

Abstract
Precisely how DNA-targeting chemotherapeutic drugs trigger cancer cell death remains unclear, as it is difficult to separate direct DNA damage from other effects in cells. Recent work on curaxins, a class of small molecule drugs with broad anticancer activity, show that they interfere with histone-DNA interactions and destabilize nucleosomes without causing detectable DNA damage. Chromatin “damage” caused by curaxins is sensed by the histone chaperone FACT, which binds unfolded nucleosomes becoming trapped in chromatin. In this study, we investigated whether classical DNA-targeting chemotherapeutic drugs also similarly disturbed chromatin to cause chromatin trapping of FACT (c-trapping). Drugs that directly bound DNA induced both chromatin damage and c-trapping. However, chromatin damage occurred irrespective of direct DNA damage and was dependent on how a drug bound DNA, specifically, in the way it bound chromatinized DNA in cells. FACT was sensitive to a plethora of nucleosome perturbations induced by DNA-binding small molecules, including displacement of the linker histone, eviction of core histones, and accumulation of negative supercoiling. Strikingly, we found that the cytotoxicity of DNA-binding small molecules correlated with their ability to cause chromatin damage, not DNA damage. Our results suggest implications for the development of chromatin-damaging agents as selective anticancer drugs.
Introduction
DNA-targeting small molecules have been widely used for cancer treatment for many years. This broad group includes chemicals with different mechanisms of action, but their toxicity was mostly explained by their ability to cause DNA damage (e.g. see rev. (1)). Many of these molecules are used for cancer treatment, since tumor cells are more vulnerable to DNA damage due to their high proliferation rate and frequently non-functional DNA-repair (2,3). Compounds target DNA via different mechanisms. Some form chemical (covalent) bonds with DNA (e.g., cross-linking agents). Others bind DNA non-covalently via either intercalation between base pairs or accommodation in DNA grooves (1). Some compounds do not stably bind DNA, but their complex with DNA is stabilized by proteins, such as topoisomerases (4,5). Finally, some compounds do not bind DNA but inhibit enzymes using DNA as a substrate, such as DNA polymerases or topoisomerases (6,7).
Eukaryotic DNA is packed into chromatin, which is a highly-ordered complex of DNA and histone proteins. The basic unit of chromatin, nucleosome, consists of a core, a complex of four pairs of histones: central H3/H4 tetramer with two H2A/H2B dimers outside, wrapped with DNA. Some nucleosomes are locked by binding the linker histone H1, which forms contacts with entering and exiting strings of DNA and the core histones (8).
The DNA-damaging effect of small molecules depends significantly on chromatin organization, e.g., some agents have a preference for linker versus nucleosomal DNA (9,10). On the other hand, there are reports that DNA-targeting small molecules perturb chromatin structure (11-14). However, how exactly they affect the chromatin and what impact chromatin alterations have on their biological activity are less studied. One of the reasons of this deficit was difficulty in separation of DNA damage from “chromatin damage” in cells.
We have previously identified small molecule, curaxin CBL0137, which has broad anti-cancer activity, and binds DNA without detectable DNA damage in mammalian cells (15). Although curaxin does not chemically modify DNA, it changes the shape of the DNA helix, which increases the inter-base-pair distance, unwinds DNA and leads to the unwrapping of DNA from the histone octamer and to nucleosome disassembly in vitro and in cells (14).
Nucleosome disassembly induced by CBL0137 is “sensed” by the histone chaperone FACT (FAcilitates Chromatin Transcription) (14), whose normal function is to control nucleosome stability during replication, transcription, and DNA repair (16). FACT consists of two subunits, Suppressor of Ty 16 (SPT16) and Structure Specific Recognition Protein 1 (SSRP1). It interacts with the nucleosome via several dynamic contacts with histone oligomers and DNA (17). Mammalian FACT binds poorly to the intact nucleosome (18,19). The weakening of DNA/histone binding provides FACT access to several binding sites hidden inside the nucleosome (18). At lower CBL0137 concentrations (1 molecule per >10-100bp), DNA is partially unwrapped from the core, leading to the dissociation of the H2A/H2B dimers and exposure of the surface of the H3/H4 tetramer (14). FACT binds the H3/H4 surface via its SPT16 subunit (14,18). At higher CBL0137 concentrations (1 molecule per 1-10bp), DNA is completely unwrapped from the nucleosome, what culminates in the disassembly of the histone core and the appearance of histones in the nucleoplasm (14). Unwrapped DNA undergoes significant negative supercoiling, which results in base unpairing and transition from the normal B-shape helix to alternative DNA structures (ADS). In cells treated with CBL0137, we detected the appearance of left-handed Z-DNA. The SSRP1 subunit binds DNA prone to the Z-DNA transition through its c-terminal intrinsically disordered domain (14). We named the massive binding of FACT to different components of disassembled chromatin in curaxin-treated cells “chromatin trapping” or c-trapping (14).
This study was based on the hypothesis that curaxins may not be unique in their effect on chromatin, and, therefore, in the ability to cause c-trapping. We proposed that other small molecules targeting DNA, may disturb chromatin and this could be detected using c-trapping assays. We selected a set of DNA-targeting agents, representing different mechanisms of action, and tested their ability to cause c-trapping and destabilize chromatin in vitro and in vivo. We tried to uncover the reasons why some compounds have stronger chromatin destabilizing effects than others and how it correlates with their cytotoxicity.
Materials and Methods
Chemicals, plasmids, antibodies and cells
Chemicals, plasmids, antibodies and cells used in the study are described Supplementary Materials and Methods and in Supplementary Table S1.
Cells
HeLa, HT1080, MCF7, HCT116 cells were purchased from ATCC, HL60/VCR were provided by R. Glazer (Georgetown University Medical Center). MCF7 cells were authenticated using short tandem repeat profiling and found to be 100% match to original ATCC cells. Other cells were not authenticated. All cells are tested for mycoplasma contamination at least once a month.
Cytotoxicity
HeLa, MCF7, HCT116 and HT1080 cells (3×103 cells/well) were seeded in 96-well plates for overnight adhesion. HL60/VCR suspension cells (5×103 cells/well) were seeded in 96-well plates and treated the same day. 50μM of 9-Aminoacridine was used as a positive control for complete cell death. Cells were treated for 48 h with a range of drug concentrations. Cell viability was determined with resazurin saline solution. Fluorescence was measured at 560Ex/590Em using Tecan Infinite 200 PRO reader.
Nascent RNA-sequencing
Nascent RNA-sequencing was done using Click-iT™ Nascent RNA Capture Kit (Invitrogen, cat#C10365). RNA labeling with EU was done in HT1080 cells for 15 minutes in duplicates. Isolation of labeled RNA was done according to manufacturer instruction. Library preparation and sequencing was done in Genomics Core of RPCI using Illumina protocols and equipment.
ChIP-sequencing
ChIP-sequencing was performed as previously described (20). HT1080 cells were treated for 1.5 hour with 3μM of CBL0137, aclacinomycin A, or 0.3μM of CBL0100. Experiment was repeated two times. The ChIP libraries prepared at UB Genomics and Bioinformatics Core using Illumina ChIP-seq kits were pair-end sequenced on an Illumina HiSeq2000 with 100bp reads. Raw reads passed quality filter from Illumina RTA are pre-processed by using FASTQC for sequencing base quality control and mapped to human reference genome (hg19) using BWA (21). MACS 2.0 (22) with default parameters for pair-end BAM files is used to identify peaks. Heatmaps and profiles under all conditions were generated using deeptools on RPKM normalized coverage data using merged bam files of biological replicates. Tag densities for RefGenes body with 3kb up and down stream are plotted. The heatmaps for all RefGenes are clustered into 4 groups using k-mean, within each cluster, the genes are ordered by mean tag density. Cluster 1 shows the greatest change of binding profiles. The different binding site status is identified by DiffBind R package (23). Heatmaps of the unexpressed and expressed genes were prepared using nascent RNA-Seq data for control untreated HT1080 cells.
Mononucleosome-based assays
Recombinant human mononucleosomes, purchased from EpiCypher (cat# 16-0009) at 0.125μM were incubated with compounds for 20 min at RT. Following the incubation, samples were run in 5% native PAGE at 120v at 4°C. Gels were stained with ethidium bromide and nucleosomal DNA were quantitated with the GelDoc-It TS imaging system equipped with a Series 6100 Camera (UVP). Experiments were repeated at least twice.
Comet assay
Comet assay was done using kit from Cell Biolabs, Inc. (cat# STA-355) in alkali conditions according to manufacturer protocol.
