

Abstract
An ascorbate-mediated production of oxidative stress has been shown to retard tumor growth. Subsequent glycolysis inhibition has been suggested. Here, we further define the mechanisms relevant to this observation. Ascorbate was cytotoxic to human neuroblastoma cells through the production of H2O2, which led to ATP depletion, inhibited GAPDH, and non-apoptotic and non-autophagic cell death. The mechanism of cytotoxicity is different when PARP-dependent DNA repair machinery is active or inhibited. Ascorbate-generated H2O2 damaged DNA, activated PARP, depleted NAD+, and reduced glycolysis flux. NAD+ supplementation prevented ATP depletion and cell death, while treatment with a PARP inhibitor, olaparib, preserved NAD+ and ATP levels but led to increased DNA double-strand breakage and did not prevent ascorbate-induced cell death. These data indicate that in cells with an intact PARP-associated DNA repair system, ascorbate-induced cell death is caused by NAD+ and ATP depletion, while in the absence of PARP activation ascorbate-induced cell death still occurs but is a consequence of ROS-induced DNA damage. In a mouse xenograft model, intraperitoneal ascorbate inhibited neuroblastoma tumor growth and prolonged survival. Collectively, these data suggest that ascorbate could be effective in the treatment of glycolysis-dependent tumors. Also, in cancers that use alternative energy metabolism pathways, combining a PARP inhibitor with ascorbate treatment could be useful.
Keywords: Ascorbate, neuroblastoma, glycolysis, DNA damage, GAPDH, poly-ADP ribose polymerase
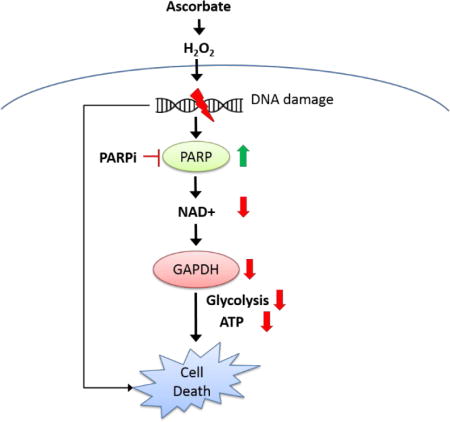
Graphical abstract
Pharmacologic ascorbate forms H2O2, damages DNA and activates PARP. Activated PARP depletes NAD+, and therefore inhibited GAPDH activity, and ATP is depleted in neuroblastoma cells, leading to cell death. In the absence of PARP activation, fatal DNA damage accumulates and leads to cell death.
INTRODUCTION
Neuroblastomas arise from neural crest cells that normally differentiate into sympathetic ganglia and the adrenal medulla. Neuroblastomas are the most common extracranial solid tumors in children, with 1/3 of these tumors diagnosed in infants under 1 year old, and 75% diagnosed by age 5 (1,2). Neuroblastomas account for 15% of all pediatric cancer deaths, despite representing only 6% of all childhood cancers. Although spontaneous regression happens in some infants, many of those affected have aggressive metastatic disease and a poor prognosis (3,4). Current therapies include combinations of multi-agent chemotherapy, surgery, radiation, myeloablative consolidation therapy with stem cell rescue and transplantation, differentiation therapy with 13-cis retinoic acid, and immunotherapy (5,6). Even with intensive multimodal therapy, only 50% of newly diagnosed high-risk neuroblastoma patients will survive, and in relapsed patients less than 10% will survive. Moreover, treatment-related toxicities can impact long term physical, social, and academic development in young children (7). Effective and safe treatments are, therefore, urgently needed.
Recent evidence suggests that high dose intravenous ascorbate (IVC) could represent a well-tolerated cancer treatment. In vitro and in vivo studies have shown that pharmacologic concentrations of ascorbate, which are achievable by intravenous administration, induce cancer cell death without harming normal cells. This has been observed when ascorbate is used alone or in combination with the first line chemotherapies or radiation therapy (8–14). Ascorbate has the benefit of being safe and free of common toxic side effects that often accompany chemotherapies. Good tolerability to IVC has now been observed in over 10 clinical trials (15). Because of its low-toxicity, IVC is worth considering for the treatment of neuroblastomas.
Delivering ascorbate intravenously bypasses the absorption barrier that occurs with oral ingestion, and can establish millimolar concentrations that are many folds higher than its normal physiological concentrations (16–18). These millimolar concentrations last for several hours until the kidneys restore ascorbate levels to physiological concentrations. The pharmacokinetic behaviors of these high ascorbate concentrations exceed those of a vitamin and resemble the use of a drug (16,19). These pharmacological concentrations of ascorbate generate hydrogen peroxide (H2O2) and downstream reactive oxygen species (ROS) through the Fenton chemistry (8,10,20,21), which causes oxidative damage that preferentially affects tumor cells (8,10,20,21). Because H2O2 and ROS have multiple effects (22), it has been difficult to identify a specific molecular mechanism that mediates the cancer cell-specific effects of ascorbate. While some studies indicated apoptosis-dependent mechanisms (23–25), others suggested apoptosis-independent mechanisms (21,26–28). Autophagy and ATP depletion were also suggested to be causing cell death in ascorbate-treated cancer cells (11,29–31). Recent studies have found that oxidative stress induced by ascorbate damaged DNA and inhibited glycolysis in susceptible cancer cells, but not in normal cells, and proposed this to be the mechanism mediating ascorbate’s selective cytotoxicity (10,32). However, it is not well understood how ascorbate depletes ATP. Here we investigated the mechanisms that mediate ascorbate-induced toxicity in neuroblastoma cells.
RESULTS
Pharmacologic ascorbate induced neuroblastoma cell death independent of apoptosis and autophagy
Two neuroblastoma cell lines originated from human metastatic neuroblastomas (SK-N-SH and SH-SY5Y) and a murine neuroblastoma line (Neuro2a) were exposed to 0-2 mM of ascorbate, which are concentrations easily achievable clinically (8). Cell viability was markedly decreased after 24 hours of treatment, with IC50 values of 0.13 – 0.45 mM (Fig. 1A). In contrast, the same treatment only minimally influenced viability of normal fibroblasts (CCD-34SK). Addition of catalase to the cell culture media, an enzyme that specifically degrades H2O2, completely reversed ascorbate-induced cell death in SH-SY5Y neuroblastoma cells (Fig. 1A, B). These data confirmed conclusions from previous studies that pharmacological concentrations of ascorbate selectively induced cell death in cancer cells versus normal cells, through formation of H2O2 (12,20,21,33). A previous study suggested that ascorbate induced cancer cell cytotoxicity by enhanced cellular uptake via its oxidized form dehydroascorbate (DHA) (32), however the study did not report data with direct DHA treatment on cancer cells. Here, we treated SH-SY5Y with DHA, using the same conditions as in ascorbate treatment. In contrast to the reported hypothesis (32), our data showed that treatment with DHA did not reduce cell viability of SH-SY5Y cells (Fig 1B). Our data here is consistent with previous data in a lymphoma cell line that DHA treatment did not influence cell viability while the same concentrations of ascorbate did (21).
FIGURE 1.
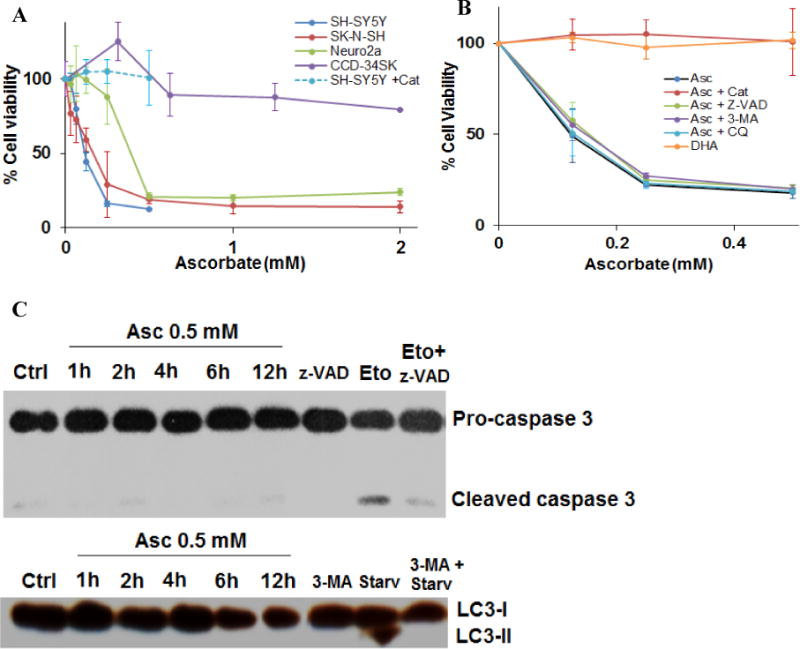
Ascorbate induced selective cytotoxicity in neuroblastoma cells. A. Dose response of neuroblastoma and normal cells to ascorbate induced cell death. Human and mouse neuroblastoma cell lines (SH-SY5Y, SK-N-SH, and Neuro2A) and a fibroblast cell line (CCD-34SK) were exposed to 0- 2 mM ascorbate, and cell viability was detected 24 hours post treatment. + Cat, pre-incubation with 600 U/mL catalase for 30 minutes. Data represents Mean ± SD of 1-3 experiments each done in triplicate. B. Dose response of SH-SY5Y cells to ascorbate induced cell death in the presence or absence of H2O2, apoptosis, and autophagy inhibitors, or to dehydroascorbate (DHA). Cells were exposed to DHA, or pre-exposed for 30 minutes to catalase (Cat, 200 U/mL), Z-VAD-FMK (Z-VAD, 25 μM), 3-methyladenine (3-MA, 5 mM), and Chloroquine (CQ, 20 μM), and then exposed to ascorbate. Viability was detected at 24 hours. Data represents Mean ± SD of 1-3 experiments each done in triplicate. C. Western blots for caspase-3 cleavage, and LC3-II formation in SH-SY5Y cells. Z-VAD, 25 μM; Eto, etoposide 40 μM; 3-MA, 5 mM; Starv, serum starvation for 3 hours.
