

Abstract
Background
Hyperglycemia is common in extremely low gestational age newborns (ELGAN) and is associated with increased mortality and morbidity, including abnormal neurodevelopment. Hippocampus-mediated cognitive deficits are common in this population, but the specific effects of hyperglycemia on the developing hippocampus are not known.
Methods
The objective of this study was to determine the acute and long-term effects of hyperglycemia on the developing hippocampus in neonatal rats using a streptozotocin (STZ)-induced model of hyperglycemia. STZ was injected on postnatal day (P) 2, and littermates in the control group were injected with an equivalent volume of citrate buffer. The acute effects of hyperglycemia on markers of oxidative stress, inflammatory cytokines, microglial activation, and reactive astrocytosis in the hippocampus were determined in the brain tissue collected on P6. The long-term effects on hippocampus-mediated behavior and hippocampal dendrite structure were determined on P90.
Results
On P6, the transcript and protein expression of markers of oxidative stress and inflammatory cytokines, including the CXCL10/CXCR3 pathway, were upregulated in the hyperglycemia group. Histological evaluation revealed microglial activation and astrocytosis. The long-term assessment on P90 demonstrated abnormal performance in Barnes maze neurobehavioral testing and altered dendrite structure in the hippocampus of formerly hyperglycemic rats.
Conclusions
Neonatal hyperglycemia induces CXCL10/CXCR3 signaling, microglial activation, and astrocytosis in the rat hippocampus and alters long-term synaptogenesis and behavior. These results may explain the hippocampus-specific cognitive deficits common in ELGAN who experience neonatal hyperglycemia.
Keywords: Prematurity, Blood glucose, Inflammation, Oxidative stress, Neurodevelopment, Hyperglycemia, ELGAN, Hippocampus
Background
Hyperglycemia (blood glucose concentration > 150 mg/dL [> 8 mmol/L]) is seen during the first 1 to 2 weeks after birth in 30–80% of extremely low gestational age newborns (ELGAN; birth at < 28 weeks of gestation) [1–3]. A combination of relative hypoinsulinism, peripheral insulin resistance, and glucose infusion for nutrition is responsible for hyperglycemia [4]. Hyperglycemia in this preterm population is associated with increased risk of morbidity and mortality in the neonatal period [5], growth deceleration, and abnormal neurodevelopment that persist until at least 2 years of life [2, 3].
The effects of hyperglycemia on the developing hippocampus are relatively unknown. The hippocampus is central to recognition memory and is vulnerable to injury during a variety of perinatal conditions [6]. Hippocampus-mediated long-term cognitive deficits are common in the ELGAN population. [7] Structural and functional hippocampal deficits have been reported in children with hyperglycemia due to early onset type 1 diabetes (T1D) [8], but whether hyperglycemia has a role in the hippocampal deficits seen in the ELGAN population is not known. Our recent study in rats demonstrates that recurrent hyperglycemia in the neonatal period leads to long-term abnormalities in hippocampal neurochemistry and synaptic architecture that are suggestive of suppressed neuronal activity [9].
In a previous study, using a neonatal rat model, we have demonstrated that recurrent episodes of hypoinsulinemic hyperglycemia upregulate poly(ADP-ribose) polymerase-1 (PARP-1) expression in the cerebral cortex [10], likely due to oxidative stress [11]. There was co-activation of nuclear factor of kappa light polypeptide gene enhancer in B cells (NF-κB) as well as microglial activation. NF-κB is a transcription factor that modulates inflammatory response at the site of injury. [12, 13] The interplay between PARP-1 and NF-κB has been implicated in the upregulation of inflammatory cytokines and microglial activation [14].
Among the chemokines in the brain, C-X-C motif chemokine ligand 10 (CXCL10) and its cognate receptor C-X-C motif chemokine receptor 3 (CXCR3) are important because of the role of CXCL10/CXCR3 signaling in neuronal function and synaptic activity [15]. Both the neurons and glia express CXCL10 and CXCR3 [16, 17]. In hippocampal cell cultures, increased CXCL10 expression alters GABA and glutamate receptor expression and synaptic network activity [18]. Increased CXCL10 levels in the CSF are associated with cognitive deficits in adult humans with CNS infections (e.g., encephalitis) and neurodegenerative conditions (e.g., Alzheimer’s disease) [17, 19–21].
CXCL10/CXCR3 signaling has been implicated in the pathogenesis of T1D, including pancreatic β cell destruction [22–24]. Serum CXCL10 levels are increased in human adults with T1D [25, 26] and in the prediabetes stage in the non-obese diabetic mouse model of T1D [27]. Conversely, hyperglycemia causes CXCL10 release from human monocytes via the Toll-like receptor (TLR) 2 and TLR 4 pathway [26]. Administration of NF-κB inhibitor abrogates CXCL10 release. Whether hyperglycemia affects CXCL10/CXCR3 signaling in the developing hippocampus is not known.
The objective of our study was to determine the acute and long-term effects of hyperglycemia on the developing hippocampus in neonatal rats. The acute effects were determined by measuring the expression of CXCL10/CXCR3, PARP-1, and NF-κB, and their downstream targets, including apoptosis-inducing factor (AIF) and B cell lymphoma 2 (Bcl-2), and NMDA receptor (NR2B) expression. The long-term effects were determined by evaluating hippocampal dendritic architecture using microtubule-associated protein (MAP-2) immunohistochemistry, postsynaptic density protein (PSD) 95 protein expression, and performance in a hippocampus-mediated behavioral test (Barnes maze) at adulthood.
Methods
Animals
Experiments were performed with the approval of the Institutional Animal Care and Use Committee of the University of Minnesota. Male and female Wistar rats were studied from postnatal day 1 (P1) to P90 (adulthood). Pregnant dams were purchased (Charles River Laboratories, Wilmington, MA) and housed under standard laboratory conditions on ad lib diet and 12:12 h light and dark cycles.
Induction of neonatal hyperglycemia
Hyperglycemia was induced using streptozotocin (STZ) (Enzo Life Sciences, Farmingdale, NY), a model that was adapted from previous studies [28–31]. STZ has relative selective cytotoxicity against pancreatic β cells and leads to hypoinsulinemic hyperglycemia. STZ administration in the neonatal period leads to transient hyperglycemia with hypoinsulinism [31], similar to the condition in the human ELGAN. Pilot studies were performed to determine the STZ dose that would cause moderate hyperglycemia comparable to that experienced by the human ELGAN (blood glucose concentration, 270 mg/dL; 15 mmol/L) [1]. Doses between 50 and 100 mg/kg were trialed. The 100-mg/kg dose led to the most consistent hyperglycemia without significant mortality. This dose was used in subsequent experiments. Immediately prior to injection, STZ was dissolved in a citrate buffer (pH 4.5) to achieve a final concentration of 15 mg/mL. Pups were separated from their dams, and 100 mg/kg of STZ was intraperitoneally injected on postnatal day (P) 2 (total volume injected = 50–60 μL). P2 was chosen to represent the ELGAN population, as this time point in rat pups corresponds approximately to the human brain development equivalent to a 24- to 26-week gestation preterm human infant [32, 33]. Littermates in the control group were injected with an equivalent volume of citrate buffer alone. Pups were returned to their dams after injection. Animals were weighed, and glucose measurements were determined in the tail vein blood samples (Accu-Chek®) daily until P6, and on P14 and at adulthood.