Extraction of soluble and chromatin-bound proteins from cells and western blotting
Cells were lysed on ice using 1× Cell Culture Lysis Reagent (Promega, cat# E1531) containing protease inhibitors (1:25, Roche, cat# 1838145). Extracts were centrifuged at 10,000 rpm for 10 min at 4°C to obtain the soluble fraction. The chromatin-bound proteins were obtained by resuspension of the remaining pellets in 1× Cell Culture Lysis Reagent followed by sonication three times for 30 sec each using the Bioruptor UCD-200, Diagenode. Protein concentrations were measured using Quick Start Bradford 1× Dye Reagent (Bio-Rad, cat# 500-0205). Equal amounts of protein were run on gradient 4-20% precast gels (Invitrogen) and transferred to Immobilon-P membrane (Millipore). Western blot analysis is described Supplementary Materials and Methods.
Immunofluorescent staining
Immunofluorescent staining are described Supplementary Materials and Methods. Fluorescent images of live and fixed cells and compound auto-fluorescence were obtained with a Zeiss Axio Observer A1 inverted microscope with N-Achroplan 100×/1.25 oil lens, Zeiss MRC5 camera, and AxioVision Rel.4.8 software.
Accession numbers
Sequencing data in the form of bed files are available at GEO: GSE107595, GSE107633.
Results
Only compounds directly binding DNA in cells induce c-trapping
If change of DNA helical shape is responsible for nucleosome destabilization then drugs can destabilize nucleosomes either via direct binding to DNA or via inhibition of topoisomerases, enzymes that prevent the accumulation of supercoiling in cells (reviewed in (24)). Drugs can either inhibit DNA cleavage by topoisomerases (supercoiling accumulates) or re-ligation (supercoiling is released due to DNA breaks). Reports from Henikoff and Neefjes groups showed that anthracyclines, which bind DNA and inhibit topoisomerases II, caused histone eviction from chromatin in cells and nucleosome disassembly under cell-free conditions (25,26). However, whether they induced c-trapping was unknown.
To better understand what causes c-trapping, we used a set of known compounds with different modes of DNA binding and inhibition of topoisomerases (Table 1): (i) direct DNA binders that inhibit primarily the cleavage activity of topoisomerases - curaxin CBL0100, aclacinomycin A, Hoechst 33342, (ii) direct DNA binders that inhibit re-ligation of topoisomerases II - doxorubicin and mitoxantrone, (iii) compounds that do not bind DNA but form a cleavable complex with DNA and topoisomerases I - SN38, or topoisomerases II - etoposide, and (iv) catalytic inhibitors of topoisomerases II that do not bind DNA – merbarone and ICRF-193. We also included an inhibitor of DNA synthesis (gemcitabine), which is unable to bind DNA or inhibit topoisomerases (negative control), and CBL0137 as a positive control for c-trapping (14,15).
Table 1.
Properties of DNA – targeting small molecules used in this study
| Drug | Mechanism of action/type | Binds DNA? | c-trapping | Cytotoxicity | |||
|---|---|---|---|---|---|---|---|
| Yes/No | Conc (μM)* | IC50 | Confidence Intervals** | ||||
| CBL0100 | Chromatin destabilizing, indirect inhibition of FACT, curaxin (this study) | yes | yes | 0.2 | 0.01 μM | 0.007 | 0.015 |
| CBL0137 | Chromatin destabilizing, indirect inhibition of FACT, curaxin | yes | yes | 0.6 | 0.1 μM | 0.07 | 0.14 |
| Doxorubicin | TOPO II poison, anthracycline | yes | yes | 1.2 | 0.55 μM | 0.44 | 0.68 |
| Aclacinomycin A | TOPO II inhibitor, anthracycline | yes | yes | 0.5 | 0.02 μM | 0.02 | 0.03 |
| Mitoxantrone | TOPO II poison, anthraquinone | yes | yes | 2.5 | 0.05 μM | 0.04 | 0.06 |
| SN-38 | TOPO I inhibitor, analog of camptothecin | no | no | NA | 0.52 μM | 0.29 | 0.93 |
| Etoposide | TOPO II poison, podophyllotoxin derivative | no | no | NA | 1.17 μM | 0.8 | 1.7 |
| Gemcitabine | Inhibitor of nucleotide synthesis, nucleoside analog | no | no | NA | NA | NA | NA |
| Merbarone | Catalytic TOPO II inhibitor | no | no | NA | Very wide | Very wide | Very wide |
| ICRF-193 | Catalytic TOPO II inhibitor | no | no | NA | 4.2 μM | 3.15 | 5.6 |
| Hoechst 33342 | TOPO I and II poison, minor groove binder | yes | yes | 0.6 | 5.3 μM | 1.8 | 15.8 |
Concentration of a compound leading to complete redistribution of FACT from soluble to chromatin fraction after 24 h of incubation based on western blotting.
95% Confidence Intervals represented for HeLa cells achieved with GraphPrismPad software.
Since c-trapping and cytotoxicity of CBL0137 occur at the same concentrations (although with a time delay: c-trapping - within a minute, cell death - around 48 hours after the start of treatment) (14,15), we identified cytotoxic concentrations of the compounds in several human cell lines (HeLa, HT1080, HCT116 and MCF7, Fig. S1-S4, Table 1) and used the range of doses (from non-toxic to maximal toxicity) to detect c-trapping. Using western blotting, we first screened all compounds for the presence of c-trapping activity at 24 hours after start of treatment in case some compounds might work slowly. FACT subunits disappeared from the soluble and accumulated in the chromatin fractions of all cell types treated with DNA-binding compounds, CBL0137, CBL0100, doxorubicin, aclacinomycin A, mitoxantrone and Hoechst 33342, while, there was no change in the distribution of FACT in any cells treated with compounds unable to bind DNA directly (Fig. 1A, S5-S7, Table 1). These data were confirmed via fluorescent imaging of live cells (HeLa or HT1080) expressing GFP-tagged SSRP1. Previously we have demonstrated using western blotting and immunofluorescent staining that tagged SSRP1 undergoes c-trapping similarly to endogenous FACT (14). In untreated cells SSRP1 is diffusely distributed in nucleoplasm and enriched in nucleoli. All compounds directly binding DNA caused significant change in the visual pattern of SSRP1 distribution in nuclei. Neither of non-DNA binding compounds had this effect (Fig. 1B, S8-S9). Thus, c-trapping is induced by the binding of small molecules to DNA rather than by inhibition of topoisomerases.
Figure 1.
