

Abstract
Highly glycolytic cervical cancers largely resist treatment by cisplatin and co-administered pelvic irradiation as the present standard of care. In this study, we investigated the effects of inhibiting glycolysis and thiol redox metabolism to evaluate them as alternate treatment strategies in these cancers. In a panel of multiple cervical cancer cell lines, we evaluated sensitivity to inhibition of glycolysis (2-DG) with or without simultaneous inhibition of glutathione and thioredoxin metabolism (BSO/AUR). Intracellular levels of total and oxidized glutathione, thioredoxin reductase activity, and indirect measures of intracellular reactive oxygen species (ROS) were compared before and after treatment. Highly radio-resistant cells were the most sensitive to 2-DG, whereas intermediate radio-resistant cells were sensitive to 2-DG plus BSO/AUR. In response to 2-DG/BSO/AUR treatment, we observed increased levels of intracellular oxidized glutatione, redox-sensitive dye oxidation and decreased glucose utilization via multiple metabolic pathways including the TCA cycle. 2-DG/BSO/AUR treatment delayed the growth of tumors composed of intermediate radio-resistant cells and effectively radio-sensitized these tumors at clinically relevant radiation doses both in vitro and in vivo. Overall, our results support inhibition of glycolysis and intracellular redox metabolism as an effective alternative drug strategy for the treatment of highly glycolytic and radio-resistant cervical cancers.
Introduction
The current standard of care for locally advanced cervical cancer is concurrent cisplatin chemotherapy with pelvic irradiation which includes the administration of both external beam radiotherapy and intracavitary brachytherapy. Despite significant advances in radiation treatment delivery, more than 30% of patients fail this treatment. The prognosis of these patients is poor as there is currently no curative treatment for metastatic cervical cancer. Complete surgical resection including total pelvic exenteration has been used to salvage limited volume local pelvic recurrences but with significant treatment–related morbidity. We have previously reported that increased uptake of 18F-fluoro-deoxy-glucose (FDG) on pretreatment positron emission tomography (PET) is prognostic for poor outcomes, and that cervical tumors with residual FDG uptake after standard of care chemo-radiation have inferior long term survival. [1, 2] Cervical tumors with persistent FDG uptake after chemo-radiation have altered expression of genes from the PI3K/AKT pathway, and increased expression of phosphorylated AKT is associated with poor outcomes after standard of care treatment. [3] AKT inhibitors can be used to reduce cervical tumor cell glucose uptake and metabolism, which results in tumor cell death. [4] Furthermore, redox signaling has been suggested to regulate both AKT activation and cervical cancer cell survival.[5]
Enhanced glutathione (GSH) and thioredoxin metabolism (Trx) are two mechanisms by which cancer cells mitigate the redox stress resulting from increased steady-state levels of reactive oxygen species (ROS) produced by disruptions in oxidative metabolism. [6–10] GSH and Trx neutralize hydroperoxides, and upregulation of their respective metabolic pathways occurs in many cancers.[8, 9, 11] Glutathione peroxidase (GPx) enzymes inactivate H2O2 and other hydroperoxides using reducing equivalents derived from the conversion of GSH to glutathione disulfide (GSSG). GSSG is then recycled back to GSH by glutathione reductase (GR), consuming reducing equivalents from NADPH in the process.[7–9, 12] The Trx system neutralizes H2O2 and hydroperoxides via the action of peroxiredoxins (Prx), which consumes reduced Trx(SH)2 and releases oxidized TrxS2 (Trx)[13] that is then reduced back to Trx(SH)2 by thioredoxin reductase (TR); this process also consumes reducing equivalents from NADPH.[7–9, 12] Given that both GSH- and Trx-dependent peroxide metabolism require NADPH as the ultimate source of electrons, these pathways are inextricably linked to glucose metabolism, because the latter is required for the production of the majority of the NADPH pool. [6, 9, 14]
Glucose deprivation and treatment with 2-DG are known to selectively induce more oxidative stress in cancer cells compared to normal cells.[8, 10, 15] 2-DG is transported into cells and phosphorylated to 2-DG-6-phosphate by hexokinase, after which it accumulates and inhibits downstream glycolytic enzymes. [16] Previous studies in head and neck cancer cells have shown that 2-DG treatment promotes shunting of glucose into the pentose phosphate pathway in an effort to combat the increase in intracellular oxidative stress. [17] Aside from inhibition of NAPDH production by targeting glucose metabolism, strategies using direct redox metabolism inhibitors such as L-buthionine-sulfoximine (BSO), a γ-glutamate cysteine ligase inhibitor, and Auranofin (AUR), a thioredoxin reductase inhibitor, alone or in combination have been shown to induce redox imbalances in several cancer cell types. [7, 8, 11, 12, 18]
Our previous work demonstrated that highly glycolytic cervical cancers are resistant to standard of care therapy (cisplatin plus pelvic irradiation). The objective of the current study is to test whether inhibition of glycolysis, in combination with simultaneous inhibition of intracellular GSH- and Trx-mediated redox metabolism, is an effective alternative drug strategy for highly glycolytic and radio-resistant cervical cancers. Because our ultimate goal is translation of these results into the clinic, we selected three drugs for preclinical studies that are already approved for human use: 1) 2-DG to inhibit glycolysis, 2) BSO to target GSH synthesis and 3) AUR to target thioredoxin reductase.
Material and Methods
Cell culture and Reagents
Cervical cancer cell lines were obtained from the American Type Culture collection (ATCC) 2009–2010. Experiments were performed on cell lines between passages 10 and 30. Cells were maintained in IMDM media (Life Technologies, CA) with 10% heat inactivated FBS and incubated at 37°C in 5% CO2. Mycoplasma testing was performed periodically by the iPSC Center at Washington University School of Medicine using MycoAlert Plus Kit (Lonza, Basel, Switzerland) to verify no infection. Last date of mycoplasma testing for cell lines used in this study was April 20, 2017. L-buthionine-sulfoximine (BSO), Auranofin (AUR), 2-Deoxy glucose (2-DG), protease and phosphatase inhibitor cocktails were purchased from Sigma (Saint Louis, MO). All drugs for cell culture were dissolved in normal saline except AUR which was dissolved in 0.05% dimethyl sulfoxide (DMSO).
Western blotting
Phosphorylation of AMPK pathway and activation of autophagy with or without 2-DG+BSO+AUR were determined by western blotting with primary antibodies against phosphorylated and total forms of Ser15p53, p53, Thr269/Ser272p62, p62, Thr389p70, p70, Ser317Ulk, Ulk, Ser62Myc, Myc, cleaved caspase 3 and PARP, Thr286CamKII, CamKII, Thr172AMPK, AMPK, Ser428LKB1, LKB1, Thr183/Tyr185 JNK, JNK (1:1000; Cell Signaling Technology, MA), for total forms of thioredoxin reductase1 and 2 (TR1 and TR2), GAPDH, MAPLC3, and Bim (1:1000, Cell Signaling Technology, MA), and for Actin from (1:1000,Santa Cruz Biotechnology, CA) and tubulin (1:10,000, Sigma, Saint Louis, MO). Blots were probed with HRP-conjugated anti-rabbit (Cell Signaling Technology, Beverly, MA) or anti-mouse polyclonal IgG secondary antibodies (Santa Cruz Biotechnology, CA) for 1h at room temperature. For detection, Amersham ECL select (GE healthcare, PA) was used according to manufacturer’s protocol. Images were acquired using Chemidoc Imaging systems (BioRad).
Cell viability and clonogenic survival assays
For cell viability assays, cells were treated with the glycolytic and redox inhibitors 2-DG (20 mM), BSO (1 mM) for 48h and AUR (100 nM) for 24h, respectively. Cell viability was tested using Alamar Blue from Life Technologies, according to manufacturer’s instructions.
