

Abstract
Background
Mutations activating the α subunit of heterotrimeric Gs protein are associated with a number of highly specific pathological molecular phenotypes. One of the best characterized is the McCune Albright syndrome. The disease presents with an increased incidence of neoplasias in specific tissues.
Main body
A similar repertoire of neoplasms can develop whether mutations occur spontaneously in somatic tissues during fetal development or after birth.
Glands are the most “permissive” tissues, recently found to include the entire gastrointestinal tract. High frequency of activating Gαs mutations is associated with precise diagnoses (e.g., IPMN, Pyloric gland adenoma, pituitary toxic adenoma). Typically, most neoplastic lesions, from thyroid to pancreas, remain well differentiated but may be a precursor to aggressive cancer.
Conclusions
Here we propose the possibility that gain-of-function mutations of Gαs interfere with signals in the microenvironment of permissive tissues and lead to a transversal neoplastic phenotype.
Keywords: GNAS, Heterotrimeric Gs protein, Activating mutation, Neoplasm, McCune Albright Syndrome, Intraductal papillary mucinous neoplasm, Fibrous dysplasia
Background
Heterotrimeric Gαβγ proteins are central to sensing a great number of extracellular stimuli. Each of the subunits is encoded by a multigene family that accommodates coupling to a huge diversity of seven transmembrane receptors. In metazoan organisms, four classes of Gα subunits, Gs, Gi, Gq and G12, couple several hundreds receptors to distinct sets of effector proteins. The Gs class of alpha subunits includes two genes GNAS and GNAL encoding Gαs and Gαolf proteins respectively that stimulate the effector protein adenylyl cyclase and regulate certain ion channels.
Whole genome analysis revealed that mutations affecting G proteins and GPCRs are more frequent than previously thought in transformed cells [1]. In a growing number of neoplasias, two hotspots on Gαs, Arg (R)201 and Gln (Q)227, are found mutated to three (Cys/His/Ser) or two (Arg/Leu) amino acids, respectively (Fig. 1a). By contrast, activating mutations in Gαolf have not been found. In Gαs, R201 mutations are more common than Q227 but substitutions at either residue inhibit intrinsic GTPase catalytic activity. The crystal structure shows these residues contribute to transition state interactions during GTP hydrolysis [2] (Fig. 2). As a consequence, mutant Gαs with substitutions at either residue remains GTP-bound, persisting in an active state that prolongs the effector protein interaction with dissociated Gαs or βγ. Cholera toxin achieves an equivalent result by ADP ribosylating R201 [3].
Fig. 1.
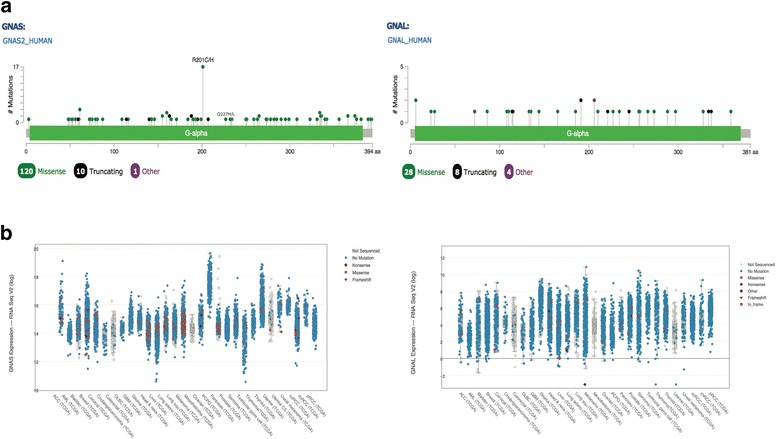
GNAS/GNAL Cross Cancer Comparison in Human Tumor Samples based on TCGA provisional data. Cbioportal was used to generate figures. a Mutational Diagrams. No activating alleles were found in GNAL. Two activating alleles were found in GNAS producing residues substitutions at R201 and Q227 (see in the text). b Expression Level Diagrams. GNAS shows higher expression level compared to GNAL in a cross cancer comparison
Fig. 2.
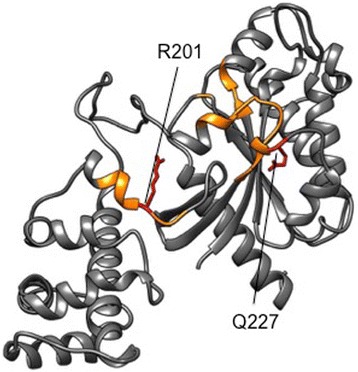
3-D model of GαS. Residues 40 to 394 of Gαs are represented based on protein model portal, template 3sn6A. The GTP binding domains are indicated in yellow, the most common gsp mutations, at R201 (the target of Cholera toxin) and Q227, are indicated in red
The TCGA database shows tissue distribution of Gαs activating mutations among 29 cancers (Table 1, Fig. 1b). An obvious indication for a cancer subtype is not evident. However, recent data point to certain highly prevalent neoplasms not included in the TCGA database (Table 2 and reference therein). The frequency of Gαs activating mutations in adenocarcinoma is comparable between the two data sets. The striking difference is the common occurrence of Gαs activating mutations in adenoma and other early neoplasia in neuroendocrine tumors. (Table 2). Gαs is associated with neoplasia and cancer but not Gαolf, probably because Gαs is ubiquitously expressed at high levels whereas Gαolf is highly expressed only in olfactory epithelium and other specialized cells (Fig. 1), and tumors from those tissues have not been well characterized. These features of effector protein regulation and expression pattern influence the oncogenic potential of Gs class genes.
Table 1.
Activating alleles of Gαs and/or Rαs in cBioPortal cancer cohorts
| Cancer Study Type | GNASb | (%c) | GNASa,b RASb | RASb | Percent | Total Samples |
|---|---|---|---|---|---|---|
| AML | 0 | 0 | 21 | (11%) | 191 | |
| Bladder | 0 | 0 | 0 | (0%) | 131 | |
| Breast Cohort 1 | 1 | 0 | 6 | (1%) | 816 | |
| Breast Cohort 2 | 1 | 0 | 3 | (1%) | 482 | |
| Renal Clear Cell Carcinoma | 0 | 0 | 0 | (0%) | 499 | |
| Colorectal Adenocarcinoma | 1 | 0 | 91 | (43%) | 212 | |
| Head and Neck Squamous Cell | 0 | 0 | 0 | (0%) | 279 | |
| Diffuse Glioma | 0 | 0 | 8 | (1%) | 794 | |
| Lung Adenocarcinoma | 2 | (2%) | 2 | 76 | (33%) | 230 |
| Lung Squamous Cell Carcinoma | 0 | 0 | 0 | (0%) | 178 | |
| Pan-Lung Cancer | 5 | (<1%) | 3 | 232 | (20%) | 1144 |
| High Grade Serous Ovarian Cancer | 0 | 0 | 4 | (1%) | 316 | |
| Pancreatic Adenocarcinoma | 7 | (5%) | 3 | 138 | (93%) | 149 |
| Prostate Adenocarcinoma | 0 | 0 | 0 | (0%) | 333 | |
| Stomach Adenocarcinoma | 3 | (1%) | 1 | 28 | (10%) | 287 |
| Papillary Thyroid Cancer | 0 | 0 | 0 | (0%) | 507 | |
| Uterine Endometrial Carcinoma | 0 | 0 | 51 | (21%) | 240 |
aCoincidence of GNAS and KRAS activating mutations. Among the cancer cohorts from TCGA in cBioPortal [100, 101], pancreatic adenocarcinoma, pan-lung, and stomach cancers had the highest frequencies of activating alleles of Gαs in the absence of KRAS mutations
bActivating alleles of GNAS and/or KRAS
c% GNAS activating alleles with two or more occurrences in the cancer cohort
Table 2.
Incidence of “gsp” in “receptive organs”
| Site | Histology (Dysplasia/Neoplasia) | Incidence % (n) | Refs |
|---|---|---|---|
| Thyroid | Toxic thyroid adenoma | 23% (65) | [102–104] |
| Non functional adenoma | 0% (31) | [102, 104] | |
| Carcinoma | 0% (18) | [104, 105] | |
| Pituitary | GH-secreting adenoma | 41% (504) | [106–126] |
| Prolactin-secreting adenoma | 0% (7) | [107] | |
| Non functioning | 3% (32) | [107] | |
| Muscle | Intramuscolar myxoma | 45% (101) | [39, 127, 128] |
| Various myxoid lesions | 0% (105) | [39, 127–130] | |
| Bone | Fibrous dysplasia (FD) | 81% (414) | [127, 131–134] |
| Low grade periosteal osteosarcoma | 0% (11) | [135] | |
| Low grade central osteosarcoma | 3% (35) | [135–137] | |
| Low grade parosteal osteosarcoma | 0% (80) | [135, 137] | |
| Osteofibrous dysplasia | 0% (13) | [132, 134, 138] | |
| Ossifying fibroma | 0% (66) | [131, 132] | |
| Blood | Hematological conditions | 0.6% (512) | [139–141] |
| Kidney | Renal cell carcinoma | 17% (30) | [142] |
| Lung | Mucinous cystoadenoma | 0.5% (208) | [24, 68, 143] |
| Pancreas | Intraduct. Papill. Mucin. Neop.(IPMN) | 58% (809) | [17, 19–21, 30, 68, 144–148] |
| Incipient IPMN | 33% (21) | [149] | |
| Intraduct. Tubulopapill. Neop. (ITPN) | 60% (15) | [146] | |
| Intraepithelial neoplasia (PanIN) | 2% (246) | [20, 150] | |
| Serous cystoadenoma (SCN) | 0% (54) | [20, 144] | |
| Mucin. Cyst.Neop. (MCN) | 0% (31) | [20, 144] | |
| Neuroendocrine tumor | 0% (52) | [20] | |
| Ductal adenocarcinoma (PDAC) | 0.4% (253) | [20, 30, 144, 145, 150] | |
| Biliary tract | IPMN of the bile duct | 23% (120) | [42, 151–153] |
| Biliary intraepithelial neoplasia | 1% (82) | [153, 154] | |
| Stomach | Pyloric gland adenoma | 48% (23) | [41] |
| Gastric adenocarc. of the fundic gland | 24% (29) | [64, 74] | |
| Hyperplastic gastric polyps | 0% (10) | [127] | |
| Foveolar type adenoma | 0% (23) | [41] | |
| Intestinal type adenoma | 0% (34) | [41] | |
| Gastric adenocarcinoma | 0% (71) | [41] | |
| Duodenum | Pyloric gland adenoma | 92% (35) | [41] |
| Gastric foveolar metaplasia | 41% (66) | [66] | |
| Gastric heterotopia | 28% (81) | [66] | |
| Adenocarcinoma | 17% (30) | [66] | |
| Gastroent. neuroen. tum. (GEP-NET) | 0% (31) | [155] | |
| Colon-rectum | Villous adenoma | 67% (55) | [155–158] |
| Tubular villous adenoma | 4% (154) | [155–157] | |
| Tubular adenoma | 0% (28) | [155, 157] | |
| Polyps | 0% (45) | [156] | |
| Adenocarcinoma | 3% (820) | [68, 155–159] | |
| Liver | Normal liver Intrahepat.cholangioc | 12%(43) | [160] |
| Advanc. Intrahepatic cholangiocarc | 3% (38) | [160] | |
| Hepatocell. Adenoma | 4% (179) | [22, 161] | |
| Hepatocell. Carcinoma | 0.8% (245) | [22] | |
| Fluke-ass. cholangiocarcinoma | 9.3% (53) | [23] | |
| Appendix | Low grade app. muc. neop. (LAMN) | 43% (84) | [31, 40, 68, 162, 163] |
| Adenocarcinoma | 46% (106) | [31, 40, 68, 69, 162–164] | |
| Gonads | Leydig cell stromal tumor | 67% (6) | [165] |
| Lobular Endocer. Glandular Hyperpl. | 28% (32) | [166] | |
| Juvenile Ovarian granul. cell tumor | 30% (30) | [167] | |
| Mucinous cystoadenoma | 9% (45) | [68, 168] | |
| Mucinous border line tumor | 4% (53) | [68, 168] | |
| Mucinous cystadenocarcinoma | 2% (45) | [68, 168] | |
| Ovarian granulosa cell tumor | 0% (25) | [169, 170] | |
| Other sex cord stromal tumors | 0% (6) | [170] | |
| Adenocarcinoma | 4% (92) | [166] | |
| Squamous cell carcinoma | 0% (43) | [166] | |
| Adrenocortical | Cortisol producing adenoma | 20% (25) | [28, 90, 171] |
| Adrenocrotical Carcinoma | 3% (40) | [80] |
An extensive list of neoplasias, flanked by gsp prevalence. Numbers in bold correspond to tumors presenting a rate over 10%. In each responsive tissue/organ, incidences in the double digits often pinpoint a single diagnosis that stands out among other virtually gsp negative tumor types. The large majority of other neoplasms are negative, for instance ref [50] analyzed 1126 cases falling within 15 diagnosis and found all negatives
Equivalent activating mutations of the Gq class proteins Gαq and Gα11 occur in virtually all blue naevi [4], and for Gαi2, in ovary and adrenal gland [5]. These activating mutations in Gα genes are analogous to oncogenic mutations in the small G protein Ras which inhibit GTP hydrolysis [6], present in about 30% of all tumors, reaching nearly 100% in specific types [7]. Such specific mutation profiles in G alpha genes, tightly linked to gain-of-function in benign neoplasia and some aggressive cancers, leaves little doubt about the selective advantage provided by the mutation to transforming cells. Yet, how this molecular mechanism contributes to the pathogenesis remains obscure.
