

Abstract
We have reported a murine model of autoimmune cholangitis, generated by altering the AU rich element by deletion of the IFN-γ 3′UTR region (coined ARE-Del−/−), that has striking similarities to human primary biliary cholangitis (PBC) with female predominance. Previously, we suggested that the gender bias of autoimmune cholangitis was secondary to intense and sustained type I and II IFN signaling. Based on this thesis, and to define the mechanisms that lead to portal inflammation, we specifically addressed the hypothesis that type I IFNs are the driver of this disease. To accomplish these goals, we crossed ARE-Del−/− mice with IFN type I receptor alpha chain (Ifnar1) knockout mice. We report herein that loss of type I IFN receptor signaling in the double construct of ARE-Del−/− Ifnar1−/− mice dramatically reduces liver pathology and abrogated gender bias. More importantly, female ARE-Del−/− mice have an increased number of germinal center (GC) B cells as well as abnormal follicular formation, sites which have been implicated in loss of tolerance. Deletion of type I IFN signaling in ARE-Del−/− Ifnar1−/− mice corrects these GC abnormalities, including abnormal follicular structure. In conclusion, our data implicate type I IFN signaling as a necessary component of the gender bias of this murine model of autoimmune cholangitis. Importantly these data suggest that drugs that target the type I IFN signaling pathway would have potential benefit in the earlier stages of PBC.
Introduction
Interferon gamma (IFN-γ) is an inflammation modulator regulating both pro- and anti-inflammatory responses and its expression is critical for the initial host innate immune response and is also essential to mount an adaptive immune response. Over expression of IFN-γ has been demonstrated as a key factor in the induction of autoimmunity. Of note, it has been found that IFN-γ also plays an essential role in the development and severity of female dominant autoimmune diseases (1, 2). Our group has produced several lines of evidence that IFN-γ is involved in the pathogenesis of both murine model and human primary biliary cholangitis (PBC) (3–7). In particular, we have taken advantage of a “designer” mouse with posttranscriptional dysregulation of IFN-γ through deletion of the IFN 3′ untranslated region (3′ UTR) AU-rich element (ARE−/−). These animals, coined ARE−/− mice, exhibit prolonged and chronic over-expression of IFN-γ and more importantly, develop a female predominant autoimmune cholangitis, with portal inflammation, liver granulomas, elevation of bile salts, elevation of sera IgM, and the presence of both anti-mitochondrial antibodies (AMAs) and antibodies to gp210 (8).
We propose that IFN-γ is critically involved with gender bias. Previous analysis of differentially expressed genes in female ARE-Del−/− mice revealed stronger type I and II IFN signaling and lymphocyte-mediated immune responses, especially CD4 T cell-mediated responses. Type I IFN receptors were detected as one of top upstream regulators next to IFN-γ in liver gene expression analysis (8). Therefore to extend these pilot observations and define a translational application to human PBC, we developed IFNα/β receptor alpha chain-deficient ARE-Del−/− animals, coined ARE-Del−/− Ifnar1−/− mice. Herein we report that depletion of type I IFN signaling significantly prevents the female-prevalent autoimmune cholangitis phenotype including portal and lobular duct inflammation, granuloma formation, bile duct damage, and elevation of total bile acids. Furthermore, these mice with deficient type I IFN signaling no longer manifest abnormal follicular T helper (Tfh) cells and germinal centers (GC) formation found in ARE-Del−/− mice. Therefore, we propose that crosstalk between interferon signaling and Tfh and B cells found in GCs are critical to loss of tolerance and female predominance (9–11). Furthermore, these data highlight potential therapeutic pathways for human PBC.
Materials and Methods
Generation of mice
ARE-Del−/− mice were generated and maintained as previously reported (8, 12). Ifnar1−/− mice were initially obtained from the Jackson laboratory and backcrossed onto the C57BL/6 background by speed congenic analysis. For generating Ifnar1−/−ARE-Del−/− mice, male ARE-Del+/− mice were mated with female Ifnar1−/−mice to obtain male ARE-Del+/− Ifnar1+/− mice, which were subsequently back-crossed with female Ifnar1−/− mice to obtain ARE-Del−/− Ifnar1−/− mice. The parental ARE-Del−/− and the derived ARE-Del−/− Ifnar1−/− mice were genotyped at 3 to 4 weeks of age to confirm the ARE-Del and KO Ifnar1 genes in their genomic DNA. Animal care was provided in accordance with the procedures outlined in the “Guide for Care and Use of Laboratory Animals” (National Research Council; 2011; National Academy Press: Washington, D.C.). At serial ages, animals were sacrificed to collect sera, spleen, and liver tissues for serological, gene expression and cellular immunological analysis. The experimental protocols were approved by NCI at Frederick and the University of California Animal Care and Use Committee. All experiments were performed in group sizes of 4–10 and replicated at least twice; the numbers in each experiment are noted in the figure legends.
Anti-mitochondrial and anti-SP100 autoantibodies
IgM and IgG anti PDC-E2 assays were performed by ELISA with data presented as O.D. ± SEM as previously described with known positive and negative controls and standardized recombinant PDC-E2 (5, 13, 14). Antibodies to SP100 were evaluated by ELISA using INOVA kits and again including known controls (15, 16).
Total bile acid analysis (TBA)
TBA was analyzed using freshly collected serum and a Total Bile Acid Enzymatic Cycling Assay Kit (Diazyme, Poway, CA), as previously described (8). Data were acquired in a kinetic scan mode using the 405 nm wavelength in 1 minute intervals. ΔA405/min was calculated for standard, control, blank (DDW with R2) and samples by subtracting the O.D. value at each interval; ΔA405/min was consistent at all intervals. The concentration of TBA was then quantitated as previously described (8)
Peptide array
Autoantigen arrays with immobilized synthetic peptides were generated by PEPperPRINT. The arrays were hybridized with serum from ARE-Del−/− mice after blocking, and the autoantibodies bound to their corresponding peptides on the array were detected with fluorophore-conjugated second antibodies against different isotypes of autoantibodies (IgG and IgM). Each microarray image was quantified by PepSlide Analyzer and statistical significance determined using the Holm-Sidak method, with alpha=5.000%
Cell isolation and flow cytometry analysis
Mononuclear cells (MNCs) were isolated from spleen(17, 18). For cell surface staining, 1×106 MNCs were re-suspended in staining buffer (0.2% BSA, 0.04% EDTA and 0.05% sodium azide in PBS), divided into 25μl aliquots, and incubated with anti-mouse FcR blocking reagent (eBioscience) for 15 minutes at 4°C. Cells were washed and stained for 30 minutes at 4°C with cocktails containing combinations of fluorochrome conjugated monoclonal antibody (mAbs) for cell surface markers CD4 (GK1.5), CD8a (53–6.7), B220 (RA3-6B2), PD-1 (29F.1A12), CD95 (FAS, Jo2), GL-7 (GL7), and CXCR5 (2G8). All reagents were purchased from BioLegend (BioLegend, San Diego, CA) and optimal dilutions used throughout with positive and negative controls.
