

Summary
The lineage-specific transcription factor (TF) MEF2C is often deregulated in leukemia. However, strategies to target this TF have yet to be identified. Here we used a domain-focused CRISPR screen to reveal an essential role for LKB1 and its Salt-Inducible Kinase effectors (SIK3, in a partially redundant manner with SIK2) to maintain MEF2C function in acute myeloid leukemia (AML). A key phosphorylation substrate of SIK3 in this context is HDAC4, a repressive cofactor of MEF2C. Consequently, targeting of LKB1 or SIK3 diminishes histone acetylation at MEF2C-bound enhancers and deprives leukemia cells of the output of this essential TF. We also found that MEF2C-dependent leukemias are sensitive to on-target chemical inhibition of SIK kinase activity. This study reveals a chemical strategy to block MEF2C function in AML, highlighting how an oncogenic TF can be disabled by targeting of upstream kinases.
eTOC
Chemical modulation of sequence-specific DNA-binding transcription factors is a central challenge in molecular medicine. Tarumoto et al. used CRISPR screening to reveal how a cancer-promoting transcription factor can be selectively disabled through chemical inhibition of a specific kinase signaling cascade.
Introduction
Diverse genetic mutations that promote the hematopoietic malignancy acute myeloid leukemia (AML) are known to deregulate the expression of a common set of lineage-specifying transcription factors (TFs) (Rosenbauer and Tenen, 2007). Myocyte Enhancer Factor 2C (MEF2C) is one such TF, which is uniquely expressed in hematopoietic, muscle, and neuronal lineages and commonly upregulated in leukemia (Cante-Barrett et al., 2014). For example, MEF2C transcription is activated by MLL fusion oncoproteins, which is vital for promoting self-renewal in vitro and AML progression in vivo (Krivtsov et al., 2006; Schwieger et al., 2009). Remarkably, normal myelopoiesis is minimally impaired in Mef2c-mutant mice (Cante-Barrett et al., 2014), yet bone marrow cells in these animals are resistant to transformation by retrovirally-expressed MLL fusion proteins (Brown et al., 2017). This highlights an elevated demand for MEF2C function in AML when compared to the normal myeloid lineage. In addition, high MEF2C expression in pediatric AML is associated with adverse-risk and poor outcome (Laszlo et al., 2015). These findings all point to MEF2C as a transcriptional dependency in AML and a potential therapeutic target in this disease.
LKB1 phosphorylates and activates 14 downstream kinases, including AMP-activated protein kinases (AMPK) and AMPK-related kinases (Jaleel et al., 2005; Lizcano et al., 2004). While LKB1 is a broadly expressed kinase with diverse cellular functions, it has been extensively characterized as a metabolic regulator (Shackelford and Shaw, 2009). Upon conditions of ATP depletion, LKB1 activates AMPK to coordinate a switch from anabolic to catabolic metabolism. LKB1 has been validated as a tumor suppressor that is genetically inactivated in solid tumors and in the Peutz-Jeghers cancer predisposition syndrome (Ollila and Makela, 2011). Loss of LKB1 in these tumors results in diminished AMPK activity and metabolic alterations that drive tumorigenesis (Kottakis et al., 2016). While LKB1 is not mutated in AML, it has been proposed that LKB1-AMPK signaling suppresses leukemia development (Green et al., 2010). Other studies however suggest that LKB1 and AMPK are essential for normal and malignant hematopoiesis (Ollila and Makela, 2011; Saito et al., 2015). These contradictions suggest that our mechanistic understanding of the LKB1 signaling pathway in normal and malignant hematopoiesis is incomplete.
Other AMPK-related kinases can also carry out functions downstream of LKB1 (Lizcano et al., 2004; Shackelford and Shaw, 2009). One subfamily of AMPK-related kinases is the Salt-Inducible Kinases (SIK1, SIK2, and SIK3), which require LKB1-mediated phosphorylation for their kinase activity (Lizcano et al., 2004). A conserved function of SIKs is in promoting energy storage in liver and adipose tissues (Horike et al., 2003; Uebi et al., 2012; Wang et al., 2011). SIK2 and SIK3 are also known to regulate cytokine production in innate immune cells (Clark et al., 2012; Darling et al., 2017; Sanosaka et al., 2015). To mediate its biological effects, SIKs control gene expression via phosphorylation of transcriptional cofactors (Shackelford and Shaw, 2009), including class IIa histone deacetylases (HDAC4, HDAC5, HDAC7 and HDAC9) and the CREB-regulated transcriptional coactivator CRTC2 (Berdeaux et al., 2007; Patel et al., 2014; van der Linden et al., 2007). To our knowledge, a role for Salt-Inducible Kinases in leukemia has not been previously reported.
Here we identify a requirement for LKB1-SIK signaling to sustain the essential transcriptional function of MEF2C in AML. This regulation occurs, at least in part, through the SIK substrate HDAC4, which functions as a repressive cofactor of MEF2C. Unlike other cancer types, we found that AML cells are sensitive to genetic or chemical inhibition of SIK kinase activity, a perturbation that we show suppresses MEF2C function. Together, our results implicate LKB1-SIK as a signaling vulnerability of MEF2C-dependent leukemias.
Results
Kinase domain-focused CRISPR screening identifies SIK3 and LKB1 as AML-biased dependencies
We previously described domain-focused CRISPR screening as a strategy for identifying therapeutic targets in cancer (Shi et al., 2015). Here, we applied this approach to kinases to identify signaling vulnerabilities in AML. A pooled library of sgRNAs targeting 482 protein kinase domains was profiled in dropout screens performed in 26 Cas9-expressing human cancer cell lines, including eight AML lines (Table S1). The performance of spike-in negative and positive controls in the library validated the overall accuracy of the approach (Figure S1A). The majority of kinase dependencies identified were essential in all 26 cancer cell lines, such as ATR, PLK1, and CHEK1 (Figure 1A), in accord with prior genome-wide CRISPR screens (Tzelepis et al., 2016; Wang et al., 2015; Wang et al., 2017). We ranked all kinase dependencies in our screen based on the degree of AML specificity. Consistent with prior observations, we found that MLL fusion AML cell lines were hypersensitive to targeting of CDK6 (Placke et al., 2014). Other AML-biased hits included FLT3, JAK2, or JAK3 in cell lines harboring oncogenic mutations of these genes (Figure 1A). In addition, AML lines harboring JAK2 mutations were hypersensitive to targeting of DYRK1A, a kinase previously implicated in Down’s Syndrome-associated AML (Malinge et al., 2012) (Figure 1A).
Figure 1. Kinase domain-focused CRISPR screening identifies LKB1 and SIK3 as AML-biased dependencies.

(A) Summary of kinase domain-focused CRISPR screens. Plotted is the log2 fold-change of sgRNA abundance during ~14 population doublings. The effect of individual sgRNAs targeting each domain were averaged. AML: acute myeloid leukemia, CML: chronic myeloid leukemia.
(B) Summary of arrayed format validation experiments of individual sgRNAs in competition-based proliferation assays in the indicated cell lines. Plotted is the fold-change (log2) of sgRNA+/GFP+ cells during 12 to 18 days in culture (average of triplicates).
(C and E) Competition-based proliferation assays in the cells infected with the indicated sgRNAs linked to GFP. ‘e’ refers to the exon # that is targeted by each sgRNA. n=3.
(D) Western blot in the cells infected with the indicated sgRNA.
(F) Competition-based proliferation assays in NOMO-1 cells co-infected with GFP-linked sgRNA and mCherry-linked sgRNA. The percentage of double mCherry+/GFP+ cells is shown. n=4.
All bar graphs represent the mean ± SEM. Neg1, Neg2: negative controls. RPA3, Rpa3: positive controls
See also Figure S1.
