

Abstract
This work introduces light-activated bioorthogonal non-canonical amino acid tagging (laBONCAT) as a method to selectively label, isolate, and identify proteins newly synthesized at user-defined regions in tissue culture. By photocaging L-azidohomoalanine (Aha), metabolic incorporation into proteins is prevented. The caged compound remains stable for many hours in culture, but can be photochemically liberated rapidly and on demand with spatial control. Upon directed light exposure, the uncaged amino acid is available for local translation, enabling downstream proteomic interrogation via bioorthogonal conjugation. Exploiting the reactive azide moiety present on Aha’s amino acid side chain, we demonstrate that newly synthesized proteins can be purified for quantitative proteomics or visualized in synthetic tissues with a new level of spatiotemporal control. Shedding light on when and where proteins are translated within living samples, we anticipate that laBONCAT will aid in understanding the progression of complex protein-related disorders.
Graphical Abstract
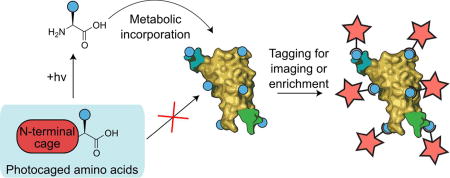
Biological processes are staggeringly dynamic and heterogeneous. Though all cells within an organism share a common genome, differential expression of genes into proteins regulate developmental processes, tissue morphogenesis and function, disease susceptibility and response, and a diverse array of signaling events governed by a wide variety of intra- and extra-cellular cues. While each protein is encoded by a gene, protein quantity and activity cannot be determined through genomic or transcriptomic analysis1,2; such techniques are blind to posttranscriptional phenomena (e.g., translational regulation, modification, protein-biomolecular interactions), necessitating strategies to directly measure protein identity and abundance3.
Recent advances in mass spectrometry and genomic sequencing have enabled high-throughput proteomic analysis, whereby the abundance, turnover, modification, and interactions of thousands of proteins can be measured in minutes4,5. From this has emerged a growing appreciation of the exceptionally dynamic nature of the proteome, which undergoes large-scale biochemical shifts during cellular proliferation, migration, and differentiation. Efforts to quantify temporal variations of the proteome have focused on the pulsed labeling of cultures with specialty amino acids that can distinguish newly synthesized from pre-existing cellular proteins.
One particularly powerful method for interrogating proteomic fluctuations in culture is known as bioorthogonal non-canonical amino acid tagging (BONCAT). In BONCAT, pulsing cells with non-canonical amino acid (ncAA) analogs yields newly synthesized proteins that bear bioorthogonal reactive groups (e.g., azides, alkynes)6–8. Metabolically labeled proteins can be covalently modified with an affinity tag that is then exploited for purification9, enabling isolation of proteins synthesized over a short window of time and providing temporally resolved proteomics.
As protein expression, degradation, translocation, and post-translational modification occur at different rates depending on cellular and subcellular location within tissues10, we sought to control BONCAT within user-defined regions of culture. Recognizing light’s unique ability to initiate chemical reactions at a time and place of interest, we developed a light-activated bioorthogonal non-canonical amino acid tagging (laBONCAT) approach (Figure 1). This strategy relies on the introduction of a molecular photocage onto the α-amine of an ncAA, preventing metabolic incorporation into proteins. Upon user-directed light exposure, the ncAA is liberated and made available for incorporation into newly translated proteins. Subsequent labeling and enrichment of these proteins is exploited for proteomic analysis.
Figure 1.

Light-activated bioorthogonal non-canonical amino acid tagging (laBONCAT). Photocaged non-canonical amino acids (ncAA) become available for stochastic incorporation into newly translated proteins following directed light exposure. Bioorthogonal handles installed by ncAAs can be exploited for protein purification prior to quantitative proteomics or fluorescent tagging for visualization.