To detect the involvement of apoptosis, a pan-caspase inhibitor Z-VAD-FMK (Z-VAD), was used. The addition of Z-VAD (25 μM) did not affect ascorbate-induced cell death at all (Fig. 1B), indicating that ascorbate-induced SH-SY5Y cell death is independent of caspase activation. Also, 3-methyladenine (3-MA, 5 mM), a Type III Phosphatidylinositol 3-kinases (PI3K) inhibitor, did not influence the cell death caused by ascorbate. Because 3-MA blocks autophagosome formation via PI3K inhibition, this indicates that ascorbate-induced SH-SY5Y cell death is not dependent on autophagy (Fig. 1B). The independence of autophagy was further confirmed by using another autophagy inhibitor chloroquine (CQ, 20 μM), which inhibits fusion of autophagosomes with lysosomes and lysosomal protein degradation by raising the lysosomal pH. CQ did not affect the ascorbate-induced cell death in SH-SY5Y cells (Fig. 1B). Moreover, we examined cleavage of caspase-3, the marker molecule for the execution of apoptosis, using western blots. As Etoposide is known to induce apoptosis (34), it was used as a positive control. Etoposide (40 μM) treatment induced caspase-3 cleavage, and the cleavage was inhibited by Z-VAD, as expected (Fig. 1C). However, caspase-3 cleavage was absent in SH-SY5Y cells treated with ascorbate (0.5 mM, 1-12 h) (Fig. 1C). Similarly, the conversion of microtubule-associated protein light chain 3-I (LC3-I) to LC3-II was not observed in ascorbate treated SH-SY5Y cells, whereas serum starvation, known to induce autophagy (35), increased LC3-II and the increase was suppressed by 3-MA (Fig. 1C). These data implicated involvement of a cell death pathway independent of apoptosis and autophagy.
Pharmacologic ascorbate reduced neuroblastoma cell glycolysis, NAD+ and ATP levels, leading to cell death
Based on the peroxide-mediated mechanisms of ascorbate’s action, it has been proposed that ascorbate induces cancer cell death by damaging DNA, inhibiting glycolysis and depleting ATP (10,11,20,32). Here, we further investigated the effect of pharmacologic ascorbate on cancer cell’s energy metabolism. A marked reduction in the extracellular acidification rate (ECAR) was detected in SH-SY5Y cells treated with ascorbate (Fig. 2A). Ascorbate at 0.5 mM and above almost completely inhibited ECAR. These same concentrations also yield a profound loss of cell viability. The inhibition in ECAR was recapitulated by H2O2 (100 μM, or 75 pmoles/cell) treatment. The reduction of ECAR indicated inhibition in glucose metabolism. As ECAR is contributed by acid production from both glycolysis and the oxidative phosphorylation (TCA cycle) (36), we also detected the cellular respiratory activity, as indicated by oxygen consumption rate (OCR) (Fig. 2B). When SH-SY5Y cells were treated with ascorbate (0.5-10 mM) there was first a transient increase and then a non-significant decrease (~0-30%) in OCR (Fig. 2B). These data indicate that the reduction in ECAR mainly suggested an inhibition in glycolysis rather than inhibition in oxidative phosphorylation. The transient increase in OCR was likely due to ascorbate oxidation that consumes O2 and forms H2O2 (37). In the presence of cells, it is also possible that the cells transiently upregulated respiration as a reflex to glycolysis inhibition (38). As a critical enzyme in the glycolysis pathway, the activity of glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was significantly inhibited by ascorbate treatment (Fig. 2C), consistent with previous report (32), while the protein level of the enzyme was unchanged (Fig. 2D). The inhibition of GAPDH activity was also induced by H2O2 (100 μM) treatment, and was reversed by catalase, an H2O2 degrading enzyme (Fig. 2C). In contrast, the treatment with DHA did not decrease GAPDH activity (Fig. 2C). These data indicate that ascorbate-induced GAPDH inhibition is also mediated by H2O2, and is likely an effect initiated by the extracellular concentrations of ascorbate. The effect of ascorbate is not related to uptake through DHA. Nicotinamide adenine dinucleotide (NAD+), the critical co-factor for GAPDH-catalyzed oxidative phosphorylation, was significantly reduced by ascorbate treatment (Fig. 2E). The reduction in NAD+ was dose dependent and time dependent to ascorbate.
FIGURE 2.
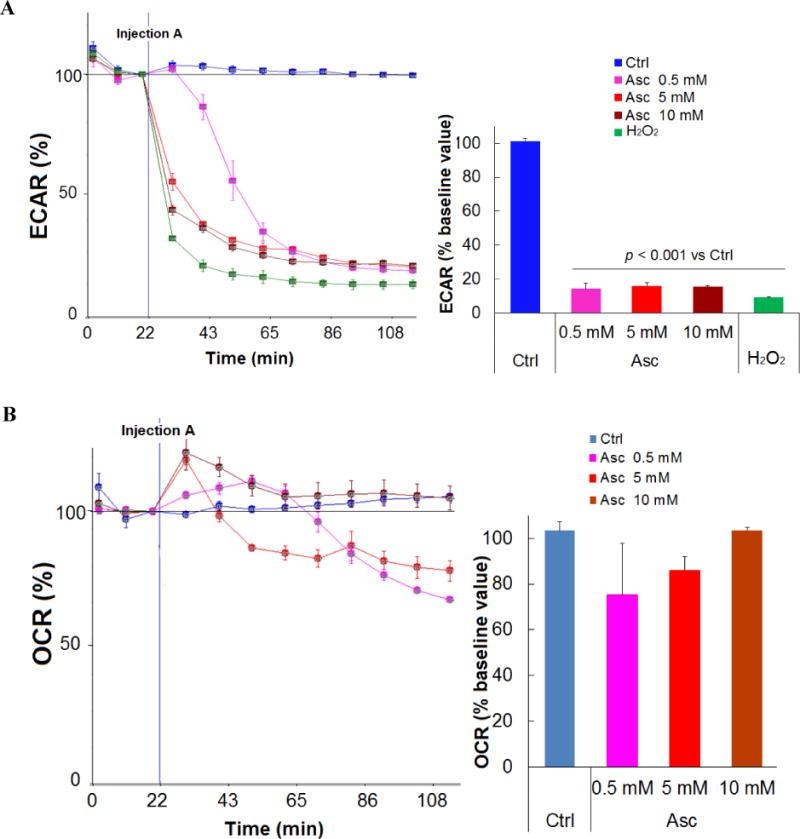
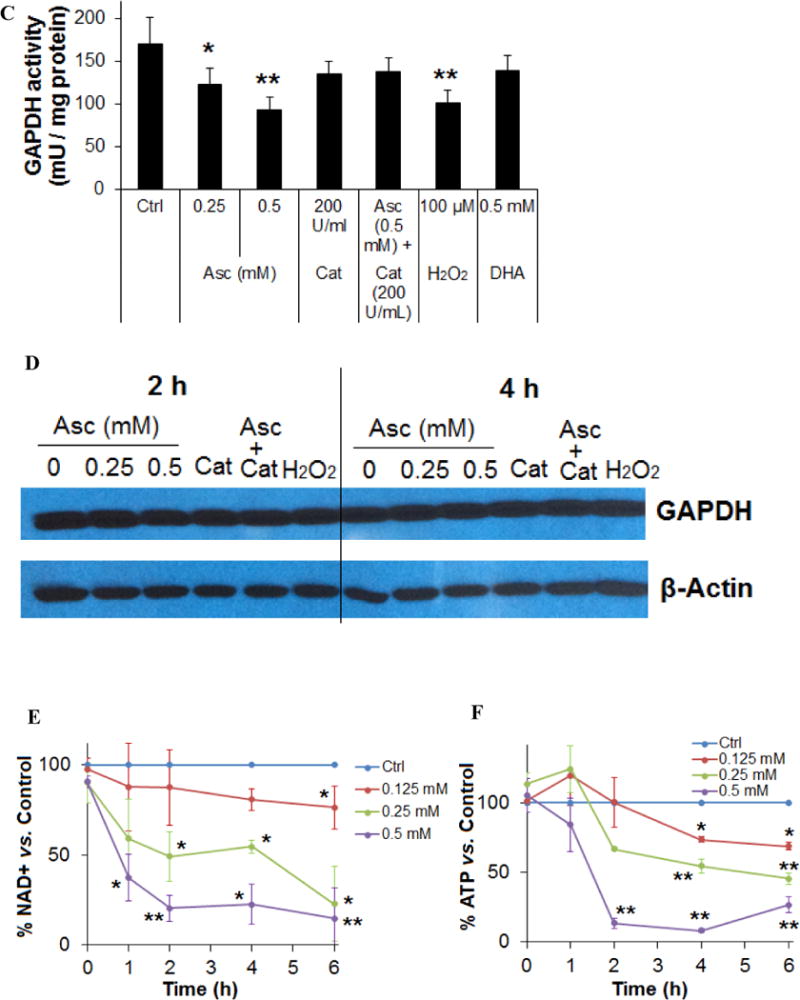
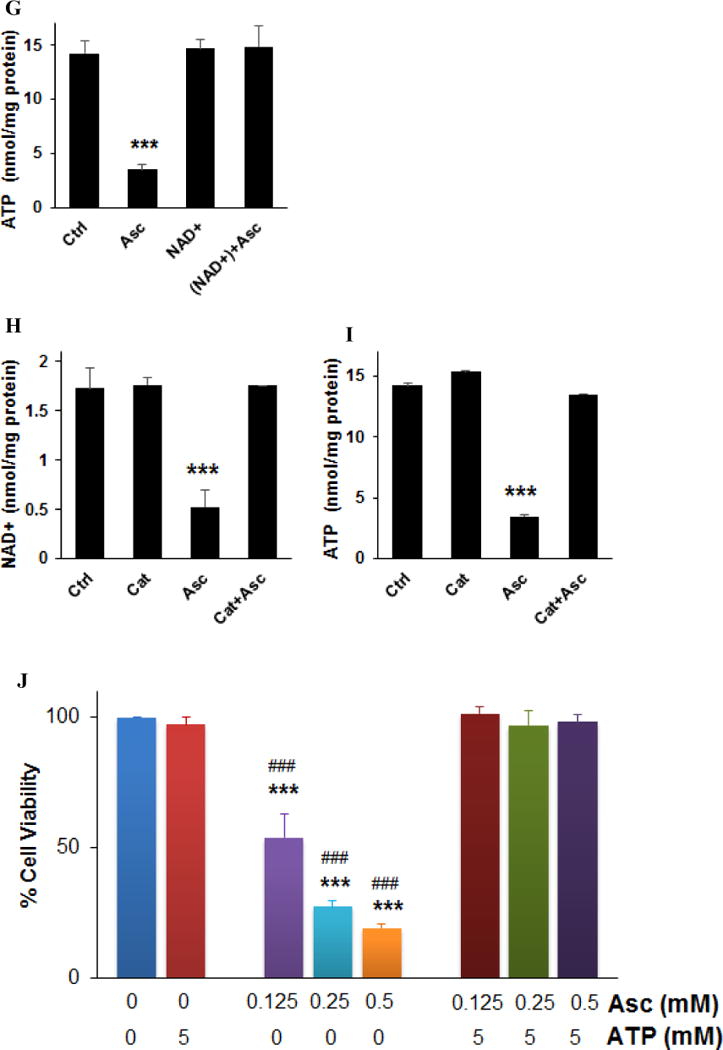
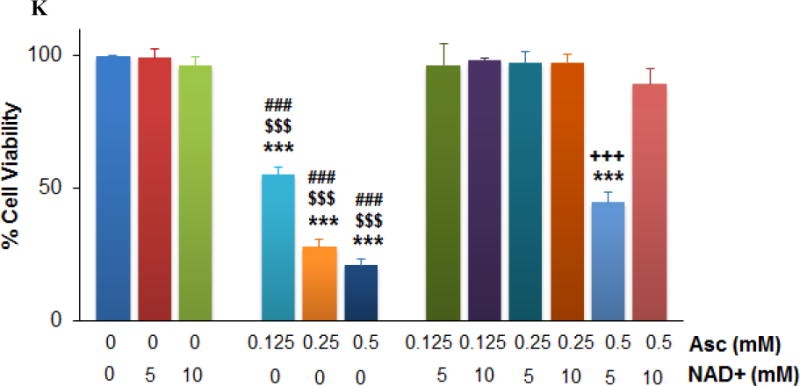
