prrt1tm1(KOMP)Vlcg] was generated by Velocigene as part of KOMP and maintained on a C57BL/6 background. Mice (males and females) were used at the indicated ages for the following: biochemistry (P14), primary hippocampal culture (P1), electrophysiology (whole-cell patch-clamp, P12–P15; fEPSP, 8–12 weeks), and behavior (3–5 months). The use and maintenance of mice were carried out according to NIH guidelines and approved by the IACUC at University of California (UC), Davis.
Biochemical Fractionation and Quantitative Immunoblotting
The rostral two-thirds of P14 mouse brains were homogenized, and biochemical fractions were analyzed as described (Chenaux et al., 2016).
Primary Culture of Dissociated Hippocampal Neurons
Neurons from hippocampi of P1 WT and SynDIG4−/− littermates were dissociated individually in papain (Worthington) and plated (12,500 cells/cm2) on poly-L-lysine-coated glass coverslips media supplemented with B-27 (Thermo Fisher Scientific). Neurons were analyzed as previously described (Kirk et al., 2016).
Electrophysiology
Intra- and extracellular recordings were performed using standard methods as previously described (Chenaux et al., 2016). Expression in Xenopus laevis oocytes and outside-out patch-clamp electrophysiological recordings were performed as described (Priel et al., 2006).
Mouse Behavior
Founder mice generated on the C57BL/6N background were backcrossed four times (N4) onto C57BL/6J to remove the rd8 mutation (Mattapallil et al., 2012). Heterozygous N4 mice devoid of rd8 were then intercrossed to produce WT and SynDIG4−/− mice.
Statistical Analyses
Data were analyzed in GraphPad Prism with the following statistical tests: two-tailed Student’s t test (immunocytochemistry); one-way ANOVA (electrophysiology in oocytes); one-way ANOVA and Bonferroni’s multiple comparison test (fEPSP), unpaired Student’s t test (mEPSC). For the Morris water maze, latency was analyzed using two-way repeated-measures ANOVA with Bonferroni’s post hoc tests. Probe trial data were analyzed using one-way ANOVA with Bonferroni post-tests. Novel object data were analyzed using paired Student’s t test. All data are shown as mean ± SEM. Significance was *p ≤ 0.5, **p ≤ 0.01, and ***p ≤ 0.001; NS indicates not significant.
Supplementary Material
Highlights.
SynDIG4 affects AMPAR biophysical properties in a subunit-dependent manner
Loss of SynDIG4 results in reduced extrasynaptic AMPAR and weaker synapses
SynDIG4 is necessary for tetanus-induced, but not theta-burst, LTP
SynDIG4 KO mice exhibit deficits in two independent cognitive behavior tasks
Acknowledgments
Funding was provided by the American Heart Association (11POST7020009 to L.M.), Brain & Behavior Research Foundation NARSAD Young Investigator (grant 20748 to L.M.), United States-Israel Binational Science Foundation (BSF; grant 2012781 to Y.S.-B.) and the United States National Science Foundation (NSF; grant 1322302 to E.D.), NIH (R01 MH097887 to J.W.H., R01 NS078792 to J.W.H., U54 HD079125 MCP to J.L.S. and J.N.C.) and NIH New Director’s Innovator Award Program (DP2 OD006479-01 to E.D.), UC Davis Academic Senate Research Program Pilot Grant (to E.D.), and Whitehall Foundation (2015-05-106 to E.D.). L.M.K. is a trainee of the UC Davis MCB T32 Training Program (award GM007377). SynDIG4 KO mice were generated using the UC Davis Mouse Biology Program.
Footnotes
Supplemental Information includes Supplemental Experimental Procedures, four figures, and four tables and can be found with this article online at https://doi.org/10.1016/j.celrep.2018.02.026.
DECLARATION OF INTERESTS
The authors declare no competing interests.