While laBONCAT methodologies can be theoretically applied to any amino acid (including stable isotopes of natural amino acids, ncAAs, and other variants useful for quantitative proteomics), we first sought to demonstrate its utility using L-Azidohomoalanine (Aha). Aha is an azide-bearing ncAA that is metabolically incorporated by endogenous cellular machinery as a methionine (Met) surrogate11–13 whose low-level incorporation does not significantly alter protein expression14,15 (Figure 2a). Aha’s azido functionality represents a useful bioorthogonal handle for subsequent labeling reactions, including the strain-promoted azide-alkyne cycloaddition16 (SPAAC) (Figure 2b). We synthesized a photocaged Aha (NPPOC-Aha, Figure 2a) through condensation of the α-amine of Aha17 with the activated ester of 2,5-dioxopyrrolidin-1-yl (2-(2-nitrophenyl)propyl) carbonate18 (NPPOC). As NPPOC-caged amines undergo irreversible β-elimination upon exposure to near-ultraviolet light18 (λ = 365 nm; Figure 2c), Aha can be photochemically generated in situ in response to mild and cytocompatible light exposure18–22. We believe this to be the first example of an amino acid (canonical or otherwise) that has been photocaged at its N-terminus to prevent translation.
Figure 2.

Chemical structures, reaction diagrams, and photokinetics. (a) Structures of L-methionine (Met), L-azidohomoalanine (Aha), and photocaged Aha (NPPOC-Aha). (b) SPAAC is a bioorthogonal reaction for labeling azides incorporated into proteins with strained cyclooctynes. (c) Upon light exposure, photolysis of NPPOC-Aha yields free Aha for incorporation into newly synthesized proteins. (d) Kinetic analysis of NPPOC-Aha photolysis demonstrates rapid uncaging suitable for biological sampling. (e) A carboxyfluorescein-labeled bicyclononyne (FAM-BCN) provides a fluorescent tag for labeling azide-functionalized proteins.
For this system to be effective, the kinetics of NPPOC uncaging should be rapid, such that the liberation of free Aha is not rate limiting compared with the biological processes under study. To determine its photolysis kinetics, NPPOC-Aha (dissolved in H2O:CH3CN, 50:50) was irradiated with collimated UV light18 (λ = 365 nm, 10 mW cm−2, 0 – 600 s exposure). Degradation products were quantitatively analyzed by HPLC, with elution fractions compositionally identified by mass spectrometry. A first-order decay constant of 0.0075 ± 0.0002 s−1 was observed for NPPOC photolysis (Figure 2d); 90% of the NPPOC cleaved after 5 minutes of mild irradiation (10 mW cm−2), a timescale suitable for many biological applications.
To demonstrate photomediated incorporation of Aha, Met-depleted HeLa cells were incubated with NPPOC-Aha (250 µM). Subsequent irradiation with UV light (λ = 365 nm, 10 mW cm−2, 5 min) yielded photoliberated Aha for metabolic incorporation. Two hours after light exposure, cells were lysed and their proteins were treated with a bicyclononyne-modified fluorescein (FAM-BCN, 100 nM, Figure 2e) to introduce a fluorescent label by SPAAC. Protein fluorescence was then used to quantify the extent of Aha incorporation following protein separation by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE). Non-irradiated samples and the Met control exhibited a similar lack of fluorescence; the UV-treated NPPOC-Aha and Aha control displayed significant fluorescent enhancement (Figure 3a), indicating successful implementation of laBONCAT.
Figure 3.

In vitro analysis of light-activated bioorthogonal non-canonical amino acid tagging (laBONCAT), stability, and isolation. (a) Analysis of fluorescently labeled protein lysate by SDS-PAGE. In vitro incorporation of free L-azidohomoalanine (Aha) and Aha after photoliberation provides azide-functionalized proteins for fluorescent tagging by carboxyfluorescein-labeled bicyclononyne (FAM-BCN). Only samples incubated with free Aha or light-treated photocaged Aha (NPPOC-Aha) are fluorescently labeled. Coomassie staining indicates near-uniform protein loading. (b) The effect of light intensity and NPPOC-Aha concentration on azide incorporation into newly synthesized proteins was investigated, based on protein fluorescence. (c) The degree of incorporation from part b normalized for expected free Aha concentration. (d) Light irradiation itself does not impact ncAA incorporation of Aha (*p<0.05 by one-way ANOVA followed by Tukey’s test). (e) A significant amount of NPPOC-Aha remains stable over several hours in media and in contact with live cells suitable for labeling new proteins in tissue culture. (f) Following photomediated Aha incorporation, newly synthesized proteins can be isolated by affinity purification for downstream proteomic analysis. Fractions from left to right: flow through 1 (F1), wash 1–5 (W1–5), and elution 1 (E1).