Histopathology
Portions of livers were excised immediately upon sacrifice, and fixed in 10% paraformaldehyde solution for 2 days at room temperature, embedded in paraffin, cut into 4-μm sections and deparaffinized for routine hematoxylin and eosin (H&E) staining (19). Whole spleens were excised and embedded in paraffin and cut into horizontal sections and deparaffinized for H&E and peanut agglutinin(PNA) staining. Each histological score means the sum of severity and frequency scores from a specimen in a blind test as described previously (8, 20, 21).
Adoptive transfer of CD4 T cells
Spleen cells were collected from 20 week old female ARE-Del−/− and wild type (WT) mice. Mononuclear cells were isolated from spleen and CD4+ T cells purified by negative selection with microbeads and MiniMacs separation columns (MiltenyiBiotec, Auburn, CA). 10-week-old female C57BL6 mice were used as recipients. Aliquot of 1×106 CD4+ T cells were transferred into recipient mice via tail vein injection as previously described (8, 22). Eight weeks after cell transfer, mice were sacrificed and splenic cells collected to analyze Tfh and GC B cells by flow cytometric analysis. Livers were collected from recipient mice for pathological analysis.
Statistical analysis
One-way ANOVA, two-tailed unpaired t test and nonparametric Mann-Whitney test were used for analysis. P values < 0.05 were considered statistically significant.
Results
Female-biased liver inflammation was ameliorated by deletion of the Ifnar1 gene in ARE-Del−/− mice
We first compared histological changes in liver samples of both gender double knock-out ARE-Del−/− Ifnar1−/− mice with ARE-Del−/− mice. Figure 1A demonstrates that ARE-Del−/− mice have distinct lymphocyte infiltration near the portal tracts with disruption of small bile ducts that are more severe in female mice. The histological scores of inflammation demonstrate the severities in specific regions including bile duct disruption, granuloma formation, and portal and lobular inflammation in the liver of each strain of mice (Figure 1B). Female ARE-Del−/− mice manifest increased inflammation in these regions compared to male ARE-Del−/− mice, and although this inflammation was significantly suppressed by the depletion of type I IFN receptor, it did not completely delete cholangitis and revert to the same levels as WT and Ifnar1−/− control littermates. However, removal of the Ifnar1 gene distinctively ameliorated the gender difference in the severity of inflammation between male and female ARE-Del−/− Ifnar1−/− mice. The reductions in liver pathology by deletion of the Ifnar1 gene were also exhibited in heterozygous ARE-Del+/− Ifnar1−/−mice and male ARE-Del mice. However, since the disease is minimal in these groups, the overall reductions are minimal (data not shown). Thus, although chronic expression of IFN-γ elicit cholangitis in both genders, type I IFN signaling is critical for gender biased PBC-like lesions. ARE-Del−/− Ifnar1−/− mice compared to ARE-Del+/− Ifnar1−/− mice, manifest less disease, implying that threshold levels of Type II IFN may overcome the loss of Type I IFN signaling.
Figure 1. Deletion of the Ifnar1 gene suppresses female-biased pathological phenotypes in ARE-Del−/− mice.
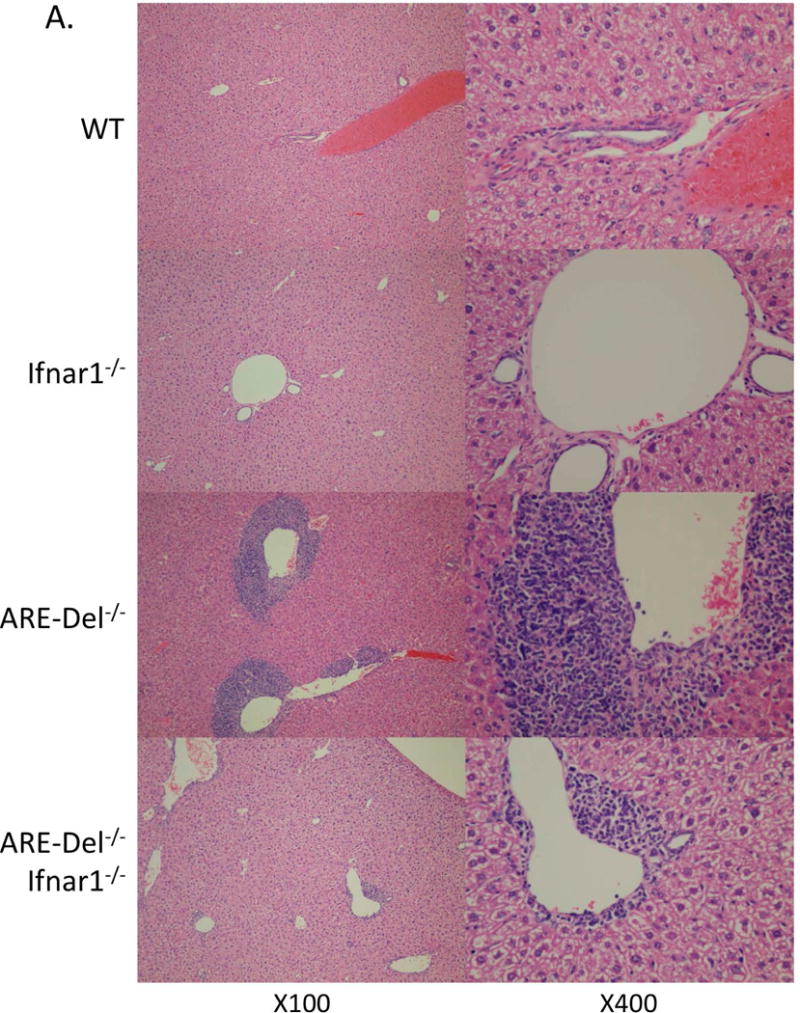
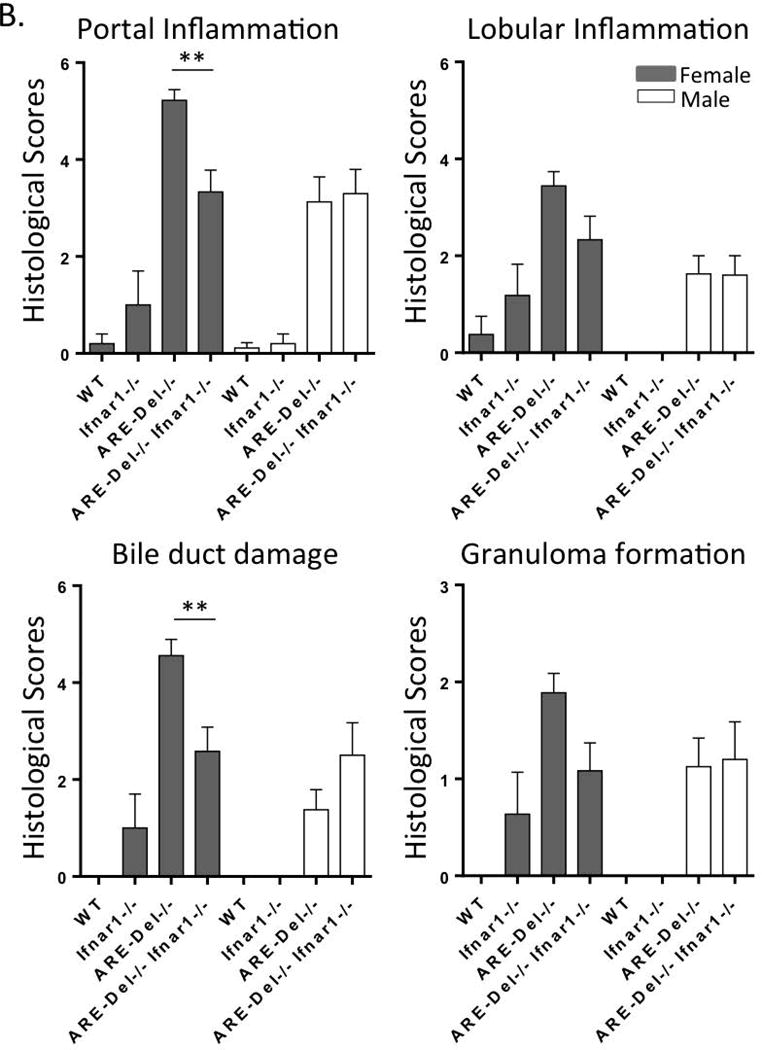
A. Representative H&E staining of female Ifnar1−/−, ARE-Del−/−, ARE-Del−/− Ifnar1−/−mice compared to control (WT) littermates. B. Pathological score of liver histology of portal inflammation, lobular inflammation, biliary duct damage and granuloma formation. Statistical analysis was performed by the nonparametric Mann Whitney test using GraphPad Prism 6.0 (mean ± SEM; n=7–8). The two-tailed p-value < 0.05 was taken as significance (* P< 0.05, ** P< 0.01, *** P< 0.001, n.s., not significant).
