

Abstract
Rare cancers pose unique challenges to research due to their low incidence. Barriers include a scarcity of tissue and experimental models to enable basic research and insufficient patient accrual for clinical studies. Consequently, an understanding of the genetic and cellular features of many rare cancer types and their associated vulnerabilities has been lacking. However, new opportunities are emerging to facilitate discovery of therapeutic targets in rare cancers. Online platforms are allowing patients with rare cancers to organize on an unprecedented scale, tumor genome sequencing is now routinely performed in research and clinical settings, and the efficiency of patient-derived model generation has improved. New CRISPR/Cas9 and small-molecule libraries permit cancer dependency discovery in a rapid and systematic fashion. In parallel, large-scale studies of common cancers now provide reference datasets to help interpret rare cancer profiling data. Together, these advances motivate consideration of new research frameworks to accelerate rare cancer target discovery.
Introduction
A cancer type is typically designated as “common” or “rare” on the basis of its incidence. While no universally accepted cutoff exists, the National Cancer Institute (NCI) defines rare cancers as those occurring at a frequency below 15 cases per 100,000 individuals per year (National Cancer Institute, 2007). According to this definition, only 11 adult cancer types are common in the US (prostate, breast, lung, colon, uterine corpus, bladder, rectum, ovary, kidney, melanoma, and non-Hodgkin lymphoma) (Greenlee et al., 2010). The remaining adult cancer types, collectively accounting for approximately 25% of adult tumors overall, are thus considered rare (Greenlee et al., 2010). All pediatric cancers are classified as rare when the above definition is applied (Ward et al., 2014).
Despite their high collective occurrence, basic biological and clinical knowledge of many individual rare cancer types is lacking. Many of the barriers to research directly stem from the low incidence rates for rare cancer types. While tens of thousands of patients may be living with a given rare cancer (Orphanet, 2016), the geographic dispersion of rare cancer patients often limits the number of cases seen at any one institution. As a result, knowledge of rare cancers is often based on single-patient case reports (Tomich et al., 2017; Simonetti et al., 2016; Panda et al., 2016; Afani et al., 2016) or single-institution retrospective analyses that describe small cohorts of patients seen over many decades (Arikan et al., 2014; Karatayli-Ozgursoy et al., 2016; Worhunsky et al., 2015). This evidence, which can be anecdotal, is in stark contrast to the multi-institutional cancer statistics studies routinely reported for common cancer types (National Cancer Institute, 2016b). Low incidence rates also make clinical trial recruitment for rare cancers extremely challenging, stalling efforts to test emerging therapeutic hypotheses (Casali et al., 2015; Tan et al., 2003). In basic research settings, a scarcity of rare tumor tissue and patient-derived models can preclude well-powered studies aimed at elucidating underlying biology (Boehm and Golub, 2015).
Disparities in the study and understanding of common versus rare cancers are perhaps best illustrated by the respective availability of effective therapeutic interventions. Selected examples can be drawn from the National Comprehensive Cancer Network Clinical Practice Guidelines in Oncology (NCCN Guidelines), an evidence-based set of guidelines for the clinical practice of adult oncology in the US (www.nccn.org). Over 20 Category 1 interventions (meaning uniform consensus that the intervention is appropriate and based on a high level of evidence) exist for breast and prostate cancer, the most common cancers affecting women and men, respectively (NCCN Guidelines, 2017b; NCCN Guidelines, 2017c; Siegel et al., 2017). By contrast, no Category 1 interventions exist for either chordoma or chondrosarcoma, bone cancer types each of which affects fewer than 1,000 individuals per year in the US (NCCN Guidelines, 2017a; McMaster et al., 2001; Orphanet, 2016).
For many patients with a rare cancer, the lack of effective treatment regimens translates to a suboptimal clinical outcome. Indeed, a study of population-based cancer registry data on European patients reported that rare cancer patients on average had worse outcomes, with estimated average 5-year relative survival rates of 47% for patients with rare cancers (defined in the study as an incidence of <6/100,000/year) versus 65% for those with common cancers (Gatta et al., 2011). In addition to a paucity of effective treatments (owing in some cases to a limited understanding of disease-modifying targets), other factors contributing to poor outcomes may include delays in obtaining a correct diagnosis, lack of treatment guidelines, fewer clinical trials dedicated to rare cancers, and less investment in drug development (Blay et al., 2016; Panageas, 2015).
With respect to knowledge of therapeutic targets in rare cancers, which is the focus herein, new advances may help overcome historical challenges. Patients with rare cancers are increasingly interconnected online and costs of genome sequencing in both basic research and clinical settings have dropped precipitously. Indeed, it is estimated that in aggregate, nearly 3,000 tumor samples corresponding to exceedingly rare cancer types have been subjected to whole-exome, whole-genome, or other next-generation sequencing (Figure 1A). Further, the success rates of generating new organoid and cell-line models have increased substantially. These foundational advances make it possible to apply new CRISPR/Cas9 technologies and small-molecule libraries to map the dependencies for each rare cancer type in the laboratory, leading to more robust therapeutic hypotheses. Here, we describe progress in each of these areas, illustrating how the convergence of these approaches provides a framework for enabling rare cancer target discovery in an increasingly systematic fashion.