Main body
GNAS and the activating mutation
Gαs is encoded by the GNAS locus. This is a highly complex gene with alternative promoters controlling the expression of multiple transcripts. Gαs is also regulated by genomic imprinting. In a few specific tissues, including the renal proximal tubule, the thyroid, the pituitary, and the gonads, one of the two alleles, usually the father’s, is silenced by methylation during development [8, 9].
In humans, germline transmission of Gαs activating mutations is not observed, suggesting they are embryonic lethal. However, postzygotic activating mutations are maintained through somatic mosaicism, causing McCune Albright Syndrome (MAS, MIM#174800). Predominant MAS symptoms are related to endocrine tumors (principally in pituitary, ovarian, adrenal and thyroid glands), skin pigmentation (cafè au lait) and polyostotic fibrous dysplasia.
When somatic mutations occur during adult life, the phenotype emerges independently in the same “permissive” tissues affected in MAS and displays analogous symptoms to postzygotic mosaicism. Over a quarter century ago, a direct link between GNAS gain-of-function mutations and cell transformation was established in growth hormone (GH)-secreting pituitary adenoma of acromegalic patients and in small subsets of other endocrine tumors [6, 10]. The constitutively active forms of GNAS (Fig. 2), generated by the hotspot mutations described in the previous paragraph, were collectively named gsp in recognition of their oncogenic potential.
Gsp, the microenvironment and precursor cell maturation
The hypothalamic hypophysiotropic hormone (GHRH) is the principal regulator of secretory and proliferative functions of somatotrophs [11]. Gαs directly couples the GHRH receptor to its downstream effector proteins. This fully explains the secretory properties of pituitary adenoma that are common to most gsp+ tumors discovered, including exocrine (mostly mucous) and endocrine subtypes. The last category could be extended to dysplasia involving the bone marrow, as it produces signals that regulate other organs in the body (i.e. osteocalcin targeting pancreas and testis or fibroblast growth factor-23 (FGF-23) targeting kidney [12]).
In fibrous dysplasia (FD) gsp prevalence is over 80% (Table 2 at the bottom). Patients affected by this uncommon bone disorder present fibrous tissue in place of normal bone with lesions reflecting the dysfunction of osteogenic progenitors. The protein product of gsp indirectly promotes transcription of the proto-oncogene c-fos specifically in the marrow of affected bones [13]. In addition, the expression of osteoblast-specific genes is inhibited while IL-6 expression is increased, thus promoting the action of osteoclasts [14]. As a result, unorganized and poorly mineralized woven bone is observed with the retraction of osteogenic cells from the bone surface and the formation of Sharpey’s fibers. Hematopoietic marrow is replaced by fibrotic marrow with characteristics of osteogenic progenitors that hyperproliferate but cannot completely differentiate into osteoblasts. Surprisingly, the mutation introduced in a murine zygote was transmissible. It did not produce endocrine neoplasm but did replicate human fibrous dysplasia [15]. In this animal model, the expression of mutant Gαs was achieved by viral transduction thus leaving the natural GNAS locus unaffected. Despite this bias, the experiment proved that functional up-regulation of Gαs, per se, preserves most functional aspects specific of stem cells or of osteoblastic progenitors but it compromises cell organization in the tissue.
In summary, by conditioning stem cell maturation, gsp dramatically compromises the hematopoietic microenvironment leading to abnormal histology [16].
An analogous picture is emerging in a number of rare and well-differentiated neoplastic forms, with gsp affecting cellular organization of permissive mature tissues. Acting early during tumor progression, gsp prevents the correct maturation of cell precursors possibly unbalancing intracellular signaling relevant to cell maturation. The list of gsp tumors dramatically extended in the last few years (Table 2) covering virtually all regions of the gastroenteric tract, from gastric to colorectal mucosa including accessory organs such as pancreas and liver.
As for bone and pituitary, the mutation is likely underlining a signaling pathway pivoting around cAMP (see below) that is sufficient to distort tissue differentiation or maintenance but only exceptionally to guide full malignant transformation.
The first report to identify gsp in the digestive tract describes a screen for biomarkers discriminating among pancreatic cysts [17]. These lesions are pockets filled with fluids. Quite common with aging, cysts are usually revealed by radiology exams. Cysts pose a serious clinical challenge, as occasionally they may be cancerous and justify surgical intervention. Yet, the final diagnosis can only be made by histological exam of the resected organ. Gsp was identified as a sensitive and specific marker of intraductal papillary mucinous neoplasm (IPMN). Although it remains debated if IPMN should be considered a direct precursor of pancreatic ductal adenocarcinoma (PDA), the treatment of choice is resection since patients with IPMN are at high risk to develop PDA [18].
Gsp is present in low grade IPMN and does not increase with the level of the dysplasia [19]. Discriminating IPMN in two subgroups, namely mucinous/intestinal and tubular subtypes, Hosoda et al. frequently found gsp in the first group, considered more indolent [20]. Tamura et al. reported analogous findings although no correlation was found between GNAS status and IPMN histologic grade or clinical characteristics, including patient postoperative outcomes [21].
IPMN substantially lacks significant symptoms. Nonetheless, there is a latent but significant impact of gsp and cAMP signaling in neoplastic transformation of selected microenviroments. Chronic inflammation is often characterized by gsp+ lesions. In pancreas, neoplastic lesions including IPMN are typically associated to chronic inflammation and fibrosis. Gastric foveolar metaplasia in the duodenum was considered a reactive process caused by inflammatory conditions before a genetic component was suggested by the higher gsp prevalence in transformed areas as compared to the surrounding healthy tissue. A correlation with the inflammatory process is observed in liver, where gsp was reported to define a rare subgroup of inflammatory tumors characterized by STAT3 activation mediated by GNAS directly upregulating target genes of the inflammatory IL-6-STAT3 pathway via Src [22].
Another puzzling link with inflammation is gsp incidence within a spectrum of somatic mutations (TP53, KRAS, SMAD4, CDKN2A, MLL3 and RNF43) shared by pancreatic tumors and cholangiocarcinoma developing in chronic inflammation induced by the parasite Opisthorchis viverrini [23]. Upregulated PKA synergizes with Wnt/β catenin to promote the slow progression of the tumor. However, the more aggressive outcome with Opisthorchis infection might be stimulated by the exogenous etiology of the inflammatory process.
Elevated Gαs activity can be accomplished by multiple mechanisms, or its consequences achieved by other activators on the same pathway [24]. For instance, in pituitary, gsp negative somatotroph tumors show loss of methylation and biallelic GNAS expression, that is likely to translate in increased wt Gαs expression [25, 26]. Cortisol-producing adenomas, another gsp+ tumor (Table 2), shows that alterations of components of Gαs downstream signaling pathway, namely a defective form of the regulatory subunit of PKA [27], could provide a surrogate of gsp activity in preventing normal cell maturation and tissue organization. Screens for somatic mutations in cortisol-producing adenomas demonstrated mutually exclusive mutations activating PKA and Gαs [28].
A better understanding of the pathogenesis triggered by gsp would likely provide important diagnostic tools to preoperatively predict the histological subtype for pancreatic lesions [29] and possibly others.
Functional consequences of gsp signaling
The classical signaling pathway portrayed downstream of Gαs depicts the activation of adenylyl cyclase and a consequent increase in cytosolic cAMP (Fig. 3). In turn, cAMP interacts with the regulatory subunits of PKA setting the catalytic subunits free to phosphorylate multiple targets. Consistently, PKA is up-regulated in gsp+ neoplasms such as IPMN [30] or appendiceal adenoma [31]. But PKA activation may not be the sole target of gsp signaling, as PKA mutations are not commonly reported in sporadic cancers of the GI tract. At least in tissues where gsp+ commonly occurs, this could suggest that gsp activates additional targets in parallel to PKA. On the other hand, the very rare Carney complex (CC, a heterozygous, autosomal dominant syndrome caused by mutations up-regulating PKA in all tissues), partially overlaps MAS symptoms including adrenocortical, pituitary, thyroid, skin tumors and pigmented lesions, myxomas (combined with FD symptoms is defined Mazabraud's syndrome), schwannomas, liver cancer and even IPMN [32]. Widespread PKA activation in all cells may phenocopy more focal lesions that contin a gsp+ mutation within susceptible tissue.
Fig. 3.
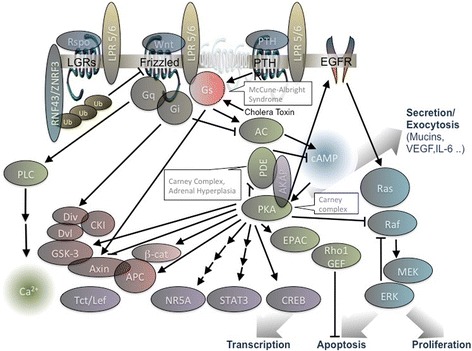
GPCR signaling is functional to almost any aspect of cell physiology including the organization of the stem cells niche. Gs coupled receptors like LGR and PTHR cooperate with FZD producing very articulated downstream signalling that is also modulated by single transmembrane domain co-receptors. Here a highly simplified scheme representing signalling intermediates described in the text. Multiple arrows indicate indirect activation. In the callout, congenital diseases associated to mutations upregulating the downstream pathway
In cortical cells renewing adrenal cortex, GNAS and PKA mutations produce benign lesions that lead to Cushing disease. Nonetheless, there are some distinctions: gsp produces micronodular disease in MAS or ACTH-independent macronodular hyperplasia, and, occasionally cortisol producing adenoma; PKA mutations produce primary pigmented nodular adrenocortical disease [33].
The molecular consequences of the upregulation of the Gs->cAMP->PKA axe on different tissue microenvironments remains to be clarified. Essential targets for activation in gsp neoplasias have not yet been identified but candidates include PKA and its substrates, such as AKAP and other scaffolding proteins, ion channels, receptor tyrosine kinases, and cAMP response element-binding (CREB) protein that drives transcription of cAMP-responsive element-containing genes [34, 35], PKA functional and structural interaction with the EGF receptor [36] could be particularly relevant. Gαs could promote cell proliferation by PKA-dependent cross-talk with the EGF pathway at multiple levels, but in particular at the level of KRAS. KRAS is one of the most frequently mutated genes in human tumors [37] and its simultaneous expression with gsp is found in certain tumor types.
Thyroid carcinoma express activating mutations of Gαs (12.5%), most commonly with activating mutations of the KRAS-paralog NRAS (8.5%). The co-occurrence of mutations simultaneously activating Gαs and KRAS is not rare in tumors like IPMN but may be coincidental, perhaps resulting from contamination of samples with cells from two independent origins.
However, in other tumor types, like pituitary [38] or muscle [37, 39], activating mutations of Ras family members are extremely rare and associated with malignant features, likely representing a late event in tumor progression. In any case, activating alleles of GNAS certainly occur in cancer independently of activating alleles of KRAS (Table 1).
Although synergy between GNAS and KRAS is not obvious, multiple studies analyzed the simultaneous presence of both oncogenes. Whereas some cases found a positive correlation [40, 41], others observed different frequencies of mutations affecting KRAS and GNAS as in colloid vs. tubular subtypes of invasive IPMN. This may suggest two separate progression pathways [19] supporting previous findings in papillary neoplasms of the bile duct [42, 43].
Cooperative signaling between Gαs and Kras proteins was recently demonstrated in a mouse model of IPMN. Conditional expression of gsp increased intracellular cAMP concentration and fibrosis, but mice developed IPMN-like lesions, with PKA activation and mucin over-production, as in human, only when mutant KRAS was co-expressed. [44] Gsp may provide a selective advantage to cell precursors that, instead of undergoing normal differentiation, become an indolent neoplasm usually described by a relatively rare and fine-focused diagnosis showing precise histologic characteristics. Retrospective analysis reported hepatobiliary and pancreatic neoplasms in about 30% of MAS patients [45–47]. Cell autonomous signals produced by the activation of both pathways are likely to converge as cAMP controls MAPK signaling at multiple levels.
Field effect (i.e. the existence of histologically abnormal microfoci within apparently normal tissue that predisposes to the occurrence of simultaneous and independent primary tumors) [43, 48] may also contribute to disease progression. For example, gsp is present in about half of IPMN samples but TCGA identified activating alleles of GNAS with wild type KRAS in only 3% of PDA. By contrast, about half of IPMN samples and upwards of 92% of PDA have activating alleles of KRAS but wild type GNAS (Table 1). This suggests gsp may contribute to chronic pancreatic disease but secondary mutations, such as KRAS activation, are necessary for transformation and tumor progression [43, 49]. Mosaic analysis is required to determine whether cell autonomous signaling or field effect explain the apparent interaction between Kras and Gαs proteins.