To control the extent of ncAA incorporation into newly synthesized proteins, we varied NPPOC-Aha concentration (0 – 250 µM) and the intensity of light irradiation (0 – 10 mW cm−2) employed during photo-uncaging. As expected, Aha incorporation increased with NPPOC-Aha concentration for a given exposure condition; for low NPPOC-Aha concentrations, metabolic labeling increased with light intensity (Figure 3b). When the extent of incorporation was normalized for the expected concentration of liberated Aha, based on values predicted by the photokinetics and assuming no side reactions accompanying photolysis, the result was a smooth, continuous curve that plateaus above ~50 µM free Aha (Figure 3c). To determine the potential effects of UV irradiation on metabolic incorporation, labeled protein fluorescence was compared for samples treated with Aha (50 µM) +/− light (10 mW cm−2, 5 min) (Figure 3d). Finding no statistical difference in protein labeling following UV irradiation, we compared incorporation between Aha and irradiated NPPOC-Aha cultures (each at 100 µM). NPPOC-Aha + light gave rise to slightly less incorporation than Aha alone, which we attribute to incomplete photoconversion of NPPOC-Aha to Aha. This is supported by data that protein labeling does not depend on whether NPPOC-Aha is irradiated separately or during incubation with cells (Figure 3d).
To assess its in vitro stability, NPPOC-Aha (100 µM) was incubated in media with HeLa cells for 0 – 4 hr prior to light exposure (10 mW cm−2, 5 min) and subsequent metabolic labeling (2 hr). Aha incorporation was observed for all irradiated samples, though its extent decreased over time. This was attributed to unknown cellular processing of NPPOC-Aha; simple hydrolysis yielding free Aha does not explain this behavior, as non-irradiated samples do not show increased incorporation over time. While the >4 hours of working time is likely sufficient for many applications, we anticipate that different photocages and/or ncAAs may exhibit increased long-term stability. Such stability is useful in sampling biological systems, as it decouples media swaps from proteome labeling, allowing researchers to standardize their experimental conditions.
After demonstrating the ability to label newly synthesized proteins, we sought to extend the laBONCAT methodologies to their affinity purification. After NPPOC-Aha uncaging and metabolic incorporation of the ncAA, proteins were biotinylated via SPAAC with a dibenzocyclooctyne-modified biotin probe. Biotinylated proteins were captured on a streptavidin resin prior to protein elution by streptavidin denaturation. Eluents were subjected to SDS-PAGE and silver stained for visualization (Figure 3f). Results highlight the capability to selectively isolate newly synthesized proteins from irradiated samples.
Building on the capability to fluorescently tag cellular lysates as well as isolate species of interest via laBONCAT, we turned our efforts towards the technique’s unique ability to label newly synthesized proteins in vitro with spatial control; we anticipate that this method could be applied to the isolation of proteins transcribed at user-specified times and locations from heterogeneous features in tissue culture, especially tissue slices. HeLa cells were treated with NPPOC-Aha (100 µM) and exposed to light (λ = 365 nm, 10 mW cm−2, 5 min). Two hours after exposure, cells were fixed, permeabilized, and treated with FAM-BCN (100 nM) to introduce a fluorescent label via SPAAC. The extent of fluorescent labeling of cells treated in this method was similar to free Aha, while NPPOC-Aha in the absence of light exhibited low-level labeling similar to Met (Figure 4).
Figure 4.

Irradiation of photocaged L-azidohomoalanine (NPPOC-Aha) yields free Aha for ncAA in vitro incorporation. (a) Fluorescent tagging of fixed cells demonstrates similar fluorescent labeling (green) of Aha and irradiated NPPOC-Aha, but not L-methionine (Met) and unexposed NPPOC-Aha. (scale bar = 100 µM). (b) Metabolic labeling of cells in synthetic tissue is confined to regions near user-defined exposure (hashed area). Actin staining (red) remains uniform across sample (scale bar = 1 mm).
Next, we demonstrated the ability to control Aha incorporation spatially within synthetic tissues. Cells were encapsulated in oxime-based poly(ethylene glycol) hydrogels (7 wt%), treated with NPPOC-Aha and selectively irradiated through a slitted photomask. Cells were fixed and fluorescently labeled with FAM-BCN, phalloidin, and Hoechst. The observed cellular FAM signal was localized near exposed regions, corresponding to patterned Aha incorporation. FAM fluorescence decreased exponentially away from exposed regions in a diffusion-predicted manner. Actin and DNA staining lack substantial patterning (Figure 4, Supplementary Figure 6).