Figure 1. Progress toward the Genomic Characterization of Rare Cancers.
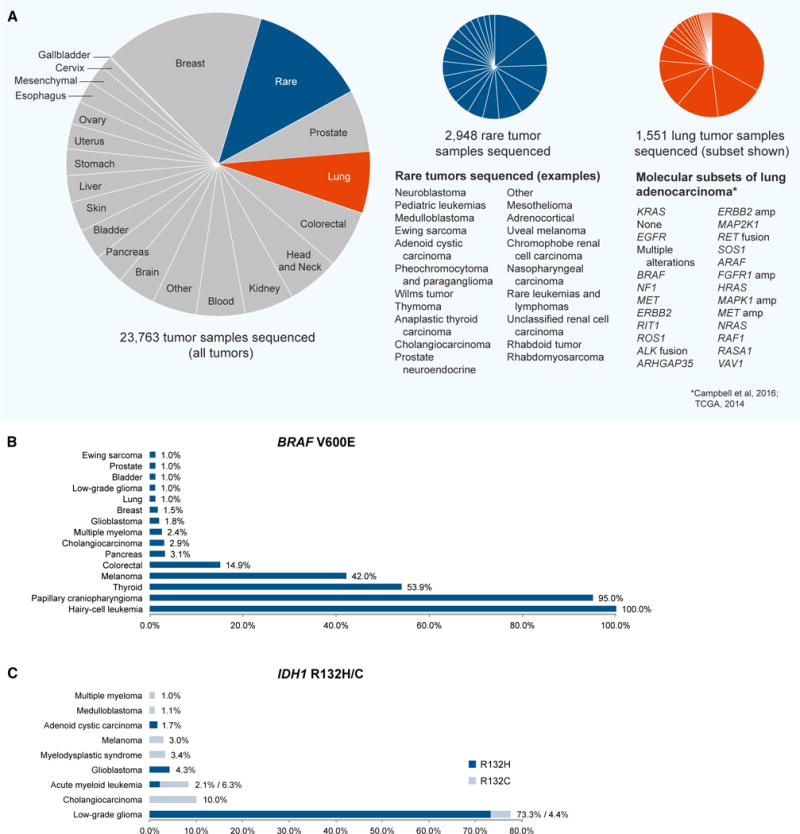
(A) Cancer types and subtypes that have been analyzed using whole-exome, whole-genome, or other next-generation sequencing to date. Left, all tumor samples by lineage (source data: www.cbioportal.org; https://gdc.cancer.gov/; Brastianos et al., 2014; Tiacci et al., 2011). Center, examples of rare tumor sequencing studies performed to date; “rare” is used in the figure to describe exceedingly rare cancers with an estimated incidence below ~1/100,000 in a population. Due to the evolving taxonomy of cancers, some subjective decisions were made with respect to classifying unusual subtypes of common cancers (incidence below ~1/100,000) as “rare” in this pie chart. Rare cancer types are listed in order of decreasing study size. Right, molecular subsets of lung adenocarcinoma. Molecular subtypes were identified using a cohort of 227 lung adenocarcinoma cases with complete data and expert histological review originally reported by Cancer Genome Atlas Research Network (2014) and re-analyzed by Campbell et al. (2016). The list of known and putative receptor tyrosine kinase/Ras/Raf pathway driver genes was derived by Campbell et al. (2016). Subtypes are listed in order of decreasing frequency.
(B) Frequency of BRAF V600E mutations across rare and common tumor types (source data: www.cbioportal.org).
(C) Frequency of IDH1 R132H/C mutations across rare and common tumor types (source data: www.cbioportal.org). cBioPortal and GDC datasets were accessed Feb–March 2017.
Overcoming Biospecimen Scarcity via Online Platforms
While patients with a rare tumor type may be geographically segregated, many have recently leveraged the Internet to virtually convene, organize, and engage in research. This online aggregation now facilitates rare cancer research on a scale that had once been challenging to achieve.
In some cases, the virtual organization of rare cancer patients has been mediated by consortia or foundations expressly dedicated to advancing rare disease research. Umbrella organizations such as the National Organization for Rare Disorders and the Rare Cancer Research Foundation provide online tools and infrastructure to disease-specific organizations, in turn enabling rare cancer patients to participate in clinical and basic research. This foundation-assisted engagement of rare cancer patients via online platforms has in several cases boosted study sample sizes and thereby enabled large-scale studies. For example, the Leiomyosarcoma (LMS) Direct Research Foundation (LMSDR) engaged LMS patients online in order to curate 300 archival samples, resulting in the discovery of new prognostic biomarkers and molecular subtypes (Edris et al., 2012; Beck et al., 2010; Leiomyosarcoma Direct Research Foundation, 2016). Similarly, the Multiple Myeloma Research Foundation (MMRF) has leveraged a strong online community to complete the sequencing and clinical annotation of 190 and 420 patient tumors, respectively (Multiple Myeloma Research Foundation, 2015).