Ascorbate inhibited energy metabolism in neuroblastoma cells. A. Reduction of extracellular acidification rate (ECAR). B. Changes in oxygen consumption rate (OCR). SH-SY5Y cells were exposed to 0.5 mM (pink), 5 mM (red), or 10 mM (brown) ascorbate, or H2O2 (100 μM, green), and ECAR and OCR were measured by Seahorse. Injection A indicated the start point of each treatment. Bar graph shows the % change of ECAR (A) or OCR (B) at the highest inhibition (at about 105 min). C. Ascorbate but not DHA inhibited GAPDH activity in an ROS-dependent way. SH-SY5Y cells were treated for 4 hours. GAPDH activity was detected using a colorimetric assay from Abcam, and the units were normalized to protein content of each sample. Cat, catalase. Data are Mean ± SD (n = 3). *, p < 0.05 versus untreated cells (Ctrl) by ANOVA tests. D. Western blots of GAPDH protein expression level in SH-SY5Y cells. Cat, catalase 200 U/mL. H2O2, 200 μM. β-actin was a loading control. E and F. Decrease of NAD+ (E) and ATP (F) in SH-Sy5y cells with ascorbate treatment. ATP and NAD+ were analyzed by an HPLC assay, normalized first to the total cellular protein in each sample, and then compared to the untreated cells (Ctrl). The absolute values of ATP and NAD+ in nmoles/mg protein for the control groups were equivalent to those shown in H and I. Data are means ± SD (n = 3). *, p < 0.05; **, p < 0.01 versus control by ANOVA tests. G. Supplementation of NAD+ reversed ATP depletion. Asc, ascorbate 0.5 mM. NAD+ (10 mM) was pre-incubated with cells 30 minutes before ascorbate treatment. ATP was detected by HPLC at 4 hours of ascorbate treatment. ***, p < 0.001 versus control by ANOVA. H and I. Depletion of NAD+ (H) and ATP (I) by ascorbate was reversed by catalase. Ctrl, untreated SH-Sy5y cells; Cat, catalase 100 U/mL; Asc, 0.5 mM ascorbate. ATP and NAD+ were detected by HPLC at 4 hours of treatment. Data are means ± SD (n = 3). ***, p < 0.001 versus control by ANOVA. J and K. Supplementation of ATP (J) or NAD+ (K) reversed ascorbate-induced cell death. SH-SY5Y cells were pre-incubated with ATP or NAD+ for 30 minutes, and then were treated with ascorbate. Cell viability was detected 24 hours post-treatment. Data are means ± SD (n = 3). ***, p < 0.001 versus untreated cells; ###, p < 0.001 versus cells supplemented with 5 mM of ATP or NAD+; $$$, p < 0.001 versus cells supplemented with 10 mM NAD+; +++, p < 0.001 versus cells treated with 0.5 mM ascorbate; all by ANOVA tests.
Known as Warburg Effect (39), cancer cells depend more on glycolysis for ATP production while normal cells dependent more on oxidative phosphorylation. As a related finding, previous studies have shown that mitochondria were unlikely to play an important role in the cell’s sensitivity to ascorbate-induced death (11). Therefore, we predicted that ATP levels would substantially decrease subsequent to the glycolysis inhibition caused by NAD+ reduction and GAPDH inhibition in neuroblastoma cells. Indeed, in SH-SY5Y cells ATP was significantly decreased by ascorbate in a dose and time dependent manner (Fig. 2F). Supplementing NAD+ to the ascorbate-treated neuroblastoma cells completely rescued ATP levels (Fig. 2G), indicating reduction in NAD+ is likely responsible for ascorbate-induced GAPDH inhibition and glycolysis inhibition. Catalase completely protected NAD+ and ATP levels when added to ascorbate treatment (Fig. 2H, I), indicating H2O2 mediated these effects.
Lose of ATP resulted in cell death (40–42), and supplementation of ATP (5 mM) to SH-SY5Y cells treated with ascorbate (0.125-0.5 mM) restored cell viability (Fig. 2J). Supplementation of NAD+ to the cells also restored the cell viability (Fig. 2K) because of protection in ATP (Fig. 2G). Taken together, these data indicate that by formation of H2O2 (20), ascorbate reduces cellular NAD+ and therefore inhibits GAPDH, which leads to glycolysis inhibition. Glycolysis inhibition in turn causes ATP crisis in neuroblastoma cells that results in cell death.
Pharmacologic ascorbate activated poly-ADP-ribose polymerase (PARP) and therefore depleted NAD+
DNA double-strand breakage has been reported in ascorbate-treated cancer cells, which represented a consequence from H2O2 and ROS formation (10,20). Here, we detected phosphorylation of markers of DNA double-strand damage in ascorbate-treated neuroblastoma cells. Phosphorylation of ataxia telangiectasia mutated (ATM) and histone 2AX (H2AX) were increased in ascorbate-treated SH-SY5Y cells, as was the phosphorylation of checkpoint kinase 2 (Chk2) (Fig. 3A), a key component of the DNA damage response. Consistent with reported data (10), H2O2 treatment mimicked the effects of ascorbate, whereas catalase prevented the phosphorylation of ATM, H2AX, and Chk2 (Fig. 3A). The DNA repair enzyme PARP was activated by ascorbate treatment, which was reflected by increased levels of poly-ADP-ribose (PAR) (Fig. 3B). Because activated PARP utilizes NAD+ as a substrate (43), significant decrease in NAD+ levels was found in ascorbate-treated cells (Fig. 2D and Fig. 3C), and subsequently decrease in ATP levels (Fig. 2E and Fig. 3D). A PARP inhibitor, olaparib (Fig. 3B), preserved NAD+ and ATP levels (Fig. 3C, D). This suggests in neuroblastoma cells PARP activation drives ascorbate-induced NAD+ and ATP depletion.
FIGURE 3.
