

SUMMARY
Kinetochores connect centromeric nucleosomes with mitotic-spindle microtubules through conserved, cross-interacting protein subassemblies. In budding yeast, the heterotetrameric MIND complex (Mtw1, Nnf1, Nsl1, Dsn1), ortholog of the metazoan Mis12 complex, joins the centromere-proximal components, Mif2 and COMA, with the principal microtubule-binding component, the Ndc80 complex (Ndc80C). We report the crystal structure of Kluyveromyces lactis MIND and examine its partner interactions, to understand the connection from a centromeric nucleosome to a much larger microtubule. MIND resembles an elongated, asymmetric Y; two globular heads project from a coiled-coil shaft. An N-terminal extension of Dsn1 from one head regulates interactions of the other head, blocking binding of Mif2 and COMA. Dsn1 phosphorylation by Ipl1/Aurora B relieves this autoinhibition, enabling MIND to join an assembling kinetochore. A C-terminal extension of Dsn1 recruits Ndc80C to the opposite end of the shaft. The structure and properties of MIND show how it integrates phospho-regulatory inputs for kinetochore assembly and disassembly.
eTOC blurb
The yeast MIND complex acts as a Y-shaped adaptor linking the centromeric nucleosome to the much larger microtubule.
INTRODUCTION
Kinetochores are multi-protein assemblies that attach eukaryotic chromosomes to spindle microtubules and mediate chromosome segregation in mitosis. Genomic stability depends on tight coordination of kinetochore assembly, orientation, spindle attachment and disassembly with cell cycle progression.
In budding yeast, kinetochores assemble on “point” centromeres and connect to a single spindle microtubule. Yeast centromeric DNA has a defined sequence of approximately 150 base pairs, which specifies kinetochore assembly by associating with a centromere-specific nucleosome core (Biggins, 2013). In metazoans, kinetochores assemble on “regional” centromeres of up to several megabases, and establish attachments to multiple microtubules (Przewloka and Glover, 2009; Verdaasdonk and Bloom, 2011). Nonetheless, structural and functional conservation of kinetochore components shows that central elements of kinetochore architecture and mechanism are conserved across eukaryotic phyla (Cheeseman and Desai, 2008; Joglekar et al., 2009; Joglekar et al., 2008; Santaguida and Musacchio, 2009). Moreover, the copy number of kinetochore proteins in point and regional centromeres is proportional to the number of kinetochore-microtubule attachments, not to the length of centromeric DNA, suggesting that kinetochores of multi-cellular eukaryotes are effectively parallel repeats of the single kinetochore-microtubule assembly found in budding yeast (Joglekar et al., 2009; Zinkowski, 1991).
The conserved core of a kinetochore comprises approximately 35 different protein species, most of them present in multiple copies, giving a total assembly of more than 250 protein subunits, not including transiently attached regulatory components (Biggins, 2013; De Wulf et al., 2003; Joglekar et al., 2006; Westermann et al., 2003). The core components form well-defined subassemblies, usually classified as inner, centromere-associated, and outer, microtubule-associated protein complexes. Electron micrographs of kinetochores isolated from budding yeast show a dense, central hub, approximately 37 nm in diameter, surrounded by 5–7 projecting modules, believed to contain the proteins that contact taxol-stabilized microtubules (Gonen et al., 2012).
The heterotetrameric MIND complex of budding yeast, comprising Mtw1, Nnf1, Nsl1 and Dsn1, is essential for kinetochore bi-orientation and proper chromosome segregation (Cheeseman et al., 2006; Hornung et al., 2014; Kline et al., 2006; Pagliuca et al., 2009). It is a conserved structural element that links inner-layer kinetochore components, Mif2 and COMA, with outer-layer components, Ndc80 complex (Ndc80C) and Spc105 (Biggins, 2013; De Wulf et al., 2003). The MIND complex is thus the crucial bridge between chromatin-associated and microtubule-associated subassemblies and a central regulatory target. “Mis12 complex” (Mis12C) is the designation of its close homolog in fission yeast and metazoans.
As seen by negative-stain electron microscopy (EM), MIND/Mis12C is a 200 Å long, rod-like structure with a globular region at one end (Cho, 2011; Hornung et al., 2011; Hornung et al., 2014; Maskell et al., 2010; Petrovic et al., 2014; Petrovic et al., 2010; Screpanti et al., 2011). The narrower end binds the microtubule-associated Ndc80C and Spc105 (Maskell et al., 2010; Petrovic et al., 2014). Ndc80C, a 550 Å long rod, defines the axis of a kinetochore. It is a heterotetramer, with a globular module at each end of an α-helical, coiled-coil stalk. Subunits Ndc80 and Nuf2 form the larger, microtubule-binding globular end and most of the stalk; Spc24 and Spc25, the remainder of the stalk and the smaller, MIND-binding globular end (Ciferri et al., 2008; Wei et al., 2006). Ndc80C also binds the 10-protein DASH/Dam1 complex, an essential, yeast-specific, outer kinetochore component crucial for microtubule attachment (Cheeseman et al., 2001; De Wulf et al., 2009).
Ipl1/Aurora B, a principal regulatory kinase for mitosis, promotes kinetochore stability, corrects microtubule attachment, and recruits spindle assembly checkpoint proteins. Lack of bi-orientation, and thus absence of tension between sister centromeres, leads to Ipl1/Aurora B phosphorylation of Ndc80C and DASH/Dam1, activation of the spindle checkpoint, and kinetochore-microtubule detachment and reattachment during anaphase delay (Biggins and Murray, 2001; Cheeseman, 2002; Krenn and Musacchio, 2015; Tien et al., 2010). Ipl1/Aurora B is also required to assemble functional kinetochores. Phosphorylation of Dsn1 by Ipl1/Aurora B promotes recruitment of MIND to centromeres and strengthens the association of inner- and outer-kinetochore components (Akiyoshi et al., 2013; Emanuele et al., 2008; Kim and Yu, 2015; Yang et al., 2008). Two serines on Dsn1, conserved in the human and S. cerevisiae proteins, are sites of Ipl1/Aurora B phosphorylation (Akiyoshi et al., 2013; Welburn et al., 2010; Yang et al., 2008). Regulation at these sites thus appears to be conserved, despite differences between yeast and metazoans in the timing of kinetochore reassembly and microtubule reattachment.
We present here the x-ray crystal structure of the budding-yeast MIND complex. An accompanying paper (Petrovic et. al.) describes the very similar structure of human Mis12C. They are heterotetrameric coiled-coil rods with two centromere-proximal globular domains. We map the binding of Mif2 and COMA subunit Ame1 to one globular head of MIND and show that an N-terminal segment of Dsn1 is an auto-inhibitory regulator of these interactions. We present a model for relief of auto-inhibition by Ipl1/Aurora B phosphorylation of Dsn1, allowing Mif2 and COMA to recruit MIND to kinetochores. We have also determined the structure of the C-terminal, microtubule-proximal tail of Dsn1 bound to the Spc24/Spc25 subcomplex of Ndc80C; it occupies a conserved cleft previously shown to also bind Cnn1 (Malvezzi et al., 2013). The structural data presented here enable us to propose a molecular model for the central kinetochore assembly in point-centromere yeast, explaining the role of MIND in kinetochore organization and regulation through its association with Mif2, COMA and Ndc80C.
RESULTS
A Stable MIND Core Complex
The yeast MIND complex is a heterotetramer, with one chain each of Mtw1, Nnf1, Nsl1, and Dsn1 (De Wulf et al., 2003). Stable subcomplexes of Mtw1/Nnf1 and Dsn1/Nsl1 can also be prepared as recombinant products (Hornung et al., 2011). We expressed (in bacteria) and purified the K. lactis MIND heterotetramer, which has less extensive regions of apparently unstructured polypeptide chain than does its otherwise very similar S. cerevisiae ortholog (29 to 44% identity and 59 to 77% amino-acid similarity for the four subunits). Secondary-structure prediction suggests the presence of extended coiled-coil regions in each of the four components (Data D1). We analyzed by mass spectrometry both trypsin-treated and spontaneously proteolyzed MIND complexes and found proteolytically sensitive segments at the N and C termini of Dsn1 (residues 1-229 and 480-491) and at the C-terminus of Mtw1 (residues 234-288). We therefore made a truncated heterotetrameric MIND core, comprising Dsn1 (230-479), Mtw1 (1-233), and full length Nsl1 (1-216) and Nnf1 (1-205). We call this construct “MIND complex 1” (MIND-C1) (Figure S1). Crystallization of MIND-C1 ultimately led to the structures described here.
X-ray Crystallographic Structure Determination
MIND-C1 crystallized in four different space groups, but weak and anisotropic data required an iterative strategy for structure determination (Experimental Procedures, Figure S2 and Table S1). An initial low-resolution structure showed us how to prepare a heterodimeric “MN complex 2” (MN-C2) containing Mtw1 (1-120) and Nnf1 (1-102). Crystals of MN-C2 yielded high-resolution data (Table S2) and a structure that contributed to phase determination for MIND-C1. We traced residues Mtw1 1-233, Nnf1 8-179, 186-203, Nsl1 5-150, 167-216 and Dsn1 253-310, 354-405, 419-479 and validated the amino acid sequence assignment by anomalous difference Fourier analysis (Figure S2B); loop 311-353 in Dsn1 is partially modeled as poly-alanine (311-343, 349-353). Some segments connecting α-helices are in poor density, and the chain trace in those regions is approximate. The beginning and end of each segment and a string of broken density define the overall configuration of the segment in question, and we have therefore kept the poorly defined residues in the model. We list these segments in the Experimental Procedures and provide details in Figure S6. We refined the final MIND-C1 coordinates with data to minimum Bragg spacings of 3.2, 4.1, and 3.4 Å along a*, b* and c*, respectively (Rfree/Rwork = 36.4/32.5%) (Table S1).
Molecular Structure of the MIND Complex
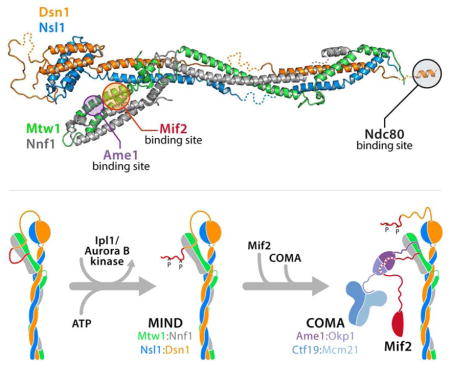
MIND-C1 is an asymmetric “Y”, with a coiled-coil rod and two globular modules, of which one (“head II”) caps one end of the rod and the other (“head I”) branches off about one-third of the way toward the other end (Figure 1A). Its general outline thus agrees with published images from negative-stain EM. Head I contains N-proximal segments of Mtw1 and Nnf1; head II, N-proximal segments of Dsn1 and Nsl1 (Figure 1A). These N-proximal segments are helical hairpins, with various local elaborations; the two hairpins in each head pack onto each other to create four-helix bundles. Head I, which includes Mtw1 residues 1-120 and Nnf1 residues 1-102 (MN-C2), has docking sites on its surface for peptidic segments of interacting partners, as we show later. Head II includes Dsn1 residues 253-303 and Nsl1 residues 1-90 (Figure 1B). Dsn1 residues 230-252, at the N-terminus of the Dsn1 polypeptide chain in our crystals, are disordered. A long loop (residues 303-363) at the C-terminal end of the Dsn1 helical hairpin projects away from the globular core and folds back along it, connecting into the coiled-coil rod. Although we can trace this loop in the electron density map, most of its structure derives from contacts with neighboring molecules in the crystal lattice. The connection of the helical hairpin of Nsl1 with the coiled-coil is through a short helix α3, (Figure 1A and C), which reinforces the core bundle of head II, and a short peptide that interacts with the C-terminal end of the Dsn1 loop. In the various crystal forms we have characterized, head II swivels 20° about the axis of the coiled-coil, while the angle at which head I projects from the coiled-coil backbone of the complex appears to vary less.
Figure 1. Structure of the K. lactis MIND Complex.

(A) Cartoon representation of the x-ray crystal structure of K. lactis MIND-C1 containing Mtw12-233, Nnf18-203, Nsl15-216 and Dsn1253-471 (top) and orthogonal view (bottom), with globular domains labeled head I (Mtw11-115/Nnf11-120) and head II (Dsn1230-362/Nsl11-91). Grey bars define three distinct coiled coil (CC) modules connecting through junctions 1 (J1) and 2 (J2). Disordered residues of Nnf1180-185, Nsl1151-166, and Dsn1344-348 and 406-418 not traceable in the electron density map shown as dotted lines (see panel (B)). All structure figures generated with PyMol (http://www.pymol.org).
(B) Schematic of Mtw1, Nnf1, Nsl1 and Dsn1 sequences, showing residues in MIND heads I and II and coiled-coil modules; narrow lines show regions deleted from the MIND-C1 complex (Mtw1234-288 and Dsn11-229 and 480-491). White dashed lines indicate residues not in the model (too disordered to trace) but confirmed present by mass-spectrometry.
