

Abstract
In-cell NMR spectroscopy was used to screen for drugs that disrupt the interaction between prokaryotic ubiquitin like protein, Pup, and mycobacterial proteasome ATPase, Mpa. This interaction is critical for Mycobacterium tuberculosis resistance against nitric oxide (NO) stress; interruption of this process was proposed as a mechanism to control latent infection. Three compounds isolated from the NCI Diversity set III library rescued the physiological proteasome substrate from degradation suggesting that the proteasome degradation pathway was selectively targeted. Two of the compounds bind to Mpa with sub-micromolar to nanomolar affinity, and all three exhibit potency toward mycobacteria comparable to antibiotics currently available on the market, inhibiting growth in the low micromolar range.
Graphical abstract
The multidrug resistance of bacterial pathogens places a strong demand on developing new antibacterial agents including those selected by using high throughput screening (HTS).1,2 Typical target-based HTS programs utilize a combination of cell-based and biochemical assays to select potent binders to a target molecule or molecular complex from a large compound library.3 Cell-based assays, such as reporter-based or phenotypic assays, provide indirect readout of binding events, and biochemical assays interrogate binding directly by measuring purified protein activities (Figure 1). HTS is a mainstay of drug discovery, even though the attrition of drug candidates selected by HTS in clinical trials remains high.3
Figure 1.
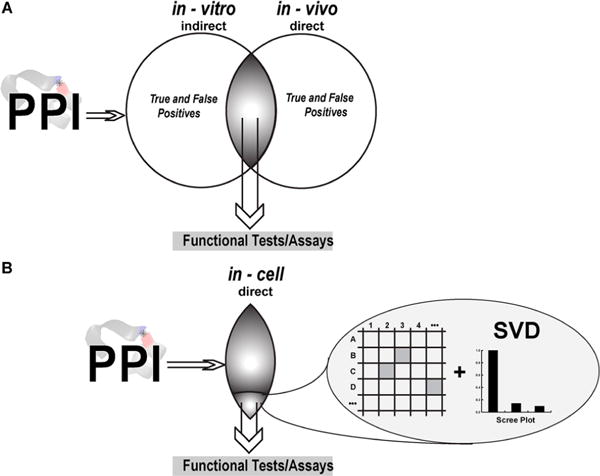
High throughput screening using in-cell NMR. (A) Screening for compounds that disrupt protein–protein interactions (PPIs) in vitro and in vivo produces hits with positive or negative responses. (B) Examining PPI targets by using in-cell NMR simultaneously affords the benefits of both in vitro and in vivo studies by providing amino acid residue specific information while the system remains in a physiologically relevant environment. Spectral data are analyzed by singular value decomposition (SVD) to ensure that the observed changes are real and not a result of an altered cellular environment.
Protein–protein interaction (PPI) targets, when one of the proteins is an intrinsically disordered protein (IDP), have traditionally been challenging for HTS, despite the therapeutic relevance and untapped abundance of IDPs. Inhibition of large PPI surfaces by small molecules is difficult, and in vitro biochemical assays may not identify the physiologically relevant PPIs present in-cell.4 As a result, potential hits from HTS contain a large number of false positives (Figure 1A) that complicate identifying the most potent molecules and justify the need for new HTS approaches to alleviate the difficulties in selecting biologically active compounds.4
In-cell NMR-based technology,5–12 in particular STructural INTeractions by in-cell NMR, STINT-NMR,13,14 was developed to study protein interactions in live cells at amino acid residue resolution.5,15–19 The method entails sequentially expressing two (or more) proteins within a single cell in a time-controlled manner and monitoring their interactions by using in-cell NMR spectroscopy. A series of in-cell NMR spectra collected at different interactor protein concentrations are analyzed by using singular value decomposition, SVD.20,21 The analysis objectively identifies the amino acid residues involved in the principal binding mode of a target protein with its interactor (Figure 1B).
STINT-NMR presents a unique tool for drug screening against PPI targets22 by providing a physiological environment to study protein–protein complexes and identifying the binding epitope of a small molecule on the target protein. The method differs from previously described in-cell NMR screening schemes in which small molecules rather than protein targets are observed.23,24 STINT-NMR thus combines the strength of in vitro and in vivo screening and significantly increases the chances of finding biologically potent compounds, including allosteric inhibitors of PPI targets (Figure 1B).
Here we developed STINT-NMR based screening (Supplementary Table 1) to identify library compounds that disrupt the proteasome system in mycobacteria. The proteasome system is a potential target for controlling latent Mycobacterium tuberculosis, (Mtb) infection.25 Disruption of the system by small molecule inhibitors of the mycobacterial proteasome,26 such as peptide aptamers targeting Pup16 and deletion mutations of proteasome proteins,25 results in killing of mycobacteria under nitric oxide stress.
We specifically targeted PPIs between mycobacterial ATPase, Mpa, and prokaryotic ubiquitin like protein, Pup. Mpa is a part of the proteasome system and functions to unfold substrates prior to proteasome degradation.27 Pup is covalently attached to the proteasome substrates that are tagged for degradation.28 Unlike ubiquitin, Pup is an IDP and folds upon binding to Mpa.29,30
RESULTS
In-Cell NMR-Based Library Screening
To prepare the in-cell [U-15N]Pup–Mpa complex, two compatible plasmids, pASK-Pup and pRSF-Mpa, were cotransformed into E. coli strain BL(DE3) Codon+ for sequential overexpression.17 [U-15N]Pup was overexpressed for 2 h, and the cells were pelleted, washed in unlabeled M9, and resuspended in LB medium. Overexpression of unlabeled Mpa proceeded for an additional 16 h. Changes in the in-cell NMR spectrum of [U-15N]Pup due to sequential overexpression of Mpa indicated the formation of a Pup–Mpa complex31 (Figure 2A).
Figure 2.
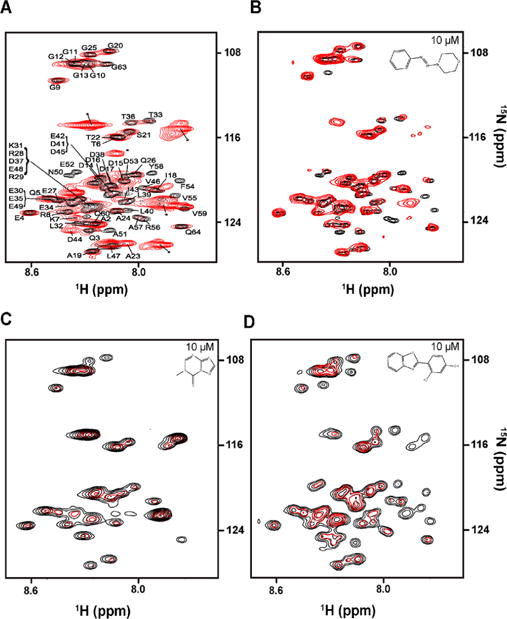
Screening by in-cell STINT-NMR. (A) Overlay of in-cell 1H–15N HSQC spectra of [U-15N]Pup overexpressed for 2 h (black) and [U-15N]Pup–Mpa complex (red) following 8 h of Mpa overexpression. Peaks labeled with an asterisk are metabolites. (B) Overlay of in-cell 1H–15N HSQC spectra of the [U-15N]Pup–Mpa complex containing 0.1% v/v DMSO (black) and 10 μM MTBA (red). (C) Overlay of in-cell 1H–15N HSQC spectra of the [U-15N]Pup–Mpa complex containing 0.1% v/v DMSO (black) and 10 μM MTBB (red). (D) Overlay of in-cell 1H–15N HSQC spectra of the [U-15N]Pup–Mpa complex containing 0.1% v/v DMSO (black) and 10 μM MTBC (red).
