

Abstract
PURPOSE
To conduct a Phase I trial of a Modified Vaccinia Ankara vaccine delivering wild type human p53 (p53MVA) in combination with gemcitabine chemotherapy in patients with platinum-resistant ovarian cancer.
EXPERIMENTAL DESIGN
Patients received gemcitabine on days 1 and 8 and p53MVA vaccine on day 15, during the first 3 cycles of chemotherapy. Toxicity was classified using the NCI Common Toxicity Criteria and clinical response assessed by CT scan. Peripheral blood samples were collected for immunophenotyping and monitoring of anti-p53 immune responses.
RESULTS
11 patients were evaluated for p53MVA/gemcitabine toxicity, clinical outcome and immunological response. Toxicity: There were no DLTs but 3/11 patients came off study early due to gemcitabine-attributed adverse events (AEs). Minimal AEs were attributed to p53MVA vaccination. Immunological and Clinical Response: Enhanced in vitro recognition of p53 peptides was detectable after immunization in both the CD4+ and CD8+ T cell compartments in 5/11 and 6/11 patients respectively. Changes in peripheral T regulatory cells (Tregs) and myeloid derived suppressor cells (MDSC) did not correlate significantly with vaccine response or progression free survival (PFS). Patients with the greatest expansion of p53-reactive T cells had significantly longer PFS than patients with lower p53-reactivity post therapy. Tumor shrinkage or disease stabilization occurred in 4 patients.
CONCLUSION
p53MVA was well tolerated, but gemcitabine without steroid pre-treatment was intolerable in some patients. However, elevated p53-reactive CD4+ and CD8+T cell responses post therapy correlated with longer PFS. Therefore, if responses to p53MVA could be enhanced with alternative agents, superior clinical responses may be achievable.
Keywords: immunotherapy, p53, clinical trial, ovarian cancer, gemcitabine
Introduction
More than 70% of patients with ovarian cancer present with advanced disease (stage III/IV)[1] and undergo aggressive surgical staging and cytoreduction, followed by systemic chemotherapy. Around 60-80% of patients initially respond to platinum-based chemotherapy (cisplatin/carboplatin) in combination with paclitaxel[2, 3]. However, the vast majority later relapse and eventually develop chemoresistant disease. Hence, new treatments are being actively pursued. The discovery that some cytotoxic agents can interfere with immunosuppressive pathways has led to novel immunotherapy agents being combined with conventional chemotherapy.
Standard of care options for platinum-resistant ovarian cancer include single agent gemcitabine chemotherapy. Intracellular metabolism of gemcitabine produces the active diphosphate (dFdCDP) and triphosphate (dFdCTP) nucleosides, which inhibit DNA synthesis. Similar to other agents used in the setting of platinum resistant disease, gemcitabine has relatively low efficacy in extending survival [4]. In patients with platinum and paclitaxel-resistant ovarian cancer, the response rate to single agent gemcitabine ranges from 13 to 17% with PFS of 3-5 months [5–7]. However, gemcitabine may induce beneficial effects which could help overcome immune suppression and enhance responses to immunotherapy. Although gemcitabine therapy often induces neutropenia, it has shown positive immunomodulatory effects in pancreatic cancer patients, including increased frequency of peripheral CD3+CD45RO+ memory T cells [8], decrease of Tregs[9, 10], reduced MDSC[10] and increased monocytes and dendritic cells [11]. Proposed mechanisms for immunomodulation in cancer patients include inhibition of STAT3 phosphorylation in myeloid cells[10] and preventing the conversion of effector T cells to Tregs[12]. In addition, reduced TGF-β and arginase-1 in cancer patients plasma has been reported post gemcitabine therapy[10].
There is clinical evidence that gemcitabine may enhance responses to a variety of immunotherapies, including antigen pulsed DC (dendritic cells)[13], peptides[14–16] and monoclonal antibodies [17]. However, most of these studies were conducted in pancreatic cancer and there is little published data on this approach in ovarian cancer patients. Dijkraaf et al compared combinations of synthetic long p53 peptides, IFN-α and gemcitabine to treat patients with platinum-resistant ovarian cancer. Gemcitabine significantly reduced the frequency of MDSC and all patients vaccinated with the p53 peptide vaccine demonstrated strong vaccine-induced p53-specific T cell responses[18]. Therefore it seemed logical to combine gemcitabine chemotherapy with our p53-based vaccine.
Wild type p53 protein maintains normal cell division and mutations in this gene are present in the majority of solid tumors[19]. p53 gene mutations result in the accumulation of high levels of oncogenic p53 protein within tumor cells. In contrast, the concentration of normal p53 in healthy cells is low, making p53 an attractive target for immunotherapy of a wide range of malignancies. Because p53 is an autoantigen widely expressed throughout development, immunological tolerance could limit the effectiveness of p53 directed immunotherapy. wild-type p53 is not presented on the surface of parenchymal cells in healthy adults[20], however humans can mount anti-p53 immune responses when p53 is presented as an antigen. The detection of endogenous anti-p53 reactive T cells in cancer patients, suggests a lack of p53 specific tolerance at the T cell level [21]. Several groups have generated human CTL against wild-type p53 peptides in vitro[22, 23]. Therefore, p53 immunotherapy aims to exploit the incomplete tolerance and stimulate immune mediated killing of p53 over expressing tumor cells. If dominant mutations in p53 occurred which generated tumor specific antigens, this would provide ideal targets for a cancer vaccine. Unfortunately, the sites of p53 mutations are highly variable among tumor types and patients, and do not correspond to immunogenic T-cell epitopes[24]. Therefore p53 based vaccines commonly utilize the wt sequence.
Clinical trials targeting p53 by administration of synthetic peptides have yielded promising results. Administration of the canarypox virus ALVAC to patients with unresectable colorectal cancer demonstrated that 2/5 patients receiving the highest vaccine dose developed p53 specific responses [25]. Ad-p53 pulsed dendritic cells have been evaluated in in cancer patients with advanced malignancies [26]. The majority of patients generated potent p53-specific immunity and evidence of clinical benefit was demonstrated. No evidence of autoimmunity or serious adverse events were reported. Leffers et al reported that a p53-synthetic long peptide vaccine was well-tolerated and induced p53-specific T cell responses in patients with recurrent ovarian cancer [27]. When the vaccine was combined with cyclophosphamide, a reduction in Tregs was observed and 2/10 patients showed stable disease [28]. Another study utilizing combinations of p53 peptides, montanide, cytokines and dendritic cells in ovarian cancer patients also reported the induction of p53-specific immune responses[29].
We have developed a strategy using the genetically engineered version of the MVA virus (Modified Vaccinia Ankara) to immunize patients with the wild type p53 antigen (p53MVA). Using a viral vector to deliver full-length p53 has the potential to generate sustained antigen expression and the presentation of numerous antigenic determinants on different HLA molecules. MVA has a demonstrated safety record, being used in numerous clinical trials with only mild side-effects. Despite its inability to propagate in most mammalian cells, MVA still efficiently expresses viral and exogenous recombinant genes making it a potent antigen delivery platform. Furthermore, due to the inactivation of immune evasion genes, MVA vectors demonstrate useful adjuvant properties [30]. MVA vectors are taken up by antigen presenting cells such as dendritic cells, allowing cross presentation of transgene encoded antigens and priming of specific T cell responses [30].