Total bile salts and serological markers of PBC were suppressed by deletion of the Ifnar1 gene in female ARE-Del−/− mice
Increased levels of total bile acids (TBA) and AMAs are the characteristic serological markers for PBC. To determine the role of type I IFN in the expression of these markers, we first measured serum TBA in ARE-Del−/− Ifnar1−/− compared to ARE-Del−/− mice (Figure 2). Consistent with our previous report (8), female ARE-Del−/− mice have higher levels of serum TBA compared to male ARE-Del−/− mice. However, depletion of Ifnar1 in female ARE-Del−/− mice significantly reduced serum TBA levels, resulting in the disappearance of the significant gender difference.
Figure 2. Serum level of TBA and AMA in ARE-Del−/−Ifnar1−/− mice.
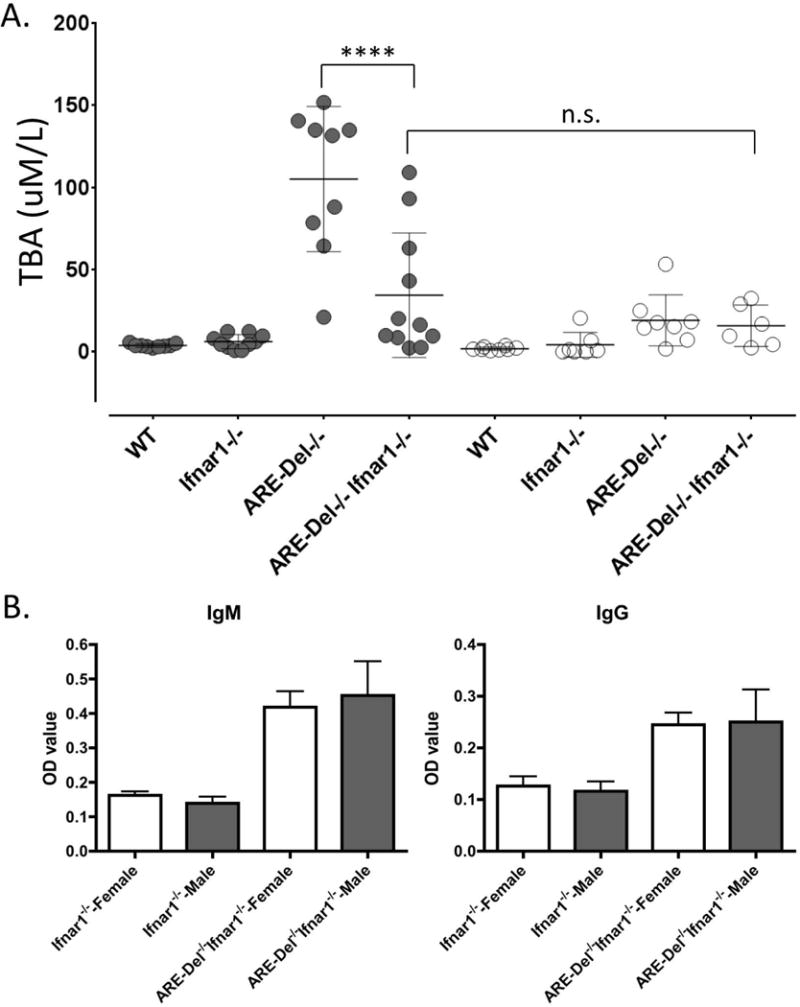
A. Serum TBA levels at age 20 (±2) weeks in female and male Ifnar1−/−, ARE-Del−/−, ARE-Del−/− Ifnar1−/− mice compared to WT littermates (n=7–8). B. Serum anti-PDC-E2 antibodies (IgM and IgG) were detected by the standard enzyme-linked immune-sorbent assay (ELISA) against recombinant proteins of the pyruvate dehydrogenase complex-E2 subunit (PDC-E2). Data represents mean ± SEM. Statistical analysis was performed by one-way ANOVA. **** P< 0.001, n.s., not significant.
Elevated levels of serum IgM in ARE-Del−/− mice were more pronounced in female than male mice (Supplemental figure 1). Furthermore and consistent with our previous report, IgM anti-PDC-E2 and IgM anti-SP100 were elevated in female ARE-Del−/− mice (Supplemental Figure 1). This gender biased IgM anti-PDC-E2 and anti-SP100 expression in ARE-Del−/− mice was no longer present in ARE-Del−/− Ifnar1−/− mice. Indeed, the serum levels of both IgM and IgG to PDC-E2 were not significantly different between male and female ARE-Del−/− Ifnar1−/− mice (Figure 2B).