AUTHOR CONTRIBUTIONS
Methodology, L.M., L.M.K., G.C., Y.S.-B., J.N.C., J.W.H., and E.D.; Investigation, L.M., L.M.K., G.C., D.J.S., K.R.P., M.C.P., M.Q., T.H., and K.E.P.; Formal Analysis, L.M., L.M.K., G.C., D.J.S., M.Q., Y.S.-B., J.L.S., J.W.H., and E.D.; Writing, L.M., L.M.K., G.C., D.J.S., and E.D.; Funding Acquisition, L.M., Y.S.-B., J.W.H., and E.D.; Supervision, J.W.H. and E.D.
References
- Bourne JN, Harris KM. Coordination of size and number of excitatory and inhibitory synapses results in a balanced structural plasticity along mature hippocampal CA1 dendrites during LTP. Hippocampus. 2011;21:354–373. doi: 10.1002/hipo.20768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brielmaier J, Matteson PG, Silverman JL, Senerth JM, Kelly S, Genestine M, Millonig JH, DiCicco-Bloom E, Crawley JN. Autism-relevant social abnormalities and cognitive deficits in engrailed-2 knockout mice. PLoS ONE. 2012;7:e40914. doi: 10.1371/journal.pone.0040914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Chetkovich DM, Petralia RS, Sweeney NT, Kawasaki Y, Wenthold RJ, Bredt DS, Nicoll RA. Stargazin regulates synaptic targeting of AMPA receptors by two distinct mechanisms. Nature. 2000;408:936–943. doi: 10.1038/35050030. [DOI] [PubMed] [Google Scholar]
- Chen N, Pandya NJ, Koopmans F, Castelo-Székelv V, van der Schors RC, Smit AB, Li KW. Interaction proteomics reveals brain region-specific AMPA receptor complexes. J Proteome Res. 2014;13:5695–5706. doi: 10.1021/pr500697b. [DOI] [PubMed] [Google Scholar]
- Chenaux G, Matt L, Hill TC, Kaur I, Liu XB, Kirk LM, Speca DJ, McMahon SA, Zito K, Hell JW, Díaz E. Loss of SynDIG1 reduces excitatory synapse maturation but not formation in vivo. eNeuro. 2016;3 doi: 10.1523/ENEURO.0130-16.2016. ENEURO.0130-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen SJ, Munchow AH, Rios LM, Zhang G, Asgeirsdóttir HN, Stackman RW., Jr The rodent hippocampus is essential for nonspatial object memory. Curr Biol. 2013;23:1685–1690. doi: 10.1016/j.cub.2013.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Díaz E. Regulation of AMPA receptors by transmembrane accessory proteins. Eur J Neurosci. 2010;32:261–268. doi: 10.1111/j.1460-9568.2010.07357.x. [DOI] [PubMed] [Google Scholar]
- Erlenhardt N, Yu H, Abiraman K, Yamasaki T, Wadiche JI, Tomita S, Bredt DS. Porcupine controls hippocampal AMPAR levels, composition, and synaptic transmission. Cell Rep. 2016;14:782–794. doi: 10.1016/j.celrep.2015.12.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granger AJ, Shi Y, Lu W, Cerpas M, Nicoll RA. LTP requires a reserve pool of glutamate receptors independent of subunit type. Nature. 2013;493:495–500. doi: 10.1038/nature11775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huganir RL, Nicoll RA. AMPARs and synaptic plasticity: the last 25 years. Neuron. 2013;80:704–717. doi: 10.1016/j.neuron.2013.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson AC, Nicoll RA. The expanding social network of ionotropic glutamate receptors: TARPs and other transmembrane auxiliary subunits. Neuron. 2011;70:178–199. doi: 10.1016/j.neuron.2011.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan BA, Fernholz BD, Boussac M, Xu C, Grigorean G, Ziff EB, Neubert TA. Identification and verification of novel rodent postsynaptic density proteins. Mol Cell Proteomics. 2004;3:857–871. doi: 10.1074/mcp.M400045-MCP200. [DOI] [PubMed] [Google Scholar]