Tissue and cell specificity
Although Gαs is ubiquitously expressed, gsp only produces significant consequences in “permissive organs” [1, 50]. As mentioned in the first paragraph, in almost all endocrine tissues GNAS is transcribed from the maternal allele [51], therefore a predominance of gsp vs WT GNAS caused by tissue specific imprinting could partially explain why penetrance is so low elsewhere.
In adult pituitary, Gαs is monoallelically expressed from the maternal allele and indeed Hayward et al. identified the mutation in the maternal allele in 21 out of 22 GH-secreting adenoma analyzed. Yet, imprinting was relaxed for GNAS while still fully in place for the other genes belonging to the same locus (NESP55 and XLαs) [52]. In bone, the paternal allele is not differentially methylated and both alleles are expressed as in other pluripotent stem cells [9]. Imprinting by itself is therefore insufficient to explain tissue specificity.
Even in the same tissue, gsp oncogenic effects can be highly lineage cell specific and prevent, rather than promote, proliferation/survival. [53] In pituitary, the mutation emerges as a proliferatory stimulus only in GH-secreting cells while in other pituitary cells derived from common progenitors (gonadotroph or lactotroph derived) cAMP accumulation inhibits growth [26].
In the skin, gsp has never been associated with neoplastic growth. On the contrary, in an animal model of basal-cell carcinogenesis the Gs–PKA axis was recently shown to play an important tumor suppressive role. By controlling s-HH and Hippo signaling PKA defined the size of the stem cell compartment limiting self-renewal. Upregulating its signaling by exogenous gsp expression caused hair follicle stem cells exhaustion [54]. Consistently, in MAS the presence of gsp does not promote melanoma but only causes hyperpigmentation of melanocytes by upregulating tyrosinase gene [55].
Several issues should be addressed before we understand the basis of such tissue- and cell-specificity. Among them, cell lineage specific expression of Gαs effector isoforms (that include 10 adenylyl cyclases isoforms, 2 PKA catalytic subunits [26]) or the mechanisms opposing the increased cAMP levels (i.e. 11 subfamilies of phosphodiesterases [56]).
Tissue specific expression of different transcripts and alternative compartmentalization of Gαs may also differentiate the response by restricting signaling at precise subcellular locations [57, 58]. Traditionally, heterotrimeric G protein signaling has been described only at the plasma membrane. Acylated Gαs is anchored to the plasma membrane, but the post-translational modification is reversible and Gαs is also found cytosolic, particularly in the constitutively active form. [59] Recently, it became clear that upon GPCR activation, internalized receptors produce intracellular signaling by upregulating adenylyl cyclase located in the endosomal compartment [60, 61].
cAMP is a diffusible signaling molecule but is concentrated in local microdomains due to the action of phosphodiesterases [62]. Another potential mechanism to determine “cell specific” responses comes from the subcellular localization of PKA [33] mediated by AKAP that scaffolds several components of the pathway mentioned above [63] (Fig. 3). The list could continue, but summarizing, the expression of a constitutively active mutant is expected to affect the signaling network at multiple districts and unlikely to entirely mimic the stimulus of a Gs coupled receptor, nor to produce a generalized cAMP increase. The implications on cell physiology are unpredictable but may explain the different phenotypes observed in MAS vs Carney complex and the absence of phenotype in most tissues of the organism. More work is required to identify the critical steps in neoplastic transformation, beginning with the most relevant Gs mediated pathways in each tissue specific cell type (Fig. 4).
Fig. 4.
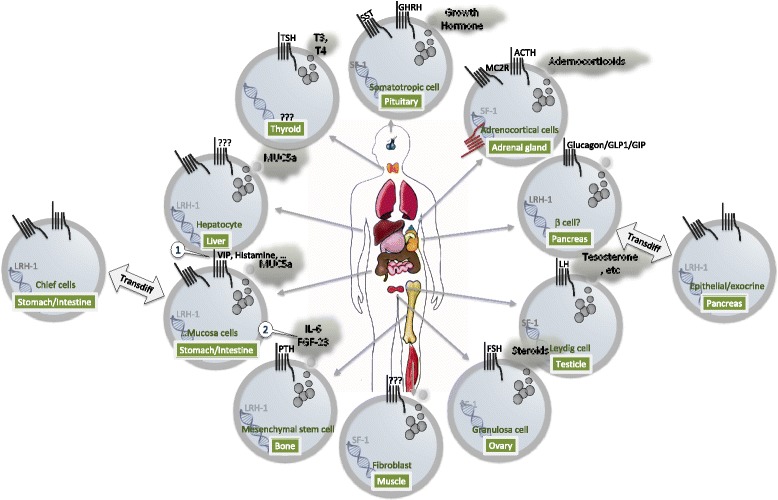
GPCR dependent production of intracellular cAMP determines secretory function in specialized cells of different exocrine organs. Among GPCRs determinant for the differentiation and function of tissues displaying the gsp+ phenotype, are secretin family receptors (glucagon, GIP, secretin, VIP, GHRH) and LGR receptors (LGR 1-8). The same pathways instrumental to zonation and differentiation are also likely acting on transcriptional programs related to the differentiation stage reached by the cells. NR5A master regulators are present in cells displaying the gsp+ phenotype (thyroid [95], osteoblast [93], somatotropic, pancreatic cells [98], hepatocytes, intestinal crypt [91], adrenocortical, Leydig and granulosa cells [99])
A GPCR perspective of the niche microenvironment
The analysis of GNAS in neoplasias of the gastrointestinal tract is shedding new light on the significance of the mutation. Gastric adenocarcinoma and most gastric tumors are gsp negative (Table 2). Conversely, gastric neoplasia of fundic gland and pyloric gland adenoma are gsp+. Both subtypes are rare and share neck cell/chief cell lineage phenotype [64]. In the normal gastric mucosa, the digestive-enzymes secreting chief cells differentiate from mucous neck cells via trans-differentiation [65]. Neoplastic cells possess characteristics of immature chief cells transitioning from mucous neck cells to serous chief cells. Probably, the same diversion from advanced phases of the differentiation program occurs in the intestine where a potential histogenic link is observed between these gastric lesions and analogous lesions appearing as gastric foveolar epithelium but commonly present in duodenal biopsy specimens [66]. Depending on the absence or presence of oxyntic glands, these lesions are classified into gastric foveolar metaplasia or gastric heterotopia. Microdissection-based analysis of three gastric heterotopia lesions identified common GNAS mutations in both components of the gastric mucosa: foveolar epithelium and oxyntic glands suggesting that the initial mutation occurred in stem cells, before they differentiated into both epithelial components and that the mutation did not cause detectable morphological changes in oxyntic glands [66]. This scenario is reminiscent of osteoblasts maturation described above.
In pituitary, gsp emerges only in somatotrophs that can also transdifferentiate from closely-related secretory cells. The emerging picture is that GNAS activating mutations allow cell survival only after a specific commitment has been undertaken and confer growth advantage only to precise lineages.
Stem cell self-renewal is critical in embryos and in adult to repopulate tissues undergoing continuous renovation. Cross-talk from multiple ligands evoking Gs-signaling orchestrates self-renewal: Wnt, sHH and R-spondins being among the best-described examples.
GPCR signaling is also involved in the differentiation of immature progenitors during cell migration. Leucine-rich repeat-containing G-protein coupled receptors (LGRs) regulate maturation of adult stem cells during renewal of papillary intestine mucosa or the patterning of hepatic lobules. In other tissues analogous processes coordinately regulate maturation, migration, and proliferation. For instance, adrenal cortex undergoes constant cell renewal as proliferation in the outer cortex continues with centripetal cell migration and differentiation according to cell location along the cortex medulla axis. Angiotensin II and ACTH signaling are crucial for modulating the size of each zone [67].
The same GPCR acting during development or in adult stem cell niches may also regulate the terminally differentiated cell. Examples include ACTH signaling to MC2R in zona fasciculata differentiation of adrenal cortex, PTH in osteoblasts, and LH gonadotropin in Leydig cells. In these cells, trophic hormones evoke Gαs-cAMP dependent growth and hormone release. Many exocrine or endocrine organs are among those affected by constitutively active Gs protein (see Table 2 and Fig. 4). Consistent with the clinical features presented by affected patients [68], glyco-proteins are secreted in IPMN [21, 42], IPNN of the bile duct [42], duodenum [66], appendix [69], cervical mucosa [70] by glands trans-differentiated towards a gastric phenotype, and hormones are secreted in pituitary (GH), thyroid (TSH), and gonads (LH and FSH).
Hence, multiple examples support the idea that gsp may bypass GPCR mediated signals to subvert differentiation and promote proliferation in progenitor cells rather than terminally differentiated cells.
cAMP and Wnt signaling pathways
Wnt has been implicated in the oncogenesis of all “permissive tissues” of the gsp mutation (thyroid [71], bone [72], pituitary [73], stomach [74], intestine [75], colon, pancreas, adrenocortical [76]). In adrenal cortex, gene expression analysis aimed at understanding cAMP tumorigenic activity indicated cell cycle and Wnt signaling as the most affected pathways [33].
Concerted cAMP and Wnt signaling is likely to represent a hallmark of gsp+ neoplasias. Cross-talk could occur at any level of the signaling cascade. Wnt activates intracellular signaling by simultaneously interacting with two co-receptors: a lipoprotein receptor-related protein (LRP5/6) and one out of ten members of the Frizzled family (FZD1-10) characterized by conserved cysteine-rich domain and seven transmembrane domains. In addition to coupling Gs and other heterotrimeric G proteins, FZDs interact with the scaffolding protein disheveled. [77] As a result, FZDs act upstream of three principal signaling mechanisms: the ‘canonical’, “planar cell polarity” and ‘calcium’ pathways [78].
Interestingly, in addition to FZDs, several classical GPCRs interact with LRP6 and activate downstream signaling. These include prostaglandin E2 and F2, M1 acetylcholine muscarinic, lysophosphatidic acid, gonadotrophin-releasing hormone and PTH type 1 (PTH1R) receptors.
In the canonical pathway, FZDs activates β-catenin by disassembly of an intracellular inhibitory complex formed by GSK3, adenomatosis polyposis coli (APC), axin and casein kinase Iα (CKIα). Wnt stimulation prevents the formation of the complex, cytoplasmic β-catenin is stabilized, and translocates to the nucleus to associate with transcription factors.
Hashimoto et al. analyzed a cohort of 20 patients affected by familial adenomatous polyposis with inherited APC mutations. Out of 6 cases with associated pyloric gland adenomas of the stomach, 5 were gsp+, all carrying APC mutations and high nuclear β-catenin expression levels [79]. All other patients and other types of lesions (foveolar adenoma and fundic gland polyps) were gsp negative.
The cooperative effect of gsp and APC inactivation was also demonstrated in animal models of intestinal tumor formation [75]. A correlation between the two pathways is supported by animal models in which increased PKA activity led to high prostaglandin E2 levels and activated Wnt signaling [33, 80].
In colon cancer without functional APC, cell proliferation is stimulated by the proinflammatory metabolite prostaglandine E2, an agonist for the Gs-coupled EP2. Under these circumstances, Gαs-GTP, activated either by the ligand or by mutation, directly interacts with the RGS homology (RH) domain of axin. The interaction sets GSK-3β free from the complex and leads to the stabilization and nuclear translocation of β-catenin [81]. Analogously, PTH1R was shown to promote the direct association of Gαs-GTP with axin [82].
Other critical points of convergence of the two pathways may involve less well characterized signaling intermediates. An emerging finding shared by ovarian development and digestive mucosa or adrenal cortex regeneration mediated by adult stem cells [83], is the involvement of a protein complex module featuring (Fig. 3):
Wnt binding to FZD and LRPs
a family of four cysteine rich proteins R-spondins (Rspo1-4) binding to LGR 4, 5 and 6
E3 ubiquitin ligases ZNRF3 and RNF43.
RNF43 and the closely related homolog ZNFR43 act as co-receptors to transduce signaling across the plasma membrane, they target cytosolic loops of FZD promoting its ubiquitination, internalization and degradation [84]. Mutations inactivating RNF43/ZNRF43 are expected to reduce FZD ubiquitination and to upregulate Wnt signaling. Similar genetic anomalies are observed in approximately 90% of colorectal cancers as well as in other cancer types, such as hepatocellular carcinomas or gastric cancers [33] including most gsp+ neoplasias: liver fluke associate cholangiocarcinoma, IPMN, ovary [85], adrenocortical carcinomas [80, 86]. In addition to mutations, epigenetics or other regulatory mechanisms reducing RNF43 expression could play an analogous effect synergizing with GNAS in the formation of IPMN and other neoplasia in the pancreas [87].