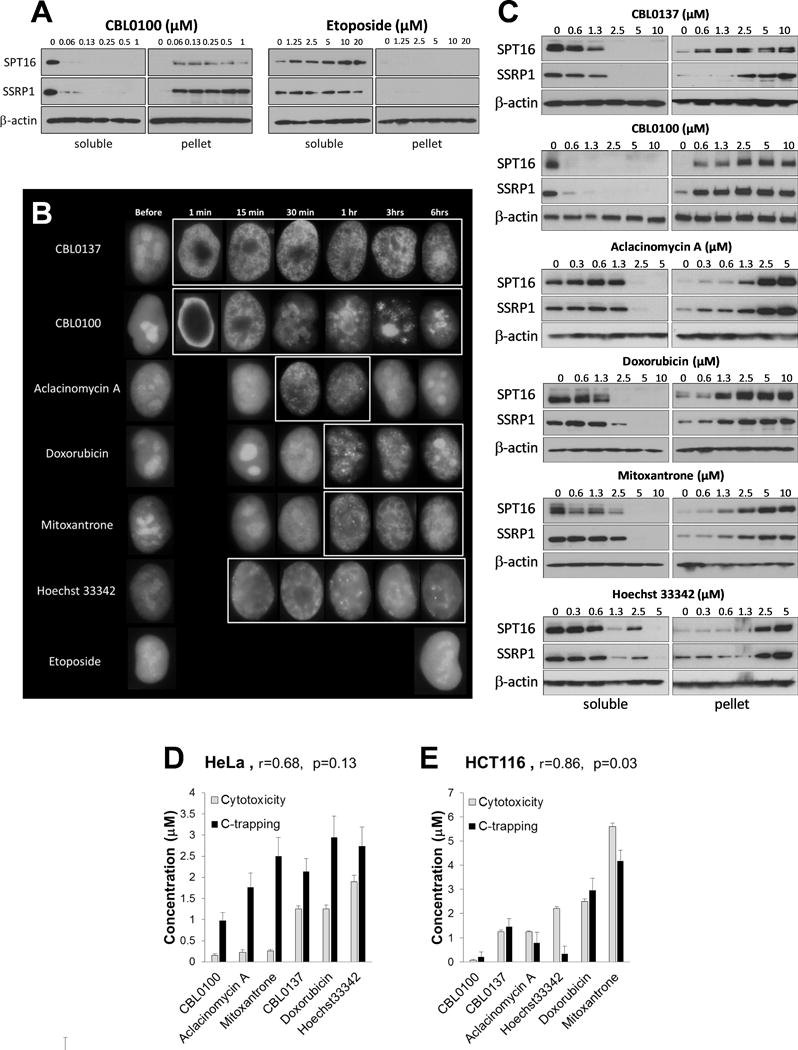
Testing of the ability of DNA-targeting compounds to induce c-trapping. (A) Examples of screening of the compounds for the presence of c-trapping upon treatment of HeLa cells with either CBL0100 or Etoposide. Representative immunoblots of soluble protein extracts and chromatin pellets of HeLa cells treated for 24 hours. Results for other drugs and cells are shown on Fig. S5-7. (B) Comparison of the kinetic of c-trapping in HT1080 cells expressing C-GFP-tagged SSRP1 via live cell imaging. Photographs of typical cell nuclei similar to >90% of cells in population. All compounds were used at 2.5μM. White frames are used to indicate nuclei where c-trapping is observed. Missing time-points for some drugs did not show any difference from untreated cells. For multiple cell images see Supplementary Figures S8-S9. (C) Comparison of potency of DNA-binding compounds to induce c-trapping in HeLa cells upon short time treatment, selected based on the kinetic of c-trapping defined in B. Representative immunoblots of soluble protein fractions and chromatin pellets of HeLa cells treated for 30 min with CBL0137, CBL0100, Aclacinomycin A and Hoechst 33342, or for 60 min with Doxorubicin and Mitoxantrone. Similar data for HCT116 cells are shown in Supplementary Fig. S13. (D and E) Analyses of correlations between cytotoxicity of the compounds and their potency in inducing c-trapping for HeLa (D) and HCT116 (E) cells. Cytotoxicity is the dose causing killing of 90% of cells upon 48 treatment (IC90%). Bars are mean of IC90% for three replicates within experiment ±SD. C-trapping is the dose causing redistribution of 50% of FACT from soluble to pellet fractions defined by quantitation of immunoblots (EC50%). Bars are mean of EC50% for SSRP1 and SPT16 subunits defined separately for soluble and protein fractions ±SD. r - Pearson correlation coefficient, p-value of correlations for n=6.
Live cell imaging revealed that DNA-binding compounds induced c-trapping with different kinetics, some of them within minutes, others required 1-2 hours (Fig. 1B). Different kinetic was confirmed using western blotting (Fig. S10). This kinetic correlated with the compound accumulation in cell nuclei, monitored by auto-fluorescence of the drugs (Fig. S11) and with whether compounds are substrate of multi-drug transporters or not (Fig. S12). Using these observations we selected optimal time for measurement of c-trapping, caused by six active compound to identify dose causing redistribution of 50% of SSRP1 and SPT16 from nucleoplasm to chromatin in two cell lines, HeLa and HCT116 (Fig. 1C and S13). This dose positively correlated with concentration of each drug which kills 90% of cells (IC90%) 48 hours later (Fig. 1D, E), suggesting that the mechanism underlying c-trapping contributes to the death of tumor cells.
Correlation between c-trapping and nucleosome destabilization
Previously we established that c-trapping occurs in response to chromatin disassembly in cells (14). To test effect of the compounds on nucleosome stability we incubated recombinant mononucleosomes with different concentrations of the drugs. All compounds able to induce c-trapping, except Hoechst 33342, caused complete disassembly of the mononucleosome. Hoechst 33342 destabilize but did not disassemble nucleosome (Fig. 2A, B).
Figure 2.
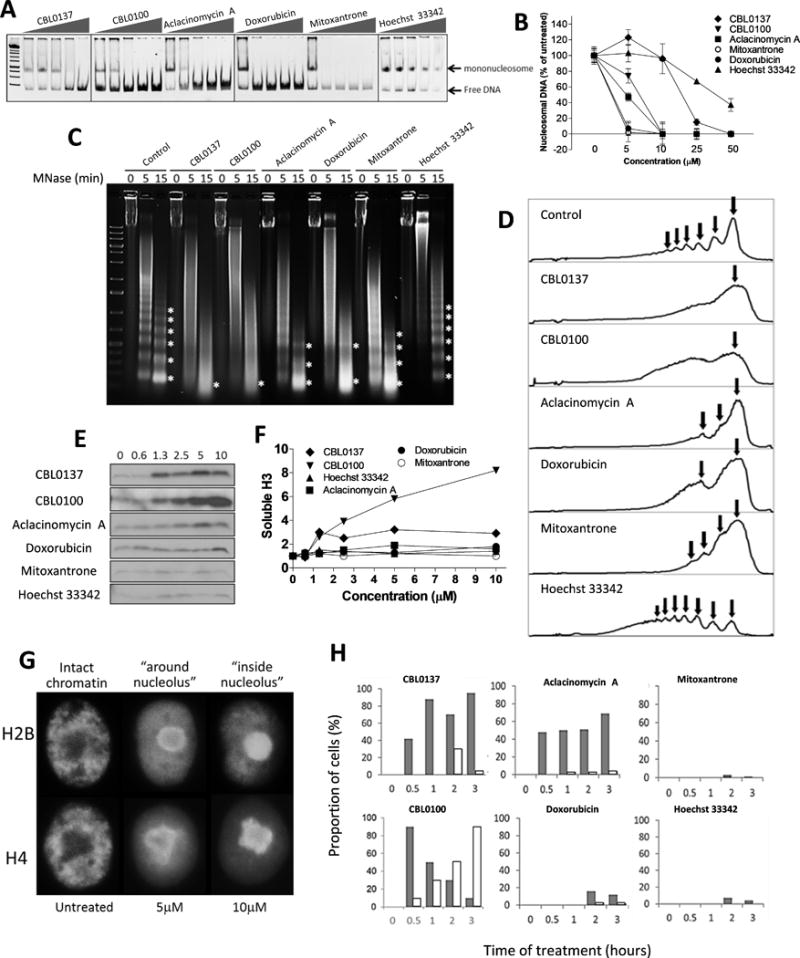
Effect of the compounds on nucleosome stability. (A-B) Effect of the drugs (0, 5, 10, 25 and 50μM) on the stability of mononucleosome in cell-free conditions upon 15 min incubation at room temperature. Then reaction mixture was run in 5% native polyacrylamide gel. (A) Images of representative runs. (B) Quantitation of several experiments via densitometry of nucleosomal DNA. Mean % of control ± SD. (C-D). Effect of the drugs on the sensitivity of chromatin in HeLa cells to micrococcal nuclease (MNase) digestion. Drugs (100μM, equivalent of 10μM in cell-based experiments based on DNA amount) were incubated for 15 minutes with nuclei of HeLa cells, followed by MNase digestion for 5 or 15 minutes, DNA purification and agarose gel electrophoresis. (C) Images of representative runs. (D) Profiles of light intensity of lanes corresponding to 15 minutes of MNase treatment obtained using Image J. Asterisks on panel C correspond to arrows on panel D and indicate nucleosome protected bands. All direct DNA-binding compounds used in the study are known to inhibit nucleases, including MNase (27), therefore the resulting effect is seen as loss of nucleosome protected bands (effect of the drugs on chromatin) and appearance of higher molecular weight smear (under-digested naked nuclear DNA – effect of the drugs on MNase). Effect of Hoechst 33342 cannot be assessed due to complete inhibition of MNase. We compared the results by examining the loss of a regular nucleosome ladder, rather than by the degree of digestion. (E, F) Effect of the compounds on the distribution of histones in cells. (E) Immunoblotting of soluble protein extracts of HeLa cells treated with the indicated doses of the compounds for 1.5 hours and probed with antibody to histone H3. (F) Quantitation of data shown in E. (G) Accumulation of mCherry-tagged histone H2B and mOrange-tagged histone H4 around (CBL0137, 5μM) and inside nucleoli (CBL0137, 10μM). HeLa cells were treated for 1 hour. (H) Quantitation of histones H2B eviction from chromatin in HeLa cells treated with 10μM of different drugs, for the indicated periods of time. Grey bars – proportion of cells with histones around nucleoli, white bars – inside nucleoli. Similar data for histone H4 is shown in Supplementary Figure S14.