In other cases, patients have self-organized online and disseminated knowledge of their own disease (Call et al., 2010; Hauber et al., 2011). Illustrative examples include the role of patients with gastrointestinal stromal tumor (GIST) or chronic myeloid leukemia (CML) in the development and application of therapies targeting the KIT and Bcr-Abl oncoproteins (see Box 1). New social media platforms, such as Facebook and Twitter, now enable such virtual patient aggregation on an even larger scale (Seltzer et al., 2014; Katz et al., 2016). For example, Facebook groups exist for the majority of rare cancer types, and these groups may currently include over 1,000 members (Figure 2). In the future, these social media platforms may accelerate clinical trial enrollment for patients with rare tumors. Today, these proof-of-concept examples demonstrate that tumor profiling and clinical studies that were once prohibitively large for rare cancer types now can be achieved through direct engagement with patients online.
Box 1. Leveraging the Internet to Accelerate Rare Cancer Research: The Case of Imatinib.
The therapeutic development and ultimate FDA approval of imatinib for chronic myelogenous leukemia (CML), a rare cancer with approximately 9,000 new cases per year in the US (Siegel et al., 2017), is an example of the successful use of the Internet to accelerate translational rare cancer research. In parallel, patients with another rare cancer in which imatinib is effective, gastrointestinal stromal tumor (GIST), successfully leveraged online resources to have an impact on the course of their disease.
In 1999, CML patients who were enrolled in a phase I study testing the targeted inhibition of the oncogenic Bcr-Abl fusion protein in CML were experiencing disease remission following treatment with the Bcr-Abl inhibitor STI571 (later named imatinib) (Druker et al., 2001; The New York Times, 2009). Prior to publication of the study findings, patients treated with this drug as part of the clinical trial began disseminating news of their responses in online patient chat rooms. While knowledge of these anecdotal clinical responses motivated other CML patients to seek inclusion in a trial, the pharmaceutical company Novartis had not made sufficient quantities of imatinib to enable a large-scale phase II trial. Consequently, a patient with CML drafted a petition, addressed to the then-CEO of Novartis, requesting that Novartis scale production of the drug. Using the Internet, this patient identified at least 2,000 additional patients to sign the petition. In response, Novartis increased production of imatinib, permitting the enrollment of many additional patients as part of a phase II trial (The New York Times, 2001). Imatinib was approved for CML shortly thereafter (Cohen et al., 2002).
In addition to its use for the treatment of CML, imatinib was approved for the treatment of GIST in 2001 (Dagher et al., 2002). In this cancer type, the anticancer activity of imatinib is mediated through inhibition of another protein target, KIT, which is commonly activated by gene mutation in GIST tumors (Demetri, 2001). Historically, a subset of GIST patients have been misdiagnosed with leiomyosarcoma (LMS), which is typically treated with chemotherapies largely ineffective against GIST (Price et al., 2007). From 1998 to 2000, prior to the approval of imatinib for GIST, hundreds of patients misdiagnosed with LMS joined an active LMS online community, part of the Association of Cancer Online Resources website, and were advised by other group members to request testing for the KIT mutation. These patients were told that their physicians might not have been familiar with this new test. Through these online exchanges, hundreds of misdiagnosed patients requested testing for the KIT mutation, and those who were KIT positive were ultimately diagnosed with GIST. Obtaining a correct diagnosis and thus qualifying for treatment with imatinib is expected to have dramatically improved clinical outcomes for many of these patients (Frydman, 2009).
Furthermore, an online support group, the Life Raft Group (LRG) (www.liferaftgroup.org), emerged in 2000 from these engaged GIST patients. Using online resources, GIST patients receiving imatinib as part of a trial shared symptoms and side effects of treatment (The Life Raft Group, 2000). The patient-driven database maintained by LRG has enabled long-term retrospective analyses on KIT-positive GIST patients treated with imatinib (Call et al., 2012).
Figure 2. Aggregating Patients with Rare Cancers Via Social Media Platforms.
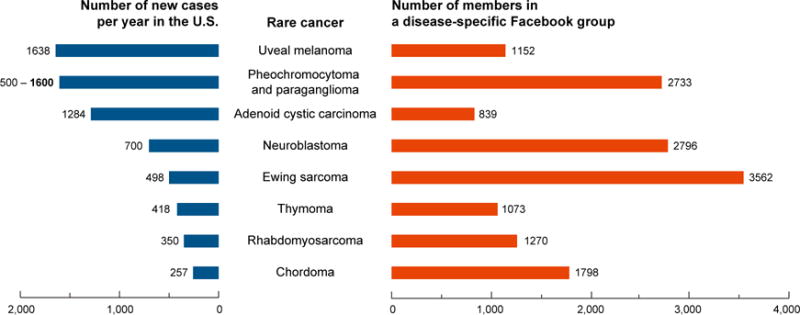
Social media platforms, such as Facebook, are a vehicle to connect patients with rare cancers. Depicted are eight cancer types that are exceedingly rare, typically precluding well-powered studies at any one institution. However, there now exists at least one Facebook group relevant to each cancer type (right, in orange). Membership in these Facebook groups sometimes exceeds the number of new cases of the disease per year in the US (left, in blue). While members may not all be patients living with disease, these groups reflect disease-specific communities that are coalescing via online platforms and may translate to increased access to patients for research studies. Number of new cases per year in the US is based on the current estimated US population (321 million). Facebook data were accessed on July 2, 2017; group numbers are subject to change.