PKA was shown to phosphorylate and upregulate β-catenin signaling [88, 89] possibly in conjunction with other transcription factors. In adrenal tumors autonomously producing cortisol, gain of function mutations in β-catenin and in either Gαs or PKA were reported as mutually exclusive [90]. Other transcription factors regulated by cAMP are two nuclear receptors binding to the same consensus sequences and named steroidogenic factor 1 (SF-1/NR5A1) and liver receptor homologue 1 (LRH-1/NR5A2).
NR5A2 has an important role in the gastrointestinal system regulating functions such as bile acid and pancreatic fluid biosynthesis and secretion, glucose sensing and cell renewal in the crypt. The latter aspect is mediated by the interaction with CREB and β-catenin [91]. In addition to being expressed in the intestinal crypt cells [92], NR5A2 is also expressed by osteoblasts [93]. NR5A1 expression profile is restricted to adrenal cortex, gonads, spleen, pituitary, gonadotropes, hypothalamic VMN [94]. NR5A1 target genes are implicated at every level of the hypothalamic-pituitary-gonadal axis and gonadal or adrenal steroidognesis [94].
An interplay between Wnt and ACTH->cAMP->PKA pathways within the process of renewal and lineage conversion has been suggested in zona fasciculata development of the adrenal cortex. The mechanism portrays PKA phosphorylation of NR5A1. A subtle balance between Wnt and PKA activation determines functional zonation titrating NR5A1 and β-catenin. By this means, Wnt and Adrenocorticotropric Hormone (ACTH) stimulation determine and maintain cortex renewal. [67]
NR5A1 regulation by cAMP is also reported to direct functional differentiation of thyroid progenitor cells and to be involved in adenoma development [95]. NR5A2 was shown to play a role in intestine tumorigenesis controlling enterocytes cell cycle and inflammatory cytokines. NRA5A2 is a susceptibility locus for human pancreatic cancer. Intriguingly, in an animal model of KRAS driven neoplasia, NRA5A2 function constrains tumor initiation [96].
By compromising the correct tuning of Wnt signaling, gsp would thus distort the cellular response to surrounding signals. However, since an optimal level of Wnt/βcatenin signaling is essential to tumor formation [97], a constitutive activation of Gαs may not necessarily allow full transformation [43] explaining why the oncogenic effect is manifest only under specific circumstances in permissive tissues.
In summary, loss-of-function mutations in the Wnt signalosome that inhibit β-catenin may synergize with gsp, suggesting that.constitutive Gαs activity lowers Wnt signaling activation threshold.
Conclusions
Gsp is emerging as an oncogene acting in multifactorial transformation processes in low-grade or benign neoplasia. In the digestive tract, it is often associated with papillary morphology and high mucin secretion, reminiscent of previously described endocrine tumors. High Gαs activity may interfere with signaling in immature stages but is not sufficient to progress to invasive carcinoma. Therefore, gsp could be a marker for differential diagnosis of early neoplasia.
Acknowledgements
Not applicable.
Funding
We are grateful to AIRC for supporting grant IG 17132 to C. Bassi (to cover for fellowships, overheads and reagents).
Availability of data and materials
The results in Fig. 1 are based upon data generated by the TCGA Research Network: http://cancergenome.nih.gov/.
Abbreviations
- ACTH
Adrenocorticotropric hormone
- APC
Adenomatosis polyposis coli
- CKIα
Casein kinase Iα
- CREB
cAMP response element-binding
- FD
Fibrous dysplasia
- GH
Growth hormone
- GHRH
hypothalamic hypophysiotropic hormone/GR Releasing hormone
- IPMN
Intraductal papillary mucinous neoplasia
- LRG
Leucine-rich repeat-containing G-protein coupled receptors
- LRH-1/NR5A2
Liver receptor homologue 1
- MAS
McCune Albright Syndrome
- PDA
Pancreatic ductal adenocarcinoma
- SF-1/NR5A1
Steroidogenic factor 1
Authors’ contributions
GI, CB and TMW conceived the topic. GI, TMW and HSK prepared the figures. LG, LDC, MTV, MP, DM contributed to different paragraphs and to interpret the relevant literature. GI, TMW wrote the manuscript. All authors read, revised and approved the final manuscript.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Giulio Innamorati, Email: giulio.innamorati@univr.it.
Thomas M. Wilkie, Email: Thomas.Wilkie@UTSouthwestern.edu
Havish S. Kantheti, Email: hxk15043O@utdallas.edu
Maria Teresa Valenti, Email: mariateresa.valenti@univr.it.
Luca Dalle Carbonare, Email: luca.dallecarbonare@univr.it.
Luca Giacomello, Email: luca.giacomello@univr.it.
Marco Parenti, Email: marco.parenti@unimib.it.
Davide Melisi, Email: davide.melisi@univr.it.
Claudio Bassi, Email: claudio.bassi@univr.it.
References
- 1.O'Hayre M, Vazquez-Prado J, Kufareva I, Stawiski EW, Handel TM, Seshagiri S, Gutkind JS: The emerging mutational landscape of G proteins and G-protein-coupled receptors in cancer. Nat Rev. 2013;13(6):412–24. 10.1038/nrc3521. [DOI] [PMC free article] [PubMed]
- 2.Rasmussen SG, DeVree BT, Zou Y, Kruse AC, Chung KY, Kobilka TS, Thian FS, Chae PS, Pardon E, Calinski D, et al. Crystal structure of the beta2 adrenergic receptor-Gs protein complex. Nature. 2011;477(7366):549–555. doi: 10.1038/nature10361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Birnbaumer L. Expansion of signal transduction by G proteins. The second 15 years or so: from 3 to 16 alpha subunits plus betagamma dimers. Biochimica et biophysica acta. 2007;1768(4):772–793. doi: 10.1016/j.bbamem.2006.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Van Raamsdonk CD, Griewank KG, Crosby MB, Garrido MC, Vemula S, Wiesner T, Obenauf AC, Wackernagel W, Green G, Bouvier N, et al. Mutations in GNA11 in uveal melanoma. New Eng J Med. 2010;363(23):2191–2199. doi: 10.1056/NEJMoa1000584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lyons J, Landis CA, Harsh G, Vallar L, Grunewald K, Feichtinger H, Duh QY, Clark OH, Kawasaki E, Bourne HR, et al. Two G protein oncogenes in human endocrine tumors. Science. 1990;249(4969):655–659. doi: 10.1126/science.2116665. [DOI] [PubMed] [Google Scholar]
- 6.Landis CA, Masters SB, Spada A, Pace AM, Bourne HR, Vallar L. GTPase inhibiting mutations activate the alpha chain of Gs and stimulate adenylyl cyclase in human pituitary tumours. Nature. 1989;340(6236):692–696. doi: 10.1038/340692a0. [DOI] [PubMed] [Google Scholar]
- 7.di Magliano MP, Logsdon CD. Roles for KRAS in pancreatic tumor development and progression. Gastroenterology. 2013;144(6):1220–1229. doi: 10.1053/j.gastro.2013.01.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mantovani G, Lania AG, Spada A. GNAS imprinting and pituitary tumors. Mol Cell Endocrinol. 2010;326(1-2):15–18. doi: 10.1016/j.mce.2010.04.009. [DOI] [PubMed] [Google Scholar]
- 9.Grybek V, Aubry L, Maupetit-Mehouas S, Le Stunff C, Denis C, Girard M, Linglart A, Silve C. Methylation and transcripts expression at the imprinted GNAS locus in human embryonic and induced pluripotent stem cells and their derivatives. Stem Cell Rep. 2014;3(3):432–443. doi: 10.1016/j.stemcr.2014.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vallar L, Spada A, Giannattasio G. Altered Gs and adenylate cyclase activity in human GH-secreting pituitary adenomas. Nature. 1987;330(6148):566–568. doi: 10.1038/330566a0. [DOI] [PubMed] [Google Scholar]
- 11.Spada A, Lania A, Mantovani G. Hormonal signaling and pituitary adenomas. Neuroendocrinology. 2007;85(2):101–109. doi: 10.1159/000100440. [DOI] [PubMed] [Google Scholar]
- 12.Long F. Building strong bones: molecular regulation of the osteoblast lineage. Nat Rev Mol Cell Biol. 2012;13(1):27–38. doi: 10.1038/nrm3254. [DOI] [PubMed] [Google Scholar]
- 13.Candeliere GA, Glorieux FH, Prud'homme J, St-Arnaud R. Increased expression of the c-fos proto-oncogene in bone from patients with fibrous dysplasia. New Eng J Med. 1995;332(23):1546–1551. doi: 10.1056/NEJM199506083322304. [DOI] [PubMed] [Google Scholar]
- 14.Riminucci M, Kuznetsov SA, Cherman N, Corsi A, Bianco P, Gehron Robey P. Osteoclastogenesis in fibrous dysplasia of bone: in situ and in vitro analysis of IL-6 expression. Bone. 2003;33(3):434–442. doi: 10.1016/S8756-3282(03)00064-4. [DOI] [PubMed] [Google Scholar]
- 15.Saggio I, Remoli C, Spica E, Cersosimo S, Sacchetti B, Robey PG, Holmbeck K, Cumano A, Boyde A, Bianco P, et al. Constitutive expression of Gsalpha(R201C) in mice produces a heritable, direct replica of human fibrous dysplasia bone pathology and demonstrates its natural history. J Bone Miner Res. 2014;29(11):2357–2368. doi: 10.1002/jbmr.2267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Riminucci M, Robey PG, Saggio I, Bianco P. Skeletal progenitors and the GNAS gene: fibrous dysplasia of bone read through stem cells. J Mol Endocrinol. 2010;45(6):355–364. doi: 10.1677/JME-10-0097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dal Molin M, Matthaei H, Wu J, Blackford A, Debeljak M, Rezaee N, Wolfgang CL, Butturini G, Salvia R, Bassi C, et al. Clinicopathological correlates of activating GNAS mutations in intraductal papillary mucinous neoplasm (IPMN) of the pancreas. Ann Surg Oncol. 2013;20(12):3802–3808. doi: 10.1245/s10434-013-3096-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Adsay V, Mino-Kenudson M, Furukawa T, Basturk O, Zamboni G, Marchegiani G, Bassi C, Salvia R, Malleo G, Paiella S, et al. Pathologic evaluation and reporting of intraductal papillary mucinous neoplasms of the pancreas and other tumoral intraepithelial neoplasms of pancreatobiliary tract: recommendations of verona consensus meeting. Ann Surg. 2016;263(1):162–177. doi: 10.1097/SLA.0000000000001173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tan MC, Basturk O, Brannon AR, Bhanot U, Scott SN, Bouvier N, LaFemina J, Jarnagin WR, Berger MF, Klimstra D, et al. GNAS and KRAS Mutations Define Separate Progression Pathways in Intraductal Papillary Mucinous Neoplasm-Associated Carcinoma. J Am Coll Surg. 2015;220(5):845–854. doi: 10.1016/j.jamcollsurg.2014.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hosoda W, Sasaki E, Murakami Y, Yamao K, Shimizu Y, Yatabe Y. GNAS mutation is a frequent event in pancreatic intraductal papillary mucinous neoplasms and associated adenocarcinomas. Virchows Arch. 2015;466(6):665–74. doi: 10.1007/s00428-015-1751-6. [DOI] [PubMed] [Google Scholar]
- 21.Tamura K, Ohtsuka T, Matsunaga T, Kimura H, Watanabe Y, Ideno N, Aso T, Miyazaki T, Ohuchida K, Takahata S, et al. Assessment of clonality of multisegmental main duct intraductal papillary mucinous neoplasms of the pancreas based on GNAS mutation analysis. Surgery. 2015;157(2):277–284. doi: 10.1016/j.surg.2014.09.013. [DOI] [PubMed] [Google Scholar]
- 22.Nault JC, Fabre M, Couchy G, Pilati C, Jeannot E, Tran Van Nhieu J, Saint-Paul MC, De Muret A, Redon MJ, Buffet C, et al. GNAS-activating mutations define a rare subgroup of inflammatory liver tumors characterized by STAT3 activation. J Hepatol. 2012;56(1):184–191. doi: 10.1016/j.jhep.2011.07.018. [DOI] [PubMed] [Google Scholar]