For the clonogenic cell survival assays, cells were treated with or without drugs alone or in combination using the following concentrations: 2-DG (20 mM), cisplatin (500 nM), BSO (1 mM) for 48h and AUR (100 nM) for 24h. For radiation experiments 10 mM-2-DG, 500 uM-BSO and 50 nM-AUR were added simultaneously, incubated for 5 hrs in the presence of drugs, then irradiated using an RS2000 160kV X-ray Irradiator using a 0.3 mm copper filter (Rad Source Technologies, Suwanee, GA). Cells were harvested after 24 h by trypsinization including all floating cells and 500 cells each were plated in a 6-well dish. The colonies were counted 10 days later after staining with crystal violet.
FDG uptake assays
FDG uptake assay was performed as described previously. [3] Briefly, cells were seeded 48 hours prior to 18F–FDG labeling and maintained in standard tissue culture conditions. Cells were incubated in glucose free media for 30 minutes prior to the addition of 18F–FDG. 18F–FDG (20 μCi) was added to the glucose free medium for 1h. Cells were washed, harvested, and counted on a gamma counter. Data was normalized to total cell count.
Glutathione and Thioredoxin reductase Assay
Cells were grown in 100 mm dishes and treated with or without drugs as described above then harvested by scraping and frozen as a dry pellet. Cell pellets were lysed in 50 mM potassium phosphate buffer pH 7.8 containing 1.34 mM diethylenetriaminepenta-acetic acid, centrifuged at 5000 rpm for 5 minutes and then the supernatant was assayed using a TR assay kit (Sigma-Aldrich). Cells were processed as for the GSH assay by scraping into 150 μL of 5% 5-sulfosalicylic acid (Sigma-Aldrich). Total GSH and GSSG content was determined spectrophotometrically by NADPH recycling assay as described previously.[7]
Reactive oxygen species (ROS) quantification
Levels of pro-oxidants were determined using the oxidation-sensitive (CDCFH2, 10 μg/ml) and oxidation-insensitive (CDCF, 10 μg/ml) fluorescent dyes obtained from Molecular Probes. The cells were washed once with PBS and labelled with florescent dyes for 10 mins at 37°C in PBS. At the end of the incubation, plates were read at Ex/Em: 492–495/517–527 nm. The mean fluorescent intensity (MFI) was plotted after correction for auto-fluorescence from unlabeled cells. Steady-state levels of superoxide were estimated using oxidation of the fluorescent dye, DHE (dihydroethidine), obtained from Molecular Probes (Eugene, OR, U.S.A.). Briefly cells were incubated with DHE (10 μM) at 37 °C for 30 min in culture media and suspended in PBS. Samples were analyzed using a flow cytometer (λex=488nm and λemission=585nm band-pass filter). The MFI (mean fluorescence intensity) was analyzed in each sample and corrected for auto-fluorescence from unlabeled cells.[10]
Manipulation of cellular antioxidants
For experiments using the thiol antioxidant, N-acetylcysteine (NAC), cells were treated with 10 mM NAC for 5 hours following 2-DG and BSO for 24h followed by 2-DG, BSO and AUR for another 24h. For overexpression of catalase, cells were infected with 100 MOI of adenovirus expressing catalase (AdCAT) or empty virus (AdEmpty). After 48h, virus was removed and the drugs were added followed by colony forming assay as described above. Over expression of catalase was confirmed by catalase activity assay as previously described.[19] Activity was expressed in mκunits/milligram protein.
Stable isotope-based metabolomics
Stable isotope labeling of cells was performed by substituting the culture media with labeling media consisting of glucose- and glutamine-free DMEM (Gibco A1443001) supplemented with 10% dialyzed FBS, 2 mM glutamine, and 10 mM uniformly labeled 13C glucose (Cambridge Isotopes). 2DG, BSO, and AUR were added to the labeling media at the time of substitution, and labeling and drug treatment were performed for 18 h. Cells were then processed for liquid chromatography/mass spectrometry (LC/MS)-based metabolomic profiling according to the protocol described in.[20] Briefly, cells were washed with PBS and milliQ water, and metabolism was quenched with cold methanol. Cells were scraped and transferred to microfuge tubes. The methanol was evaporated using a speed-vac, leaving a dried cell pellet that was extracted with 2:2:1 methanol:acetonitrile:water (1 mL per 100-mm plate surface area equivalent). Metabolite extracts were concentrated using a speed-vac to remove the extraction solvent and reconstituted in 1:1 acetonitrile:water in a 10:1 original:final volume ratio. Five μL of each concentrated metabolite extract were injected onto a 3 μm, 150mm × 1mm Luna NH2 column (Phenomenex) operated in hydrophilic interaction liquid chromatography (HILIC) mode on an Agilent 1260 high performance liquid chromatography system. The mobile phases were A: 20 mM NH4OAc/NH4OH at pH 9.5 in 95% H2O/5% acetonitrile and B: 95% acetonitrile/5% H2O. A gradient from 100% B to 100% A was run over 40 min. Column eluate was injected into an electrospray source operating in negative ionization mode and mass analyzed in an Agilent 6530 or 6540 quadrupole time-of-flight mass spectrometer. Data were processed using MSConvert [21], XCMS [22], X13CMS [20], and Graphpad Prism 7.
Tumor Growth Delay DG, BSO, AUR with and without tumor directed irradiation
All the in vivo studies were conducted according to the protocols approved by Washington University Division of Comparative Medicine and Institutional Animal Care and Use Committee. 3.5 million SiHa and or CaSki cells were injected subcutaneously into the left flank of 6–8 week old, female nude mice in a half matrigel, half serum-free IMDM mixture. After two weeks, initial tumor sizes were recorded using calipers and mice were grouped (N=5 per treatment group) with 5 mm starting tumor volumes. For tumor growth delay studies in Caski tumors with drugs alone, treatment groups included: sham injection, DG alone, BSO + AUR (BA), and DG + BSO + AUR (DBA). Mice were injected three times per week with 400mg/kg 2-DG, 200mg/kg BSO and 1.5mg/kg AUR over a period of 35 days. For tumor growth delay studies in SiHa tumors with drug treatment and concurrent tumor directed radiation, single fraction radiation doses of 2 or 4 Gy were delivered after 1 week of treatment with DBA. Targeted radiation delivery was performed using the Xstrahl Small Animal Radiation Research Platform (SARRP) 200 (Xstrahl Life Sciences, Suwannee, GA). Mice were placed on the irradiation platform one at a time and fitted with a nose cone for isofluorane anesthesia. CT images imported into Muriplan were used to select an isocenter. The tumor was then irradiated using anterior-posterior opposed beams using the 10 mm × 10 mm collimator at a dose rate of 3.9 Gy/min. DBA treatment was continued for 1 week after radiation treatment.
Tumor measurements were taken by caliper on a weekly basis. At the end of the experiment, animals were sacrificed, tumors were excised and weighed. Intra-tumoral GSH, GSSG and TR was quantified as described above.
Results
Radio-resistant and highly glycolytic cervical cancer cells are sensitive to the glycolytic inhibitor 2-DG
To establish baseline sensitivities for cervical cancer cell lines to treatment, we tested the sensitivity of a panel of cervical cancer cell lines to radiation and cisplatin monotherapy (Figure 1). Radio-sensitivity was determined by colony forming assay (CFA) 48 hours after treatment with a single fraction of 2, 4 and 6 Gy radiation. CaSki was the most radio-resistant cell line, while C33A was the most sensitive (Figure 1A). Cisplatin sensitivity was determined by CFA 48 hrs after treatment with 500 nM cisplatin. CaSki was also the most resistant to cisplatin, and C33A was the most sensitive (Figure 1B).
Figure 1. Analysis of sensitivity of cervical cancer cells to radiation, cisplatin and 2-DG monotherapy.