In a first-in-human, single agent trial, a dose of 5.6 × 108pfuof p53MVA was well tolerated and immunogenic, but no clinical responses were apparent[31]. Therefore, we designed a study which combined the vaccine with the immunomodulatory chemotherapy agent gemcitabine. The primary study objectives were to assess the immunogenicity and tolerability of the combined treatment. The secondary objectives were to assess any changes in the frequency of immune suppressive cells in the periphery and clinical responses to therapy. Here we report the findings of this study in regard to safety, clinical response and immunological endpoints.
Methods
Patients and Eligibility
Participants were women with recurrent epithelial ovarian, peritoneal or fallopian tube cancer with evidence of tumoral p53 overexpression by immunohistochemistry (≥10% of cells within the tumor staining positive) or p53 mutational analysis. The Institutional Review Board approved the conduct of the study (ClinicalTrials.gov Identifier NCT02275039), which was in accordance with the Declaration of Helsinki and Good Clinical Practice. Prior to treatment, all patients signed the informed consent. Patients with platinum resistant disease i.e. those showing recurrence or progression within 0-6 months following platinum-based chemotherapy were eligible. Patients with borderline platinum sensitivity, i.e. those with documented disease recurrence or progression within 6-12 months following platinum-based therapy were also eligible. Other eligibility criteria included: ≤ 2 chemotherapy lines for recurrent disease, 18 years of age or older, Karnofsky Performance Status of 80-100 and a life expectancy of greater than 3 months. Adequate bone marrow, renal and liver functions were required. Prior exposure to gemcitabine was not allowed. Patients with brain metastasis, or those receiving any additional investigational agents, radiation therapy or systemic corticosteroids were excluded. In addition, patients with immunodeficiency (including HIV and organ graft related), a history of autoimmune disease, severe environmental allergies, or myopericarditis were ineligible.
Study Design
This was a single institution, dose de-escalating Phase I study of p53MVA vaccination in combination with gemcitabine, in women with recurrent ovarian cancer and tumoral p53 overexpression. The primary endpoints were safety and the frequency of circulating p53-responsive T cells. Secondary endpoints were PFS and the frequency of circulating Tregs and MDSC. As systemic corticosteroids were prohibited from this study, pre-treatment with Dexamethasone was not given prior to chemotherapy. A three vaccination schedule was employed, as previously evaluated in our single agent study. Gemcitabine was given prior to each p53MVA vaccination to reduce immuno suppression and enhance the vaccine response. Gemcitabine was administered at a dose of 1000 mg/m2 as a 30-min intravenous (IV) infusion according to a modified standard of care schedule, on days 1 and 8 every 21 days. The p53MVA vaccine was administered at a dose of 5.6 × 108 pfu p53MVA, intramuscularly on day 15 of the first 3 cycles of gemcitabine. The safety and immunogenicity of this p53MVA dose was established by our previous single agent, Phase I study conducted in patients with gastrointestinal malignancies[31]. All subjects were monitored for one hour in the clinic after each immunization for temperature changes and local reactions at the injection site. All subjects were contacted 24 and 48 hours after each immunization in order to record any vaccine related complications. In the case of drug related adverse events, dose de-escalation was employed, with reduced doses of 800 mg/m2 gemcitabine and 1.0 × 108 pfu of p53MVA permitted. Only one dose reduction was allowed for both study agents. After completing p53MVA vaccination, patients continued single agent gemcitabine on days 1 and 8, every 21 days until confirmed disease progression or unacceptable toxicity. See Figure 1 for the complete treatment schedule.
Figure 1.
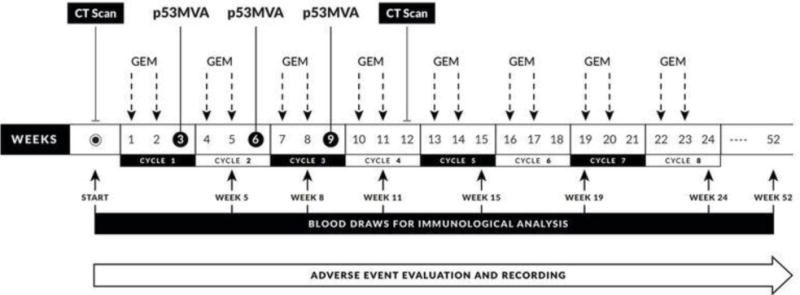
Study schema showing the trial schedule of imaging, gemcitabine administration, p53MVA vaccination and phelbotomy for immunological analysis. After three cycles of combined p53MVA and gemcitabine therapy, single agent gemcitabine was administered according to a modified standard of care regime untill week 52.
p53MVA Vaccine Formulation
p53MVA is a Modified Vaccinia Ankara vector expressing the full length, wild type human p53 gene. The p53MVA vaccine product was manufactured at the Center for Biomedicine and Genetics at City of Hope using GMP-grade materials and the final formulation was diluted in phosphate-buffered saline (PBS) and 7.5% lactose. The vaccine was frozen in high dose and low dose vials, and was thawed and administered as previously described [31].
Toxicity Evaluation and Response Assessment
Dose limiting toxicity (DLT) and adverse events (AE) were graded by the NCI Common Terminology Criteria of Adverse Events (CTCAE) version 4.3. This was a de-escalating Phase I study. The maximum tolerable dose (MTD) was defined as less than 2 patients out of 6 with a DLT, and required at least 6 patients to be treated at that dose. DLT was assessed during the first 2 cycles of treatment. To be evaluable for DLT, patients must have received at least 3 out of 4 doses of gemcitabine and 2 doses of p53MVA, or must have experienced a DLT. Patients who required a dose reduction for gemcitabine or p53MVA were evaluable, provided they received the number of doses specified above. Unevaluable patients were replaced to allow the target accrual of 6-12 evaluable patients to be reached. DLTs were defined as any of the following events during the first 2 cycles of treatment and attributable to study procedures: any ≥ Grade 3 hematologic toxicity which did not improve to baseline within 2 weeks, any ≥ Grade 3 non-hematologic toxicity, any ≥ Grade 2 autoimmune, allergic reactions (including bronchospasm or generalized urticaria) and any ≥ Grade 2 myocarditis thought to be related to p53MVA. All patients underwent baseline radiologic evaluation (CT or PET/CT). Restaging scans were obtained every 12 weeks until progressive toxicity, intolerable toxicity, or patient’s request to discontinue treatment. Clinical response was determined by irRECIST (immune-related criteria).
In Vitro Assessment of p53 T Cell Response
A library of overlapping p53 peptides were synthesized in the laboratory as previously described [32] and frozen in 100% DMSO at a concentration of 1 mg/ml. A human CMV pp65 peptide pool (NIH AIDS Reagent Program, Division of AIDS, NIAID, NIH)) was used as a positive control. Peripheral blood samples were collected from patients by venipuncture in ACD tubes and processed within an hour. PBMCs were purified by density gradient separation using Ficoll-Paque Plus (GE Healthcare). PBMC were cultured in serum free conditions in X-VIVO 20 medium (Lonza) and tested for reactivity to p53 peptides as previously described [31]. In vitro stimulation was carried out for 24 hours in the presence of the following peptides: p53 (10 μg/ml), CMV pp65 (1 μg/ml), or a negative control (DMSO alone). After 24 hours, CD4+ and CD8+ T cells were analysed for the expression of the activation marker CD137 by flow cytometry. The CD137 assay has been validated for both CD8+ and CD4+ responses, and there is evidence that it detects a broader repertoire of antigen-specific T cells than cytokines such as IFN-γ[33]. Cells from each in vitro stimulation were co-stained with anti-CD4, anti-CD8 and anti CD137-PE or a PE-labelled isotype control. CD137 positivity was assessed relative to the isotype control PE signal. Flow cytometric analyses were carried out using a Gallios™ (Beckman Coulter) flow cytometer. All data was analysed with Kaluza software. Patients showing an increase in p53 responsive T cells above baseline levels, that reached a minimum of 0.5% of the CD4+ or CD8+ gate, were defined as T cell responders.