To confirm that the effects are due to hyperglycemia and not the administered STZ, a separate set of rats was subjected to recurrent hypoinsulinemic hyperglycemia twice daily from P3 to P12 using a previously described model from our lab [10]. Briefly, the pups were pretreated with octreotide (100 μg/kg, s.c.) to block insulin release from the pancreas, followed by 30% dextrose in a dose of 3 g/kg s.c. Littermates in the control group were injected with octreotide followed by an equivalent volume of 0.9% saline [10]. This model results in hyperglycemia (mean blood glucose ~ 220 mg/dL) of 2-h duration [10].
Tissue preparation
Rats subjected to STZ-induced hyperglycemia and control rats were killed on P6 or P90 using sodium pentobarbital (100 mg/kg, i.p.). Animals subjected to the recurrent hyperglycemia were killed on P13. The brain was removed, and the hippocampus was dissected on ice, then flash-frozen using liquid nitrogen, and stored at − 80 °C until analysis. Rats used for histochemical analysis underwent in situ transcardial perfusion-fixation with paraformaldehyde before brain removal as in our previous studies [10, 34].
Quantitative real-time PCR
Transcript expression in the hippocampus was determined using commercial primers (TaqMan, Applied Biosystems, Carlsbad, CA) and a gene expression kit (TaqMan, Applied Biosystems, Carlsbad, CA) as previously described from our lab [34]. Samples (n = 6–12/group) were assayed in duplicate using ribosomal protein s18 as control. Some experiments were repeated using GAPDH as control to confirm the observed results.
Western blot analysis
Protein concentrations of CXCR3 and PSD95 in the hippocampus were determined using 20 μg of hippocampal homogenate and primary antibodies against CXCR3 and PSD95 (1:1000; Abcam, Cambridge, MA) and β-actin (1:5000; Abcam) using previously published methods (n = 4–6/group) [34]. Following incubation with corresponding fluorescent secondary antibodies, the membranes were imaged (Odyssey Infrared Imaging System; LI-COR Biosciences, Lincoln, NE). The density of the target protein relative to β-actin was determined.
Immunohistochemistry
The coronal brain sections (20 μm) corresponding to 0.8 to 2.6 mm anterior to the interaural line in an age-appropriate rat brain atlas [35] were obtained using a cryostat. The choice was based on our recent study demonstrating neurochemical and structural changes in the hippocampus of the corresponding region following recurrent hyperglycemia [9]. On P6, microglial activation in the hippocampus was determined using CD-11 immunohistochemistry (1:300; Abcam) as previously described [10]. A chromagen kit (Vector Laboratories) was used to visualize the protein/antibody complex, and all CD11 cells in the hippocampus were counted at × 10 magnification using ImageJ software (ImageJ, US National Institutes of Health, Bethesda, MD, USA; https://imagej.nih.gov/ij/) and the total number of cells/mm2 was determined (n = 6 per group, two slides per animal). Anti-S100β protein (1:200; Abcam) was used to quantify immunoreactive astrocytes in the hippocampus, which were counted in a similar manner at × 20 magnification (n = 6 per group, two slides per animal) as the microglia.
The cellular origin of CXCL10 (Bioss 1:100) was determined by colocalizing with GFAP, NeuN, and Iba1. The cellular origin of CXCR3 (Novus 1:100) was determined by colocalizing with NeuN (neuron, Millipore 1:100), Iba1 (microglia, Wako 1:1000), and GFAP (astrocyte, Novus 1:200) using previously described methods [34].
On P90, the dendritic structure of the hippocampus was determined by staining for microtubule-associated protein-2 (MAP-2) as previously described [36]. The mean integrated density of MAP-2 fluorescence in the CA1 region was determined using a software program (Adobe Photoshop, San Jose, CA) as described in our previous study [37].
Neurobehavioral testing
Barnes maze
STZ (n = 18) and control (n = 21) rats were tested for hippocampal function on P90 for a total of 7 days during the light phase (6 am to 6 pm) on the Barnes maze [38]. The elevated platform was circular, black, 0.95 m in diameter, with 18 evenly spaced 10-cm holes placed around the edge. A black escape chamber served as the goal location and was placed under one of the holes (see Fig. 1). This goal location remained in the same position in relation to the spatial cues of the room throughout the entire experiment. All training and probe trials were video recorded, and the animal was tracked using a software program (AnyMaze, Stoelting Co. Wooddale, IL).
Fig. 1.
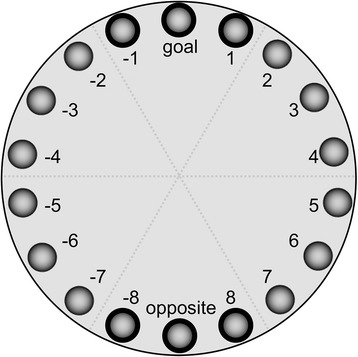
Barnes maze schematic demonstrating 18 evenly spaced holes around the perimeter of the platform with a goal location indicated at the top and an opposite hole at the bottom of the maze
The rats were habituated to the maze prior to testing, during which time they were allowed to explore the maze and were guided to the goal location to enter the escape box. Rats were trained on the Barnes maze for 3 days followed by 1 day of probe testing (probe 1). Then they were trained for an additional 2 days followed by probe 2. The rats in both groups were trained for three trials per day with an inter-trial period of 5 min. The maze and goal location chamber were cleaned with 70% ethanol between each trial to eliminate odor cues. Each trial began with the lights off in the room and the animal placed in the center of the maze beneath an opaque container.
To begin the trial, bright overhead lights were turned on to serve as an aversive stimulus, the opaque container was removed, and the rat was given 3 min to explore the maze and find the goal location [39]. If the rat did not reach the goal location during the allotted 3 min, it was guided to enter the goal location after the trial period has ended. Once it entered the dark chamber, the lights were turned off and the animal was allowed to stay in the goal location while the maze was being cleaned for the next trial. Probe testing was a single 90-s trial similar to the previous training days, except the goal location was covered (instead of the hole leading to the dark chamber). Head pokes into each of the 18 holes as the animal explored the maze were measured. During probe testing, head pokes into the goal hole and the holes adjacent to the goal hole (goal hole, hole 1, and hole − 1; Fig. 1) were grouped together and categorized as “good guesses” while those near the opposite side (opposite hole, hole 8, and hole − 8; Fig. 1) were categorized as “bad guesses.” For each animal and each probe session, error rates were calculated as (good guess pokes)/(total head pokes) or (bad guess pokes)/(total head pokes) in order to capture polar spatial memory retention.