- 23.Ong CK, Subimerb C, Pairojkul C, Wongkham S, Cutcutache I, Yu W, McPherson JR, Allen GE, Ng CC, Wong BH, et al. Exome sequencing of liver fluke-associated cholangiocarcinoma. Nat Genet. 2012;44(6):690–693. doi: 10.1038/ng.2273. [DOI] [PubMed] [Google Scholar]
- 24.Kan Z, Jaiswal BS, Stinson J, Janakiraman V, Bhatt D, Stern HM, Yue P, Haverty PM, Bourgon R, Zheng J, et al. Diverse somatic mutation patterns and pathway alterations in human cancers. Nature. 2010;466(7308):869–873. doi: 10.1038/nature09208. [DOI] [PubMed] [Google Scholar]
- 25.Picard C, Silvy M, Gerard C, Buffat C, Lavaque E, Figarella-Branger D, Dufour H, Gabert J, Beckers A, Brue T, et al. Gs alpha overexpression and loss of Gs alpha imprinting in human somatotroph adenomas: association with tumor size and response to pharmacologic treatment. Int J Cancer. 2007;121(6):1245–1252. doi: 10.1002/ijc.22816. [DOI] [PubMed] [Google Scholar]
- 26.Peverelli E, Mantovani G, Lania AG, Spada A. cAMP in the pituitary: an old messenger for multiple signals. J Mol Endocrinol. 2014, 52(1):R67–77. [DOI] [PubMed]
- 27.Mantovani G, Lania AG, Bondioni S, Peverelli E, Pedroni C, Ferrero S, Pellegrini C, Vicentini L, Arnaldi G, Bosari S, et al. Different expression of protein kinase A (PKA) regulatory subunits in cortisol-secreting adrenocortical tumors: relationship with cell proliferation. Exp Cell Res. 2008;314(1):123–130. doi: 10.1016/j.yexcr.2007.08.024. [DOI] [PubMed] [Google Scholar]
- 28.Thiel A, Reis AC, Haase M, Goh G, Schott M, Willenberg HS, Scholl UI. PRKACA mutations in cortisol-producing adenomas and adrenal hyperplasia: a single-center study of 60 cases. Eur J Endocrinol. 2015;172(6):677–685. doi: 10.1530/EJE-14-1113. [DOI] [PubMed] [Google Scholar]
- 29.Kanda M, Knight S, Topazian M, Syngal S, Farrell J, Lee J, Kamel I, Lennon AM, Borges M, Young A, et al. Mutant GNAS detected in duodenal collections of secretin-stimulated pancreatic juice indicates the presence or emergence of pancreatic cysts. Gut. 2013;62(7):1024–1033. doi: 10.1136/gutjnl-2012-302823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Furukawa T, Kuboki Y, Tanji E, Yoshida S, Hatori T, Yamamoto M, Shibata N, Shimizu K, Kamatani N, Shiratori K. Whole-exome sequencing uncovers frequent GNAS mutations in intraductal papillary mucinous neoplasms of the pancreas. Sci Rep. 2011;1:161. doi: 10.1038/srep00161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Alakus H, Babicky ML, Ghosh P, Yost S, Jepsen K, Dai Y, Arias A, Samuels ML, Mose ES, Schwab RB, et al. Genome-wide mutational landscape of mucinous carcinomatosis peritonei of appendiceal origin. Genome Med. 2014;6(5):43. doi: 10.1186/gm559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gaujoux S, Tissier F, Ragazzon B, Rebours V, Saloustros E, Perlemoine K, Vincent-Dejean C, Meurette G, Cassagnau E, Dousset B, et al. Pancreatic ductal and acinar cell neoplasms in Carney complex: a possible new association. J Clin Endocrinol Metabol. 2011;96(11):E1888–E1895. doi: 10.1210/jc.2011-1433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Almeida MQ, Stratakis CA. How does cAMP/protein kinase A signaling lead to tumors in the adrenal cortex and other tissues? Mol Cell Endocrinol. 2011;336(1-2):162–168. doi: 10.1016/j.mce.2010.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bossis I, Voutetakis A, Bei T, Sandrini F, Griffin KJ, Stratakis CA. Protein kinase A and its role in human neoplasia: the Carney complex paradigm. Endocr Relat Cancer. 2004;11(2):265–280. doi: 10.1677/erc.0.0110265. [DOI] [PubMed] [Google Scholar]
- 35.Pearce LR, Komander D, Alessi DR. The nuts and bolts of AGC protein kinases. Nat Rev Mol Cell Biol. 2010;11(1):9–22. doi: 10.1038/nrm2822. [DOI] [PubMed] [Google Scholar]
- 36.Tortora G, Ciardiello F. Protein kinase A as target for novel integrated strategies of cancer therapy. Ann N Y Acad Sci. 2002;968:139–147. doi: 10.1111/j.1749-6632.2002.tb04332.x. [DOI] [PubMed] [Google Scholar]
- 37.Cox AD, Fesik SW, Kimmelman AC, Luo J, Der CJ. Drugging the undruggable RAS: mission possible? Nat Rev Drug Discov. 2014;13(11):828–851. doi: 10.1038/nrd4389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Spada A, Lania A. Growth factors and human pituitary adenomas. Mol Cell Endocrinol. 2002;197(1-2):63–68. doi: 10.1016/S0303-7207(02)00279-4. [DOI] [PubMed] [Google Scholar]
- 39.Willems SM, Mohseny AB, Balog C, Sewrajsing R, Briaire-de Bruijn IH, Knijnenburg J, Cleton-Jansen AM, Sciot R, Fletcher CD, Deelder AM, et al. Cellular/intramuscular myxoma and grade I myxofibrosarcoma are characterized by distinct genetic alterations and specific composition of their extracellular matrix. J Cell Mol Med. 2009;13(7):1291–1301. doi: 10.1111/j.1582-4934.2009.00747.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Singhi AD, Davison JM, Choudry HA, Pingpank JF, Ahrendt SA, Holtzman MP, Zureikat AH, Zeh HJ, Ramalingam L, Mantha G, et al. GNAS is frequently mutated in both low-grade and high-grade disseminated appendiceal mucinous neoplasms but does not affect survival. Human Pathol. 2014;45(8):1737–1743. doi: 10.1016/j.humpath.2014.04.018. [DOI] [PubMed] [Google Scholar]
- 41.Matsubara A, Sekine S, Kushima R, Ogawa R, Taniguchi H, Tsuda H, Kanai Y. Frequent GNAS and KRAS mutations in pyloric gland adenoma of the stomach and duodenum. J Pathol. 2013;229(4):579–587. doi: 10.1002/path.4153. [DOI] [PubMed] [Google Scholar]
- 42.Sasaki M, Matsubara T, Nitta T, Sato Y, Nakanuma Y. GNAS and KRAS mutations are common in intraductal papillary neoplasms of the bile duct. PloS one. 2013;8(12):e81706. doi: 10.1371/journal.pone.0081706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Innamorati G, Valenti MT, Giacomello L, Dalle Carbonare L, Bassi C. GNAS Mutations: drivers or co-pilots? yet, promising diagnostic biomarkers. Trends Cancer. 2016;2(6):282–285. doi: 10.1016/j.trecan.2016.05.005. [DOI] [PubMed] [Google Scholar]
- 44.Taki K, Ohmuraya M, Tanji E, Komatsu H, Hashimoto D, Semba K, Araki K, Kawaguchi Y, Baba H, Furukawa T. GNAS and Kras cooperate to promote murine pancreatic tumorigenesis recapitulating human intraductal papillary mucinous neoplasm. Oncogene. 2016;35(18):2407–12. doi: 10.1038/onc.2015.294. [DOI] [PubMed] [Google Scholar]
- 45.Gaujoux S, Salenave S, Ronot M, Rangheard AS, Cros J, Belghiti J, Sauvanet A, Ruszniewski P, Chanson P. Hepatobiliary and pancreatic neoplasms in patients with McCune-Albright syndrome. J Clin Endocrinol Metabol. 2014;99(1):E97–E101. doi: 10.1210/jc.2013-1823. [DOI] [PubMed] [Google Scholar]
- 46.Parvanescu A, Cros J, Ronot M, Hentic O, Grybek V, Couvelard A, Levy P, Chanson P, Ruszniewski P, Sauvanet A, et al. Lessons from McCune-Albright syndrome-associated intraductal papillary mucinous neoplasms: : GNAS-activating mutations in pancreatic carcinogenesis. JAMA Surg. 2014;149(8):858–862. doi: 10.1001/jamasurg.2014.535. [DOI] [PubMed] [Google Scholar]
- 47.Zacharin M, Bajpai A, Chow CW, Catto-Smith A, Stratakis C, Wong MW, Scott R. Gastrointestinal polyps in McCune Albright syndrome. J Med Genet. 2011;48(7):458–461. doi: 10.1136/jmg.2010.086330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chai H, Brown RE. Field effect in cancer-an update. Ann Clin Lab Sci. 2009;39(4):331–337. [PubMed] [Google Scholar]
- 49.Pea A, Yu J, Rezaee N, Luchini C, He J, Dal Molin M, Griffin JF, Fedor H, Fesharakizadeh S, Salvia R, et al. Targeted DNA sequencing reveals patterns of local progression in the pancreatic remnant following resection of intraductal papillary mucinous neoplasm (IPMN) of the pancreas. Ann Surg. 2017;266(1):133–41. doi: 10.1097/SLA.0000000000001817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Je EM, An CH, Chung YJ, Yoo NJ, Lee SH. GNAS mutation affecting codon 201 is rare in most human tumors. Pathol Oncol Res. 2015;21(3):859–60. doi: 10.1007/s12253-015-9919-6. [DOI] [PubMed] [Google Scholar]
- 51.Mantovani G, Ballare E, Giammona E, Beck-Peccoz P, Spada A. The gsalpha gene: predominant maternal origin of transcription in human thyroid gland and gonads. J Clin Endocrinol Metabol. 2002;87(10):4736–4740. doi: 10.1210/jc.2002-020183. [DOI] [PubMed] [Google Scholar]
- 52.Hayward BE, Barlier A, Korbonits M, Grossman AB, Jacquet P, Enjalbert A, Bonthron DT. Imprinting of the G(s)alpha gene GNAS1 in the pathogenesis of acromegaly. J Clin Invest. 2001;107(6):R31–R36. doi: 10.1172/JCI11887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.NWAS OFFERMANNS. Mammalian G proteins and their cell type specific functions. Physiol Rev. 2005;85:1159–1204. doi: 10.1152/physrev.00003.2005. [DOI] [PubMed] [Google Scholar]
- 54.Iglesias-Bartolome R, Torres D, Marone R, Feng X, Martin D, Simaan M, Chen M, Weinstein LS, Taylor SS, Molinolo AA, et al. Inactivation of a Galpha(s)-PKA tumour suppressor pathway in skin stem cells initiates basal-cell carcinogenesis. Nat Cell Biol. 2015;17(6):793–803. doi: 10.1038/ncb3164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kim IS, Kim ER, Nam HJ, Chin MO, Moon YH, Oh MR, Yeo UC, Song SM, Kim JS, Uhm MR, et al. Activating mutation of GS alpha in McCune-Albright syndrome causes skin pigmentation by tyrosinase gene activation on affected melanocytes. Horm Res. 1999;52(5):235–240. doi: 10.1159/000023467. [DOI] [PubMed] [Google Scholar]
- 56.Lania A, Persani L, Ballare E, Mantovani S, Losa M, Spada A. Constitutively active Gs alpha is associated with an increased phosphodiesterase activity in human growth hormone-secreting adenomas. J Clin Endocrinol Metabol. 1998;83(5):1624–1628. doi: 10.1210/jcem.83.5.4814. [DOI] [PubMed] [Google Scholar]
- 57.Irannejad R, von Zastrow M. GPCR signaling along the endocytic pathway. Curr Opin Cell Biol. 2014;27:109–116. doi: 10.1016/j.ceb.2013.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Aydin C, Aytan N, Mahon MJ, Tawfeek HA, Kowall NW, Dedeoglu A, Bastepe M, Extralarge XL. (Alpha)s (XXL(alpha)s), a variant of stimulatory G protein alpha-subunit (Gs(alpha)), is a distinct, membrane-anchored GNAS product that can mimic Gs(alpha) Endocrinology. 2009;150(8):3567–3575. doi: 10.1210/en.2009-0318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Levis MJ, Bourne HR. Activation of the alpha subunit of Gs in intact cells alters its abundance, rate of degradation, and membrane avidity. J Cell Biol. 1992;119(5):1297–1307. doi: 10.1083/jcb.119.5.1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cabrera-Vera TM, Vanhauwe J, Thomas TO, Medkova M, Preininger A, Mazzoni MR, Hamm HE. Insights into G protein structure, function, and regulation. Endocr Rev. 2003;24(6):765–781. doi: 10.1210/er.2000-0026. [DOI] [PubMed] [Google Scholar]