Immunophenotyping
Flow cytometry analysis of cell surface molecules on PBMC was conducted using antibodies from Becton Dickinson or eBioscience. Cells were washed and stained with antibody for 30 minutes at room temperature, in the dark, in the presence of 1% FBS. For regulatory T cell (Treg) intra-nuclear FOXP3 analysis, permeabilization was performed using the eBioscience anti-FOXP3 staining set, according to the manufacturer’s instructions. Tregs were identified as CD3+CD4+CD25+CD127low/−FOXP3+ and expressed as a percentage of CD4+ cells. Myeloid-derived suppressor cells (MDSC) were identified as HLADR−LIN1low/−CD33+CD11b+and expressed as a % of the live PBMC population. T cell phenotyping included analysis of the frequency of PD-1+ T cells within the CD4+ and CD8+ compartments. Flow cytometry gating was carried out as previously described [31]. Blood samples from age and sex matched healthy donor controls were obtained through the City of Hope Michael Amini Transfusion Medicine Center.
Data Analysis
We evaluated baseline % p53-reactive T cells, MDSCs and Tregs along with their nadir, peak, maximum increase (or decrease), and defined a % p53-reactive CD4 and CD8 vaccine response as an increase from baseline greater than 0.5%. This 0.5% increase corresponded to defining CD4 responders as patients whose peak CD4 counts were above the median for the study patients. Statistical comparison of PFS based on single factors were evaluated by the log-rank test with the median PFS estimates and Kaplan-Meier method. Multivariate Cox regression was used to adjust for age, stage and prior lines of therapy, along with step-wise forward model selection, and were summarized with a hazard ratio (HR), the 95% confidence interval and used Wald statistics. Values ≤ 0.05 were considered significant. Correlation structure of the variables was evaluated with Pearson’s correlation coefficient. Comparison of the frequency of Tregs, MDSC and PD-1+ T cells between patients and healthy donors were carried out by Wilcoxon Two-Sided exact test, as were differences in baseline MDSCs, Tregs, or T cell subsets between stage 3 and stage 4 patients or between relevant variables and vaccine responders and non-responders (e.g. MDSCs and Tregs). Statistical calculations were carried out using R version 3.4.2 and SAS version 9.4.
Results
Patient Characteristics
Participants were recruited from ovarian cancer patients attending Medical Oncology clinics at the City of Hope National Medical Center between December 2014 and November 2016. 12 patients began study treatment, but 1 patient came off study prior to p53MVA vaccination due to gemcitabine-related adverse events. The remaining 11 patients (median age 59, range 41-76) were evaluated for toxicity of p53MVA/gemcitabine, clinical outcome and immunological response. The characteristics of all patients treated with the combination are summarized in Table 1. Ten of the vaccinated patients were platinum resistant; this included one patient who relapsed at 6 months and was considered platinum resistant. The prior lines of previous therapy for recurrent disease ranged from 0 to 2. One patient ended participation after one cycle of treatment due to rapid progression of disease. One patient withdrew after two cycles of therapy and 2 patients after only one cycle, due to gemcitabine toxicity.
Table 1.
Demographic and clinical characteristics of patients treated with the p53MVA vaccine and gemcitabine chemotherapy.
| p53MVA vaccine in combination with Gemcitabine n=11 | |
|---|---|
|
| |
| Age at study enrollment (median and range) | 59 (range 41-76) |
|
| |
| Race/ethnicity (no. patients): | |
| Asian | 3 (27%) |
| Caucasian | 7 (64%) |
| Hispanic | 1 (9%) |
|
| |
| Performance status (no. patients): | 4 (36%) |
| 0 | |
| 1 | 6 (55%) |
| 2 | 1 (9%) |
|
| |
| Histology (no. patients): | |
| High grade serous carcinoma(Patients 2,3,4,8,9,10) | 6 (54%) |
| Low grade serous carcinoma (Patients 1 and 11) | 2 (36%) |
| Adenocarcinoma, NOS (Patient 6) | 1 (9%) |
| Carcinosarcoma (Patient 5) | 1 (9%) |
| Clear cell (Patient 7) | 1 (9%) |
|
| |
| Stage (no. patients): | |
| III | 4 (36%) |
| IV | 7 (64%) |
|
| |
| Platinum sensitivity (no. patients): | |
| platinum refractory1 | 9 (81%) |
| borderline platinum sensitive | 2 (18%) |
|
| |
| Prior lines of therapy for recurrent disease2 | |
| 0 lines | 2(18%) |
| 1 line | 7(63%) |
| 2 lines | 2(18%) |
|
| |
| Off treatment reason (no. patients) | |
| Disease progression | 6 (55%) |
| Toxicity | 4 (36%) |
| Patient Refusal | 1 (9%) |
|
| |
| Number of study drug courses completed (median and range) | 4 (1, 11) |
|
| |
| Follow up (median months and range) | 17.5 (2.1 – 18.7) |
One patient relapsed at 6 months and was considered platinum-resistant.
adjuvant or neoadjuvant chemotherapy not included; also non-chemotherapy lines of therapy for recurrent disease not included (PARP inhibition × 1 patient; hormonal therapy × 1 patient).
Safety and Tolerability
Table 1 details the adverse events that were related to therapy. No DLTs were seen on this study. Injection site reaction (ISR) was the most commonly reported side-effect of p53MVA vaccination. The most common adverse events attributed to gemcitabine were neutropenia and skin rash. p53MVA immunization in combination with gemcitabine was well tolerated in the majority of patients, but 3/11 patients came off study within 2 months due to gemcitabine attributed adverse events. Gemcitabine without steroid pretreatment was intolerable in some patients. This included 1 patient with a allergic skin reaction, one with grade 2 pneumonitis and 1 patient with a grade 2 CNS adverse event (reversible posterior leukoencephalopathy syndrome) which resolved rapidly after cessation of therapy and a short course of steroids.
Clinical Outcome
In a total of 11 patients receiving a minimum of one treatment cycle, no complete responses were observed. Three patients demonstrated stable disease (SD) by irRECIST criteria. A partial response (PR) was seen in 1 patient on the second post therapy CT scan (64% reduction according to irRECIST criteria). This patient (Patient 4), showed a dramatic CA-125 decline from 2637 to 150 over a 6 month period. The PFS in treated patients ranged from 0.95 - 9.2 months (median 3 months). Protocol pre-specified follow-up of patients was insufficient for an overall survival assessment.
Immune Response
p53-specific T cell responses were evaluated by quantification of CD137+ T cells after stimulation with p53 peptides. The CD137 marker has been validated for measuring both CD8+ and CD4+responses [33–35], with the suggestion that this assay may detect a broader repertoire of antigen-specific T cells than measurement of IFN-γ [33]. Representative flow cytometry plots from Patient 3 are shown in Figures 2A and 3A. The fluorescence from the negative control stimulation was used to set the negative gates. Figures 2B and 3B show the longitudinal frequency of p53-reactive T cells pre- and post-vaccination in the CD4+ and CD8+ compartments in all 11 patients. Only one post-vaccine blood draw was available from Patients 2, 5 and 10 due to early withdrawal. In two responders, the expansion of p53-reactive T cells was greatest after the first vaccination, however in other responders the peak responses occurred after two or three rounds of p53MVA therapy.