Depletion of type I IFN receptor prevents female-prevalent excessive GC formation in ARE-Del−/− mice
As shown in Figure 3A, the lymph follicles are diffusely distributed and follicular size was homogeneous in splenic sections of both gender of WT mice. In contrast, various sized follicles were observed in spleen of ARE-Del−/− mice. Immunohistochemistry PNA staining reflected that ARE-Del−/− mice had more PNA positive GCs than males, indicating enhanced GC formation; the numbers of lymphoid follicles per section was increased in female ARE-Del−/− compared to males (Figure 3B). We next examined GC B cells in ARE-Del−/− mice. GC B cells typically display PNA lectin and express CD95, therefore the splenic B220+PNA+CD95+B cells were analyzed by flow cytometry. As shown in Figure 3C and 3D, the frequency of PNA+CD95+ cells was significantly increased in ARE-Del−/− mice; deleting the type I IFN receptor ameliorated the accumulation of GC B cells in spleens of ARE-Del−/− mice. Moreover, in ARE Del−/− mice, there is a more diffuse nature of the GCs as well as loss of the marginal zone B cells. Both of these features were also rescued by deletion of Ifnar1 (Supplemental Figure 2). These data suggest that gender biased GC formation in ARE-Del−/− mice is dependent on type I IFN in ARE-Del−/− mice.
Figure 3. Female-prevalent GC formation in ARE-Del−/− mice.
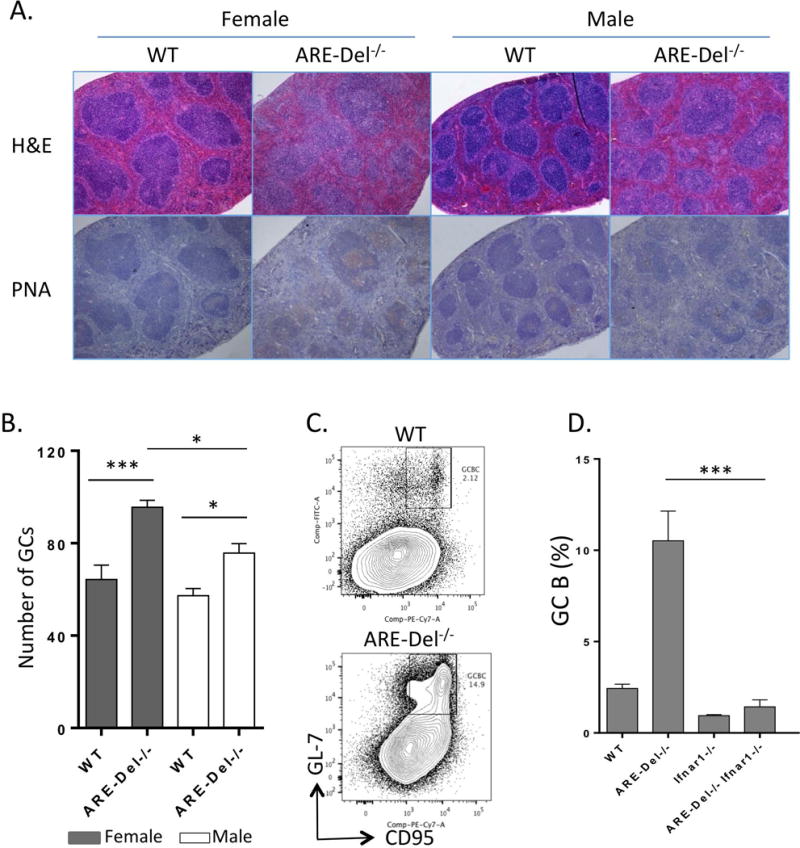
A. Representative H&E and PNA staining of spleen from ARE-Del−/− mice of both genders at age 20 (±2) weeks. B. GC numbers was counted on each entire spleen cross section with largest wide and longest diameter, estimating a same size of each specimen (mean ± SEM, n=4–5). C. Representative image of CD95hiPNAhi GC B cells analyzed by flow cytometry. The gate of B cells was the B220+ population. The image from female ARE-Del−/− mice was compared to WT littermate. D. Percentages of B220+CD95hiPNAhiGC B cells in female Ifnar1−/−, ARE-Del−/−, ARE-Del−/− Ifnar1−/− mice compared to control littermates (mean ± SEM, n=4–5). Statistical analysis was performed by one-way ANOVA. * P< 0.05, ** P< 0.01, *** P< 0.001, n.s., not significant.
Distinct Tfh and GC responses gender bias and Type I IFN
GC B cell differentiation and responses are regulated by Tfh cells (23, 24), and IFN-γ has the potential for activation of Tfh cells. We therefore analyzed the Tfh subpopulation of T cells in the spleen in ARE-Del+/− mice to determine whether lower expression of IFN-γ could enhance the reaction and accumulation of Tfh cells in heterozygous female mice. As shown in Figure 4A, there are clearly distinct populations of pre-Tfh and Tfh cells in ARE-Del+/− mice compared to WT mice. The frequency of Tfh cells was more pronounced in female compared to male ARE-Del+/−mice (Figure 4B). However, the frequency of PNA+CD95+ GC B cells was not strongly increased in ARE-Del+/− mice and GC abnormalities were less than observed in ARE-Del−/− mice. We next analyzed the frequency of both Tfh cells and GC B cells in ARE-Del+/− Ifnar1−/− mice compared to type I IFN receptor-sufficient ARE-Del+/− mice. As shown in Figure 4B, deletion of the Ifnar1 gene suppressed the accumulation of both Tfh and GC B cells in ARE-Del+/− mice; i.e. the frequency of these cells was not significantly different between female and male ARE-Del+/− Ifnar1−/− mice.
Figure 4. Deletion of the Ifnar1 gene suppresses female-biased induction of Tfh cells in the spleen of heterozygous ARE-Del+/− mice.
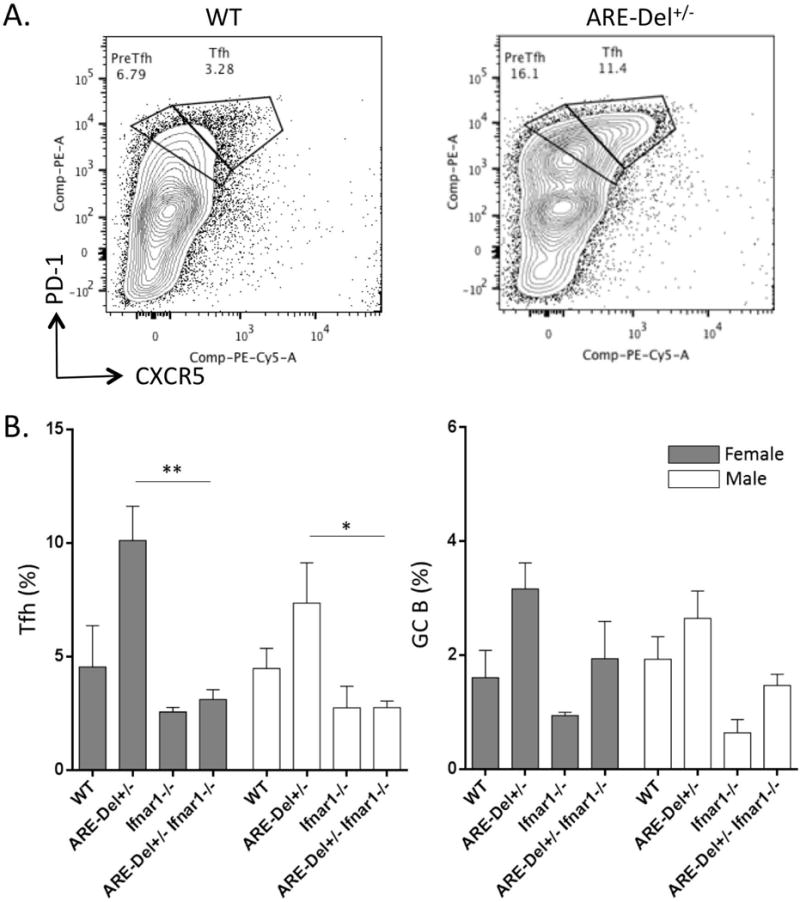
A. Representative image of flow cytometric analysis of isolated splenocyte populations for Tfh cells (CD4+CXCR5hiPD-1hi) in female ARE-Del+/− mice compared to control littermates. The percentages of splenic CD4+CXCR5hiPD-1hi Tfh cells (B) and B220+CD95hiPNAhi GC B cells (C) were analyzed by flow cytometry from female and male Ifnar1−/−, ARE-Del−/−, ARE-Del−/− Ifnar1−/− mice compared to control littermates (mean ± SEM, n=4–5). Statistical analysis was performed by one-way ANOVA. * P< 0.05, ** P< 0.01, n.s., not significant.