(C) Folding diagrams of Mtw1, Nnf1, Nsl1 and Dsn1, showing MIND components as structural paralogs. Dashed lines: residues not traced in the density map.
The two heterodimeric sub-complexes alternate in contributing to the coiled-coil that defines the axis of the MIND complex. The Dsn1-Nsl1 coiled-coil (CC1) that emanates from head II terminates at the junction (J1) from which head I projects (Figure 1A). A long, Mtw1-Nnf1 coiled-coil (CC2), roughly coaxial with CC1, extend from this junction; a continuation of the Nsl1 chain as a third coiled-coil element may help ensure this approximate coaxial orientation and further restrict any tendency to bend. Dsn1 continues as a partly disordered extended chain and then reinforces the Mtw1-Nnf1 coiled-coil toward the C-terminal end of CC2. The third part of the rod (CC3) has contributions from all four chains, creating a transition to a short coiled-coil segment that pairs Dsn1 and Mtw1. The C-terminal peptides of these two chains are not part of the MIND-C1 construct (Figure 1B); as we show below, they connect MIND with the Spc24/Spc25 end of Ndc80C.
We can consider all four chains in the MIND complex as “structural paralogs” -- related polypeptide-chain organization and similar three-dimensional conformation -- because each has a helical hairpin near its N-terminus linked to a long, segmented α-helical stem (Figure 1C). The Dsn1-Nsl1 and Mtw1-Nnf1 pairs are also structural paralogs, and in the same sense, because of the interchain contacts within each heterodimeric sub-complex.
Comparison of structures in several crystal forms suggests that head I has a relatively fixed orientation with respect to the rod axis, and that while head II can rotate by up to 20° about the coiled coil axis, a more substantial hinge point is between CC2 and CC3 (Figure S5C). In crystals with a nanobody bound on head II, the axis of the rod bends by about 60° at the CC2-CC3 junction. The bend must be imposed by packing in the crystal, as it is about 160 Å distant from the nanobody contact.
Mif2 and Ame1 Interact with MIND Head I
An N-terminal region of human CENP-C1-71 and its S. cerevisiae ortholog Mif21-38 share a sequence motif sufficient for high affinity binding with the Mis12C/MIND complex (Hornung et al., 2014; Screpanti et al., 2011). We prepared Mtw11-120/Nnf11-102 and Dsn1230-397/Nsl11-118 and showed by size-exclusion chromatography that K. lactis Mif21-41 co-elutes with the former but not with the latter (Figure 2A). We purified the resulting complex, crystallized it, and determined the structure at 2.9 Å resolution by molecular replacement with MN-C2 as the search model (Figure 2B and Table S2). We could build Mif2 residues 5-36 into continuous density. The peptide wraps around the domain, with residues 5-15 contacting Mtw1 α2, α3 and residues 28-36 contacting Mtw1 α3 and Nnf1 α1 (Figure 2B). The Mif2 residues in these interfaces are conserved in all budding yeast (Figure 2C). Those in the C-terminal part of the peptide are also conserved in metazoans, as are the contacting residues in Mtw1 and Nnf1 (Data D1). The accompanying paper shows that the interaction of the human Mis12C with CENP-C resembles the one shown here, particularly for Mif2 residues 28-36 and CENP-C residues 35-43 (Petrovic et al, 2016).
Figure 2. Mif2 Interaction with MIND Head I.

(A) Mif21-41 binds MIND head I (Mtw11-120/Nnf11-102). Coomassie-stained SDS-PAGE gels of elution fractions from size exclusion chromatography (SEC) testing interaction of Mif21-41 peptide with head I and head II D230L. In top two gels, lane “a”: Mtw11-120/Nnf11-102; lane “b”: Mif21-41; lane “c”: head II D230L (Dsn1230-397/Nsl11-118). Grey boxes at left display graphic representations of gel input consistent with figures used throughout this paper.
(B) Crystal structure of MIND head I (Mtw11-120/Nnf11-102) with Mif21-41 shown in red. Mif2 peptide contacts two distinct surfaces on head I: residues 5-15 contact Mtw1 α2, α3 (binding site I: BS I); residues 28-36 contact Mtw1 α3 and Nnf1 α1 (BS II) (top). Mif2 residues 1-4 and 37-41 are flexible and cannot be traced in density map. Bottom panels: close up view of the two binding sites. In BS I, an extensive hydrogen-bond and salt-bridge network connects R11 and K12 of Mif2 with D25, E84, D77 and D81 of Mtw1 (bottom right). In BS II, a hydrophobic cleft between Mtw1 (V23 and K26) and Nnf1 (Y36 and K82) frames Mif2 residues L31, F34 and F35 and hydrogen bonds link Mif2 E29 with R89, N90 and Y36 of Mtw1 (bottom left).
(C) Sequence alignment of N-terminus of Mif2. Residues contacting Mtw1 and Nnf1 marked with green and gray dots, respectively. Sequence conservation shown in Clustalx color scheme of Jalview (Waterhouse et al., 2009).
The heterotetrameric COMA (Ctf19, Okp1, Mcm21, Ame1) complex associates tightly with MIND, as do its orthologs in metazoans (Hornung et al., 2014). The Ame1-Okp1 heterodimeric sub-complex is essential for MIND recruitment to kinetochores and for viability in budding yeast (Hornung et al., 2014). Negative-stain electron microscopy and binding experiments suggest that Ame1-Okp1 interacts with the Mtw1-Nnf1 globular domain (head I), through a conserved, 15-residue N-terminal motif (Hornung et al., 2014). In experiments analogous to those in the preceding section, we showed that this N-terminal Ame1 peptide indeed associates with head I.
Mapping of conserved residues in an alignment of Mtw11-120/Nnf11-102 sequences from 10 fungal species onto the structure of MN-C2 yields three discrete surface patches (Figure 3A and Data D1). One of these (site I) is the Mif2 binding site. Mif2 and COMA do not compete for interaction with MIND (Hornung et al., 2014). We can therefore eliminate any substantial part of site I as an Ame1 contact. After confirming by a size-exclusion experiment that Ame1 (1-24) indeed binds Mtw11-120/Nnf11-102, we tested a panel of head I constructs mutated in sites II and III, to determine the likely interaction surface. The experiments in Figure 3B show that site II is necessary for COMA binding with MIND.
Figure 3. Ame1 Binds a Conserved Surface Patch on MIND Head I.

(A) Surface representation of MIND head I bound with Mif2 peptide showing three conserved surface patches. Mif2 binding site I in red (as in Figure 2B); site II in blue; site III in magenta.
(B) Mtw1 point mutations N32A, Y36A, E73A, D77A disrupt Ame1 binding. Top, overlay of size exclusion chromatograms testing binding of His6-GST-tagged Ame11-24 peptide with MIND head I wild-type (Mtw11-120/Nnf11-102) and mutants (Site II mut: Mtw11-120: N32A, Y36A, E73A, D77A/Nnf11-102 and Site III mut: Mtw11-120/Nnf11-102: E76A, E83A). Bottom, coomassie stained SDS-PAGE gels with marked fractions corresponding to chromatogram at top.
Dsn1 Interaction with Spc24/Spc25
Deletion of 28 residues at the C-terminus of S. cerevisiae Dsn1 perturbs binding to the Spc24/Spc25 components of Ndc80 and generates a lethal phenotype in yeast (Malvezzi et al., 2013). A related segment from Cnn1 also binds Spc24/Spc25, and we expect from the structure of that complex that neither the Cnn1 nor the Dsn1 segment will be tightly folded in the absence of its target (Malvezzi et al., 2013; Schleiffer et al., 2012). Our limited proteolysis experiments indeed led us to delete the C-terminal residues (480-491) of K. lactis Dsn1 when designing MIND-C1. Moreover, deletion of these residues without truncating Mtw1 made the Mtw1 C-terminus very protease sensitive, with aggregation of the residual MIND, so our design for MIND-C1 had a 55-residue C-terminal deletion in Mtw1 as well as the 12-residue C-terminal deletion in Dsn1 (Figure 1B).
The MIND-C1 structure shows close approach of the trimmed C-termini of Dsn1 and Mtw1. We therefore tested whether the C-terminal region of Mtw1 contributes, either directly or indirectly, to the interaction of Dsn1 with the Ndc80 complex. We synthesized S. cerevisiae Dsn1560-576 and two conserved peptides from the C-terminus of Mtw1 (Mtw1230-262 and Mtw1274-289) and labeled them with fluorescein isothiocyanate (FITC) for fluorescence polarization binding assays. Spc24/Spc25 bound Dsn1560-576 with KD ~ 12 μM; Mtw1230-262 and Mtw1274-289 did not bind (Figure 4A). We tested whether addition of either of the Mtw1 peptides would increase the affinity of Dsn1560-576 for Spc24/Spc25 but did not detect a measurable change (data not shown). Thus, Dsn1560-576 is necessary and sufficient for binding Spc24/Spc25.
Figure 4. Dsn1 Interacts with Spc24/Spc25.

(A) Fluorescence polarization data for binding of ScSpc24/Spc25 to FITC-Dsn1556-572, FITC-Dsn1556-572:S2D, FITC-Mtw1230-262 and FITC-Mtw1274-289 peptides. Concentrations of FITC peptides at 50 nM were mixed with increasing concentrations of ScSpc24/Spc25 (0 - 50 μM). Fluorescence polarization plotted as mean ± standard error from 6 replicates. See Experimental Procedures for data fitting.
(B) Top, enlargements of C-terminal MIND Dsn1 peptide in complex with Spc24/Spc25. Dsn1 (residues 560-571 QQLLKGLSLSFS, in orange) shown bound with Spc24155-213 (purple) and Spc25132-221 (green). Conserved hydrophobic Dsn1 residues in contact with Spc24/Spc25 shown as sticks. Bottom, zoomed view of the hydrophobic cleft of Spc24/Spc25 binding Dsn1.
(C) Sequence alignment of the Spc24/Spc25 binding motif of Dsn1 showing conserved hydrophobic residues represented as sticks in the structure in (B) and conservation in other budding yeast of S. cerevisiae Dsn1 S567 and S569 (in green).
Structure of Spc24/Spc25 Bound with the C-terminal Segment of Dsn1
Dsn1 and Cnn1 compete for binding with Spc24/Spc25 (Schleiffer et al., 2012). Cnn160-84 binds by insertion of a short α-helix into a hydrophobic cleft on the Spc24/Spc25 heterodimer (Malvezzi et al., 2013). Mps1-dependent phosphorylation of Ser74 on Cnn1 regulates the association. We crystallized S. cerevisiae Spc24155-213/Spc25132-221 with bound Dsn1560-572 and determined the structure at a resolution of 1.8 Å (Table S2). An α helix, Dsn1 residues 560-571 lies in the cleft previously identified from the Cnn1 structure, with conserved Dsn1 serine residues (S567 and S569) at the interface (Figure 4B and C). We further showed that a peptide with both these serines mutated to aspartic acid, Dsn1560-576:S2D, failed to bind Spc24/Spc25, consistent with previous data showing that phosphorylation of Cnn1 blocks its interaction with Spc24/Spc25 (Figure 4A) (Malvezzi et al., 2013).
Dsn1 N-terminal Arm Interacts with MIND Head I
Ipl1/Aurora B regulates the interaction of CENP-C/Mif2 with Mis12C/MIND by phosphorylating two conserved serine residues on Dsn1 (S100 and S109 in humans, S240 and S250 in S. cervisiae) (Akiyoshi et al., 2013; Yang et al., 2008). In human cells that express phospho-mimetic Dsn1S100E, S109E or that have a deletion in Dsn1 of residues 92-113, CENP-C binds more tightly with Mis12C than it does in wild-type controls (Kim and Yu, 2015). The Ipl1 phospho-targeted residues in K. lactis Dsn1, S213 and S223, are in an unstructured region (residues 205-255), part of which is in our crystallized complex (Figure 1B and Data D1). These data suggest that phosphorylation displaces the Dsn1 segment bearing the serines from an autoinhibitory site elsewhere on MIND, opening up the complex for recruitment by CENP-C/Mif2.
We could not examine this potential mechanism directly in the MIND-C1 structure, because our crystals of K. lactis MIND-C1 lack Dsn1 residues 1-229 and because electron density of the N terminus of the truncated protein is uninterpretable from Q230 to S252 (Figure 1 and Data D1). Nonetheless, the structure of the complex, inspection of crystal contacts, and oligomerization properties of various constructs prompted us to determine whether Dsn1205-255 binds the globular, head I domain of Mtw1/Nnf1.