Mapping Rare Cancer Genomes
Identifying rational interventions for each rare cancer type will likely be aided by genomic and cellular characterization of these tumors. Ideally, such information might include a full map of germline and somatic variation, together with a survey of the malignant, stromal, and immune cell types and states present in each rare tumor.
Mapping the germline basis of rare cancers can provide insight into causative variants as well as therapeutic opportunities. The completion of reduced-representation haplotype maps of the human genome (International HapMap 3 Consortium et al., 2010) made large-scale genome-wide association studies (GWAS) possible. In parallel, the recognition that germ-line susceptibility loci in the DNA-repair pathway occur across tumor types and render tumors sensitive to PARP inhibition (Pritchard et al., 2016; Matulonis et al., 2016; Kaufman et al., 2015) provided proof of concept that mapping the germline basis of tumors may lead to new inclusion criteria for basket trials (in which patients are selected based on the molecular, as opposed to histological, features of their tumors). However, while in aggregate over 800 germline cancer-susceptibility SNPs have now been discovered (NHGRI-EBI GWAS Catalog; MacArthur et al., 2017; Burdett et al., 2017), the statistical power required to map predisposition associations initially necessitated a focus on common cancer types or familial syndromes.
However, new integrated strategies are shedding light on rare cancer susceptibility despite smaller cohort sizes. For instance, the initial observation that LMO1 variation led to neuroblastoma susceptibility was bolstered by somatic copy number analysis and RNAi-based validation (Wang et al., 2011). Similarly, the insight that variation at the NFIB transcription factor locus predisposed to metastatic osteosarcoma was strengthened by a transposon-based genetic screen (Mirabello et al., 2015). A complementary approach leverages the divergent incidences of rare tumors across populations. For instance, while nasopharyngeal carcinoma is rare in the US, it is more common in parts of China, enabling appropriately powered association studies in this population (Cui et al., 2016; Chang and Adami, 2006).
New advances promise to further accelerate the pace of discovery of the germline basis of rare tumors. Large-scale efforts in data sharing under the auspices of The Global Alliance for Genomics and Health are aiming to create international frameworks for responsible data sharing (Global Alliance for Genomics and Health, 2016). The recognition that a large number of small, siloed rare disease datasets impede discovery propelled the launch of a Matchmaker Exchange (www.matchmakerexchange.org). This project provides a federated platform to connect rare disease genotypes and phenotypes through a common application programming interface (Philippakis et al., 2015). Remarkably, over 120,000 human exomes and 15,000 genomes have now been sequenced and aggregated (Lek et al., 2016; Genome Aggregation Database, 2017), providing an essential reference dataset for rare cancer predisposition studies to come.
In parallel to these advances in germline genomics, over the last decade there has been considerable progress in the development and implementation of new technologies to characterize somatic alterations in tumor genomes. New second-generation sequencing technologies and applications (reviewed in Meyerson et al., 2010) have permitted investigators to detect somatic genomic and epigenomic variation from individual tumors with increased efficiency and resolution. In 2008, the report of the application of whole-genome sequencing to a single acute myeloid leukemia tumor sample (Ley et al., 2008) and the first report from The Cancer Genome Atlas (TCGA) project, focused on glioblastoma (Cancer Genome Atlas Research Network, 2008), launched the era of somatic cancer genomics in earnest.
The progress over the last decade has propelled our understanding of the somatic genomic basis of both common and rare cancers. Over 140 such sequencing studies have now been reported and deposited into the NCI’s Genomic Data Commons (GDC) and/or other public databases (https://gdc.cancer.gov/; dcc.icgc.org/; www.cbioportal.org), totaling over 23,000 tumor samples subjected to whole-exome, whole-genome, or other next-generation sequencing (Figure 1A). It is estimated that nearly 3,000 of these tumor samples represent exceedingly rare cancer types (Figure 1A).
The pursuit of a genomic understanding of common and rare cancers has been mutually beneficial. In one direction, a suite of technological advances, initially developed and applied to common cancers, has now enabled genomic analysis of rare cancers. Reciprocally, in some cases, rare tumors exhibit striking genomic homogeneity (for instance, near universal SMARCB1 loss in malignant rhabdoid tumors [Lee et al., 2012]; BRAF mutations in papillary craniopharyngiomas [Brastianos et al., 2014] and hairy-cell leukemia [Tiacci et al., 2011]; and IDH1 mutations in low-grade gliomas [Yan et al., 2009]), enabling genomic discoveries with small sample cohort sizes that are pertinent to common and rare cancers alike (Figure 1B). In this regard, rare cancer types have the potential to provide disproportionate insight into tumor biology in spite of their low incidence. Still, for many rare cancer types, a genomic understanding remains incomplete or absent.