In-cell STINT-NMR was used to screen a total of 1597 compounds from the NIH/NCI Diversity Set III (dtp.cancer.gov). The library was derived from 140 000 compounds by selecting for a diversity of pharmacophores (hydrogen bond donor or acceptor, positive charge, aromatic, hydrophobic, acid, or base) and by removing pharmacologically undesirable features, such as obvious leaving groups, weakly bonded heteroatoms, organometallics, and polycyclic aromatic hydrocarbons. The library compounds are stable in human plasma. Because NMR chemical shifts are exquisitely sensitive to protein–protein and protein–compound binding, any compound that passes through the bacterial cell wall and plasma membrane and binds to the Pup–Mpa complex will cause changes in the in-cell NMR spectrum of [U-15N]Pup–Mpa (Figure 2A).
Since an in-cell NMR experiment requires about an hour to collect a spectrum, it is impractical to screen one molecule at a time. Instead, we used a previously developed matrix method22 to screen the library. Matrix samples of the library compounds were prepared by combining all of the compounds located in a particular column or row of a plate for a total of 18 samples per 10 × 8 matrix plate. This resulted in a 4-fold reduction in the number of samples tested in the primary screening.
An in-cell STINT-NMR spectrum was collected for each matrix sample mixture. Two control spectra per plate were acquired to account for the different DMSO concentrations in the rows and columns. All spectra were individually overlaid with the controls to detect changes in chemical shifts or peak intensities. If two samples from a particular row or a column exhibited similar spectral changes, it was assumed that the active compound was located at the intersection between the column and the row.22 Thirty compounds were identified and individually screened to verify that the observed in-cell NMR spectral changes match those exhibited by the mixtures. Three compounds showed the same changes in the in-cell NMR spectra as the mixtures and were selected for the secondary screening. One candidate, NSC 136065 (MTBA), elicited chemical shift changes (Figure 2B), while two others, NSC 145180 (MTBB) and NSC 33005 (MTBC), resulted in changes to peak intensities (Figure 2C,D).
Secondary Screening Confirms Dose Response of Candidate Compounds
The candidate compounds identified in the first round of screening were individually titrated in-cell against the Pup–Mpa complex to establish a dose response relationship with the changes observed in the [U-15N]Pup–Mpa STINT-NMR spectrum. Spectra were acquired at 10 μM, 20 μM, and 60 μM concentrations of each compound and analyzed by using SVD.32 The analysis differentiates between concentration-dependent and concentration-independent changes in chemical shifts and peak intensities. The candidate compounds did not perturb the in vitro NMR spectrum of Pup alone (Supplemental Figure 1) confirming that direct binding of the candidate compound requires a Pup–Mpa interaction.
The results of SVD analysis for the 3 compounds are shown in Figure 3. An abrupt drop in the singular values of Scree plots following the first binding mode indicates the presence of a single principal binding mode (Figure 3A–C). Pup residues that comprise the principal binding mode with MTBA, MTBB, and MTBC are indicated in Figure 3D–F. Mapping these residues onto the Pup–Mpa complex shows that the α-helical segment and C-terminal tail of Pup participate in the interaction between the selected compounds and the PPI target (Figure 3G,H). Of the 30 compounds subjected to secondary screening, 15 exhibited a single principal binding mode and were further tested for activity in vivo and in vitro.
Figure 3.
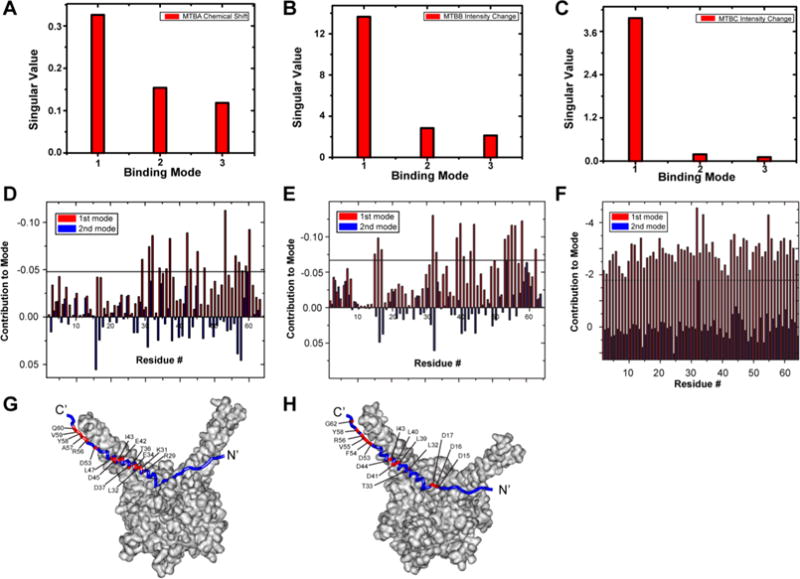
SVD analysis of selected compounds identify Pup residues involved in small molecule binding. (A, B, C) Scree plots of SVD analysis of matrices of change in [U-15N]-Pup chemical shifts due to MTBA (A), changes in [U-15N]-Pup peak intensities due to MTBB (B), and changes in peak intensities due to MTBC (C). (D, E, F) The weighted contribution of each Pup residue to the first (red) and second (blue) binding modes in response to MTBA (D), MTBB (E), and MTBC (F). There are two bars per residue. The largest weighted contribution from the second binding mode was used as a threshold to highlight the amino acids most strongly implicated in the binding. (G, H) Pup residues involved in MTBA (G) and MTBB (H) (red) binding are mapped onto the Pup–Mpa complex. Some labeled residues are obscured due to image orientation. The Pup–Mpa structure (PDB code 3M9D) was constructed by using Accelrys Discovery Studio 2.5.
Selected Compounds Inhibit Mycobacterial Growth under Nitric Oxide Stress
The ability of the 3 candidate compounds to inhibit cell growth under nitric oxide stress was examined using the nonvirulent Bacillus Calmette–Guérin, BCG, strain of Mycobacterium bovis. BCG cells were exposed to 100 μM of candidate compounds. To determine the viability of the cell culture, cell dilutions were spot plated onto solid mycobacterial agar. Rifampicin, which kills Mtb by inhibiting RNA polymerase33 and is used in treating Mtb infection, and the commercially available proteasome inhibitor bortezomib, Btz, which is used in cancer clinical trials,34 were used as positive controls. Three out of the 15 compounds tested, MTBA, MTBB, and MTBC, exhibited activity against BCG (Figure 4A). Inhibition of BCG growth by these compounds was strictly dependent on the presence of nitric oxide stress (Supplemental Figure 2).
Figure 4.
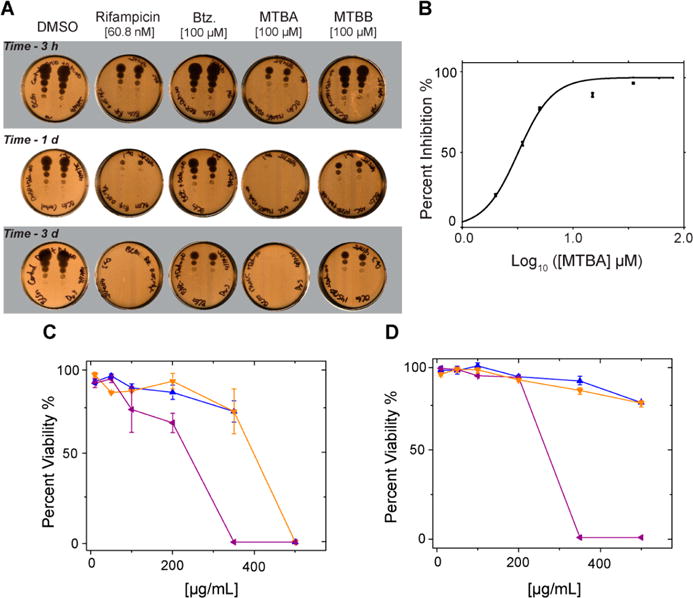
Activity assay secondary screening. (A) Cell viability spot plate assay. Cells were treated with DETA-NO and either DMSO, Rifampicin, Btz, MTBA, or MTBB, for up to 6 days. Culture samples were collected at different times, diluted, plated, and incubated for 2–3 weeks prior to scanning and analysis: 3 h, 1 day, and 3 day time points are shown. (B) Inhibition of BCG cell growth under nitric oxide stress by MTBA. Colony forming unit (CFU) plate assay yields an IC50 of 8 μM. (C, D) Cytotoxic effects of MTBA (blue), MTBB (orange), and MTBC (purple) after 24 h exposure to A549 epithelial cells (C) and mouse macrophage cell line J774.16 (D).