To test the effect of the compounds on nucleosome stability within chromatin we incubated HeLa cell nuclei with the drugs, followed by assessment of chromatin sensitivity to digestion with micrococcal nuclease (MNase), preferentially digesting protein-free DNA. All DNA-binding drugs, except Hoechst 33342 caused loss of nucleosome ladder. Hoechst 33342 inhibits nuclease activity (27) to the extent that its effect cannot be assessed. Curaxins were stronger than anthraquinones, although doxorubicin caused effects similar to curaxins. (Fig. 2C, D).
To assess chromatin integrity in cells we monitored appearance of chromatin-free histones in extracts of cells incubated with the compounds, able to cause c-trapping. A minimal amount of canonical core histones is detected in the soluble fraction of untreated cells (Fig. 2E). With increasing concentrations of the compounds, histone H3 appeared in extracts of cells treated with the curaxins and aclacinomycin A. A weak, but reproducible, increase in histone H3 was observed upon treatment with 10μM doxorubicin. No free H3 was detected in the lysates of cells treated with mitoxantrone or Hoechst 33342 (Fig. 2E, F). In addition, we monitored the distribution of the outer H2B and inner H4 histones fused with fluorescent tags in cell nuclei via live cell imaging. Excess of free histones, which are not incorporated into chromatin, is accumulated in the nucleoli, first concentrated around and later inside nucleoli ((14,28), Fig. 2G). Consistent with western blotting, curaxins and aclacinomycin A caused eviction of both outer and inner histones (Fig. 2H and S14). Doxorubicin caused visible nucleoli accumulation of histones only at high concentrations (≥10μM), while no significant difference from untreated cells was seen in case of mitoxantrone and Hoechst 33342 in line with cell fractionation data (Fig. 2H and S14). Similar response of endogenous H3 was observed via immunofluorescent staining (Fig. S15).
The unexpected finding was absence of nucleosome disassembly in cells treated with agents that clearly cause c-trapping, mitoxantrone and Hoechst 33342. In cells multiple nucleosomes are stabilized in a “closed” position by the histone H1 (8). Importantly, H1 has a very fast exchange rate (8). We proposed that some of the compounds may have limited access to DNA within a closed nucleosome, but gain access to it if the nucleosomes lack H1. In this case, H1 may not be able to reattach to the nucleosome. We tested this hypothesis by studying the dose and time dependent effect of the drugs on the nuclear distribution of the human histone H1 tagged with mCherry. All drugs, inducing c-trapping caused H1 accumulation in nucleoli (Fig. 3A, B). The dose and time for histone H1 eviction positively correlated with c-trapping, suggesting that c-trapping may occur due to partial opening of the nucleosome without loss of the core histones.
Figure 3.
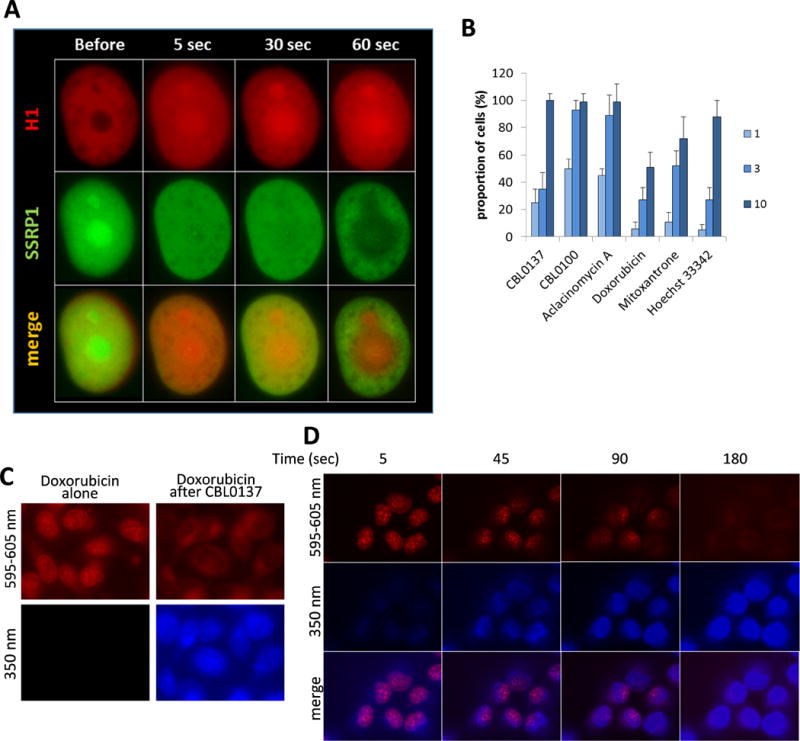
Effect of the compounds on the distribution of histone H1 in cells (A, B) and competition of curaxins and Doxorubicin for binding to DNA in cells (C and D). (A) Time-dependent effect of 3μM of CBL0137 on the distribution of mCherry tagged H1 (H1.5) and GFP-tagged SSRP1 in HT1080 cell. (B) Effect of the different doses of the compounds (in μM) on the distribution of histone H1 in HT1080 cells treated for 3 hours. Bars - proportion of cells in population with nucleoli-accumulated H1, ±SD between two experiments. (C-D) Imaging of live cells with filters corresponding to autofluorescence of the compounds: 595-605nm for Doxorubicin (red), and 350nm for curaxins (blue). HT1080 cells were incubated with 1μM of Doxorubicin for 3 hours, then 1μM of CBL0137 (C) or CBL0100 (D) was added for 10 min (C) or for the indicated times (D).
So far we observed that curaxins were the strongest in all cell-based assays, while their effect on nucleosome in cell-free conditions is weaker than anthraquinones. We proposed that among tested compounds, curaxins have the highest capacity to bind chromatinized DNA in cells. To test this, we incubated cells with doxorubixin for 3 hours to allow the drug to bind nuclear DNA and then added the curaxins at the same concentration. Since doxorubicin and curaxins have different fluorescence spectra, we were able to monitor each drug separately. The addition of both curaxins quickly displaced doxorubicin from the cell nuclei (Fig. 3C,D). A similar experiment cannot be done with the other compounds because aclacinomycin A and mitoxantrone do not have-nuclear fluorescence and Hoechst 33342 has a similar fluorescence spectrum to the curaxins.
Differences in c-trapping between curaxins and other compounds
In cells treated with CBL0137, FACT is trapped in chromatin by two distinct mechanisms: (i) binding of the SPT16 subunit to the surface of the H3/H4 tetramer, which is exposed by the detachment of the H2A/H2B dimer (“n-trapping”) and (ii) binding of the SSRP1 subunit to alternative DNA structures (e.g., Z-DNA (“z-trapping”)) (14). SSRP1 can also bind different alternative DNA structures (ADS), such as bent or cruciform DNA, via the HMG domain (29-32). We tested whether other compounds, similarly to CBL0137, induced “n-trapping” and “z-trapping”. Separating these two processes in cells is difficult since SSRP1 and SPT16 are always in a complex and dissociation of the dimer makes both subunits unstable (33). Previously, we used SSRP1 deletion mutants to distinguish these two phenomena. SSRP1 lacking the c-terminus DNA-binding domains (HMG and CID) can only undergo c-trapping when bound to SPT16 (n-trapping), while SSRP1 deletion mutants consisting of only the c-terminal domains bind DNA, either in the Z-form (CID domain, z-trapping) or as cruciform/bent DNA (HMG domain) (14). CBL0137 and CBL0100 caused both n- and z-trapping (Fig. 4A, B). Surprisingly, none of other compounds changed the distribution of either HMG or CID constructs, suggesting that none of them caused z-trapping. However, all of them caused “n-trapping” (Fig. 4A, B). The only exception was that aclacinomycin A did not have an effect on SSRP1 that lacked the HMG domain, suggesting that this domain may be involved in c-trapping by aclacinomycin A.
Figure 4.