We created three head II constructs containing different portions of the Dsn1 N-terminal region: head IID205 contains Dsn205-313/Nsl11-83; head IID230 contains Dsn1230-313/Nsl11-83; and head IID256 contains Dsn1256-313/Nsl111-83 (Figure S1 for MIND constructs). We systematically tested each head II construct by size exclusion chromatography for binding with head I (Mtw11-102/Nnf11-120) (Figure 5). Head IID256 did not bind head I (Figure 5A), but head IID230 and head IID205 both bound strongly enough for co-elution (Figure 5B and C). Only the latter bound MIND-C1, however (Figure 5E). We interpret these results to indicate that intramolecular binding of the Dsn1 N-terminal segment (residues 230-255) present in MIND-C1 masks the site for association of an exogenous Dsn1230-255 peptide (here supplied on head IID230), but that it does not mask additional parts of the complete site accessible to an exogenous Dsn1205-255 peptide (supplied by head IID205). (It is likely that binding of head IID205 also displaces the partial intramolecular inhibition from residues 230-255, but our experiments are insensitive to that possibility.) We take these data as direct evidence for intramolecular interaction between the N-terminal arm of Dsn1 and head I.
Figure 5. N-terminal Arm of Dsn1 (Residues 205-255) Binds MIND Head I.

Elution fractions from SEC resolved on Coomassie stained SDS-PAGE gels (chromatograms not shown). Gel lanes labeled as load (L), followed by fraction number. Grey boxes left of each gel include cartoon representations of proteins injected onto the column (input); right boxes show elution complex(es) (output). Each panel has three gels: top two, controls with elution profiles from each component alone; bottom, the binding experiment.
(A) Head II D256: Dsn1256-313/Nsl111-83 does not bind head I: Mtw11-120/Nnf11-102.
(B) Head II D230: Dsn1230-397/Nsl11-118 binds head I: Mtw11-120/Nnf11-102.
(C) Head I (fraction a): Mtw11-120/Nnf11-102 binds head II D205 (fraction b): Dsn1205-397/Nsl11-118. Fraction (M): molecular weight markers at 10, 15, 20, 25, 30 kilodalton (kDa).
(D) MIND-C1 Mtw11-233/Nnf11-205/Nsl11-206/Dsn1230-479 does not bind head II D230.
(E) MIND-C1 binds head II D205.
(F) MIND-C1 does not bind phospho-mimetic head II-D205-S2D: Dsn1205-397:S213D and S223D/Nsl11-118.
Why is the intramolecular interaction of Dsn1 residues 230-255 with head I not present in our crystals? Our best ordered crystals grew from an optimized mother liquor that included 0.2 M KSCN and 0.2 M KBr -- i.e., from mildly chaotropic conditions, presumably required to avoid aggregation and poor crystal growth (Table S3). An occasional intermolecular association through the Dsn1 N-terminal arm, at the high protein concentration used for crystallization, could account for aggregation. We have shown that in 0.2 M KSCN, 0.2 M KBr, head IID230 does not bind head I (Figure S3). That is, the conditions needed for well-ordered crystals precluded formation of the contact.
Evidence that Dsn1 Phosphorylation Relieves Autoinhibition
We tested whether phosphorylation of S213 and S223 might affect the Dsn1 interaction with head I and found that phospho-mimetic version of head IID205 (designated head IID205-2D), with aspartic acid instead of serine at positions 213 and 223, failed to bind MIND-C1. The phospho-mimetic substitutions evidently weakened or eliminated any binding from the first part of the arm (residues 205-230), and internal competition from Dsn1 residues 230-255 presumably blocked interaction with the second part (Figure 5F). These observations support our inference that the consequence of Ipl1/Aurora B phosphorylation of serines 213 and 223 is to relieve autoinhibition -- specifically by dissociating the N-terminal extension of head II from the surface of head I, allowing CENP-C/Mif2 to bind instead.
We also tested directly, whether Mif2 and the arm of Dsn1 compete, using bacterially expressed K. lactis Mif2, MIND-C1 and head IID205. Mif2 bound stably to MIND-C1 as shown by a shift in the peak elution volume in analytical size exclusion chromatography (Figure 6A). A mixture of Mif2, MIND-C1 and head IID205 gave two distinct elution peaks: peak 1 (fractions 2–5) containing Mif2 bound to MIND-C1 and peak 2 (fractions 8–10) containing MIND-C1 bound to head IID205 (Figure 6A). The two ligands are thus mutually exclusive in their association with MIND-C1. We further investigated this competition by measuring the affinity of synthesized FITC-Mif21-41 with different MIND complexes in a fluorescence polarization experiment. The affinity of Mif21-41 for MIND-C1:D205 was negligible when compared with a KD of 1.8 μM for the phospho-mimetic MIND-C1:D205:S2D (Figure 6B and C). Furthermore, deleting the Dsn1 arm (residues 205-229) led to a ten-fold increase in the affinity of Mif2 for MIND-C1 (KD ~ 0.17 μM). These measurements are consistent with the Dsn1 autoinhibition mechanism proposed above.
Figure 6. N-terminal Arm of Dsn1 Competes with Mif2 and Ame1 for Binding with MIND Head I.

(A) SEC showing Mif2 and Dsn1205-230 compete for binding with MIND head I. Top, overlay of chromatograms using recombinant, full length His6-MBP-Mif2 and MIND-C1 alone and in the presence of head II D205: Dsn1205-397/Nsl11-118. Bottom, coomassie stained SDS-PAGE gels with elution fractions corresponding to chromatogram, above. Top gel: MIND-C1 binds either full length Mif2 or head II D205. Bottom three gels: control experiments with MIND-C1, Mif2, and MIND-C1:Mif2 complex.
(B) Fluorescence polarization data for binding of different MIND constructs to FITC-Mif21-41 (top) and FITC-Ame11-25 (bottom) peptides. FITC peptides at 50 nM mixed with increasing concentrations of MIND (0 -10 μM). Fluorescence polarization plotted as mean ± standard error from 6 replicates.
(C) Table of calculated KD values from experiments in (B). See Experimental Procedures for data fitting methods (n.b.: negligible binding.)
(D) Schematic of phospho-Dsn1 binding Mif2 and COMA. Left, unphosphorylated N-terminal arm of MIND Dsn1205-255 binds two distinct sites on head I. Middle, phosphorylation of Dsn1 at serines 213 and 223 disassociates the N-terminal region of Dsn1 arm from head I. Right, phosphorylation of Dsn1S213, S223 relieves MIND auto-inhibition, permitting binding of Mif21-41 and Ame11-24 (COMA), which together recruit MIND to kinetochores. Dashed red Mif2 segment indicates uncertainty concerning interaction between COMA and Mif2 preceding MIND recruitment.
Dsn1 and Ame1 Compete for Binding Mtw1/Nnf1 Head I
Ame11-24 and Mif21-41 both bind head I. The results of the previous section show that the Mif2 site overlaps the site for association of the Dsn1 segment comprising residues 205-230. One model for autoinhibition is that part of the Dsn1 arm (residues 205-229) competes with Mif2 for one site on head I and part of the arm (residues 230-255) competes with Ame1 for another site. The results in Figure S3B (panels 4 and 5) are consistent with this simple picture; they show that head IID230, which contains only one part of the Dsn1 arm, binds with wild-type head I but not with head Imut1, which bears mutations that likewise prevent Ame1 from binding (Figure S3B).
We also examined the effect of the Ame11-24 peptide on the association of head I with head IID205. Panels 1 and 2 of Figure S3C show that head I and head IID205 associated with 1:1 stoichiometry, even when the latter had the two phospho-mimetic mutations (IID205-S2D). Addition of Ame1 peptide to these complexes interfered with their heterodimeric association. Figure S3C, panels 3 and 4, shows that His6-GST-Ame1 (1-24) shifted more of head I (Mtw11-120/Nnf11-102) into fractions 2 and 3 than it did of head II D205 (Dsn1205-313/Nsl11-83). The phospho-mimetic mutant of head II, which associates less strongly with head I than does wild-type, was essentially absent from fraction 2 and only weakly present in fraction 3. Using fluorescence polarization experiments, we also measured the affinity of Ame11-25 for different MIND constructs. Like Mif2, Ame1 bound strongly to MIND-C1 (KD ~ 0.26 μM) and to the phospho-mimetic MIND-C:D205-2D (KD ~ 1.5 μM), but did not associate with wild-type MIND-C1:D205, which includes the unmodified Dsn1 arm (residues 205-255) (Figure 6B and C). We conclude that not only do Ame1 and Dsn1 interact with a common site on head I but that Ame1 can exclude Dsn1230-255 in the absence of the additional Dsn1:head I contacts made by the 205-229 part of the Dsn1 arm -- i.e., the contacts eliminated by phosphorylation of Ser213 and Ser223. Thus, the same Ipl1 phosphorylation event can in principle stimulate recruitment and stronger binding of MIND by both Mif2 and COMA.
DISCUSSION
Kinetochores of point-centromere yeast expand by about fivefold, both in dimension and in molecular multiplicity, in bridging a single, centromeric nucleosome with a single, kinetochore microtubule. The dimensional expansion is from ~100 Å for the nucleosome to ~500 Å for the Ndc80C-DASH/Dam1 “corona” surrounding an end-on linked microtubule; the molecular multiplicity expansion is from two, for Cse4 and Mif2, to 8-10 for Ndc80C and a still larger number for DASH/Dam1 ring (Joglekar et al., 2006). The MIND complex links these two levels, connecting Cse4 and Mif2, bound at its globular end, with Ndc80C, bound by its narrow end. The molecular structure of MIND and the characteristics of its contacts with partner proteins together suggest how it can expand the kinetochore and show how it integrates phospho-regulatory inputs for both assembly and disassembly. Its dimensions and apparent oligomerization properties fit a hub-and-spokes model for EM images of negatively stained, isolated yeast kinetochores (Gonen et al., 2012).
Mechanism of Phospho-Dsn1 Promoted Kinetochore Assembly
The N-terminal segment of Dsn1 (residues 1-255, of which only residues 230-255 are present in our crystals), probably unstructured, projects from head II. Inner-kinetochore components Mif2 and Ame1 bind noncompetitively to head II at adjacent, but distinct interfaces (Figures 2 and 3). Both interactions are through flexible peptide segments at termini of the respective proteins, minimizing steric exclusion and allowing two relatively large proteins to contact a single, small domain on MIND. The positions of the two heads of MIND and the relatively flexible connection between head II and the coiled-coil stalk enable the extended, N-terminal arm of Dsn1 to associate with head I. Amino-acid residues in Dsn1 between positions 205 and 255 bind head I and block the sites for Mif2 and Ame1 (Figure 6D). Phosphorylation of serine residues within this region (Ser213 and Ser223, known targets for the Ipl1/Aurora B protein kinase) eliminates the contact and exposes the sites, so that Mif2 and Ame1 can associate instead. Dsn1 residues between 205 and 230 contact the Mif2 site; residues between 230 and 255, the Ame1 site. Although serines 213 and 223 are both in the former segment, Ame1 binds tightly enough to displace Dsn1230-255, unless the arm is also “clamped down” by binding of 205-230 to the Mif2 site. Differential affinities, the high effective concentration of a locally attached ligand, and Dsn1 phosphorylation are thus components of a MIND-based, regulatory switch in kinetochore assembly.
Significance of Small Interfaces and Flexible Linkers for Efficient Kinetochore Assembly and Disassembly
MIND binds its partners, Mif2, Ame1, and Spc24/Spc25, through 33, 23 and 12-residue peptides that project from the ends of even longer, unstructured, flexible linkers, of between 12 and 200 amino-acid residues. The characteristics of these linkers facilitate efficient capture and localization of other kinetochore components. Moreover, the positions of these linkers on the rod reflect a likely order of kinetochore assembly. The linkers from Mif2 and Ame1 recruit MIND through contacts at its globular end; a linker from the opposite end of MIND recruits Ndc80C. Thus, each extended linker is distal to the interface established by the preceding step.
Interfaces of folded, globular domains with short peptides have several advantages for regulated assembly and disassembly. (1) As noted above for Mif2 and Ame1, one small, globular domain can bind two (or more), much larger partner proteins or protein complexes, if the contacts are with peptide segments that extend away from the bulky, folded domains of the partners. (2) Peptides are much better substrates for modification than are surfaces of folded domains. For example, full activity of a protein kinase requires that several residues adjacent to the residue to be modified fit into specificity pockets on the surface of the enzyme. A residue on an unfolded peptide is clearly a more available substrate than one on a stably folded, domain surface. Similar arguments apply to other post-translational modifications. (3) Even a tightly bound ligand will dissociate (and reassociate) many times during the course of a cell cycle. Thus even an assembled interface can be subject to modification by a suitably localized enzyme. The interfaces between Mif2 and Mtw1/Nnf1, Ame1 and Mtw1/Nnf1, Dsn1 and Spc24/Spc25 all have conserved serine and threonine residues, and phospho-regulated disruption of kinetochore interfaces at conserved, modifiable residues (e.g., serines 567 and 569 in the C-terminal segment of S. cerevisiae Dsn1, leading to loss of contact with Spc24/Spc25) is a potential mechanism for disassembling a kinetochore at the end of anaphase. Cell-cycle dependent activation or recruitment of the relevant kinase can provide the required temporal control. (4) Flexible connections between more rigid structural members allows an assembly to adapt in shape to distinct functional demands and to respond to external force without rupturing. In the case of a yeast kinetochore, for example, side-on and end-on attachment to microtubules have very different geometric requirements. The compliance of such interactions, both in extension (if the interacting segment is part of a longer, flexible linker) and direction, may be relevant when a kinetochore is under tension in metaphase and anaphase.