MTBA reduced cell viability within 3 h and exhibited a maximum effect 24 h post-treatment. Importantly, 100 μM MTBA reduced cell viability comparable to that of rifampicin, more than 0.05 μg/mL (~60 nM) of rifampicin after 24 h, and significantly more than 100 μM Btz (Figure 4A). Colony forming unit (CFU) plating assay determined the IC50 for MTBA to be 8 ± 2 μM against BCG under nitric oxide stress (Figure 4B). The IC50 of Btz and rifampicin against BCG are 50 μM26 and 0.3 μM,35 respectively.
MTBB reduced cell viability after 24 h with the maximum effect occurring 48 h post-treatment. At 4 days post-treatment, 100 μM MTBB was more effective than 100 μM Btz at inhibiting cell growth. MTBC inhibited the growth of BCG after 8 days (Supplemental Figure 3). Since by this time the nitric oxide in the cultures was exhausted, it is likely that the mechanism of MTBC activity is different from that of MTBA and MTBB.
The cytotoxicity of the three compounds against mammalian cells was measured by using human epithelial A549 and murine macrophage J774.16 cells (Figure 4C,D). Compounds MTBA and MTBB were well tolerated by both cell lines up to and beyond 200 μg/mL, which is more than ten times the IC50 for these compounds. MTBC was well tolerated below 200 μg/mL and precipitated at greater than 200 μg/mL concentration.
Selected Compounds Inhibit Mycobacterial Proteasome Pathway
Two approaches were used to verify that the selected compounds specifically target the mycobacterial proteasomal pathway. In the first, the physiological disposition of the proteasome substrate FabD was monitored in the presence or absence of the selected compounds by using BCG that expresses FLAG-fabD from a chromosomally integrated vector. FLAG-fabD is constitutively expressed in mycobacteria at very low levels and is readily detected when the mycobacterial proteasome is inactive.16,36 Western blotting showed that all three compounds, MTBA, MTBB, and MTBC, rescued FabD from proteasome degradation as effectively as the proteasome inhibitor Btz (Figure 5A). These results confirm that the selected compounds render the proteasome inactive.
Figure 5.
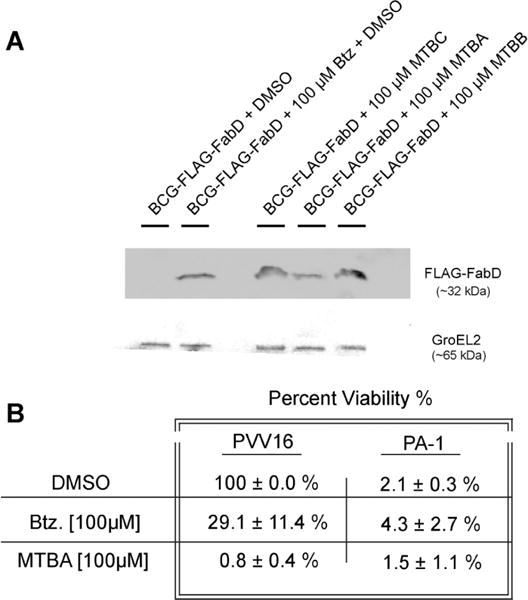
Selected compounds specifically target the mycobacterial proteasome pathway. (A) BCG proteasome is inactivated by selected compounds. Western blot of samples collected at time of maximal effect (refer to text and Figure 4A): DMSO, 6 day; Btz, 4 day; MTBC, 6 day; MTBA, 1 day; MTBB, 4 day. (B) Percent viability of BCG cells containing empty vector, BCG–pVV16, or BCG–pVV16-PA1 were treated with DETA-NO and either DMSO, Btz, or MTBA for 4 days.
In the second approach, the extent to which the most potent selected compound, MTBA, inhibited mycobacterial growth via other cellular pathways was examined. This was accomplished by using peptide aptamer PA1, which binds specifically to Pup.16,37 PA1 potently inhibits the function of the BCG proteasome,16 and the PA1 interaction surface overlaps with the Mpa helical binding surface on Pup.17,29 PA1 was cloned into a constitutively active shuttle vector, pVV16, which confers kanamycin and hygromycin resistance (BEI Resources). Previously we showed that BCG transformed with plasmid expressing PA1, BCG–pVV16-PA1, experienced strong growth inhibition under nitric oxide stress compared to cells transformed with empty vector pVV16.15 CFU plating was used to quantify the effect of 100 μM MTBA on the BCG–pVV16 and BCG–pVV16-PA1 cells under nitric oxide. Expression of PA1 reduced viability of vehicle control, DMSO, cells by 98%, and the addition of 100 μM Btz did not further affect overall cell viability (Figure 5B). BCG– pVV16 treated with Btz also showed ~70% growth inhibition relative to the vehicle control. This was expected since both Btz and PA1 affect the proteasomal pathway. BCG–pVV16-PA1 cells treated with 100 μM MTBA under nitric oxide stress also showed no additional growth inhibition. The results suggest that MTBA inhibits growth through the proteasomal pathway.
Selected Compounds Exhibit Nanomolar to Sub-micromolar Affinities for Mtb Proteasome ATPase, Mpa
Changes in the in-cell NMR spectrum of [U-15N]Pup relied on the formation of a Pup–Mpa complex.17 The selected compounds, however, may bind to either Pup or Mpa or both. Adding 100 μM MTBA, MTBB, or MTBC to purified 10 μM [U-15N]Pup did not change the 1H–15N heteronuclear single quantum coherence, HSQC, NMR spectra (Supplemental Figure 1). To determine if MTBA and MTBB bind to Mpa, tryptophan fluorescence titrations were performed. Only MTBA and MTBB were used in these experiments due to the limited solubility of MTBC. Both compounds bound tightly to Mpa, with resolved dissociation constants, Kd, of 1.0 ± 0.1 nM for MTBA and 0.4 ± 0.1 μM for MTBB (Figures 6A,B). These results suggest that the compounds bind to Mpa and that Pup serves as a reporter of this interaction.
Figure 6.