- Kalashnikova E, Lorca RA, Kaur I, Barisone GA, Li B, Ishimaru T, Trimmer JS, Mohapatra DP, Díaz E. SynDIG1: an activity-regulated, AMPA- receptor-interacting transmembrane protein that regulates excitatory synapse development. Neuron. 2010;65:80–93. doi: 10.1016/j.neuron.2009.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapus G, Székely JI, Durand J, Ruiz A, Tarnawa I. AMPA receptor antagonists, GYKI 52466 and NBQX, do not block the induction of long-term potentiation at therapeutically relevant concentrations. Brain Res Bull. 2000;52:511–517. doi: 10.1016/s0361-9230(00)00288-4. [DOI] [PubMed] [Google Scholar]
- Kaur I, Yarov-Yarovoy V, Kirk LM, Plambeck KE, Barragan EV, Ontiveros ES, Díaz E. Activity-dependent palmitoylation controls SynDIG1 stability, localization, and function. J Neurosci. 2016;36:7562–7568. doi: 10.1523/JNEUROSCI.4859-14.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirk LM, Ti SW, Bishop HI, Orozco-Llamas M, Pham M, Trimmer JS, Díaz E. Distribution of the SynDIG4/proline-rich transmembrane protein 1 in rat brain. J Comp Neurol. 2016;524:2266–2280. doi: 10.1002/cne.23945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee KF, Soares C, Béïque JC. Tuning into diversity of homeostatic synaptic plasticity. Neuropharmacology. 2014;78:31–37. doi: 10.1016/j.neuropharm.2013.03.016. [DOI] [PubMed] [Google Scholar]
- Lovero KL, Blankenship SM, Shi Y, Nicoll RA. SynDIG1 promotes excitatory synaptogenesis independent of AMPA receptor trafficking and biophysical regulation. PLoS ONE. 2013;8:e66171. doi: 10.1371/journal.pone.0066171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu W, Shi Y, Jackson AC, Bjorgan K, During MJ, Sprengel R, Seeburg PH, Nicoll RA. Subunit composition of synaptic AMPA receptors revealed by a single-cell genetic approach. Neuron. 2009;62:254–268. doi: 10.1016/j.neuron.2009.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mattapallil MJ, Wawrousek EF, Chan CC, Zhao H, Roychoudhury J, Ferguson TA, Caspi RR. The Rd8 mutation of the Crb1 gene is present in vendor lines of C57BL/6N mice and embryonic stem cells, and confounds ocular induced mutant phenotypes. Invest Ophthalmol Vis Sci. 2012;53:2921–2927. doi: 10.1167/iovs.12-9662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J, Chávez AE, Mineur YS, Morimoto-Tomita M, Lutzu S, Kim KS, Picciotto MR, Castillo PE, Tomita S. CaMKII phosphorylation of TARPγ-8 is a mediator of LTP and learning and memory. Neuron. 2016;92:75–83. doi: 10.1016/j.neuron.2016.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Priel A, Selak S, Lerma J, Stern-Bach Y. Block of kainate receptor desensitization uncovers a key trafficking checkpoint. Neuron. 2006;52:1037–1046. doi: 10.1016/j.neuron.2006.12.006. [DOI] [PubMed] [Google Scholar]
- Romberg C, Raffel J, Martin L, Sprengel R, Seeburg PH, Rawlins JN, Bannerman DM, Paulsen O. Induction and expression of GluA1 (GluR-A)-independent LTP in the hippocampus. Eur J Neurosci. 2009;29:1141–1152. doi: 10.1111/j.1460-9568.2009.06677.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sällman Almén M, Bringeland N, Fredriksson R, Schiöth HB. The dispanins: a novel gene family of ancient origin that contains 14 human members. PLoS ONE. 2012;7:e31961. doi: 10.1371/journal.pone.0031961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwenk J, Harmel N, Zolles G, Bildl W, Kulik A, Heimrich B, Chisaka O, Jonas P, Schulte U, Fakler B, Klöcker N. Functional proteomics identify cornichon proteins as auxiliary subunits of AMPA receptors. Science. 2009;323:1313–1319. doi: 10.1126/science.1167852. [DOI] [PubMed] [Google Scholar]