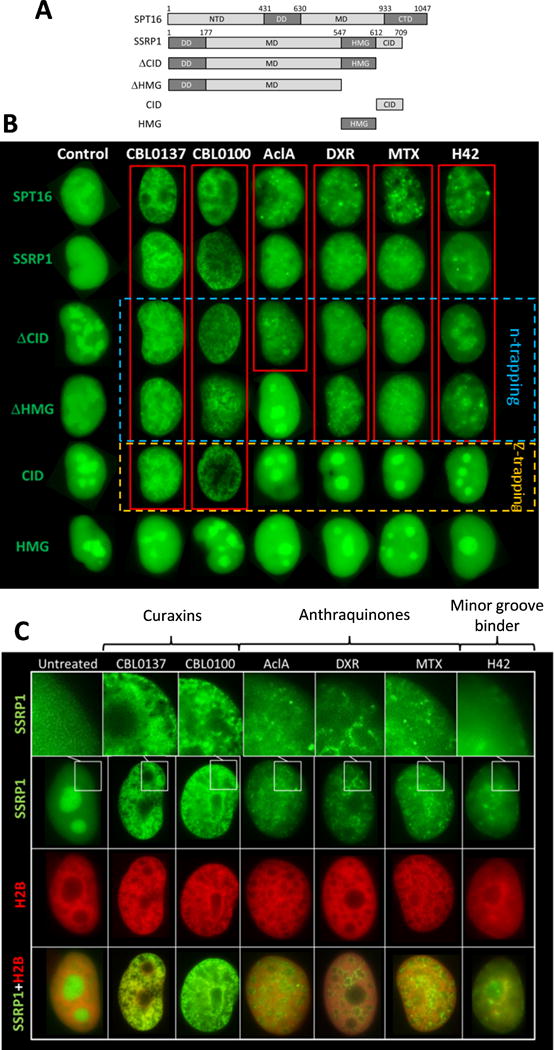
Differences in c-trapping between CBL0137, CBL0100, Aclacinomycin A (AclA), Doxorubicin (DXR), Mitoxantrone (MTX) and Hoechst 33342 (H42). (A, B) Comparison of the ability of the compounds to cause c-trapping of SSRP1, SPT16 and SSRP1 deletion mutants. (A) Scheme of SSRP1 expression constructs used in the study. All constructs are fused with GFP provided with nuclear localization signal on N-termini. (B) Photographs of typical nuclei (present in >90% of cells in population) of HT1080 cells transduced with the indicated constructs. Cells were treated with 3μM of the compounds for 1.5 hours. Red frames are used to indicate nuclei where c-trapping is observed. ΔHMG and ΔCID construct lack c-termini DNA binding domains (HMG or CID) and therefore can cause c-trapping only via SPT16 subunit (“n-trapping” – dashed blue frame). CID domain construct binds Z-DNA (“z-trapping” – dashed orange frame). HMG domain binds bent or kinked DNA. Neither of compounds tested caused c-trapping via HMG domain. (C) Different patterns of SSRP1 distribution in the process of c-trapping caused by different drugs. Photographs of typical (present in more than 90% of cells) nuclei of HT1080 cells expressing GFP-tagged SSRP1 and mCherry-tagged histone H2B. Cells were treated with 3μM of all compounds, except CBL0100, which was used at 0.3μM, for 1.5 hours.
Thus, while c-trapping looks very similar via immunoblotting, experiments with FACT deletion mutants demonstrated that the compounds induce c-trapping via different FACT subunits. Moreover, they induce different microscopic patterns of FACT distribution in cell nuclei (Fig. 4C). Taking into account difference in the shapes and sizes of nuclei, these patterns were characteristic for all cells in populations (Fig.S8-S9). Curaxins pattern was the most contrasting (Fig. 4C). Anthraquinones caused more subtle changes, and Hoechst 33342 - the mildest: transitions between bright and dim zones without clear boundaries. Since the differences were observed even at concentrations of the compounds that caused complete transition of FACT from soluble to chromatin-bound fractions (10μM, Fig. S16), they could not be explained by different amounts of bound and free FACT. We hypothesized that these patterns reflect binding of FACT to different chromatin regions. To test this, we compared genome-wide FACT-chromatin binding in cells treated with 3μM CBL0137, 0.3μM CBL0100 (the same visual pattern as CBL0137) and 3μM aclacinomycin A (different pattern) using chromatin immunoprecipitation (ChIP) with SSRP1 antibody followed by deep sequencing (seq).
In control cells SSRP1 peaks coincide with coding regions of transcribed genes (Fig. 5A). Both curaxins induced appearance of multiple new sharp peaks genome-wide, absent in control cells (Fig. 5A). Curaxin-induced peaks were located at regions, which were previously observed in CBL0137-treated cells and identified as mini-satellites, tandem dinucleotide purine/pyrimidine repeats, prone to Z-DNA transition ((14) and Fig. 5B, S17). Much more rarely new peaks appeared in aclacinomycin A samples. In most cases they overlap curaxin-induced peaks (Fig. 5A), however aclacinomycin-specific peaks appeared at regions annotated as G-quadruplex by Non-B DNA Database (34) (Fig. 5B). Although anthracyclines were reported to bind G-quadruplex DNA in cells and cell-free conditions (35,36), there is no data of FACT binding to this type of DNA. FACT peaks in curaxins treated cells, in contrast to aclacinomycin A, demonstrated absence of colocalization of with G-quadruplex regions (Fig. S17).
Figure 5.
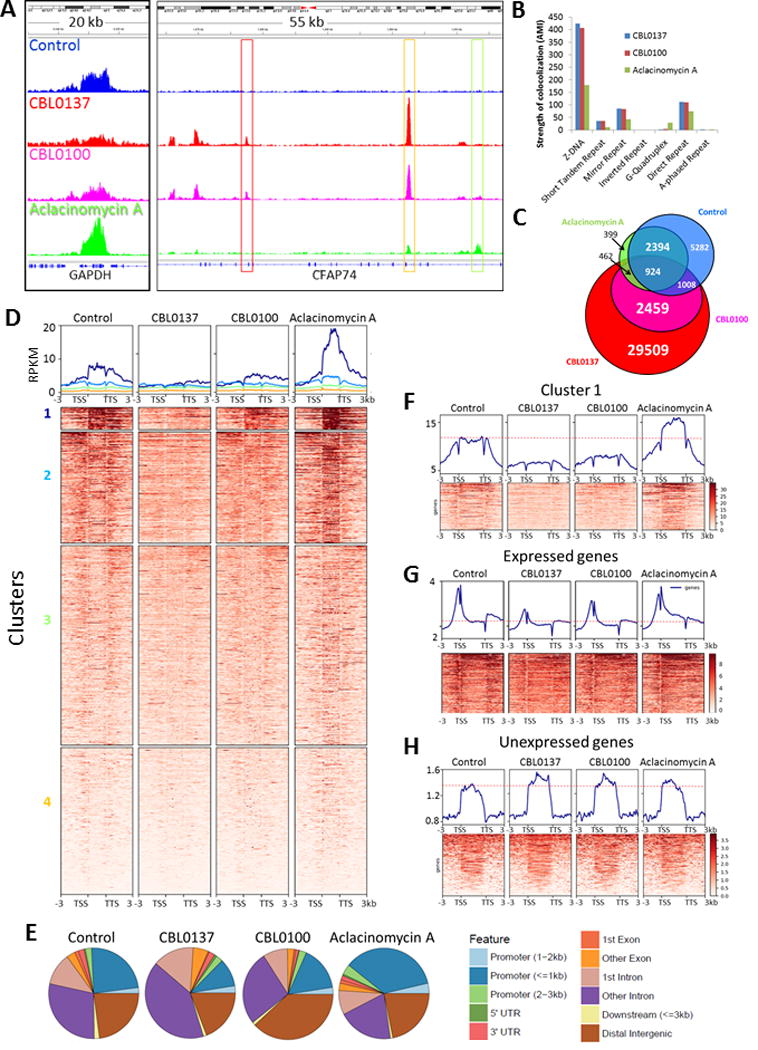
Curaxins and Aclacinomycin A differ in the effect on FACT distribution genome-wide. ChIP-seq with SSRP1 antibody in HT1080 cells control or treated with 3μM of CBL0137 and Aclacinomycin A, or 0.3μM of CBL0100 for 1 hour. (A) Integrated genome views of the selected regions of human genome, demonstrating FACT enrichment at GAPDH gene in different conditions (left panel), and appearance of new peaks in treated cells (right panel). Red frames show curaxin-specific peaks, green frame – Aclacinomycin A specific peak and yellow – peak present in all treated cells. (B) Analysis of colocalization of SSRP1 peaks and regions prone to non-B DNA transitions defined by non-B DNA Database using ColoWeb. Bars are AMI (Above Median Integral) indices. All shown indices are highly significant with p<0.01. Details of the analysis and data for the absence of colocalization are shown on Supplementary Figure S17. (C) Venn-diagram showing distribution of SSRP1 peaks (score greater than 50 from MACS2) between control and treated cells. Numbers – number of peaks, specific and common for different conditions. (D) Heatplots and average gene profiles of SSRP1 distribution over genes which showed change in SSRP1 binding (fold change > 1.5, p-value < 0.05 in any treatment vs control). (E) Distribution of SSRP1 peaks in control and treated cells in relation to genome annotation features. (F) Similar analysis as in D done for all genes. Only cluster 1 is shown. All clusters as well as data for non-coding RNAs and miRNAs are shown on Supplementary Figures S18-S20. (G) Profiles of SSRP1 distribution over coding regions of genes transcribed in basal conditions in HT1080 cells based on the data of nascent RNA-seq. (H) Profiles of SSRP1 distribution over coding regions of genes which are not transcribed in untreated HT1080 cells. On panels F-H horizontal red-dashed lines are placed on top of FACT enrichment profile at coding regions in untreated cells for the comparison of FACT enrichment in different conditions.