We focused our subsequent experiments on Salt-Inducible Kinase 3 (SIK3), which scored strongly in MOLM-13 and MV4-11 cells and in a more intermediate fashion in other AML cell lines (Figure 1A). Importantly, SIK3 sgRNAs had a minimal impact on the growth of non-AML cell lines tested in our screen. We also identified LKB1 (also known as STK11) as displaying an AML-biased pattern of dependence, albeit in a less selective pattern than SIK3 (Figure 1A). The known requirement of LKB1 to activate SIK3 raised the possibility that these two dependencies were linked to one another (Lizcano et al., 2004). In arrayed format sgRNA competition assays performed in 17 cell lines, we confirmed the AML-biased pattern of LKB1 and SIK3 dependence observed in the pooled screens (Figures 1B–C). Western blotting and cDNA rescue experiments validated that these proliferation-arrest phenotypes were a consequence of on-target LKB1 or SIK3 mutagenesis (Figures 1D and S1B–G).
The two AML lines with the strongest SIK3 dependence (MOLM-13 and MV4-11) both possess MLL fusions and an activating mutation (internal tandem duplication) of FLT3 (Figure 1A). We confirmed the association between SIK3-dependence and MLL fusion status using two different genetically engineered AML cell lines. First, sgRNAs targeting Sik3 in murine MLL-AF9/NrasG12D AML cells led to a severe growth arrest, whereas NIH3T3 immortalized fibroblasts were unaffected by Sik3 targeting (Figure 1E). Second, MLL-AF9-transformed human CD34+ cells were also sensitive to SIK3 inactivation, irrespective of whether FLT3ITD or NRASG12D were introduced as a cooperating oncogene (Figure S1H) (Wei et al., 2008; Wunderlich et al., 2013). The prior description of viable Sik3-mutant mice would suggest that this kinase is dispensable for normal myelopoiesis (Darling et al., 2017; Sanosaka et al., 2015; Sasagawa et al., 2012).
Since SIK3 is homologous to SIK1 and SIK2, we evaluated whether redundancy conceals a broader requirement for SIKs in cancer. By performing dual targeting of each SIK gene combination in the NOMO-1 AML line, we found a partially redundant relationship between SIK2 and SIK3 (Figure 1F). While no AML cell lines are affected by targeting SIK1 or SIK2 alone (Figure 1A), the effects of targeting SIK3 became more pronounced when co-targeted with SIK2 (Figure 1F). By expanding this co-targeting experiment to 17 cell lines, we observed a broad AML-specific requirement for SIK2+SIK3 resembling the pattern of LKB1-dependence and with a bias for lines with MLL fusions (Figure 1B). To simplify our mechanistic evaluation of SIK3, we performed our subsequent experiments primarily in MOLM-13 cells, since the degree of redundancy with SIK2 was less apparent in this context.
SIK3 functions downstream of LKB1 and upstream of HDAC4 to support AML proliferation
LKB1 phosphorylates the t-loop of the SIK3 kinase domain at threonine 221 (Lizcano et al., 2004). Using the aforementioned cDNA rescue assay, we found that the T221A mutation of SIK3 was unable to support the proliferation of MOLM-13 cells (Figures 2A–C and S2A–B). We also observed that expressing a phosphomimetic allele (T221E) of SIK3 bypassed the proliferation arrest caused by inactivating LKB1 (Figures 2A–B, 2D, S2A and S2C) (Lizcano et al., 2004). These results suggest that the essential function of LKB1 in AML is to phosphorylate and activate SIKs.
Figure 2. Genetic experiments establish an essential LKB1-SIK3-HDAC4 pathway in AML.

(A) SIK3 mutants used in this study.
(B) Western blot in the cells transduced with empty vector or the indicated SIK3 cDNA.
(C and D) Competition-based proliferation assays in the cells transduced with empty vector or the indicated SIK3 cDNA and infected with the indicated GFP-linked sgRNA..
(E) Chemical structure of HG-9-91-01.
(F – H) Relative growth of the indicated cells, harboring empty vector, SIK3, or SIK3T142Q cDNA, upon HG-9-91-01 treatment. Normalized relative luminescence unit (RLU) was shown after 3 days culture with DMSO (0.1%) or HG-9-91-01 at the indicated concentrations. The mean ± SEM (n=3) and four-parameter dose-response curves are plotted.
(I) Competition-based proliferation assays in the cells co-infected with GFP-linked sgRNA and mCherry-linked sgRNA. The percentage of double mCherry+/GFP+ cells is shown.
(J) Western blot in the cells infected with the indicated sgRNA.
(K) Western blot in the cells harboring empty vector, SIK3, or SIK3T142Q cDNA, followed by treatment with DMSO (0.1%) or 100 nM HG-9-91-01 for 2 hrs.
All bar graphs represent the mean ± SEM (n=3). Neg1, Neg2: negative controls.
See also Figure S2.
The results above suggest that the catalytic function of SIK3 is critical in AML. We directly verified this by using a kinase-inactive allele (K95M) of SIK3 in the cDNA rescue assay (Figures 2A–C and S2A–B) (Katoh et al., 2006). To further validate the SIK requirement for AML expansion we made use of HG-9-91-01, a small molecule inhibitor of SIK kinase activity (Figure 2E) (Clark et al., 2012). Since this molecule is known to inhibit other kinases with comparable efficiency to SIKs (e.g. SRC and YES1) (Clark et al., 2012), we used a gatekeeper allele of SIK3 (T142Q), which is resistant to HG-9-91-01-mediated inhibition (Figures 2A and S2A) (Patel et al., 2014). Importantly, SIK3T142Q behaves like the wild-type allele in the cDNA rescue assay (Figures 2B–C and S2B). We found that the proliferation of SIK-dependent AML lines (MOLM-13 and MV4-11) and a SIK-independent line (K562) cells was similarly sensitive to HG-9-91-01 (GI50 ~ 100–200 nM) (Figures 2F–H). However, expression of SIK3T142Q rendered MOLM-13 and MV4-11 cells 10-fold less sensitive to HG-9-91-01, while expressing this allele in K562 had no effect on compound sensitivity (Figures 2F–H and S2D). This indicates that the sensitivity of SIK-dependent AML lines to HG-9-91-01 occurs via an on-target inhibition of SIK kinase activity, whereas the sensitivity of SIK-independent cell lines occurs via an off-target mechanism. While SIK3 is known to perform kinase-independent functions in certain contexts (Walkinshaw et al., 2013), our experiments demonstrate that AML cells rely on SIK kinase activity to proliferate.
To identify a critical downstream target of SIK3 in AML, we focused on its previously reported substrates, class IIa HDACs and CRTC2 (Berdeaux et al., 2007; Patel et al., 2014; van der Linden et al., 2007). Since phosphorylation by SIK3 (as well as other kinases) negatively regulates the function of these transcriptional cofactors by promoting nuclear exclusion, we reasoned that genetically inactivating the critical substrate would alleviate the degree of SIK3 dependence for AML proliferation. When we performed dual CRISPR targeting of SIK3 with each of class IIa HDACs or CRTC2, we only observed a rescue upon co-targeting of HDAC4 (Figures 2I and S2E–H). Western blotting revealed a reduction of HDAC4 phosphorylation upon genetic targeting of SIK3 or chemical inhibition of SIK with HG-9-91-01 (Figures 2J and 2K). These findings suggest that inhibition of HDAC4 is one of the key functions of SIK3 in supporting AML proliferation.
LKB1 and SIK3 are critical to maintain histone acetylation at MEF2C-bound enhancer elements
The known role of HDAC4 as a transcriptional cofactor led us to hypothesize that regulation of AML-promoting TFs might underlie the unique dependence on LKB1-SIK pathway in AML. To evaluate this, we mapped the genome-wide pattern of histone H3 lysine 27 acetylation (H3K27ac) in LKB1- and SIK3-deficient cells, reasoning that DNA motifs associated with LKB1/SIK3-dependent H3K27ac might point us to relevant TFs functioning downstream in this pathway. Chromatin immunoprecipitation was performed followed by deep sequencing (ChIP-seq) to measure H3K27ac enrichment, which revealed decreases in H3K27ac at specific cis-elements in a concordant pattern in LKB1- and SIK3-targeted cells (Figures 3A, 3B and S3A). We observed a loss of H3K27ac at similar sites following a 2-hour exposure to 100 nM HG-9-91-01, and this effect was rescued by expressing the SIK3 gatekeeper mutant (Figures 3A, 3C and S3B).