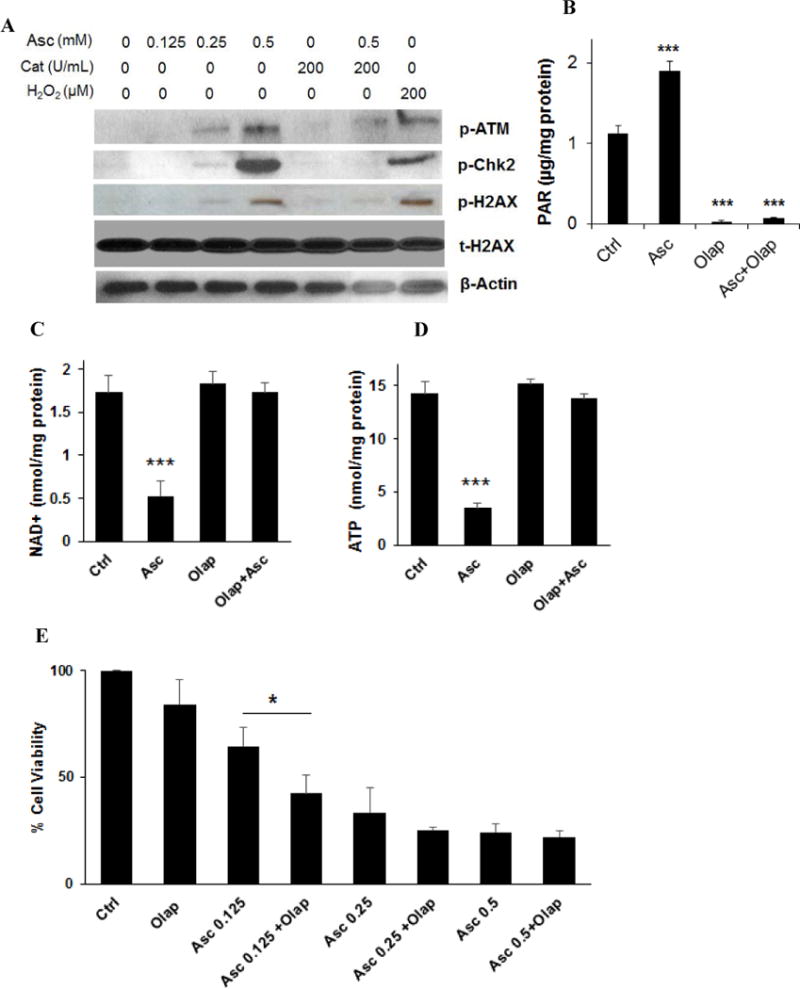
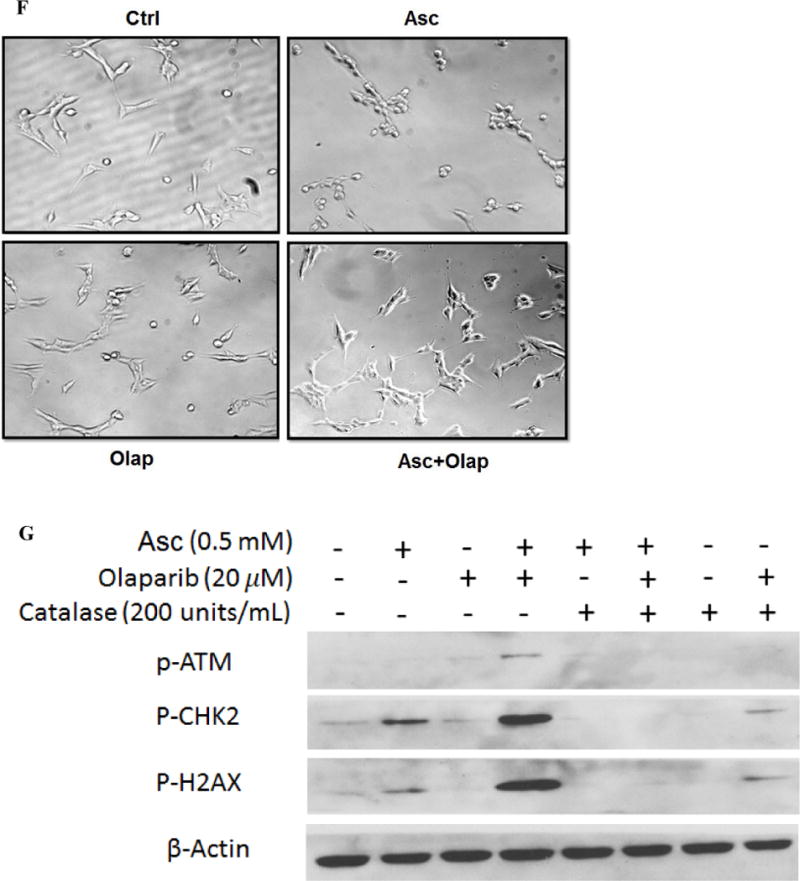
Ascorbate depleted NAD+ through PARP activation. A. Ascorbate-generated ROS induced phosphorylation of markers of DNA damage. SH-Sy5y cells were treated for 2 hours. Western blots were performed to detect phosphorylated ATM (p-ATM), Chk2 (p-Chk2), and H2AX (p-H2AX). Asc, ascorbate; Cat, catalase. β-actin was a loading control. B. PARP activation measured by PAR levels. SH-SY5Y cells were treated under indicated conditions. PAR levels were measured using an HT PARP in vivo Pharmacodynamic Assay II kit from Trevigen, at 2 hours of treatment, and then normalized to protein content of each sample. AA, ascorbate 0.5 mM; Olap, olaparib 20 μM. Data are Mean ± SD (n = 3). C and D. Reverse of NAD+ (C) and ATP (D) levels in ascorbate-treated SH-SY5Y cells by PARP inhibitor. Asc, ascorbate 0.5 mM; Olap, olaparib 20 μM. Data are Mean ± SD (n = 3). E. PARP inhibitor did not reverse cell death induced by ascorbate. SH-SY5Y cells were exposed to olaparib (Olap, 20 μM), and/or ascorbate (Asc) at the indicated concentrations for 24 hours. Data are Mean ± SD (n = 3). *, p < 0.05; ***, p < 0.001 by ANOVA. F. Reverse of SH-SY5Y morphological changes by PARP inhibitor at 2 hours of treatment. Asc, ascorbate 0.5 mM; Olap, olaparib 20 μM. G. Phosphorylation of DNA double strain damage markers. SH-SY5Y cells were treated for 2 hours.
Interestingly, although PARP inhibition preserved NAD+ and ATP levels, it did not prevent ascorbate-induced cell death (Fig. 3E). Olaparib appeared to only mitigate morphological changes of SH-SY5Y cells at 2 hours of ascorbate treatment (Fig. 3F), but did not change cell viability at 24 hours in addition to ascorbate (Fig. 3E). At a lower concentration of ascorbate (0.125 mM), cytotoxicity was even enhanced by the PARP inhibitor (Fig. 3E). This indicates that under conditions of PARP inhibition, mechanisms independent of NAD+ and ATP depletion can also mediate ascorbate-induced cell death. We postulate that PARP inhibition leads to accumulation of DNA damage, which leads to cell death despite that NAD+ and ATP were protected. Consistent with this hypothesis, phosphorylation of H2AX was greatly enhanced when olaparib was combined with ascorbate, so were phosphorylation of ATM and Chk2 (Fig. 3G).
Pharmacologic ascorbate inhibited neuroblastoma growth in a mouse model
The impact of ascorbate in neuroblastoma tumor growth was tested in SH-SY5Y tumor xenografts. The tumor growth was significantly reduced by 4 g/kg daily ascorbate given by intraperitoneal (IP) injection (Fig. 4A), which is equivalent to intravenous (IV) dose of ~1.3 g/kg (20). The survival of the tumor bearing mice was significantly improved by ascorbate treatment, even when the treatment stopped at day-38 (Fig. 4B). To validate the mechanisms in vivo, we detected GAPDH expression and activity in tumor tissues. Ascorbate treatment did not alter GAPDH expression (Fig. 4C), only significantly decreased GAPDH activity in tumors (Fig. 4D), consistent with in vitro data. We further detected the DNA damage markers p-ATM, p-Chk2 and p-H2AX. At least two of the three markers p-Chk2 and p-H2AX showed increase in ascorbate-treated tumors versus control tumors (Fig. 4E). Because ATM phosphorylation is an early-time response to DNA damage usually peaks within 60 minutes of exposure to ROS stress (44), we did not detect obvious increase of p-ATM with the in vivo tumor samples presumably due to the samples were collected >24 hours after the last ascorbate treatment. No adverse effects were observed and no differences in the body weight were found between the treated and untreated mice (Fig. 4E). IP injection of 4 g/kg ascorbate increased blood ascorbate concentrations to ~15 mM after 30 min of injection (Fig. 4F), consistent with previous reports with similar administrations (8,11), and are relevant to clinical use in humans (8,10,19).
FIGURE 4.