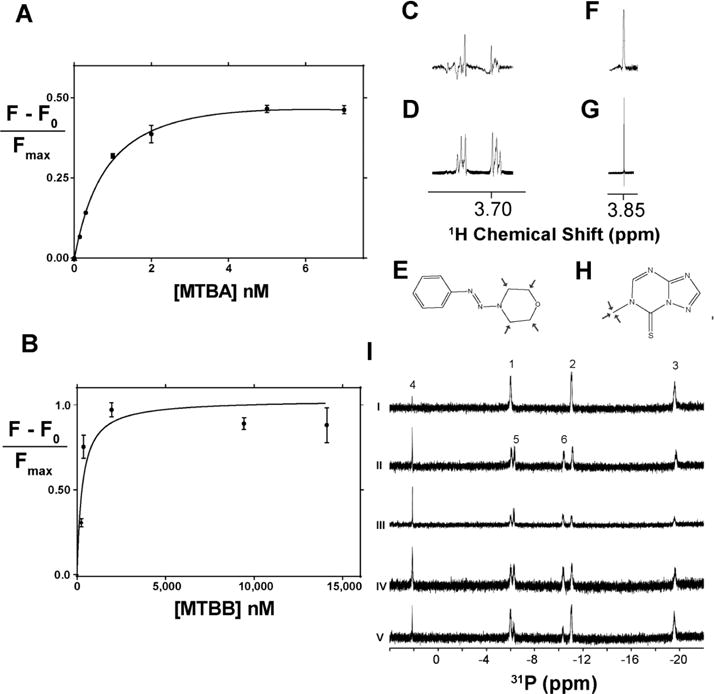
Binding properties of selected compounds. (A, B) Binding isotherms for MTBA (top) and MTBB (bottom) generated from tryptophan fluorescent titrations of Mpa. Dissociation constants were resolved for MTBA, 1.0 +/− 0.1 nM (R2 = 0.99), and MTBB, 0.4 ± 0.1 μM (R2 = 0.92). (C) STD NMR spectral peak of 100 μM MTBA with 10 μM Mpa showing saturation of the morpholine ring signal at 3.7 ppm. (D) 1H peak arising from the morpholine ring of MTBA. (E) Structure of MTBA with arrows highlighting the binding epitope, as determined by STD NMR. (F) STD NMR spectral peak of 100 μM MTBB with 10 μM Mpa showing saturation of the methyl group signal at 3.85 ppm. (G) 1H peak arising from the methyl group of MTBB. (H) Structure of MTBB with arrows highlighting the binding epitope, as determined by STD NMR. Due to extreme hydrophobicity, MTBC was not titrated. (I)31P NMR ATPase assay shows that selected compounds do not inhibit ATP hydrolysis. 1, 2, and 3 refer to γ-, α-, and β-ATP phosphates, respectively, and 4 is an inorganic phosphate signal. Upon hydrolysis, β- and α-ADP phosphate signals, 5 and 6, respectively, arise as ATP is hydrolyzed. (i) ATP in ATPase buffer; (ii) ATP and Mpa,; (iii) Mpa, ATP, and 100 μM MTBA; (iv) Mpa, ATP, and 100 μM MTBB; (v) Mpa, ATP, and 100 μM MTBC.
Saturation transfer difference NMR (STD NMR) was used to determine the epitope of the compounds used to bind to Mpa. In STD NMR, protons on candidate compounds that are located within 5 Å from those on Mpa exhibit STD NMR peaks (Figure 6C,F). Proton peaks from the morpholine group of MTBA (3.7 ppm) and the methyl group of MTBB (3.85 ppm) correlated with STD NMR signal (Figure 6E,H). These results suggest that both polar and hydrophobic moieties are important for Mpa binding.
Phosphorus NMR was used to examine if the compounds affect the ATPase function of Mpa. The31P NMR spectrum of ATP exhibits three peaks corresponding to α, β, and, γ phosphates (Figure 6I). Adding 10 μM of Mpa to 1 mM ATP results in the appearance of α and β phosphate peaks from ADP and a concomitant disappearance of ATP phosphate peaks. Adding 100 μM of MTBA, MTBB, or MTBC to Mpa and ATP did not prevent the appearance of ADP peaks. These results confirm that the compounds do not inhibit Mpa ATPase function and do not block ATP binding sites on Mpa.
DISCUSSION
In-cell NMR presents a unique opportunity for screening library compounds against PPIs by directly observing in-cell protein NMR chemical shift changes due to compound binding (Figure 1).38 Importantly, in-cell NMR based screening is not affected by common pan-assay interference compounds,39 or PAINS, which plague screening programs based on fluorescence readout. PAINS interact with the entire proteome and are not expected to cause specific effects on the in-cell NMR spectra of target molecules.
Primary screening using in-cell NMR selects for compounds with highly desirable cell penetration properties since only compounds that pass through the cell wall and membrane can influence PPIs.22,40 The success rate of in-cell NMR screening was similar to that of typical HTS, which is ~1%. The screening throughput was medium to low, ~100 compounds/day, suggesting that only very focused fragment based libraries can be used for primary in-cell NMR screening. To increase the throughput, mixtures of compounds can be screened by STINT-NMR. In contrast, traditional in-cell NMR based small molecule screening requires testing one compound at a time.23,24
Library screening against the mycobacterial proteasome target Pup–Mpa complex identified three compounds, MTBA, MTBB, and MTBC, that affect in-cell Pup–Mpa binding, inhibit degradation of proteasome substrates, and result in mycobacterial cell death under nitric oxide stress conditions. Importantly, the selected compounds are nontoxic to mammalian cells. SVD analysis of in-cell NMR spectra identified interaction surfaces on Pup–Mpa that are affected by the binding of each compound. MTBA and MTBB affect the C-terminal half of Pup, which was previously identified as a binding surface for Mpa.17,20,29
Since only selected Pup residues along the Pup–Mpa binding interface are affected (Figure 3G,H), we hypothesize that MTBA and MTBB do not directly compete with Pup for Mpa but bind to an allosteric site located away from the Pup–Mpa interface. MTBC binding affected all Pup residues (Figure 3F), possibly because of strengthened Pup–Mpa interactions. Importantly, none of these compounds changed the enzymatic activity of Mpa (Figure 6C).
All three compounds rescued the physiological proteasome substrate from degradation suggesting that the proteasome degradation pathway was targeted (Figure 5). The selectivity of compound targeting was assessed by using a previously identified peptide aptamer, PA1, that binds to Pup and inhibits mycobacterial growth under nitric oxide stress16 by precisely targeting the proteasome degradation pathway. The simultaneous expression of PA1 with exposure to the candidate compounds allowed us to test the specificity of each compound’s mode of action. Since combining MTBA, MTBB, or MTBC and PA1 did not further inhibit mycobacterial growth compared to PA1 alone, we assumed that the MTBA, MTBB, and MTBC mode of action is limited to disrupting the Pup–Mpa complex.
In vitro characterization of binding to purified Pup and Mpa revealed that MTBA and MTBB bind specifically to Mpa (Figure 6). The low solubility of MTBC prevented characterization of this compound in vitro. By using STD NMR, the binding epitopes of MTBA and MTBB, which will be used in a subsequent medicinal chemistry program to develop effective therapeutic agents against Mtb, were identified. The binding of MTBB to Mpa was sub-micromolar, and the binding of MTBA to Mpa was sub-nanomolar, which is more than 1000-fold lower than the IC50 of the compound, 8 μM. The discrepancy between binding affinity measured in vitro and estimated from IC50 in vivo suggests that in the mycobacterial cytosol Mpa may undergo a conformational change that alters its binding affinity for MTBA.10,18,41,42 This result highlights the importance of in-cell screening of the compounds and possible pitfalls in overemphasizing in vitro data to rationalize functional results.
Importantly, both MTBB and MTBC exhibit potency toward mycobacteria comparable to antibiotics currently available on the market. The described in-cell NMR based screening technology is broadly applicable to select a new generation of potent antibacterial agents against multidrug resistant pathogens.
METHODS
Reagents and Chemicals
All chemicals used were Molecular Biology grade or better.