Figure 3. LKB1 and SIK3 are critical to maintain histone acetylation at MEF2C-bound enhancer elements.

(A) ChIP-seq profiles of H3K27ac and MEF2C at the indicated genomic loci, chosen because H3K27ac is decreased upon SIK3 or LKB1 knockout in MOLM-13 cells. Cells were harvested on day 5 after sgRNA infection, or after 2-hrs treatment of DMSO (0.1%) or 100 nM HG-9-91-01 for analysis of H3K27ac.
(B) Comparison of fold-change of H3K27ac tags upon SIK3 or LKB1 knockout. Each dot represents a single peak of H3K27ac identified in the control sample.
(C) Venn diagram depicting the overlap H3K27ac peaks that are decreased upon SIK3 knockout and following HG-9-91-01 treatment.
(D) Motif analysis of SIK3-dependent acetylation sites.
(E) Density plot of 568 SIK3-dependent enhancers, plotting H3K27ac and MEF2C enrichment at these sites. Enhancers are ranked by fold-change of H3K27ac upon SIK3 knockout.
(F) A meta-profile of MEF2C occupancy at 568 SIK3-dependent enhancer elements as compared to a randomly chosen set of 568 H3K27ac enriched sites.
(G and H) ChIP-qPCR with the indicated antibody at select MEF2C-bound elements in MOLM-13 cells treated with DMSO or 100 nM HG-9-91-01 for 2 hrs. (n=3 (MEF2C) or n=5 (HDAC4)).
Bar graphs represent the mean ± SEM.
See also Figure S3.
Over 98% of the SIK3-dependent H3K27ac sites were outside of proximal promoter regions, and hence were likely to represent distal enhancers (Figure S3C). Gene ontology analysis suggested that these cis-elements were located near genes that control hematopoietic development and cell proliferation/differentiation (Figure S3D). We performed motif analysis to identify over-represented TF binding sites at the LKB1/SIK3-dependent enhancers. While several motifs were identified in this analysis, the outlier with the lowest p-value corresponded to the binding motif of the MEF2 TF family (Figures 3D and S3E–H). Among the members of this family, MEF2C was a logical candidate as it is an established dependency in MLL fusion AML and known to be repressed via a direct interaction with HDAC4 (Krivtsov et al., 2006; Lu et al., 2000). To examine this, we performed MEF2C ChIP-seq and confirmed that LKB1/SIK3-dependent H3K27ac coincided with sites of MEF2C occupancy (Figures 3E–F). MEF2C protein levels were not altered by LKB1/SIK3 knockout or following HG-9-91-01 treatment (Figures S3I–J). In addition, MEF2C occupancy on chromatin did not change upon HG-9-91-01 treatment or HDAC4 knockout, whereas HG-9-91-01 exposure led to increased HDAC4 binding to MEF2C-bound sites (Figures 3G–H and S3K). This epigenomic analysis suggests that LKB1-SIK signaling is critical in AML to prevent HDAC4 from inactivating the function of MEF2C on chromatin.
Overlapping LKB1, SIK, and MEF2C dependencies in human AML cell lines
MEF2C is known to be essential in MLL fusion mouse models (Brown et al., 2017; Krivtsov et al., 2006), however the role of this TF as a dependency in human AML cells has not been previously established. To evaluate the correlation among LKB1, SIK, and MEF2C dependence, we employed TF DNA-binding domain-focused CRISPR screening as described above. When ranked based on AML bias, MEF2C was #12 and, as expected, was a strong dependency in the MLL fusion lines (Figure 4A). Importantly, MEF2C dependence overlapped with LKB1 and SIK3 dependence (compare Figures 1A and 4A). This pattern is distinct from other TFs, such as MYB and CBFB, which are broader dependencies across leukemia lines (Figure 4A). We verified the overall similarity of MEF2C, SIK3, and SIK2+SIK3 dependence in the leukemia cell lines using competition assays of individual sgRNAs (Figures 4B–D and S4A). Importantly, MLL fusion leukemia lines expressed MEF2C and HDAC4 at higher levels than leukemia lines that lacked MLL-rearrangements (Figures 4E and S4B), consistent with prior findings (Gaal et al., 2017; Krivtsov et al., 2006).
Figure 4. LKB1, SIK, and MEF2C are linked dependencies in acute myeloid leukemia.

(A) Summary of pooled TF DNA binding domain-focused CRISPR screens. Plotted is the log2 fold-change of sgRNA abundance during ~14 population doublings. The effect of individual sgRNAs targeting each domain was averaged.
(B) Competition-based proliferation assays in MOLM-13 cells infected with control (Neg1 or RPA3) or MEF2C sgRNA. Bar graphs represent the mean ± SEM (n=3).
(C) Western blot in the cells infected with the indicated sgRNA.
(D) Scatter plot depicting average log10 fold-change of MEF2C or SIK2+SIK3 sgRNAs for the indicated cell lines in competition-based proliferation assays. The data for SIK2+SIK3 sgRNAs are the same as in Figure 1B (average of triplicates).
(E) Western blot of the indicated proteins in leukemia lines.
(F and G) GSEA plot of the MEF2C signature upon SIK3 or LKB1 knockout. Normalized enrichment score (NES) and family-wise error rate (FWER) p-value are shown.
(H) Unbiased GSEA using all signatures in the Molecular Signature Database v5.2 (MSigDB) together with the MEF2C signature. NES and FWER p-value are plotted for 12,996 gene sets, each represented as a single dot.
(I) GSEA plot of the MEF2C gene signature upon 2-hrs treatment of HG-9-91-01. NES and FWER p-value are shown.
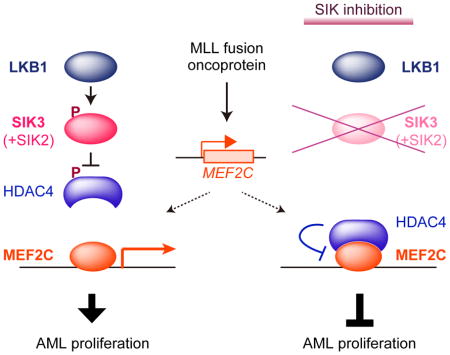
(J) Model.
See also Figure S4.
The transcriptional output of MEF2C is suppressed by LKB1-SIK inhibition
To evaluate the importance of LKB1-SIK in MEF2C function, we first CRISPR-targeted MEF2C in MOLM-13 cells and used RNA-seq to define the top 200 down-regulated genes as a MEF2C transcriptional signature, which includes many essential genes needed for MOLM-13 proliferation (Wang et al., 2017) (Figure S4C, Table S3, and data not shown). We then performed RNA-seq analysis in MOLM-13 cells after targeting LKB1 or SIK3, and found that both kinase perturbations significantly suppressed the MEF2C signature (Figures 4F and 4G). As a control for the specificity of this result, we interrogated a MYB transcriptional signature (Xu et al., 2018), and observed no suppression of this pathway (Figure S4D). Of note, the effect of SIK3 inactivation on the MEF2C signature was stronger in magnitude than any of the 12,995 signatures in the Molecular Signatures Database (Figure 4H). Furthermore, a 2-hour exposure to 100 nM HG-9-91-01 was sufficient to suppress the MEF2C signature and this effect was alleviated by expressing the SIK3 gatekeeper mutant (Figures 4I and S4E). We found that the effect of SIK3 targeting on transcription was attenuated if performed in cells deficient in HDAC4 (Figures S4F and S4G). Moreover, we failed to identify changes in H3K27ac enrichment using ChIP-seq upon inactivating LKB1 or SIK3 in HEL cells, a leukemia cell line with low MEF2C and HDAC4 expression (Figures 4E and S4H–I). Taken together, these findings suggest that LKB1-SIK3 signaling selectively supports the transcriptional output of MEF2C through inhibition of HDAC4 (Figure 4J).