Statistical analysis
All data are reported as mean ± SEM. The control and hyperglycemia groups were compared using two-tailed unpaired t tests (GraphPad Prism, La Jolla, CA). Barnes maze data were analyzed using mixed-model multifactorial ANOVA with day and treatment as independent factors and rats as a random event variable. Statistical significance was set at alpha < 0.05.
Results
STZ induces moderate neonatal hyperglycemia and growth restriction in rats
Hyperglycemia was observed by 24 h after the STZ injection with a mean blood glucose concentration of 265.4 ± 14.5 mg/dL on P3. Hyperglycemia was sustained until P6, at which time the mean glucose value was 267.1 ± 27.6 mg/dL. The mean blood glucose concentration from P3 to P6 was 144.1 ± 2.5 mg/dL in the control group. All these intergroup differences were statistically significant, p < 0.01. By P14, the difference between STZ and control animals’ blood glucose values were less pronounced but still significant (185.0 ± 4.5 mg/dL vs. 163.0 ± 2.3 mg/dL, p = 0.002).
By chance, the pups randomized to the STZ group were 12% lighter on P2 compared with those randomized to the control group (p < 0.05). Subsequently, the animals in the STZ group continued to remain 20–24% lighter until P6 (Table 1).
Table 1.
Mean daily weights comparing control and STZ groups
| Postnatal day | Control group mean body weight (g) | STZ group mean body weight (g) | Difference between groups (%) |
|---|---|---|---|
| 2 | 8.3 ± 0.14 | 7.3 ± 0.14 | 12.0 |
| 3 | 10.3 ± 0.27 | 7.8 ± 0.19 | 24.3 |
| 4 | 12.2 ± 0.24 | 9.8 ± 0.24 | 19.7 |
| 5 | 13.8 ± 0.26 | 11.0 ± 0.25 | 20.3 |
| 6 | 15.9 ± 0.34 | 12.2 ± 0.23 | 23.3 |
Values are mean ± SEM, n = 21–65/group
On P90, there was no difference between the control and STZ groups in blood glucose levels after overnight fasting (123.0 ± 7.5 mg/dL vs. 142.4 ± 9.3 mg/dL, p = 0.1). However, the STZ group had higher blood glucose levels compared with the control group in the fed state (286.1 ± 68.5 mg/dL vs. 152.5 ± 3.1 mg/dL, p < 0.05). Male rats in the STZ group had a lower body weight on P90 compared with the control group (404.5 ± 13.9 g vs. 485.8 ± 5.7 g, p = 0.0002). This difference was not present for female rats (248.2 ± 23.4 g vs. 271.8 ± 4.4 g, p = 0.37).
The mean blood glucose concentration in the recurrent hypoinsulinemic hyperglycemia model was 217 ± 17 mg/dL (vs. control, 128 ± 2 mg/dL, p < 0.05).
Neonatal hyperglycemia upregulates markers of oxidative stress and inflammation in the hippocampus on P6
The transcript expression of PARP-1 was upregulated by 58% and that of NF-κB was upregulated by 276% in the STZ group relative to the control group (p < 0.05). Transcript expression of AIF was not altered, while Bcl2 was upregulated 60% and NR2B mRNA expression was downregulated 40% in the STZ group (Table 2).
Table 2.
Relative mRNA expression of upstream and downstream effectors of CXCL10/CXCR3 signaling on P6
| Transcript name | Gene ID | Description | Control group | STZ group |
|---|---|---|---|---|
| PARP-1 (n = 6) | Parp1 | Poly(ADP-ribose) polymerase 1 | 1.0 ± 0.17 | 1.6 ± 0.13* |
| NF-κB (n = 6) | Nfkb1 | Nuclear factor of κ light polypeptide gene enhancer in B cells 1 | 1.0 ± 0.10 | 2.8 ± 0.18* |
| BCL2 (n = 12) | Bcl2 | B cell leukemia/lymphoma 2 | 1.0 ± 0.06 | 1.6 ± 0.07* |
| AIF (n = 7) | Aifm1 | Apoptosis-inducing factor | 1.0 ± 0.10 | 1.1 ± 0.12 |
| NR2B (n = 6) | Grin2b | Glutamate receptor, ionotropic, NMDA2B | 1.0 ± 0.10 | 0.6 ± 0.05* |
Values are mean ± SEM, n = 6–12
*p < 0.05
Compared with the control group, the expression of CXCL10 and its receptor CXCR3 mRNA transcripts were upregulated in the STZ group (Fig. 2a, b). Repeating the CXCR3 qPCR experiments using GAPDH as a reference gene demonstrated a similar degree of upregulation (STZ group, 6.0 ± 1.5 a.u.; control group, 1.0 ± 0.32 a.u., p < 0.05). CXCR3 protein expression was 10% higher in the STZ group (Fig. 2c). CXCL10 colocalized with neurons, astrocytes, and microglia (Fig. 3). CXCR3 strongly colocalized with neurons and microglia, but not with astrocytes (Fig. 3).
Fig. 2.
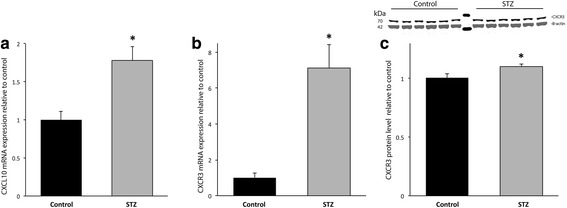
The acute effect of STZ-induced hyperglycemia on the expression of CXCL10 and its receptor CXCR3 on P6. Black, control group; gray, hyperglycemia group, *p < 0.05. a CXCL10 mRNA expression was increased by 78%. b CXCR3 mRNA expression was increased by sevenfold in the hyperglycemia group, relative to the control group. c CXCR3 protein expression was increased by 10%
Fig. 3.
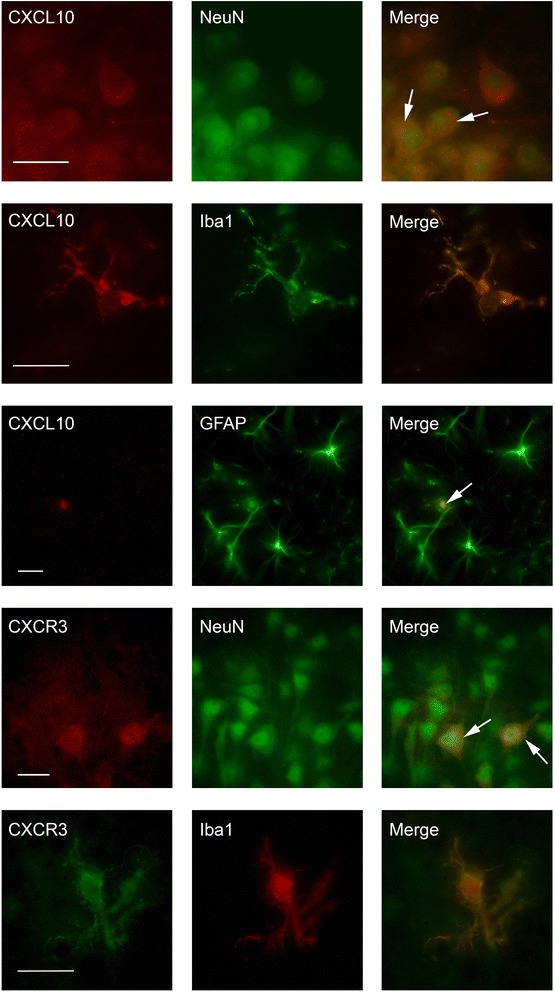
CXCL10 colocalizes with neurons, microglia, and astrocytes; CXCR3 colocalizes with neurons and microglia. Brain sections from the hippocampal CA1 region positive for CXCL10 and NeuN (with merge image), CXCL10 and Iba1 (with merge image), CXCL10 and GFAP (with merge image), CXCR3 and NeuN (with merge image); and CXCR3 and Iba1 (with merge image). Scale bars = 25 μm
CXCL10 and CXCR3 transcript expression were upregulated by 97% (1.97 ± 0.36 a.u. vs. 1.0 ± 0.11 a.u., p < 0.05) and 442% (4.42 ± 1.06 a.u. vs. 1.0 ± 0.08 a.u., p < 0.05), respectively, in the recurrent hyperglycemia group compared with the control group.