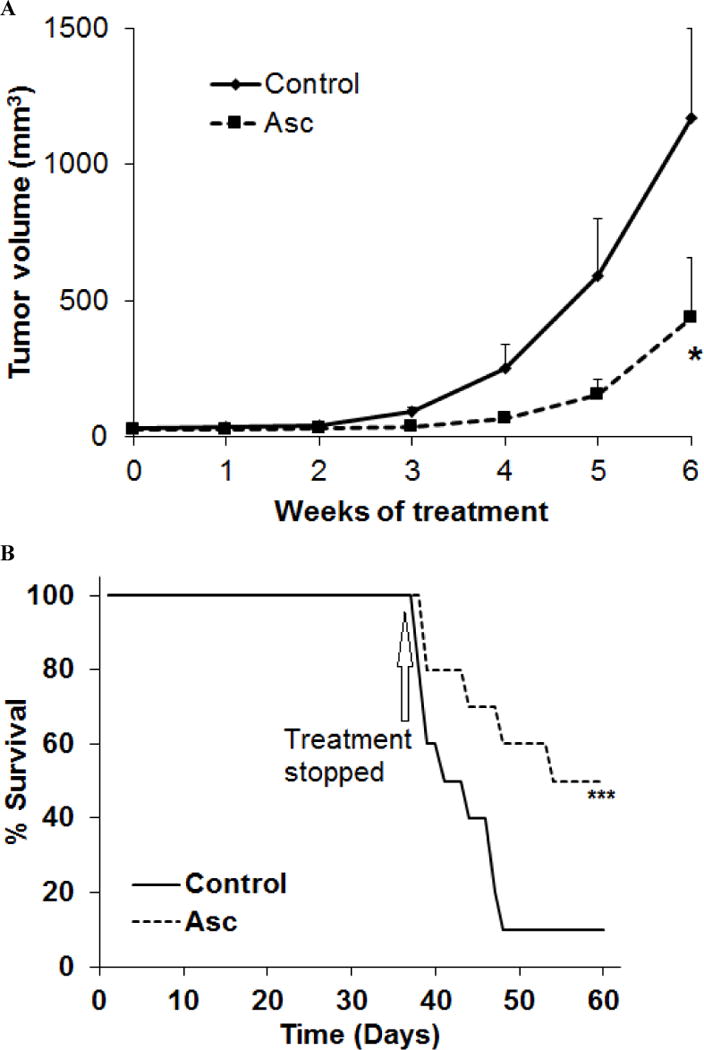
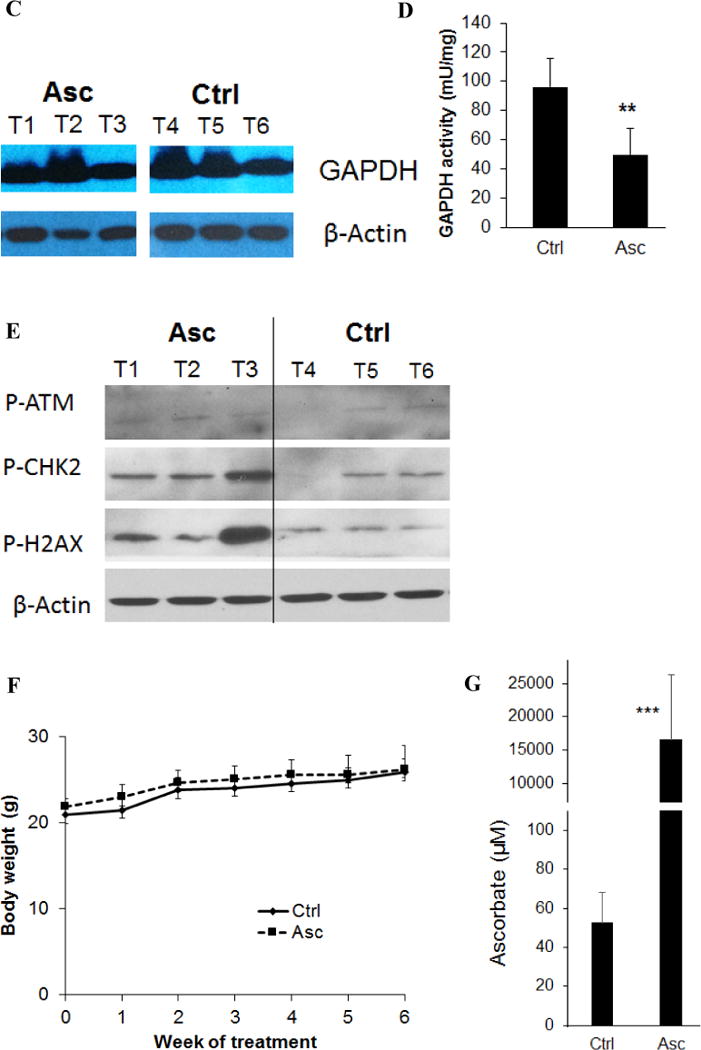
Ascorbate inhibited neuroblastoma growth and improved survival in a mouse xenograft model. A. Growth curve of SH-SY5Y tumor in nude mice (n=10 per group). Asc, ascorbate 4 g/kg body weight, daily intraperitoneal injection. Control mice received saline by daily intraperitoneal injection. *, p < 0.05 by one way ANOVA. B. Kaplan-Meier survival curve (n = 10 per group). Arrow marks day-38 when treatment stopped. ***, p <0.001 by log-rank test. n =10 for each group. C. Western blots for GAPDH protein levels in tumor tissues of mice treated or untreated with ascorbate. β-actin was a loading control. T, tumor tissues. n = 3 for each group. D. GAPDH activity in tumor tissues of mice treated or untreated with ascorbate. Data are Mean ± SD of 3 mice in each group. **, p < 0.01 versus control, by t-test. E. Western blots for DNA damage markers p-ATM, p-Chk2 and p-H2AX in tumor tissues of mice treated or untreated with ascorbate. β-actin was a loading control. T, tumor tissues. n = 3 for each group. F. Body weight of mice in each group. G. Blood ascorbate concentrations 30 min after ascorbate administration. ***, p < 0.001 versus control, by t-test.
DISCUSSION
Recent studies found that ascorbate-induced oxidative stress caused DNA damage and inhibited glycolysis in susceptible cancer cells but not in normal cells, and attributed this selectivity to fundamental differences of energy metabolism between cancer and normal cells known as Warburg Effect (10,20,32,39). Relative to normal cells, cancer cells rely to a greater extent on glycolysis for their ATP production. Compared to mitochondrial oxidative phosphorylation, the efficiency of ATP production from glycolysis is much less (2 ATPs vs 36 ATPs from a glucose molecule). Glycolysis inhibition, therefore, would predictably have a greater impact on cancer cell ATP levels than it would have on non-cancer cell ATP levels (10). We postulated earlier that the ascorbate-generated H2O2 induces DNA damage and activates PARP. Activated PARP depletes NAD+ and therefore inhibits glycolysis (20). Data in this study provide unequivocal evidence to support the hypothesis (Fig. 5). Depletions of NAD+ inhibits GAPDH activity, and this in turn reduces ATP levels in neuroblastoma cells. Supplementation of NAD+ preserved ATP levels in the cells, and protected them from ascorbate-induced death. Interestingly, PARP inhibitor did not prevent ascorbate-induced cell death despite that PARP inhibition preserved NAD+ and ATP levels. This is presumably because PARP inhibition increases fatal DNA double-strand damages. Consistent with our observation, another study showed that PARP inhibition does not prevent oxidant-induced cell death, but instead changes the mechanism of cell death (45). These different but related mechanisms could each play an important role in cancer cell’s responses to ascorbate. If a cancer cell repairs DNA at the cost of NAD+, a reduced glycolysis flux and ATP depletion result. If the PARP-dependent DNA repair machinery is inhibited, although ATP and NAD+ are preserved, excessive DNA damage could accumulate and lead to cell death. Ideally, complementary but distinct cytotoxic mechanisms could reduce the emergence of treatment resistance. Also, in cancers that use alternative energy metabolism pathways other than glucose, combining PARP inhibitor with ascorbate treatment could enhance effectiveness. In that case, the dose of PARP inhibitor could be reduced to minimize associated toxicities. Ascorbate is also reported to have synergistic effects with other chemotherapeutic agents and radiation therapy (9,10,13,14,46). Regardless, further studies of ascorbate cytotoxicity in cancer cells that lack PARP but have PARP-independent DNA repair mechanisms are warranted.
FIGURE 5.
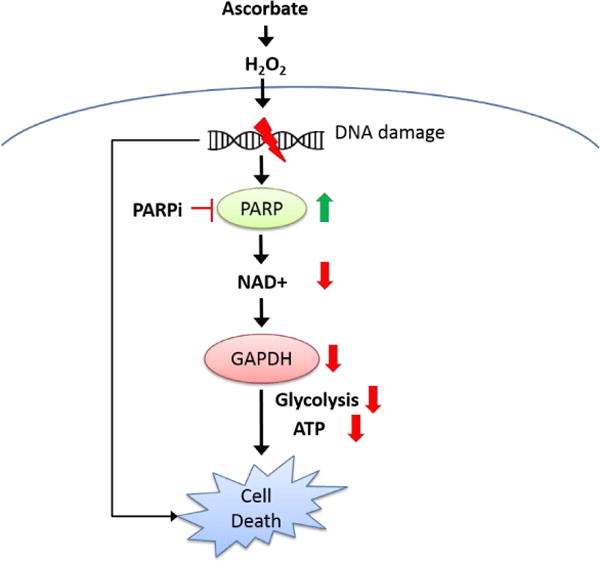
A simplified scheme for the mechanisms of pharmacologic ascorbate-induced cell death. Through oxidative stress, ascorbate damages DNA and activates PARP. Activated PARP depletes NAD+, therefore GAPDH activity is inhibited, and ATP is depleted in neuroblastoma cells, leading to cell death. In the absence of PARP activation, fatal DNA damage accumulates and leads to cell death. PARPi, PARP inhibitor.
The ascorbate-induced loss of GAPDH activity has two possible explanations. First, GAPDH activity is directly influenced by NAD+ levels. Second, ascorbate may oxidize the GAPDH active-site (cysteine 152, C152), which is known to lower GAPDH activity. Interestingly, a previous study showed that C152 of GAPDH underwent S-glutathionylation with ascorbate treatment, which is reversible. However the irreversible oxidized form, Cys-SO3H, was not detected (32). Glutathionylation effects on protein activity are not completely known, but one of its roles is that it protects the cysteine amino acids from becoming further oxidized. Here, we show that supplementation of NAD+ prevents ascorbate-induced ATP loss in cancer cells. Together, these data support the possibility that GAPDH activity loss in ascorbate treated cells is likely to be mediated by loss of NAD+.
Our current study does not rule out a role for mitochondria in ascorbate-induced cancer cell death. Nevertheless, mitochondrial contributions are likely to be quite limited, as previous studies have shown that mitochondrial DNA-depleted cancer cells (ρ0 cells) that lack functional respiratory chains have the same sensitivity to ascorbate as the mitochondria intact cells (11). Also, transfection and overexpression of mitochondria-targeted catalase did not rescue the cell death under ascorbate treatment (11).
A recent study (32) suggested that oncogenic KRAS/BRAF sensitize colon cancer cells to ascorbate treatment, and suggested the underlying reason is because these mutations upregulate expression of GLUT1, and therefore enhance uptake of vitamin C via its oxidized form dehydroascorbate (DHA), and then reducing of DHA inside the cells on the expense of glutathione induced oxidative stress. This hypothesis emphasizes a role of DHA in inducing intracellular oxidative stress. Our data here do not support this hypothesis because treatment with DHA did not cause cell death, and did not reduce GAPDH activity, as treatment with the same concentration of ascorbate did. Previously we published data treating lymphoma cells with 2 mM of ascorbate, or 2 mM of DHA (21). Whereas DHA was up taken into the cells and reduced to ascorbate, the DHA treatment did not induce any cell death, while 2 mM ascorbate killed ~100% of the cancer cells. Moreover, addition of catalase into culture media reverses the GAPDH inhibition. As catalase (232 kDa) supplemented to culture media does not enter cells, the data indicate that H2O2 outside of the cells likely initiate the damage. A recent study (47) indicated that ascorbate influenced hypoxia induced factor (HIF) activity and reduced GLUT1 expression while DHA failed to do so. A study used DHA, but called it vitamin C, failed to have anti-cancer effects (48). Other studies also showed that H2O2 formation from ascorbate oxidation happens outside of the cells (49), and that pancreatic cancer cell with wildtype KRAS showed no difference in sensitivity to ascorbate than those harboring KRAS mutations (11).
Our in vivo data showed neuroblastoma xenograft tumors were sensitive to high dose parenteral ascorbate. This raises the potential for using high dose intravenous ascorbate (IVC) treatment in children with high-risk metastatic neuroblastoma, who face a poor prognosis and many adverse effects from current treatments. The safety and low-toxicity of IVC has been proved by a number of clinical trials in adult oncologic patients (8,10,12,19,50–52). However these data are not available for the pediatric population. Further pharmacokinetic and safety studies in children are clearly needed to fully evaluate this translational opportunity.