This technique provides an inherent improvement to traditional BONCAT by allowing for precise timing in metabolic incorporation. In addition, the photochemical nature of the system provides a degree of spatial control for the investigation of heterogeneous biological systems. While not the focus of this manuscript, we anticipate that laBONCAT could be readily combined with strategies for pulsed stable isotope labeling by amino acids in cell culture23 (pSILAC) to purify, identify, and quantify proteins expressed at user-defined regions in culture. This newfound ability is expected to prove particularly useful in the investigation of heterogeneous protein-related disease (e.g., Alzheimer’s), potentially yielding new diagnostic markers and therapeutic targets.
In this manuscript, we have demonstrated the ability to control protein translation spatiotemporally through laBONCAT. While the method has been initially validated using NPPOC-Aha responsive to near-UV light, facile extension of the technique to control incorporation of different amino acids using different photocage/light combinations is expected. We believe that this strategy shows unique promise towards the visualization and characterization of newly synthesized proteins at user-defined time and places of interest.
METHODS
Detailed methods and experimental conditions are described in the Supporting Information.
Supplementary Material
Acknowledgments
Marvin was used for drawing, displaying, and characterizing chemical structures, substructures and reactions, Marvin 17.2.27.0, 2017, ChemAxon (http://www.chemaxon.com). We acknowledge the support from the NIH to the University of Washington (UW) W. M. Keck Microscopy Center (S10 OD016240). This work was supported by the Mary Gates Endowment (P.E.F.), the NASA Space Grant Consortium (NNX15AJ98H, P.E.F.), a Washington Research Foundation Fellowship (P.E.F), a UW Faculty Startup Grant (C.A.D.) and a UW Royalty Research Fund Grant (A112554, C.A.D.).
Footnotes
ASSOCIATED CONTENT
The Supporting Information is available free of charge on the ACS Publications website at DOI: XXX.
This includes full experimental procedures, synthesis and characterization of all small molecules and functional polymers, control experiments for affinity purification of newly synthesized proteins, and full analysis of spatially labeled synthetic tissues.
Author Contributions
For this manuscript, S.M.A and C.A.D. conceived and designed the experiments; S.M.A, E.R.R., P.E.F., and J.V.W. performed the experiments; S.M.A. and C.A.D. analyzed the data and prepared the figures; S.M.A. and C.A.D. wrote the paper.
The authors declare no competing financial interest.
References
- 1.Taniguchi Y, Choi PJ, Li G, Chen H, Babu M, Hearn J, Emili A, Xie XS. Quantifying E. coli Proteome and Transcriptome with Single-Molecule Sensitivity in Single Cells. Science. 2010;329:533–538. doi: 10.1126/science.1188308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Vogel C, Marcotte EM. Insights into the regulation of protein abundance from proteomic and transcriptomic analyses. Nat. Genet. Rev. 2012;13:227–232. doi: 10.1038/nrg3185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pandey A, Mann M. Proteomics to study genes and genomes. Nature. 2000;405:837–846. doi: 10.1038/35015709. [DOI] [PubMed] [Google Scholar]
- 4.Aebersold R, Mann M. Mass spectrometry-based proteomics. Nature. 2003;422:198–207. doi: 10.1038/nature01511. [DOI] [PubMed] [Google Scholar]
- 5.Domon B, Aebersold R. Mass spectrometry and protein analysis. Science. 2006;312:212–217. doi: 10.1126/science.1124619. [DOI] [PubMed] [Google Scholar]
- 6.Dieterich DC, Link AJ, Graumann J, Tirrell DA, Schuman EM. Selective identification of newly synthesized proteins in mammalian cells using bioorthogonal noncanonical amino acid tagging (BONCAT) Proc. Natl. Acad. Sci. U. S. A. 2006;103:9482–9487. doi: 10.1073/pnas.0601637103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dieterich DC, Lee JJ, Link AJ, Graumann J, Tirrell DA, Schuman EM. Labeling, detection and identification of newly synthesized proteomes with bioorthogonal non-canonical amino-acid tagging. Nat. Protoc. 2007;2:532–540. doi: 10.1038/nprot.2007.52. [DOI] [PubMed] [Google Scholar]