Excitingly, building on the success of the Therapeutically Applicable Research to Generate Effective Treatments (TARGET) initiative for pediatric cancers (Mullighan et al., 2009; Pugh et al., 2013; Chun et al., 2016), a series of ten rare adult tumor types is now being analyzed as part of TCGA, leading to new insights into these diseases. For example, despite an extremely low incidence, 173 cases of neuroendocrine-based pheochromocytoma (PCC) and paraganglioma (PGL) tumors have been collected and analyzed, leading to the discovery of novel WNT-activating MAML3 and CSDE1 alterations and the convergence of germline and somatic alterations in the RET, NF1, and VHL genes (Fishbein et al., 2017). Sequencing of chromophobe renal cell carcinoma (ChRCC) revealed novel structural rearrangements at the TERT promoter and novel mtDNA mutations (Davis et al., 2014). As another example, a relationship between whole-genome doubling and aggressive disease was discovered in adrenocortical carcinoma (Zheng et al., 2016), pointing to the importance of genome structure in the etiology of certain rare tumors. In pediatric cancers, the discovery of novel fusion proteins is providing clear mechanistic hypotheses, such as the discovery of the MYB-QKI protein driving pediatric angiocentric gliomas (Bandopadhayay et al., 2016).
In many cases, these rare cancer genome studies point directly to new cancer targets that had previously been overlooked. For instance, the observation that mutations in chromatin-regulating genes occur across cancer types (Garraway and Lander, 2013) has now been extended to a series of rare tumor types. Examples include BAP1 mutations in uveal melanoma and cholangiocarcinoma (Harbour et al., 2010; Jiao et al., 2013); MLL3 mutations in cholangiocarcinoma (Ong et al., 2012); and SMARCA2, KDM6A, and ARID1A mutations in adenoid cystic carcinoma (Stephens et al., 2013; Ho et al., 2013; Ross et al., 2014). Further, potentially targetable mutations in the G-protein alpha subunit are characteristic of uveal melanoma (Van Raamsdonk et al., 2009). In some cases, direct possibilities for therapeutic intervention have emerged, such as the discovery that PCC/PGL tumors can harbor mutations in genes (including BRAF and FGFR1) for which approved therapies already exist (Fishbein et al., 2017). Similarly, mutations in the FGF-IGF-PI3K pathway, including in the FGFR2 gene itself, were found to be harbored by up to 30% of adenoid cystic carcinomas (Stephens et al., 2013; Ho et al., 2013).
Looking ahead, in addition to bulk tumor measurements, recent advances in plate- and droplet-based single-cell sequencing have now made it possible to profile tumors at unprecedented cellular resolution (Tirosh et al., 2016a, 2016b). In these studies, the malignant, stromal, and immune composition of each tumor can be resolved. As single-cell genomic studies increasingly point toward causative mechanisms (Tanay and Regev, 2017), the implementation of such advanced technology may soon be routine in clinical settings and applied to rare tumors.
More generally, these and other cancer genome studies point to eroding distinctions between rare and common tumors and to their complementarity. For example, genomic analysis of common cancer types, such as lung adenocarcinoma, suggests that molecular subcategorization into collections of rare, molecularly distinct “tumor types” (sharing a common etiology or histology) has high clinical relevance (Figure 1A). Reciprocally, now that sample collection bottlenecks are beginning to be overcome for rare cancers, in some cases it may be possible to further leverage their homogeneity to power the analysis of cancer genomes and discover genes and variants inducing tumor fitness, with smaller sample numbers than might otherwise be needed.
Finally, alongside the generation of cancer genome data by individual laboratories and centers, infrastructure to support data sharing, controlled access, and advanced analysis across cancer genome studies may further enable appropriately powered studies of rare tumors. The recent launch of the NCI’s GDC (https://gdc.cancer.gov/) establishes a unified data repository that harmonizes existing cancer genome data and thereby permits direct comparison of datasets generated by different large-scale research programs. Further, the GDC supports submission of data from other community- and investigator-led sequencing projects, including those studying rare tumor types. For example, the MMRF is depositing its genomic and clinical data on multiple myeloma in the GDC (National Cancer Institute, 2016a). In the future, a shift to cloud-based data platforms may increase the flexibility, reliability, and security of genomic analyses, as well as reduce costs, through harnessing the elastic compute capacity of commercial clouds such as Google, Amazon, or Microsoft (Stein et al., 2015). The NCI’s Cancer Genomics Cloud Pilots program is helping to hasten this transition and democratize access to cancer genome data.
Advances in Clinical Tumor Sequencing
In parallel to the cohort-based research studies described above, tumor sequencing is increasingly being used in clinical settings to guide therapeutic decision making for individual patients (Abrams et al., 2014). Historically, pathologists relied primarily on the histology of tumor tissue to subcategorize cancers. Over the last several decades, the clinical implementation of flow cytometry, multiplex ligation-dependent probe amplification, reverse transcription (RT)-PCR, and fluorescence in situ hybridization allowed pathologists to further subcategorize cancers, leading to changes in clinical management. As one illustrative example, the pediatric tumor type formerly treated uniformly as “small-round-blue-cell” tumor is now subclassified according to the presence of specific fusion transcripts (e.g., EWS-FLI1 versus PAX3-FKHR), leading to distinct treatment regimens (Lewis et al., 2007).