- Schwenk J, Harmel N, Brechet A, Zolles G, Berkefeld H, Müller CS, Bildl W, Baehrens D, Hüber B, Kulik A, et al. High-resolution proteomics unravel architecture and molecular diversity of native AMPA receptor complexes. Neuron. 2012;74:621–633. doi: 10.1016/j.neuron.2012.03.034. [DOI] [PubMed] [Google Scholar]
- Schwenk J, Baehrens D, Haupt A, Bildl W, Boudkkazi S, Roeper J, Fakler B, Schulte U. Regional diversity and developmental dynamics of the AMPA-receptor proteome in the mammalian brain. Neuron. 2014;84:41–54. doi: 10.1016/j.neuron.2014.08.044. [DOI] [PubMed] [Google Scholar]
- Shanks NF, Savas JN, Maruo T, Cais O, Hirao A, Oe S, Ghosh A, Noda Y, Greger IH, Yates JR, 3rd, Nakagawa T. Differences in AMPA and kainate receptor interactomes facilitate identification of AMPA receptor auxiliary subunit GSG1L. Cell Rep. 2012;1:590–598. doi: 10.1016/j.celrep.2012.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sumioka A, Brown TE, Kato AS, Bredt DS, Kauer JA, Tomita S. PDZ binding of TARPγ-8 controls synaptic transmission but not synaptic plasticity. Nat Neurosci. 2011;14:1410–1412. doi: 10.1038/nn.2952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomita S, Chen L, Kawasaki Y, Petralia RS, Wenthold RJ, Nicoll RA, Bredt DS. Functional studies and distribution define a family of transmembrane AMPA receptor regulatory proteins. J Cell Biol. 2003;161:805–816. doi: 10.1083/jcb.200212116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrigiano GG. The self-tuning neuron: synaptic scaling of excitatory synapses. Cell. 2008;135:422–435. doi: 10.1016/j.cell.2008.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turrigiano G. Homeostatic synaptic plasticity: local and global mechanisms for stabilizing neuronal function. Cold Spring Harb Perspect Biol. 2012;4:a005736. doi: 10.1101/cshperspect.a005736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valente P, Castroflorio E, Rossi P, Fadda M, Sterlini B, Cervigni RI, Prestigio C, Giovedì S, Onofri F, Mura E, et al. PRRT2 is a key component of the Ca(2+)-dependent neurotransmitter release machinery. Cell Rep. 2016;15:117–131. doi: 10.1016/j.celrep.2016.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valenzuela DM, Murphy AJ, Frendewey D, Gale NW, Economides AN, Auerbach W, Poueymirou WT, Adams NC, Rojas J, Yasenchak J, et al. High-throughput engineering of the mouse genome coupled with high-resolution expression analysis. Nat Biotechnol. 2001;21:652–659. doi: 10.1038/nbt822. [DOI] [PubMed] [Google Scholar]
- Vogel-Ciernia A, Wood MA. Examining object location and object recognition memory in mice. Curr Protoc Neurosci. 2014;69:8.31:8.31.1–8.31.17. doi: 10.1002/0471142301.ns0831s69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Engelhardt J, Mack V, Sprengel R, Kavenstock N, Li KW, Stern-Bach Y, Smit AB, Seeburg PH, Monyer H. CKAMP44: a brain-specific protein attenuating short-term synaptic plasticity in the dentate gyrus. Science. 2010;327:1518–1522. doi: 10.1126/science.1184178. [DOI] [PubMed] [Google Scholar]
- Yang M, Lewis FC, Sarvi MS, Foley GM, Crawley JN. 16p11.2 deletion mice display cognitive deficits in touchscreen learning and novelty recognition tasks. Learn Mem. 2015;22:622–632. doi: 10.1101/lm.039602.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.