- 8.Sletten EM, Bertozzi CR. Bioorthogonal chemistry: fishing for selectivity in a sea of functionality. Angew. Chemie Int. Ed. 2009;48:6974–6998. doi: 10.1002/anie.200900942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Szychowski J, Mahdavi A, Hodas JJL, Bagert JD, Ngo JT, Landgraf P, Dieterich DC, Schuman EM, Tirrell Da. Cleavable biotin probes for labeling of biomolecules via azide-alkyne cycloaddition. J. Am. Chem. Soc. 2010;132:18351–18360. doi: 10.1021/ja1083909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Larance M, Lamond AI. Multidimensional proteomics for cell biology. Nat. Rev. Mol. Cell Biol. 2015;16:269–280. doi: 10.1038/nrm3970. [DOI] [PubMed] [Google Scholar]
- 11.Kiick KL, Saxon E, Tirrell DA, Bertozzi CR. Incorporation of azides into recombinant proteins for chemoselective modification by the Staudinger ligation. Proc. Natl. Acad. Sci. 2002;99:19–24. doi: 10.1073/pnas.012583299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Beatty KE, Fisk JD, Smart BP, Lu YY, Szychowski J, Hangauer MJ, Baskin JM, Bertozzi CR, Tirrell DA. Live-cell imaging of cellular proteins by a strain-promoted azide-alkyne cycloaddition. Chembiochem. 2010;11:2092–2095. doi: 10.1002/cbic.201000419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dieterich DC, Hodas JJ, Gouzer G, Shadrin IY, Ngo JT, Triller A, Tirrell DA, Schuman EM. In situ visualization and dynamics of newly synthesized proteins in rat hippocampal neurons. Nat. Neurosci. 2010;13:897–905. doi: 10.1038/nn.2580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Howden AJM, Geoghegan V, Katsch K, Efstathiou G, Bhushan B, Boutureira O, Thomas B, Trudgian DC, Kessler BM, Dieterich DC, Davis BG, Acuto O. QuaNCAT: quantitating proteome dynamics in primary cells. Nat. Methods. 2013;10:343–346. doi: 10.1038/nmeth.2401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bagert JD, Xie YJ, Sweredoski MJ, Qi Y, Hess S, Schuman EM, Tirrell DA. Quantitative, time-resolved proteomic analysis by combining bioorthogonal noncanonical amino acid tagging and pulsed stable isotope labeling by amino acids in cell culture. Mol. Cell. Proteomics. 2014;13:1352–8. doi: 10.1074/mcp.M113.031914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Baskin JM, Prescher JA, Laughlin ST, Agard NJ, Chang PV, Miller IA, Lo A, Codelli JA, Bertozzi CR. Copper-free click chemistry for dynamic in vivo imaging. Proc. Natl. Acad. Sci. U. S. A. 2007;104:16793–16797. doi: 10.1073/pnas.0707090104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Roth S, Drewe WC, Thomas NR. A concise and scalable route to L-azidohomoalanine. Nat. Protoc. 2010;5:1967–1973. doi: 10.1038/nprot.2010.164. [DOI] [PubMed] [Google Scholar]
- 18.DeForest CA, Tirrell DA. A photoreversible protein-patterning approach for guiding stem cell fate in three-dimensional gels. Nat. Mater. 2015;14:523–531. doi: 10.1038/nmat4219. [DOI] [PubMed] [Google Scholar]
- 19.Bryant SJ, Nuttelman CR, Anseth KS. Cytocompatibility of UV and visible light photoinitiating systems on cultured NIH/3T3 fibroblasts in vitro. J. Biomater. Sci. Polym. Ed. 2000;11:439–457. doi: 10.1163/156856200743805. [DOI] [PubMed] [Google Scholar]
- 20.DeForest CA, Polizzotti BD, Anseth KS. Sequential click reactions for synthesizing and patterning three-dimensional cell microenvironments. Nat. Mater. 2009;8:659–664. doi: 10.1038/nmat2473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.DeForest CA, Anseth KS. Cytocompatible click-based hydrogels with dynamically tunable properties through orthogonal photoconjugation and photocleavage reactions. Nat. Chem. 2011;3:925–931. doi: 10.1038/nchem.1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wong DY, Ranganath T, Kasko AM. Low-Dose, Long-Wave UV Light Does Not Affect Gene Expression of Human Mesenchymal Stem Cells. PLoS One. 2015;10:1–21. doi: 10.1371/journal.pone.0139307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schwanhäusser B, Gossen M, Dittmar G, Selbach M. Global analysis of cellular protein translation by pulsed SILAC. Proteomics. 2009;9:205–209. doi: 10.1002/pmic.200800275. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.