Adoptive transfer of CD4 T cells from ARE-Del−/− mice leads to serum bile acid secretion with increased GC responses
We recently reported that transfer of CD4 T cells, but not CD8 T cells from ARE-Del−/− to B6/Rag1−/− mice induces PBC-like pathological changes in their livers (8). RNA-sequencing of female-specific gene expression suggested that CD4 T cells are a distinct factor in the pathology observed in female mice compared to male ARE-Del−/− mice (8). A recent report suggests that IFN-γ stimulates GC formation and development, a process thought to be critical for autoimmune disease development (10). Importantly, IFN-γ signaling in intrinsic B cells was shown to be required for spontaneous GC formation and autoantibody generation (11). We thus examined whether chronic overexpression of IFN-γ in ARE-Del−/− mice promotes excessive GC responses. To test the role of ARE-Del−/− CD4 T cells in GC formation, especially induction of GC B cell abnormal responses, CD4 T cells from female ARE-Del−/− mice were transferred to WT mice. The Tfh and GC B cells in recipient mice were evaluated by flow cytometric analysis 8 weeks after cell transfer. Adaptive transfer of CD4 T cells from ARE-Del−/− mice notably increased GC B cells in the recipient mice (Figure 5A). Although the increase of Tfh cells in spleen of ARE-Del−/− transfer group did not reach statistical significance (Figure 5A), there were similar phenotypes of Tfh cells from ARE-Del mice with high expression of PD-1 in recipient mice. C57BL/6 mice are Th1 biased and our data has shown that some wild type mice have a mild induction of Tfh cells, a distinct population of cells with PD-1 expression was not as clearly distinct as observed in ARE-Del mice. Thus, the data also supports the hypothesis that the IFN-γ activated Tfh cells regulate differentiation and responses of GC B cells in ARE-Del−/− mice. Moreover, TBA secretion was significantly upregulated in recipient mice upon adoptive transfer of CD4 T cells from ARE-Del−/− mice (Figure 5B). In addition, adoptive transfer of CD4 T cells from ARE-Del+/− mice to recipient WT mice also increased liver infiltration of T cells but interestingly, an increase in TBA secretion was not detected (data not shown). These data suggest that Tfh cells generated from transferred autoreactive CD4 T cells induce excessive GC formation and GC B cell responses. We propose that this is the initial step in the pathogenesis of disease in ARE-Del−/− mice.
Figure 5. Adoptive transfer of ARE-Del−/− CD4+ T cells enhances GC responses and TBA secretion in recipient mice.
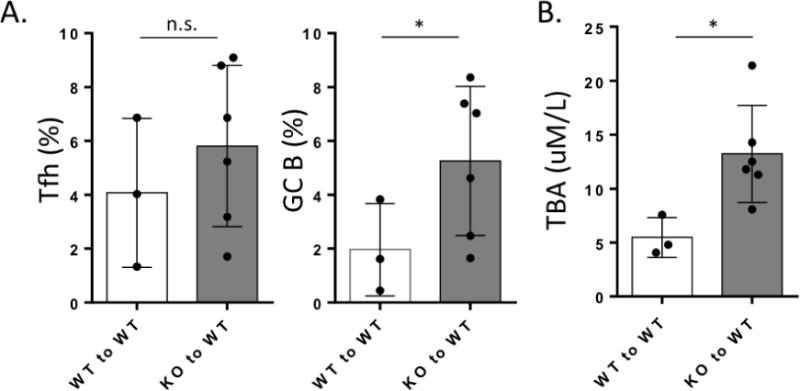
Splenic CD4+ T cells from ARE-Del−/− mice (KO) (n=6) or from control littermates (WT) (n=3) were adoptively transferred to WT mice. After 8 weeks, splenocyte populations were isolated and Tfh and GC B cells were analyzed by flow cytometry. A. Percentages of CD4+CXCR5hiPD-1hi Tfh cells and B220+CD95hiPNAhi GC B cells in spleen from KO to WT mice compared to control groups. B. Serum TBA levels were measured from KO to WT mice compared to control groups. Data represent mean ± SD. At least two independent experiments were performed. Statistical analysis was performed by the unpaired Student’s t-test. * P< 0.05, ** P< 0.01, *** P< 0.001, n.s., not significant.
Discussion
Female bias is one of the hallmarks of autoimmunity and remains one of the critical enigmatic issues in autoimmunity, including PBC (25, 26). Recently, we have provided evidence that chronic overexpression of IFN-γ leads to gender bias autoimmune cholangitis, which mimics the characteristics of PBC (8). However, IFN-γ is promiscuous with a pleiotropic mode of action (27). We reasoned, based on our previous data in ARE-Del−/− mice, that study of type I and type II IFNs interplay was essential to understand our data and our thesis that such interplay was essential for female sex bias. Second we propose that if our thesis is correct, then it leads logically to the possible use of specific therapeutic agents to treat human PBC. Herein we demonstrate that in ARE-Del−/− mice, the pathologic effects of overexpression of IFN-γ and gender bias is dependent on activation of type I IFN signaling. Indeed we report that blocking the IFN-α/β receptor signaling pathway using ARE-Del−/− Ifnar1−/− (i.e. double knockout) mice corrects cholangitis with direct therapeutic implications.