Model of the Core Kinetochore Complex
A yeast kinetochore connects a ~150 base pair centromere, marked by a centromeric nucleosome, with a single microtubule. It thus adapts a ~100 Å structure with (approximate) twofold symmetry to a ~250 Å diameter tube with 13 parallel protofilaments (Figure 7A). This progressive expansion in scale from inner to outer kinetochore is both in dimension and in stoichiometry, and MIND appears to be a critical spatial and stoichiometric adaptor. Quantitative fluorescence microscopy suggests that a yeast centromeric nucleosome recruits one Mif2 dimer (perhaps clamped across its dyad) and that the attached microtubule has ~8 Ndc80 complexes (Joglekar et al., 2006). The estimates for COMA and MIND are 2–3 and 6–7, respectively. Electron micrographs of negatively-stained kinetochore particles isolated from yeast cells show a hub with 5–7 projections, about 400 Å long (Gonen et al., 2012). Although the microscopy did not allow assignment of molecular identity, our results suggest that interactions of the MIND complex could determine the observed organization of these particles. Moreover, assuming that the estimates of molecular ratios are correct, what we know about the various components makes MIND the only plausible candidate. Taking these data into account, we propose a model for kinetochore assembly in which oligomeric clustering of MIND creates a core that recruits outer-kinetochore components and defines their copy number (Figure 7B).
Figure 7. Kinetochore Model.

(A) Schematic cross section through a point-centromere kinetochore, illustrating the dimensional expansion from a centromeric nucleosome (bottom) to a microtubule (top). Various conserved molecular complexes are labeled and color-coded. MIND occupies a central position in the hierarchy extending from nucleosome to Ndc80C/DASH.
(B) Surface rendering of a 3-D view similar to the cross section in (A), but omitting the nucleosome-proximal components. The choice of a sixfold structure is arbitrary -- EM images appear to show 5,6, and 7 (Gonen et al, 2012). We have use head I - head II interactions seen in all our crystal forms to construct this model; most of its features do not depend on assuming that those particular contacts are present.
(C) Comparison of kinetochore model with experimental data. EM images adapted from Macmillan Publisher Ltd: [Nature Structural & Molecular Biology] (Gonen et al., 2012), copyright (2012). End-on view of the surface rendering, with nucleosome-proximal components omitted along with the microtubule and DASH, shows proposed head I to head II interactions that generate the central core. Perspective view on the right includes DASH and the microtubule from (B).
We have reported here various observations supporting the notion that MIND can oligomerize even in the absence of COMA or Mif2. In each of the four crystal forms we have examined, we find the same crystallographic packing interface between heads I and II of neighboring MIND-C1 molecules (Figure S4 and S5A). Although recombinant MIND is predominantly monomeric at low concentration in solution, at concentrations greater than about 10 mg/mL, we detect formation of a discrete dimer (Figure S5B). The contact between complexes is evidently not sufficient to create a larger, long-lived oligomer, but further contributions from COMA, Mif2, or post-translational modification could reinforce the interaction we have detected. We can model 5, 6 and 7-fold rings of MIND complexes, with contacts between heads of adjacent complexes that closely resemble the conserved interface in our crystals (Figure 7, Figure S5D). Interaction of two of the MIND complexes in the ring with the N termini of the two chains in the nucleosome-bound Mif2 dimer will stabilize a ring-like MIND oligomer, as will the contacts with Ame1, since COMA also binds Mif2 (Hornung et al., 2014).
The structure of the MIND complex also suggests an interpretation of published images from negative-stain EM of isolated kinetochores (Gonen et al., 2012) (Figure 7C). The size of the hub-and-spokes assembly in these images is consistent with the diameter expected for a radially displayed MIND structure (Figure 7C and Figure S5D). The inner-kinetochore components CBF3, Cse4 nucleosome, COMA and Mif2 would contribute further to the density at the center of the particles seen in the micrographs (Gonen et al., 2012). The organization of these isolated kinetochore assemblies does not depend on microtubules or DASH/Dam1, as neither are present in the preparation (Gonen et al., 2012). Our molecular interpretation of the micrographs is compatible with the head-head contacts and MIND oligomerization proposed in the preceding paragraph and chosen in constructing Figure 7B; small adjustments in the contacts and observed flexibility in the connection between head II and CC1 could accommodate the observed variation in number of spokes. The same molecular picture is equally compatible, however, with any other set of interactions that clusters the heads, provided that it generates a hub-and-spokes assembly having the observed dimensions.
The model in Figure 7 illustrates that the structure of a kinetochore has both the regulatory properties derived from flexible peptide links to relatively small interfaces and the tensile strength required to resist the forces exerted by microtubule depolymerization. Although any one interface would not withstand the force needed to move a chromosome, a series of parallel interfaces can readily do so. Further reinforcement, for specificity as well as stability, comes from crosslinking through interactions of COMA with both MIND and Mif2. The open framework, generated by the long, coiled-coil rods that are common features of both Ndc80C and MIND, is necessary to allow rapid escape of dissociating tubulin heterodimers, while the flexibility of the hinge-like linkages between subcomplexes (Mif2 to MIND, MIND to Ndc80C, COMA to MIND, and presumably similar connections with additional kinetochore components) imparts compliance in response to both lateral and longitudinal forces.
The structure and interactions of MIND are thus appropriate for its function as the adaptor between a centromere anchoring subassembly and a microtubule-contacting framework that can track during depolymerization.
STAR METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for reagents may be directed to, and will be fulfilled by Stephen C. Harrison (harrison@crystal.harvard.edu)
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Expression plasmids contained cDNA sequences from Kluyveromyces lactis and Saccharomyces cerevisiae. Recombinant proteins were overexpressed in Escherichia coli Rosetta2 pLysS cells (Novagen) in 2xYT broth (Yeast Extract Tryptone from Research Products International) or M9, minimal salts media (Sigma) for selenomethionine-derivatized proteins. Cells were grown at 37 °C at 220 rpm in 2 L shaking flasks until A600 reached 0.6 then immediately induced by addition of isopropyl β-D-1-thiogalactopyranoside (IPTG from GoldBio) to a final concentration of 0.25–0.5 mM. After induction, cells were grown for 16–18 hours at 18 °C.
METHOD DETAILS
Protein Expression and Purification
Production of MIND complex components
Kluyveromyces lactis (K. lactis) genes for Mtw1, Nnf1, Nsl1 and Dsn1 proteins were cloned by PCR amplification from genomic DNA into a pET3aTR vector modified for ligation-independent cloning (LIC) (Schmitzberger and Harrison, 2012; Tasto, 2001). All constructs were designed for poly-cistronic co-expression as heterotetramer or heterodimer with a tobacco etch virus (TEV) protease cleavable hexahistidine (His6)-tag at the N terminus of each protein in the following order: Dsn1, Mtw1, Nnf1 and Nsl1 for MIND-C1 and Mtw1, Nnf1 and Dsn1, Nsl1 for head I and head II constructs (see Figure S1 for full list of constructs). All MIND constructs were overexpressed in Escherichia coli (E. coli) Rosetta2 pLysS cells (Novagen) as described above and harvested by centrifugation at 4000 rpm in a J6-MI centrifuge for 30 min (Beckman Coulter). Cell pellets were resuspended in lysis buffer made of 50 mM HEPES (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid), pH 7.6, 600 mM NaCl, 10 mM imidazole, 1 mM tris(2-carboxyethyl)-phosphine (TCEP), 10% glycerol, supplemented with protease inhibitors (1 mM phenylmethanesulfonyl fluoride, 2 μg/ml aprotinin, 1 μg/ml leupeptin, 1 μg/ml pepstatin, all from Roche). Cells were lysed, and spun down at 19000 rpm for 45 min in a JA-20 fixed angle rotor (Avanti J-E series centrifuge, Beckman Coulter) to remove debris. The sample was immobilized on Co2+ resin (TALON Metal Affinity Resin, Clontech) and incubated for 1 hour at 4 °C while rotating. The immobilized protein was washed with 15 bed volumes of buffer containing 50 mM HEPES, pH 7.5, 600 mM NaCl, 10 mM imidazole, 1 mM TCEP, 10% glycerol, and eluted in 50 mM HEPES, pH 7.5, 50 mM NaCl, 400 mM imidazole, 1 mM TCEP, 10% glycerol. Protein samples were immediately loaded on HiTrap Q HP (GE Healthcare) anion exchange column followed by size exclusion chromatography (SEC) on a preparative Superdex-S200 HiLoad 16/60 column (GE Healthcare). All proteins were concentrated with Amicon Ultra centrifugal filters (Millipore) and immediately used for crystallization trials or frozen and kept at -80 °C for biochemical experiments. All MIND constructs, except head I (Mtw11-120/Nnf11-102) were stored in SEC buffer from the final Superdex 200 run including: 30 mM HEPES, pH 7.5, 150 mM NaCl, 1 mM TCEP and 50 mM CHES, pH 9.0, 150 mM NaCl, 1 mM TCEP for head I (Mtw11-120/Nnf11-102).
Production of Mif2 and Ame1
K. lactis full-length Mif2 and Mif21-41 peptide were cloned with a cleavable TEV protease site after the N-terminal His6-maltose binding protein (MBP)-tag, expressed and purified as described above. Full-length Mif2 was used without removing the His6-MBP-tag in 30 mM HEPES, pH 7.5, 150 mM NaCl, 1 mM TCEP. The elution fractions from Superdex 200 of His6-MBP-Mif21-41 were combined and concentrated to less than 2 ml using Amicon Ultra 10K centrifugal filters and incubated with TEV while rotating overnight at 4 °C. After cleaving the tag, the sample was processed with an Amicon Ultra 10K centrifugal filter, separating the His6-MBP-tag and TEV protease as retentate from the Mif21-41 peptide as filtrate. The Mif21-41 peptide was added to different MIND constructs and concentrated together to test binding and for crystallization experiments with Mtw11-120/Nnf11-102.
K. lactis Ame11-24 was cloned into a single expression LIC vector with an N-terminal His6-glutathione S-transferase (GST)-tag as described above. His6-GST-Ame11-24 peptide was purified the same way as the MIND constructs, stored and used for binding experiments in 50 mM CHES, pH 9.0, 150 mM NaCl, 1 mM TCEP.
Production of Spc24/Spc25 and Dsn1560-572
The coding region for Spc24 and Spc25 were amplified by PCR from isolated S. cerevisiae genomic DNA and cloned into a pET3aTR vector encoding an N-terminal His6-tag with TEV protease cleavage site added to Spc25. The heterodimer Spc24 (residues 155–213)/Spc25 (residues132–221) was recombinantly expressed and purified following a protocol similar to the one described above for the production of MIND constructs. After the Co2+-resin affinity purification step, TEV protease was added to the eluted sample to cleave the His6-tag and the product was passed again over Co2+-resin to clear sample of free His6-tag and His6-TEV protease. Spc24155-213/Spc25132-221 was concentrated and loaded on gel filtration Superdex-S200 HiLoad 16/60 column (GE Healthcare) equilibrated in gel filtration (GF) buffer (20 mM bis-tris propane, pH 7.6, 300 mM NaCl, 5 mM dithiothreitol).
The S. cerevisiae Dsn1 peptide fragment (residues 560–572: QQLLKGLSLSFSK) was produced by Tufts University Core Facility (Boston, MA), and resuspended in GF buffer.
Generation of Nanobodies
Nanobodies against the MIND-C1 complex were raised in alpacas according to established protocols in the laboratory of Hidde Ploegh at the Whitehead Institute for Biomedical Research. Briefly, recombinantly produced MIND-C1 was injected into an alpaca at 2 mg/mL to elicit an immune response. After two rounds of immunization, total RNA was isolated and used to synthesize the variable fragments of heavy chain antibodies. Nanobodies were selected for binding MIND-C1 by phage display as described in detail in (Li et al., 2016).
Production and screening of Nanobodies
Eight nanobodies with a C-terminal His6-tag were recombinantly expressed in Rosetta2 pLysS cells as described above. Cells were harvested following the protocol for MIND complexes described above. Purification of the nanobodies included gravity flow Co2+-affinity column, concentration of the sample eluted in 30 mM HEPES, pH 7.5, 200 mM NaCl and 400 mM imidazole, followed by size exclusion chromatography using a Superdex-S200 HiLoad 16/60 column in buffer containing 30 mM HEPES, pH 7.5, 150 mM NaCl and 1 mM EDTA.
To test binding, each nanobody (at 20 μM concentration) was incubated for 30 min with MIND-C1 (at 2 μM) and loaded on an analytical Superdex-S200 Increase 10/300 GL column (GE Healthcare). One of the eight nanobodies tested formed a complex with MIND-C1 that was stable throughout the chromatography run and was used for crystallization trials.