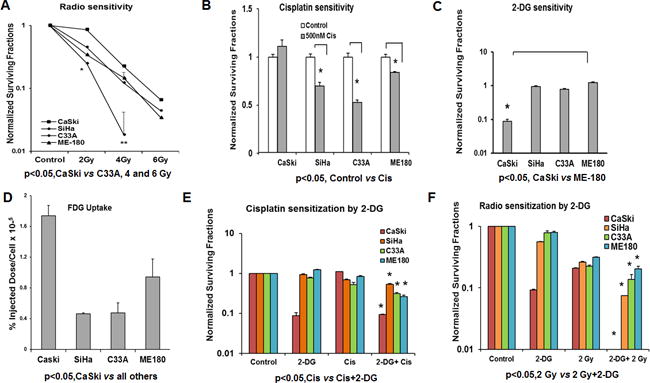
(A–C) Cells were seeded on to 48 well plates and treated with increasing doses of radiation (2, 4 and 6 Gy), 500nM cisplatin and 20mM 2-DG for 48hrs. Cells were trypsinized, counted and plated 500 cells per well on a 6 well plate for ten days and counted after crystal violet staining. Clonogenic survival data was normalized to untreated controls. (D) Cells were incubated in glucose free medium for 30 minutes prior to incubation with 18F-fluoro-deoxy-glucose as described in the methods section. Data was normalized to total cell count. (E-F) Cells were treated with 500nM cisplatin and 20mM 2-DG for 48hrs and cells were irradiated using 2 Gy along with 20mM 2-DG and clonogenic survival assay performed as described above. Statistical analysis: Error bars represent ± SD of N=3 experiments performed on different days. (A) *p<0.05, CaSki vs C33A, 4 and 6 Gy (B) *Control vs Cis SiHa, C33A and ME-180 p<0.05 (C) *CaSki vs all cell lines p<0.05 (D) p<0.05, CaSki vs all others, *p<0.003 CaSki vs SiHa, p<0.0002 CaSki vs C33A and p<0.01 CaSki vs ME-180 (E) *Cis vs Cis+2-DG p<0.0001 for all the cell lines (F) *2-DG or 2Gy vs 2-DG+2-Gy, p<0.05 for ME-180 and C33A and p<0.01 for CaSki and SiHa cell lines.
To test for the sensitivity to the glycolytic inhibitor 2-deoxyglucose (2-DG) a CFA was performed 24hrs after treatment with 20mM 2-DG (Figure 1C). Radio- and chemo-resistant CaSki cells were exquisitely sensitive to 2-DG monotherapy compared to the other cell lines. To further explore whether 2-DG sensitivity correlated with glucose uptake, we performed an FDG uptake assay (Figure 1D). Consistent with the increased 2DG sensitivity, CaSki had the highest FDG uptake in vitro followed by ME-180, C33A and SiHa.
We next tested whether pretreatment with 2-DG would enhance the effects of cisplatin and radiation treatment (Figure 1E–F). 2-DG enhanced the effects of cisplatin only minimally (Figure 1E). In contrast, 2-DG significantly sensitized all cell lines tested to radiation, with the most dramatic effects observed in CaSki (p< 0.0001) after treatment with 2-DG and 2 Gy radiation (Figure 1F).
2-DG induced cell death is enhanced by BSO and AUR through disruption of GSH and Trx metabolism
BSO and AUR have been shown to inhibit GSH synthesis and inhibit TR activity. [7, 23, 24] To characterize baseline levels of thiol redox pathway intermediates, intracellular GSH and GSSG levels were quantified in all cell lines tested (Figure 2A–D). The total GSH content was high in CaSki and SiHa cells, while GSSG and % GSSG (% of total cellular GSH present in the form of GSSG) were found to be significantly higher in CaSki, consistent with a higher baseline level of oxidative stress in that cell line. Interestingly, thioredoxin reductase (TR) activity was significantly higher in SiHa versus all other cells tested (Figure 2D).
Figure 2. Effects of combined treatment with 2-DG, BSO and AUR.
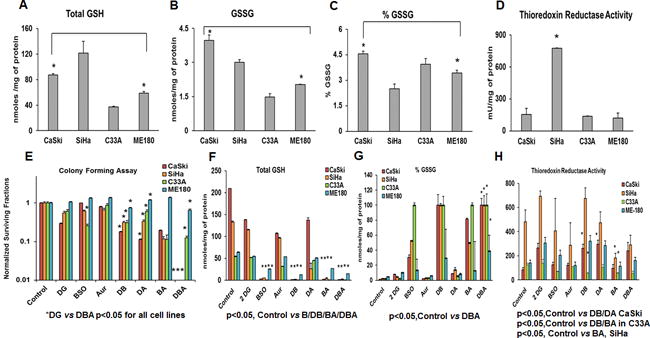
(A–D) Baseline levels of GSH and TR activity of CaSki, SiHa, C33A and ME-180 were determined as described in Materials and methods section. (E) Cells were seeded on to 48 well plates and treated with 20mM 2-DG and 1mM BSO for 48h and 100nM AUR for 24h. Clonogenic survival data was normalized to untreated controls. (F–H) Cells were harvested post treatment and GSH levels, %GSSG and TR levels were quantified Statistical Analysis: Error bars represent ± SD of N=3 experiments performed on different days. (A–C)*CaSki vs ME-180 p<0.05 for Total GSH, GSSG and % GSSG (D) *p<0.05, SiHa vs all other cell lines (E) *DG vs DBA p<0.05 for all cell lines, p< 0.05 DG vs DB for all cell lines, p < 0.05 DG vs DA for CaSki, SiHa and ME180. (F) *p<0.05 Control vs B, DB, BA, and DBA for all cell lines (G) p<0.05, Control vs DBA for all cell lines (H) p<0.05 SiHa Control vs BA, CaSki Control vs DB and DA, Control vs DB and BA in C33A.
To examine if GSH depletion and TR inhibition would enhance the toxicity induced by 2-DG treatment, cells were treated with 20 mM 2-DG and 1 mM BSO for 48h with 100 nM AUR added for the last 24 h. CaSki and SiHa cells were significantly more sensitive to 2-DG+BSO+AUR (DBA) treatment than ME-180 and C33A, with significant decreases in clonogenic survival compared to treatment with 2-DG alone (Figure 2E). In addition, C33A was more sensitive to BSO monotherapy compared to other cell lines (Figure 2E).
To examine whether BSO and AUR treatment altered intracellular redox pools, levels of GSH, %GSSG and TR activity were measured before and after treating cells with 2-DG+/−BSO+/−AUR (Figure 2F–H). BSO monotherapy was effective in reducing total GSH content in all cell lines tested with residual GSH detected after BSO treatment in ME-180 cells (Figure 2F). 2-DG+BSO (DB), BSO+ AUR (BA) and 2-DG+BSO+AUR (DBA) significantly decreased total GSH content in all cell lines tested (Figure 2F). Percent (%) GSSG (% of total cellular GSH present in the form of GSSG) levels were significantly increased after BSO containing combination treatments in all cell lines tested, with the most significant increases in %GSSG seen in CaSki, SiHa and C33A cells. (Figure 2G). Increases in %GSSG in response to treatment were more modest in ME-180, consistent with post-treatment residual GSH pools in that cell line. We next tested whether baseline levels of GSH, %GSSG and TR activity correlated with response to DBA treatment. There was a linear correlation between total GSH levels and sensitivity to DBA (Pearson’s r 0.963) while % GSSG levels and sensitivity to DBA showed a linear negative correlation (Pearson’s r-0.982). These results show that BSO was having the desired effect of depleting total GSH in all cervical carcinoma cells tested, with maximum increase in intracellular oxidative stress (estimated by increase in % GSSG) achieved with DB, BA and DBA combinations. Overall, the simultaneous manipulations of GSH and TR using BSO and AUR combined with 2DG were most effective at increasing metabolic oxidative stress in cervical cancer cell lines.
