

Abstract
Cloaking its carboxyl groups with a hydrophobic moiety is shown to enable a protein to enter the cytosol of a mammalian cell. Diazo compounds derived from (p-methylphenyl)glycine were screened for the ability to esterify the green fluorescent protein (GFP) in an aqueous environment. Esterification of GFP with 2-diazo-2-(p-methylphenyl)-N,N-dimethylacetamide was efficient. The esterified protein entered the cytosol by traversing the plasma membrane directly, like a small-molecule prodrug. As with prodrugs, the nascent esters are substrates for endogenous esterases, which regenerate native protein. Thus, esterification could provide a general means to deliver native proteins to the cytosol.
Graphical Abstract
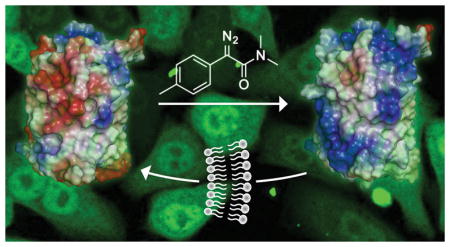
Approximately 20% of the drugs in today’s pharmacopeia are proteins.1 Essentially all of those proteins act on extracellular targets. This limitation arises from an intrinsic inability of proteins to enter the cytosol.2 Although viral vectors can be used to deliver DNA that encodes a protein of interest, this genetic approach lacks regulation and can induce stress responses, carcinogenesis, or immunogenicity.3 In contrast, the direct delivery of proteins into cells would enable temporal control over cellular exposure and minimize deleterious off-target effects.4
Proteins can be delivered into cells by using site-directed mutagenesis,5 irreversible chemical modification,6 conjugation of transduction domains (such as cell-penetrating peptides, CPPs),7 cationic lipid carriers,8 or electroporation.9 Many of these strategies show promise but also pose problems,2,4 such as inefficient escape from endosomes or inapplicability in an animal.
To cross the plasma membrane, proteins must overcome two barriers: Coulombic repulsion from the anionic glycocalyx and exclusion from the hydrophobic environment of the lipid bilayer.10 Natural and synthetic systems suggest means to overcome these barriers. For example, mammalian ribonucleases are capable of cytosolic entry that is mediated by clusters of positively charged residues.11 Cellular uptake can also be enhanced by exogenous hydrophobic moieties.12 For example, noncovalent complexation with pyrene butyrate enables the cytosolic delivery of a green fluorescent protein (GFP) conjugate to a cationic CPP.13 Additionally, several natural and synthetic protein transduction domains (e.g., penetratin, TP10, and pVEC) consist of cationic and hydrophobic residues, which impart an amphipathic character.7e,7f,14 Their hydrophobic residues are crucial for mediating membrane translocation.
We envisioned a different strategy—one that invokes a chemoselective reaction that remodels the protein surface to become less anionic and more hydrophobic. The surface of proteins displays cationic groups (i.e., guanidinium, ammonium, and imidazolium) and anionic groups (carboxylates). We hypothesized that the esterification of its carboxyl groups could endow a protein with the ability to access the cytosol. In particular, by cloaking negative charges with a hydrophobic moiety, we might increase the nonpolar surface area while enabling endogenous positive charges to manifest favorable Coulombic interactions with anionic cell-surface components. The ensuing mode-of-action would resemble that of small-molecule prodrugs, which have been in the pharmacopoeia for decades.15
To effect our strategy, we employed diazo compounds derived from (p-methylphenyl)glycine. We had shown previously that the basicity of such diazo compounds enables the efficient esterification of carboxylic acids in an aqueous environment.16 Now, we exploited the modular nature of this scaffold. Specifically, we deimidogenated azide precursors16,17 to access diazo compounds 1–6, which span a range of hydrophobicity (Figure 1).
Figure 1.

Bar graph showing the extent of esterification of the superfolder variant of GFP with diazo compounds 1–6 (black) and the internalization of the ensuing esterified GFPs into CHO-K1 cells (green). Values (± SD) were determined by mass spectrometry and flow cytometry, respectively. Parenthetical logP values were calculated with software from Molinspiration (Slovensky Grab, Slovak Republic).
Then, we screened solution conditions for maximal protein esterification by our scaffold. We were aware that the mechanism of esterification requires a protonated carboxyl group,18 which is encouraged by a low pH and an organic cosolvent. Using GFP and diazo compound 3, we found that an aqueous solution at pH 6.5 that contains 20% v/v acetonitrile gives a high yield of esters (Figure S1). These conditions should be tolerable by most proteins.