To assess quantitatively FACT redistribution genome wide we aimed to find (i) where FACT was relocated upon treatments; and (ii) what proportion of FACT molecules underwent re-localization. For the first task, we identified peaks in each of the conditions and compared them with control and between treatment groups (Fig. 5C). In line with visual impression peaks induced by CBL0100 almost completely overlap with peaks induced by CBL0137, i.e. both curaxins induced similar redistribution of FACT from coding regions to tandem dinucleotide purine/pyrimidine repeats. In contrast, regions bound by FACT in aclacinomycin A treated cells were not significantly different from FACT-enriched regions in control cells. Novel sites of FACT binding appeared in aclacinomycin A treated cells were mostly shared with curaxins (Fig. 5C). From the comparison of regions of FACT enrichment we concluded, that curaxins impose FACT to leave regions where it was present in control cells and to bind new regions, while what happened with FACT in aclacinomycin A treated cells was unclear.
Quantitating average FACT enrichment at coding regions of all genes, where FACT presence was changed upon any of treatment, we have found that curaxins significantly reduced FACT presence over coding regions, while aclacinomycin A increased (Fig. 5A, D). Proportion of FACT in aclacinomycin A treated cells were also significantly increased at promoter regions (Fig. 5E). Similar patterns were clearly seen when we did this analyses for all genes, long non-coding RNAs and miRNAs (Fig. S18-S20). Clustering of the genes, based on the degree of changes, up or down, demonstrated, that the strongest change was observed for the genes with the highest SSRP1 enrichment in untreated cells (Fig. 5D, F). Since, FACT is expected to be enriched at coding regions of transcribed genes; we used data of nascent RNA-sequencing from the same cells to compare SSRP1 distribution at coding regions of expressed vs unexpressed genes (Fig. 5G, H). Curaxins treatment led to the reduction of FACT presence at coding regions and promoter of transcribed genes, but in opposite to the increase of FACT presence at non-transcribed genes, while aclacinomycin A treatment has opposite effects (Fig. 5G, H).
In line with different microscopic pattern of c-trapping, curaxins cause redistribution of FACT from transcribed regions to mini-satellites, while aclacinomycin A induced further accumulation of FACT at coding regions of transcribed genes. These observations suggested that curaxins induced appearance of de-novo sites for FACT binding and thus lead to redistribution of FACT, while aclacinomycin A makes existing sites more attractive for FACT binding.
Correlation of chromatin- and DNA-damaging activities of the compounds and their cytotoxicity
To understand the role of “chromatin damage” and c-trapping in the cytotoxic activity of the tested compounds we quantitated their effects on FACT and chromatin in cells and cell-free systems and ranked them based on these activities to run correlation analysis. Since DNA damage was traditionally seen as a reason of cell death upon treatment with some of these compounds, we also measured their DNA – damaging activity using γH2AX staining and comet assay in the same cells (HeLa and HT1080) and at the same concentration range at which c-trapping was observed (Fig. S21). This analysis showed that c-trapping positively correlated with cytotoxicity as well as with eviction of core histones, positive correlation with the eviction of H1 was also observed, but not significant. Most importantly, cytotoxicity within this group of compounds significantly correlated with c-trapping, but not with DNA-damaging activity of the compounds (Fig. 6 and S22). This observation suggests that ability of compounds directly binding DNA in cells, to cause “chromatin damage” has much stronger input in their cytotoxicity, than their ability to cause DNA damage. Another important observation is that there is poor correlation between compounds effects on chromatin in cells (c-trapping and histone eviction) and their effects on nucleosome in cell-free conditions (Fig. 6 and S22), most probably due to their different ability to reach and bind genomic DNA in cells.
Figure 6.
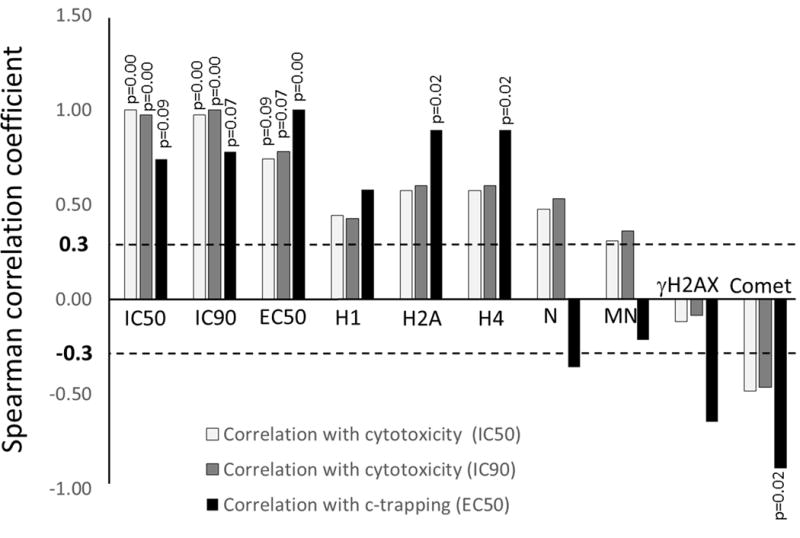
Analyses of correlations between different parameters of drug activities in cells and cell-free conditions. HeLa cells data were used for all cell based assays. The following parameters were used: IC50 and IC90 – cytotoxicity, concentration of a drug killing 50% or 90% of cells in population upon 48 hours incubation; EC50 - c-trapping, dose causing redistribution of 50% of FACT from soluble to pellet fractions during 30 (CBL0137, CBL100, Aclacinomycin A, Hoechst 33342) or 60 (Doxorubicin, Mitoxantrone) minutes incubation; H1 – eviction of histone H1, concentration of a drug causing maximal accumulation of H1 inside nucleoli upon 2 hours of incubation; H2B and H4 – eviction of core histones, concentration of a drug at which accumulation of these histones around nucleoli started to be observed upon 2 hours of incubation. N – destabilization of mononucleosome in cell free conditions, concentration of a drug causing 50% reduction of nucleosomal DNA; MN – effect of a drug on sensitivity of chromatin to MNase digestion; γH2AX – proportion of cells in population with positive staining; Comet – proportion of cells in population with tail moment > than in control cells. All these parameters were ranked (see details on Supplementary Figure S22) and Spearman correlation coefficients (Pearson correlation on ranked variables) were calculated between each parameter and cytotoxicity or c-trapping. Bars – Spearman correlation coefficients. P-values are shown for significant correlations with n=6 at p<0.1.
Discussion
Our study suggests that many known and some novel molecules exert their biological activity via destabilization of chromatin, i.e “chromatin damage”. Effects of different small molecules on chromatin in cells were previously noticed in multiple studies (e.g. (9,25,26)). We made an attempt to understand why some compounds do this and some not, even if they belong to the similar functional groups, e.g. TOPO inhibitors. We cannot extrapolate our finding to all small molecules, but our data suggest that compounds able to bind genomic DNA in cells, cause “chromatin damage”. Importantly, ability of a compound to bind naked or nucleosomal DNA in cell-free conditions is not directly translated into their ability to bind genomic DNA in cells. Many direct DNA-binders don’t reach nuclear DNA in live cells, e.g. propidium iodide. Some are distributed between nuclei and other organelles (e.g. lysosomes), or emitted from cells by multi-drug transporters and therefore amount of a drug bound to DNA varies. Unfortunately, there are limited possibilities to measure binding of small molecule to DNA in cells. If on a larger cohort of compounds with well-established ability to bind nuclear DNA, it will be confirmed that they cause c-trapping and histone eviction, then these relatively simple assays may serve as a surrogate markers of compound binding to genomic DNA in cells.