Consistent with these findings, the TR activity was significantly reduced by DBA treatment only in C33A cells (Figure 2H). TR activity was decreased by BA treatment but not completely eliminated in SiHa cells (Figure 2H). In contrast, DA and DB combination treatment increased TR activity in CaSki, the most sensitive cell line to 2-DG monotherapy. There was no significant difference in TR activity in ME-180 cells in response to BSO, AUR, DA and DBA treatments (Figure 2H). To test the specificity of Auranofin in target inhibition we combined 2-DG along with TR1 and TR1 knockdown via siRNAs. A colony forming assay was performed after CaSki (Supplementary Figure 1A–B) and SiHa (Supplementary Figure 1C–D) were transfected with TR1, TR2 and TR1+TR2 for 48h followed by 2-DG treatment for 24hrs. TR1, TR2 and TR1+TR2 knockdown decreased clonogenic survival in both CaSki (p<0.01 Control vs TR1, p<0.01 Control vs TR2, p<0.01 Control vs TR1+TR2) and SiHa (p<0.001 Control vs TR1/2, p<0.01 Control vs TR1+TR2) (Supplementary Figure 1A–D).
Treatment with 2DG, BSO and AUR increases intracellular reactive oxygen species as determined by changes in oxidation sensitive dye oxidation
To determine the potential role of pro-oxidants in DBA induced cell death, we quantified intracellular oxidation before and after treatment using 2′, 7′ –dichlorofluorescin diacetate (DCFDA) oxidation and dihydroethidium (DHE) as previously described. [10] There was significantly higher DCFDA oxidation in SiHa and C33A after 2-DG+BSO+AUR treatment (Figure 3A). When the cells were stained with CDCF, the oxidation insensitive analogue of CDCFH2, there was no significant difference in any of the cells confirming that the differences observed were indeed due to changes in levels of dye oxidation and not due to changes in cell size, probe influx, probe efflux or changes in ester cleavage (Figure 3B). Only SiHa and ME-180 cells displayed significant increases in DHE oxidation after DBA treatment (Figure 3C). These results suggest that in cervical cancer cells, treatment with DBA induces accumulation of pro-oxidants, presumably superoxide and hydroperoxides.
Figure 3. Analysis of pro-oxidant and superoxide levels before and after 2-DG, BSO and AUR treatment.
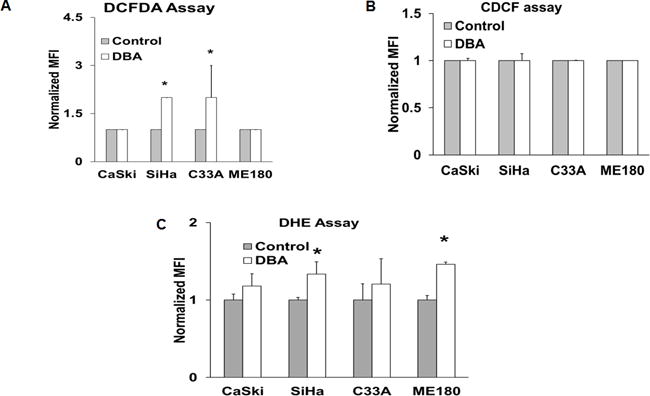
(A–C) Cells were grown in 6 well plates and labelled with 10 μM DHE, 10 μg/ml CDCFH2 or 10 μg/ml CDCF and analyzed by flow cytometry. Values are expressed as the MFI. Statistical Analysis: Error bars represent ±SD of three separate experiments. *Control vs treated SiHa and C33A, p<0.05, N=3.
NAC, a thiol antioxidant, was used to determine whether 2-DG+BSO+AUR induced cytotoxicity was related to thiol oxidation and depletion of intracellular thiol pools. NAC significantly inhibited the decrease in cell viability seen in SiHa cells following exposure to DBA whereas no inhibition of toxicity was seen in similarly treated CaSki cells (Supplementary Figure 2 A–B). NAC significantly inhibited BSO+AUR (BA) treatment-related toxicity in both CaSki and SiHa cells. Furthermore, the addition of 2-DG to BSO+AUR increases cytotoxicity in a fashion that cannot be reversed by simply restoring intracellular reduced thiol pools with NAC. Interestingly, the addition of NAC also did not inhibit 2-DG associated toxicity in CaSki cells, the most sensitive cell line to 2-DG monotherapy. We also performed clonogenic cell survival assays after adenoviral mediated over-expression of the anti-oxidant enzyme catalase in presence or absence of 2-DG+BSO+AUR (Supplementary Figure 3 A–B). Catalase expression in mκunits/milligram in the order of Ad empty and Ad-catalase is as follows (SiHa 8.0/316) CaSki (4/76) C33A (6.0/116). Over expression of Catalase partially restored survival in response to DG, BSO, DB and DBA treatments in SiHa cells, whereas effects of catalase overexpression were not significant in similarly treated CaSki cells (Supplementary Figure 3). The varying ability of catalase overexpression to inhibit toxicity suggests cell line specific differences in the relative abundance of hydroperoxides versus superoxide (Figure 3A–C).
2-DG+BSO+AUR (DBA) treatment decreases tricarboxylic acid (TCA) cycle activity and lactate production in DBA-sensitive cell lines
To determine effects of DBA treatment on glucose metabolic pathways, we performed metabolomic analysis of C33A, CaSki, ME-180, and SiHa cells grown in 10 mM uniformly labeled 13C-glucose. Each untreated cell line showed similar rates of glucose entry into the tricarboxylic acid (TCA) cycle, as represented by citrate labeling (Figure 4A). DBA treatment resulted in significant (p < 0.05) decrease in citrate labeling in CaSki, ME180, and SiHa cells (Figure 4A). Upon DBA treatment, SiHa showed the lowest labeling in citrate (46%), followed by CaSki (57%), ME-180 (66%), and C33A (69%); the differences between SiHa and ME-180 or C33A were significant (p < 0.05) in pairwise comparisons. This ordering of citrate labeling parallels that of cell survival after DBA treatment, with SiHa and CaSki being exquisitely sensitive to the combined drugs and ME-180 and C33A being relatively resistant (Figure 2A). Other evidence of decreased TCA cycle activity in DBA-sensitive cells after DBA treatment can be seen in the labeling of glutamate, which is derived from the TCA cycle intermediate 2-oxoglutarate (Figure. 4B; labeled fraction of glutamate in SiHa and CaSki is significantly lower than in either C33A or ME180), as well as the labeling pattern of the hexosamine biosynthesis pathway product UDP-N-acetylglucosamine (Figure 4C), whose M+7, M+8, and M+13 isotopologues reflect the addition of a labeled cytosolic acetyl-CoA derived from labeled mitochondrial citrate to the M+5 (labeled ribose moiety), M+6 (labeled glucosamine moiety), and M+11 (both ribose and glucosamine moieties labeled) isotopologues, respectively. The fraction of UDP-GlcNAc in the M+7, M+8, and M+13 isotopologue forms decreased upon DBA treatment in all cell lines, but the decrease was greatest in SiHa and CaSki, indicating decreased availability of labeled acetyl-CoA and thus also decreased TCA cycle activity. DBA treatment also resulted in decreased ratio of the +10 to +5 isotopologue in NAD representing decreased flux from glucose to ribose of NAD in the sensitive cell lines CaSki and SiHa (Figure 4D) and decreased lactate production (Figure 4E).
Figure 4. Analysis of glucose carbon utilization before and after 2-DG, BSO and AUR treatment using uniformly labelled 13C glucose.