Neonatal hyperglycemia causes microglial activation and astrocytosis in the hippocampus on P6
Relative to the control group, more CD11-positive cells, representing microglia, were present in the hippocampal CA1 region in the STZ group (control group, 68.62 ± 11.37 cells/mm2; STZ group, 110.28 ± 15.12 cells/mm2, p < 0.05; Fig. 4). In a similar manner, the number of S100β immunoreactive astrocytes was increased over threefold in the STZ group, relative to the control group (control group, 21.55 ± 4.88 cells/mm2; STZ group, 67.44 ± 10.77 cells/mm2, p < 0.05; Fig. 4).
Fig. 4.
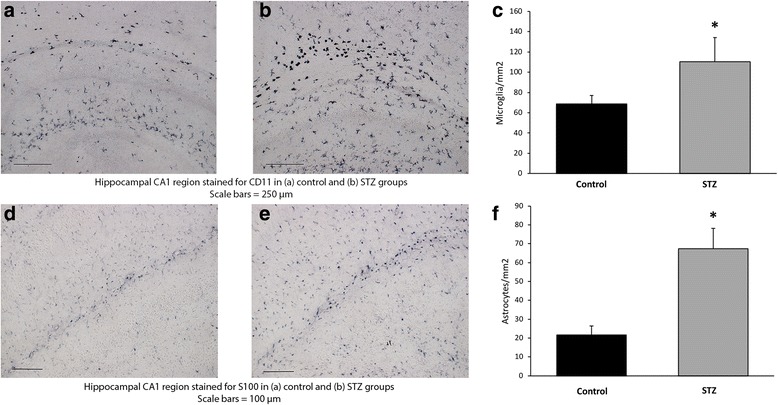
The acute effect of STZ-induced hyperglycemia on microgliosis and astrocytosis on P6. Representative photomicrographs of the hippocampal CA1 region at 10× stained for CD-11 from a control and b hyperglycemia groups. Scale bars = 250 μm. The number of CD-11-positive microglia was significantly increased in the hyperglycemia group (gray) relative to control (black), *p < 0.05 (c). Representative photomicrographs of the hippocampal CA1 region at 20× stained for S100β astrocytes from d control and e hyperglycemia groups. Scale bars = 100 μm. The number of S100β positive astrocytes was significantly increased in the hyperglycemia group (gray) relative to the control (black), p < 0.05 (f)
Neonatal hyperglycemia leads to long-term hippocampus-based deficits in learning and memory
Analysis of performance in the Barnes maze demonstrated a significant overall main effect of guess type (good guess vs. bad guess) (F(1, 39) = 163.92, p < 0.0001) after collapsing across both probe sessions. While there was no significant main effect of hyperglycemia on guess rate (F(1, 39) = 0.49, p = 0.48), there was a significant interaction between hyperglycemia and guess type (F(1, 1) = 9.31, p < 0.01). Rats in the STZ group made fewer “good guesses” and more “bad guesses” (Fig. 5).
Fig. 5.
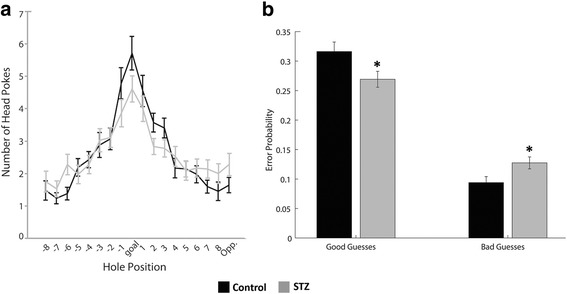
The effect of STZ-induced hyperglycemia on long-term hippocampal function as measured by the Barnes maze behavioral test on P90. Hole pokes made by animal on the maze during probe trials (schematic is shown in Fig. 1), with goal location represented in the center and subsequent holes moving progressively away from the goal (a). Quantified “good guesses” represented by hole pokes nearest the goal location compared with “bad guesses” represented by hole pokes furthest from the goal location, demonstrating that hyperglycemia group (gray) made fewer “good” and more “bad” guesses compared with the control group (black) (b), *p < 0.05
Neonatal hyperglycemia leads to decreased dendritic density in the adult hippocampus
The effect of neonatal hyperglycemia on the dendritic structure in the CA1 region of the adult hippocampus is shown in Fig. 6. Representative micrographs demonstrate qualitatively disorganized and truncated dendrites in a formerly hyperglycemic rat hippocampus compared with a control rat (Fig. 6a, b). Quantitatively, there was a 16% decrease in integrated MAP-2 density in the STZ group compared with the control group (p < 0.05; Fig. 6c). The protein expression of PSD95 was decreased by 11% in the STZ group vs. the control group (p < 0.05; Fig. 6d).
Fig. 6.
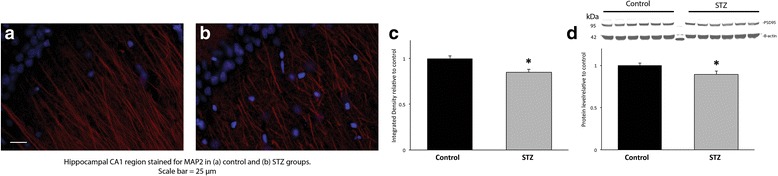
The long-term effect of STZ-induced hyperglycemia on synaptogenesis on P90. Representative fluorescent photomicrographs of the hippocampal CA1 region immunostained for MAP2 (red) in a control and b hyperglycemia groups. Scale bars = 25 μm. Representative micrographs demonstrate qualitatively disorganized and truncated dendrites in the formerly hyperglycemic hippocampus compared with the hippocampus of the control rat. c The quantified average integrated density of fluorescent staining was decreased by 16% in the hyperglycemia group (gray) relative to the control group (black), *p < 0.05. d Protein expression of PSD95 in the hippocampus on P90 was decreased by 11% in the hyperglycemia group (gray) relative to the control group (black), *p < 0.05
Discussion
Understanding how high glucose levels affect the developing brain is important because it impacts clinical decision-making. Currently, treatment of hyperglycemia in preterm infants is highly variable and controversial and involves restricting glucose infusion and/or insulin therapy [40]. Better evidence of how hyperglycemia impacts neurodevelopmental outcomes will help neonatal providers make better-informed treatment decisions.