Mycobacterium Growth
The attenuated vaccine (ΔRD1) Bacillus Calmette–Guerin (BCG), Pasteur strain of Mycobacterium bovis was grown as described previously.16 BCG-FLAG-FabD, which expresses FLAG-labeled proteasomal substrate malonyl Co-A acyl carrier protein transacylase (FLAG-FabD) from a chromosomally integrated vector was previously prepared.15 BCG and BCG-FLAG-FabD were grown in mycomedia (Middlebrook 7H9 medium (Difco), supplemented with 0.5% (v/v) glycerol, 10% (v/v) oleic acid–albumin–dextrose–catalase (OADC), and 0.05% (v/v) Tween 80).43 BCG cells were plated on Middlebrook 7H10 (Difco) plates supplemented with 10% OADC, 0.01% (w/v) cycloheximide (CX). Plates and 7H9 media were supplemented with 25 μg/mL of kanamycin when culturing BCG cells expressing peptide aptamers and empty vector pVV16.
Plasmids
Control vector pVV16 was from BEI Resources. All other plasmids used were previously constructed in our lab.16,17 pASK-Pup17 expresses Pup-GGQ from the tetracycline (tet) promoter/operator, which is induced by tetracycline or anhydrotetracycline. This plasmid confers ampicillin resistance and contains an f1 origin and the tet gene, which codes for Tet repressor. pRSF-Mpa17 expresses Mycobacterium tuberculosis (Mtb) Mpa from the T7 promoter/lac operator (PT7/lacOp), which is induced by isopropyl 1-thio-β-D-galactopyranoside (IPTG). This plasmid confers kanamycin resistance and contains an RSF replication origin and the lacI gene, which encodes for Lac repressor. Plasmid pVV16-PA116 expresses a peptide aptamer (PA) constrained within the active site loop of thioredoxin. The plasmid is constitutively active and confers kanamycin, and hygromycin resistance for selection in E. coli and BCG. BCG cells were transformed by electroporation with vector pVV16 and pVV16-PA1, which overexpresses peptide aptamer-1 using procedures previously described.16
Sequential Overexpression of15N-Pup and Mpa
Plasmids pASK-Pup and pRSF-Mpa were cotransformed into E. coli strain BL21(DE3) Codon+ (Novagen).17 Our method entailed over-expressing and labeling Pup followed by overexpression of unlabeled Mpa. For [U-15N] labeling, cells were grown in minimal medium (M9 salts containing 10 μM calcium chloride, 1 mM thiamine hydrochloride, 2 mM magnesium sulfate, and 0.4% glucose) containing 100 mg/L of ampicillin, 35 mg/L of kanamycin, and 1 g/L of [U-15N]ammonium chloride as the sole nitrogen source. Pup expression was induced with 0.2 mg mL−1 of anhydrotetracycline in dimethylformamide (DMF) at an OD595 of 0.7 and grown for 2 h at 37 °C. A 50 mL [U-15N]Pup control sample was collected, centrifuged, washed twice with 50 mL of NMR buffer (10 mM potassium phosphate pH 6.5), washed twice with 1 mL of NMR buffer containing 10% glycerol, and stored at −80 °C for subsequent NMR analysis. The remainder of the culture was centrifuged, washed twice with M9 salts, and resuspended in fresh unlabeled minimal medium containing 100 mg/L of ampicillin and 35 mg/L of kanamycin to a final OD595 of 0.7. Mpa expression was induced with 1 mM IPTG, and cells were grown for 10 h at 37 °C. The cells were harvested and washed twice with 50 mL of NMR buffer containing 10% glycerol. The cell pellet was resuspended to a final OD595 of 95 with NMR buffer containing 10% glycerol. Aliquots of 250 μL were centrifuged and stored at −80 °C for subsequent NMR analysis. Prior to in-cell NMR, the cryopreserved cells containing the [U-15N]Pup-Mpa complex were extensively washed in NMR buffer to remove proteins that leaked from the cells during cryostorage.
Purification of Pup and Mpa
Overexpressing cells were lysed by sonication at 30% power with 0.3 s ON, 1.0 s OFF using a Branson sonifier, and proteins were purified by Ni-NTA (Qiagen) affinity chromatography under native conditions.17 Pup was dialyzed into NMR buffer. Mpa was dialyzed into ATPase buffer, 50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 20 mM MgCl2, and 1 mM DTT.30
NMR Spectroscopy
To prepare NMR samples, frozen cell aliquots harboring [U-15N]Pup and unlabeled Mpa were thawed on ice for 10 min, washed twice with 1.5 mL of NMR buffer, and resuspended in 500 μL of NMR buffer containing 10% D2O. NMR experiments were performed on a 500 MHz Avance II NMR spectrometer (Bruker) equipped with a TCI cryoprobe. All in-cell and in vitro Pup data were collected at 298 K, which yielded high quality spectra. Spectra were acquired using a Watergate version of a 1H–15N heteronuclear single quantum coherence (HSQC) experiment, recorded with 64 transients as 512 × 64 complex points in the proton and nitrogen dimensions, respectively, apodized with a squared cosine-bell window function and zero-filled to 1024 × 128 points prior to Fourier transformation. The corresponding sweep widths were 12 and 35 ppm in the 1H and15N dimensions, respectively. Spectra were processed by using Topspin 3.2 (Bruker) and CARA.44,45
One dimensional31P NMR was used to measure the ATPase activity of Mpa. All data were collected at 298 K by using a 600 MHz Avance III NMR spectrometer (Bruker) equipped with a QCI-P cryoprobe (Bruker). Each experiment consisted of 256 scans. Spectra were obtained and processed using Topspin 3.2 (Bruker).
Saturation transfer difference (STD) NMR was used to determine the binding epitope on selected compounds.46 Samples consisted of 10 μM Mpa and 100 μM of the compound dissolved in NMR buffer, 10 mM potassium phosphate, pH 6.5, and 10% D2O.17 STD NMR spectra were collected as previously described46 by using a Bruker Avance 700 MHz spectrometer equipped with a z-gradient TXI cryoprobe. The on-resonance irradiation of the protein during the 1D STD NMR experiment was performed at a chemical shift of 1 ppm where no compound resonances were present. Off-resonance irradiation was applied at 44 ppm. A sequence of Gauss shaped pulses with a strength of 86 Hz and a length of 50 ms separated by a 1 ms delay was applied for 2 s during selective presaturation of the protein. All spectra were obtained at 298 K with 2048 scans. The NMR data were processed and analyzed using Topspin 2.1 (Bruker).
ATPase Activity Assay
To test for ATP hydrolysis, 10 μM Mpa in ATPase buffer (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 20 mM MgCl2, 1 mM DTT containing 10% D2O) was incubated for 30 min at RT with either 100 μM buffered ATP or ATP plus 100 μM of candidate compound. To serve as a control, the NMR spectrum of 100 μM buffered ATP was taken last. Upon hydrolysis α- and β-ADP signals arose at −10.3 and −6.2 ppm, respectively. A corresponding increase in the signal was attributed to the generation of inorganic phosphate at 2.3 ppm. α-, β-, and γ-ATP signals were visible at −10.8, −19.5, and −5.8 ppm relative to 85% H3PO4 in H2O.
Library Screening
The NIH/NCI Diversity Set III library consisted of 20 96-well plates each containing 80 compounds, that is, 10 columns and 8 rows. Each compound was solubilized in DMSO at 10 mM concentration and stored at −80 °C. Matrix samples of the library compounds were prepared by combining 2.5 μL of each compound located in a particular column or row of a plate for a total of 18 samples per matrix plate. To preserve the integrity of the compounds, each original 96-well plate was thawed only once to make a matrix plate and a duplicate plate. Because DMSO affects the in-cell spectrum of Pup, a control spectrum of each batch of cells was collected in the presence of 4% and 5% DMSO. These values correspond to the final concentration of DMSO in NMR samples, 500 μL total, prepared from matrix samples derived from columns (8 × 2.5 μL) and rows (10 × 2.5 μL) of the library plates. The final concentration of each compound in the matrix sample was 50 μM.