CONCLUSIONS
Neuroblastomas affect children at ages of 0-14, and account for 15% of all pediatric cancer deaths. Current treatments are intense and have many side effects. Ascorbate has been reported as a relatively safe and potentially effective treatment for a number of cancers. The advantage is that ascorbate treatment is free of major toxic side effects. In this study, we first show sensitivity of neuroblastoma to ascorbate treatment, both in vitro and in vivo. We then elucidate that through oxidative stress, ascorbate depletes NAD+, inhibits glycolysis and therefore depletes ATP in neuroblastoma cells which are reported to depend on glycolysis for energy metabolism. The study also shows that PARP activation is critical in ascorbate-induced NAD+/ATP depletion. However, in the absence of PARP activation, fatal DNA damage accumulates and leads to cell death. These studies suggest that ascorbate could be effective in the treatment of glycolysis-dependent tumors including neuroblastoma. In glycolysis-independent tumors, combining PARP inhibitor with ascorbate could be useful.
MATERIAL AND METHODS
Ascorbate, dehydroascorbic acid, cell lines and cell viability assay
L-ascorbic acid was purchased from Sigma-Aldrich (catalogue #A5960) with a purity of ≥ 99.0%. Ascorbate solution was prepared by dissolving the L-ascorbic acid in ultra-pure water with NaOH (10 M in ultra-pure water) added drop-wise to adjust pH to 7.0, and then filtered through 0.22 μM syringe filter. The process was done on ice and avoided direct light. Ascorbate solution (1.0 M) was stored in small aliquots in amber vials at −80 °C, and each aliquot was thawed for one-time immediate use. Dehydroascorbic acid (DHA) was made on ice immediately prior to utilization, using the method of bromine oxidation of freshly prepared L-ascorbic acid solution as previously described (53).
The human neuroblastoma cell line SH-SY5Y was obtained from the Alzheimer Disease Center at the University of Kansas Medical Center. Other cell lines were purchased from American Type Culture Collection (ATCC). All cells were cultured in recommended media supplemented with 10% Fetal Bovine Serum (FBS). The cytotoxic effects of ascorbate on SH-SY5Y cells was measured by the Sulforhodamine B staining assay which determines cell density based on the measurement of cellular protein content (54) (CytoScan™-SRB Cytotoxicity Assay, G-Biosciences, St. Louise, MO). The amount of dye extracted from stained cells is directly proportional to the cell mass. Briefly, cells in log-phase growth in 96 well plates (1×104 cells/well) were exposed to 100 μL culture media containing different concentrations of ascorbate, and/or other reagents as indicated. Because the specification of exposure/dose of both H2O2 and ascorbate are best presented as moles per cell (55), we provide both the initial nominal concentration and moles/cell for these reagents, as appropriate. After 24 hours of treatment, culture medium was removed and cells were fixed with the fixative reagent provided, washed and air dried. SRB dye solution (100 μL) was added and incubated for 30 minutes at room temperature in the dark. Cells were then washed with the wash solution provided and air dried. Then color was established by adding 200 μL SRB solubilization buffer, and absorbance was detected at 565 nm. Cell viability for the treatment groups was normalized to the control groups (the control group was set to 100%).
Western blots
Cells or mouse tumor tissues were lysed on ice in radioimmunoprecipitation (RIPA) assay buffer (25 mM tris-HCl (pH 7.6), 150 mM NaCl, 1 mM EDTA, 1% NP-40, 1% sodium deoxycholate, 0.1% SDS, 1×Pierce protease and phosphatase inhibitor), followed by homogenization and sonication. Samples were then centrifuged at 20,000 g for 15 minutes at 4°C. Equal amounts of protein extracts (20 μg) were separated by 8–12 % SDS-PAGE, and transferred onto polyvinylidene difluoride membranes (PVDF). The membranes were treated with specified antibodies (Cell Signaling, Danvers, MA), and then incubated with horseradish peroxidase conjugated secondary antibodies (Cell Signaling, Danvers, MA). Blots were detected using enhanced chemiluminescence Western blotting detection reagents (Thermo Scientific, Rockford, IL).
ECAR and OCR detection
Approximately 80,000 SH-SY5Y cells were plated in an XF24 cell culture microplate (Agilent Technologies/Seahorse Bioscience) using a standard manufacturer-recommended two-step seeding procedure. After plating cells, the microplate was kept overnight in a 37 °C, 5% CO2 incubator. This plating protocol yielded a monolayer cell density of ~90%. Individual wells were next placed in pyruvate-free and buffer-free DMEM (with 25 mM glucose). The plate was incubated in a 37 °C, non-CO2 incubator for 45 min and then transferred to the microplate stage of a Seahorse XF24 flux analyzer. ECAR and OCR measurements were taken using a 3 minute mix, 2 minute wait, and 3 minute measurement cycling protocol repeated three times for baseline, and nine times for post-injection A. Injection A yielded either a final concentration of: 0 mM ascorbate/H2O2 (vehicle/control), 0.5 mM ascorbate, 5 mM ascorbate, 10 mM ascorbate, or 100 μM H2O2 (equivalent to 75 pmoles/cell). Each treatment (injection A) was completed in 4 independent wells.
GAPDH activity assay
The enzyme activity of GAPDH was determined using a GAPDH Activity Assay Kit (Abcam, Cambridge, England) following the manufactures’ protocol. The assay is based on colorimetric measurement of NADH formation catalyzed by GAPDH. One million cells (106) in 10 mL medium were seeded into 35 mm petri-dish and treated with ascorbate. Cells or mouse tumor tissues were suspended in GAPDH assay buffer and subjected to homogenization and sonication. The cytosolic fraction was prepared by centrifuging the lysate at 20,000 g for 15 minutes at 4 °C, and then 20 μL were used for each reaction. The samples were incubated with GAPDH substrates and a color developer provided at 37 °C, and the OD450 nm was measured in kinetic mode for 10 minutes. For the calculation of NADH production, a standard curve was generated using NADH amount ranging from 2.5 to 12.5 nmol/reaction. Two time points in the liner range of the sample curve were used for the calculation. A positive control was provided in the kit. Background controls were samples incubated with the color developer but without GAPDH substrate. GAPDH activity (U) was calculated as the amount of NADH production (nmol) in unit time (minute), and was normalized to the protein content of the whole-cell lysate detected by the bicinchoninic acid protein method (Pierce Biotechnology).
ATP, NAD+ and ascorbate detection
Cellular ATP and NAD+ were extracted by rapidly lysing cells in 0.05 M KOH and then immediately neutralizing to pH 6.0 using 0.1 M KH2PO4. The supernatant was analyzed for ATP and NAD+ using a gradient method on reversed-phase high-performance liquid chromatography (HPLC) with UV detection, as described previously (10). Blood and tissue samples from mice were processed and ascorbate detected using an HPLC method with coulometric electrochemical detection, as previous described (56). All cell and tissue values were normalized to the protein content (BCA Assay).
Mouse xenografts and treatment
All animal experiments followed a protocol approved by the Institutional Animal Care and Use Committee at the University of Kansas Medical Center. SH-SY5Y cells (2 ×106) suspended in Matrigel solution (6 mg/mL in PBS) were injected subcutaneously into the flank of female athymic mice (Ncr-nu/nu ages 6 weeks) (Envigo, Indianapolis, IN). When tumors became palpable, mice were divided into two groups (10 mice per group) based on tumor size to have equivalent average tumor burden in each group. Treatment commenced with intraperitoneal injection of 4 g ascorbate/kg daily. Ascorbate was prepared as 1.0 M solution in water with pH adjusted to 7.0 with NaOH. Control group was injected with same volume of saline with equal osmolality. Longitudinal tumor volume was measured weekly by caliper, and calculated using volume = (length) × (width)2 × 0.5. Treatment stopped at day-38 when tumor volume in the control group began to reach preset criteria for euthanasia (1,500 cm3). Mice were monitored daily to remove any mouse that reached the tumor volume or became moribund. Survival was recorded as the reverse of the mouse removal rate. Frozen tumor tissues were subjected to Western blot and GAPDH activity analysis (as descried in prior sections).
Statistical Analysis
SYSTAT 11 software was used for student T-test for comparison between 2 groups, and for ANOVA when comparison involved more than 2 groups. Log-rank test was used for survival. A difference was considered significant at the p < 0.05 level. Correlation analysis used the standard Pears Tests.
Acknowledgments
This work was supported by a grant from the University of Kansas Endowment provided by the L. Charles Hilton Family Foundation. The Kansas University Alzheimer’s Disease Center is supported by P30 AG035982.We thank former graduate students Kishore Polireddy and Lezi E, and current graduate student Ruochen Dong for exploratory work on this project.
Abbreviations
- 3-MA
3-methyladenine
- Asc
ascorbate
- ATM
ataxia telangiectasia mutated
- ATP
adenosine triphosphate
- BRAF
murine sarcoma viral oncogene homolog B
- Cat
catalase
- Chk2
checkpoint kinase 2
- CQ
chloroquine
- Cys-SO3H
cysteine S-sulfate
- DHA
dehydroascorbate
- ECAR
extracellular acidification rate
- Eto
etoposide
- FBS
fetal bovine serum
- GAPDH
glyceraldehyde 3-phosphate dehydrogenase
- GLUT1
glucose transporter 1
- H2AX
histone 2AX
- H2O2
hydrogen peroxide
- HIF
hypoxia induced factor
- IVC
intravenous ascorbate
- KRAS
Kirsten rat sarcoma viral oncogene
- LC3-I and II
microtubule-associated protein light chain 3-I and II
- NAD+/NADH
nicotinamide adenine dinucleotide oxidized/reduced form
- OCR
oxygen consumption rate
- Olap
olaparib
- PAR
poly-ADP-ribose
- PARP
poly-ADP-ribose polymerase
- PARPi
PARP inhibitor
- PI3K
phosphatidylinositol 3-kinases
- ROS
reactive oxygen species
- SD
standard deviation
- SRB
Sulforhodamine B
- starv
serum starvation
- TCA cycle
tricarboxylic acid cycle
- Z-VAD
Z-Valine-Alanine-Aspartic acid fluoromethyl ketone
Footnotes
AUTHOR CONTRIBUTIONS
QC formed the hypothesis and designed the study. EM participated in experimental design and conducted most of the experiments and collected data. QC and EM analyzed the results, and wrote the first manuscript draft. PC conducted the HPLC analysis of ATP, NAD+ and ascorbate, and part of the cytotoxicity experiments. HMW conducted the Seahorse experiments. TW conducted part of the western blots experiments and data analysis. RHS participated in study design, trouble shooting, data analysis, and oversaw the Seahorse experiments. All authors contributed to manuscript preparation.