Figure 2. Expansion of anti-p53 CD4+ T cells in patients treated with p53MVA and gemcitabine correlates with longer progression free survival (PFS).
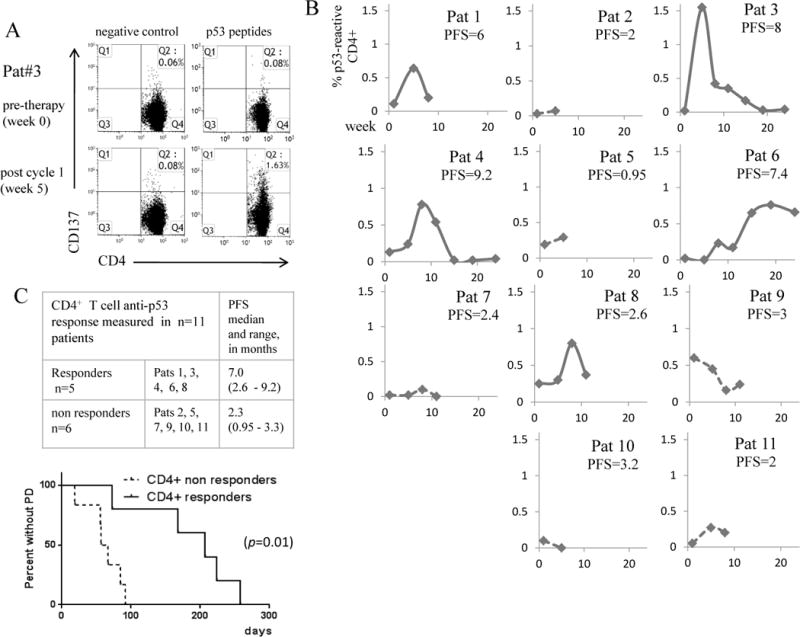
PBMC collected from patients pre and post therapy were assessed for p53 reactivity in vitro by 24 hour stimulation with a p53 peptide library or controls (positive control = CMV pp65 peptide, negative control = DMSO only). The T cell activation marker CD137 was used as a measure of T cell reactivity and was quantified by flow cytometry. Panel A shows the FACS plots from Patient 3, the highest immunological responder. Panel B shows the % of p53 reactive CD4+ T cells detected pre and post vaccination in all 11 patients. To control for background reactivity, % CD137+ cells in response to p53 peptide - %CD137+ T cells in the negative control stimulation were plotted. ‘CD4+ responders’ (solid lines) were defined as those showing a raise in p53-reactive CD4+ T cells above baseline, reaching a minimum of 0.5% reactive cells. Patients not reaching this level were defined as ‘CD4+ non-responders’ (broken lines). Panel C shows the patients in each group with PFS median and range. The accompanying Kaplan Meier curve shows that the difference in PFS between the two groups was statistically significant (p=0.01).
Figure 3. Expansion of anti-p53 CD8+ T cells in patients treated with p53MVA and gemcitabine correlates with longer progression free survival (PFS).
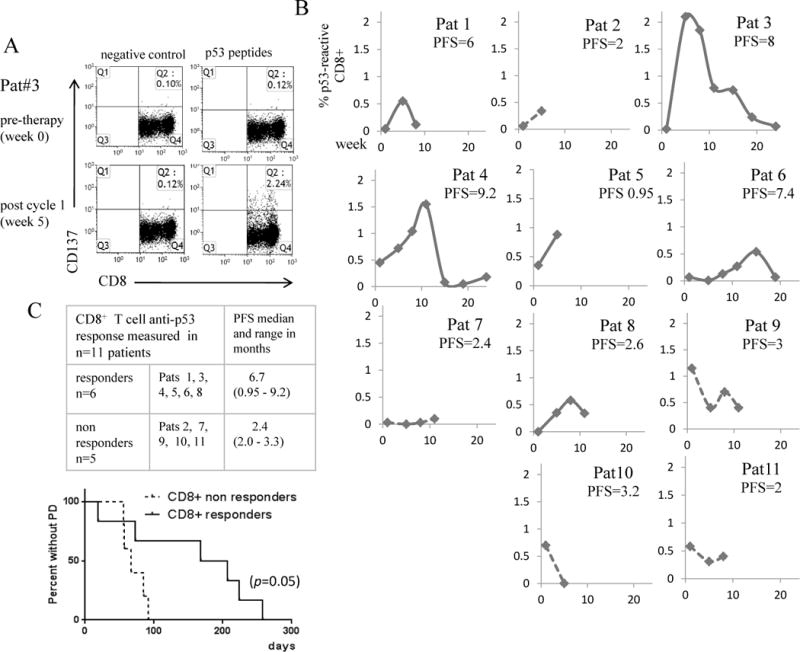
PBMC collected from patients pre and post therapy were assessed for p53 reactivity in vitro by 24 hour stimulation with a p53 peptide library or controls (positive control = CMV pp65 peptide, negative control = DMSO only). The T cell activation marker CD137 was used as a measure of T cell reactivity and was quantified by flow cytometry. Panel A shows the FACS plots from Patient 3, the highest immunological responder. Panel B shows the % of p53 reactive CD8+ T cells detected pre and post vaccination in all 11 patients. To control for background reactivity, % CD137+ cells in response to p53 peptide - %CD137+ T cells in the negative control stimulation were plotted. ‘CD8+ responders’ (solid lines) were defined as those showing a rise in p53-reactive CD4+ T cells above baseline, reaching a minimum of 0.5% reactive cells. Patients not reaching this level were defined as ‘CD8+ non-responders’ (broken lines). Panel C shows the patients in each group and with PFS median and range. The accompanying Kaplan Meier curve shows that the difference in PFS between the two groups was statistically significant (p=0.05).
Frequency of Immunosuppressive Cell Subsets
The frequency of Tregs and MDSC detected in the periphery did not differ significantly from healthy control samples, and did not decline significantly after one cycle of therapy when analyzed as a group (Figure 4A). A decline in Tregs and MDSC after therapy was detected in 7/11 and 6/11 patients respectively (Figure S1 and S2) although in some cases these changes were small and transient. The frequency of PD-1+ T cells at baseline was higher in patients compared to healthy controls, with this reaching statistical significance in the CD8+ compartment (Figure 4B).
Figure 4. Peripheral blood Tregs and MDSC did not differ significantly to healthy controls and did not decline post therapy in the majority of patients. The frequency of baseline PD-1+CD8+T cells differs significantly from healthy donors.