Limited Proteolysis and Mass Spectrometry
To identify a stable MIND heterotetramer suitable for crystallization trials, we performed limited proteolysis with trypsin using full-length recombinantly produced K. lactis MIND containing Mtw1, Nnf1, Nsl1 and Dsn1. Full-length MIND at 2 mg/mL was treated with trypsin at 1:1,000, 1:500, 1:250 and 1:100 ratio (w/w) for 30 min at room temperature in 100 μl reaction volumes. Trypsin digestion was stopped by adding crystalline powder of guanidine hydrochloride (Sigma) until saturation and submitted for ion trap mass spectrometry analysis at the Howard Hughes Medical Institute Mass Spectrometry Laboratory at University of California, Berkeley. The most stable MIND-C1 complex had two Dsn1 deletions (residues 1-229 and 480-491) and a Mtw1 C-terminal deletion (residues 234-288).
Protein Crystallization and Data Collection
All crystals of MIND-C1, MIND-C1-nanobody, MN-C2, MN-C2-Mif21-41 and Spc24/Spc25–Dsn1560-572 were grown in 48 well plates at 18 °C by hanging-dr op vapor diffusion with 200 μl reservoir solution and a drop of 1–2 μl each, protein and reservoir solution at different ratios. Details of protein concentration, sample buffer, crystallization condition and cryo-protection solution for each of the crystallized complexes are provided in Table S3.
All datasets except selenomethionine-labeled MN-C2 were collected at the Advanced Photon Source at Argone National Laboratory on NE-CAT beamlines 24ID-C and 24ID-E. The datasets for labeled MN-C2 were collected at beamline 8.2.2 at the Advanced Light Source. We screened 3046 crystals, collected and processed more than 400 promising datasets to build the final K. lactis MIND-C1 structure.
X-ray Structure Determination
MIND-C1
MIND-C1 native and mercury-derivative crystals are both in space group P21, with one complex per asymmetric unit. Soaking with ethylmercurithiosalicylic acid (EMTS) made the derivate non-isomorphous, however, and not useful for phasing by isomorphous replacement. Anomalous signal from selenomethionine-substituted and tantalum bromide-derivatized crystals was too weak to be used for de novo phasing, but proved useful for structure validation (see below and also Figure S2). We therefore obtained starting phases from the mercury anomalous signal, relying on high redundancy for adequate signal (Akey, 2014).
We processed with XDS (Kabsch, 2010) the single anomalous dispersion (SAD) data from 37 mercury derivative crystals. We only refined crystal orientation and unit cell dimensions in the INTEGRATE step but carried out full post-refinement of experimental parameters in CORRECT. We observed substantial fluctuations in data quality in frames recorded along the goniometer rotation, presumably because of the small crystal size (10-15 μm) and the difficulty in mounting single ones. We therefore excluded frames for which the mosaicity exceeded a heuristically determined threshold. After comparing individual crystals to a reference dataset (cross R factors), we selected 33 and scaled them together in XSCALE. The merged data used for phasing have over 30-fold redundancy and anomalous signal to about 5 Å resolution (Table S1).
We found 6 mercury sites using SHELXD (Sheldrick, 2010), refined them in SHARP (Vonrhein, 2007) and calculated phase probabilities. Initial maps were obtained after statistical density modification using RESOLVE (54% solvent content) (Terwilliger, 2000) (Figure S2A).
Phases for the native, two selenomethionine-substituted and a tantalum-derivatized crystal forms were determined by molecular replacement using PHASER (McCoy et al., 2007). For this, we flooded the initial map with dummy atoms and carved out individual domains by inspection in PyMol (Schrödinger, LLC). To extract density, we used masks calculated from the dummy atom domain models. As we know a posteriori, the domains are head I, head II, coiled-coil 1 (CC1), coiled-coil 2 (CC2) and the third part of the rod (CC3). Molecular replacement solutions were difficult to obtain when we used density of the whole complex as a search model. We attribute this difficulty to the differences between crystal forms in bending or twisting along the rod of MIND-C1, leading to an overall poor match due to the extended shape of the complex. We therefore tested different combinations of domains as search models and found solutions when head I, CC1 and CC2 were defined as a single rigid group. We subsequently could place head II and CC3 (plus a nanobody in case of SeMet II and Tantalum). In the native crystal form, MIND-C1 is shifted about 5 Å in the a/c-plane with respect to the crystallographic origin with respect to its position in the mercury derivative, confirming non-isomorphism.
To obtain better phases, we used the MN-C2 head I structure (see below) as partial model for SAD phasing in PHASER and multi-crystal density averaging with DMMULTI (Cowtan, 1994) and PHENIX multi_crystal_average (Adams et al., 2010), applying the previously determined domain masks and transformation matrices obtained from the molecular replacement solutions. The visual appearance of the resulting maps convinced us that we had obtained reasonable phase information to approximately 4.5 Å resolution (Figure S2A), but phase extension to the diffraction limit in the native crystal form worked poorly, because of the lower resolution limits for the other crystal forms and the native-crystal solvent content of only 54%.
We built poly-alanine models for the helical parts of the complex manually and with PHENIX find_helices_strands. We matched the lengths and positions of observed helices to secondary structure predictions (www.predictprotein.org) (Yachdav et al., 2014), thereby obtaining a consistent picture of all subunit connectivities. We assigned the amino-acid sequence with the help of cysteine and methionine positions observed in anomalous difference Fourier maps calculated from the mercury- and the two selenomethionine-substituted crystal forms of MIND-C1, respectively. The anomalous difference Fourier analysis confirmed the position of 6 out of 7 cysteines and 10 out of 17 methionines (Figure S2B). We completed the model by iterations of interactive model building in O (Jones, 1991) with refinement.
Native diffraction data were processed as described above for the mercury derivative. Because of strong anisotropy, we ellipsoidally truncated native intensities at Bragg spacings of 3.2, 4.1, 3.4 Å along the crystallographic a*, b* and c* axis, respectively, using the anisotropy server (Figure S2C) (Strong, 2006). After conversion of intensities to structure factor amplitudes, we applied an overall isotropic Wilson B factor of 80 Å2.
We refined the model in PHENIX with individually restrained coordinates and B factors, applying rotamer and secondary structure restraints throughout the refinement, and analyzed the final model with MolProbity (Chen et al., 2010). Refinement and model statistics are in Table S1. The final model includes Mtw1 (residues 1-233), Nnf1 (8-203), Nsl1 (5-216) and Dsn1 (253-471). Flexible residues that have not been traced in the electron density map include Nnf1 (180-185), Nsl1 (151-166), and Dsn1 (230-252, 344-348, 406-418 and 472-479). We modeled all but five omitted residues in the long loop of Dsn1, residues 311-353, as poly-alanine (311-343, 349-353). Electron density for the segments Mtw1 44-58 and 169-188 is poor and did not allow confident assignment of backbone stereochemistry, and Cα positions will have larger errors than in the rest of the model. However, the former segment is well defined in the MN-C2 structure.
The final R factors with Rwork/Rfree of 32.2/36.3 % are higher than expected for a 3.2 Å resolution structure, because of the strongly anisotropic diffraction (Figure S2C). If we calculate R factors with data cut off at 3.8 Å resolution, at which completeness drops below 84% and thus is a more realistic number for the overall resolution for the electron density map, Rwork/Rfree is 31.3/34.8 % and within the acceptable range of R factors observed for structures in the Protein Data Bank (PDB) determined at similar resolution.
The native unit-cell dimensions, with a and c almost equal in length (Table S1), could in principle allow pseudo-merohedral twinning in space group P21. Native moments and the L-test (Padilla and Yeates, 2003) calculated from intensities or amplitudes with phenix.xtriage do not indicate twinning (the expected values for untwinned and perfectly twinned crystals, respectively, are given in parenthesis): <I2>/<I>2 = 2.04 (2.0, 1.5), <F>2/<F2> = 0.797 (0.785, 0.885), <|E2-1|>=0.716 (0.736, 0.541), <|L|> = 0.471 (0.500, 0.375), <L2> = 0.301 (0.333, 0.200). We also excluded twinning by test refinement with “l, -k, h” as twin law, obtaining Rwork/Rfree equal to 31.0/36.3 %, with a twin fraction of 0.11.
MN-C2
Native and selenomethionine-labeled MN-C2 (Mtw11-120/Nnf11-102) complex both crystallized in space group P62. We indexed and integrated the data with XDS and used 13 SAD datasets from 11 selenomethionine-labeled crystal of MN-C2 for phase determination. We located the heavy atom sites with SHELXD (Sheldrick, 2010) and used them as input in PHENIX AutoSol wizard (Terwilliger et al., 2009) for phase calculation with PHASER and solvent flattening with RESOLVE (Terwilliger, 2000). We completed an initial model from PHENIX AutoBuild by iterative rounds of manual building in Coot (Emsley et al., 2010) and refined it with phenix.refine (Adams et al., 2010) (Table S2). The final model includes Mtw1 residues 3-98 and Nnf1 residues 13-102.
MN-C2-Mif21-41 complex
Rod-shaped crystals of the MN-C2 (Mtw11-120/Nnf11-102)-Mif21-41 complex were in space group P212121 (Table S2). Diffraction was anisotropic and required ellipsoidal truncation of the intensities at Bragg spacings of 3.1, 2.4, 2.9 Å along the crystallographic a*, b* and c* axis, respectively, using the anisotropy server (Strong, 2006). We determined the structure of the complex by molecular replacement with PHASER (McCoy et al., 2007) using MN-C2 as a search model (two molecules per asymmetric unit with 51% solvent content). We built the Mif2 peptide into difference maps with Coot and O and refined the final model (Figure 2B), including Mif2 residues 5-38, with phenix.refine (Afonine et al., 2012).
Spc24/Spc25 - Dsn1 complex
Crystals of S. cerevisiae Spc24155-213/Spc25133-221 - Dsn1560-572 complex grew in space group I4122 with one molecule per asymmetric unit. We indexed and integrated reflections using XDS and scaled the data using the CCP4-suite program Aimless (Winn et al., 2011). We determined phases by molecular replacement in PHASER with the crystal structure of Spc24/Spc25 as a search model (PDB accession code: 2FTX) (McCoy et al., 2007) and used difference Fo-Fc maps from Phaser and composite omit maps calculated with PHENIX to build the peptide (plus two N-terminal residues inherited from the TEV protease cleavage site, Ser-2 and Asn-1) with Coot (Emsley et al., 2010). We refined the model with maximum likelihood targets in phenix.refine (Afonine et al., 2012)(Table S2). The final model includes the entire sequences of Spc24155-213 and Spc25133-221 and residues 560-571 of Dsn1.
Binding Studies by Size Exclusion Chromatography
Analytical size exclusion chromatography (SEC) experiments at 4 °C were on a Superdex-S200 Increase 10/300 GL column (GE Healthcare). Protein samples were mixed at different concentrations, specified in the figure legends for each experimental complex, and incubated for 30 min on ice before injecting onto the column in a final volume of 200 μl. The SEC buffer for binding experiments with S. cerevisiae Spc24155-213/Spc25133-221 included 30 mM HEPES, pH 7.5, 150 mM NaCl, 1 mM TCEP. The rest of the experiments testing the interaction of MIND-C1, head I and head II mutants with different binding partners (Figures 2, 3, 5 and 6) were with an SEC buffer containing 50 mM CHES, pH 9.0, 150 mM NaCl, 1 mM TCEP. Elution fractions were resolved on 4–20% SDS-PAGE gels and stained with Coomassie blue.
Binding Studies by Fluorescence Polarization
We carried out binding experiments in low-volume 384 well plates (Corning) on a SpectraMax M5e instrument (Molecular Devices) at 23 °C (excitation wavelength, 484 nm; emission wavelength, 525 nm). FITC-Mif21-41 and FITC-Ame11-25 peptides were synthesized by the Tufts University Core Facility. Each reaction contained FITC-labeled peptide at 50 nM and concentrations of MIND-C1, MIND-C1:D205 and MIND-C1:D205-2D (see Figure S1 for MIND construct details) ranging from 0 – 10 μM. The experiments were done in 50 mM CHES, pH 9, 150 mM NaCl and 1 mM TCEP in a final volume of 20 μl.
We also carried out binding experiments of ScSpc24/Spc25 with Dsn1 and Mtw1 peptides in conditions similar to the protocol described above. FITC-Dsn1560-572, FITC-Mtw1230-262 and FITC-Mtw1274-289 peptides were used at 50 nM and mixed with ScSpc24/Spc25 at concentrations from 0 – 50 μM in a final volume of 20 μl. The buffer for these reactions contained 30 mM HEPES, pH 7.5, 150 mM NaCl and 1 mM TCEP. The reactions were analyzed after incubating for 15 minutes at room temperature.
Size Exclusion Chromatography – Multi-Angle Light Scattering
Size Exclusion Chromatography – Multi-Angle Light Scattering (SEC-MALS) was carried out with a Dawn Heleos-II Multi-Angle Light Scattering (MALS) detector (Wyatt) and the Optilab T-Rex refractive index detector (Agilent Technologies) using a Superose 6 PC 3.2/30 GL column (GE Healthcare Life Sciences). We injected 100 μl of MIND-C1 at 15 mg/mL in buffer containing 30 mM HEPES, pH 7.5, 150 mM NaCl, 1 mM TCEP. Molar mass calculations were done with Astra software using a dn/dc value of 0.179.