Next, we evaluated diazo compounds 1–6 for their ability to esterify a protein and facilitate its internalization into a mammalian cell. We found that more polar diazo compounds alkylated more carboxyl groups than did less polar compounds (Figures 1 and S2). Then, we treated live cells with esterified proteins and quantified internalization with flow cytometry. We discovered that the level of cellular internalization parallels the number of labels per protein (Figure 1), which suggests that simply masking anionic groups is advantageous. Moreover, cellular fluorescence increases in a time-dependent manner (Figure S3), as expected for a process based on vectorial diffusion from the outside to the inside.
Of the six diazo compounds, compound 1 was the most effective in engendering cellular uptake and was selected for further study. On average, 11 of the 32 carboxyl groups in GFP were masked as neutral esters by diazo compound 1 (Figure S2). Although the esterification of 11 carboxyl groups in GFP could produce 32C11 = 1.2 × 108 different molecules, esters are most likely to form with solvent-accessible carboxyl groups that have a high pKa value (Table S1).18 That trend was apparent in tandem mass spectrometry data (Figure S4). This selectivity has fortuitous consequences. An aspartate or glutamate residue within a hydrophobic patch is a likely target for esterification, which would extend the size of the patch. Clustered anionic residues likewise have high pKa values, and their esterification would overcome a strong deterrent to cellular uptake. In contrast, an aspartate or glutamate residue within a salt bridge is unlikely to be esterified, but a salt bridge manifests less Coulombic repulsion with anionic cell-surface components than do isolated or clustered anionic residues.
We used confocal microscopy to visualize the uptake of GFP by live mammalian cells. For calibration, we compared the uptake of GFP with that of a “super-charged” variant in which site-directed mutagenesis was used to replace anionic residues with arginine (Figure S5).5a Unmodified GFP did not enter cells (Figure 2). Super-charged GFP did enter cells, but produced a punctate pattern of fluorescence that is suggestive of endosomal localization. At 4 °C, which is a temperature that precludes endocytosis,19 the fluorescence from super-charged GFP was scant and localized to the plasma membrane.
Figure 2.

Images of the cellular internalization of GFP and its super-charged and esterified variants. CHO-K1 cells were incubated with protein (15 μM) for 2 h at 37 or 4 °C. Cells were then washed, stained with Hoechst 33342 and wheat germ agglutinin (WGA)–Alexa Fluor 647, and imaged by confocal microscopy (Hoechst 33342: ex. 405 nm, em. 450 nm; WGA–Alexa Fluor 647: ex. 647 nm, em. 700 nm; GFP: ex. 488 nm, em. 525 nm). Scale bars: 25 μm.
Images of cells treated with GFP–1 were in marked contrast to those treated with unmodified GFP or super-charged GFP. At 37 °C, treatment with GFP–1 elicited diffuse fluorescence, suggestive of cytosolic localization (Figure 2). Most remarkably, this pattern persisted at 4 °C, indicating that uptake does not rely on endocytosis. In other words, GFP–1 appears to enter cells by passing directly through the plasma membrane, like a small-molecule prodrug.15
To enter the nucleus, a protein must pass through the cytosol. To verify cytosolic entry, we reiterated a known GFP variant bearing a nuclear localization signal (nlsGFP; Figure S5)20 and esterified that variant with compound 1 (Figure S6). We then treated live cells with either nlsGFP or esterified nlsGFP (nlsGFP–1) and visualized the cells with confocal microscopy. In the ensuing images (Figure 3), nlsGFP colocalizes with membrane stain (Pearson’s r = 0.21) and is excluded from the nucleus (r = −0.12). This result is expected, as GFP is impermeant but a nuclear localization signal is cationic and can form salt bridges with the anionic glycocalyx. In contrast, nlsGFP–1 not only exhibits diffuse staining like GFP–1 (Figure 2), but also colocalizes with a nuclear stain (r = 0.51) to an extent expected for this particular variant.20 These data indicate that nlsGFP–1 accesses the nucleus and, thus, the cytosol.
Figure 3.

Images of the nuclear internalization of a protein that contains a nuclear localization signal and its esterified variant. CHO-K1 cells were incubated with nlsGFP or nlsGFP–1 (15 μM) for 2 h at 37 °C. Cells were then washed, stained with Hoechst 33342 and WGA–Alexa Fluor 647, and imaged by confocal microscopy (Hoechst 33342: ex. 405 nm, em. 450 nm; WGA–Alexa Fluor 647: ex. 647 nm, em. 700 nm; GFP: ex. 488 nm, em. 525 nm). Scale bars: 25 μm.