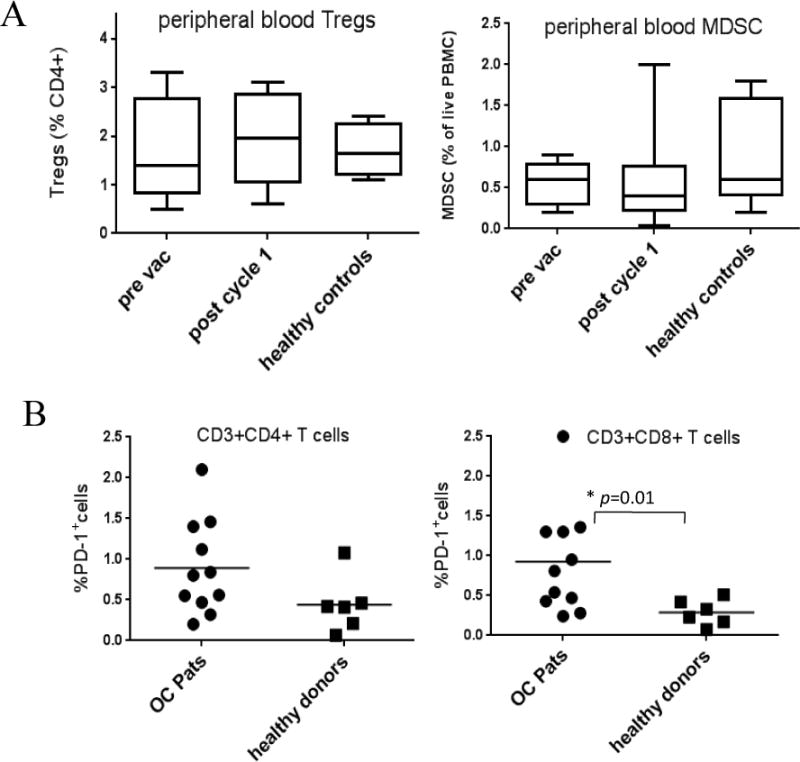
The frequency of peripheral blood Tregs and MDSC pre and post therapy and the percentage of baseline PD-1+ T cells was assessed by flow cytometry in patients, with age and sex matched controls. Panel A shows box plots with the frequency of Tregs (left) and MDSC (right) pre vaccination and after one cycle of therapy compared to healthy controls. Plots represent the mean and standard deviation of 11 patients or 5 healthy donors. Panel B shows the frequency of PD-1+ T cells in the CD4+ T cell population (left) and CD8+ T cell population (right). All 11 patients (OC Pats) are represented by circles and the 6 healthy controls by squares. The frequency of PD-1+CD8+T cells was significantly lower in the healthy controls compared to patients (p=0.0099).
Correlation of immunological and clinical variables
An objective measure of immunological response was applied to the data set. ‘T cell responders’ were defined as patients showing an increase in p53 responsive T cells after therapy ≥ 0.5% of the CD4+ or CD8+ population. According to this criteria, 5/11 patients showed a CD4+ response and 6/11 showed a CD8+ T cell response. p53-responders showed longer PFS than non-p53 responders (Figures 2C and 3C), with this association reaching statistical significance. Of note, the one patient who was a CD8+ responder, but not a CD4+ responder had poor PFS. CD4+ peak frequency, CD4+ peak frequency above median (equivalent to a CD4+ responder in this patient population), also were associated with better PFS (see Supplemental Table 1, ST1). Maximum increase in p53-reactive CD4+ and CD4+ T cells, and peak p53-reactive CD8+ T cells also had a marginally significant, positive impact on PFS.
Patients with stage 4 disease showed inferior PFS, and baseline MDSC had a marginally negative impact on PFS (see ST1). Interestingly, stage 4 patients had a lower expansion of p53-reactive CD4+ T cells (mean of 0.21 vs 0.78, p<0.05), and higher MDSC frequencies (mean of 0.72 vs 0.28, p<0.05) than stage 3 patients (see Supplemental Table 2 for correlations). Combined with small sample size, this correlational structure limits the interpretation of multivariate analysis. As a result, while adjusting for age, stage and number of prior lines of therapy (0 vs 1 or 2) we did not see any of the variables (including adjusted variables) achieve statistical significance (see ST1). Consequently, the forward stepwise regression final model included only CD4 response. CD8 response was less predictive and we cannot conclude that it is an important prognostic feature independent from CD4 response. There was no significant difference in baseline, peak and lowest frequency of Tregs and MDSC between immunological responders and non responders (Figure S1B and S2B). The difference in p53 vaccine response or PFS between patients that did or did not show a fall in Tregs or MDSC was not statistically significant (Figure S1C and S2C).
Discussion
In this report we describe the phase I clinical trial of a p53MVA vaccine in combination with gemcitabine chemotherapy in platinum-resistant ovarian cancer. We observed expected toxicities related to gemcitabine in the absence of steroid premedication. p53MVA toxicity appeared limited to injection site reactions. Clinical efficacy was not a primary endpoint for this study but we observed durable disease control in 4/11 patients. One partial response yields a response rate of 9%, which is inferior to published reports of single agent gemcitabine. However, four out of eleven patients are alive at the time of this submission, two showing overall survival exceeding two years. The effect of p53MVA and gemcitabine on survival is difficult to assess however, as many patients received additional chemotherapy agents after leaving the study and protocol-defined follow-up was limited to 12 months. Therefore, PFS was taken as the primary indicator of clinical activity. The median PFS from all 11 patients was 3 months (range 0.95 - 9.2 months).
An expansion of p53-reactive CD4+ and CD8+ T cells post vaccination was seen in 5/11 patients, approximately half the participants. Patients were defined as ‘immunological responders’ if they showed a rise in p53-reactive T cells above baseline and reached a minimum of 0.5% of the positive CD4+ or CD8+ T cell gate (as defined by cytometry). Patients not reaching this level were defined as ‘immunological non-responders’. Some responses were low and in 3/11 patients only one post therapy blood draw was available. However, despite these limitations, a statistically significant relationship between immunological and clinical response was apparent. The continuous measures of peak values and change, as well as dichotomizing patients based on the peak CD4+ percentage supported this conclusion. Patients with an anti-p53 T cell response i.e. those defined as ‘immunological responders’ showed significantly longer PFS than ‘immunological non-responders’. This is similar to other immunotherapy clinical trials demonstrating improved clinical outcome associated with immunological response, in a range of tumor types [36–39].
One of the primary aims of combining the vaccine with gemcitabine chemotherapy was to reduce the immunosuppression from Tregs and MDSC and enhance the immunological and clinical response.. Surprisingly, both the peripheral Treg and MDSC frequencies detected were in the same range as healthy donor samples (Figure 4A). Peripheral blood measurements may not reflect the frequency of Tregs and MDSC within the tumor microenvironment. Studies in tumor bearing mice report variable frequencies of spleen, lung, tumor, lymph node and peripheral blood MDSC. Furthermore, there is no consensus on which compartment depletion of MDSCs is most important for enhancing antitumor immune responses [40].
During the treatment phase 7/11 and 6/11 of our patients showed a decline in Tregs and MDSC respectively (Figures S1A and S2A). There was no statistically significant effect of baseline, lowest or peak Treg or MDSC between p53 ‘responders and ‘non-responders’ (Figure S1B and S2B). The anti-p53 vaccine response did not show a significant relationship with decline in Tregs or MDSC (Figures S1C and S2C). Patients showing a decline in Tregs had a higher mean PFS than patients with no detectable Treg decline, but this did not reach statistical significance (Figure S1C). Factors such as disease stage, chemotherapy dose, administration schedule, time and location of blood sampling may contribute to variable MDSC levels reported by independent studies[40]. A study in pancreatic cancer patients showed that blood MDSCs were highly variable pre and post gemcitabine treatment. However, when patients with higher baseline MDSCs showed a fall in MDSC, this was associated with greater immunological response[41]. In our study, individual patient’s MDSC decline was not associated with improved response, however, all our patients had baseline MDSC within the range for healthy donors. Perhaps below a certain level, MDSC decline has minimal impact on anti tumor immunity. However, our study did show that baseline MDSCs were marginally associated with worse outcome, were more prevalent in stage 4 patients, and were marginally associated with CD4 and CD8 changes post vaccination.