Sequence Alignments and Figure Preparation
Sequences were aligned with T-Coffee (Notredame, 2000) and displayed with ESPript (Robert and Gouet, 2014). We prepared the figures with PyMol (Schrödinger, LLC) and matplotlib (Hunter, 2007).
QUANTIFICATION AND STATISTICAL ANALYSIS
Fluorescence polarization is plotted using the Prism7 software as mean ± standard error from 6 replicates and shown as a function of protein concentration. We calculated the dissociation constant (KD) from a non-linear least squares fit with a single site-specific binding model.
DATA AND SOFTWARE AVAILABILITY
Atomic coordinates have been deposited in the Protein Data Bank with accession numbers 5T58 (MIND-C1), 5T51 (MN-C2), 5T59 (MN-C2 – Mif21-41), 5T6J (Spc24/Spc25-Dsn1560-572).
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, Peptides, and Recombinant Proteins | ||
| Dsn1560-572 QQLLKGLSLSFSK |
Tufts University Core Facility | N/A |
| FITC-Dsn1560-572 FITC-AHA-QQLLKGLSLSFSK |
Tufts University Core Facility | N/A |
| Mtw1230-262 KDFRTRYIDIRTNNVLRKLGLLGDKEDEKQSAK |
Tufts University Core Facility | N/A |
| FITC-Mtw1230-262 FITC-AHA-KDFRTRYIDIRTNNVLRKLGLLGDKEDEKQSAK |
Tufts University Core Facility | N/A |
| Mtw1274-289 SIDIEEPQLDLLDDVL |
Tufts University Core Facility | N/A |
| FITC-Mtw1274-289 FITC-AHA- SIDIEEPQLDLLDDVL |
Tufts University Core Facility | N/A |
| FITC-Mif21-41 MDYMNLGVKSRKTGLTVNKTVQKDEYSMENLNDFFKDEQDS |
Tufts University Core Facility | N/A |
| FITC-Ame11-25 MDALKQRHLKLLYRQRGSASRTIDY |
Tufts University Core Facility | N/A |
| PEG 550 MME | Hampton Research | HR2-611 |
| PGA-LM | Molecular Dimensions | MD2-250-108 |
| Ethylmercurithiosalicylic acid, sodium salt Heavy Atom Screen M1 | Hampton Research | HR2-448 |
| L(+)-Selenomethionine | Acros Organics | 3211-76-5 |
| PEG 8000 | Sigma | 81268-1kg |
| Tantalum Cluster Derivatization Kit | Jena Bioscience | PK-103 |
| PEG 3000 | Rigaku | 1008057 |
| Deposited Data | ||
| Atomic coordinates, MIND-C1 structure | Protein Data Bank | PDB: 5T58 |
| Atomic coordinates, MN-C2 structure | Protein Data Bank | PDB: 5T51 |
| Atomic coordinates, MN-C2-Mif21-41 structure | Protein Data Bank | PDB: 5T59 |
| Atomic coordinates, Spc24/Spc25-Dsn1560-572 structure | Protein Data Bank | PDB: 5T6J |
| Experimental Models: Organisms/Strains | ||
| E. coli Rosetta 2 (DE3) | EMD | #71400 |
| Recombinant DNA | ||
| pLIC_M42 H6-TEV- *MIND-C1 | This study | N/A |
| pLIC_M147 H6-TEV- *MIND-C1:D205 | This study | N/A |
| pLIC_M147_mut2 H6-TEV- *MIND-C1:D205-2D | This study | N/A |
| pLIC_M142 H6-TEV- *MIND-C1:D367 | This study | N/A |
| pLIC_M89 H6-TEV- *head I MN-C2 | This study | N/A |
| pLIC_M89_mut4 H6-TEV- *head I Site II mut. Mtw1 N32A/Y36A/E73A/D77A |
This study | N/A |
| pLIC_M89_mut5 H6-TEV- *head I Site III mut Nnf1 E76A/E83A |
This study | N/A |
| pLIC_M110 H6-TEV- *head II D256 | This study | N/A |
| pLIC_M100 H6-TEV- *head II D230 | This study | N/A |
| pLIC_M136 H6-TEV- *head II D205 | This study | N/A |
| pLIC_M136_mut2 H6-TEV- *head II D205-2D | This study | N/A |
| pLIC_M107 H6-TEV- *head II D230L | This study | N/A |
| pLIC_Mf6 H6-MBP-TEV- *Mif2-FL | This study | N/A |
| pLIC_Mf3 H6-MBP-TEV- *Mif2 (1-41) | This study | N/A |
| pLIC_C6 H6-GST-TEV- *Ame1 (1-24) | This study | N/A |
| pLIC_S08 H6-TEV- *Spc24/Spc25 | This study | N/A |
| pET30b-VHH3- *nanobody | This study | N/A |
| Software and Algorithms | ||
| XDS | (Kabsch, 2010) | xds.mpimfheidelberg.mpg.de |
| SHELXD | (Sheldrick, 2010) | shelx.uniac.gwdg.de/SHELX/ |
| SHARP | (Vonrhein, 2007) | www.globalphasing.com/sharp/ |
| RESOLVE | (Terwilliger, 2000) | https://solve.lanl.gov/Resolve/resolve.html |
| PHASER | (McCoy et al., 2007) | wwwstructmed.cimr.cam.ac.uk/phaser_obsolete/ |
| PyMol | Schrödinger, LLC | www.pymol.org |
| DMMULTI | (Cowtan, 1994) | www.ccp4.ac.uk/html/dmmulti.html |
| PHENIX | (Adams et al., 2010) | www.phenix-online.org |
| O | (Jones, 1991) | Xray.bmc.uu.se/alwyn/A-Z_of_O/AZ_frameset.html |
| anisotropy server | (Strong, 2006) | https://services.mbi.ucla.edu/anisoscale/ |
| Secondary structure prediction | (Yachdav et al., 2014) | www.predictprotein.org |
| MolProbity | (Chen et al., 2010) | Molprobity.biochem.duke.edu |
| Coot | (Emsley et al., 2010) | www2.mrclmb.cam.ac.uk/personal/pemsley/coot/ |
| Prism7 | GraphPad | www.graphpad.com |
| T-Coffee | (Notredame, 2000) | tcoffee.crg.cat |
| matplotlib | (Hunter, 2007) | matplotlib.org |
| ESPript | (Robert and Gouet, 2014) | espript.ibcp.fr/ESPript/ESPript/ |
Supplementary Material
Figure S1. MIND Constructs and Cartoon Drawings Used in this Study, Related to Figure 1
(A) The mercury derivative electron density map at different stages of phase problem solution: initial map obtained after single anomalous dispersion (SAD) phasing (left), after statistical density modification using RESOLVE (middle), and after including partial head I structure factors from MN-C2 and multi-crystal density averaging (right).
(B) Sequence assignment validation by anomalous Fourier difference analysis. Peak heights in standards deviations above the mean are shown from anomalous Fourier maps calculated with model phases and observed anomalous differences from the mercury- (left) and two selenomethione-derivatized (middle and right) crystals. Peaks that map to mercury-derivatized cysteine or selenomethionine residues are annotated with an arrow and the corresponding subunit name and residue number. The inset structure shows the location of the mapped peaks as spheres.
(C) Native diffraction data and final electron density maps. Left, analysis of anisotropic diffraction using the CCP4 program truncate (French and Wilson, 1978). Mean F/σ along the three crystallographic axes is plotted against resolution. Resolution cutoffs of 3.2, 4.1 and 3.4 Å were applied during ellipsoidal truncation using the anisotropy server (Michael Strong, 2006). Middle, final composite omit map calculated with phenix.composite_omit_map. Right, final σA-weighted 2mFo-DFc electron density map calculated with phenix.refine.
(A) MIND Head I Does Not Bind Head II D230 in the Presence of Mild Chaotropic Salts. Superdex 200 size exclusion chromatography testing the interaction between MIND head I (Mtw11-120/Nnf11-102, fraction A) and head II (Dsn1230-313/Nsl11-83, fraction B) in the presence of 0.2 M KBr and 0.2 M KSCN. The elution profiles are overlaid in the chromatogram on top and the fractions are resolved on the Coomassie stained SDS-PAGE gels.
(B) SEC experiments as in (A) show that Dsn1230-255 contacts MIND head I at the conserved Ame1-binding site containing Mtw1 residues N32, Y36, E73 and E77. Bottom two gels show that MIND head II D230 (Dsn1230-397/Nsl11-118) binds head I (Mtw11-120/Nnf11-102), but does not bind head I Site II mut (Mtw11-120:N32A, Y36A, E73A, E77A/Nnf11-102). Top three gels are control experiments with elution profiles of each construct alone.
(C) Binding experiments showing that Dsn1 competes with Ame1 for binding MIND head I. Elution fractions from the top two gels show that MIND head I (Mtw11-120/Nnf11-102), fraction a, forms stoichiometric complexes with both head II D205 (Dsn1205-397/Nsl11-118), fraction b, and head II D205-S2D (Dsn1205-397:S213D, S223D/Nsl11-118), fraction c. Bottom two gels show that His6-GST-Ame11-24 breaks the stoichiometric complex and binds head I more tightly in the presence of head II D205-S2D (gel 4) than in the presence of head II D205 (gel 3). Fraction M: molecular-mass marker bands for 10, 15, 20, 25, 30, 40, 50, 60 kDa.
Common to all MIND-C1 crystal forms is how head II binds head I of an adjacent MIND molecule related by crystallographic symmetry. On the left, three MIND molecules are shown for each native (A), SeMet I (B), SeMet II (C) and tantalum-derivatized (D) crystal form. Unit cells are drawn in black and the distance between molecules is given in angstrom (Å). On the right, the packing is shown for each crystal, viewed along the three crystallographic axes.
(A) Close-up views of the head II-head I interface observed in four crystal forms (Figure S4).
(B) Left, detection of a discrete MIND dimer by SEC-MALS. Right, oligomerization depends on the presence of head II, because a construct MIND-C1:D367 does not show a shoulder in the elution profile at higher molecular weight as seen for MIND-C1 (blue curve, indicated with an arrow). Moreover, oligomerization does not require the N-terminal Dsn1 arm, as a construct MIND-C1:D261, lacking the arm, shows the same elution profile (red curve) as MIND-C1.
(C) Superposition MIND from four crystal forms reveals hinge regions. We superimposed MIND-C1 from the crystal shown in Figure S4 on CC1 and CC2. Movements of the head I, head II and CC3 domains are roughly indicated by arrows.
(D) Schematic illustrating how a 6-fold ring of MIND complexes is generated, where contacts between heads of adjacent complexes closely resemble the conserved interface shown in (A).
(A) Ribbon representation of the refined MIND-C1 model to show the B factor distribution, where ribbon thickness is proportional to the B factor of the residues’ Cα atom and colored in a gradient from dark blue (low B factors) to orange (high B factors). Residue numbers for the termini and chain breaks of the four subunits are also shown.
(B) Real space correlation coefficients (RSCC) for each MIND-C1 residue were calculated with phenix.real_space_correlation. Residue ranges are highlighted as follows: dotted line, residues included in the crystallization construct; solid line, residues modeled in structure; black bar, residues modeled as poly-alanine; gray bar, residues modeled in poor electron density, where the chain trace is approximate.
Table S1. MIND-C1 Data Collection, Phasing and Refinement Statistics, Related to Experimental Procedures
Table S2. MN-C2, MN-C2-Mif2 and Spc24/Spc25-Dsn1 Data Collection, Phasing and Refinement Statistics, Related to Experimental Procedures
Table S3. Protein Crystallization Conditions for MIND-C1, MN-C2, MN-C2-Mif2 and Spc24-Spc25-Dsn1 Complexes, Related to Experimental Procedures
Supplemental Movie 1. Kinetochore Model, Related to Figure 7.
Amino acid sequences from six fungal and four metazoan species were aligned. Sequence accession numbers are as follows, where the numbers for each species correspond to Mtw1, Nnf1, Nsl1 and Dsn1, respectively: Kluyveromyces lactis (k_lac: XM_455186, XM_454208, XM_452912, XM_453754), Saccharomyces cerevisiae (s_cer: NM_001178179, NM_001181770, NM_001184047, NM_001179532), Eremothecium gossypii (e_gos: NM_209816, NM_208451, NM_210764, NM_211975), Candida dubliniensis (c_dub: XM_002418821, XM_002421686, XM_002421339, XM_002416733), Candida albicans (c_alb: XM_709448, XM_713094, XM_883711, XM_713294), Lachancea thermotolerans (l_the: XM_002552753, XM_002553514, XM_002555972, XM_002553655), Homo sapiens (h_sap: XM_005256798, NM_007221, NM_015471, NM_001145316), Mus musculus (m_mus: XM_006537204, NM_025928, NM_198654, NM_025853), Gallus gallus (g_gal: NM_001044677, NM_001044676, NM_001044646, NM_001044660), Xenopus laevis (x_lae: NM_001092335, NM_001091138, XM_012969245, NM_001166438). Secondary structure is annotated at the top, predicted in black and observed in the crystal structure in blue. Residues built in the crystal structure are shown with a solid bar at the bottom of the alignment in color corresponding to the MIND subunit (Mtw1, green; Nnf1, gray; Nsl1, blue, Dsn1, orange). The orange dashed line in Dsn1 indicates the long loop 311-353 partially modeled as poly-alanine. Dsn1 residues 205-252 involved in regulating Mif2 and Ame1 binding are shown as open green ovals. S. cerevisiae Dsn1 residues 560-572, crystallized with Spc24-Spc25, are boxed orange. Specific residues mentioned in the text are highlighted as solid ovals.