Our study demonstrates that hyperglycemia comparable in severity to that experienced by the ELGAN is associated with growth deceleration and evidence of oxidative stress and inflammation in the hippocampus in the neonatal period, as well as long-term negative effects on hippocampal dendrite development and function in adulthood in rats. These findings are consistent with clinical data reporting restriction of somatic and brain growth [2] and impaired neurodevelopment in preterm infants with a history of hyperglycemia in the neonatal period [3]. The data are also consistent with previous studies showing an association between recurrent hyperglycemia and oxidative stress and microgliosis in the cerebral cortex and hippocampus of neonatal rats [10, 11].
We sought to use an animal model of hyperglycemia that was similar in etiology, severity, and duration to that seen in the ELGAN population. Although hyperglycemia experienced by these extremely preterm infants is multifactorial in nature [1], our STZ model addresses a major component of this phenomenon, namely functional hypoinsulinism. The neonatal STZ model takes advantage of the drug’s relative selectivity for pancreatic β cells via uptake by the glucose transporter 2 (GLUT2) at the cell membrane. Extra-pancreatic effects of systemically administered STZ are limited to the liver and kidney, which also express GLUT2. [41] There are no direct CNS effects since the blood-brain barrier does not express GLUT2 [42]. Therefore, we are confident that the hippocampal structural and functional effects are due to hyperglycemia and not STZ itself. Similarities between the results of the STZ model in the present study and those in our previous [10] and current non-STZ recurrent hyperglycemia model corroborate this contention. By inducing hyperglycemia between P2 and P6, we were able to probe the effects on the preterm brain, as the rat pup brain on P2 is developmentally equivalent to a human preterm brain at approximately 24 to 26 weeks gestation and the P6 pup brain being a developmental approximate to a 32-week gestation human brain [32, 33].
Our previous study demonstrated that hyperglycemia is associated with PARP-1 and NF-κB upregulation and microgliosis in the cerebral cortex [10], likely in response to oxidative stress [11]. AIF expression was not altered, while Bcl2 expression was upregulated, suggesting lack of cell death. Our present study confirms a similar response in the hippocampus and demonstrates the novel finding that hyperglycemia upregulates the CXCL10/CXCR3 signaling pathway. Although this pathway is implicated in the pancreatic β cell destruction in T1D and serum CXCL10 is considered a biomarker of T1D in adult humans [43], to our knowledge, this is the first demonstration of CXCL10/CXCR3 upregulation in the hippocampus due to hyperglycemia. Upregulation of these markers in the recurrent hyperglycemia model rules out STZ as the primary cause of CXCL10/CXCR3 upregulation. CXCL10/CXCR3 upregulation was likely mediated through NF-kB. Consistent with this possibility, a previous study demonstrated that blockage of NF-kB with IkB abrogates hyperglycemia-induced CXCL10 release from monocytes [26]. However, a feed-forward effect of CXCL10/CXCR3 on PARP1 and NF-kB expression cannot be ruled out [44]. Our evidence that CXCR3 is expressed in the neurons and microglia, and CXCL10 in all three cell types (neurons, astrocytes, and microglia) during the acute phase is consistent with previous studies [16–18, 45], and suggests that all three cell types are the likely source of CXCL10 and that neurons and microglia are the likely targets of CXCL10 action. The increased number of microglia in the STZ group in the absence of an upregulation of apoptotic factors suggests that microglia may play a neuroprotective role in facilitating tissue repair and preventing further injury [46]. The increased number of astrocytes in the STZ group may represent a developmental difference in astrocyte response to dysglycemia, as some of the adult diabetes models have observed a decrease in activated astrocytes in the setting of hyperglycemia [47].
Abnormal CXCL10/CXCR3 signaling may be responsible for the decreased synaptic density and abnormal performance in Barnes maze at adulthood, likely by altering NMDA receptor expression [18] and glutamatergic neurotransmission. NR2B, a subunit of NMDA receptors, plays an important role in synaptogenesis and learning and memory performance in the hippocampus [48]. NR2B antagonists alter long-term potentiation (LTP) and long-term depression (LTD) in the hippocampi of mice [49]. NR2B transcript expression was 40% lower in the STZ group on P6. In support of abnormal glutamatergic neurotransmission, we have recently demonstrated that recurrent hyperglycemia in neonatal rats is associated with long-term impairments in glutamate-glutamine cycling between neuron and glia in the hippocampus [9].
Animals in the STZ group had higher blood glucose concentration in the fed state at adulthood. This is consistent with previous data in this model [50]. While hyperglycemia in adulthood may explain some of the structural and functional effects [51], it does not negate the significance of our results in the context of ELGAN population who are also at increased risk for metabolic disease including impaired insulin sensitivity as adults [52]. The evidence for insulin resistance appears as early as 2 years of age [53].
Although our model was well suited for assessing the effects of hyperglycemia, it did not account for the other components of neonatal hyperglycemia in human preterm infants, including dextrose infusion and treatment with insulin. In adults with diabetes, glucose fluctuations have a greater triggering effect on oxidative stress than chronic sustained hyperglycemia [54], although the data have been equivocal in preterm infants [55]. Another limitation of the study was the difficulty in determining the sequence of changes among the various transcript and protein expressions. However, in a chronic hypoxia injury model, data support CXCL10 expression is downstream of NF-κB signaling [56]. An exact pathway analysis without appropriate knock-out models, blocking agents, or cell culture experiments cannot be determined. Probing additional markers of oxidative stress would have also strengthened this study and supported this pathway analysis, although conventional markers of oxidative stress have previously been reported in the STZ and recurrent hyperglycemia models. Rosa et al. used a neonatal STZ model of hyperglycemia and demonstrated increased production of superoxide anion and NADPH oxidase activity and an increase in lipid peroxidation in the neonatal rat brain following STZ-induced hyperglycemia [57]. Similarly, Tayman et al. demonstrated increased total oxidant status, xanthine oxidase, and malondialdehyde, and decreased total antioxidant status in the brain tissue of rats after recurrent neonatal hyperglycemia [11].
Finally, this study focused on the hippocampus and hippocampus-mediated learning. However, other functional deficits are also common in extremely preterm infants, for example, deficits in executive function [58, 59]. As mentioned previously, we have demonstrated that recurrent hyperglycemia leads to oxidative stress (PARP-1, NF-κB upregulation) and microgliosis in the cerebral cortex, the brain region important for executive function [10]. Future studies are necessary to explore the full extent of hyperglycemia’s effect on the brain regions.