Type I and type II IFNs have distinct roles in immune responses through the interaction with specific cell surface receptors and activation of classical JAK-STAT signaling (28). Each receptor is composed of two chains; type I for IFNAR1 and IFNAR2, and type II receptor for IFNGR1 and IFNGR2. When they bind to their receptors, Type I IFN regulates transcription by the STAT2-STAT1 heterodimer complex while type II IFN requires STAT1 homodimer formation. It is well-known that there are defined different consequences of altering interferon production and metabolism, i.e. blockade leads to clinically significant cellular responses. These data are derived by use of both neutralizing antibodies and, in mice specific receptor gene deletion (29–31).For example, protective anti-viral effects of IFN-γ are reduced or eliminated in Ifnar1 knockout fibroblasts, i.e. by preventing type I IFN priming (29). Our previous data revealed that increased expression of IFN-γ by replacement of the AU rich region potentially activates type I interferon signaling (9). In our study deletion of the type I IFN receptor in ARE-Del−/− mice dramatically reduces cholangitis, suggesting that IFN-γ activated type I IFNs synergize with IFN-γ-mediated cellular responses.
Although the molecular and cellular mechanisms for the crosstalk of type I and II IFNs are not well understood, the STAT1-mediated signaling pathway is thought to be a regulating factor because low levels of type I interferon primes IFN-γ-mediated immune responses by modulating sufficient STAT1 expression in mouse fibroblasts (29, 32). For this reason, we first compared STAT1 expression in male and female ARE-Del−/− mice, expecting that female ARE-Del−/− mice would have stronger STAT1 expression than male mice. However, we did not see significant gender differences of STAT1 protein expression in the liver of ARE-Del−/− mice (data not shown). Based on liver gene expression profiles in ARE-Del−/− mice, the earliest events in PBC may lead to up-regulation of both type I and II IFN and consequently modulate MHC class II expression in both male and female mice. However, female ARE-Del−/− mice would have enhanced IFN signaling that is likely involved in CD4 helper T cell-mediated cellular responses. Hence, it can be reasoned that STAT1 is a central modulator for the MHC class II expression in disease progression, and importantly, STAT1-independent pathways would be required for female gender bias.
Type I and type II IFN signaling regulates several X-chromosome encoded immune-related genes, which may subsequently influence the immune response in a sex-dependent manner (33, 34). Thus, we compared X-chromosome encoded immune-related genes with our hepatic gene expression data in female ARE-Del−/− mice (8). Interestingly, female specific DEGs overlapped with many X-chromosome encoded immune-related genes including CXCR3, TLR7, TLR8, IL2G, G6PD, GAB3, ARHGEF6, ARHGAP6, PFC, CSF2RA and IL3RA. Further pathway analysis demonstrated that plasmacytoid dendritic cells (pDC), a major source of IFN-α, is a potential cellular target. Several studies have noted that female biased production of IFN-α by plasmacytoid dendritic cells (pDC) may lead to gender-biased infection and autoimmunity (35–38). Therefore, female biased induction of IFN-α by plasmacytoid dendritic cells (pDC) in ARE-Del mice may play a role in this gender-biased PBC like disease.
Deletion of B cell-intrinsic IFN-γ receptor and TLR7 dramatically suppressed spontaneous GCs and the production of pathogenic autoantibodies in a murine model of lupus (39). In our preliminary data, deletion of TLR7 rescued GC formation and pathological phenotypes, which correlated with deletion of the type I IFNAR chain (Supplemental Figure 2). Importantly, IFNAR signals can potentially block TLR7 tolerance in both murine and human B cells. This regulation of TLR7 tolerance and activation is dependent on activation of the PI3K/Akt/mTOR signaling pathway in B cells (40). Considering that STAT1-independent distinct signaling pathways, i.e. mitogen-activated protein kinase (MAPK) p38 and the PI3K/AKT/mTOR, are indispensable for type I and II IFNs responses (28), we propose that future studies should examine signaling cascades and gender differences. Given that IFN-γ distorts the lysosomal localization of mTOR inhibiting its activation by i.e. PI3K-AKT upstream signaling (41), altered PI3K/AKT/mTOR signaling pathways may critically affect IFN-γ mediated cellular and molecular responses in specific cell types. Within this view, our previous hepatic gene expression between male and female ARE-Del−/− mice at 20 weeks of age indicated that females manifest activated mitogen-activated protein kinase (MAPK) p38 and the PI3K/AKT/mTOR signaling pathways (8) (Supplemental Figure 4). Moreover, SOCS inhibition was found only in female mice indicating that Stat-Jak signaling was inhibited by negative feedback in female ARE-Del−/− mice. Hence we propose that drugs that inhibit Jak-Stat signaling pathways have potential utility in treating PBC during stages of activated T cell infiltration.
Of note, the gender bias of Tfh and GC responses in both homo- and heterozygotes were type I IFN dependent, but homozygote ARE-Del mice have overall higher GC responses than heterozygotes. It is possible that the GC B cell reaction is the initial step in loss of B cell tolerance, as total B cells were clearly suppressed in ARE-Del−/− mice but not ARE-Del+/− mice compared to control littermates. Furthermore a more significant difference in female compared to male ARE-Del mice was observed (Supplemental Figure 3). Based on our previous report, homozygotes have autoantibodies to nuclear DNA and higher immunoglobulin class switching (12), indicating that homozygotes have a more specific GC response than heterozygotes. Importantly, macrophage depletion in heterozygotes by clodronate-containing liposomes generates autoantibodies to nuclear DNA, which implies that not only autoreactive CD4 T cells but impaired clearance of apoptotic cells by macrophages in GC may play critical roles in an enhanced GC response in homozygotes (42, 43).
Our previous cell transfer results support the thesis that CD4 T cells are critical for progression of PBC and suggest that IFN-γ-induced Th1 responses via CD4 T cell activation drives the gender-biased progression of PBC (8). In this study we demonstrated that transfer of ARE-Del−/− CD4 T cells leads to appearance of abnormal Tfh cells, which have been shown to have the potential to change GC to excessively respond and elicit autoreactive GC B cells. Conversely, B cell-intrinsic IFN-γ receptor signaling is also required for spontaneous GC formation and pathogenic Tfh cell development (11). Therefore, it is logical to conclude that IFN-γ cross-talk between B and Tfh cells in the GC plays a role in loss of B cell tolerance. It should be noted nonetheless that serum TBA levels in serum were statistically elevated only in female ARE-Del−/− mice, indicating that a threshold level of IFN-γ may be a requisite for abnormal TBA secretion. On the other hand, serum bile acid levels may only reflect the extent of biliary damage. Homozygous ARE-Del mice also develop higher levels of autoantibodies than heterozygous ARE-Del mice, and female ARE-Del−/− mice have more IgM reactivity. It has been proposed that leakage of hydrophobic bile acids into the periductal area may be cytopathic for biliary epithelial cells (44). Therefore, the higher level seen herein of bile acids may become a secondary factor in the perpetuation of disease in ARE-Del−/− mice.
Supplementary Material
Acknowledgments
We thank Megan Karwan for conducting animal experiments; Charlotte Hanson for help with technical assistance; Seohyun Kim for analyzing the peptide array data. We also thank Dr. Wolfgang Kastenmüller for consultation on this project. This research was supported by the Intramural Research Program of the NIH, Cancer and Inflammation Program, Center for Cancer Research, National Cancer Institute. The authors declare no competing financial interests.