Data Analysis
NMR spectral data was analyzed as previously reported.10,16,20 To reassign the in-cell Mpa–[U-15N]Pup peaks that changed position from in vitro assignments, minimum chemical shift changes were assumed to occur in both the proton and nitrogen dimensions.47 Chemical shift changes were calculated as Δ = (δH2 + (δN/4)2)1/2, where δH(N) represents the change in hydrogen and nitrogen chemical shifts in the absence and presence of a small molecule. Changes in intensity were calculated by using ΔI = (I/Iref)bound − (I/Iref)free, where (I/Iref)free is the scaled intensity of an individual peak in the in-cell spectrum of Mpa–[U-15N]Pup in the absence of a small molecule, (I/Iref)bound is the scaled intensity of individual peaks in the in-cell spectrum of Mpa–[U-15N]Pup in the presence of a small molecule, and Iref is a glutamine side chain peak at 7.45 and 112.5 ppm in the proton and nitrogen dimensions, respectively, that does not shift in the presence of a small molecule. The data consisting of either the chemical shift or intensity changes due to adding small molecule compounds were compiled into matrix M. Matrix M was assembled in Excel (Microsoft, Inc.), exported as an ASCII text file, and read into MATLAB (Mathworks, Inc.). Singular value decomposition, SVD, of matrix M was accomplished by using the [U, S, V] = svd[M] command. The output matrices generated, U, S, and V, are the left singular vectors, the singular value matrix, and right singular vectors, respectively. Scree plots of singular values were used to visualize the contribution of each binding mode to M.16 The threshold to determine the amino acids involved in the changes in quinary interactions was set to the maximum contribution of the second binding mode.
Spot Plate Assay
A spot plate assay was used to determine the efficacy of selected compounds to inhibit cell growth. BCG, BCG–pVV16, BCG–pVV16-PA1, and BCG–pVV16-PA3 cells were grown in mycomedium43 at 37 °C for 7–10 days to late log (stationary) phase. To prepare samples, the cell culture was gently sonicated in a cup horn to remove clumps, and the concentration was adjusted to OD600 = 1.25 using mycomedium. Two milliliter aliquots were transferred to each well of a 6-well plate. DMSO (1%), DETA-NO (250 μM) or water, and selected compounds (100 μM) or rifampicin (50 ng/mL) or Btz (100 μM) were added to each well, and the plates were incubated at 37 °C for 4 days. Expression of PA1 and PA3 from pVV16 is constitutive. Samples (150 μL) were removed at 3 h (zero tp) and 1, 2, 3, 4, 5, and 6 days, briefly sonicated, and serially diluted into mycomedium to produce dilutions from 10−1 to 10−7. Five microliter spots were plated in duplicate on Middlebrook 7H11 plates utilizing a multichannel pipet and incubated for 2–3 weeks at 37 °C. Plates were scanned (Microtek) to digitize the images for analysis.
Cytotoxicity of Compounds against A549 and J774.16 Cells
J774.16 mouse BALB/c monocyte macrophage cells were grown in DMEM supplemented 20% FBS, 5% NCTC-109, and 2 mM L-glutamine. A549 human epithelial cells were grown in EMEM supplemented with 10% FBS and 2 mM L-glutamine. Cell lines were grown to 90% confluence in 5% CO2 at 37 °C using a water jacketed incubator before being split and plated into 96-well plates (Corning). Plated cells were incubated for 24 h with either Triton X-100, which served as a killing control, DMSO as a vehicle control, or serial dilutions up to 500 μg/mL of MTBA, MTBB, or MTBC. Triton X-100 killed all cells at 0.1%. Trypan blue (0.4% in PBS) was added to each well. Individual wells of stained cells were counted under an Axiovert 25 Phase Contrast Inverted Microscope (Zeiss). Percent viability was determined by dividing the number of live cells by the total number of cells and multiplying by 100%. Counts were performed in triplicate, where the number of cells was divided by the sample volume. Data was plotted using OriginLab 9 (OriginLab).
Determination of IC50
A dose–response assay was performed to determine the half maximal inhibitory concentration, IC50, of MTBA against BCG. BCG cells were grown for 7–10 days and diluted to an OD600 of 1.25. Two milliliter aliquots of the cell suspension were portioned into a 6-well plate and exposed to 250 μM DETA-NO and MTBA at concentrations of 0, 2, 3.5, 5, 15, 35, 60, and 80 μM. After 24 h of exposure, BCG cells were collected, gently sonicated to remove clumps, and sequentially diluted 100-fold to create 100, 10−2, 10−4, 10−6, and 10−8 dilutions. Dilutions were plated by spreading 10 or 100 μL, in duplicate, onto 7H11 plates supplemented with CX. After 3 weeks, CFUs were counted, and the CFU/mL for each concentration of MTBA was averaged and compared to untreated BCG cells.
Percent inhibition was determined by taking the difference between untreated (0 μM MTBA) cells and treated cells (2–80 μM MTBA) and dividing the difference by the untreated CFU/mL average. To resolve a value for IC50, a plot of percent inhibition, Y, versus MTBA concentration was fit to
by using GraphPad Prism 6.
Western Blotting
BCG-FLAG-FabD cultures were grown in mycomedia43 at 37 °C for 7–10 days to late log (stationary) phase for Western blot analyses. To inhibit the BCG-FLAG-FabD proteasome, cultures were diluted to an OD600 of 1.25, 100 μM Btz was added, and the cultures were grown for an additional 4 days.
Two milliliters of culture was collected, washed with phosphate buffered saline (PBS) containing protease inhibitor, resuspended in Tris-sodium dodecyl sulfate (Tris/SDS) buffer (50 mM Tris-HCl, pH 8.0, 0.3% (w/v) SDS) to 0.25 mg/μL by weighing the cell pellet, and sonicated.16 Sonicated samples were frozen and thawed 10 times, and the sonication was repeated to lyse the bacteria. SDS (1%) and β-mercaptoethanol (0.0125%) were added, and the lysates were heated at 37 °C for 5 min, clarified by centrifugation and stored at −80 °C for subsequent analysis.
Thawed lysates were assayed for total protein by using the RC DC Protein Assay (Bio-Rad) and a SynergyH1 microplate reader (BioTek) to ensure even loading of samples. Lysates were immunoblotted and probed for FLAG-FabD with anti-FLAG horseradish peroxidase (HRP)-conjugated polyclonal antibody (Cell Signaling) or for GroEL2 with anti-groEL2 (Abcam) monoclonal primary antibody (Novagen) and HRP-conjugated goat anti-mouse IgG (Amersham) secondary antibody. Blots were incubated in Clarity Western ECL Substrate (Bio-Rad) for 5 min and imaged on a ChemiDoc MP System (Bio-Rad) by using Quantity One software.
Native Tryptophan Fluorescence Titrations
Native tryptophan fluorescence experiments were conducted using a Horiba Jobin Yvon Fluorolog-3–22 spectrofluorometer equipped with a 3 × 3 mm2 Spectrosil Quartz cuvette (Starna). Five nanomolar Mpa solutions were individually titrated with 0.05–50 nM MTBA or 50–15000 nM MTBB in 200 μL of ATPase buffer without DTT. The excitation and emission wavelengths were 285 and 352 nm, respectively. Dissociation constants, Kd, were estimated from the changes in peak fluorescence intensities as a function of Mpa concentration by using Prism 6 software (GraphPad). Data were fit to the equation (F − F0)/Fmax = [compound]/(Kd + [compound]) where F is the fluorescence intensity at a given Mpa concentration, F0 is the fluorescence intensity of the compound, and Fmax is the maximum fluorescence intensity of 5 nM Mpa.