Highlights.
MIND-complex crystal structure defines pivotal interactions in kinetochore assembly
One end of 200 Å-long MIND rod binds Mif2 and COMA; the other, Ndc80 complex
Phosphorylation of Dsn1 subunit relieves autoinhibition of Mif2 and COMA binding
Acknowledgments
We thank the Northeast Collaborative Access Team (NE-CAT) staff at the Advanced Photon Source (APS) sector 24 for advice and assistance in data collection; D. King (University of California, Berkeley) for mass spectrometry; K. Corbett (Ludwig Institute for Cancer Research), U.S. Cho (University of Michigan), S. Hinshaw and F. Schmitzberger for their generosity with reagents and helpful scientific discussions; H. Ploegh and J. Ingram (Whitehead Institute for Biomedical Research) for generating nanobodies; K. Arnett (Center for Macromolecular Interactions, Harvard Medical School) for assistance with binding studies; A. Morin for assistance with graphic design; K. Diederichs (Universität Konstanz), R. Keegan (STFC Rutherford Appleton Lab) for advice on data collection and processing; S. Biggins (Fred Hutchinson Cancer Center), A. Musacchio, A. Petrovic (Max-Plank Institute Dortmund) for critical reading of the manuscript and members of the Harrison and Biggins laboratories for helpful discussions. NE-CAT is funded by NIH grant P41 GM103403 and the Pilatus 6M detector on 24-ID-C by NIH-ORIP HEI grant S10-RR029205. APS is operated for the DOE Office of Science by Argonne National Laboratory under contract DE-AC02-06CH11357. The research was supported by the Howard Hughes Medical Institute (SCH).
Footnotes
AUTHOR CONTRIBUTIONS
Y.N.D. and S.C.H. designed the experiments. Y.N.D., R.V. and Y.K. cloned, expressed, purified proteins and performed crystallization trials. S.J., S.B. and R.V. helped design experiments and interpret data. S.J. developed strategies for computational data analysis and analyzed MIND-C1 data. Y.N.D. and S.J. determined the structures of MIND complexes. R.V. determined the structure of Spc24/Spc25-Dsn1. Y.N.D. and S.C.H. wrote the paper with input from S.J.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr D Biol Crystallogr. 2010;66:213–221. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Afonine PV, Grosse-Kunstleve RW, Echols N, Headd JJ, Moriarty NW, Mustyakimov M, Terwilliger TC, Urzhumtsev A, Zwart PH, Adams PD. Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr D Biol Crystallogr. 2012;68:352–367. doi: 10.1107/S0907444912001308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akey DLB, WC, Dutta S, Konwerski J, Jose J, Jurkiw TJ, DelProposto J, Ogata CM, Skiniotis G, Kuhn RJ, Smith JL. Flavivirus NS1 Structures Reveal Surfaces for Associations with Membranes and the Immune System. Science. 2014;343:5. doi: 10.1126/science.1247749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akiyoshi B, Nelson CR, Biggins S. The aurora B kinase promotes inner and outer kinetochore interactions in budding yeast. Genetics. 2013;194:785–789. doi: 10.1534/genetics.113.150839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biggins S. The composition, functions, and regulation of the budding yeast kinetochore. Genetics. 2013;194:817–846. doi: 10.1534/genetics.112.145276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biggins S, Murray AW. The budding yeast protein kinase Ipl1/Aurora allows the absence of tension to activate the spindle checkpoint. Genes Dev. 2001;15:3118–3129. doi: 10.1101/gad.934801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloom K. The Centromere Frontier: Kinetochore Components, Microtubule-Based Motility, and the CEN-Value Paradox. Cell. 1993;73:4. doi: 10.1016/0092-8674(93)90242-i. [DOI] [PubMed] [Google Scholar]
- Cheeseman IM, Brew C, Wolyniak M, Desai A, Anderson S, Muster N, Yates JR, Huffaker TC, Drubin DG, Barnes G. Implication of a novel multiprotein Dam1p complex in outer kinetochore function. J Cell Biol. 2001;155:1137–1145. doi: 10.1083/jcb.200109063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheeseman IM, Chappie JS, Wilson-Kubalek EM, Desai A. The conserved KMN network constitutes the core microtubule-binding site of the kinetochore. Cell. 2006;127:983–997. doi: 10.1016/j.cell.2006.09.039. [DOI] [PubMed] [Google Scholar]
- Cheeseman IM, Desai A. Molecular architecture of the kinetochore-microtubule interface. Nat Rev Mol Cell Biol. 2008;9:33–46. doi: 10.1038/nrm2310. [DOI] [PubMed] [Google Scholar]
- Cheeseman IM, AS, Jwa M, Green EM, Kang J-S, Yates JR, III, Chan CSM, Drubin DG, Barnes G. Phospho-Regulation of Kinetochore-Microtubule Attachments by the Aurora Kinase Ipl1p. Cell. 2002;111:9. doi: 10.1016/s0092-8674(02)00973-x. [DOI] [PubMed] [Google Scholar]
- Chen VB, Arendall WB, 3rd, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho U-S, Corbett KD, Al-Bassam J, Bellizzi JJ, III, De Wulf P, Espelin CW, Miranda JJ, Simons K, Wei RR, Sorger PK, Harrison SC. Molecular Structures and Interactions in the Yeast Kinetochore. Cold Spring Harbor Symposia on Quantitative Biology. 2011;LXXV:7. doi: 10.1101/sqb.2010.75.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciferri C, Pasqualato S, Screpanti E, Varetti G, Santaguida S, Dos Reis G, Maiolica A, Polka J, De Luca JG, De Wulf P, et al. Implications for kinetochore-microtubule attachment from the structure of an engineered Ndc80 complex. Cell. 2008;133:427–439. doi: 10.1016/j.cell.2008.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke LaCJ. Isolation of a yeast centromere and construction of functional small circular chromosomes. Nature. 1980;287:6. doi: 10.1038/287504a0. [DOI] [PubMed] [Google Scholar]
- Cowtan K. dmmulti. Joint CCP4 and ESF-EACBM Newsletter on Protein Crystallography. 1994;31:4. [Google Scholar]
- De Wulf P, McAinsh AD, Sorger PK. Hierarchical assembly of the budding yeast kinetochore from multiple subcomplexes. Genes Dev. 2003;17:2902–2921. doi: 10.1101/gad.1144403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Wulf P, Montani F, Visintin R. Protein phosphatases take the mitotic stage. Curr Opin Cell Biol. 2009;21:806–815. doi: 10.1016/j.ceb.2009.08.003. [DOI] [PubMed] [Google Scholar]
- Emanuele MJ, Lan W, Jwa M, Miller SA, Chan CS, Stukenberg PT. Aurora B kinase and protein phosphatase 1 have opposing roles in modulating kinetochore assembly. J Cell Biol. 2008;181:241–254. doi: 10.1083/jcb.200710019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonen S, Akiyoshi B, Iadanza MG, Shi D, Duggan N, Biggins S, Gonen T. The structure of purified kinetochores reveals multiple microtubule-attachment sites. Nat Struct Mol Biol. 2012;19:925–929. doi: 10.1038/nsmb.2358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornung P, Maier M, Alushin GM, Lander GC, Nogales E, Westermann S. Molecular architecture and connectivity of the budding yeast Mtw1 kinetochore complex. J Mol Biol. 2011;405:548–559. doi: 10.1016/j.jmb.2010.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hornung P, Troc P, Malvezzi F, Maier M, Demianova Z, Zimniak T, Litos G, Lampert F, Schleiffer A, Brunner M, et al. A cooperative mechanism drives budding yeast kinetochore assembly downstream of CENP-A. J Cell Biol. 2014;206:509–524. doi: 10.1083/jcb.201403081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter JD. Matplotlib: A 2D graphics environment. Computing In Science & Engineering. 2007;9:5. [Google Scholar]
- Joglekar AP, Bloom K, Salmon ED. In vivo protein architecture of the eukaryotic kinetochore with nanometer scale accuracy. Curr Biol. 2009;19:694–699. doi: 10.1016/j.cub.2009.02.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joglekar AP, Bouck D, Finley K, Liu X, Wan Y, Berman J, He X, Salmon ED, Bloom KS. Molecular architecture of the kinetochore-microtubule attachment site is conserved between point and regional centromeres. J Cell Biol. 2008;181:587–594. doi: 10.1083/jcb.200803027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joglekar AP, Bouck DC, Molk JN, Bloom KS, Salmon ED. Molecular architecture of a kinetochore-microtubule attachment site. Nat Cell Biol. 2006;8:581–585. doi: 10.1038/ncb1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones TA, Zou J-Y, Cowan SW, Kjeldgaard M. Improved methods for building protein models in electron density maps and the location of errors in these models. Acta Crystallogr A. 1991;A47:9. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- Kabsch W. Xds. Acta Crystallogr D Biol Crystallogr. 2010;66:125–132. doi: 10.1107/S0907444909047337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Yu H. Multiple assembly mechanisms anchor the KMN spindle checkpoint platform at human mitotic kinetochores. J Cell Biol. 2015;208:181–196. doi: 10.1083/jcb.201407074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kline SL, Cheeseman IM, Hori T, Fukagawa T, Desai A. The human Mis12 complex is required for kinetochore assembly and proper chromosome segregation. J Cell Biol. 2006;173:9–17. doi: 10.1083/jcb.200509158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krenn V, Musacchio A. The Aurora B Kinase in Chromosome Bi-Orientation and Spindle Checkpoint Signaling. Front Oncol. 2015;5:225. doi: 10.3389/fonc.2015.00225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Park E, Ling J, Ingram J, Ploegh H, Rapoport TA. Crystal structure of a substrate-engaged SecY protein-translocation channel. Nature. 2016;531:395–399. doi: 10.1038/nature17163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malvezzi F, Litos G, Schleiffer A, Heuck A, Mechtler K, Clausen T, Westermann S. A structural basis for kinetochore recruitment of the Ndc80 complex via two distinct centromere receptors. EMBO J. 2013;32:409–423. doi: 10.1038/emboj.2012.356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maskell DP, Hu XW, Singleton MR. Molecular architecture and assembly of the yeast kinetochore MIND complex. J Cell Biol. 2010;190:823–834. doi: 10.1083/jcb.201002059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McEwen BF, Heagle AB, Cassels GO, Buttle KF, Rieder CL. Kinetochore Fiber Maturation in PtK1 Cells and Its Implications for the Mechanisms of Chromosome Congression and Anaphase Onset. The Journal of Cell Biology. 1997;137:14. doi: 10.1083/jcb.137.7.1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray BMSaAW. Lack of tension at kinetochores activates the spindle checkpoint in budding yeast. 2001;11:6. doi: 10.1016/s0960-9822(01)00451-1. [DOI] [PubMed] [Google Scholar]
- Notredame CH, DG, Heringa J. T-Coffee: A novel method for fast and accurate multiple sequence alignment. J Mol Biol. 2000;302:205–217. doi: 10.1006/jmbi.2000.4042. [DOI] [PubMed] [Google Scholar]
- Padilla JE, Yeates TO. A statistic for local intensity differences: robustness to anisotropy and pseudo-centering and utility for detecting twinning. Acta Crystallographica Section D. 2003;59:1124–1130. doi: 10.1107/s0907444903007947. [DOI] [PubMed] [Google Scholar]
- Pagliuca C, Draviam VM, Marco E, Sorger PK, De Wulf P. Roles for the conserved spc105p/kre28p complex in kinetochore-microtubule binding and the spindle assembly checkpoint. PLoS One. 2009;4:e7640. doi: 10.1371/journal.pone.0007640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrovic A, Mosalaganti S, Keller J, Mattiuzzo M, Overlack K, Krenn V, De Antoni A, Wohlgemuth S, Cecatiello V, Pasqualato S, et al. Modular assembly of RWD domains on the Mis12 complex underlies outer kinetochore organization. Mol Cell. 2014;53:591–605. doi: 10.1016/j.molcel.2014.01.019. [DOI] [PubMed] [Google Scholar]
- Petrovic A, Pasqualato S, Dube P, Krenn V, Santaguida S, Cittaro D, Monzani S, Massimiliano L, Keller J, Tarricone A, et al. The MIS12 complex is a protein interaction hub for outer kinetochore assembly. J Cell Biol. 2010;190:835–852. doi: 10.1083/jcb.201002070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Przewloka MR, Glover DM. The kinetochore and the centromere: a working long distance relationship. Annu Rev Genet. 2009;43:439–465. doi: 10.1146/annurev-genet-102108-134310. [DOI] [PubMed] [Google Scholar]
- Robert X, Gouet P. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 2014;42:W320–324. doi: 10.1093/nar/gku316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santaguida S, Musacchio A. The life and miracles of kinetochores. EMBO J. 2009;28:2511–2531. doi: 10.1038/emboj.2009.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schleiffer A, Maier M, Litos G, Lampert F, Hornung P, Mechtler K, Westermann S. CENP-T proteins are conserved centromere receptors of the Ndc80 complex. Nat Cell Biol. 2012;14:604–613. doi: 10.1038/ncb2493. [DOI] [PubMed] [Google Scholar]