We propose that the binding of almost any small molecules to DNA interferes with nucleosome stability in several ways. Intercalators change the shape of the DNA helix, make DNA less flexible and disrupt the contacts of amino-acids and nucleotides important for nucleosome stability. Additionally, many of these compounds are positively charged and therefore their binding neutralizes negative charge of DNA, which plays important role in nucleosome stability. Situation with minor groove binder is less clear: although Hoechst 33342 caused c-trapping in cells, it neither disassembled nucleosomes in vitro nor in cells. It is known from crystal structure of nucleosome, that the strongest contacts are formed between the octamer and minor groove of DNA at several regions of nucleosome (37). We can speculate that although Hoechst 33342 may have difficulties in getting to the minor groove at these regions, however if it gets there, for example as a result of nucleosome breathing, the strong contact of histone with DNA will not be restored. Such nucleosome may not be fully disassembled, since other core/DNA contacts are preserved, but it should have part of DNA uncoiled, what facilitates access of FACT to nucleosome and obstructs histone H1 binding. These phenomena we observed in cells treated with Hoechst 33342.
However, some of these compounds are less capable of binding DNA in cells in the context of chromatin for reasons currently unknown. Since the structure of mammalian chromatin is very complex and cannot be easily visualized at a molecular level, some intermediate system, such as topologically constrained nucleosomal arrays in vitro, may be required to gain an understanding of this phenomenon.
Curaxins happened to be the strongest inducers of “chromatin damage” in cells among tested compounds most probably due to the combination of structural and biological characteristics. First, they are extremely efficient in reaching nuclear DNA since they are not substrate of multi-drug transporters. They bind DNA with high affinity (14,15), yet being relatively small comparing with anthraquinones. The small size may facilitate binding to bent nucleosomal DNA. However, too small size may make intercalation unstable, since compound may “enter” and “exit” DNA too easily without prolonged residence. That is why many planar carbazole compounds are weak DNA binders, i.e. form complex with DNA only at high concentrations. Presence of “ears” in curaxins, two symmetrical carbazole side-chains protruding into major groove, are critical for biological activity of curaxins most probably due to anchoring of them between bases. Finally, relatively small and flexible “tail” – side chain filling minor groove with positively charged nitrogen add to the stability of binding.
Although, all these speculations are based on computer modeling and structural studies of different compounds binding to naked and nucleosomal DNA are needed, we believe that the difference between compounds in their potency to cause chromatin damage and c-trapping may be explained by their binding to different genomic regions in cells. Therefore, we propose the following model. Curaxins can bind both, naked and nucleosomal DNA, what leads to destabilization of chromatin genome-wide and occurrence of opened accessible to FACT nucleosomes at multitude of locations, as well as regions of DNA without nucleosomes, undergoing transition to Z-DNA. The latter may be facilitated by underlying sequence of DNA or chromatin structure. Alternatively, regions of nucleosome loss may occur randomly and just those, which are composed of tandem dinucleotide purine/pyrimidine repeats, have higher probability of transition to Z-DNA. It is likely that FACT binding to the new regions is stronger than to the regions which it binds in untreated cells, what leads to redistribution of FACT. Alternative explanation is that FACT enrichment at transcribed regions is reduced because curaxins creates many more sites for FACT binding and this leads to the “dilution” of FACT at transcribed regions. However, previously we showed that much higher salt concentrations are needed to extract FACT from chromatin in curaxin treated cells than in control cells (15), suggesting that affinity of binding is increased upon curaxin treatment.
Doxorubicin, mitoxantrone and aclacinomycin A share many properties and induce similar pattern of FACT redistribution, what suggests that they have similar effect on chromatin in cells. We propose that these compounds have more difficulties getting to nuclear DNA than curaxins, and therefore much higher concentrations of these compounds are needed to cause effects similar to curaxins in cells, although in cell-free conditions they cause stronger nucleosome destabilization. Base on literature they also prefer binding linker versus nucleosomal DNA (9). This should lead to the preferential binding to promoters or coding regions of highly transcribed genes. It was shown by Yang that doxorubicin causes the strongest histone eviction from promoter regions which are also depleted in nucleosomes (25). Even at these regions they likely do not cause complete nucleosome disassembly, since we do not observe significant eviction of core histones and experiments with FACT mutants demonstrated that anthraquinones induces only n-trapping, or binding of FACT to destabilized nucleosomes, but no z-trapping or FACT binding to naked DNA.
Based on this we propose the following model for these molecules. It is known that in untreated cells passage of RNA polymerase partially uncoils nucleosomal DNA, making core available for FACT binding, while recoiling of DNA releases FACT from chromatin. Therefore in untreated cells FACT binding to these regions is temporal. Anthraquinones via binding to DNA stabilize uncoiled state of nucleosome creating permanent sites for FACT binding and therefore increase total amount of binding sites and residence time of FACT at these sites leading to FACT enrichment.
In conclusion, our findings confirm the previously observed effects of anthracyclines on chromatin and expand these observations towards the proposition that any DNA binding compound should destabilize nucleosomes in vitro and in cells if they can bind DNA in the context of chromatin and accumulate in the nucleus. FACT is sensitive to a plethora of perturbations of the nucleosomes induced by the binding of the small molecule to DNA, including displacement of the linker histone, eviction of the core histones, and accumulation of negative supercoiling. Most importantly, we suspect that the “chromatin-damaging” effect of small molecules plays a more important role in the toxicity of DNA binding molecules than their DNA-damaging activity. We do not question the role of DNA damage in the cytotoxic activity of many other molecules, however, “chromatin damage” caused by direct DNA binders may outweigh DNA damage in its influence on cell functioning and viability. The exact mechanism of cell death upon “chromatin damage” still needs to be established.
Supplementary Material
Acknowledgments
We would like to thank Bruce Specht for the administrative help, Catherine Burkhart from Burkhart Document Solutions for critical review and editing of the manuscript. This work was supported by Incuron LLC to K.G.; by National Cancer Institute [R01CA197967 to K.G. and P30CA016056 to Roswell Park Cancer Center]; and by Komen Foundation award [CCR13264604 to K.G.].
Abbreviations
- ADS
Alternative DNA Structures
- ChIP
Chromatin ImmunoPrecipitation
- CID
C-terminus Intrinsically Disordered
- c-trapping
chromatin trapping
- FACT
Facilitates Chromatin Transcription
- HMG
High Mobility Group
- MNase
Micrococcal Nuclease
- n-trapping
nucleosome trapping
- seq
next generation sequencing
- SPT16
Suppressor of Ty 16
- SSRP1
Structure Specific Protein 1
- z-trapping
Z-DNA trapping
Footnotes
Conflict of interest disclosure statement: K.V. Gurova is co-author of patent application 61/102,913, filed on October 6, 2008. “Carbazole compounds and therapeutic uses of the compounds” that describes structure and anticancer properties of Curaxins”. K.V. Gurova had a research grant from Incuron and is also a consultant of Incuron.