Murine studies suggest that MDSC frequency vary depending on the interval between chemotherapy administration and blood sampling. MDSC levels may initially increase post chemotherapy, with a fall in MDSC not detectable until several weeks later [42]. Other studies show an initial fall post chemotherapy, followed by a rebound of MDSC in the spleen and tumor[43]. A study of pancreatic cancer patients showed that after a resting period, peripheral MDSC, Tregs and plasma TGFβ-1 reverted to pre-gemcitabine levels [10]. It’s possible that Treg and MDSC frequencies in our patients were dynamic during treatment, and our blood drawing schedule was not optimal to detect gemcitabine induced declines. Additionally, we did not measure factors such as arginase-1 or TGFβ-1 in the plasma, which could have improved the detection of immunomodulation in our responders.
It is unclear why approximately half the patients responded immunologically to p53 vaccination and half did not. The p53MVA/gemcitabine combination was not well tolerated in some patients, resulting in early withdrawal of 3 patients. This could be due to the fact that steroids were not permitted on this study. Dexamethasone is commonly used as pre-medication prior to gemcitabine at baseline, or at the onset of mild allergic reactions to chemotherapy. The fact that patients on this trial did not receive steroid pre-medication could account for the higher than expected rate of allergic reactions to gemcitabine that caused early withdrawal. If these patients had been able to complete combination treatment, this may have resulted in better immunological and clinical responses. However, 4 of the patients that did complete combination therapy did not respond clinically, with progressive disease detected on the first post-treatment scan.
None of the patients on this study had received more than two lines of platinum based chemotherapy for recurrent disease. Considering that multiple lines of chemotherapy are common in this patient population, our study participants would not be considered heavily pre-treated. However, even though the inclusion criteria allowed a platinum free interval of ≤ 12 months, the majority of patients (9/11) had platinum resistant disease. Of the two patients with borderline platinum sensitivity, one patient had low grade serous carcinoma who had a rising CA 125 on Carboplatin and Taxol but did not meet the GOG criteria for biochemical progression. This patient was labelled as borderline platinum sensitive since radiographic progression was documented at 11 months after completing platinum therapy. This likely reflected the natural course of disease rather than borderline sensitivity to platinum agents.
Immune based therapies that have induced durable responses in melanoma and lung cancer patients have shown limited success in ovarian cancer patients, probably because of the ability of ovarian malignancies to block the development of anti-tumor immune responses[44]. Analysis of ten clinical trials with 2285 ovarian patients showed disappointing results to various immunotherapy agents. Meta-analysis of six studies showed that there was no significant difference in overall survival and recurrence-free survival between patients treated with immunotherapy compared to controls[45]. To our knowledge, there is only one other published report of p53 immunotherapy combined with gemcitabine in platinum-resistant ovarian cancer patients. In a three arm trial, Dijkgraaf et al treated 6 patients with a combination of p53 peptides, gemcitabine and IFN-α. This combination achieved disease control in 3/6 of these patients (two partial responses and one stable disease) post therapy[18].
Expansion of p53-reactive T cells after p53MVA/gemcitabine therapy was associated with a small but statistically significant extension of PFS. This is encouraging, especially in a patient population with such poor prognosis. If this p53 response could be induced in all vaccinated patients, and the magnitude and duration of the response expanded further, greater clinical benefit may be achievable. This might be possible if the vaccine was coupled to an immunomodulatory agent with greater stimulatory activity than gemcitabine. Strategies which have shown potential in ovarian cancer, and hence could be combined with the p53MVA vaccine, include TGFβ and IDO blockade. Anti TGFβ has shown potential in ovarian murine models[46]. Elevated IDO expression is associated with poor prognosis in ovarian cancer patients [47], making the IDO inhibitor, epacadostat, another candidate. The mesothelin cancer vaccine in combination with epacadostat is being evaluated in platinum-resistant ovarian cancer[48]. Another potential agent is Ontak, a Treg depletion agent, is currently being investigated with a DC vaccine in ovarian cancer patients[48].
Other immuno modulatory agents that could be combined with the p53MVA vaccine include antibodies that block PD-1/PD-L1 and CTLA-4. Hodi et al reported that anti-CTLA-4 combined with a tumor cell vaccine stimulated a four year remission in an ovarian cancer patient [49]. These agents are being intensely evaluated in numerous tumor types. However, ovarian cancer response rates to single agent checkpoint inhibition have been low. Combinations of these agents may be more effective[48]. Our immunophenotyping data revealed that in contrast to Treg and MDSC levels, the frequency of PD-1+CD8+ T cells were higher in our trial participants compared to healthy controls (Figure 4B). This is in agreement with our immunophenotyping data from patients with gastrointestinal malignancies[31] and raises the possibility of combining PD-1/PD-L1 blockade with the p53MVA vaccine in this patient population. We recently reported the outcome of a triple negative breast cancer patient treated with p53MVA and the anti-PD-1 antibody, pembrolizumab. The patient experienced a complete remission, accompanied by a p53-specific T cell response[50]. Therefore we are evaluating the potential of this combination of agents in patients with platinum-resistant ovarian cancer.
Supplementary Material
Table 2.
Summary of all toxicities Grade 2 and above attributed to study treatment.
| Gem Alone (before p53MVA) n=11 | During p53MVA (possibly attributed to p53MVA or Gem) n=11 | Gem Alone (after p53MVA) n=3 | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Adverse Event | Grade 2 | Grade 3 | Grade 4 | Grade 2 | Grade 3 | Grade 4 | Grade 2 | Grade 3 | Grade 4 |
| Abdominal pain | 1 (33%) | ||||||||
| Allergic reaction | 1 (9%) | ||||||||
| Anemia | 2 (18%) | 2 (18%) | 2 (18%) | 3 (27%) | |||||
| Back pain | 1 (9%) | ||||||||
| Creatinine increased | 2 (18%) | ||||||||
| Dyspnea | 1 (9%) | 1 (33%) | |||||||
| Edema | 1 (9%) | 1 (33%) | |||||||
| Fatigue | 2 (18%) | 4 (36%) | 1 (33%) | ||||||
| Fever & Chills | 2 (18%) | ||||||||
| Headache | 1 (9%) | ||||||||
| Hypertension | 1 (9%) | 3 (27%) | |||||||
| Hypophosphatemia | 1 (9%) | 1 (9%) | |||||||
| Hypoxia | 1 (9%) | ||||||||
| Infection | 1 (9%) | ||||||||
| Injection site reaction | 2 (18%) | ||||||||
| Lymphocyte count decreased | 1 (9%) | 1 (9%) | 1 (9%) | ||||||
| Nausea & Vomiting | 1 (9%) | ||||||||
| Neutrophil count decreased | 1 (9%) | 1 (9%) | 1 (9%) | 2 (18%) | 3 (27%) | 1 (33%) | |||
| Platelet count decreased | 1 (9%) | ||||||||
| Pleuritic pain | 1 (33%) | ||||||||
| Pneumonitis | 1 (9%) | ||||||||
| Rash maculo-papular | 2 (18%) | ||||||||
| Reversible posterior leukoencephalopathy | 1 (9%) | ||||||||
| Sore throat | 1 (9%) | ||||||||
Translational Relevance.
Levels of wild type p53 protein are low in normal cells, however p53 mutations result in high levels of intratumoral p53, making p53 an attractive target for immunotherapy. p53 gene mutations are present in the majority of ovarian tumors and are associated with poor prognosis. Endogenous anti-p53 T cell responses have been demonstrated in ovarian cancer patients, however the ovarian cancer microenvironment is highly immunosuppressive, making immunotherapy challenging. We evaluated a p53 cancer vaccine (p53MVA) in combination with gemcitabine in patients with platinum-resistant ovarian cancer. The rationale being that the immunomodulatory effects of gemcitabine would enhance the action of the p53MVA vaccine. The combination was generally well tolerated, but in some patients gemcitabine toxicity limited treatment duration. In 5/11 participants, the frequency of p53-reactive CD4+ and CD8+ T cells increased post vaccination. These ‘immunological responders’ showed significantly longer progression free survival (PFS) compared to ‘immunological non-responders’.