Discussion
Our findings lead us to propose a model in which oncoprotein-driven addiction of leukemia cells to MEF2C imposes an essential requirement for LKB1-SIK signaling. Since MEF2C expression is highly lineage-specific (Cante-Barrett et al., 2014), this model can explain why two broadly-expressed kinases are critical for sustaining the proliferation of leukemia, while being dispensable in the many cancers that lack MEF2C expression. This work highlights a powerful capability of CRISPR-based screening to expose linkages between transcriptional regulators and signaling pathways in maintaining the growth of cancer cells.
The role of SIKs in regulating MEF2C through phosphorylation of HDACs is not without precedent, since SIK-HDAC-MEF2C pathway has been described previously in C. elegans neurons and in mammalian myocytes and chondrocytes (Berdeaux et al., 2007; Sasagawa et al., 2012; van der Linden et al., 2007). The regulation of MEF2C in these lineages may explain why SIK3-deficient mice develop aging-associated defects in skeletal development (Darling et al., 2017; Sasagawa et al., 2012). Other phenotypes in SIK3-deficient mice include protection from obesity, defects in gluconeogenesis (Uebi et al., 2012), and hypersensitivity to endotoxic shock (Sanosaka et al., 2015), which might also occur via HDAC4-MEF2C regulation (Han et al., 1997; Xu et al., 2015), or may involve modulation of other TFs.
Our study implicates LKB1-SIK signaling as having therapeutic significance in AML. First, the potency and selectivity of AML growth-arrest following LKB1 or SIK2+SIK3 targeting resembles the effects of targeting other validated kinase oncogenes in AML, such as FLT3 and JAK2. Moreover, the on-target sensitivity of AML lines to HG-9-91-01 compares favorably to the sensitivity of cancer cell lines to other approved kinase inhibitors used in oncology, although additional optimization of these small-molecules will be required for therapeutic investigation (Sundberg et al., 2016; Wein et al., 2016). Our genetic experiments suggest that dual inhibition of SIK2 and SIK3 would be the ideal strategy to achieve potent MEF2C inhibition in AML, while preserving the homeostatic and tumor-protective functions of LKB1 in other cell types (Ollila and Makela, 2011). Another attractive feature of SIK inhibition in AML is the possibility that this intervention could be evaluated in the subset of AML patients in which MEF2C is upregulated, (Laszlo et al., 2015; Schwieger et al., 2009). Since MEF2C is deregulated in lymphoid malignancies, it is likely that LKB1-SIK signaling will be important in other hematopoietic cancer contexts as well (Homminga et al., 2011). Importantly, our screens validate several other actionable kinase dependencies in MLL fusion leukemia (e.g. CDK6), which could be evaluated as additional targets for combination therapy. Collectively, our results provide a rationale for further optimization and evaluation of potent and selective small molecules that inhibit SIK kinase activity as therapeutics in pre-clinical AML models.
TFs are commonly deregulated in the pathogenesis of cancer and represent a rich source of cancer cell dependencies (Bhagwat and Vakoc, 2015). However, the activating regions and DNA-binding domains of TFs are challenging targets for drug development (Koehler, 2010). This obstacle has motivated a widespread interest in targeting TFs by indirect means, such as by targeting TF cofactors (Kwiatkowski et al., 2014; Zuber et al., 2011b). Our study points to a broader potential for kinase inhibition as a strategy for disabling select TFs that promote cancer. We anticipate that expanded CRISPR screening efforts that correlate TF and kinase dependencies in cancer cell line panels will avail additional opportunities to broaden this concept to other untreatable malignancies.
STAR Methods
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Christopher Vakoc (vakoc@cshl.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell lines
MOLM-13, MV4-11, NOMO-1, THP-1, HEL, SET-2, U937 (acute myeloid leukemia, AML), K562 (chronic myeloid leukemia, CML), ASPC1, CFPAC-1, BXPC-3, MIAPACA-2, SUIT-2 (pancreatic cancer), DMS114, NCI-H446, NCI-H526, NCI-H82 (lung cancer), SNU449, SNU475, SNU387, SNU423 (liver cancer), RH30 (sarcoma) and murine RN2 (MLL-AF9/NRasG12D AML) (Zuber et al., 2011a) cells were cultured in RPMI supplemented with 10% FBS. KASUMI-1 (AML) cells were cultured in RPMI with 20% FBS. OCI-AML3 (AML) cells were cultured in alpha-MEM with 20% FBS. MA9 (AML) cells were cultured in IMDM supplemented with 20% FBS, 10 ng/ml SCF, 10 ng/ml TPO, 10 ng/ml FLT3L, 10 ng/ml IL-3, 10 ng/ml IL-6, and their derivatives MA9-ITD and MA9-RAS cells were cultured in IMDM with 20% FBS. A549 (lung cancer), HEPG2, HUH1 (liver cancer), RD (sarcoma) and HEK293T cells were cultured in DMEM with 10% FBS. HEP3B2 (liver cancer) cells were cultured in EMEM with 10% FBS. Murine NIH3T3 cells were cultured in DMEM with 10% bovine calf serum. Penicillin/streptomycin was added to all media. All cell lines were cultured at 37°C with 5% CO2, and were periodically tested mycoplasma negative.
METHODS DETAILS
Plasmid construction and sgRNA cloning
The sgRNA lentiviral expression vector with optimized sgRNA scaffold backbone (LRG2.1) and the lentiviral Cas9 vector will be described in a separate study (Grevet et al, submitted). Briefly, the Cas9 expression cassette was cloned by PCR. The 5′-3xFLAG-tagged human-codon optimized Cas9 cDNA from Streptococcus pyogenes (Addgene: #49535)(Shalem et al., 2014) was PCR amplified and ligated it into a lentiviral EFS-Cas9-P2A-Puro expression vector using In-Fusion cloning system (Takara Bio). This Cas9 expression vector was termed LentiV_Cas9_puro. The LRG2.1 vector was derived from a lentiviral U6-sgRNA-EFS-GFP expression vector (LRG, Addgene: #65656) by replacing the original wild-type sgRNA scaffold with our optimized version (LRG2.1 will be available through Addgene). LRCherry2.1 was derived from LRG2.1 by replacing GFP with mCherry CDS. All the sgRNAs were cloned into the LRG2.1 vector using a BsmBI restriction site. In this study, LRG2.1 or LRCherry2.1 were used for expressing sgRNA in human cell lines, while LRG (Addgene: 65656) was used for sgRNA expression in murine cell lines.
LentiV_Neo vector was derived from LentiV_Cas9_puro vector by removing Cas9 and replacing a puromycin-resistant gene with a neomycin-resistant gene. A partial SIK3 cDNA (GE Dharmacon, Clone ID: 40081639) was cloned into LentiV_Neo vector using In-Fusion cloning system, and N-terminus of SIK3 cDNA (1–138 bp from translation start site) was added to obtain SIK3 cDNA corresponding to NM_001281749. For construction of SIK3 mutants, the following base substitutions were introduced into the cDNA; CRISPR-resistant, G501->C; K95M (kinase-dead), A284->T; T221A, A661->G; T221E, A661CC->GAG; T142Q, A424CA->CAA. Flag-tagged LKB1 cDNA (addgene: #8590) (Shaw et al., 2004) was cloned into LentiV_Neo vector. For a CRISPR-resistant mutant, C333->A substitution was introduced. Sequences of all sgRNAs used for arrayed experiments are provided in the supplemental table (Table S3. sgRNA, qPCR primer, MEF2C signature. Related to all Figures)
Construction of domain-focused sgRNA library
The kinase domain-focused sgRNA library was designed based on the human kinase gene list from a previous study (Manning et al., 2002). The kinase enzymatic domain and TF DNA-binding domain information were retrieved from NCBI database conserved domain annotation. Six independent sgRNAs were designed for targeting each individual domain regions. All the sgRNAs were designed using the same design principle reported previously and the sgRNAs with the prediction of high off-target effect were excluded (Hsu et al., 2013). Domain targeting and positive/negative control sgRNAs were synthesized in duplicate or triplicate in a pooled format on an array platform (Twist Bioscience) and then PCR cloned into the Bsmb1-digested LRG2.1 vector using Gibson Assembly kit (NEB). To ensure the representative and identity of sgRNA in the pooled lentiviral plasmids, a deep-sequencing analysis was performed on a MiSeq instrument (Illumina) and verified that 100% of the designed sgRNAs were cloned in the LRG2.1 vector and the abundance of >95% of the sgRNA constructs was within 5-fold of the mean (data not shown).