Financial Support: Funding supported in part by National Institutes of Health grant, DK090019 (MEG) and the National Cancer Institute intramural research program.
List of Abbreviations
- 3′-UTR
3′ Untranslated region
- AMAs
Antimitochondrial autoantibodies
- ARE
AU rich element
- GC
Germinal centers
- H&E
Hemaotoxylin and eosin
- IFN-γ
Interferon gamma
- mAbs
Monoclonal antibodies
- PBC
Primary biliary cholangitis
- PNA
peanut agglutinin
- TBA
Total bile acids
- Tfh
Follicular T helper
- WT
Wild type
References
- 1.Rubtsova K, Marrack P, Rubtsov AV. TLR7, IFNgamma, and T-bet: their roles in the development of ABCs in female-biased autoimmunity. Cell Immunol. 2015;294:80–83. doi: 10.1016/j.cellimm.2014.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Webb GJ, Siminovitch KA, Hirschfield GM. The immunogenetics of primary biliary cirrhosis: A comprehensive review. Journal of autoimmunity. 2015 doi: 10.1016/j.jaut.2015.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kawata K, Tsuda M, Yang GX, Zhang W, Tanaka H, Tsuneyama K, Leung P, et al. Identification of potential cytokine pathways for therapeutic intervention in murine primary biliary cirrhosis. PloS one. 2013;8:e74225. doi: 10.1371/journal.pone.0074225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yang CY, Ma X, Tsuneyama K, Huang S, Takahashi T, Chalasani NP, Bowlus CL, et al. IL-12/Th1 and IL-23/Th17 biliary microenvironment in primary biliary cirrhosis: implications for therapy. Hepatology. 2014;59:1944–1953. doi: 10.1002/hep.26979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang J, Yang GX, Tsuneyama K, Gershwin ME, Ridgway WM, Leung PS. Animal models of primary biliary cirrhosis. Seminars in liver disease. 2014;34:285–296. doi: 10.1055/s-0034-1383728. [DOI] [PubMed] [Google Scholar]
- 6.Shimoda S, Nakamura M, Ishibashi H, Hayashida K, Niho Y. HLA DRB4 0101-restricted immunodominant T cell autoepitope of pyruvate dehydrogenase complex in primary biliary cirrhosis: evidence of molecular mimicry in human autoimmune diseases. J Exp Med. 1995;181:1835–1845. doi: 10.1084/jem.181.5.1835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kita H, Matsumura S, He XS, Ansari AA, Lian ZX, Van de Water J, Coppel RL, et al. Quantitative and functional analysis of PDC-E2-specific autoreactive cytotoxic T lymphocytes in primary biliary cirrhosis. J Clin Invest. 2002;109:1231–1240. doi: 10.1172/JCI14698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bae HR, Leung PS, Tsuneyama K, Valencia JC, Hodge DL, Kim S, Back T, et al. Chronic expression of interferon-gamma leads to murine autoimmune cholangitis with a female predominance. Hepatology. 2016;64:1189–1201. doi: 10.1002/hep.28641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ueno H, Banchereau J, Vinuesa CG. Pathophysiology of T follicular helper cells in humans and mice. Nat Immunol. 2015;16:142–152. doi: 10.1038/ni.3054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee SK, Silva DG, Martin JL, Pratama A, Hu X, Chang PP, Walters G, et al. Interferon-gamma excess leads to pathogenic accumulation of follicular helper T cells and germinal centers. Immunity. 2012;37:880–892. doi: 10.1016/j.immuni.2012.10.010. [DOI] [PubMed] [Google Scholar]
- 11.Domeier PP, Chodisetti SB, Soni C, Schell SL, Elias MJ, Wong EB, Cooper TK, et al. IFN-gamma receptor and STAT1 signaling in B cells are central to spontaneous germinal center formation and autoimmunity. J Exp Med. 2016;213:715–732. doi: 10.1084/jem.20151722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hodge DL, Berthet C, Coppola V, Kastenmuller W, Buschman MD, Schaughency PM, Shirota H, et al. IFN-gamma AU-rich element removal promotes chronic IFN-gamma expression and autoimmunity in mice. Journal of autoimmunity. 2014;53:33–45. doi: 10.1016/j.jaut.2014.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shuai Z, Wang J, Badamagunta M, Choi J, Yang G, Zhang W, Kenny TP, et al. The fingerprint of antimitochondrial antibodies and the etiology of primary biliary cholangitis. Hepatology. 2017 doi: 10.1002/hep.29059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hsueh YH, Chang YN, Loh CE, Gershwin ME, Chuang YH. AAV-IL-22 modifies liver chemokine activity and ameliorates portal inflammation in murine autoimmune cholangitis. J Autoimmun. 2016;66:89–97. doi: 10.1016/j.jaut.2015.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yang CY, Leung PS, Yang GX, Kenny TP, Zhang W, Coppel R, Norman GL, et al. Epitope-specific anti-nuclear antibodies are expressed in a mouse model of primary biliary cirrhosis and are cytokine-dependent. Clin Exp Immunol. 2012;168:261–267. doi: 10.1111/j.1365-2249.2012.04577.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tana MM, Shums Z, Milo J, Norman GL, Leung PS, Gershwin ME, Noureddin M, et al. The Significance of Autoantibody Changes Over Time in Primary Biliary Cirrhosis. Am J Clin Pathol. 2015;144:601–606. doi: 10.1309/AJCPQV4A7QAEEFEV. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yang GX, Wu Y, Tsukamoto H, Leung PS, Lian ZX, Rainbow DB, Hunter KM, et al. CD8 T cells mediate direct biliary ductule damage in nonobese diabetic autoimmune biliary disease. J Immunol. 2011;186:1259–1267. doi: 10.4049/jimmunol.1001597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Oertelt S, Lian ZX, Cheng CM, Chuang YH, Padgett KA, He XS, Ridgway WM, et al. Anti-mitochondrial antibodies and primary biliary cirrhosis in TGF-beta receptor II dominant-negative mice. Journal of immunology. 2006;177:1655–1660. doi: 10.4049/jimmunol.177.3.1655. [DOI] [PubMed] [Google Scholar]
- 19.Leung PS, Yang GX, Dhirapong A, Tsuneyama K, Ridgway WM, Gershwin ME. Animal models of primary biliary cirrhosis: materials and methods. Methods in molecular biology. 2012;900:291–316. doi: 10.1007/978-1-60761-720-4_14. [DOI] [PubMed] [Google Scholar]