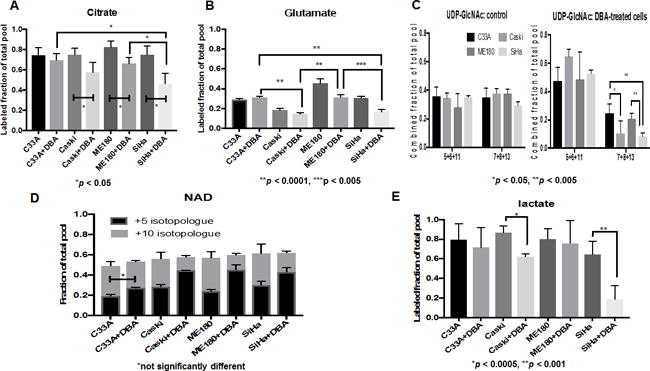
(A–E) Samples were prepared as described in the methods section. A, B, E: Labelled fraction (all isotopologues containing one or more labeled carbons) of the total pool of each metabolite is plotted. C, D: Fraction of the specified isotopologue or combination of isotopologues is plotted. All comparisons marked are pairwise t-tests.
2-DG+BSO+AUR induced oxidative stress results in autophagic cell death via AMPK activation
To determine whether DBA treatment stimulated nutrient stress induced cell signaling pathways, a series of Western blots were performed. DBA treatment induced AMPK and JNK activation as evidenced by increases in pAMPK and pJNK expression in DBA-treated CaSki and SiHa cells (Figure 5A). This effect was not dependent on expression of LKB1 as SiHa cells are LKB1 negative (Figure 5A). DBA treatment was also found to promote accumulation of pro-cell death BH3 only protein Bim (Figure 5A). DBA treatment induced autophagy in CaSki cells but not SiHa cells as evidenced by MAPLC3 cleavage (Figure 5A). CaSki cells have higher Myc and Ser62 Myc levels at baseline and after DBA treatment (Figure 5B).
Figure 5. Effects of 2-DG, BSO and AUR on cell signaling via the AMPK and autophagy pathways.
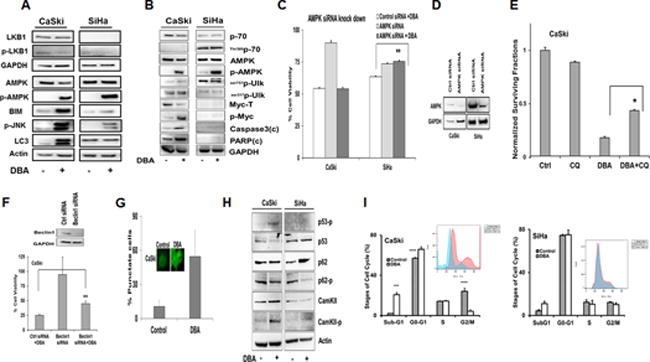
(A) Cells were treated with 20mM 2-DG, 1mM BSO and 100nM AUR for 24h and lysates were prepared for western blotting as described. (B) Cells were treated with 20mM 2-DG, 1mM BSO and 100nM AUR for 24h and lysates were prepared for western blotting as described. (C-D) CaSki and SiHa cells were transfected with scrambled and AMPK siRNA and protein levels were determined by Western blot. (E) CaSki cells were treated with 10mM 2-DG, 500μM BSO and 50nM AUR for 24h with or without CQ (60μM) and CFA was performed. (F) CaSki cells were transfected with control and Beclin1 siRNA and after 48h, treated with 20mM 2-DG, 1mM BSO and 100nM AUR for 24h and cell viability was determined by Alamar Blue and Western Blot of Beclin 1. (G) Immunofluorescence was performed on CaSki cells after treatment with 10mM 2-DG, 500nM BSO and 50nM AUR for 18h in a chamber slide. Total number of stained cells and punctate cells were counted in 6 different fields and plotted. (H) CaSki and SiHa cells were treated with 20mM 2-DG, 1mM BSO and 100nM AUR for 24h and Western blotting was performed. (I) CaSki and SiHa cells were treated with 10mM 2-DG, 500μM BSO and 50nM AUR for 24h and stained with propidium iodide and analyzed by flow cytometry. Statistical Analysis: (C) * p <0.05 Control siRNA + DBA vs AMPK siRNA + DBA SiHa. (E) *DBA vs DBA+CQ, p<0.05, N=3 (F) **Control siRNA vs Beclin 1 siRNA, P<0.05, N=3 (I) *p<0.05 Control vs DBA *p<0.01
Knockdown via siRNA was used to test whether AMPK signaling was required for DBA-induced cell death in CaSki and SiHa cells (Figure 5C–D). Interestingly, AMPK knockdown rescued DBA-induced cell death in LKB1 negative SiHa cells (Figure 5C), but not in LKB1 intact CaSki cells. To test whether cell death induced by DBA treatment was due to autophagic cell death in CaSki cells, we used an orthogonal strategy with drugs and siRNA knockdown. Chloroquine, an autophagy inhibitor, significantly inhibited DBA induced cell death in CaSki confirmed by CFA and cell viability (Figure 5E and Supplementary Figure 3C). Beclin1 knock down also rescued CaSki cells significantly from DBA induced cell death, (DBA alone =27%, siRNABeclin1+DBA=45%, p<0.0003) (Figure 5F). The number of punctate CaSki cells (cleaved LC3) after DBA treatment was 50% compared to control cells (Figure 5G), and this punctate pattern was not observed in SiHa cells as evidenced by no cleaved fragment of LC3 (Figure 5A). DBA treatment activated AMPK autophagic cell pathway in CaSki as seen by ser317Ulk phosphorylation in contrast to the ser757Ulk in SiHa (Figure 5B). AMPK activates p62 which is a marker for autophagic cell death.[25] DBA treated CaSki cells displayed activation of p62 but similar results were not observed in SiHa (Figure 5H). DBA treatment induced PARP and caspase 3 cleavages in CaSki but not in SiHa cells (Figure 5B). We observed DBA treatment induced p53 phosphorylation (Figure 5H) resulting in G0-G1 cell cycle arrest in CaSki cells and cell death as evidenced by higher sub-G1 fraction (Figure 5I). Taken together, these results suggest that oxidative stress in DBA-sensitive CaSki cells results in AMPK, p53 and JNK activation, BIM accumulation, and activation of the caspase and PARP dependent autophagic form of cell death. In contrast, in SiHa cells this effect is LKB1 independent, AMPK dependent, non-autophagic form of cell death.
2-DG+BSO+AUR delays SiHa and CaSki tumor growth in vivo
To determine whether our in vitro observations with 2-DG, BSO and AUR treatment could be verified in in vivo, mice with CaSki xenografts were treated with 2-DG, BSO+AUR and 2-DG+BSO+AUR for 35 days three times a week. Drug treatment did not result in weight loss or behavioral changes during the period of study. CaSki tumor growth delay in response to 2-DG only was observed after 28 days (Figure 6A). Both BA and DBA treatments were effective in reducing CaSki tumor volumes (Figure 6A). To assess whether 2-DG, BSO and AUR combination altered GSH/GSSG levels in vivo, tumors were harvested at the end of the experiment and GSH/GSSG levels, %GSSG and TR activity were quantified. Similar to our in vitro results, % GSSG increased in response to 2-DG, and BA treatments (Figure 6B). 2-DG and BA treatments decreased GSH levels and increased TR activity in CaSki tumors (Figure 6C–D). In order to test the radiosensitizing potential of our new drug strategy, CaSki cells were treated with 2-DG, BSO+AUR, and 2-DG+BSO+AUR and after 24 h were irradiated using single fraction radiation dose of 2 Gy. 2-DG, BA and DBA radio-sensitized CaSki cells as evidenced by reduced surviving fraction in Figure 6E.
Figure 6. Effects 2-DG, BSO and AUR treatment on CaSki and xenografts.