In more recent years, the somatic sequencing of individual rare cancer types, driven largely by cost reductions (Figure 3), has revealed additional key diagnostic classifications that are likely to inform prognosis and targeting strategies. For instance, analysis of ChRCC confirmed that this rare tumor is a distinct disease entity from the more common clear cell renal carcinoma, suggesting against the direct implementation of clear cell RCC therapies for such tumors (Davis et al., 2014). As another example, adenoid cystic carcinomas of the breast were found to lack mutations and copy number alterations characteristic of other triple-negative breast cancers (Martelotto et al., 2015). The genomics-based subclassification of pediatric medulloblastoma or leukemia facilitates risk stratification (Jones et al., 2012; Mullighan, 2013). Finally, cholangiocarcinoma genomics revealed distinct mutational patterns between cases induced (versus those not induced) by intrahepatic Opisthorchis viverrini infection, suggesting distinct therapeutic approaches might be needed within the same rare tumor type based on the causative etiology (Chan-On et al., 2013).
Figure 3. Advances in the Development of Patient-Derived Models.
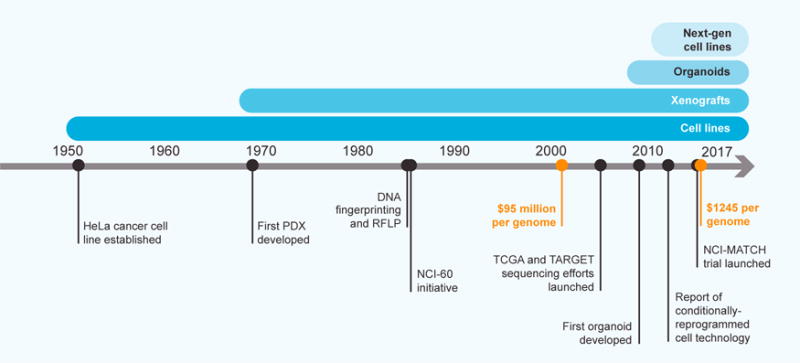
Timeline describing key developments in the generation of patient-derived models. Declines in sequencing costs (orange points); and the emergence of model types, including cell lines, xenografts, organoids, and “next-generation” cell lines, are highlighted (source data: Rygaard and Povlsen, 1969; Jeffreys et al., 1985; The Cancer Genome Atlas, 2005; Wetterstrand, 2016; and references cited in the main text).
In some instances, technologies have made it possible to identify novel oncogenic drivers in rare cancers with potential benefits to current patients. To provide several illustrative examples, as outlined above, improved outcomes with imatinib treatment in gastrointestinal stromal tumor (GIST) were achieved based on the discovery of mutations in the KIT tyrosine kinase gene (see Box 1; Heinrich et al., 2008; Tuveson et al., 2001). In addition, the discovery of NTRK fusions across adult and pediatric cancers (e.g., thyroid cancers and infantile fibrosarcoma) has led to clinical trials with NTRK-targeted agents in adults and children (Stransky et al., 2014; Knezevich et al., 1998; NCT02576431).
The cohort-based genomic discoveries made as part of the TCGA, International Cancer Genome Consortium, and TARGET initiatives have aided the design of more cost-effective sequencing panels, facilitating the testing of larger numbers of rare cancer samples in clinical settings. Individual hospitals (Wagle et al., 2012; Cheng et al., 2015; Roychowdhury et al., 2011), for-profit companies (www.foundationmedicine.com), or consortia (www.rarecancergenome.org) now routinely offer such panel tests to patients and clinicians to guide individual treatment options or in certain cases have performed extensive sequencing (e.g., whole-genome, whole-exome, and transcriptome) on patient tumors.
Proof-of-concept studies determining the feasibility of such studies are now complete. For example, for rare pediatric tumors, initial CLIA sequencing efforts confirmed that many parents and children wish to have such testing performed, results could be returned over a clinically practical time frame, and that “actionable” gene mutations with matched therapies could be identified in a subset of patients (Harris et al., 2016; Parsons et al., 2016; Mody et al., 2015). These initial feasibility studies provided rationale for the NCI-Molecular Analysis for Therapy Choice (NCI-MATCH) trial and the NCI-Children’s Oncology Group (NCI-COG) Pediatric MATCH trial, nationwide trials to test the hypothesis that matching a tumor’s genomic profile with potential targeted agents will improve outcomes in adults and children, respectively (NCT02465060; NCT03155620).
Accordingly, large-scale precision oncology data-sharing projects are emerging, such as the AACR Project Genomics Evidence Neoplasia Information Exchange (GENIE), which is coordinating the aggregation and release of clinical cancer sequencing and patient outcome datasets across multiple institutions. Similarly, in support of the National Cancer Moonshot initiative, Foundation Medicine, Inc. has now made approximately 18,000 clinical sequencing results available, including many from rare tumors (Foundation Medicine, 2016). Together, these technologies, datasets, and sharing initiatives should accelerate the path toward precision oncology for both common and rare cancers (reviewed in Hyman et al., 2017).