- 61.Marrari Y, Crouthamel M, Irannejad R, Wedegaertner PB. Assembly and trafficking of heterotrimeric G proteins. Biochemistry. 2007;46(26):7665–7677. doi: 10.1021/bi700338m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Baillie GS. Compartmentalized signalling: spatial regulation of cAMP by the action of compartmentalized phosphodiesterases. FEBS J. 2009;276(7):1790–1799. doi: 10.1111/j.1742-4658.2009.06926.x. [DOI] [PubMed] [Google Scholar]
- 63.Zaccolo M. Spatial control of cAMP signalling in health and disease. Curr Opin Pharmacol. 2011;11(6):649–655. doi: 10.1016/j.coph.2011.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kushima R, Sekine S, Matsubara A, Taniguchi H, Ikegami M, Tsuda H. Gastric adenocarcinoma of the fundic gland type shares common genetic and phenotypic features with pyloric gland adenoma. Pathol Int. 2013;63(6):318–325. doi: 10.1111/pin.12070. [DOI] [PubMed] [Google Scholar]
- 65.Ota H, Yamaguchi D, Iwaya M, Kobayashi M, Tateiwa N, Iwaya Y, Momose M, Uehara T. Principal cells in gastric neoplasia of fundic gland (chief cell predominant) type show characteristics of immature chief cells. Pathol Int. 2015;65(4):202–204. doi: 10.1111/pin.12244. [DOI] [PubMed] [Google Scholar]
- 66.Matsubara A, Ogawa R, Suzuki H, Oda I, Taniguchi H, Kanai Y, Kushima R, Sekine S. Activating GNAS and KRAS mutations in gastric foveolar metaplasia, gastric heterotopia, and adenocarcinoma of the duodenum. Br J Cancer. 2015;112(8):1398–1404. doi: 10.1038/bjc.2015.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Drelon C, Berthon A, Mathieu M, Martinez A, Val P. Adrenal cortex tissue homeostasis and zonation: a WNT perspective. Mol Cell Endocrinol. 2015;408:156–164. doi: 10.1016/j.mce.2014.12.014. [DOI] [PubMed] [Google Scholar]
- 68.Nishikawa G, Sekine S, Ogawa R, Matsubara A, Mori T, Taniguchi H, Kushima R, Hiraoka N, Tsuta K, Tsuda H, et al. Frequent GNAS mutations in low-grade appendiceal mucinous neoplasms. Br J Cancer. 2013;108(4):951–958. doi: 10.1038/bjc.2013.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Sio TT, Mansfield AS, Grotz TE, Graham RP, Molina JR, Que FG, Miller RC. Concurrent MCL1 and JUN amplification in pseudomyxoma peritonei: a comprehensive genetic profiling and survival analysis. J Hum Genet. 2014;59(3):124–128. doi: 10.1038/jhg.2013.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ota H, Harada O, Uehara T, Hayama M, Ishii K. Aberrant expression of TFF1, TFF2, and PDX1 and their diagnostic value in lobular endocervical glandular hyperplasia. Am J Clin Pathol. 2011;135(2):253–261. doi: 10.1309/AJCPQMAO3PW4OGOF. [DOI] [PubMed] [Google Scholar]
- 71.Sastre-Perona A, Santisteban P. Role of the wnt pathway in thyroid cancer. Front Endocrinol (Lausanne) 2012;3:31. doi: 10.3389/fendo.2012.00031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Regard JB, Cherman N, Palmer D, Kuznetsov SA, Celi FS, Guettier J-M, Chen M, Bhattacharyya N, Wess J, Coughlin SR, et al. Wnt/beta-catenin signaling is differentially regulated by Galpha proteins and contributes to fibrous dysplasia. Proc Nat Acad Sci U S A. 2011;108(50):20101–20106. doi: 10.1073/pnas.1114656108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Gueorguiev M, Grossman AB. Pituitary gland and beta-catenin signaling: from ontogeny to oncogenesis. Pituitary. 2009;12(3):245–255. doi: 10.1007/s11102-008-0147-x. [DOI] [PubMed] [Google Scholar]
- 74.Nomura R, Saito T, Mitomi H, Hidaka Y, Lee SY, Watanabe S, Yao T. GNAS mutation as an alternative mechanism of activation of the Wnt/beta-catenin signaling pathway in gastric adenocarcinoma of the fundic gland type. Hum Pathol. 2014;45(12):2488–2496. doi: 10.1016/j.humpath.2014.08.016. [DOI] [PubMed] [Google Scholar]
- 75.Wilson CH, McIntyre RE, Arends MJ, Adams DJ. The activating mutation R201C in GNAS promotes intestinal tumourigenesis in Apc(Min/+) mice through activation of Wnt and ERK1/2 MAPK pathways. Oncogene. 2010;29(32):4567–4575. doi: 10.1038/onc.2010.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Tissier F, Cavard C, Groussin L, Perlemoine K, Fumey G, Hagnere AM, Rene-Corail F, Jullian E, Gicquel C, Bertagna X, et al. Mutations of beta-catenin in adrenocortical tumors: activation of the Wnt signaling pathway is a frequent event in both benign and malignant adrenocortical tumors. Cancer Res. 2005;65(17):7622–7627. doi: 10.1158/0008-5472.CAN-05-0593. [DOI] [PubMed] [Google Scholar]
- 77.Nichols AS, Floyd DH, Bruinsma SP, Narzinski K, Baranski TJ. Frizzled receptors signal through G proteins. Cell Signal. 2013;25(6):1468–1475. doi: 10.1016/j.cellsig.2013.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Koval A, Purvanov V, Egger-Adam D, Katanaev VL. Yellow submarine of the Wnt/Frizzled signaling: submerging from the G protein harbor to the targets. Biochem Pharmacol. 2011;82(10):1311–1319. doi: 10.1016/j.bcp.2011.06.005. [DOI] [PubMed] [Google Scholar]
- 79.Hashimoto T, Ogawa R, Matsubara A, Taniguchi H, Sugano K, Ushiama M, Yoshida T, Kanai Y, Sekine S: Familial adenomatous polyposis-associated and sporadic pyloric gland adenomas of the upper gastrointestinal tract share common genetic features. Histopathology. 2015;67(5):689-98. 10.1111/his.12705. [DOI] [PubMed]
- 80.Juhlin CC, Goh G, Healy JM, Fonseca AL, Scholl UI, Stenman A, Kunstman JW, Brown TC, Overton JD, Mane SM, et al. Whole-exome sequencing characterizes the landscape of somatic mutations and copy number alterations in adrenocortical carcinoma. J Clin Endocrinol Metabol. 2015;100(3):E493–E502. doi: 10.1210/jc.2014-3282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Castellone MD, Teramoto H, Williams BO, Druey KM, Gutkind JS. Prostaglandin E2 promotes colon cancer cell growth through a Gs-axin-beta-catenin signaling axis. Science. 2005;310(5753):1504–1510. doi: 10.1126/science.1116221. [DOI] [PubMed] [Google Scholar]
- 82.Wan M, Li J, Herbst K, Zhang J, Yu B, Wu X, Qiu T, Lei W, Lindvall C, Williams BO, et al. LRP6 mediates cAMP generation by G protein-coupled receptors through regulating the membrane targeting of Galpha(s) Sci Signal. 2011;4(164):ra15. doi: 10.1126/scisignal.2001464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Chassot AA, Gillot I, Chaboissier MC. R-spondin1, WNT4, and the CTNNB1 signaling pathway: strict control over ovarian differentiation. Reproduction. 2014;148(6):R97–110. doi: 10.1530/REP-14-0177. [DOI] [PubMed] [Google Scholar]
- 84.Zebisch M, Jones EY. ZNRF3/RNF43 - A direct linkage of extracellular recognition and E3 ligase activity to modulate cell surface signalling. Prog Biophys Mol Biol. 2015;118(3):112–8. doi: 10.1016/j.pbiomolbio.2015.04.006. [DOI] [PubMed] [Google Scholar]
- 85.de Lau W, Peng WC, Gros P, Clevers H. The R-spondin/Lgr5/Rnf43 module: regulator of Wnt signal strength. Genes Dev. 2014;28(4):305–316. doi: 10.1101/gad.235473.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Assie G, Letouze E, Fassnacht M, Jouinot A, Luscap W, Barreau O, Omeiri H, Rodriguez S, Perlemoine K, Rene-Corail F, et al. Integrated genomic characterization of adrenocortical carcinoma. Nat Genet. 2014;46(6):607–612. doi: 10.1038/ng.2953. [DOI] [PubMed] [Google Scholar]
- 87.Sakamoto H, Kuboki Y, Hatori T, Yamamoto M, Sugiyama M, Shibata N, Shimizu K, Shiratori K, Furukawa T. Clinicopathological significance of somatic RNF43 mutation and aberrant expression of ring finger protein 43 in intraductal papillary mucinous neoplasms of the pancreas. Mod Pathol. 2015;28(2):261–267. doi: 10.1038/modpathol.2014.98. [DOI] [PubMed] [Google Scholar]
- 88.Taurin S, Sandbo N, Qin Y, Browning D, Dulin NO. Phosphorylation of beta-catenin by cyclic AMP-dependent protein kinase. J Biol Chem. 2006;281(15):9971–9976. doi: 10.1074/jbc.M508778200. [DOI] [PubMed] [Google Scholar]
- 89.Hino S, Tanji C, Nakayama KI, Kikuchi A. Phosphorylation of beta-catenin by cyclic AMP-dependent protein kinase stabilizes beta-catenin through inhibition of its ubiquitination. Mol Cell Biol. 2005;25(20):9063–9072. doi: 10.1128/MCB.25.20.9063-9072.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Goh G, Scholl UI, Healy JM, Choi M, Prasad ML, Nelson-Williams C, Kunstman JW, Korah R, Suttorp AC, Dietrich D, et al. Recurrent activating mutation in PRKACA in cortisol-producing adrenal tumors. Nat Genet. 2014;46(6):613–617. doi: 10.1038/ng.2956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Botrugno OA, Fayard E, Annicotte JS, Haby C, Brennan T, Wendling O, Tanaka T, Kodama T, Thomas W, Auwerx J, et al. Synergy between LRH-1 and beta-catenin induces G1 cyclin-mediated cell proliferation. Mol Cell. 2004;15(4):499–509. doi: 10.1016/j.molcel.2004.07.009. [DOI] [PubMed] [Google Scholar]
- 92.Mueller M, Cima I, Noti M, Fuhrer A, Jakob S, Dubuquoy L, Schoonjans K, Brunner T. The nuclear receptor LRH-1 critically regulates extra-adrenal glucocorticoid synthesis in the intestine. J Exp Med. 2006;203(9):2057–2062. doi: 10.1084/jem.20060357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Riancho JA, Liu Y, Sainz J, Garcia-Perez MA, Olmos JM, Bolado-Carrancio A, Valero C, Perez-Lopez J, Cano A, Yang T, et al. Nuclear receptor NR5A2 and bone: gene expression and association with bone mineral density. Eur J Endocrinol. 2012;166(1):69–75. doi: 10.1530/EJE-11-0571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Ruggiero C, Doghman M, Lalli E. How genomic studies have improved our understanding of the mechanisms of transcriptional regulation by NR5A nuclear receptors. Mol Cell Endocrinol. 2015;408:138–144. doi: 10.1016/j.mce.2014.10.022. [DOI] [PubMed] [Google Scholar]
- 95.Suzuki M, Egashira N, Kajiya H, Minematsu T, Takekoshi S, Tahara S, Sanno N, Teramoto A, Osamura RY. ACTH and alpha-subunit are co-expressed in rare human pituitary corticotroph cell adenomas proposed to originate from ACTH-committed early pituitary progenitor cells. Endocr Pathol. 2008;19(1):17–26. doi: 10.1007/s12022-008-9014-6. [DOI] [PubMed] [Google Scholar]
- 96.von Figura G, JPt M, Wright CV, Hebrok M. Nr5a2 maintains acinar cell differentiation and constrains oncogenic Kras-mediated pancreatic neoplastic initiation. Gut. 2014;63(4):656–664. doi: 10.1136/gutjnl-2012-304287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Krausova M, Korinek V. Wnt signaling in adult intestinal stem cells and cancer. Cell Signal. 2014;26(3):570–579. doi: 10.1016/j.cellsig.2013.11.032. [DOI] [PubMed] [Google Scholar]
- 98.Hale MA, Swift GH, Hoang CQ, Deering TG, Masui T, Lee YK, Xue J, MacDonald RJ. The nuclear hormone receptor family member NR5A2 controls aspects of multipotent progenitor cell formation and acinar differentiation during pancreatic organogenesis. Development. 2014;141(16):3123–3133. doi: 10.1242/dev.109405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Yazawa T, Imamichi Y, Miyamoto K, Khan MR, Uwada J, Umezawa A, Taniguchi T. Regulation of Steroidogenesis, development, and cell differentiation by steroidogenic factor-1 and liver receptor homolog-1. Zoolog Sci. 2015;32(4):323–330. doi: 10.2108/zs140237. [DOI] [PubMed] [Google Scholar]