Conclusion
In summary, we found that hyperglycemia in newborn rats induces CXCL10/CXCR3 signaling, microglial activation, and astrocytosis, and alters long-term synaptogenesis and function in the hippocampus. Therefore, efforts at minimizing neonatal hyperglycemia should be considered in the ELGAN, so as to reduce the risk of adverse neurodevelopment, in a patient population that is already vulnerable to brain injury from additional comorbidities.
Acknowledgements
The authors would like to acknowledge Motaz Nashawaty and Saher Fatima for their assistance with the experiments.
Funding
This study was supported by the Viking Children’s Fund; Grant number: UMF0015965. This funding body did not play a role in the design of the study; the collection, analysis, or interpretation of the data; or in writing the manuscript.
Availability of data and materials
All data presented in this study are included in the manuscript.
Abbreviations
- AIF
Apoptosis-inducing factor
- Bcl-2
B cell lymphoma 2
- CXCL10
C-X-C motif chemokine ligand 10
- CXCR3
C-X-C motif chemokine receptor 3
- ELGAN
Extremely low gestational age newborns
- GLUT2
Glucose transporter 2
- IBA1
Ionized calcium-binding adapter molecule
- LTD
Long-term depression
- LTP
Long-term potentiation
- MAP-2
Microtubule-associated protein
- NF-κB
Nuclear factor of kappa light polypeptide gene enhancer in B cells
- NR2B
NMDA receptor
- P(x)
Postnatal day x
- PARP-1
Poly(ADP-ribose) polymerase-1
- PSD
Postsynaptic density protein
- STZ
Streptozotocin
- T1D
Type 1 diabetes
- TLR
Toll-like receptor
Authors’ contributions
KS and RR formulated the concept and designed the study. KS, KE, TM, JC, LH, and AM performed the studies including the animal experiments, qPCR, Western blot, immunohistochemistry, and behavioral assessment. KS, BS, and RR analyzed the data and interpreted the results. KS wrote the paper. All authors read and approved of the final manuscript.
Ethics approval and consent to participate
All experiments were performed with the approval of the Institutional Animal Care and Use Committee of the University of Minnesota.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Katherine M. Satrom, Phone: 763-229-8578, Email: ksatrom@umn.edu
Kathleen Ennis, Email: enni0012@umn.edu.
Brian M. Sweis, Email: sweis001@umn.edu
Tatyana M. Matveeva, Email: matve004@umn.edu
Jun Chen, Email: chen3713@umn.edu.
Leif Hanson, Email: hansonl@stolaf.edu.
Akhil Maheshwari, Email: akhilm@health.usf.edu.
Raghavendra Rao, Email: raghurao@umn.edu.
References
- 1.Beardsall K, Vanhaesebrouck S, Ogilvy-Stuart AL, Vanhole C, Palmer CR, Ong K, vanWeissenbruch M, Midgley P, Thompson M, Thio M, et al. Prevalence and determinants of hyperglycemia in very low birth weight infants: cohort analyses of the NIRTURE study. J Pediatr. 2010;157:715–719 e711–715–719 e713. doi: 10.1016/j.jpeds.2010.04.032. [DOI] [PubMed] [Google Scholar]
- 2.Ramel SE, Long JD, Gray H, Durrwachter-Erno K, Demerath EW, Rao R. Neonatal hyperglycemia and diminished long-term growth in very low birth weight preterm infants. J Perinatol. 2013;33:882–886. doi: 10.1038/jp.2013.77. [DOI] [PubMed] [Google Scholar]
- 3.van der Lugt NM, Smits-Wintjens VE, van Zwieten PH, Walther FJ. Short and long term outcome of neonatal hyperglycemia in very preterm infants: a retrospective follow-up study. BMC Pediatr. 2010;10:52. doi: 10.1186/1471-2431-10-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hays SP, Smith EO, Sunehag AL. Hyperglycemia is a risk factor for early death and morbidity in extremely low birth-weight infants. Pediatrics. 2006;118:1811–1818. doi: 10.1542/peds.2006-0628. [DOI] [PubMed] [Google Scholar]
- 5.Kao LS, Morris BH, Lally KP, Stewart CD, Huseby V, Kennedy KA. Hyperglycemia and morbidity and mortality in extremely low birth weight infants. J Perinatol. 2006;26:730–736. doi: 10.1038/sj.jp.7211593. [DOI] [PubMed] [Google Scholar]
- 6.Perlman JM. Neurobehavioral deficits in premature graduates of intensive care—potential medical and neonatal environmental risk factors. Pediatrics. 2001;108:1339–1348. doi: 10.1542/peds.108.6.1339. [DOI] [PubMed] [Google Scholar]
- 7.Aanes S, Bjuland KJ, Skranes J, Lohaugen GC. Memory function and hippocampal volumes in preterm born very-low-birth-weight (VLBW) young adults. NeuroImage. 2015;105:76–83. doi: 10.1016/j.neuroimage.2014.10.023. [DOI] [PubMed] [Google Scholar]
- 8.Rao R, Hershey T. The impact of hypoglycemia on the developing brain. Transl Endocrinol Met. 2012;3:23. [Google Scholar]
- 9.Rao R, Nashawaty M, Fatima S, Ennis K, Tkac I. Neonatal hyperglycemia alters the neurochemical profile, dendritic arborization and gene expression in the developing rat hippocampus. NMR Biomed. In press [DOI] [PMC free article] [PubMed]
- 10.Gisslen T, Ennis K, Bhandari V, Rao R. Recurrent hypoinsulinemic hyperglycemia in neonatal rats increases PARP-1 and NF-kappaB expression and leads to microglial activation in the cerebral cortex. Pediatr Res. 2015;78:513–519. doi: 10.1038/pr.2015.136. [DOI] [PubMed] [Google Scholar]
- 11.Tayman C, Yis U, Hirfanoglu I, Oztekin O, Goktas G, Bilgin BC. Effects of hyperglycemia on the developing brain in newborns. Pediatr Neurol. 2014;51:239–245. doi: 10.1016/j.pediatrneurol.2014.04.015. [DOI] [PubMed] [Google Scholar]
- 12.Baker RG, Hayden MS, Ghosh S. NF-kappaB, inflammation, and metabolic disease. Cell Metab. 2011;13:11–22. doi: 10.1016/j.cmet.2010.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang T, Zhang X, Li JJ. The role of NF-kappaB in the regulation of cell stress responses. Int Immunopharmacol. 2002;2:1509–1520. doi: 10.1016/S1567-5769(02)00058-9. [DOI] [PubMed] [Google Scholar]