References
- 1.Park JR, Bagatell R, London WB, Maris JM, Cohn SL, Mattay KK, Hogarty M, Committee COGN. Children’s Oncology Group’s 2013 blueprint for research: neuroblastoma. Pediatr Blood Cancer. 2013;60:985–993. doi: 10.1002/pbc.24433. [DOI] [PubMed] [Google Scholar]
- 2.Cheung NK, Zhang J, Lu C, Parker M, Bahrami A, Tickoo SK, Heguy A, Pappo AS, Federico S, Dalton J, Cheung IY, Ding L, Fulton R, Wang J, Chen X, Becksfort J, Wu J, Billups CA, Ellison D, Mardis ER, Wilson RK, Downing JR, Dyer MA, St Jude Children’s Research Hospital-Washington University Pediatric Cancer Genome, P Association of age at diagnosis and genetic mutations in patients with neuroblastoma. JAMA. 2012;307:1062–1071. doi: 10.1001/jama.2012.228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nakagawara A. Molecular basis of spontaneous regression of neuroblastoma: role of neurotrophic signals and genetic abnormalities. Hum Cell. 1998;11:115–124. [PubMed] [Google Scholar]
- 4.Board P. D. Q. P. T. E. PDQ Cancer Information Summaries. National Cancer Institute (US); Bethesda (MD): 2002. Neuroblastoma Treatment (PDQ(R)): Health Professional Version. [Google Scholar]
- 5.Cheung NK, Dyer MA. Neuroblastoma: developmental biology, cancer genomics and immunotherapy. Nat Rev Cancer. 2013;13:397–411. doi: 10.1038/nrc3526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Maris JM. Recent advances in neuroblastoma. N Engl J Med. 2010;362:2202–2211. doi: 10.1056/NEJMra0804577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Knight KR, Kraemer DF, Neuwelt EA. Ototoxicity in children receiving platinum chemotherapy: underestimating a commonly occurring toxicity that may influence academic and social development. J Clin Oncol. 2005;23:8588–8596. doi: 10.1200/JCO.2004.00.5355. [DOI] [PubMed] [Google Scholar]
- 8.Chen Q, Espey MG, Sun AY, Pooput C, Kirk KL, Krishna MC, Khosh DB, Drisko J, Levine M. Pharmacologic doses of ascorbate act as a prooxidant and decrease growth of aggressive tumor xenografts in mice. Proc Natl Acad Sci U S A. 2008;105:11105–11109. doi: 10.1073/pnas.0804226105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Espey MG, Chen P, Chalmers B, Drisko J, Sun AY, Levine M, Chen Q. Pharmacologic ascorbate synergizes with gemcitabine in preclinical models of pancreatic cancer. Free Radic Biol Med. 2011;50:1610–1619. doi: 10.1016/j.freeradbiomed.2011.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ma Y, Chapman J, Levine M, Polireddy K, Drisko J, Chen Q. High-dose parenteral ascorbate enhanced chemosensitivity of ovarian cancer and reduced toxicity of chemotherapy. Science translational medicine. 2014;6:222ra218. doi: 10.1126/scitranslmed.3007154. [DOI] [PubMed] [Google Scholar]
- 11.Du J, Martin SM, Levine M, Wagner BA, Buettner GR, Wang SH, Taghiyev AF, Du C, Knudson CM, Cullen JJ. Mechanisms of ascorbate-induced cytotoxicity in pancreatic cancer. Clin Cancer Res. 2010;16:509–520. doi: 10.1158/1078-0432.CCR-09-1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schoenfeld JD, Sibenaller ZA, Mapuskar KA, Wagner BA, Cramer-Morales KL, Furqan M, Sandhu S, Carlisle TL, Smith MC, Abu Hejleh T, Berg DJ, Zhang J, Keech J, Parekh KR, Bhatia S, Monga V, Bodeker KL, Ahmann L, Vollstedt S, Brown H, Shanahan Kauffman EP, Schall ME, Hohl RJ, Clamon GH, Greenlee JD, Howard MA, Shultz MK, Smith BJ, Riley DP, Domann FE, Cullen JJ, Buettner GR, Buatti JM, Spitz DR, Allen BG. O2- and H2O2-Mediated Disruption of Fe Metabolism Causes the Differential Susceptibility of NSCLC and GBM Cancer Cells to Pharmacological Ascorbate. Cancer Cell. 2017 doi: 10.1016/j.ccell.2017.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Verrax J, Calderon PB. Pharmacologic concentrations of ascorbate are achieved by parenteral administration and exhibit antitumoral effects. Free Radic Biol Med. 2009;47:32–40. doi: 10.1016/j.freeradbiomed.2009.02.016. [DOI] [PubMed] [Google Scholar]
- 14.Du J, Cieslak JA, 3rd, Welsh JL, Sibenaller ZA, Allen BG, Wagner BA, Kalen AL, Doskey CM, Strother RK, Button AM, Mott SL, Smith B, Tsai S, Mezhir J, Goswami PC, Spitz DR, Buettner GR, Cullen JJ. Pharmacological Ascorbate Radiosensitizes Pancreatic Cancer. Cancer Res. 2015;75:3314–3326. doi: 10.1158/0008-5472.CAN-14-1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen Q, Polireddy K, Chen P, Dong R. The unpaved journey of vitamin C in cancer treatment. Can J Physiol Pharmacol. 2015;93:1–9. doi: 10.1139/cjpp-2014-0509. [DOI] [PubMed] [Google Scholar]
- 16.Graumlich JF, Ludden TM, Conry-Cantilena C, Cantilena LR, Jr, Wang Y, Levine M. Pharmacokinetic model of ascorbic acid in healthy male volunteers during depletion and repletion. Pharm Res. 1997;14:1133–1139. doi: 10.1023/a:1012186203165. [DOI] [PubMed] [Google Scholar]
- 17.Levine M, Conry-Cantilena C, Wang Y, Welch RW, Washko PW, Dhariwal KR, Park JB, Lazarev A, Graumlich JF, King J, Cantilena LR. Vitamin C pharmacokinetics in healthy volunteers: evidence for a recommended dietary allowance. Proc Natl Acad Sci U S A. 1996;93:3704–3709. doi: 10.1073/pnas.93.8.3704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Padayatty SJ, Sun H, Wang Y, Riordan HD, Hewitt SM, Katz A, Wesley RA, Levine M. Vitamin C pharmacokinetics: implications for oral and intravenous use. Ann Intern Med. 2004;140:533–537. doi: 10.7326/0003-4819-140-7-200404060-00010. [DOI] [PubMed] [Google Scholar]
- 19.Hoffer LJ, Levine M, Assouline S, Melnychuk D, Padayatty SJ, Rosadiuk K, Rousseau C, Robitaille L, Miller WH., Jr Phase I clinical trial of i.v. ascorbic acid in advanced malignancy. Ann Oncol. 2008;19:1969–1974. doi: 10.1093/annonc/mdn377. [DOI] [PubMed] [Google Scholar]
- 20.Chen Q, Espey MG, Sun AY, Lee JH, Krishna MC, Shacter E, Choyke PL, Pooput C, Kirk KL, Buettner GR, Levine M. Ascorbate in pharmacologic concentrations selectively generates ascorbate radical and hydrogen peroxide in extracellular fluid in vivo. Proc Natl Acad Sci U S A. 2007;104:8749–8754. doi: 10.1073/pnas.0702854104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen Q, Espey MG, Krishna MC, Mitchell JB, Corpe CP, Buettner GR, Shacter E, Levine M. Pharmacologic ascorbic acid concentrations selectively kill cancer cells: action as a pro-drug to deliver hydrogen peroxide to tissues. Proc Natl Acad Sci U S A. 2005;102:13604–13609. doi: 10.1073/pnas.0506390102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Levine M, Violet PC. Data Triumph at C. Cancer Cell. 2017;31:467–469. doi: 10.1016/j.ccell.2017.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lin SY, Lai WW, Chou CC, Kuo HM, Li TM, Chung JG, Yang JH. Sodium ascorbate inhibits growth via the induction of cell cycle arrest and apoptosis in human malignant melanoma A375.S2 cells. Melanoma Res. 2006;16:509–519. doi: 10.1097/01.cmr.0000232297.99160.9e. [DOI] [PubMed] [Google Scholar]
- 24.Carosio R, Zuccari G, Orienti I, Mangraviti S, Montaldo PG. Sodium ascorbate induces apoptosis in neuroblastoma cell lines by interfering with iron uptake. Mol Cancer. 2007;6:55. doi: 10.1186/1476-4598-6-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hong SW, Jin DH, Hahm ES, Yim SH, Lim JS, Kim KI, Yang Y, Lee SS, Kang JS, Lee WJ, Lee WK, Lee MS. Ascorbate (vitamin C) induces cell death through the apoptosis-inducing factor in human breast cancer cells. Oncol Rep. 2007;18:811–815. [PubMed] [Google Scholar]
- 26.Kang JS, Cho D, Kim YI, Hahm E, Yang Y, Kim D, Hur D, Park H, Bang S, Hwang YI, Lee WJ. L-ascorbic acid (vitamin C) induces the apoptosis of B16 murine melanoma cells via a caspase-8-independent pathway. Cancer Immunol Immunother. 2003;52:693–698. doi: 10.1007/s00262-003-0407-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Verrax J, Cadrobbi J, Marques C, Taper H, Habraken Y, Piette J, Calderon PB. Ascorbate potentiates the cytotoxicity of menadione leading to an oxidative stress that kills cancer cells by a non-apoptotic caspase-3 independent form of cell death. Apoptosis. 2004;9:223–233. doi: 10.1023/B:APPT.0000018804.26026.1a. [DOI] [PubMed] [Google Scholar]