Advances in Generating Patient-Derived Models from Rare Cancers
While clinical trials provide one path to confirm relationships between the genetic basis of rare tumors and their associated therapeutic vulnerabilities, a complementary approach relies on the generation and application of laboratory-based models so as to inform future clinical testing. Historically, few or zero models existed for most rare cancer types (Barretina et al., 2012). Now, a diversity of model types, including patient-derived xenografts (PDXs), organoid cultures, and “next-generation” cell-line models (as defined below), coupled with clinical information, is being developed for many rare cancers (Figure 3).
Cancer cell lines have been used for nearly three-quarters of a century to identify biological mechanisms and test novel therapeutic agents. In 1951, the first human cancer cell line, HeLa, was established from a cervical tumor (Scherer et al., 1953), and this proof-of-concept demonstration laid the groundwork for the development of additional cell lines. Over the next four decades, efforts to establish additional cancer cell lines from patients achieved modest success, culminating with the aggregation of a panel of 60 common cancer cell models representing nine tumor types as part of the National Cancer Institute-60 (NCI-60) collection in the late 1980s (Shoemaker, 2006) and approximately 1,000 cell-line models by 2012 (Barretina et al., 2012; Garnett et al., 2012).
In aggregate, these efforts yielded approximately 10–20 cell lines annually. Several key bottlenecks prevented progress for most cancer types, including rare cancers, including suboptimal ex vivo propagation technologies (<1% success rates overall), undetected mycoplasma contamination (Callaway, 2014), and extensive cross-contamination that was later detected (Yu et al., 2015) (reviewed in American Type Culture Collection Standards Development Organization Workgroup ASN-0002, 2010). Furthermore, the lack of clinical annotation and heterogeneity retained by even the successful models remained suboptimal. Despite these limitations, recent work suggests that some findings in cell lines have indeed been recapitulated in patient tumors and vice versa (Sharifnia et al., 2014; Gazdar et al., 2010; Masters, 2000).
Technological advances are now facilitating the generation of high-quality laboratory models representing many rare tumor types. In particular, new technologies now make it possible to generate patient-derived cancer cell lines with substantially higher success rates than historical approaches (Ince et al., 2015; Liu et al., 2012; Oie et al., 1996) (Figure 3). For example, the use of a Rho kinase inhibitor (Y-27632) along with conditioned media from 3T3-J2 murine feeder cells has enabled the generation of models from tumor epithelial cells (Liu et al., 2012). While traditional cell lines have been passaged for many decades, do not have coupled clinical annotation, and tend to lack the ability to recapitulate tumor histology when implanted into animals, many of these “next-generation” cell lines overcome these barriers. As one example of the utility of such cell lines, the aforementioned Rho kinase inhibitor-based approach facilitated the generation of the first model of a recurrent respiratory papillomatosis, together with matched normal tissue. This advance led to the discovery of an effective therapeutic intervention for the same patient (Yuan et al., 2012).
Other applications demonstrate new opportunities to model rare tumor types or subtypes. Soon after the discovery of the DNAJB1-PRKACA fusion transcript driving fibrolamellar hepatocellular carcinoma (Honeyman et al., 2014), a transplantable tumor line of this rare cancer was developed (Oikawa et al., 2015). The recent development of a cell-line model from a rare undifferentiated pediatric sarcoma demonstrated how the derivation of a single rare cancer model can be used in high-throughput screens to drive new therapeutic discoveries (Hong et al., 2016). In other applications, cell culture models were generated from individual lung cancer patients that had progressed after EGFR- or ALK-targeted therapies, or from the circulating tumor cells of advanced breast cancer patients, and were subjected to drug testing (Crystal et al., 2014; Yu et al., 2014). As the genomic and clinical annotation of patient tumors and derived models is now cost-effective and feasible, the quality and utility of each successful cell-line model is improved. Overall, the use of such models to nominate effective therapeutic options for relapsed patients in real time represents an exciting, but challenging future direction (recently reviewed in Friedman et al., 2015). Validating the clinical utility of such ex vivo precision functional genomic diagnostic tests remains a high priority for the field.
Importantly, some aspects of cellular physiology and heterogeneity may be inadequately retained in cell-line cultures. The recent development and application of “organoid” technology (Sato et al., 2009) provides a new opportunity to model cancer ex vivo while retaining some important physiology (Figure 3). Briefly, a cocktail of cytokines and growth factors is used in conjunction with Matrigel to propagate tumor cells in a three-dimensional matrix, retaining aspects of tissue architecture (reviewed in Clevers, 2016). Some cancers and cell types for which establishing standard cell-line models has been challenging, including prostate cancers and nonneoplastic pancreatic cells, can instead be propagated as organoids (Gao et al., 2014; Boj et al., 2015). The use of patient-derived tumor organoid cultures for drug screening has enabled researchers to identify personalized therapeutic hypotheses (van de Wetering et al., 2015).