Peptide Aptamer Experiment
BCG cells containing empty vector pVV16 and peptide aptamer, PA1, were grown in mycomedium at 37 °C overnight. Cells were plated on Middlebrook 7H10 (Difco) plates supplemented with 10% OADC, 0.01% (w/v) cycloheximide, and 25 μg/mL of kanamycin and incubated for 3 weeks at 37 °C. Colonies were grown in 5 mL of mycomedium for 5–7 days at 37 °C for PCR screening and seed stocks. pVV16-PA1 was expressed in BCG under 25 μg/mL of kanamycin selection, as previously described.16
To monitor the growth of BCG during PA expression and compound treatment, cultures were grown for 4 days with 100 μM of MTBA, MTBB, MTBC, Btz, or DMSO, 25 μg/mL of kanamycin, and 250 μM DETA-NO. Colony forming unit plating was carried out on cells containing PA1 or empty vector. Percent viability was determined by the equation
where untreated BCG cells were exposed to DMSO and DETA-NO alone and treated BCG cells were exposed to selected compounds. The data were compiled and graphed using OriginPro 9.0.
Supplementary Material
Acknowledgments
This work was supported by NIH Grant 5R01GM085006 to A.S.
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acschembio.7b00879.
Spectra of Pup alone with compounds, requirement for NO stress in MTBA- and MTBB- mediated BCG cell toxicity, maintenance of BCG growth inhibition post-treatment with MTBC, table of small molecule screening data, and table of compounds that advanced to in vivo screening (PDF)
ORCID
Alexander Shekhtman: 0000-0003-2649-2675
Notes
The authors declare no competing financial interest.
References
- 1.Nikaido H. Multidrug resistance in bacteria. Annu Rev Biochem. 2009;78:119–146. doi: 10.1146/annurev.biochem.78.082907.145923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Macarron R, Banks MN, Bojanic D, Burns DJ, Cirovic DA, Garyantes T, Green DV, Hertzberg RP, Janzen WP, Paslay JW, Schopfer U, Sittampalam GS. Impact of high-throughput screening in biomedical research. Nat Rev Drug Discovery. 2011;10:188–195. doi: 10.1038/nrd3368. [DOI] [PubMed] [Google Scholar]
- 3.Bleicher KH, Bohm HJ, Muller K, Alanine AI. Hit and lead generation: beyond high-throughput screening. Nat Rev Drug Discovery. 2003;2:369–378. doi: 10.1038/nrd1086. [DOI] [PubMed] [Google Scholar]
- 4.Scott DE, Bayly AR, Abell C, Skidmore J. Small molecules, big targets: drug discovery faces the protein-protein interaction challenge. Nat Rev Drug Discovery. 2016;15:533–550. doi: 10.1038/nrd.2016.29. [DOI] [PubMed] [Google Scholar]
- 5.Serber Z, Keatinge-Clay AT, Ledwidge R, Kelly AE, Miller SM, Dotsch V. High-resolution macromolecular NMR spectroscopy inside living cells. J Am Chem Soc. 2001;123:2446–2447. doi: 10.1021/ja0057528. [DOI] [PubMed] [Google Scholar]
- 6.Selenko P, Serber Z, Gadea B, Ruderman J, Wagner G. Quantitative NMR analysis of the protein G B1 domain in Xenopus laevis egg extracts and intact oocytes. Proc Natl Acad Sci U S A. 2006;103:11904–11909. doi: 10.1073/pnas.0604667103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Theillet FX, Binolfi A, Bekei B, Martorana A, Rose HM, Stuiver M, Verzini S, Lorenz D, van Rossum M, Goldfarb D, Selenko P. Structural disorder of monomeric alpha-synuclein persists in mammalian cells. Nature. 2016;530:45–50. doi: 10.1038/nature16531. [DOI] [PubMed] [Google Scholar]
- 8.Sakakibara D, Sasaki A, Ikeya T, Hamatsu J, Hanashima T, Mishima M, Yoshimasu M, Hayashi N, Mikawa T, Walchli M, Smith BO, Shirakawa M, Guntert P, Ito Y. Protein structure determination in living cells by in-cell NMR spectroscopy. Nature. 2009;458:102–105. doi: 10.1038/nature07814. [DOI] [PubMed] [Google Scholar]
- 9.Banci L, Barbieri L, Bertini I, Luchinat E, Secci E, Zhao Y, Aricescu AR. Atomic-resolution monitoring of protein maturation in live human cells by NMR. Nat Chem Biol. 2013;9:297–299. doi: 10.1038/nchembio.1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Majumder S, Xue J, DeMott CM, Reverdatto S, Burz DS, Shekhtman A. Probing protein quinary interactions by in-cell nuclear magnetic resonance spectroscopy. Biochemistry. 2015;54:2727–2738. doi: 10.1021/acs.biochem.5b00036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ogino S, Kubo S, Umemoto R, Huang S, Nishida N, Shimada I. Observation of NMR Signals from Proteins Introduced into Living Mammalian Cells by Reversible Membrane Permeabilization Using a Pore-Forming Toxin, Streptolysin O. J. Am. Chem Soc. 2009;131:10834–10835. doi: 10.1021/ja904407w. [DOI] [PubMed] [Google Scholar]
- 12.Inomata K, Ohno A, Tochio H, Isogai S, Tenno T, Nakase I, Takeuchi T, Futaki S, Ito Y, Hiroaki H, Shirakawa M. High-resolution multi-dimensional NMR spectroscopy of proteins in human cells. Nature. 2009;458:106–109. doi: 10.1038/nature07839. [DOI] [PubMed] [Google Scholar]
- 13.Burz DS, Dutta K, Cowburn D, Shekhtman A. Mapping structural interactions using in-cell NMR spectroscopy (STINT-NMR) Nat Methods. 2006;3:91–93. doi: 10.1038/nmeth851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Burz DS, Dutta K, Cowburn D, Shekhtman A. In-cell NMR for protein-protein interactions (STINT-NMR) Nat Protoc. 2006;1:146–152. doi: 10.1038/nprot.2006.23. [DOI] [PubMed] [Google Scholar]
- 15.Burz DS, Shekhtman A. In-cell Biochemistry using NMR spectroscopy. PLoS One. 2008;3:e2571. doi: 10.1371/journal.pone.0002571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cobbert JD, DeMott C, Majumder S, Smith EA, Reverdatto S, Burz DS, McDonough KA, Shekhtman A. Caught in action: selecting peptide aptamers against intrinsically disordered proteins in live cells. Sci Rep. 2015;5:9402. doi: 10.1038/srep09402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Maldonado AY, Burz DS, Reverdatto S, Shekhtman A. Fate of Pup inside the Mycobacterium proteasome studied by in-cell NMR. PLoS One. 2013;8:e74576. doi: 10.1371/journal.pone.0074576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.DeMott CM, Majumder S, Burz DS, Reverdatto S, Shekhtman A. Ribosome mediated quinary interactions modulate in-cell protein activities. Biochemistry. 2017;56:4117–4126. doi: 10.1021/acs.biochem.7b00613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Luchinat E, Secci E, Cencetti F, Bruni P. Sequential protein expression and selective labeling for in-cell NMR in human cells. Biochim Biophys Acta, Gen Subj. 2016;1860:527–533. doi: 10.1016/j.bbagen.2015.12.023. [DOI] [PubMed] [Google Scholar]