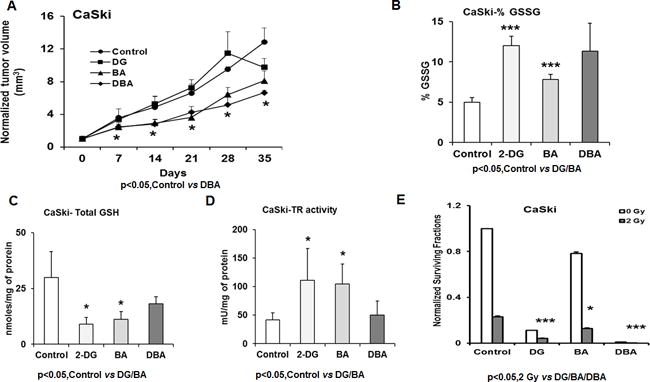
(A) CaSki xenografts were treated with 2-DG, BSO and AUR as described in materials and methods. The normalized average tumor volumes were plotted against the number of days as line graph. (B–D) The tumors were harvested from each group and GSH, GSSG and TR assays performed as described in materials and methods section. (E) CaSki cells were treated with 10mM 2-DG, 500μM BSO and 50nM AUR for 24h with or without 2 Gy radiation and CFA was performed. Statistical Analysis: (A)*Control vs DBA group p<0.05 (B) Control vs DG, BA and DBA, p<0.05 (C–D) Control vs DG and BA, p< 0.05 (E) *p, 0.05 2 Gy vs 2 Gy+2-DG/BA/DBA.
2-DG+BSO+AUR radio-sensitizes SiHa tumors at clinically relevant radiation doses in vitro and in vivo
In order to test whether targeting glycolysis and intracellular redox metabolism with DBA could function as a radio-sensitizer, we performed colony forming assays after 2 Gy radiation +/− DBA with the SiHa cell line (Figure 7A). Radio-resistant SiHa cells were sensitized to 2 Gy by the addition of DBA. To determine whether radio-sensitization could be observed in vivo, we treated SiHa xenografts with DBA for two weeks with concurrent single fraction dose of 2 and 4 Gy tumor-directed irradiation using the SARRP system. (Figure 7 B–C). We observed that DBA treatment significantly reduced tumor growth after single fraction doses of both 2 and 4 Gy (*DBA vs 2Gy + DBA, p<0.01, DBA vs 4Gy + DBA, p<0.0001). The combination of 4Gy and DBA treatment was most effective at limiting SiHa tumor growth in vivo (Figure 7 C–D).
Figure 7. 2-DG, BSO and AUR enhances radiation efficacy in vitro and in vivo.
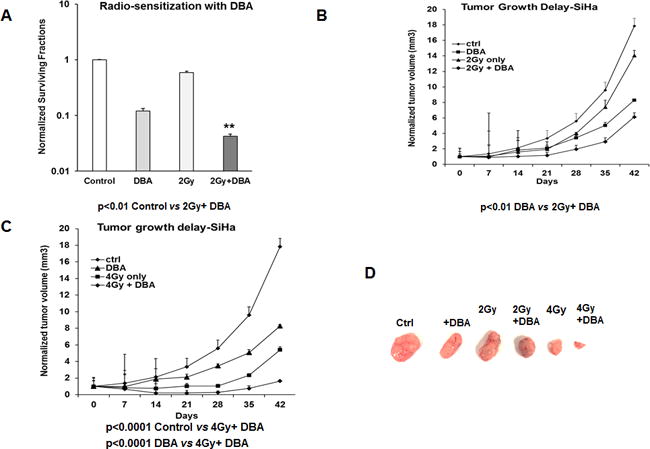
(A) SiHa cells were seeded on to 48 well plates and treated with DBA then irradiated with 2 Gy as described in the Methods. Clonogenic survival data was normalized to untreated controls. (B–D) SiHa xenografts were generated and treated with 2-DG+BSO+AUR with and without single fraction radiation doses of 2 and 4 Gy. The average tumor volumes were plotted against the number of days as line graphs Representative tumor sizes from each group at the end of the experiment. Statistical Analysis: (A) *DBA vs DBA+2Gy, p<0.05. (B–C) *DBA vs 2Gy+DBA, p<0.01, DBA vs 4Gy+DBA, p<0.0001.
Discussion
In this preclinical study we tested whether inhibition of glycolysis, with or without simultaneous inhibition of intracellular redox metabolism, was an effective treatment strategy for highly glycolytic and radio-resistant cervical cancers. 2-DG was used to inhibit glycolysis, BSO to target GSH synthesis and AUR to target thioredoxin reductase. BSO and AUR have been studied in other cancer types as a means to inhibit intracellular redox metabolism [26], [7], [12]. Our results show that the cisplatin-resistant and radio-resistant cervical cell line CaSki is highly sensitive to inhibition of glycolysis with 2-DG monotherapy. Addition of BSO and AUR sensitized SiHa cells to 2-DG treatment indicating that the redox potential generated by glycolysis was dampened by inhibition of glutathione and thioredoxin pathways. [27] Using detailed analysis of glutathione and thioredoxin activity, we were able to demonstrate direct inhibition of the redox pathways by BSO and AUR treatment. Due to the high baseline TR activity, SiHa survived better after 2-DG+AUR and BSO+AUR only treatment groups.
Although the in vitro evidence for our strategy is compelling, we wanted to test whether our results could be validated in vivo. In support of our in vitro observations, we found that CaSki xenografts responded well to drug treatment alone with metabolic inhibitors 2DG, BSO+AUR, and 2DG+BSO+AUR. Tumors samples collected at the end of the experiment demonstrated inhibition of glutathione and thioredoxin pathways by BSO and AUR respectively in CaSki xenografts. The drug strategy was well tolerated in animals, supporting the possibility of transitioning this approach to the clinic. We wanted to test whether DBA treatment could enhance radio-sensitivity and potentially be integrated or replace cisplatin treatment as part of the standard of care. Importantly, we were able to demonstrate radio-sensitization of SiHa with DBA treatment both in vitro and in vivo. In vivo, DBA treatment resulted in significant tumor growth delay at clinically relevant radiation doses, suggesting that this drug strategy could be feasible and effective at radiation doses currently used in the clinic. 2-DG and BSO have already been used in humans to treat other malignancies [28],[29] and AUR is an FDA approved drug [30], [31]. Our preclinical results form a strong rationale for testing 2DG+BSO+AUR as an alternative drug strategy for highly glycolytic, radio-resistant cervical tumors.
Mechanistically, we wanted to determine whether DBA treatment was stimulating cell signaling pathways known to sense nutrient stress. Oxidative stress is known to activate AMPK in cancer cells [32], and we observed high levels of activated AMPK activation after DBA treatment in DBA-sensitive cells. [33] We also observed DBA induced phosphorylation of JNK in DBA-sensitive cell lines, and these data support the finding that JNK induces AMPK activation under conditions of glucose deprivation. [34] This was true in both LKB1 null [35] and intact cell lines [36] suggesting that AMPK may be activated by DBA treatment via LKB1 independent pathways.[37–39] Concannon et al reported that AMPK activates BH3-only protein Bim during stress induced cell death [40], and we observed Bim accumulation in DBA-sensitive cells. Intriguingly, activation of AMPK in DBA sensitive cell lines was not associated with an increase in glucose-derived mitochondrial energy production, as entry of glucose into the TCA cycle was decreased in CaSki and SiHa upon 2-DG+BSO+AUR treatment (Figure 4A). This suggests that AMPK modulates these cells’ response to drugs through a PGC-(peroxisome proliferator-activated receptor gamma coactivator) 1α-independent mechanism. [41, 42] DBA induced LC3 cleavage and p62 phosphorylation (Figure 5A and 5H) in CaSki cells suggesting cell death may be occurring via autophagic cell death. [43] Chloroquine, a known inhibitor of autophagy, and knockdown of pro-autophagy gene BECLIN1 partially rescued CaSki cells from 2-DG+BSO+AUR induced cell death. [44]
Sustained high level AMPK activation was observed in both CaSki and SiHa in response to DBA treatment, but with different downstream effects. CaSki is MYC transformed and SiHa is not (Figure 5B). Myc is known to promote lipid, nucleotide and protein synthesis by utilizing the citric acid cycle to serve biosynthetic processes, which simultaneously leads to ATP production and the activation of cellular energy sensing protein, AMP-activated protein kinase (AMPK). Cells with normal growth control can stop cell proliferation to replenish ATP reservoirs whereas MYC expression prevents a similar break by blocking the cell cycle exit. The relentless cell cycle activation, accompanied by sustained metabolic stress and AMPK activity, switches the energy-saving AMPK to pro-apoptotic AMPK. Previously Jones et al have demonstrated that glucose deprivation induced AMPK activation directly phosphorylates N-terminal Ser15 of p53 leading to initiation of G0-G1 cell cycle arrest. [45] Our study supports this finding that AMPK leads to Ser15 p53 phosphorylation leading to a G0-G1 cell cycle arrest in CaSki cells (Figure 5H–I). The persistent AMPK activity in CaSki ultimately results in caspase and PARP dependent autophagic cell death (Figure 5A–I). MYC transformed CaSki cells experience energy crisis due to persistent cell cycle activation which results in activation of energy sensor AMPK ultimately leading to autophagic cell death.[46] This could be one of the reasons that CaSki was rescued by autophagy inhibitors and not by AMPK knock down. In contrast in SiHa cells which are not MYC transformed, DBA treatment results in a caspase/PARP and p53 independent non-autophagic cell death.