Acknowledgments
We thank Dr. Bernard Moss (NIAID) for providing MVA 1974/NIH clone 1. We also thank the following City of Hope staff and departments: the Investigational Drug Service, the Bio-Specimen coordinators, the Center for Biomedicine and Genetics and the Office of IND Development and Regulatory Affairs. Research reported in this publication included work performed in the Analytical Cytometry, Biostatistics and Pathology Core facilities. These facilities are supported by the National Cancer Institute of the National Institutes of Health (NIH) under award number P30CA033572. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
This work was supported by the City of Hope Chairs Discretionary Fund and utilized City of Hope Cancer Center funded Core Facilities (award number P30CA033572).
Footnotes
The authors declare no potential conflicts of interests.
References
- 1.Fleming G, Seidman J, Lengyel E. In: Epithelial ovarian cancer: Principles and practice of gynecologic oncology. 6th. Barakat RR, Markman M, Randall M, editors. Philadelphia: Wolters Kluwer Health/Lippincott Williams & Wilkins; 2013. [Google Scholar]
- 2.Vasey PA, et al. Phase III randomized trial of docetaxel-carboplatin versus paclitaxel-carboplatin as first-line chemotherapy for ovarian carcinoma. J Natl Cancer Inst. 2004;96(22):1682–91. doi: 10.1093/jnci/djh323. [DOI] [PubMed] [Google Scholar]
- 3.McGuire WP, et al. Cyclophosphamide and cisplatin compared with paclitaxel and cisplatin in patients with stage III and stage IV ovarian cancer. N Engl J Med. 1996;334(1):1–6. doi: 10.1056/NEJM199601043340101. [DOI] [PubMed] [Google Scholar]
- 4.Yoshino K, et al. Salvage chemotherapy using gemcitabine for taxane/platinum-resistant recurrent ovarian cancer: a single institutional experience. Anticancer Res. 2012;32(9):4029–33. [PubMed] [Google Scholar]
- 5.Shapiro JD, et al. Activity of gemcitabine in patients with advanced ovarian cancer: responses seen following platinum and paclitaxel. Gynecol Oncol. 1996;63(1):89–93. doi: 10.1006/gyno.1996.0284. [DOI] [PubMed] [Google Scholar]
- 6.D’Agostino G, et al. Phase II study of gemcitabine in recurrent platinum-and paclitaxel-resistant ovarian cancer. Gynecol Oncol. 2003;88(3):266–9. doi: 10.1016/s0090-8258(03)00011-8. [DOI] [PubMed] [Google Scholar]
- 7.Markman M, et al. Phase 2 trial of single-agent gemcitabine in platinum-paclitaxel refractory ovarian cancer. Gynecol Oncol. 2003;90(3):593–6. doi: 10.1016/s0090-8258(03)00399-8. [DOI] [PubMed] [Google Scholar]
- 8.Plate JM, et al. Effect of gemcitabine on immune cells in subjects with adenocarcinoma of the pancreas. Cancer Immunol Immunother. 2005;54(9):915–25. doi: 10.1007/s00262-004-0638-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Homma Y, et al. Changes in the immune cell population and cell proliferation in peripheral blood after gemcitabine-based chemotherapy for pancreatic cancer. Clin Transl Oncol. 2014;16(3):330–5. doi: 10.1007/s12094-013-1079-0. [DOI] [PubMed] [Google Scholar]
- 10.Eriksson E, et al. Gemcitabine reduces MDSCs, tregs and TGFbeta-1 while restoring the teff/treg ratio in patients with pancreatic cancer. J Transl Med. 2016;14(1):282. doi: 10.1186/s12967-016-1037-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Soeda A, et al. Regular dose of gemcitabine induces an increase in CD14+ monocytes and CD11c+ dendritic cells in patients with advanced pancreatic cancer. Jpn J Clin Oncol. 2009;39(12):797–806. doi: 10.1093/jjco/hyp112. [DOI] [PubMed] [Google Scholar]
- 12.Kan S, et al. Suppressive effects of cyclophosphamide and gemcitabine on regulatory T-cell induction in vitro. Anticancer Res. 2012;32(12):5363–9. [PubMed] [Google Scholar]
- 13.Hirooka Y, et al. A combination therapy of gemcitabine with immunotherapy for patients with inoperable locally advanced pancreatic cancer. Pancreas. 2009;38(3):e69–74. doi: 10.1097/MPA.0b013e318197a9e3. [DOI] [PubMed] [Google Scholar]
- 14.Kaida M, et al. Phase 1 trial of Wilms tumor 1 (WT1) peptide vaccine and gemcitabine combination therapy in patients with advanced pancreatic or biliary tract cancer. J Immunother. 2011;34(1):92–9. doi: 10.1097/CJI.0b013e3181fb65b9. [DOI] [PubMed] [Google Scholar]
- 15.Middleton GW, V J, Wadsley J, Propper D, Coxon FY, Ross PJ. A Phase III randomized trial of chemoimmunotherapy comprising gemcitabine and capecitabine with or without telomerase vaccine GV1001 in patients with locally advanced or metastatic pancreatic cancer. Journal of Clinical Oncology. 2013;31 (suppl; abstr LBA4004) [Google Scholar]
- 16.Miyazawa M, et al. Phase I clinical trial using peptide vaccine for human vascular endothelial growth factor receptor 2 in combination with gemcitabine for patients with advanced pancreatic cancer. Cancer Sci. 2010;101(2):433–9. doi: 10.1111/j.1349-7006.2009.01416.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Beatty GL, et al. A phase I study of an agonist CD40 monoclonal antibody (CP-870,893) in combination with gemcitabine in patients with advanced pancreatic ductal adenocarcinoma. Clin Cancer Res. 2013;19(22):6286–95. doi: 10.1158/1078-0432.CCR-13-1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dijkgraaf EM, et al. A phase 1/2 study combining gemcitabine, Pegintron and p53 SLP vaccine in patients with platinum-resistant ovarian cancer. Oncotarget. 2015;6(31):32228–43. doi: 10.18632/oncotarget.4772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hainaut P, Hollstein M. p53 and human cancer: the first ten thousand mutations. Adv Cancer Res. 2000;77:81–137. doi: 10.1016/s0065-230x(08)60785-x. [DOI] [PubMed] [Google Scholar]
- 20.Bueter M, et al. Influence of p53 on anti-tumor immunity (review) Int J Oncol. 2006;28(2):519–25. [PubMed] [Google Scholar]
- 21.van der Burg SH, et al. Long lasting p53-specific T cell memory responses in the absence of anti-p53 antibodies in patients with resected primary colorectal cancer. Eur J Immunol. 2001;31(1):146–55. doi: 10.1002/1521-4141(200101)31:1<146::aid-immu146>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- 22.Nikitina EY, et al. Dendritic cells transduced with full-length wild-type p53 generate antitumor cytotoxic T lymphocytes from peripheral blood of cancer patients. Clin Cancer Res. 2001;7(1):127–35. [PubMed] [Google Scholar]
- 23.Chikamatsu K, et al. Generation of anti-p53 cytotoxic T lymphocytes from human peripheral blood using autologous dendritic cells. Clin Cancer Res. 1999;5(6):1281–8. [PubMed] [Google Scholar]
- 24.Wiedenfeld EA, et al. Evidence for selection against human lung cancers bearing p53 missense mutations which occur within the HLA A*0201 peptide consensus motif. Cancer Res. 1994;54(5):1175–7. [PubMed] [Google Scholar]