- 20.Wakabayashi K, Lian ZX, Leung PS, Moritoki Y, Tsuneyama K, Kurth MJ, Lam KS, et al. Loss of tolerance in C57BL/6 mice to the autoantigen E2 subunit of pyruvate dehydrogenase by a xenobiotic with ensuing biliary ductular disease. Hepatology. 2008;48:531–540. doi: 10.1002/hep.22390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yang GX, Sun Y, Tsuneyama K, Zhang W, Leung PS, He XS, Ansari AA, et al. Endogenous interleukin-22 protects against inflammatory bowel disease but not autoimmune cholangitis in dominant negative form of transforming growth factor beta receptor type II mice. Clin Exp Immunol. 2016;185:154–164. doi: 10.1111/cei.12806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yang GX, Lian ZX, Chuang YH, Moritoki Y, Lan RY, Wakabayashi K, Ansari AA, et al. Adoptive transfer of CD8(+) T cells from transforming growth factor beta receptor type II (dominant negative form) induces autoimmune cholangitis in mice. Hepatology. 2008;47:1974–1982. doi: 10.1002/hep.22226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Weinstein JS, Herman EI, Lainez B, Licona-Limon P, Esplugues E, Flavell R, Craft J. TFH cells progressively differentiate to regulate the germinal center response. Nat Immunol. 2016;17:1197–1205. doi: 10.1038/ni.3554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.King C, Tangye SG, Mackay CR. T follicular helper (TFH) cells in normal and dysregulated immune responses. Annu Rev Immunol. 2008;26:741–766. doi: 10.1146/annurev.immunol.26.021607.090344. [DOI] [PubMed] [Google Scholar]
- 25.Sun Y, Haapanen K, Li B, Zhang W, Van de Water J, Gershwin ME. Women and primary biliary cirrhosis. Clinical reviews in allergy & immunology. 2015;48:285–300. doi: 10.1007/s12016-014-8449-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu K, Kurien BT, Zimmerman SL, Kaufman KM, Taft DH, Kottyan LC, Lazaro S, et al. X Chromosome Dose and Sex Bias in Autoimmune Diseases: Increased Prevalence of 47,XXX in Systemic Lupus Erythematosus and Sjogren’s Syndrome. Arthritis Rheumatol. 2016;68:1290–1300. doi: 10.1002/art.39560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen K, Liu J, Cao X. Regulation of type I interferon signaling in immunity and inflammation: A comprehensive review. J Autoimmun. 2017 doi: 10.1016/j.jaut.2017.03.008. [DOI] [PubMed] [Google Scholar]
- 28.Platanias LC. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat Rev Immunol. 2005;5:375–386. doi: 10.1038/nri1604. [DOI] [PubMed] [Google Scholar]
- 29.Takaoka A, Mitani Y, Suemori H, Sato M, Yokochi T, Noguchi S, Tanaka N, et al. Cross talk between interferon-gamma and -alpha/beta signaling components in caveolar membrane domains. Science. 2000;288:2357–2360. doi: 10.1126/science.288.5475.2357. [DOI] [PubMed] [Google Scholar]
- 30.Muller U, Steinhoff U, Reis LF, Hemmi S, Pavlovic J, Zinkernagel RM, Aguet M. Functional role of type I and type II interferons in antiviral defense. Science. 1994;264:1918–1921. doi: 10.1126/science.8009221. [DOI] [PubMed] [Google Scholar]
- 31.Stifter SA, Bhattacharyya N, Pillay R, Florido M, Triccas JA, Britton WJ, Feng CG. Functional Interplay between Type I and II Interferons Is Essential to Limit Influenza A Virus-Induced Tissue Inflammation. PLoS Pathog. 2016;12:e1005378. doi: 10.1371/journal.ppat.1005378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gough DJ, Messina NL, Hii L, Gould JA, Sabapathy K, Robertson AP, Trapani JA, et al. Functional crosstalk between type I and II interferon through the regulated expression of STAT1. PLoS Biol. 2010;8:e1000361. doi: 10.1371/journal.pbio.1000361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Miettinen M, Sareneva T, Julkunen I, Matikainen S. IFNs activate toll-like receptor gene expression in viral infections. Genes Immun. 2001;2:349–355. doi: 10.1038/sj.gene.6363791. [DOI] [PubMed] [Google Scholar]
- 34.Zarember KA, Godowski PJ. Tissue expression of human Toll-like receptors and differential regulation of Toll-like receptor mRNAs in leukocytes in response to microbes, their products, and cytokines. J Immunol. 2002;168:554–561. doi: 10.4049/jimmunol.168.2.554. [DOI] [PubMed] [Google Scholar]
- 35.Laffont S, Seillet C, Guery JC. Estrogen Receptor-Dependent Regulation of Dendritic Cell Development and Function. Front Immunol. 2017;8:108. doi: 10.3389/fimmu.2017.00108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rowland SL, Riggs JM, Gilfillan S, Bugatti M, Vermi W, Kolbeck R, Unanue ER, et al. Early, transient depletion of plasmacytoid dendritic cells ameliorates autoimmunity in a lupus model. J Exp Med. 2014;211:1977–1991. doi: 10.1084/jem.20132620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Scott JL, Wirth JR, EuDaly JG, Gilkeson GS, Cunningham MA. Plasmacytoid dendritic cell distribution and maturation are altered in lupus prone mice prior to the onset of clinical disease. Clin Immunol. 2017;175:109–114. doi: 10.1016/j.clim.2016.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sozzani S, Del Prete A, Bosisio D. Dendritic cell recruitment and activation in autoimmunity. J Autoimmun. 2017 doi: 10.1016/j.jaut.2017.07.012. [DOI] [PubMed] [Google Scholar]
- 39.Soni C, Wong EB, Domeier PP, Khan TN, Satoh T, Akira S, Rahman ZS. B cell-intrinsic TLR7 signaling is essential for the development of spontaneous germinal centers. J Immunol. 2014;193:4400–4414. doi: 10.4049/jimmunol.1401720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Poovassery JS, Bishop GA. Type I IFN receptor and the B cell antigen receptor regulate TLR7 responses via distinct molecular mechanisms. J Immunol. 2012;189:1757–1764. doi: 10.4049/jimmunol.1200624. [DOI] [PubMed] [Google Scholar]
- 41.Su X, Yu Y, Zhong Y, Giannopoulou EG, Hu X, Liu H, Cross JR, et al. Interferon-gamma regulates cellular metabolism and mRNA translation to potentiate macrophage activation. Nat Immunol. 2015;16:838–849. doi: 10.1038/ni.3205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rahman ZS. Impaired clearance of apoptotic cells in germinal centers: implications for loss of B cell tolerance and induction of autoimmunity. Immunol Res. 2011;51:125–133. doi: 10.1007/s12026-011-8248-4. [DOI] [PubMed] [Google Scholar]
- 43.Rahman ZS, Shao WH, Khan TN, Zhen Y, Cohen PL. Impaired apoptotic cell clearance in the germinal center by Mer-deficient tingible body macrophages leads to enhanced antibody-forming cell and germinal center responses. J Immunol. 2010;185:5859–5868. doi: 10.4049/jimmunol.1001187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fickert P, Fuchsbichler A, Marschall HU, Wagner M, Zollner G, Krause R, Zatloukal K, et al. Lithocholic acid feeding induces segmental bile duct obstruction and destructive cholangitis in mice. Am J Pathol. 2006;168:410–422. doi: 10.2353/ajpath.2006.050404. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.