Lentivirus Transduction
Lentivirus was produced in HEK293T cells by transfecting plasmids with helper plasmids (VSVG and psPAX2 (addgene # 12260)) using Polyethylenimine (PEI 25000). For HEK293T cells in 10 cm dish, 10 μg of plasmid DNA, 5 μg of VSVG, 7.5 μg of psPAX2, and 32 μl of 1 mg/ml PEI were mixed. Media was changed to fresh one at 6 hr post-transfection, and lentivirus-containing supernatant was collected at 48, 72, and 96 hr post-transfection and pooled together. For lentivirus infection, target cells were mixed with the virus and 4 μg/ml polybrene, and then centrifuged at 1,700 rpm for 15 ~ 40 min. Media was changed at 24 hr post-infection, and antibiotics (1 ~ 4 μg/ml puromycin and/or 1 mg/ml G418) was added at 48 hr post-infection if selection is needed.
Pooled CRISPR Screening
Cas9-expressing cells were established by introduction of LentiV_Cas9_puro vector. Lentivirus of pooled sgRNA library for kinase domain or DNA-binding domain was produced as described above. Virus titer was measured by infection of cells with serially diluted virus. For transduction of single sgRNA per cell, multiplicity of infection (MOI) was set to 0.3 ~ 0.4. To maintain the representation of sgRNAs during screen, the number of cells was kept 1000 times more than sgRNA number in the library. Cells were harvested at initial (day 3 post-infection) and final (12 or more doubling times after the initial) time points. Genomic DNA was extracted using QIAamp DNA mini kit (QIAGEN).
Sequencing library was constructed basically as described previously (Shi et al., 2015). Genomic DNA fragment (~200 bp) containing sgRNA was amplified by PCR, followed by end-repair with T4 DNA polymerase (NEB), DNA Polymerase I, Large (Klenow) Fragment (NEB) and T4 polynucleotide kinase (NEB), and addition of 3′ A-overhang with Klenow Fragment (3′–5′ exo-) (NEB). This DNA fragment was ligated with diversity-increased custom barcodes (Shi et al., 2015), and PCR-amplified. Barcoded libraries were pooled and analyzed by paired-end sequencing using Miseq (Illumina) with MiSeq Reagent Kit v3 (Illumina).
The sequence data were de-multiplexed and trimmed to contain only the sgRNA sequence, and subsequently mapped to the reference sgRNA library without allowing any mismatches using a similar method as previously described (Shi et al., 2015). The read counts were calculated for each individual sgRNA. The total read counts were normalized to each sample. Average log2 fold-change in the abundance of all sgRNAs targeting a given domain (CRISPR score) was calculated, as described (Wang et al., 2015). The domains with average log2 fold-change was less than −1.5 in at least 20 among 26 cell lines in the kinase domain-focused screen, or 15 among 19 cell lines in DNA binding domain-focused screen, were considered as pan-essential. AML-specific dependency was determined by subtracting average of CRISPR score in non-AML cell lines from average of CRISPR score in AML cell lines after removal of pan-essential domains, and that score was ranked in ascending order.
The Kinase and TF CRISPR screening data is provided in the supplemental tables (Table S1. Average log2 FC in kinase domain-focused screen. Related to Figure 1. Table S2. Average log2 fold-change in DNA binding domain-focused screen. Related to Figure 4.)
Competition-based cell proliferation assay
Cas9-expressing human cell lines were infected with sgRNA linked with GFP or mCherry (LRG2.1 or LRCherry2.1 vector). Percentage of GFP- and/or mCherry-positive cells was measured every 3 days from day 3 to day 21 post-infection using Guava easyCyte Flow Cytometers (Millipore). In some of leukemia cells (MOLM-13, MV4-11, HEL, OCI-AML3, U937, K562, MA9, MA9-ITD, MA9-RAS) and murine cell lines (RN2, NIH3T3), percentage of GFP- and/or mCherry-positive cells was measured every 2 days from day 3 to day 15, or from day 2 to day 12, post-infection considering their growth rate to adjust similar doubling times during experiment. Final GFP% was divided by initial GFP% to calculate fold-change of GFP. Average of fold-change of 2 independent sgRNAs or 2 sets of sgRNAs were used for calculation of SIK3, LKB1, MEF2C, and SIK2+SIK3 dependencies.
Western Blot
For knockout experiment, cells were harvested on day 4 post-infection of sgRNA. Cell pellets were suspended in Laemli sample buffer (Bio-Rad) containing 2-mercaptoethanol, and boiled for 10 min. These whole cell extracts were separated by SDS-PAGE, followed by transfer to nitrocellulose membrane and immunoblotting.
HG-9-91-01 Treatment
To test cell growth upon HG-9-91-01 treatment, 1,000 cells were plated in each well of opaque-walled 96-well plate, and mixed with serially-diluted concentration of HG-9-91-01 (Cayman Chemical) or 0.1% DMSO as control. After 72 hr incubation, the number of viable cells was measured using CellTiter Glo Luminescent Cell Viability Assay kit (Promega) with SpectraMax plate reader (Molecular Devices) following the manufacture’s protocol. For western blot, ChIP-seq and RNA-seq experiments, cells were treated with DMSO (0.1% final conc.) or 100 nM HG-9-91-01 for 2 hr, and harvested.
ChIP-qPCR and ChIP-seq
For knockout experiment, cells were harvested on day 5 post-infection of sgRNA. 5 × 106 cells were harvested, crosslinked with 1% formaldehyde for 10 min at room temperature, and quenched with 0.125 M Glycine for 10 min at room temperature. After washing with PBS, cells were incubated in 1 ml Cell Lysis Buffer (10 mM Tris-HCl pH 8.0, 10 mM NaCl, 0.2% NP-40) with protease inhibitor for 15 min on ice. Nucleus was isolated by centrifugation at 4200 rpm for 30 sec, incubated in 500 μl Nuclei Lysis Buffer (50 mM Tris-HCl pH 8.0, 10 mM EDTA 1% SDS) with protease inhibitor for 10 min on ice, and sonicated using a Bioruptor (Diagenode) in the following settings; low amplitude, On 30s, Off 30s, 10 cycles. The chromatin was centrifuged at 13,000 rpm for 15 min at 4°C. The supernatant was mixed with 3.5 ml IP Dilution Buffer (20 mM Tris-HCl pH 8.0, 2 mM EDTA, 150 mM NaCl, 1% Triton X-100, 0.01% SDS), and incubated with the indicated antibody and 25 μl of Protein A magnetic beads (Dynabeads, Thermo) at 4°C overnight. The beads were washed with IP Wash 1 Buffer (20 mM Tris-HCl pH 8.0, 2 mM EDTA, 50 mM NaCl, 1% Triton X-100, 0.1% SDS) once, High Salt Buffer (20 mM Tris-HCl pH 8.0, 2 mM EDTA, 500 mM NaCl, 1% Triton X-100, 0.01% SDS) twice, IP Wash 2 Buffer (10 mM Tris-HCl pH 8.0, 1 mM EDTA, 250 mM LiCl, 1% NP-40, 1% sodium deoxycholate) once, and TE pH 8.0 twice. Chromatin DNA was eluted in 200 μl Elution Buffer (50 mM Tris-HCl pH 8.0, 10 mM EDTA, 1% SDS) by incubation at 65°C for 15 min, and then reverse-crosslinked with 12 μl of 5 M NaCl and 1 μg/ml RNase A at 65°C overnight. DNA was treated with 4 μg/ml Proteinase K at 42°C for 2 hr, and purified using QIAquick PCR purification kit (QIAGEN). ChIP DNA was quantified using 7900HT Fast Real-Time PCR system (Applied Biosystems) with Power SYBR Green Master Mix (Thermo Fisher). The MEF2C peak loci near the decreased H3K27ac loci in LKB1/SIK3 knockout cells obtained from ChIP-seq experiments, were used for qPCR analysis. Primer sequences and their chromosomal positions used for ChIP-qPCR are provided in Table S3.