- 20.Majumder S, DeMott CM, Burz DS, Shekhtman A. Using singular value decomposition to characterize protein-protein interactions by in-cell NMR spectroscopy. ChemBioChem. 2014;15:929–933. doi: 10.1002/cbic.201400030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Burz DS, DeMott CM, Aldousary A, Dansereau S, Shekhtman A. Quantitative Determination of Interacting Protein Surfaces in Prokaryotes and Eukaryotes by Using In-Cell NMR Spectroscopy. Methods Mol Biol. 2018;1688:423–444. doi: 10.1007/978-1-4939-7386-6_20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xie J, Thapa R, Reverdatto S, Burz DS, Shekhtman A. Screening of small molecule interactor library by using in-cell NMR spectroscopy (SMILI-NMR) J Med Chem. 2009;52:3516–3522. doi: 10.1021/jm9000743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ma J, McLeod S, MacCormack K, Sriram S, Gao N, Breeze AL, Hu J. Real-time monitoring of New Delhi metallo-beta-lactamase activity in living bacterial cells by 1H NMR spectroscopy. Angew Chem, Int Ed. 2014;53:2130–2133. doi: 10.1002/anie.201308636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wen H, An YJ, Xu WJ, Kang KW, Park S. Real-time monitoring of cancer cell metabolism and effects of an anticancer agent using 2D in-cell NMR spectroscopy. Angew Chem, Int Ed. 2015;54:5374–5377. doi: 10.1002/anie.201410380. [DOI] [PubMed] [Google Scholar]
- 25.Darwin KH, Ehrt S, Gutierrez-Ramos JC, Weich N, Nathan CF. The proteasome of Mycobacterium tuberculosis is required for resistance to nitric oxide. Science. 2003;302:1963–1966. doi: 10.1126/science.1091176. [DOI] [PubMed] [Google Scholar]
- 26.Lin G, Li D, de Carvalho LP, Deng H, Tao H, Vogt G, Wu K, Schneider J, Chidawanyika T, Warren JD, Li H, Nathan C. Inhibitors selective for mycobacterial versus human proteasomes. Nature. 2009;461:621–626. doi: 10.1038/nature08357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Darwin KH, Lin G, Chen Z, Li H, Nathan CF. Characterization of a Mycobacterium tuberculosis proteasomal ATPase homologue. Mol Microbiol. 2005;55:561–571. doi: 10.1111/j.1365-2958.2004.04403.x. [DOI] [PubMed] [Google Scholar]
- 28.Cerda-Maira FA, Pearce MJ, Fuortes M, Bishai WR, Hubbard SR, Darwin KH. Molecular analysis of the prokaryotic ubiquitin-like protein (Pup) conjugation pathway in Mycobacterium tuberculosis. Mol Microbiol. 2010;77:1123–1135. doi: 10.1111/j.1365-2958.2010.07276.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang T, Darwin KH, Li H. Binding-induced folding of prokaryotic ubiquitin-like protein on the Mycobacterium proteasomal ATPase targets substrates for degradation. Nat Struct Mol Biol. 2010;17:1352–1357. doi: 10.1038/nsmb.1918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Striebel F, Hunkeler M, Summer H, Weber-Ban E. The mycobacterial Mpa-proteasome unfolds and degrades pupylated substrates by engaging Pup’s N-terminus. EMBO J. 2010;29:1262–1271. doi: 10.1038/emboj.2010.23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Burz DS, Shekhtman A. The STINT-NMR method for studying in-cell protein-protein interactions. Curr Protoc Protein Sci. 2010;17:17.11.1. doi: 10.1002/0471140864.ps1711s61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Golub GH, Van Loan CF. Matrix Computations. 4th. The Johns Hopkins University Press; Baltimore, USA: 2012. [Google Scholar]
- 33.Lin W, Mandal S, Degen D, Liu Y, Ebright YW, Li S, Feng Y, Zhang Y, Mandal S, Jiang Y, Liu S, Gigliotti M, Talaue M, Connell N, Das K, Arnold E, Ebright RH. Structural Basis of Mycobacterium tuberculosis Transcription and Transcription Inhibition. Mol Cell. 2017;66:169–179. doi: 10.1016/j.molcel.2017.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Manasanch EE, Orlowski RZ. Proteasome inhibitors in cancer therapy. Nat Rev Clin Oncol. 2017;14:417–433. doi: 10.1038/nrclinonc.2016.206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Suo J, Chang CE, Lin TP, Heifets LB. Minimal inhibitory concentrations of isoniazid, rifampin, ethambutol, and streptomycin against Mycobacterium tuberculosis strains isolated before treatment of patients in Taiwan. Am Rev Respir Dis. 1988;138:999–1001. doi: 10.1164/ajrccm/138.4.999. [DOI] [PubMed] [Google Scholar]
- 36.Pearce MJ, Mintseris J, Ferreyra J, Gygi SP, Darwin KH. Ubiquitin-like protein involved in the proteasome pathway of Mycobacterium tuberculosis. Science. 2008;322:1104–1107. doi: 10.1126/science.1163885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Reverdatto S, Rai V, Xue J, Burz DS, Schmidt AM, Shekhtman A. Combinatorial Library of Improved Peptide Aptamers (CLIPs) to inhibit RAGE signal transduction in mammalian cells. PLoS One. 2013;8:e65180. doi: 10.1371/journal.pone.0065180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Maldonado AY, Burz DS, Shekhtman A. In-cell NMR spectroscopy. Prog Nucl Magn Reson Spectrosc. 2011;59:197–212. doi: 10.1016/j.pnmrs.2010.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Baell JB, Holloway GA. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J Med Chem. 2010;53:2719–2740. doi: 10.1021/jm901137j. [DOI] [PubMed] [Google Scholar]
- 40.Serber Z, Corsini L, Durst F, Dotsch V. In-cell NMR spectroscopy. Methods Enzymol. 2005;394:17–41. doi: 10.1016/S0076-6879(05)94002-0. [DOI] [PubMed] [Google Scholar]
- 41.Inomata K, Kamoshida H, Ikari M, Ito Y, Kigawa T. Impact of cellular health conditions on the protein folding state in mammalian cells. Chem Commun (Cambridge, U K) 2017;53:11245–11248. doi: 10.1039/c7cc06004a. [DOI] [PubMed] [Google Scholar]
- 42.Lipinski C, Hopkins A. Navigating chemical space for biology and medicine. Nature. 2004;432:855–861. doi: 10.1038/nature03193. [DOI] [PubMed] [Google Scholar]
- 43.Florczyk MA, McCue LA, Purkayastha A, Currenti E, Wolin MJ, McDonough KA. A family of acrcoregulated Mycobacterium tuberculosis genes shares a common DNA motif and requires Rv3133c (dosR or devR) for expression. Infect Immun. 2003;71:5332–5343. doi: 10.1128/IAI.71.9.5332-5343.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Masse JE, Keller R, Pervushin K. SideLink: automated side-chain assignment of biopolymers from NMR data by relative-hypothesis-prioritization-based simulated logic. J Magn Reson. 2006;181:45–67. doi: 10.1016/j.jmr.2006.03.012. [DOI] [PubMed] [Google Scholar]
- 45.Wuthrich K. NMR of proteins and nucleic acids. John Wiley &Sons; New York: 1986. [Google Scholar]
- 46.Mayer M, Meyer B. Group epitope mapping by saturation transfer difference NMR to identify segments of a ligand in direct contact with a protein receptor. J Am Chem Soc. 2001;123:6108–6117. doi: 10.1021/ja0100120. [DOI] [PubMed] [Google Scholar]
- 47.Farmer BT, 2nd, Constantine KL, Goldfarb V, Friedrichs MS, Wittekind M, Yanchunas J, Jr, Robertson JG, Mueller L. Localizing the NADP+ binding site on the MurB enzyme by NMR. Nat Struct Biol. 1996;3:995–997. doi: 10.1038/nsb1296-995. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.