- 14.d'Avila JC, Lam TI, Bingham D, Shi J, Won SJ, Kauppinen TM, Massa S, Liu J, Swanson RA. Microglial activation induced by brain trauma is suppressed by post-injury treatment with a PARP inhibitor. J Neuroinflammation. 2012;9:31. doi: 10.1186/1742-2094-9-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nelson TE, Gruol DL. The chemokine CXCL10 modulates excitatory activity and intracellular calcium signaling in cultured hippocampal neurons. J Neuroimmunol. 2004;156:74–87. doi: 10.1016/j.jneuroim.2004.07.009. [DOI] [PubMed] [Google Scholar]
- 16.Biber K, Dijkstra I, Trebst C, De Groot CJ, Ransohoff RM, Boddeke HW. Functional expression of CXCR3 in cultured mouse and human astrocytes and microglia. Neuroscience. 2002;112:487–497. doi: 10.1016/S0306-4522(02)00114-8. [DOI] [PubMed] [Google Scholar]
- 17.Xia MQ, Bacskai BJ, Knowles RB, Qin SX, Hyman BT. Expression of the chemokine receptor CXCR3 on neurons and the elevated expression of its ligand IP-10 in reactive astrocytes: in vitro ERK1/2 activation and role in Alzheimer’s disease. J Neuroimmunol. 2000;108:227–235. doi: 10.1016/S0165-5728(00)00285-X. [DOI] [PubMed] [Google Scholar]
- 18.Cho J, Nelson TE, Bajova H, Gruol DL. Chronic CXCL10 alters neuronal properties in rat hippocampal culture. J Neuroimmunol. 2009;207:92–100. doi: 10.1016/j.jneuroim.2008.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Klein RS, Lin E, Zhang B, Luster AD, Tollett J, Samuel MA, Engle M, Diamond MS. Neuronal CXCL10 directs CD8+ T-cell recruitment and control of West Nile virus encephalitis. J Virol. 2005;79:11457–11466. doi: 10.1128/JVI.79.17.11457-11466.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Riemer C, Schultz J, Burwinkel M, Schwarz A, Mok SW, Gultner S, Bamme T, Norley S, van Landeghem F, Lu B, et al. Accelerated prion replication in, but prolonged survival times of, prion-infected CXCR3−/− mice. J Virol. 2008;82:12464–12471. doi: 10.1128/JVI.01371-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liu L, Callahan MK, Huang D, Ransohoff RM. Chemokine receptor CXCR3: an unexpected enigma. Curr Top Dev Biol. 2005;68:149–181. doi: 10.1016/S0070-2153(05)68006-4. [DOI] [PubMed] [Google Scholar]
- 22.Ahmadi Z, Arababadi MK, Hassanshahi G. CXCL10 activities, biological structure, and source along with its significant role played in pathophysiology of type I diabetes mellitus. Inflammation. 2013;36:364–371. doi: 10.1007/s10753-012-9555-1. [DOI] [PubMed] [Google Scholar]
- 23.Rosa JS, Mitsuhashi M, Oliver SR, Ogura M, Flores RL, Pontello AM, Galassetti PR. Ex vivo TCR-induced leukocyte gene expression of inflammatory mediators is increased in type 1 diabetic patients but not in overweight children. Diabetes Metab Res Rev. 2010;26:33–39. doi: 10.1002/dmrr.1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tanaka S, Aida K, Nishida Y, Kobayashi T. Pathophysiological mechanisms involving aggressive islet cell destruction in fulminant type 1 diabetes. Endocr J. 2013;60:837–845. doi: 10.1507/endocrj.EJ13-0222. [DOI] [PubMed] [Google Scholar]
- 25.Tanaka S, Nishida Y, Aida K, Maruyama T, Shimada A, Suzuki M, Shimura H, Takizawa S, Takahashi M, Akiyama D, et al. Enterovirus infection, CXC chemokine ligand 10 (CXCL10), and CXCR3 circuit: a mechanism of accelerated beta-cell failure in fulminant type 1 diabetes. Diabetes. 2009;58:2285–2291. doi: 10.2337/db09-0091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Devaraj S, Jialal I. Increased secretion of IP-10 from monocytes under hyperglycemia is via the TLR2 and TLR4 pathway. Cytokine. 2009;47:6–10. doi: 10.1016/j.cyto.2009.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shigihara T, Oikawa Y, Kanazawa Y, Okubo Y, Narumi S, Saruta T, Shimada A. Significance of serum CXCL10/IP-10 level in type 1 diabetes. J Autoimmun. 2006;26:66–71. doi: 10.1016/j.jaut.2005.09.027. [DOI] [PubMed] [Google Scholar]
- 28.Bonner-Weir S, Trent DF, Honey RN, Weir GC. Responses of neonatal rat islets to streptozotocin: limited B-cell regeneration and hyperglycemia. Diabetes. 1981;30:64–69. doi: 10.2337/diab.30.1.64. [DOI] [PubMed] [Google Scholar]
- 29.Kermorvant-Duchemin E, Pinel AC, Lavalette S, Lenne D, Raoul W, Calippe B, Behar-Cohen F, Sahel JA, Guillonneau X, Sennlaub F. Neonatal hyperglycemia inhibits angiogenesis and induces inflammation and neuronal degeneration in the retina. PLoS One. 2013;8:e79545. doi: 10.1371/journal.pone.0079545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Portha B, Levacher C, Picon L, Rosselin G. Diabetogenic effect of streptozotocin in the rat during the perinatal period. Diabetes. 1974;23:889–895. doi: 10.2337/diab.23.11.889. [DOI] [PubMed] [Google Scholar]
- 31.Takada J, Machado MA, Peres SB, Brito LC, Borges-Silva CN, Costa CE, Fonseca-Alaniz MH, Andreotti S, Lima FB. Neonatal streptozotocin-induced diabetes mellitus: a model of insulin resistance associated with loss of adipose mass. Metabolism. 2007;56:977–984. doi: 10.1016/j.metabol.2006.05.021. [DOI] [PubMed] [Google Scholar]
- 32.Semple BD, Blomgren K, Gimlin K, Ferriero DM, Noble-Haeusslein LJ. Brain development in rodents and humans: identifying benchmarks of maturation and vulnerability to injury across species. Prog Neurobiol. 2013;106-107:1–16. doi: 10.1016/j.pneurobio.2013.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hagberg H, Ichord R, Palmer C, Yager JY, Vannucci SJ. Animal models of developmental brain injury: relevance to human disease. A summary of the panel discussion from the Third Hershey Conference on Developmental Cerebral Blood Flow and Metabolism. Dev Neurosci. 2002;24:364–366. doi: 10.1159/000069040. [DOI] [PubMed] [Google Scholar]
- 34.Rao R, Sperr D, Ennis K, Tran P. Postnatal age influences hypoglycemia-induced poly(ADP-ribose) polymerase-1 activation in the brain regions of rats. Pediatr Res. 2009;66:642–647. doi: 10.1203/PDR.0b013e3181bbce69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sherwood NM, Timiras PS: A steriotaxic Atlas of the Developing Rat Brain. Berkeley: University of California Press; 1970.