For ChIP-seq, 3~10 ChIP samples were pooled. ChIP-seq library was prepared using the TruSeq ChIP Sample prep kit (Illumina) following manufacture’s protocol. Briefly, ChIP DNA was end-repaired, 3′-adenylated, and then ligated with indexed adaptor. These DNA was size selected (200–400 bp) via agarose gel electrophoresis, and PCR amplified. After purification of products using AMPure XP beads (Beckman Coulter), its quality was checked using Bioanalyzer with high sensitivity DNA chip (Agilent). ChIP-seq library was pooled and analyzed by single-end sequencing using NextSeq (Illumina).
ChIP-seq data analysis
Sequencing reads were mapped into reference human genome hg19 using Bowtie2 (Langmead and Salzberg, 2012). Peakcalling was done using MACS2 FDR cut off 5% with broad peak and narrow peak option for H3K27ac and MEF2C, respectively (Feng et al., 2012). The H3K27ac peaks in knockout experiment or HG-9-91-01 treatment experiment were merged with corresponding control ChIP-seq using BEDtools (Quinlan and Hall, 2010). Normalized tag count was calculated using Bamliquidator package (https://github.com/BradnerLab/pipeline). H3K27ac regions with peak intensity fold-change (log2) of −1 to control sample, were considered as decreased. AnnotatePeaks tool from HOMER suite was used to annotate H3K27ac peaks with the nearest expressed gene (Heinz et al., 2010). Gene ontology analysis of annotated regions was done using Metascape (http://metascape.org/gp/index.html#/main/step1) (Tripathi et al., 2015). For transcriptional factor binding motif analysis of 500 bp around the center of the decreased H3K27ac regions, the TRAP web tool (http://trap.molgen.mpg.de/cgi-bin/home.cgi) was used (Thomas-Chollier et al., 2011). DNA sequences of the same number of increased H3K27ac and random H3K27ac peaks were served as controls for the motif analysis. ChIP-seq tracks were generated using UCSC genome browser (Kent et al., 2002). Heat maps density plot and metagene plots were generated using ± 5 kb around each center of the decreased H3K27ac peaks or the same number of random H3K27ac peaks with 50 bp binning size.
RNA-seq
For knockout experiment, cells were harvested on day 5 post-infection of sgRNA. Total RNA was extracted using TRIzol (Thermo) following the manufacture’s protocol, and suspended in RNase-free water. RNA-seq library was prepared using TruSeq sample prep kit v2 (Illumina) according to the manufacture’s protocol. Briefly, polyA RNA was selected and fragmented enzymatically. First strand of cDNA was synthesized using Super Script II reverse transcriptase, and then second strand was synthesized. Double-stranded cDNA was end-repaired, 3′-adenylated, ligated with indexed adaptor, and then PCR-amplified. RNA-seq library was pooled and analyzed by single-end sequencing using NextSeq (Illumina).
RNA-seq data analysis
Sequencing reads were mapped into reference human genome hg19 using TopHat2 (Kim et al., 2013). Differentially expressed genes were analyzed using a Cufflinks tool, Cuffdiff (Trapnell et al., 2013) by masking structural RNAs. The genes with RPKM of more than 2 in the control were considered as expressed and used in the subsequent analysis. Fold-change of RPKM was calculated as the ratio of average RPKM of two independent sgRNA samples (MEF2C (e2.1, e3.3), SIK3 (e1, e4), LKB1 (e2, e3) or SIK3+HDAC4 (e1+e19, e4+e20)) to average RPKM of control sgRNA samples (Neg2, Neg3). Custom gene signatures for GSEA were prepared from top 200 down-regulated genes in MEF2C knockout (provided in Table S3) or MYB knockout cells (Xu et al., 2018). GSEA was performed according to the instructions using all available signatures in the Molecular Signature Database v5.2 (MSigDB) together with the custom signatures (Subramanian et al., 2005).
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical significance was evaluated by p-value from unpaired Student t test using Prism software.
DATA AND SOFTWARE AVAILABILITY
The ChIP-seq and RNA-seq data in this study is available in GEO database with accession number GSE109493
Supplementary Material
Table S1. Average log2 FC in kinase domain-focused screen. Related to Figure 1
Table S2. Average log2 fold-change in DNA binding domain-focused screen. Related to Figure 4
Table S3. sgRNA and primer sequences, and MEF2C signature. Related to STAR Methods
Highlights.
LKB1 and Salt-inducible kinases 2/3 are essential in MEF2C+ acute myeloid leukemia
LKB1-SIK signaling prevents HDAC4-mediated inhibition of MEF2C
AML cells are sensitive to chemical inhibition of SIK kinase activity
Salt-inducible kinases may have therapeutic significance in MEF2C+ AML
Acknowledgments
We would like to thank all members of the Vakoc laboratory for helpful discussions and suggestions throughout the course of this study. We thank Ross L. Levine for SET-2 cells, Robert Roeder for Cas9-expressing KASUMI-1 cells, James C. Mulloy for MA9 cells and its derivatives, David A. Tuveson for SUIT-2 cells, CSHL Cancer Center DNA Sequencing Shared Resource for deep-sequencing, CSHL Bioinformatics Shared Resource for proving Galaxy data analysis platform. This work was supported by Cold Spring Harbor Laboratory NCI Cancer Center Support grant 5P30CA045508. Additional funding was provided by the Forbeck Foundation, the Pershing Square Sohn Cancer Research Alliance, National Institutes of Health grant NCI RO1 CA174793, NCI 5P01CA013106-Project 4, and a Leukemia & Lymphoma Society Scholar Award. Y.T. is supported by the Lauri Strauss Leukemia Foundation.
Footnotes
Author Contributions
Conceptualization, Y.T., J.S. and C.R.V.; Methodology, Y.T., J.S. and C.R.V.; Investigation, Y.T., J.S., B.L., T.D.D.S, Y.H., J.P.M, and X.S.W.; Resources, O.E.D. and O.K.; Writing – Original Draft, Y.T. and C.R.V.; Writing – Review & Editing, Y.T., J.S. and C.R.V; Funding Acquisition, J.S. and C.R.V.