- Schmitzberger F, Harrison SC. RWD domain: a recurring module in kinetochore architecture shown by a Ctf19-Mcm21 complex structure. EMBO Rep. 2012;13:216–222. doi: 10.1038/embor.2012.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Screpanti E, De Antoni A, Alushin GM, Petrovic A, Melis T, Nogales E, Musacchio A. Direct binding of Cenp-C to the Mis12 complex joins the inner and outer kinetochore. Curr Biol. 2011;21:391–398. doi: 10.1016/j.cub.2010.12.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheldrick GM. Experimental phasing with SHELXC/D/E: combining chain tracing with density modification. Acta Crystallogr D Biol Crystallogr. 2010;66:479–485. doi: 10.1107/S0907444909038360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strong M, Sawaya MR, Wang S, Phillips M, Cascio D, Eisenberg D. Toward the structural genomics of complexes: Crystal structure of a PE/PPE protein complex from Mycobacterium tuberculosis. 2006;103:6. doi: 10.1073/pnas.0602606103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tasto JJ, Carnahan RH, McDonald WH, Gould KL. Vectors and gene targeting modules for tandem affinity purification in Schizosaccharomyces pombe. Yeast. 2001;18:5. doi: 10.1002/yea.713. [DOI] [PubMed] [Google Scholar]
- Terwilliger TC. Maximum-likelihood density modification. Acta Crystallographica Section D. 2000;D56:7. doi: 10.1107/S0907444900005072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terwilliger TC, Adams PD, Read RJ, McCoy AJ, Moriarty NW, Grosse-Kunstleve RW, Afonine PV, Zwart PH, Hung LW. Decision-making in structure solution using Bayesian estimates of map quality: the PHENIX AutoSol wizard. Acta Crystallogr D Biol Crystallogr. 2009;65:582–601. doi: 10.1107/S0907444909012098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tien JF, Umbreit NT, Gestaut DR, Franck AD, Cooper J, Wordeman L, Gonen T, Asbury CL, Davis TN. Cooperation of the Dam1 and Ndc80 kinetochore complexes enhances microtubule coupling and is regulated by aurora B. J Cell Biol. 2010;189:713–723. doi: 10.1083/jcb.200910142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verdaasdonk JS, Bloom K. Centromeres: unique chromatin structures that drive chromosome segregation. Nat Rev Mol Cell Biol. 2011;12:320–332. doi: 10.1038/nrm3107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vonrhein C, Blanc E, Roversi P, Bricogne G. Automated Structure Solution With autoSHARP. Methods in Molecular Biology. 2007;364:5. doi: 10.1385/1-59745-266-1:215. [DOI] [PubMed] [Google Scholar]
- Waterhouse AM, Procter JB, Martin DM, Clamp M, Barton GJ. Jalview Version 2--a multiple sequence alignment editor and analysis workbench. Bioinformatics. 2009;25:1189–1191. doi: 10.1093/bioinformatics/btp033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei RR, Schnell JR, Larsen NA, Sorger PK, Chou JJ, Harrison SC. Structure of a central component of the yeast kinetochore: the Spc24p/Spc25p globular domain. Structure. 2006;14:1003–1009. doi: 10.1016/j.str.2006.04.007. [DOI] [PubMed] [Google Scholar]
- Wei RR, Sorger PK, Harrison SC. Molecular organization of the Ndc80 complex, an essential kinetochore component. PNAS. 2005;102:4. doi: 10.1073/pnas.0501168102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welburn JP, Vleugel M, Liu D, Yates JR, 3rd, Lampson MA, Fukagawa T, Cheeseman IM. Aurora B phosphorylates spatially distinct targets to differentially regulate the kinetochore-microtubule interface. Mol Cell. 2010;38:383–392. doi: 10.1016/j.molcel.2010.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westermann S, Cheeseman IM, Anderson S, Yates JR, 3rd, Drubin DG, Barnes G. Architecture of the budding yeast kinetochore reveals a conserved molecular core. J Cell Biol. 2003;163:215–222. doi: 10.1083/jcb.200305100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AG, McCoy A, et al. Overview of the CCP4 suite and current developments. Acta Crystallogr D Biol Crystallogr. 2011;67:235–242. doi: 10.1107/S0907444910045749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yachdav G, Kloppmann E, Kajan L, Hecht M, Goldberg T, Hamp T, Honigschmid P, Schafferhans A, Roos M, Bernhofer M, et al. PredictProtein--an open resource for online prediction of protein structural and functional features. Nucleic Acids Res. 2014;42:W337–343. doi: 10.1093/nar/gku366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Wu F, Ward T, Yan F, Wu Q, Wang Z, McGlothen T, Peng W, You T, Sun M, et al. Phosphorylation of HsMis13 by Aurora B kinase is essential for assembly of functional kinetochore. J Biol Chem. 2008;283:26726–26736. doi: 10.1074/jbc.M804207200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zinkowski RP, Meyne J, Brinkley BR. The Centromere-Kinetochore Complex: A Repeat Subunit Model. Journal of Cell Biology. 1991;113:20. doi: 10.1083/jcb.113.5.1091. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. MIND Constructs and Cartoon Drawings Used in this Study, Related to Figure 1
(A) The mercury derivative electron density map at different stages of phase problem solution: initial map obtained after single anomalous dispersion (SAD) phasing (left), after statistical density modification using RESOLVE (middle), and after including partial head I structure factors from MN-C2 and multi-crystal density averaging (right).
(B) Sequence assignment validation by anomalous Fourier difference analysis. Peak heights in standards deviations above the mean are shown from anomalous Fourier maps calculated with model phases and observed anomalous differences from the mercury- (left) and two selenomethione-derivatized (middle and right) crystals. Peaks that map to mercury-derivatized cysteine or selenomethionine residues are annotated with an arrow and the corresponding subunit name and residue number. The inset structure shows the location of the mapped peaks as spheres.
(C) Native diffraction data and final electron density maps. Left, analysis of anisotropic diffraction using the CCP4 program truncate (French and Wilson, 1978). Mean F/σ along the three crystallographic axes is plotted against resolution. Resolution cutoffs of 3.2, 4.1 and 3.4 Å were applied during ellipsoidal truncation using the anisotropy server (Michael Strong, 2006). Middle, final composite omit map calculated with phenix.composite_omit_map. Right, final σA-weighted 2mFo-DFc electron density map calculated with phenix.refine.
(A) MIND Head I Does Not Bind Head II D230 in the Presence of Mild Chaotropic Salts. Superdex 200 size exclusion chromatography testing the interaction between MIND head I (Mtw11-120/Nnf11-102, fraction A) and head II (Dsn1230-313/Nsl11-83, fraction B) in the presence of 0.2 M KBr and 0.2 M KSCN. The elution profiles are overlaid in the chromatogram on top and the fractions are resolved on the Coomassie stained SDS-PAGE gels.
(B) SEC experiments as in (A) show that Dsn1230-255 contacts MIND head I at the conserved Ame1-binding site containing Mtw1 residues N32, Y36, E73 and E77. Bottom two gels show that MIND head II D230 (Dsn1230-397/Nsl11-118) binds head I (Mtw11-120/Nnf11-102), but does not bind head I Site II mut (Mtw11-120:N32A, Y36A, E73A, E77A/Nnf11-102). Top three gels are control experiments with elution profiles of each construct alone.
(C) Binding experiments showing that Dsn1 competes with Ame1 for binding MIND head I. Elution fractions from the top two gels show that MIND head I (Mtw11-120/Nnf11-102), fraction a, forms stoichiometric complexes with both head II D205 (Dsn1205-397/Nsl11-118), fraction b, and head II D205-S2D (Dsn1205-397:S213D, S223D/Nsl11-118), fraction c. Bottom two gels show that His6-GST-Ame11-24 breaks the stoichiometric complex and binds head I more tightly in the presence of head II D205-S2D (gel 4) than in the presence of head II D205 (gel 3). Fraction M: molecular-mass marker bands for 10, 15, 20, 25, 30, 40, 50, 60 kDa.
Common to all MIND-C1 crystal forms is how head II binds head I of an adjacent MIND molecule related by crystallographic symmetry. On the left, three MIND molecules are shown for each native (A), SeMet I (B), SeMet II (C) and tantalum-derivatized (D) crystal form. Unit cells are drawn in black and the distance between molecules is given in angstrom (Å). On the right, the packing is shown for each crystal, viewed along the three crystallographic axes.
(A) Close-up views of the head II-head I interface observed in four crystal forms (Figure S4).
(B) Left, detection of a discrete MIND dimer by SEC-MALS. Right, oligomerization depends on the presence of head II, because a construct MIND-C1:D367 does not show a shoulder in the elution profile at higher molecular weight as seen for MIND-C1 (blue curve, indicated with an arrow). Moreover, oligomerization does not require the N-terminal Dsn1 arm, as a construct MIND-C1:D261, lacking the arm, shows the same elution profile (red curve) as MIND-C1.
(C) Superposition MIND from four crystal forms reveals hinge regions. We superimposed MIND-C1 from the crystal shown in Figure S4 on CC1 and CC2. Movements of the head I, head II and CC3 domains are roughly indicated by arrows.
(D) Schematic illustrating how a 6-fold ring of MIND complexes is generated, where contacts between heads of adjacent complexes closely resemble the conserved interface shown in (A).
(A) Ribbon representation of the refined MIND-C1 model to show the B factor distribution, where ribbon thickness is proportional to the B factor of the residues’ Cα atom and colored in a gradient from dark blue (low B factors) to orange (high B factors). Residue numbers for the termini and chain breaks of the four subunits are also shown.
(B) Real space correlation coefficients (RSCC) for each MIND-C1 residue were calculated with phenix.real_space_correlation. Residue ranges are highlighted as follows: dotted line, residues included in the crystallization construct; solid line, residues modeled in structure; black bar, residues modeled as poly-alanine; gray bar, residues modeled in poor electron density, where the chain trace is approximate.
Table S1. MIND-C1 Data Collection, Phasing and Refinement Statistics, Related to Experimental Procedures
Table S2. MN-C2, MN-C2-Mif2 and Spc24/Spc25-Dsn1 Data Collection, Phasing and Refinement Statistics, Related to Experimental Procedures
Table S3. Protein Crystallization Conditions for MIND-C1, MN-C2, MN-C2-Mif2 and Spc24-Spc25-Dsn1 Complexes, Related to Experimental Procedures
Supplemental Movie 1. Kinetochore Model, Related to Figure 7.
Amino acid sequences from six fungal and four metazoan species were aligned. Sequence accession numbers are as follows, where the numbers for each species correspond to Mtw1, Nnf1, Nsl1 and Dsn1, respectively: Kluyveromyces lactis (k_lac: XM_455186, XM_454208, XM_452912, XM_453754), Saccharomyces cerevisiae (s_cer: NM_001178179, NM_001181770, NM_001184047, NM_001179532), Eremothecium gossypii (e_gos: NM_209816, NM_208451, NM_210764, NM_211975), Candida dubliniensis (c_dub: XM_002418821, XM_002421686, XM_002421339, XM_002416733), Candida albicans (c_alb: XM_709448, XM_713094, XM_883711, XM_713294), Lachancea thermotolerans (l_the: XM_002552753, XM_002553514, XM_002555972, XM_002553655), Homo sapiens (h_sap: XM_005256798, NM_007221, NM_015471, NM_001145316), Mus musculus (m_mus: XM_006537204, NM_025928, NM_198654, NM_025853), Gallus gallus (g_gal: NM_001044677, NM_001044676, NM_001044646, NM_001044660), Xenopus laevis (x_lae: NM_001092335, NM_001091138, XM_012969245, NM_001166438). Secondary structure is annotated at the top, predicted in black and observed in the crystal structure in blue. Residues built in the crystal structure are shown with a solid bar at the bottom of the alignment in color corresponding to the MIND subunit (Mtw1, green; Nnf1, gray; Nsl1, blue, Dsn1, orange). The orange dashed line in Dsn1 indicates the long loop 311-353 partially modeled as poly-alanine. Dsn1 residues 205-252 involved in regulating Mif2 and Ame1 binding are shown as open green ovals. S. cerevisiae Dsn1 residues 560-572, crystallized with Spc24-Spc25, are boxed orange. Specific residues mentioned in the text are highlighted as solid ovals.