Here we demonstrate that four different cervical cancer cell lines with varying mutation states in the PI3K/Akt/PTEN pathway all exhibit similar rates of glucose flux into lactate (Figure 4D–E). However, treatment with DBA differentially decreases this flux in DBA-sensitive cells (SiHa and CaSki). Furthermore, these DBA-sensitive cells are unable to process glucose through the TCA cycle (Figure 4A), with subsequent downregulation of pathways that are dependent on TCA cycle products such as glutamate and UDP-GlcNAc synthesis (Figure 4B–C). Overall, the metabolic effects of DBA treatment in DBA-sensitive MYC transformed cells is one of activation of AMPK and autophagy which may be a consequence of their decreased glucose metabolism and exhaustion of ATP reservoirs.
Supplementary Material
Significance.
This study suggests a simple metabolic approach to strike at an apparent Achilles’ heel in highly glycolytic, radio-resistant forms of cervical cancers, possibly with broader applications in cancer therapy.
Acknowledgments
This work was supported by NIH R01CA181745 (to JK Schwarz), Resident Research Seed Grant 531448 from the American Society for Radiation Oncology (to JM Floberg), Research Medical Student Grants from the Radiological Society of North America (RSNA) RMS1612 (to X Huang) and RMS1408 (to AE Elhammali), NIH R01ES022181 and R21CA191097 (to GJ Patti). The Small Animal Radiation Research Platform was purchased with the assistance of NIH S10 OD020136 (to D Hallahan). The work at the University of Iowa done by the Radiation and Free Radical Research Core Lab was partially supported by NIH R01 CA182804 and NIH P30 CA086862 (to DR Spitz). We would like to thank Michael Zahner and Cedric Mpoy for technical assistance.
Abbreviations
- 2-DG
2-deoxyglucose
- ATCC
American Type Culture Collection
- AUR
Auranofin
- BA
BSO (L-buthionine-sulfoximine)+AUR (Auranofin)
- BSO
L-buthionine-sulfoximine
- CAT
catalase
- CFA
colony forming assay
- CQ
chloroquine
- DB
2-DG (2-deoxyglucose)+BSO (L-buthionine-sulfoximine)
- DBA
2-DG (2-deoxyglucose)+BSO (L-buthionine-sulfoximine)+AUR(Auranofin)
- DCFDA
2′, 7′ –dichlorofluorescin diacetate
- DHE
dihydroethidine
- DMSO
dimethyl sulfoxide
- FDG
18F-fluoro-deoxy-glucose
- GPX
glutathione peroxidase
- GR
glutathione reductase
- GSH
glutathione
- GSSG
glutathione disulfide
- LC/MS
liquid chromatography/mass spectrometry
- MFI
mean fluorescence intensity
- NAC
N-acetylcysteine
- PET
positron emission tomography
- Prx
peroxiredoxins
- ROS
reactive oxygen species
- TCA
tricarboxylic acid
- TR
thioredoxin reductase
- Trx
thioredoxin
Footnotes
Conflict of Interest Statement:
RR, XH, JMF, AEE, MM, DRS and JKS report no conflicts of interest related to this work. GJP is a scientific advisory board member for Cambridge Isotope Laboratories and a recipient of the Agilent Early Career Award.
References
- 1.Kidd EA, et al. The standardized uptake value for F-18 fluorodeoxyglucose is a sensitive predictive biomarker for cervical cancer treatment response and survival. Cancer. 2007;110(8):1738–44. doi: 10.1002/cncr.22974. [DOI] [PubMed] [Google Scholar]
- 2.Schwarz JK, et al. Association of posttherapy positron emission tomography with tumor response and survival in cervical carcinoma. JAMA. 2007;298(19):2289–95. doi: 10.1001/jama.298.19.2289. [DOI] [PubMed] [Google Scholar]
- 3.Schwarz JK, et al. Pathway-specific analysis of gene expression data identifies the PI3K/Akt pathway as a novel therapeutic target in cervical cancer. Clin Cancer Res. 2012;18(5):1464–71. doi: 10.1158/1078-0432.CCR-11-2485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rashmi R, et al. AKT inhibitors promote cell death in cervical cancer through disruption of mTOR signaling and glucose uptake. PLoS One. 2014;9(4):e92948. doi: 10.1371/journal.pone.0092948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sen T, et al. OGDHL is a modifier of AKT-dependent signaling and NF-kappaB function. PLoS One. 2012;7(11):e48770. doi: 10.1371/journal.pone.0048770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Spitz DR, et al. Glucose deprivation-induced oxidative stress in human tumor cells. A fundamental defect in metabolism? Ann N Y Acad Sci. 2000;899:349–62. doi: 10.1111/j.1749-6632.2000.tb06199.x. [DOI] [PubMed] [Google Scholar]
- 7.Fath MA, et al. Enhancement of carboplatin-mediated lung cancer cell killing by simultaneous disruption of glutathione and thioredoxin metabolism. Clin Cancer Res. 2011;17(19):6206–17. doi: 10.1158/1078-0432.CCR-11-0736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li L, et al. Combined inhibition of glycolysis, the pentose cycle, and thioredoxin metabolism selectively increases cytotoxicity and oxidative stress in human breast and prostate cancer. Redox Biol. 2015;4:127–35. doi: 10.1016/j.redox.2014.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhou D, Shao L, Spitz DR. Reactive oxygen species in normal and tumor stem cells. Adv Cancer Res. 2014;122:1–67. doi: 10.1016/B978-0-12-420117-0.00001-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Aykin-Burns N, et al. Increased levels of superoxide and H2O2 mediate the differential susceptibility of cancer cells versus normal cells to glucose deprivation. Biochem J. 2009;418(1):29–37. doi: 10.1042/BJ20081258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rodman SN, et al. Enhancement of Radiation Response in Breast Cancer Stem Cells by Inhibition of Thioredoxin- and Glutathione-Dependent Metabolism. Radiat Res. 2016;186(4):385–395. doi: 10.1667/RR14463.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Scarbrough PM, et al. Simultaneous inhibition of glutathione- and thioredoxin-dependent metabolism is necessary to potentiate 17AAG-induced cancer cell killing via oxidative stress. Free Radic Biol Med. 2012;52(2):436–43. doi: 10.1016/j.freeradbiomed.2011.10.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Song JJ, Lee YJ. Differential role of glutaredoxin and thioredoxin in metabolic oxidative stress-induced activation of apoptosis signal-regulating kinase 1. Biochem J. 2003;373(Pt 3):845–53. doi: 10.1042/BJ20030275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Simons AL, et al. Glucose deprivation-induced metabolic oxidative stress and cancer therapy. J Cancer Res Ther. 2009;5(Suppl 1):S2–6. doi: 10.4103/0973-1482.55133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ahmad IM, et al. Mitochondrial O2*- and H2O2 mediate glucose deprivation-induced stress in human cancer cells. J Biol Chem. 2005;280(6):4254–63. doi: 10.1074/jbc.M411662200. [DOI] [PubMed] [Google Scholar]
- 16.Weindruch R, et al. Caloric restriction mimetics: metabolic interventions. J Gerontol A Biol Sci Med Sci. 2001;56(Spec No 1):20–33. doi: 10.1093/gerona/56.suppl_1.20. [DOI] [PubMed] [Google Scholar]