- 100.Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6(269):pl1. doi: 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2(5):401–404. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Tonacchera M, Vitti P, Agretti P, Ceccarini G, Perri A, Cavaliere R, Mazzi B, Naccarato AG, Viacava P, Miccoli P, et al. Functioning and nonfunctioning thyroid adenomas involve different molecular pathogenetic mechanisms. J Clin Endocrinol Metabol. 1999;84(11):4155–4158. doi: 10.1210/jcem.84.11.6157. [DOI] [PubMed] [Google Scholar]
- 103.Russo D, Arturi F, Wicker R, Chazenbalk GD, Schlumberger M, DuVillard JA, Caillou B, Monier R, Rapoport B, Filetti S, et al. Genetic alterations in thyroid hyperfunctioning adenomas. J Clin Endocrinol Metabol. 1995;80(4):1347–1351. doi: 10.1210/jcem.80.4.7714109. [DOI] [PubMed] [Google Scholar]
- 104.O'Sullivan C, Barton CM, Staddon SL, Brown CL, Lemoine NR. Activating point mutations of the gsp oncogene in human thyroid adenomas. Mol Carcinog. 1991;4(5):345–349. doi: 10.1002/mc.2940040503. [DOI] [PubMed] [Google Scholar]
- 105.Pauws E, Tummers RF, Ris-Stalpers C, de Vijlder JJ, Voute T. Absence of activating mutations in ras and gsp oncogenes in a cohort of nine patients with sporadic pediatric thyroid tumors. Med Pediatr Oncol. 2001;36(6):630–634. doi: 10.1002/mpo.1140. [DOI] [PubMed] [Google Scholar]
- 106.Park C, Yang I, Woo J, Kim S, Kim J, Kim Y, Sohn S, Kim E, Lee M, Park H, et al. Somatostatin (SRIF) receptor subtype 2 and 5 gene expression in growth hormone-secreting pituitary adenomas: the relationship with endogenous srif activity and response to octreotide. Endocrine J. 2004;51(2):227–236. doi: 10.1507/endocrj.51.227. [DOI] [PubMed] [Google Scholar]
- 107.Kim HJ, Kim MS, Park YJ, Kim SW, Park DJ, Park KS, Kim SY, Cho BY, Lee HK, Jung HW, et al. Prevalence of Gs alpha mutations in Korean patients with pituitary adenomas. J Endocrinol. 2001;168(2):221–226. doi: 10.1677/joe.0.1680221. [DOI] [PubMed] [Google Scholar]
- 108.Yasufuku-Takano J, Takano K, Morita K, Takakura K, Teramoto A, Fujita T. Does the prevalence of gsp mutations in GH-secreting pituitary adenomas differ geographically or racially? Prevalence of gsp mutations in Japanese patients revisited. Clin Endocrinol. 2006;64(1):91–96. doi: 10.1111/j.1365-2265.2005.02423.x. [DOI] [PubMed] [Google Scholar]
- 109.Fougner SL, Casar-Borota O, Heck A, Berg JP, Bollerslev J. Adenoma granulation pattern correlates with clinical variables and effect of somatostatin analogue treatment in a large series of patients with acromegaly. Clin Endocrinol. 2012;76(1):96–102. doi: 10.1111/j.1365-2265.2011.04163.x. [DOI] [PubMed] [Google Scholar]
- 110.Larkin S, Reddy R, Karavitaki N, Cudlip S, Wass J, Ansorge O. Granulation pattern, but not GSP or GHR mutation, is associated with clinical characteristics in somatostatin-naive patients with somatotroph adenomas. Eur J Endocrinol. 2013;168(4):491–499. doi: 10.1530/EJE-12-0864. [DOI] [PubMed] [Google Scholar]
- 111.Barlier A, Gunz G, Zamora AJ, Morange-Ramos I, Figarella-Branger D, Dufour H, Enjalbert A, Jaquet P. Pronostic and therapeutic consequences of Gs alpha mutations in somatotroph adenomas. J Clin Endocrinol Metabol. 1998;83(5):1604–1610. doi: 10.1210/jcem.83.5.4797. [DOI] [PubMed] [Google Scholar]
- 112.Goto Y, Kinoshita M, Oshino S, Arita H, Kitamura T, Otsuki M, Shimomura I, Yoshimine T, Saitoh Y. Gsp mutation in acromegaly and its influence on TRH-induced paradoxical GH response. Clin Endocrinol. 2014;80(5):714–719. doi: 10.1111/cen.12336. [DOI] [PubMed] [Google Scholar]
- 113.Yoshimoto K, Iwahana H, Fukuda A, Sano T, Itakura M. Rare mutations of the Gs alpha subunit gene in human endocrine tumors. Mutation detection by polymerase chain reaction-primer-introduced restriction analysis. Cancer. 1993;72(4):1386–1393. doi: 10.1002/1097-0142(19930815)72:4<1386::AID-CNCR2820720439>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- 114.Yang I, Park S, Ryu M, Woo J, Kim S, Kim J, Kim Y, Choi Y. Characteristics of gsp-positive growth hormone-secreting pituitary tumors in Korean acromegalic patients. Eur J Endocrinol. 1996;134(6):720–726. doi: 10.1530/eje.0.1340720. [DOI] [PubMed] [Google Scholar]
- 115.Johnson MC, Codner E, Eggers M, Mosso L, Rodriguez JA, Cassorla F. Gps mutations in Chilean patients harboring growth hormone-secreting pituitary tumors. J Pediatr Endocrinol Metabol. 1999;12(3):381–387. doi: 10.1515/JPEM.1999.12.3.381. [DOI] [PubMed] [Google Scholar]
- 116.Hosoi E, Yokogoshi Y, Hosoi E, Horie H, Sano T, Yamada S, Saito S. Analysis of the Gs alpha gene in growth hormone-secreting pituitary adenomas by the polymerase chain reaction-direct sequencing method using paraffin-embedded tissues. Acta Endocrinol (Copenh) 1993;129(4):301–306. doi: 10.1530/acta.0.1290301. [DOI] [PubMed] [Google Scholar]
- 117.Freda PU, Chung WK, Matsuoka N, Walsh JE, Kanibir MN, Kleinman G, Wang Y, Bruce JN, Post KD. Analysis of GNAS mutations in 60 growth hormone secreting pituitary tumors: correlation with clinical and pathological characteristics and surgical outcome based on highly sensitive GH and IGF-I criteria for remission. Pituitary. 2007;10(3):275–282. doi: 10.1007/s11102-007-0058-2. [DOI] [PubMed] [Google Scholar]
- 118.Metzler M, Luedecke DK, Saeger W, Grueters A, Haberl H, Kiess W, Repp R, Rascher W, Doetsch J. Low prevalence of Gs alpha mutations in somatotroph adenomas of children and adolescents. Cancer Genet Cytogenet. 2006;166(2):146–151. doi: 10.1016/j.cancergencyto.2005.11.001. [DOI] [PubMed] [Google Scholar]
- 119.Mendoza V, Sosa E, Espinosa-de-Los-Monteros AL, Salcedo M, Guinto G, Cheng S, Sandoval C, Mercado M. GSPalpha mutations in Mexican patients with acromegaly: potential impact on long term prognosis. Growth Horm IGF Res. 2005;15(1):28–32. doi: 10.1016/j.ghir.2004.10.001. [DOI] [PubMed] [Google Scholar]
- 120.Kan B, Esapa C, Sipahi T, Nacar C, Ozer F, Sayhan NB, Kaynar MY, Sarioglu AC, Harris PE. G protein mutations in pituitary tumors: a study on Turkish patients. Pituitary. 2003;6(2):75–80. doi: 10.1023/B:PITU.0000004797.89592.5e. [DOI] [PubMed] [Google Scholar]
- 121.Corbetta S, Ballare E, Mantovani G, Lania A, Losa M, Di Blasio AM, Spada A. Somatostatin receptor subtype 2 and 5 in human GH-secreting pituitary adenomas: analysis of gene sequence and mRNA expression. Eur J Clin Invest. 2001;31(3):208–214. doi: 10.1046/j.1365-2362.2001.00786.x. [DOI] [PubMed] [Google Scholar]
- 122.Buchfelder M, Fahlbusch R, Merz T, Symowski H, Adams EF. Clinical correlates in acromegalic patients with pituitary tumors expressing GSP oncogenes. Pituitary. 1999;1(3-4):181–185. doi: 10.1023/A:1009905131334. [DOI] [PubMed] [Google Scholar]
- 123.Ballare E, Mantovani S, Lania A, Di Blasio AM, Vallar L, Spada A. Activating mutations of the Gs alpha gene are associated with low levels of Gs alpha protein in growth hormone-secreting tumors. J Clin Endocrinol Metabol. 1998;83(12):4386–4390. doi: 10.1210/jcem.83.12.5354. [DOI] [PubMed] [Google Scholar]
- 124.Adams EF, Brockmeier S, Friedmann E, Roth M, Buchfelder M, Fahlbusch R. Clinical and biochemical characteristics of acromegalic patients harboring gsp-positive and gsp-negative pituitary tumors. Neurosurgery. 1993;33(2):198–203. doi: 10.1227/00006123-199308000-00003. [DOI] [PubMed] [Google Scholar]
- 125.Drews RT, Gravel RA, Collu R. Identification of G protein alpha subunit mutations in human growth hormone (GH)- and GH/prolactin-secreting pituitary tumors by single-strand conformation polymorphism (SSCP) analysis. Mol Cell Endocrinol. 1992;87(1-3):125–129. doi: 10.1016/0303-7207(92)90240-7. [DOI] [PubMed] [Google Scholar]
- 126.Landis CA, Harsh G, Lyons J, Davis RL, McCormick F, Bourne HR. Clinical characteristics of acromegalic patients whose pituitary tumors contain mutant Gs protein. J Clin Endocrinol Metabol. 1990;71(6):1416–1420. doi: 10.1210/jcem-71-6-1416. [DOI] [PubMed] [Google Scholar]
- 127.Walther I, Walther BM, Chen Y, Petersen I. Analysis of GNAS1 mutations in myxoid soft tissue and bone tumors. Pathol Res Pract. 2014;210(1):1–4. doi: 10.1016/j.prp.2013.09.003. [DOI] [PubMed] [Google Scholar]
- 128.Delaney D, Diss TC, Presneau N, Hing S, Berisha F, Idowu BD, O'Donnell P, Skinner JA, Tirabosco R, Flanagan AM. GNAS1 mutations occur more commonly than previously thought in intramuscular myxoma. Mod Pathol. 2009;22(5):718–724. doi: 10.1038/modpathol.2009.32. [DOI] [PubMed] [Google Scholar]
- 129.Mantovani G, Bondioni S, Corbetta S, Menicanti L, Rubino B, Peverelli E, Labarile P, Dall'Asta C, Ambrosi B, Beck-Peccoz P, et al. Analysis of GNAS1 and PRKAR1A gene mutations in human cardiac myxomas not associated with multiple endocrine disorders. J Endocrinol Invest. 2009;32(6):501–504. doi: 10.1007/BF03346496. [DOI] [PubMed] [Google Scholar]
- 130.DeMarco L, Stratakis CA, Boson WL, Jakbovitz O, Carson E, Andrade LM, Amaral VF, Rocha JL, Choursos GP, Nordenskjold M, et al. Sporadic cardiac myxomas and tumors from patients with Carney complex are not associated with activating mutations of the Gs alpha gene. Hum Genet. 1996;98(2):185–188. doi: 10.1007/s004390050187. [DOI] [PubMed] [Google Scholar]
- 131.Shi RR, Li XF, Zhang R, Chen Y, Li TJ. GNAS mutational analysis in differentiating fibrous dysplasia and ossifying fibroma of the jaw. Mod Pathol. 2013;26(8):1023–1031. doi: 10.1038/modpathol.2013.31. [DOI] [PubMed] [Google Scholar]
- 132.Tabareau-Delalande F, Collin C, Gomez-Brouchet A, Decouvelaere AV, Bouvier C, Larousserie F, Marie B, Delfour C, Aubert S, Rosset P, et al. Diagnostic value of investigating GNAS mutations in fibro-osseous lesions: a retrospective study of 91 cases of fibrous dysplasia and 40 other fibro-osseous lesions. Mod Pathol. 2013;26(7):911–921. doi: 10.1038/modpathol.2012.223. [DOI] [PubMed] [Google Scholar]
- 133.Weinstein LS, Shenker A, Gejman PV, Merino MJ, Friedman E, Spiegel AM. Activating mutations of the stimulatory G protein in the McCune-Albright syndrome. N Eng J Med. 1991;325(24):1688–95. [DOI] [PubMed]
- 134.Carter JM, Inwards CY, Jin L, Evers B, Wenger DE, Oliveira AM, Fritchie KJ. Activating GNAS mutations in parosteal osteosarcoma. Am J Surg Pathol. 2014;38(3):402–409. doi: 10.1097/PAS.0000000000000144. [DOI] [PubMed] [Google Scholar]
- 135.Salinas-Souza C, De Andrea C, Bihl M, Kovac M, Pillay N, Forshew T, Gutteridge A, Ye H, Amary MF, Tirabosco R et al: GNAS mutations are not detected in parosteal and low-grade central osteosarcomas. Mod Pathol. 2015;28(10):1336–42. 10.1038/modpathol.2015.91. [DOI] [PubMed]
- 136.Pollandt K, Engels C, Kaiser E, Werner M, Delling G. Gsalpha gene mutations in monostotic fibrous dysplasia of bone and fibrous dysplasia-like low-grade central osteosarcoma. Virchows Arch. 2001;439(2):170–175. doi: 10.1007/s004280100453. [DOI] [PubMed] [Google Scholar]
- 137.Tabareau-Delalande F, Collin C, Larousserie F, Bouvier C, Gomez-Brouchet A, Aubert S, Guinebretiere JM, Decouvelaere AV, de Muret A, Pages JC, et al. Comments on Carter et al's “activating GNAS mutations in parosteal osteosarcoma”. Am J Surg Pathol. 2015;39(7):1010–1013. doi: 10.1097/PAS.0000000000000461. [DOI] [PubMed] [Google Scholar]