- 25.van der Burg SH, et al. Induction of p53-specific immune responses in colorectal cancer patients receiving a recombinant ALVAC-p53 candidate vaccine. Clin Cancer Res. 2002;8(5):1019–27. [PubMed] [Google Scholar]
- 26.Antonia SJ, et al. Combination of p53 cancer vaccine with chemotherapy in patients with extensive stage small cell lung cancer. Clin Cancer Res. 2006;12(3 Pt 1):878–87. doi: 10.1158/1078-0432.CCR-05-2013. [DOI] [PubMed] [Google Scholar]
- 27.Leffers N, et al. Immunization with a P53 synthetic long peptide vaccine induces P53-specific immune responses in ovarian cancer patients, a phase II trial. Int J Cancer. 2009;125(9):2104–13. doi: 10.1002/ijc.24597. [DOI] [PubMed] [Google Scholar]
- 28.Vermeij R, et al. Potentiation of a p53-SLP vaccine by cyclophosphamide in ovarian cancer: A single-arm phase II study. Int J Cancer. 2011 doi: 10.1002/ijc.27388. [DOI] [PubMed] [Google Scholar]
- 29.Rahma OE, et al. A gynecologic oncology group phase II trial of two p53 peptide vaccine approaches: subcutaneous injection and intravenous pulsed dendritic cells in high recurrence risk ovarian cancer patients. Cancer Immunol Immunother. 2012;61(3):373–84. doi: 10.1007/s00262-011-1100-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Antoine G, et al. The complete genomic sequence of the modified vaccinia Ankara strain: comparison with other orthopoxviruses. Virology. 1998;244(2):365–96. doi: 10.1006/viro.1998.9123. [DOI] [PubMed] [Google Scholar]
- 31.Hardwick NR, et al. p53MVA therapy in patients with refractory gastrointestinal malignancies elevates p53-specific CD8+ T-cell responses. Clin Cancer Res. 2014;20(17):4459–70. doi: 10.1158/1078-0432.CCR-13-3361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Song GY, et al. An MVA vaccine overcomes tolerance to human p53 in mice and humans. Cancer Immunol Immunother. 2007;56(8):1193–205. doi: 10.1007/s00262-006-0270-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wehler TC, et al. Rapid identification and sorting of viable virus-reactive CD4(+) and CD8(+) T cells based on antigen-triggered CD137 expression. J Immunol Methods. 2008;339(1):23–37. doi: 10.1016/j.jim.2008.07.017. [DOI] [PubMed] [Google Scholar]
- 34.Litjens NH, et al. Activation-induced CD137 is a fast assay for identification and multi-parameter flow cytometric analysis of alloreactive T cells. Clin Exp Immunol. 2013;174(1):179–91. doi: 10.1111/cei.12152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jing L, et al. CD4 T-cell memory responses to viral infections of humans show pronounced immunodominance independent of duration or viral persistence. J Virol. 2013;87(5):2617–27. doi: 10.1128/JVI.03047-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wheeler CJ, et al. Vaccination elicits correlated immune and clinical responses in glioblastoma multiforme patients. Cancer Res. 2008;68(14):5955–64. doi: 10.1158/0008-5472.CAN-07-5973. [DOI] [PubMed] [Google Scholar]
- 37.Kirkwood JM, et al. Immunogenicity and antitumor effects of vaccination with peptide vaccine+/-granulocyte-monocyte colony-stimulating factor and/or IFN-alpha2b in advanced metastatic melanoma: Eastern Cooperative Oncology Group Phase II Trial E1696. Clin Cancer Res. 2009;15(4):1443–51. doi: 10.1158/1078-0432.CCR-08-1231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Barth RJ, Jr, et al. A randomized trial of ex vivo CD40L activation of a dendritic cell vaccine in colorectal cancer patients: tumor-specific immune responses are associated with improved survival. Clin Cancer Res. 2010;16(22):5548–56. doi: 10.1158/1078-0432.CCR-10-2138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yoshitake Y, et al. Phase II clinical trial of multiple peptide vaccination for advanced head and neck cancer patients revealed induction of immune responses and improved OS. Clin Cancer Res. 2015;21(2):312–21. doi: 10.1158/1078-0432.CCR-14-0202. [DOI] [PubMed] [Google Scholar]
- 40.Wang Z, Till B, Gao Q. Chemotherapeutic agent-mediated elimination of myeloid-derived suppressor cells. Oncoimmunology. 2017;6(7):e1331807. doi: 10.1080/2162402X.2017.1331807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Annels NE, et al. The effects of gemcitabine and capecitabine combination chemotherapy and of low-dose adjuvant GM-CSF on the levels of myeloid-derived suppressor cells in patients with advanced pancreatic cancer. Cancer Immunol Immunother. 2014;63(2):175–83. doi: 10.1007/s00262-013-1502-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Angulo I, et al. Nitric oxide-producing CD11b(+)Ly-6G(Gr-1)(+)CD31(ER-MP12)(+) cells in the spleen of cyclophosphamide-treated mice: implications for T-cell responses in immunosuppressed mice. Blood. 2000;95(1):212–20. [PubMed] [Google Scholar]
- 43.Ding ZC, et al. Immunosuppressive myeloid cells induced by chemotherapy attenuate antitumor CD4+ T-cell responses through the PD-1-PD-L1 axis. Cancer Res. 2014;74(13):3441–53. doi: 10.1158/0008-5472.CAN-13-3596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chae CS, Teran-Cabanillas E, Cubillos-Ruiz JR. Dendritic cell rehab: new strategies to unleash therapeutic immunity in ovarian cancer. Cancer Immunol Immunother. 2017 doi: 10.1007/s00262-017-1958-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Alipour S, et al. Specific immunotherapy in ovarian cancer: a systematic review. Immunotherapy. 2016;8(10):1193–204. doi: 10.2217/imt-2016-0034. [DOI] [PubMed] [Google Scholar]
- 46.Vanpouille-Box C, et al. TGFbeta Is a Master Regulator of Radiation Therapy-Induced Antitumor Immunity. Cancer Res. 2015;75(11):2232–42. doi: 10.1158/0008-5472.CAN-14-3511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Inaba T, et al. Role of the immunosuppressive enzyme indoleamine 2,3-dioxygenase in the progression of ovarian carcinoma. Gynecol Oncol. 2009;115(2):185–92. doi: 10.1016/j.ygyno.2009.07.015. [DOI] [PubMed] [Google Scholar]
- 48.Martin Lluesma S, et al. Cancer Vaccines in Ovarian Cancer: How Can We Improve? Biomedicines. 2016;4(2) doi: 10.3390/biomedicines4020010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hodi FS, et al. Immunologic and clinical effects of antibody blockade of cytotoxic T lymphocyte-associated antigen 4 in previously vaccinated cancer patients. Proc Natl Acad Sci U S A. 2008;105(8):3005–10. doi: 10.1073/pnas.0712237105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yuan Y, et al. Complete regression of cutaneous metastases with systemic immune response in a patient with triple negative breast cancer receiving p53MVA vaccine with pembrolizumab. OncoImmunology. 2017:00–00. doi: 10.1080/2162402X.2017.1363138. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.