Declaration of Interests
C.R.V. is an advisor to KSQ Therapeutics and receives research funding from Boehringer-Ingelheim.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Berdeaux R, Goebel N, Banaszynski L, Takemori H, Wandless T, Shelton GD, Montminy M. SIK1 is a class II HDAC kinase that promotes survival of skeletal myocytes. Nat Med. 2007;13:597–603. doi: 10.1038/nm1573. [DOI] [PubMed] [Google Scholar]
- Bhagwat AS, Vakoc CR. Targeting Transcription Factors in Cancer. Trends Cancer. 2015;1:53–65. doi: 10.1016/j.trecan.2015.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown F, Still E, Cifani P, Takao S, Reed C, Ficarro S, Koche R, Romanienko P, Mark W, O’Donnell C, et al. MEF2C phosphorylation is required for chemotherapy resistance in acute myeloid leukemia. biorRxiv. 2017 doi: 10.1158/2159-8290.CD-17-1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cante-Barrett K, Pieters R, Meijerink JP. Myocyte enhancer factor 2C in hematopoiesis and leukemia. Oncogene. 2014;33:403–410. doi: 10.1038/onc.2013.56. [DOI] [PubMed] [Google Scholar]
- Clark K, MacKenzie KF, Petkevicius K, Kristariyanto Y, Zhang J, Choi HG, Peggie M, Plater L, Pedrioli PG, McIver E, et al. Phosphorylation of CRTC3 by the salt-inducible kinases controls the interconversion of classically activated and regulatory macrophages. Proc Natl Acad Sci U S A. 2012;109:16986–16991. doi: 10.1073/pnas.1215450109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darling NJ, Toth R, Arthur JS, Clark K. Inhibition of SIK2 and SIK3 during differentiation enhances the anti-inflammatory phenotype of macrophages. Biochem J. 2017;474:521–537. doi: 10.1042/BCJ20160646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng J, Liu T, Qin B, Zhang Y, Liu XS. Identifying ChIP-seq enrichment using MACS. Nat Protoc. 2012;7:1728–1740. doi: 10.1038/nprot.2012.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaal Z, Olah E, Rejto L, Erdodi F, Csernoch L. Strong Correlation between the Expression Levels of HDAC4 and SIRT6 in Hematological Malignancies of the Adults. Pathol Oncol Res. 2017;23:493–504. doi: 10.1007/s12253-016-0139-5. [DOI] [PubMed] [Google Scholar]
- Green AS, Chapuis N, Maciel TT, Willems L, Lambert M, Arnoult C, Boyer O, Bardet V, Park S, Foretz M, et al. The LKB1/AMPK signaling pathway has tumor suppressor activity in acute myeloid leukemia through the repression of mTOR-dependent oncogenic mRNA translation. Blood. 2010;116:4262–4273. doi: 10.1182/blood-2010-02-269837. [DOI] [PubMed] [Google Scholar]
- Han J, Jiang Y, Li Z, Kravchenko VV, Ulevitch RJ. Activation of the transcription factor MEF2C by the MAP kinase p38 in inflammation. Nature. 1997;386:296–299. doi: 10.1038/386296a0. [DOI] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, Glass CK. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell. 2010;38:576–589. doi: 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homminga I, Pieters R, Langerak AW, de Rooi JJ, Stubbs A, Verstegen M, Vuerhard M, Buijs-Gladdines J, Kooi C, Klous P, et al. Integrated transcript and genome analyses reveal NKX2-1 and MEF2C as potential oncogenes in T cell acute lymphoblastic leukemia. Cancer Cell. 2011;19:484–497. doi: 10.1016/j.ccr.2011.02.008. [DOI] [PubMed] [Google Scholar]
- Horike N, Takemori H, Katoh Y, Doi J, Min L, Asano T, Sun XJ, Yamamoto H, Kasayama S, Muraoka M, et al. Adipose-specific expression, phosphorylation of Ser794 in insulin receptor substrate-1, and activation in diabetic animals of salt-inducible kinase-2. J Biol Chem. 2003;278:18440–18447. doi: 10.1074/jbc.M211770200. [DOI] [PubMed] [Google Scholar]
- Hsu PD, Scott DA, Weinstein JA, Ran FA, Konermann S, Agarwala V, Li Y, Fine EJ, Wu X, Shalem O, et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat Biotechnol. 2013;31:827–832. doi: 10.1038/nbt.2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaleel M, McBride A, Lizcano JM, Deak M, Toth R, Morrice NA, Alessi DR. Identification of the sucrose non-fermenting related kinase SNRK, as a novel LKB1 substrate. FEBS Lett. 2005;579:1417–1423. doi: 10.1016/j.febslet.2005.01.042. [DOI] [PubMed] [Google Scholar]
- Katoh Y, Takemori H, Lin XZ, Tamura M, Muraoka M, Satoh T, Tsuchiya Y, Min L, Doi J, Miyauchi A, et al. Silencing the constitutive active transcription factor CREB by the LKB1-SIK signaling cascade. FEBS J. 2006;273:2730–2748. doi: 10.1111/j.1742-4658.2006.05291.x. [DOI] [PubMed] [Google Scholar]
- Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, Haussler D. The human genome browser at UCSC. Genome Res. 2002;12:996–1006. doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 2013;14:R36. doi: 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koehler AN. A complex task? Direct modulation of transcription factors with small molecules. Curr Opin Chem Biol. 2010;14:331–340. doi: 10.1016/j.cbpa.2010.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kottakis F, Nicolay BN, Roumane A, Karnik R, Gu H, Nagle JM, Boukhali M, Hayward MC, Li YY, Chen T, et al. LKB1 loss links serine metabolism to DNA methylation and tumorigenesis. Nature. 2016;539:390–395. doi: 10.1038/nature20132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krivtsov AV, Twomey D, Feng Z, Stubbs MC, Wang Y, Faber J, Levine JE, Wang J, Hahn WC, Gilliland DG, et al. Transformation from committed progenitor to leukaemia stem cell initiated by MLL-AF9. Nature. 2006;442:818–822. doi: 10.1038/nature04980. [DOI] [PubMed] [Google Scholar]
- Kwiatkowski N, Zhang T, Rahl PB, Abraham BJ, Reddy J, Ficarro SB, Dastur A, Amzallag A, Ramaswamy S, Tesar B, et al. Targeting transcription regulation in cancer with a covalent CDK7 inhibitor. Nature. 2014;511:616–620. doi: 10.1038/nature13393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laszlo GS, Alonzo TA, Gudgeon CJ, Harrington KH, Kentsis A, Gerbing RB, Wang YC, Ries RE, Raimondi SC, Hirsch BA, et al. High expression of myocyte enhancer factor 2C (MEF2C) is associated with adverse-risk features and poor outcome in pediatric acute myeloid leukemia: a report from the Children’s Oncology Group. J Hematol Oncol. 2015;8:115. doi: 10.1186/s13045-015-0215-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lizcano JM, Goransson O, Toth R, Deak M, Morrice NA, Boudeau J, Hawley SA, Udd L, Makela TP, Hardie DG, et al. LKB1 is a master kinase that activates 13 kinases of the AMPK subfamily, including MARK/PAR-1. EMBO J. 2004;23:833–843. doi: 10.1038/sj.emboj.7600110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, McKinsey TA, Nicol RL, Olson EN. Signal-dependent activation of the MEF2 transcription factor by dissociation from histone deacetylases. Proc Natl Acad Sci U S A. 2000;97:4070–4075. doi: 10.1073/pnas.080064097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malinge S, Bliss-Moreau M, Kirsammer G, Diebold L, Chlon T, Gurbuxani S, Crispino JD. Increased dosage of the chromosome 21 ortholog Dyrk1a promotes megakaryoblastic leukemia in a murine model of Down syndrome. J Clin Invest. 2012;122:948–962. doi: 10.1172/JCI60455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S. The protein kinase complement of the human genome. Science. 2002;298:1912–1934. doi: 10.1126/science.1075762. [DOI] [PubMed] [Google Scholar]
- Ollila S, Makela TP. The tumor suppressor kinase LKB1: lessons from mouse models. J Mol Cell Biol. 2011;3:330–340. doi: 10.1093/jmcb/mjr016. [DOI] [PubMed] [Google Scholar]
- Patel K, Foretz M, Marion A, Campbell DG, Gourlay R, Boudaba N, Tournier E, Titchenell P, Peggie M, Deak M, et al. The LKB1-salt-inducible kinase pathway functions as a key gluconeogenic suppressor in the liver. Nat Commun. 2014;5:4535. doi: 10.1038/ncomms5535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Placke T, Faber K, Nonami A, Putwain SL, Salih HR, Heidel FH, Kramer A, Root DE, Barbie DA, Krivtsov AV, et al. Requirement for CDK6 in MLL-rearranged acute myeloid leukemia. Blood. 2014;124:13–23. doi: 10.1182/blood-2014-02-558114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbauer F, Tenen DG. Transcription factors in myeloid development: balancing differentiation with transformation. Nat Rev Immunol. 2007;7:105–117. doi: 10.1038/nri2024. [DOI] [PubMed] [Google Scholar]