- 138.Liang Q, Wei M, Hodge L, Fanburg-Smith JC, Nelson A, Miettinen M, Foss RD, Wang G. Quantitative analysis of activating alpha subunit of the G protein (Gsalpha) mutation by pyrosequencing in fibrous dysplasia and other bone lesions. J Mol Diagn. 2011;13(2):137–142. doi: 10.1016/j.jmoldx.2010.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Bejar R, Stevenson K, Abdel-Wahab O, Galili N, Nilsson B, Garcia-Manero G, Kantarjian H, Raza A, Levine RL, Neuberg D, et al. Clinical effect of point mutations in myelodysplastic syndromes. N Eng J Med. 2011;364(26):2496–2506. doi: 10.1056/NEJMoa1013343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Konoplev S, Yin CC, Kornblau SM, Kantarjian HM, Konopleva M, Andreeff M, Lu G, Zuo Z, Luthra R, Medeiros LJ, et al. Molecular characterization of de novo Philadelphia chromosome-positive acute myeloid leukemia. Leuk Lymphoma. 2013;54(1):138–144. doi: 10.3109/10428194.2012.701739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Baker BW, Norton JD. Analysis of mutations in the Gs protein alpha subunit gene in human leukaemia. Leuk Res. 1992;16(5):485–489. doi: 10.1016/0145-2126(92)90174-6. [DOI] [PubMed] [Google Scholar]
- 142.Kalfa N, Lumbroso S, Boulle N, Guiter J, Soustelle L, Costa P, Chapuis H, Baldet P, Sultan C. Activating mutations of Gsalpha in kidney cancer. J Urol. 2006;176(3):891–895. doi: 10.1016/j.juro.2006.04.023. [DOI] [PubMed] [Google Scholar]
- 143.Qiu T, Guo H, Zhao H, Wang L, Zhang Z. Next-generation sequencing for molecular diagnosis of lung adenocarcinoma specimens obtained by fine needle aspiration cytology. Sci Rep. 2015;5:11317. doi: 10.1038/srep11317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Wu J, Matthaei H, Maitra A, Dal Molin M, Wood LD, Eshleman JR, Goggins M, Canto MI, Schulick RD, Edil BH, et al. Recurrent GNAS mutations define an unexpected pathway for pancreatic cyst development. Sci Transl Med. 2011;3(92):92ra66. doi: 10.1126/scitranslmed.3002543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Takano S, Fukasawa M, Maekawa S, Kadokura M, Miura M, Shindo H, Takahashi E, Sato T, Enomoto N. Deep sequencing of cancer-related genes revealed GNAS mutations to be associated with intraductal papillary mucinous neoplasms and its main pancreatic duct dilation. PloS one. 2014;9(6):e98718. doi: 10.1371/journal.pone.0098718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Yamaguchi H, Kuboki Y, Hatori T, Yamamoto M, Shimizu K, Shiratori K, Shibata N, Shimizu M, Furukawa T. The discrete nature and distinguishing molecular features of pancreatic intraductal tubulopapillary neoplasms and intraductal papillary mucinous neoplasms of the gastric type, pyloric gland variant. J Pathol. 2013;231(3):335–341. doi: 10.1002/path.4242. [DOI] [PubMed] [Google Scholar]
- 147.Ideno N, Ohtsuka T, Matsunaga T, Kimura H, Watanabe Y, Tamura K, Aso T, Aishima S, Miyasaka Y, Ohuchida K, et al. Clinical significance of GNAS mutation in intraductal papillary mucinous neoplasm of the pancreas with concomitant pancreatic ductal adenocarcinoma. Pancreas. 2015;44(2):311–320. doi: 10.1097/MPA.0000000000000258. [DOI] [PubMed] [Google Scholar]
- 148.Kuboki Y, Shimizu K, Hatori T, Yamamoto M, Shibata N, Shiratori K, Furukawa T. Molecular biomarkers for progression of intraductal papillary mucinous neoplasm of the pancreas. Pancreas. 2015;44(2):227–235. doi: 10.1097/MPA.0000000000000253. [DOI] [PubMed] [Google Scholar]
- 149.Matthaei H, Wu J, Dal Molin M, Shi C, Perner S, Kristiansen G, Lingohr P, Kalff JC, Wolfgang CL, Kinzler KW, et al. GNAS Sequencing Identifies IPMN-specific Mutations in a Subgroup of Diminutive Pancreatic Cysts Referred to as “Incipient IPMNs”. Am J Surg Pathol. 2014;38(3):360–363. doi: 10.1097/PAS.0000000000000117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Kanda M, Matthaei H, Wu J, Hong S-M, Yu J, Borges M, Hruban RH, Maitra A, Kinzler K, Vogelstein B, et al. Presence of somatic mutations in most early-stage pancreatic intraepithelial neoplasia. Gastroenterology. 2012;142(4):730–733. doi: 10.1053/j.gastro.2011.12.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Matthaei H, Wu J, Dal Molin M, Debeljak M, Lingohr P, Katabi N, Klimstra DS, Adsay NV, Eshleman JR, Schulick RD, et al. GNAS codon 201 mutations are uncommon in intraductal papillary neoplasms of the bile duct. HPB. 2012;14(10):677–683. doi: 10.1111/j.1477-2574.2012.00504.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Tsai JH, Yuan RH, Chen YL, Liau JY, Jeng YM. GNAS Is frequently mutated in a specific subgroup of intraductal papillary neoplasms of the bile duct. Am J Surg Pathol. 2013;37(12):1862–1870. doi: 10.1097/PAS.0b013e3182986bb5. [DOI] [PubMed] [Google Scholar]
- 153.Schlitter AM, Born D, Bettstetter M, Specht K, Kim-Fuchs C, Riener MO, Jeliazkova P, Sipos B, Siveke JT, Terris B, et al. Intraductal papillary neoplasms of the bile duct: stepwise progression to carcinoma involves common molecular pathways. Mod Pathol. 2014;27(1):73–86. doi: 10.1038/modpathol.2013.112. [DOI] [PubMed] [Google Scholar]
- 154.Hsu M, Sasaki M, Igarashi S, Sato Y, Nakanuma Y. KRAS and GNAS mutations and p53 overexpression in biliary intraepithelial neoplasia and intrahepatic cholangiocarcinomas. Cancer. 2013;119(9):1669–1674. doi: 10.1002/cncr.27955. [DOI] [PubMed] [Google Scholar]
- 155.Walther BM, Walther I, Chen Y, Petersen I. GNAS1 mutation analysis in gastrointestinal tumors. Folia Histochem Cytobiol. 2014;52(2):90–95. doi: 10.5603/FHC.2014.0011. [DOI] [PubMed] [Google Scholar]
- 156.Yamada M, Sekine S, Ogawa R, Taniguchi H, Kushima R, Tsuda H, Kanai Y. Frequent activating GNAS mutations in villous adenoma of the colorectum. J Pathol. 2012;228(1):113–118. doi: 10.1002/path.4012. [DOI] [PubMed] [Google Scholar]
- 157.Fecteau RE, Lutterbaugh J, Markowitz SD, Willis J, Guda K. GNAS mutations identify a set of right-sided, RAS mutant, villous colon cancers. PloS one. 2014;9(1):e87966. doi: 10.1371/journal.pone.0087966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Sekine S, Ogawa R, Oshiro T, Kanemitsu Y, Taniguchi H, Kushima R, Kanai Y. Frequent lack of GNAS mutations in colorectal adenocarcinoma associated with GNAS-mutated villous adenoma. Genes Chromosomes Cancer. 2014;53(4):366–372. doi: 10.1002/gcc.22147. [DOI] [PubMed] [Google Scholar]
- 159.Idziaszczyk S, Wilson CH, Smith CG, Adams DJ, Cheadle JP. Analysis of the frequency of GNAS codon 201 mutations in advanced colorectal cancer. Cancer Genet Cytogenet. 2010;202(1):67–69. doi: 10.1016/j.cancergencyto.2010.04.023. [DOI] [PubMed] [Google Scholar]
- 160.Jang S, Chun SM, Hong SM, Sung CO, Park H, Kang HJ, Kim KP, Lee YJ, Yu E. High throughput molecular profiling reveals differential mutation patterns in intrahepatic cholangiocarcinomas arising in chronic advanced liver diseases. Mod Pathol. 2014;27(5):731–739. doi: 10.1038/modpathol.2013.194. [DOI] [PubMed] [Google Scholar]
- 161.Calderaro J, Labrune P, Morcrette G, Rebouissou S, Franco D, Prevot S, Quaglia A, Bedossa P, Libbrecht L, Terracciano L, et al. Molecular characterization of hepatocellular adenomas developed in patients with glycogen storage disease type I. J Hepatol. 2013;58(2):350–357. doi: 10.1016/j.jhep.2012.09.030. [DOI] [PubMed] [Google Scholar]
- 162.Liu X, Mody K, de Abreu FB, Pipas JM, Peterson JD, Gallagher TL, Suriawinata AA, Ripple GH, Hourdequin KC, Smith KD, et al. Molecular profiling of appendiceal epithelial tumors using massively parallel sequencing to identify somatic mutations. Clin Chem. 2014;60(7):1004–1011. doi: 10.1373/clinchem.2014.225565. [DOI] [PubMed] [Google Scholar]
- 163.Hara K, Saito T, Hayashi T, Yimit A, Takahashi M, Mitani K, Takahashi M, Yao T. A mutation spectrum that includes GNAS, KRAS and TP53 may be shared by mucinous neoplasms of the appendix. Pathol Res Pract. 2015;211(9):657–664. doi: 10.1016/j.prp.2015.06.004. [DOI] [PubMed] [Google Scholar]
- 164.Nummela P, Saarinen L, Thiel A, Jarvinen P, Lehtonen R, Lepisto A, Jarvinen H, Aaltonen LA, Hautaniemi S, Ristimaki A. Genomic profile of pseudomyxoma peritonei analyzed using next-generation sequencing and immunohistochemistry. Int J Cancer. 2015;136(5):E282–E289. doi: 10.1002/ijc.29245. [DOI] [PubMed] [Google Scholar]
- 165.Libe R, Fratticci A, Lahlou N, Jornayvaz FR, Tissier F, Louiset E, Guibourdenche J, Vieillefond A, Zerbib M, Bertherat J. A rare cause of hypertestosteronemia in a 68-year-old patient: a leydig cell tumor due to a somatic GNAS (Guanine Nucleotide-Binding Protein, Alpha-Stimulating Activity Polypeptide 1)-activating mutation. J Androl. 2012;33(4):578–584. doi: 10.2164/jandrol.111.013441. [DOI] [PubMed] [Google Scholar]
- 166.Matsubara A, Sekine S, Ogawa R, Yoshida M, Kasamatsu T, Tsuda H, Kanai Y: Lobular endocervical glandular hyperplasia is a neoplastic entity with frequent activating GNAS mutations. Am J Surg Pathol. 2014;38(3):370–6. 10.1097/PAS.0000000000000093. [DOI] [PubMed]
- 167.Kalfa N, Ecochard A, Patte C, Duvillard P, Audran F, Pienkowski C, Thibaud E, Brauner R, Lecointre C, Plantaz D, et al. Activating mutations of the stimulatory g protein in juvenile ovarian granulosa cell tumors: a new prognostic factor? J Clin Endocrinol Metabol. 2006;91(5):1842–1847. doi: 10.1210/jc.2005-2710. [DOI] [PubMed] [Google Scholar]
- 168.Ryland GL, Hunter SM, Doyle MA, Caramia F, Li J, Rowley SM, Christie M, Allan PE, Stephens AN, Bowtell DD, et al. Mutational landscape of mucinous ovarian carcinoma and its neoplastic precursors. Genome Med. 2015;7(1):87. doi: 10.1186/s13073-015-0210-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Ligtenberg MJ, Siers M, Themmen AP, Hanselaar TG, Willemsen W, Brunner HG. Analysis of mutations in genes of the follicle-stimulating hormone receptor signaling pathway in ovarian granulosa cell tumors. J Clin Endocrinol Metabol. 1999;84(6):2233–2234. doi: 10.1210/jcem.84.6.5739. [DOI] [PubMed] [Google Scholar]
- 170.Fragoso MC, Latronico AC, Carvalho FM, Zerbini MC, Marcondes JA, Araujo LM, Lando VS, Frazzatto ET, Mendonca BB, Villares SM. Activating mutation of the stimulatory G protein (gsp) as a putative cause of ovarian and testicular human stromal Leydig cell tumors. J Clin Endocrinol Metabol. 1998;83(6):2074–2078. doi: 10.1210/jcem.83.6.4847. [DOI] [PubMed] [Google Scholar]
- 171.Fragoso MC, Domenice S, Latronico AC, Martin RM, Pereira MA, Zerbini MC, Lucon AM, Mendonca BB. Cushing’s syndrome secondary to adrenocorticotropin-independent macronodular adrenocortical hyperplasia due to activating mutations of GNAS1 gene. J Clin Endocrinol Metabol. 2003;88(5):2147–2151. doi: 10.1210/jc.2002-021362. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The results in Fig. 1 are based upon data generated by the TCGA Research Network: http://cancergenome.nih.gov/.