- 28.Ohtani S, Iwamaru A, Deng W, Ueda K, Wu G, Jayachandran G, Kondo S, Atkinson EN, Minna JD, Roth JA, Ji L. Tumor suppressor 101F6 and ascorbate synergistically and selectively inhibit non-small cell lung cancer growth by caspase-independent apoptosis and autophagy. Cancer Res. 2007;67:6293–6303. doi: 10.1158/0008-5472.CAN-06-3884. [DOI] [PubMed] [Google Scholar]
- 29.Chen P, Yu J, Chalmers B, Drisko J, Yang J, Li B, Chen Q. Pharmacological ascorbate induces cytotoxicity in prostate cancer cells through ATP depletion and induction of autophagy. Anticancer Drugs. 2011 doi: 10.1097/CAD.0b013e32834fd01f. [DOI] [PubMed] [Google Scholar]
- 30.Farah IO, Lewis VL, Ayensu WK, Mahmud O. Assessing the survival of mrc-5 and a549 cell lines upon exposure to ascorbic Acid and sodium ascorbate - biomed 2011. Biomed Sci Instrum. 2011;47:201–206. [PubMed] [Google Scholar]
- 31.Verrax J, Dejeans N, Sid B, Glorieux C, Calderon PB. Intracellular ATP levels determine cell death fate of cancer cells exposed to both standard and redox chemotherapeutic agents. Biochem Pharmacol. 2011;82:1540–1548. doi: 10.1016/j.bcp.2011.07.102. [DOI] [PubMed] [Google Scholar]
- 32.Yun J, Mullarky E, Lu C, Bosch KN, Kavalier A, Rivera K, Roper J, Chio II, Giannopoulou EG, Rago C, Muley A, Asara JM, Paik J, Elemento O, Chen Z, Pappin DJ, Dow LE, Papadopoulos N, Gross SS, Cantley LC. Vitamin C selectively kills KRAS and BRAF mutant colorectal cancer cells by targeting GAPDH. Science. 2015;350:1391–1396. doi: 10.1126/science.aaa5004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Olney KE, Du J, van ‘t Erve TJ, Witmer JR, Sibenaller ZA, Wagner BA, Buettner GR, Cullen JJ. Inhibitors of hydroperoxide metabolism enhance ascorbate-induced cytotoxicity. Free Radic Res. 2013;47:154–163. doi: 10.3109/10715762.2012.755263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Karpinich NO, Tafani M, Rothman RJ, Russo MA, Farber JL. The course of etoposide-induced apoptosis from damage to DNA and p53 activation to mitochondrial release of cytochrome c. J Biol Chem. 2002;277:16547–16552. doi: 10.1074/jbc.M110629200. [DOI] [PubMed] [Google Scholar]
- 35.Chen R, Zou Y, Mao D, Sun D, Gao G, Shi J, Liu X, Zhu C, Yang M, Ye W, Hao Q, Li R, Yu L. The general amino acid control pathway regulates mTOR and autophagy during serum/glutamine starvation. J Cell Biol. 2014;206:173–182. doi: 10.1083/jcb.201403009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mookerjee SA, Goncalves RL, Gerencser AA, Nicholls DG, Brand MD. The contributions of respiration and glycolysis to extracellular acid production. Biochim Biophys Acta. 2015;1847:171–181. doi: 10.1016/j.bbabio.2014.10.005. [DOI] [PubMed] [Google Scholar]
- 37.Kramarenko GG, Hummel SG, Martin SM, Buettner GR. Ascorbate reacts with singlet oxygen to produce hydrogen peroxide. Photochem Photobiol. 2006;82:1634–1637. doi: 10.1562/2006-01-12-RN-774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Swerdlow RH, E L, Aires D, Lu J. Glycolysis-respiration relationships in a neuroblastoma cell line. Biochim Biophys Acta. 2013;1830:2891–2898. doi: 10.1016/j.bbagen.2013.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Warburg O, Wind F, Negelein E. The Metabolism of Tumors in the Body. J Gen Physiol. 1927;8:519–530. doi: 10.1085/jgp.8.6.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kosic M, Arsikin-Csordas K, Paunovic V, Firestone RA, Ristic B, Mircic A, Petricevic S, Bosnjak M, Zogovic N, Mandic M, Bumbasirevic V, Trajkovic V, Harhaji-Trajkovic L. Synergistic Anticancer Action of Lysosomal Membrane Permeabilization and Glycolysis Inhibition. J Biol Chem. 2016 doi: 10.1074/jbc.M116.752113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Eguchi Y, Shimizu S, Tsujimoto Y. Intracellular ATP levels determine cell death fate by apoptosis or necrosis. Cancer Res. 1997;57:1835–1840. [PubMed] [Google Scholar]
- 42.Tsujimoto Y. Apoptosis and necrosis: intracellular ATP level as a determinant for cell death modes. Cell Death Differ. 1997;4:429–434. doi: 10.1038/sj.cdd.4400262. [DOI] [PubMed] [Google Scholar]
- 43.Schraufstatter IU, Hinshaw DB, Hyslop PA, Spragg RG, Cochrane CG. Oxidant injury of cells. DNA strand-breaks activate polyadenosine diphosphate-ribose polymerase and lead to depletion of nicotinamide adenine dinucleotide. J Clin Invest. 1986;77:1312–1320. doi: 10.1172/JCI112436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Alexander A, Cai SL, Kim J, Nanez A, Sahin M, MacLean KH, Inoki K, Guan KL, Shen J, Person MD, Kusewitt D, Mills GB, Kastan MB, Walker CL. ATM signals to TSC2 in the cytoplasm to regulate mTORC1 in response to ROS. Proc Natl Acad Sci U S A. 2010;107:4153–4158. doi: 10.1073/pnas.0913860107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Filipovic DM, Meng X, Reeves WB. Inhibition of PARP prevents oxidant-induced necrosis but not apoptosis in LLC-PK1 cells. Am J Physiol. 1999;277:F428–436. doi: 10.1152/ajprenal.1999.277.3.F428. [DOI] [PubMed] [Google Scholar]
- 46.Cieslak JA, Strother RK, Rawal M, Du J, Doskey CM, Schroeder SR, Button A, Wagner BA, Buettner GR, Cullen JJ. Manganoporphyrins and ascorbate enhance gemcitabine cytotoxicity in pancreatic cancer. Free Radic Biol Med. 2015;83:227–237. doi: 10.1016/j.freeradbiomed.2015.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fischer AP, Miles SL. Ascorbic acid, but not dehydroascorbic acid increases intracellular vitamin C content to decrease Hypoxia Inducible Factor -1 alpha activity and reduce malignant potential in human melanoma. Biomed Pharmacother. 2016;86:502–513. doi: 10.1016/j.biopha.2016.12.056. [DOI] [PubMed] [Google Scholar]
- 48.Heaney ML, Gardner JR, Karasavvas N, Golde DW, Scheinberg DA, Smith EA, O’Connor OA. Vitamin C antagonizes the cytotoxic effects of antineoplastic drugs. Cancer Res. 2008;68:8031–8038. doi: 10.1158/0008-5472.CAN-08-1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Doskey CM, Buranasudja V, Wagner BA, Wilkes JG, Du J, Cullen JJ, Buettner GR. Tumor cells have decreased ability to metabolize H2O2: Implications for pharmacological ascorbate in cancer therapy. Redox biology. 2016;10:274–284. doi: 10.1016/j.redox.2016.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Monti DA, Mitchell E, Bazzan AJ, Littman S, Zabrecky G, Yeo CJ, Pillai MV, Newberg AB, Deshmukh S, Levine M. Phase I evaluation of intravenous ascorbic acid in combination with gemcitabine and erlotinib in patients with metastatic pancreatic cancer. PLoS One. 2012;7:e29794. doi: 10.1371/journal.pone.0029794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Welsh JL, Wagner BA, van’t Erve TJ, Zehr PS, Berg DJ, Halfdanarson TR, Yee NS, Bodeker KL, Du J, Roberts LJ, 2nd, Drisko J, Levine M, Buettner GR, Cullen JJ. Pharmacological ascorbate with gemcitabine for the control of metastatic and node-positive pancreatic cancer (PACMAN): results from a phase I clinical trial. Cancer Chemother Pharmacol. 2013;71:765–775. doi: 10.1007/s00280-013-2070-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Stephenson CM, Levin RD, Spector T, Lis CG. Phase I clinical trial to evaluate the safety, tolerability, and pharmacokinetics of high-dose intravenous ascorbic acid in patients with advanced cancer. Cancer Chemother Pharmacol. 2013;72:139–146. doi: 10.1007/s00280-013-2179-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Washko PW, Wang Y, Levine M. Ascorbic acid recycling in human neutrophils. J Biol Chem. 1993;268:15531–15535. [PubMed] [Google Scholar]
- 54.Vichai V, Kirtikara K. Sulforhodamine B colorimetric assay for cytotoxicity screening. Nat Protoc. 2006;1:1112–1116. doi: 10.1038/nprot.2006.179. [DOI] [PubMed] [Google Scholar]
- 55.Doskey CM, van ‘t Erve TJ, Wagner BA, Buettner GR. Moles of a Substance per Cell Is a Highly Informative Dosing Metric in Cell Culture. PLoS One. 2015;10:e0132572. doi: 10.1371/journal.pone.0132572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Washko PW, Hartzell WO, Levine M. Ascorbic acid analysis using high-performance liquid chromatography with coulometric electrochemical detection. Anal Biochem. 1989;181:276–282. doi: 10.1016/0003-2697(89)90243-1. [DOI] [PubMed] [Google Scholar]