- Saito Y, Chapple RH, Lin A, Kitano A, Nakada D. AMPK Protects Leukemia-Initiating Cells in Myeloid Leukemias from Metabolic Stress in the Bone Marrow. Cell Stem Cell. 2015;17:585–596. doi: 10.1016/j.stem.2015.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanosaka M, Fujimoto M, Ohkawara T, Nagatake T, Itoh Y, Kagawa M, Kumagai A, Fuchino H, Kunisawa J, Naka T, et al. Salt-inducible kinase 3 deficiency exacerbates lipopolysaccharide-induced endotoxin shock accompanied by increased levels of pro-inflammatory molecules in mice. Immunology. 2015;145:268–278. doi: 10.1111/imm.12445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasagawa S, Takemori H, Uebi T, Ikegami D, Hiramatsu K, Ikegawa S, Yoshikawa H, Tsumaki N. SIK3 is essential for chondrocyte hypertrophy during skeletal development in mice. Development. 2012;139:1153–1163. doi: 10.1242/dev.072652. [DOI] [PubMed] [Google Scholar]
- Schwieger M, Schuler A, Forster M, Engelmann A, Arnold MA, Delwel R, Valk PJ, Lohler J, Slany RK, Olson EN, et al. Homing and invasiveness of MLL/ENL leukemic cells is regulated by MEF2C. Blood. 2009;114:2476–2488. doi: 10.1182/blood-2008-05-158196. [DOI] [PubMed] [Google Scholar]
- Shackelford DB, Shaw RJ. The LKB1-AMPK pathway: metabolism and growth control in tumour suppression. Nat Rev Cancer. 2009;9:563–575. doi: 10.1038/nrc2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelson T, Heckl D, Ebert BL, Root DE, Doench JG, et al. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science. 2014;343:84–87. doi: 10.1126/science.1247005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw RJ, Kosmatka M, Bardeesy N, Hurley RL, Witters LA, DePinho RA, Cantley LC. The tumor suppressor LKB1 kinase directly activates AMP-activated kinase and regulates apoptosis in response to energy stress. Proc Natl Acad Sci U S A. 2004;101:3329–3335. doi: 10.1073/pnas.0308061100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J, Wang E, Milazzo JP, Wang Z, Kinney JB, Vakoc CR. Discovery of cancer drug targets by CRISPR-Cas9 screening of protein domains. Nat Biotechnol. 2015;33:661–667. doi: 10.1038/nbt.3235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundberg TB, Liang Y, Wu H, Choi HG, Kim ND, Sim T, Johannessen L, Petrone A, Khor B, Graham DB, et al. Development of Chemical Probes for Investigation of Salt-Inducible Kinase Function in Vivo. ACS Chem Biol. 2016;11:2105–2111. doi: 10.1021/acschembio.6b00217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas-Chollier M, Hufton A, Heinig M, O’Keeffe S, Masri NE, Roider HG, Manke T, Vingron M. Transcription factor binding predictions using TRAP for the analysis of ChIP-seq data and regulatory SNPs. Nat Protoc. 2011;6:1860–1869. doi: 10.1038/nprot.2011.409. [DOI] [PubMed] [Google Scholar]
- Trapnell C, Hendrickson DG, Sauvageau M, Goff L, Rinn JL, Pachter L. Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat Biotechnol. 2013;31:46–53. doi: 10.1038/nbt.2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tripathi S, Pohl MO, Zhou Y, Rodriguez-Frandsen A, Wang G, Stein DA, Moulton HM, DeJesus P, Che J, Mulder LC, et al. Meta- and Orthogonal Integration of Influenza “OMICs” Data Defines a Role for UBR4 in Virus Budding. Cell Host Microbe. 2015;18:723–735. doi: 10.1016/j.chom.2015.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzelepis K, Koike-Yusa H, De Braekeleer E, Li Y, Metzakopian E, Dovey OM, Mupo A, Grinkevich V, Li M, Mazan M, et al. A CRISPR Dropout Screen Identifies Genetic Vulnerabilities and Therapeutic Targets in Acute Myeloid Leukemia. Cell Rep. 2016;17:1193–1205. doi: 10.1016/j.celrep.2016.09.079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uebi T, Itoh Y, Hatano O, Kumagai A, Sanosaka M, Sasaki T, Sasagawa S, Doi J, Tatsumi K, Mitamura K, et al. Involvement of SIK3 in glucose and lipid homeostasis in mice. PLoS One. 2012;7:e37803. doi: 10.1371/journal.pone.0037803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Linden AM, Nolan KM, Sengupta P. KIN-29 SIK regulates chemoreceptor gene expression via an MEF2 transcription factor and a class II HDAC. EMBO J. 2007;26:358–370. doi: 10.1038/sj.emboj.7601479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walkinshaw DR, Weist R, Kim GW, You L, Xiao L, Nie J, Li CS, Zhao S, Xu M, Yang XJ. The tumor suppressor kinase LKB1 activates the downstream kinases SIK2 and SIK3 to stimulate nuclear export of class IIa histone deacetylases. J Biol Chem. 2013;288:9345–9362. doi: 10.1074/jbc.M113.456996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B, Moya N, Niessen S, Hoover H, Mihaylova MM, Shaw RJ, Yates JR, 3rd, Fischer WH, Thomas JB, Montminy M. A hormone-dependent module regulating energy balance. Cell. 2011;145:596–606. doi: 10.1016/j.cell.2011.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Birsoy K, Hughes NW, Krupczak KM, Post Y, Wei JJ, Lander ES, Sabatini DM. Identification and characterization of essential genes in the human genome. Science. 2015;350:1096–1101. doi: 10.1126/science.aac7041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Yu H, Hughes NW, Liu B, Kendirli A, Klein K, Chen WW, Lander ES, Sabatini DM. Gene Essentiality Profiling Reveals Gene Networks and Synthetic Lethal Interactions with Oncogenic Ras. Cell. 2017;168:890–903. e815. doi: 10.1016/j.cell.2017.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei J, Wunderlich M, Fox C, Alvarez S, Cigudosa JC, Wilhelm JS, Zheng Y, Cancelas JA, Gu Y, Jansen M, et al. Microenvironment determines lineage fate in a human model of MLL-AF9 leukemia. Cancer Cell. 2008;13:483–495. doi: 10.1016/j.ccr.2008.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wein MN, Liang Y, Goransson O, Sundberg TB, Wang J, Williams EA, O’Meara MJ, Govea N, Beqo B, Nishimori S, et al. SIKs control osteocyte responses to parathyroid hormone. Nat Commun. 2016;7:13176. doi: 10.1038/ncomms13176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wunderlich M, Mizukawa B, Chou FS, Sexton C, Shrestha M, Saunthararajah Y, Mulloy JC. AML cells are differentially sensitive to chemotherapy treatment in a human xenograft model. Blood. 2013;121:e90–97. doi: 10.1182/blood-2012-10-464677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Y, Milazzo JP, Somerville TDD, Tarumoto Y, Huang YH, Ostrander EL, Wilkinson JE, Challen GA, Vakoc CR. A TFIID-SAGA Perturbation that Targets MYB and Suppresses Acute Myeloid Leukemia. Cancer Cell. 2018;33:13–28. e18. doi: 10.1016/j.ccell.2017.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Z, Yoshida T, Wu L, Maiti D, Cebotaru L, Duh EJ. Transcription factor MEF2C suppresses endothelial cell inflammation via regulation of NF-kappaB and KLF2. J Cell Physiol. 2015;230:1310–1320. doi: 10.1002/jcp.24870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuber J, McJunkin K, Fellmann C, Dow LE, Taylor MJ, Hannon GJ, Lowe SW. Toolkit for evaluating genes required for proliferation and survival using tetracycline-regulated RNAi. Nat Biotechnol. 2011a;29:79–83. doi: 10.1038/nbt.1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuber J, Shi J, Wang E, Rappaport AR, Herrmann H, Sison EA, Magoon D, Qi J, Blatt K, Wunderlich M, et al. RNAi screen identifies Brd4 as a therapeutic target in acute myeloid leukaemia. Nature. 2011b;478:524–528. doi: 10.1038/nature10334. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Average log2 FC in kinase domain-focused screen. Related to Figure 1
Table S2. Average log2 fold-change in DNA binding domain-focused screen. Related to Figure 4
Table S3. sgRNA and primer sequences, and MEF2C signature. Related to STAR Methods