Finally, we investigated the bioreversibility of esterification. Incubation of a model protein esterified with diazo compound 1 in a mammalian cell extract resulted in the complete removal of labels (Figure S7). This finding is consistent with an inability of de-esterified GFP–1 (i.e., GFP) to escape from the cytosol and its accumulation there (Figure S3). Thus, the esters formed upon reaction with 1 are substrates for endogenous esterases, like prodrugs.15 Moreover, the alcohol product of the esterase-mediated hydrolysis is benign to mammalian cells (Figure S8).
In summary, we have demonstrated that esterification of protein carboxyl groups with a tuned diazo compound can engender delivery of the protein across the plasma membrane as if it were a small molecule. Further, this chemical modification is traceless, being removable by cellular esterases. This delivery strategy provides an unprecedented means to deliver native proteins into cells for applications in the laboratory and, potentially, the clinic.
Supplementary Material
Acknowledgments
We are grateful to Dr. K. A. Andersen for early observations, Dr. E. K. Grevstad for help with microscopy, L. B. Hyman for technical advice, Dr. T. T. Hoang for supplying FLAG–ANG, and Dr. C. L. Jenkins for contributive discussions. K.A.M. was supported by Molecular Biosciences Training Grant T32 GM007215 (NIH). J.E.L. was supported by a National Science Foundation Graduate Research Fellowship. This work was supported by grant R01 GM044783 (NIH) and made use of the National Magnetic Resonance Facility at Madison, which is supported by grant P41 GM103399 (NIH).
Footnotes
Synthetic methods, cell biological methods, and additional analytical data, including Table S1 and Figures S1–S8. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Dimitrov DS. Methods Mol Biol. 2012;899:1–26. doi: 10.1007/978-1-61779-921-1_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.(a) Pisal DS, Kosloski MP, Balu-Iyer SV. J Pharm Sci. 2010;99:2557–2575. doi: 10.1002/jps.22054. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Fu A, Tang R, Hardie J, Farkas ME, Rotello VM. Bioconjugate Chem. 2014;25:1602–1608. doi: 10.1021/bc500320j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.(a) Somia N, Verma IM. Nat Rev Genet. 2000;1:91–99. doi: 10.1038/35038533. [DOI] [PubMed] [Google Scholar]; (b) Collins M, Thrasher A. Proc Biol Sci. 2015;282:20143003. doi: 10.1098/rspb.2014.3003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Leader B, Baca QJ, Golan DE. Nat Rev Drug Discov. 2008;7:21–39. doi: 10.1038/nrd2399. [DOI] [PubMed] [Google Scholar]
- 5.(a) Fuchs SM, Raines RT. ACS Chem Biol. 2007;2:167–170. doi: 10.1021/cb600429k. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) McNaughton BR, Cronican JJ, Thompson DB, Liu DR. Proc Natl Acad Sci USA. 2009;106:6111–6116. doi: 10.1073/pnas.0807883106. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Bruce VJ, Lopez-Islas M, McNaughton BR. Protein Sci. 2016;25:1129–1137. doi: 10.1002/pro.2926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.(a) Futami J, Yamada H. Curr Pharm Biotechnol. 2008;9:180–184. doi: 10.2174/138920108784567326. [DOI] [PubMed] [Google Scholar]; (b) Ellis GA, Palte MJ, Raines RT. J Am Chem Soc. 2012;134:3631–3634. doi: 10.1021/ja210719s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.(a) Schwarze SR, Ho A, Vocero-Akbani A, Dowdy SF. Science. 1999;285:1569–1572. doi: 10.1126/science.285.5433.1569. [DOI] [PubMed] [Google Scholar]; (b) Fuchs SM, Raines RT. Protein Sci. 2005;14:1538–1544. doi: 10.1110/ps.051393805. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Stanzl EG, Trantow BM, Vargas JR, Wender PA. Acc Chem Res. 2013;46:2944–2954. doi: 10.1021/ar4000554. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Nischan N, Herce HD, Natale F, Bohlke N, Budisa N, Cardoso MC, Hackenberger CPR. Angew Chem, Int Ed. 2015;54:1950–1953. doi: 10.1002/anie.201410006. [DOI] [PubMed] [Google Scholar]; (e) LaRochelle JR, Cobb GB, Steinauer A, Rhoades E, Schepartz A. J Am Chem Soc. 2015;137:2536–2541. doi: 10.1021/ja510391n. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Qian Z, Martyna A, Hard RL, Wang J, Appiah-Kubi G, Coss C, Phelps MA, Rossman JS, Pei D. Biochemistry. 2016;55:2601–2612. doi: 10.1021/acs.biochem.6b00226. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Nagel YA, Raschle PS, Wennemers H. Angew Chem, Int Ed. 2017;56:122–126. doi: 10.1002/anie.201607649. [DOI] [PubMed] [Google Scholar]
- 8.Zuris JA, Thompson DB, Shu Y, Guilinger JP, Bessen JL, Hu JH, Maeder ML, Joung JK, Chen ZY, Liu DR. Nat Biotechnol. 2015;33:73–80. doi: 10.1038/nbt.3081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kim S, Kim D, Cho SW, Kim J, Kim JS. Genome Res. 2014;24:1012–1019. doi: 10.1101/gr.171322.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Palte MJ, Raines RT. J Am Chem Soc. 2012;134:6218–6223. doi: 10.1021/ja2106477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Turcotte RF, Lavis LD, Raines RT. FEBS J. 2009;276:4270–4281. doi: 10.1111/j.1742-4658.2009.07098.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Perrett F, Nishihara M, Takeuchi T, Futaki S, Lazar AN, Coleman AW, Sakai N, Matile S. J Am Chem Soc. 2005;127:1114–1115. doi: 10.1021/ja043633c. [DOI] [PubMed] [Google Scholar]
- 13.Takeuchi T, Kosuge M, Tadokoro A, Sugiura Y, Nishi M, Kawata M, Sakai N, Matile S, Futaki S. ACS Chem Biol. 2006;1:299–303. doi: 10.1021/cb600127m. [DOI] [PubMed] [Google Scholar]
- 14.(a) Elmquist A, Hansen M, Langel Ü. Biochim Biophys Acta. 2006;1758:721–729. doi: 10.1016/j.bbamem.2006.05.013. [DOI] [PubMed] [Google Scholar]; (b) Di Pisa M, Chassaing G, Swiecicki JM. Biochemistry. 2015;54:194–207. doi: 10.1021/bi501392n. [DOI] [PubMed] [Google Scholar]
- 15.(a) Testa B, Mayer JM. Hydrolysis in Drug and Prodrug Metabolism. Wiley–VCH; Weinheim, Germany: 2003. [Google Scholar]; (b) Liederer BM, Borchardt RT. J Pharm Sci. 2006:1177–1195. doi: 10.1002/jps.20542. [DOI] [PubMed] [Google Scholar]; (c) Huttunen KM, Raunio H, Rautio J. Pharmacol Rev. 2011;63:750–771. doi: 10.1124/pr.110.003459. [DOI] [PubMed] [Google Scholar]; (d) Redasani VK, Bari SB. Prodrug Design: Perspectives, Approaches and Applications in Medicinal Chemistry. Academic Press; New York, NY: 2015. [Google Scholar]
- 16.(a) McGrath NA, Andersen KA, Davis AKF, Lomax JE, Raines RT. Chem Sci. 2014;6:752–755. doi: 10.1039/c4sc01768d. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Mix KA, Raines RT. Org Lett. 2015;17:2358–2361. doi: 10.1021/acs.orglett.5b00840. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Mix KA, Aronoff MR, Raines RT. ACS Chem Biol. 2016;11:3233–3244. doi: 10.1021/acschembio.6b00810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Myers EL, Raines RT. Angew Chem, Int Ed. 2009;48:2359–2363. doi: 10.1002/anie.200804689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.(a) Roberts JD, Watanabe W, McMahon RE. J Am Chem Soc. 1951;73:760–765. [Google Scholar]; (b) Roberts JD, Watanabe W, McMahon RE. J Am Chem Soc. 1951;73:2521–2523. [Google Scholar]
- 19.Tomoda H, Kishimoto Y, Lee YC. J Biol Chem. 1989;264:15445–15450. [PubMed] [Google Scholar]
- 20.(a) Maier K, Wagner E. J Am Chem Soc. 2012;134:10169–10173. doi: 10.1021/ja302705v. [DOI] [PubMed] [Google Scholar]; (b) Ray M, Tang R, Jiang Z, Rotello VM. Bioconjugate Chem. 2015;26:1004–1007. doi: 10.1021/acs.bioconjchem.5b00141. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.