- 36.Raman L, Hamilton KL, Gewirtz JC, Rao R. Effects of chronic hypoxia in developing rats on dendritic morphology of the CA1 subarea of the hippocampus and on fear-potentiated startle. Brain Res. 2008;1190:167–174. doi: 10.1016/j.brainres.2007.11.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Won SJ, Yoo BH, Kauppinen TM, Choi BY, Kim JH, Jang BG, Lee MW, Sohn M, Liu J, Swanson RA, Suh SW. Recurrent/moderate hypoglycemia induces hippocampal dendritic injury, microglial activation, and cognitive impairment in diabetic rats. J Neuroinflammation. 2012;9:182. doi: 10.1186/1742-2094-9-182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Barnes CA. Memory deficits associated with senescence: a neurophysiological and behavioral study in the rat. J Comp Physiol Psychol. 1979;93:74–104. doi: 10.1037/h0077579. [DOI] [PubMed] [Google Scholar]
- 39.Sharma S, Rakoczy S, Brown-Borg H. Assessment of spatial memory in mice. Life Sci. 2010;87:521–536. doi: 10.1016/j.lfs.2010.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Beardsall K, Vanhaesebrouck S, Ogilvy-Stuart AL, Vanhole C, Palmer CR, van Weissenbruch M, Midgley P, Thompson M, Thio M, Cornette L, et al. Early insulin therapy in very-low-birth-weight infants. N Engl J Med. 2008;359:1873–1884. doi: 10.1056/NEJMoa0803725. [DOI] [PubMed] [Google Scholar]
- 41.Graham ML, Janecek JL, Kittredge JA, Hering BJ, Schuurman HJ. The streptozotocin-induced diabetic nude mouse model: differences between animals from different sources. Comp Med. 2011;61:356–360. [PMC free article] [PubMed] [Google Scholar]
- 42.Grieb P. Intracerebroventricular streptozotocin injections as a model of Alzheimer’s disease: in search of a relevant mechanism. Mol Neurobiol. 2016;53:1741–1752. doi: 10.1007/s12035-015-9132-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Antonelli A, Ferrari SM, Corrado A, Ferrannini E, Fallahi P. CXCR3, CXCL10 and type 1 diabetes. Cytokine Growth Factor Rev. 2014;25:57–65. doi: 10.1016/j.cytogfr.2014.01.006. [DOI] [PubMed] [Google Scholar]
- 44.Bajova H, Nelson TE, Gruol DL. Chronic CXCL10 alters the level of activated ERK1/2 and transcriptional factors CREB and NF-kappaB in hippocampal neuronal cell culture. J Neuroimmunol. 2008;195:36–46. doi: 10.1016/j.jneuroim.2008.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bhowmick S, Duseja R, Das S, Appaiahgiri MB, Vrati S, Basu A. Induction of IP-10 (CXCL10) in astrocytes following Japanese encephalitis. Neurosci Lett. 2007;414:45–50. doi: 10.1016/j.neulet.2006.11.070. [DOI] [PubMed] [Google Scholar]
- 46.Kreutzberg GW. Microglia: a sensor for pathological events in the CNS. Trends Neurosci. 1996;19:312–318. doi: 10.1016/0166-2236(96)10049-7. [DOI] [PubMed] [Google Scholar]
- 47.Coleman E, Judd R, Hoe L, Dennis J, Posner P. Effects of diabetes mellitus on astrocyte GFAP and glutamate transporters in the CNS. Glia. 2004;48:166–178. doi: 10.1002/glia.20068. [DOI] [PubMed] [Google Scholar]
- 48.Tang YP, Wang H, Feng R, Kyin M, Tsien JZ. Differential effects of enrichment on learning and memory function in NR2B transgenic mice. Neuropharmacology. 2001;41:779–790. doi: 10.1016/S0028-3908(01)00122-8. [DOI] [PubMed] [Google Scholar]
- 49.Fox CJ, Russell KI, Wang YT, Christie BR. Contribution of NR2A and NR2B NMDA subunits to bidirectional synaptic plasticity in the hippocampus in vivo. Hippocampus. 2006;16:907–915. doi: 10.1002/hipo.20230. [DOI] [PubMed] [Google Scholar]
- 50.Hemmings SJ, Spafford D, Neonatal STZ. Model of type II diabetes mellitus in the Fischer 344 rat: characteristics and assessment of the status of the hepatic adrenergic receptors. Int J Biochem Cell Biol. 2000;32:905–919. doi: 10.1016/S1357-2725(00)00019-4. [DOI] [PubMed] [Google Scholar]
- 51.Seaquist ER. The impact of diabetes on cerebral structure and function. Psychosom Med. 2015;77:616–621. doi: 10.1097/PSY.0000000000000207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Parkinson JR, Hyde MJ, Gale C, Santhakumaran S, Modi N. Preterm birth and the metabolic syndrome in adult life: a systematic review and meta-analysis. Pediatrics. 2013;131:e1240–e1263. doi: 10.1542/peds.2012-2177. [DOI] [PubMed] [Google Scholar]
- 53.Tinnion R, Gillone J, Cheetham T, Embleton N. Preterm birth and subsequent insulin sensitivity: a systematic review. Arch Dis Child. 2014;99:362–368. doi: 10.1136/archdischild-2013-304615. [DOI] [PubMed] [Google Scholar]
- 54.Monnier L, Mas E, Ginet C, Michel F, Villon L, Cristol JP, Colette C. Activation of oxidative stress by acute glucose fluctuations compared with sustained chronic hyperglycemia in patients with type 2 diabetes. JAMA. 2006;295:1681–1687. doi: 10.1001/jama.295.14.1681. [DOI] [PubMed] [Google Scholar]
- 55.Tottman AC, Alsweiler JM, Bloomfield FH, Pan M, Harding JE. Relationship between measures of neonatal glycemia, neonatal illness, and 2-year outcomes in very preterm infants. J Pediatr. 2017;188:115–121. doi: 10.1016/j.jpeds.2017.05.052. [DOI] [PubMed] [Google Scholar]
- 56.Rognlien AG, Wollen EJ, Atneosen-Asegg M, Saugstad OD. Increased expression of inflammatory genes in the neonatal mouse brain after hyperoxic reoxygenation. Pediatr Res. 2015;77:326–333. doi: 10.1038/pr.2014.193. [DOI] [PubMed] [Google Scholar]
- 57.Rosa AP, Jacques CE, de Souza LO, Bitencourt F, Mazzola PN, Coelho JG, Mescka CP, Dutra-Filho CS. Neonatal hyperglycemia induces oxidative stress in the rat brain: the role of pentose phosphate pathway enzymes and NADPH oxidase. Mol Cell Biochem. 2015;403:159–167. doi: 10.1007/s11010-015-2346-x. [DOI] [PubMed] [Google Scholar]
- 58.Aarnoudse-Moens CS, Smidts DP, Oosterlaan J, Duivenvoorden HJ, Weisglas-Kuperus N. Executive function in very preterm children at early school age. J Abnorm Child Psychol. 2009;37:981–993. doi: 10.1007/s10802-009-9327-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Burnett AC, Anderson PJ, Lee KJ, Roberts G, Doyle LW, Cheong JLY, Victorian Infant Collaborative Study G. Trends in executive functioning in extremely preterm children across 3 birth eras. Pediatrics. 2018;141 [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data presented in this study are included in the manuscript.