References
- 1.Palchaudhuri R, Hergenrother PJ. DNA as a target for anticancer compounds: methods to determine the mode of binding and the mechanism of action. Curr Opin Biotechnol. 2007;18:497–503. doi: 10.1016/j.copbio.2007.09.006. [DOI] [PubMed] [Google Scholar]
- 2.Bartkova J, Horejsi Z, Koed K, Kramer A, Tort F, Zieger K, et al. DNA damage response as a candidate anti-cancer barrier in early human tumorigenesis. Nature. 2005;434:864–70. doi: 10.1038/nature03482. [DOI] [PubMed] [Google Scholar]
- 3.Lord CJ, Ashworth A. The DNA damage response and cancer therapy. Nature. 2012;481:287–94. doi: 10.1038/nature10760. [DOI] [PubMed] [Google Scholar]
- 4.Suzuki H, Ikeda T, Yamagishi T, Nakaike S, Nakane S, Ohsawa M. Efficient induction of chromosome-type aberrations by topoisomerase II inhibitors closely associated with stabilization of the cleavable complex in cultured fibroblastic cells. Mutation research. 1995;328:151–61. doi: 10.1016/0027-5107(95)00005-4. [DOI] [PubMed] [Google Scholar]
- 5.Baldwin EL, Osheroff N. Etoposide, topoisomerase II and cancer. Current medicinal chemistry Anti-cancer agents. 2005;5:363–72. doi: 10.2174/1568011054222364. [DOI] [PubMed] [Google Scholar]
- 6.Larsen AK, Escargueil AE, Skladanowski A. Catalytic topoisomerase II inhibitors in cancer therapy. Pharmacol Ther. 2003;99:167–81. doi: 10.1016/s0163-7258(03)00058-5. [DOI] [PubMed] [Google Scholar]
- 7.Berdis AJ. DNA polymerases as therapeutic targets. Biochemistry. 2008;47:8253–60. doi: 10.1021/bi801179f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Flanagan TW, Brown DT. Molecular dynamics of histone H1. Biochim Biophys Acta. 2016;1859:468–75. doi: 10.1016/j.bbagrm.2015.10.005. [DOI] [PubMed] [Google Scholar]
- 9.Rabbani A, Iskandar M, Ausio J. Daunomycin-induced unfolding and aggregation of chromatin. The Journal of biological chemistry. 1999;274:18401–6. doi: 10.1074/jbc.274.26.18401. [DOI] [PubMed] [Google Scholar]
- 10.McMurray CT, Small EW, van Holde KE. Binding of ethidium to the nucleosome core particle. 2. Internal and external binding modes. Biochemistry. 1991;30:5644–52. doi: 10.1021/bi00237a002. [DOI] [PubMed] [Google Scholar]
- 11.Wojcik K, Zarebski M, Cossarizza A, Dobrucki JW. Daunomycin, an antitumor DNA intercalator, influences histone-DNA interactions. Cancer biology & therapy. 2013;14:823–32. doi: 10.4161/cbt.25328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hajihassan Z, Rabbani-Chadegani A. Studies on the binding affinity of anticancer drug mitoxantrone to chromatin, DNA and histone proteins. Journal of biomedical science. 2009;16:31. doi: 10.1186/1423-0127-16-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dellaire G, Kepkay R, Bazett-Jones DP. High resolution imaging of changes in the structure and spatial organization of chromatin, gamma-H2A.X and the MRN complex within etoposide-induced DNA repair foci. Cell cycle. 2009;8:3750–69. doi: 10.4161/cc.8.22.10065. [DOI] [PubMed] [Google Scholar]
- 14.Safina A, Cheney P, Pal M, Brodsky L, Ivanov A, Kirsanov K, et al. FACT is a sensor of DNA torsional stress in eukaryotic cells. Nucleic Acids Res. 2017;45:1925–45. doi: 10.1093/nar/gkw1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gasparian AV, Burkhart CA, Purmal AA, Brodsky L, Pal M, Saranadasa M, et al. Curaxins: anticancer compounds that simultaneously suppress NF-kappaB and activate p53 by targeting FACT. Sci Transl Med. 2011;3:95ra74. doi: 10.1126/scitranslmed.3002530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Formosa T. The role of FACT in making and breaking nucleosomes. Biochim Biophys Acta. 2013;1819:247–55. [PubMed] [Google Scholar]
- 17.Winkler DD, Muthurajan UM, Hieb AR, Luger K. Histone chaperone FACT coordinates nucleosome interaction through multiple synergistic binding events. The Journal of biological chemistry. 2011;286:41883–92. doi: 10.1074/jbc.M111.301465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tsunaka Y, Fujiwara Y, Oyama T, Hirose S, Morikawa K. Integrated molecular mechanism directing nucleosome reorganization by human FACT. Genes Dev. 2016;30:673–86. doi: 10.1101/gad.274183.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Valieva ME, Gerasimova NS, Kudryashova KS, Kozlova AL, Kirpichnikov MP, Hu Q, et al. Stabilization of Nucleosomes by Histone Tails and by FACT Revealed by spFRET Microscopy. Cancers (Basel) 2017:9. doi: 10.3390/cancers9010003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Garcia H, Miecznikowski JC, Safina A, Commane M, Ruusulehto A, Kilpinen S, et al. Facilitates chromatin transcription complex is an “accelerator” of tumor transformation and potential marker and target of aggressive cancers. Cell Rep. 2013;4:159–73. doi: 10.1016/j.celrep.2013.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 2010;26:589–95. doi: 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, et al. Model-based analysis of ChIP-Seq (MACS) Genome Biol. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ross-Innes CS, Stark R, Teschendorff AE, Holmes KA, Ali HR, Dunning MJ, et al. Differential oestrogen receptor binding is associated with clinical outcome in breast cancer. Nature. 2012;481:389–93. doi: 10.1038/nature10730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pommier Y, Sun Y, Huang SN, Nitiss JL. Roles of eukaryotic topoisomerases in transcription, replication and genomic stability. Nat Rev Mol Cell Biol. 2016;17:703–21. doi: 10.1038/nrm.2016.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang F, Kemp CJ, Henikoff S. Doxorubicin enhances nucleosome turnover around promoters. Curr Biol. 2013;23:782–7. doi: 10.1016/j.cub.2013.03.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pang B, Qiao X, Janssen L, Velds A, Groothuis T, Kerkhoven R, et al. Drug-induced histone eviction from open chromatin contributes to the chemotherapeutic effects of doxorubicin. Nat Commun. 2013;4:1908. doi: 10.1038/ncomms2921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Biebricher AS, Heller I, Roijmans RF, Hoekstra TP, Peterman EJ, Wuite GJ. The impact of DNA intercalators on DNA and DNA-processing enzymes elucidated through force-dependent binding kinetics. Nat Commun. 2015;6:7304. doi: 10.1038/ncomms8304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Musinova YR, Lisitsyna OM, Golyshev SA, Tuzhikov AI, Polyakov VY, Sheval EV. Nucleolar localization/retention signal is responsible for transient accumulation of histone H2B in the nucleolus through electrostatic interactions. Biochim Biophys Acta. 2011;1813:27–38. doi: 10.1016/j.bbamcr.2010.11.003. [DOI] [PubMed] [Google Scholar]
- 29.Bruhn SL, Housman DE, Lippard SJ. Isolation and characterization of cDNA clones encoding the Drosophila homolog of the HMG-box SSRP family that recognizes specific DNA structures. Nucleic Acids Res. 1993;21:1643–6. doi: 10.1093/nar/21.7.1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bruhn SL, Pil PM, Essigmann JM, Housman DE, Lippard SJ. Isolation and characterization of human cDNA clones encoding a high mobility group box protein that recognizes structural distortions to DNA caused by binding of the anticancer agent cisplatin. Proceedings of the National Academy of Sciences of the United States of America. 1992;89:2307–11. doi: 10.1073/pnas.89.6.2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Gariglio M, Foresta P, Sacchi C, Lembo M, Hertel L, Landolfo S. Suppression of high mobility group protein T160 expression impairs mouse cytomegalovirus replication. J Gen Virol. 1997;78(Pt 3):665–70. doi: 10.1099/0022-1317-78-3-665. [DOI] [PubMed] [Google Scholar]
- 32.Yarnell AT, Oh S, Reinberg D, Lippard SJ. Interaction of FACT, SSRP1, and the high mobility group (HMG) domain of SSRP1 with DNA damaged by the anticancer drug cisplatin. The Journal of biological chemistry. 2001;276:25736–41. doi: 10.1074/jbc.M101208200. [DOI] [PubMed] [Google Scholar]
- 33.Safina A, Garcia H, Commane M, Guryanova O, Degan S, Kolesnikova K, et al. Complex mutual regulation of facilitates chromatin transcription (FACT) subunits on both mRNA and protein levels in human cells. Cell cycle. 2013;12:2423–34. doi: 10.4161/cc.25452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cer RZ, Donohue DE, Mudunuri US, Temiz NA, Loss MA, Starner NJ, et al. Non-B DB v2.0: a database of predicted non-B DNA-forming motifs and its associated tools. Nucleic Acids Res. 2013;41:D94–D100. doi: 10.1093/nar/gks955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rivera JM, Martin-Hidalgo M, Rivera-Rios JC. An aquatic host-guest complex between a supramolecular G-quadruplex and the anticancer drug doxorubicin. Organic & biomolecular chemistry. 2012;10:7562–5. doi: 10.1039/c2ob25913c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Manet I, Manoli F, Zambelli B, Andreano G, Masi A, Cellai L, et al. Affinity of the anthracycline antitumor drugs Doxorubicin and Sabarubicin for human telomeric G-quadruplex structures. Physical chemistry chemical physics : PCCP. 2011;13:540–51. doi: 10.1039/c0cp00898b. [DOI] [PubMed] [Google Scholar]
- 37.Luger K, Mader AW, Richmond RK, Sargent DF, Richmond TJ. Crystal structure of the nucleosome core particle at 2.8 A resolution. Nature. 1997;389:251–60. doi: 10.1038/38444. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.