- 17.Sandulache VC, et al. Glucose, not glutamine, is the dominant energy source required for proliferation and survival of head and neck squamous carcinoma cells. Cancer. 2011;117(13):2926–38. doi: 10.1002/cncr.25868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sobhakumari A, et al. Susceptibility of human head and neck cancer cells to combined inhibition of glutathione and thioredoxin metabolism. PLoS One. 2012;7(10):e48175. doi: 10.1371/journal.pone.0048175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Spitz DR, et al. Oxygen toxicity in control and H2O2-resistant Chinese hamster fibroblast cell lines. Arch Biochem Biophys. 1990;279(2):249–60. doi: 10.1016/0003-9861(90)90489-l. [DOI] [PubMed] [Google Scholar]
- 20.Huang X, et al. X13CMS: global tracking of isotopic labels in untargeted metabolomics. Anal Chem. 2014;86(3):1632–9. doi: 10.1021/ac403384n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chambers MC, et al. A cross-platform toolkit for mass spectrometry and proteomics. Nat Biotechnol. 2012;30(10):918–20. doi: 10.1038/nbt.2377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Smith CA, et al. XCMS: processing mass spectrometry data for metabolite profiling using nonlinear peak alignment, matching, and identification. Anal Chem. 2006;78(3):779–87. doi: 10.1021/ac051437y. [DOI] [PubMed] [Google Scholar]
- 23.Fruehauf JP, et al. Melanin content and downregulation of glutathione S-transferase contribute to the action of L-buthionine-S-sulfoximine on human melanoma. Chem Biol Interact. 1998;111–112:277–305. doi: 10.1016/s0009-2797(97)00167-1. [DOI] [PubMed] [Google Scholar]
- 24.Gromer S, et al. Human placenta thioredoxin reductase. Isolation of the selenoenzyme, steady state kinetics, and inhibition by therapeutic gold compounds. J Biol Chem. 1998;273(32):20096–101. doi: 10.1074/jbc.273.32.20096. [DOI] [PubMed] [Google Scholar]
- 25.Ha S, et al. Phosphorylation of p62 by AMP-activated protein kinase mediates autophagic cell death in adult hippocampal neural stem cells. J Biol Chem. 2017;292(33):13795–13808. doi: 10.1074/jbc.M117.780874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kiebala M, et al. Dual targeting of the thioredoxin and glutathione antioxidant systems in malignant B cells: a novel synergistic therapeutic approach. Exp Hematol. 2015;43(2):89–99. doi: 10.1016/j.exphem.2014.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Anastasiou D, et al. Inhibition of pyruvate kinase M2 by reactive oxygen species contributes to cellular antioxidant responses. Science. 2011;334(6060):1278–83. doi: 10.1126/science.1211485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.O’Dwyer PJ, et al. Phase I trial of buthionine sulfoximine in combination with melphalan in patients with cancer. J Clin Oncol. 1996;14(1):249–56. doi: 10.1200/JCO.1996.14.1.249. [DOI] [PubMed] [Google Scholar]
- 29.Stein M, et al. Targeting tumor metabolism with 2-deoxyglucose in patients with castrate-resistant prostate cancer and advanced malignancies. Prostate. 2010;70(13):1388–94. doi: 10.1002/pros.21172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Messori L, Marcon G. Gold complexes in the treatment of rheumatoid arthritis. Met Ions Biol Syst. 2004;41:279–304. [PubMed] [Google Scholar]
- 31.Debnath A, et al. A high-throughput drug screen for Entamoeba histolytica identifies a new lead and target. Nat Med. 2012;18(6):956–60. doi: 10.1038/nm.2758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jeon SM, Chandel NS, Hay N. AMPK regulates NADPH homeostasis to promote tumour cell survival during energy stress. Nature. 2012;485(7400):661–5. doi: 10.1038/nature11066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kuhajda FP. AMP-activated protein kinase and human cancer: cancer metabolism revisited. Int J Obes (Lond) 2008;32(Suppl 4):S36–41. doi: 10.1038/ijo.2008.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yun H, et al. AMP kinase signaling determines whether c-Jun N-terminal kinase promotes survival or apoptosis during glucose deprivation. Carcinogenesis. 2009;30(3):529–37. doi: 10.1093/carcin/bgn259. [DOI] [PubMed] [Google Scholar]
- 35.Mack HI, Munger K. The LKB1 tumor suppressor differentially affects anchorage independent growth of HPV positive cervical cancer cell lines. Virology. 2013;446(1–2):9–16. doi: 10.1016/j.virol.2013.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wingo SN, et al. Somatic LKB1 mutations promote cervical cancer progression. PLoS One. 2009;4(4):e5137. doi: 10.1371/journal.pone.0005137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Woods A, et al. Ca2+/calmodulin-dependent protein kinase kinase-beta acts upstream of AMP-activated protein kinase in mammalian cells. Cell Metab. 2005;2(1):21–33. doi: 10.1016/j.cmet.2005.06.005. [DOI] [PubMed] [Google Scholar]
- 38.Hawley SA, et al. Calmodulin-dependent protein kinase kinase-beta is an alternative upstream kinase for AMP-activated protein kinase. Cell Metab. 2005;2(1):9–19. doi: 10.1016/j.cmet.2005.05.009. [DOI] [PubMed] [Google Scholar]
- 39.Hoyer-Hansen M, et al. Control of macroautophagy by calcium, calmodulin-dependent kinase kinase-beta, and Bcl-2. Mol Cell. 2007;25(2):193–205. doi: 10.1016/j.molcel.2006.12.009. [DOI] [PubMed] [Google Scholar]
- 40.Concannon CG, et al. AMP kinase-mediated activation of the BH3-only protein Bim couples energy depletion to stress-induced apoptosis. J Cell Biol. 2010;189(1):83–94. doi: 10.1083/jcb.200909166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hardie DG. AMP-activated protein kinase: an energy sensor that regulates all aspects of cell function. Genes Dev. 2011;25(18):1895–908. doi: 10.1101/gad.17420111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hardie DG, Ross FA, Hawley SA. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol. 2012;13(4):251–62. doi: 10.1038/nrm3311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tanida I, Ueno T, Kominami E. LC3 and Autophagy. Methods Mol Biol. 2008;445:77–88. doi: 10.1007/978-1-59745-157-4_4. [DOI] [PubMed] [Google Scholar]
- 44.Zou Y, et al. The autophagy inhibitor chloroquine overcomes the innate resistance of wild-type EGFR non-small-cell lung cancer cells to erlotinib. J Thorac Oncol. 2013;8(6):693–702. doi: 10.1097/JTO.0b013e31828c7210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jones RG, et al. AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Mol Cell. 2005;18(3):283–93. doi: 10.1016/j.molcel.2005.03.027. [DOI] [PubMed] [Google Scholar]
- 46.Haikala HM, Anttila JM, Klefstrom J. MYC and AMPK-Save Energy or Die! Front Cell Dev Biol. 2017;5:38. doi: 10.3389/fcell.2017.00038. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.