A complementary approach involves the derivation of PDXs from rare tumors, which may preserve aspects of tumor heterogeneity and microenvironmental influences that are lacking in organoid or cell-line cultures (Izumchenko et al., 2016; Morelli et al., 2012). In this approach, patient-derived tumor specimens are subcutaneously or orthotopically injected into mouse models. PDXs are providing researchers with tools to develop human randomized clinical trials in mice (Gao et al., 2015; Townsend et al., 2016; Bruna et al., 2016), with an ultimate goal to connect genotypes to dependencies in models with intact micro-environments. In recent years, several initiatives have scaled the development of such PDXs for both common and rare tumors, and these include the EurOPDX Consortium (www.europdx.eu), the NCI’s Pediatric Preclinical Testing Program (PPTP), or focused efforts in specific tumor types (Houghton et al., 2007; Pavia-Jimenez et al., 2014; Weroha et al., 2014). After testing over 80 agents in numerous models of childhood cancer, in 2015, the PPTP evolved to the Pediatric Preclinical Testing Consortium (www.ncipptc.org), which now uses genomically characterized pediatric cancer xenograft lines to generate robust preclinical datasets. Furthermore, for-profit companies now exist (www.championsoncology.com) that provide patients with rare cancers the opportunity to develop a PDX model directly from their tumors (Brait et al., 2017). Indeed, over 70 PDX models derived from rare cancers are now in the Champions Oncology biorepository (D. Ciznadija, Champions Oncology, Inc., personal communication).
Further enabling the application of these approaches to rare tumors, an evolution toward “living biorepositories” has occurred over the past decade (Chalfin et al., 2016; Vaught et al., 2009; Somiari and Somiari, 2015). Historically, archival samples were frozen or stored in fixatives such as paraffin, precluding the generation of live patient-derived models (ISBER, 2012). Now, new biorepository protocols include the routine, viable freezing of patient samples, such as in DMSO (Rasmussen et al., 2015). In addition, more robust Institutional Review Board consent protocols are being used to properly inform patients of the opportunity to participate in research projects in which patient-derived models are being generated. While some of these advances are focused on common cancers, once such infrastructure is established, the barriers for rare cancer samples to be similarly processed are expected to lower.
This confluence of new technologies promises to usher in a new era for the development of high-quality models from multiple rare tumor types. Looking ahead, it will be of great interest whether new online patient engagement strategies can facilitate the acquisition of fresh tumor samples for rare cancer types to further accelerate model generation.
New Functional Screening Tools to Map Rare Cancer Dependencies
Functional experiments using patient-derived models represent an important approach to help interpret the cancer genome with respect to dependencies. In some cases, individual tumors harbor large numbers of somatic mutations (Lawrence et al., 2013), rendering it difficult to determine which mutations enhance fitness during tumor initiation or maintenance. In addition, some mutations are presently challenging to modulate with small molecules, such as those in transcription factor genes or those that have loss-of-function effects. As such, the sequencing of rare cancers may be insufficient to nominate tractable therapeutic targets.
Mapping relationships between the features of rare tumors and their associated dependencies (defined here as genes or proteins required for the viability and/or proliferation of cancer cells) facilitates the identification of candidate therapeutic targets. New technological advances in genetic and small-molecule screening approaches, applied to patient-derived cellular models from rare and common cancers, now enable the generation of such “dependency maps” in a systematic fashion.
Cancer dependency discovery relies on loss-of-function methodologies, which aim to elucidate gene function by means of reducing or eliminating gene expression or protein activity. The development and application of small interfering RNA (siRNA)- and short hairpin RNA (shRNA)-based reagent libraries have permitted transient or stable RNAi in cancer cells at genome scale (Aza-Blanc et al., 2003; Moffat et al., 2006; Root et al., 2006). The recent emergence of clustered regularly inter-spaced short palindromic repeats (CRISPR)/Cas9-based technologies, which mediate gene inactivation (reviewed in Howard et al., 2016), now provide an exciting complementary approach (Wang et al., 2015). The joint application of both RNAi-based and CRISPR/Cas9-based technologies to discover rare cancer dependencies that are technology agnostic is now possible (Hong et al., 2016).
New libraries of small molecules are also facilitating the mapping of cancer dependencies. Pioneering efforts to produce small-molecule probes with novel mechanisms of action, such as those mobilized by the launch of the Molecular Libraries Program by the NIH in 2005, have expanded the repertoire of biologically active small molecules available for screening (Austin et al., 2004). In addition, new libraries have been assembled that include thousands of existing, approved drugs, with the potential to be repurposed for a new therapeutic indication (Huang et al., 2011; Corsello et al., 2017). Focused screening libraries may also include small-molecule probes that collectively cover a wide range of targets and cellular processes (Basu et al., 2013; Seashore-Ludlow et al., 2015; Yang et al., 2013).
Notably, small-molecule sensitivity profiling can provide therapeutic and biological insights not achievable with genetic perturbation studies alone. For example, because most small-molecule drugs modulate protein activity, the effects of which may not perfectly mirror those resulting from perturbation of nucleic acids, identifying a cancer dependency via a small-molecule probe can be an important proof of concept for ultimate therapeutic development (Schreiber, 2011). In addition, small molecules enable exploration of protein-protein interactions, alterations in protein stability, and inhibition of protein dimerization (Schreiber et al., 2015).
The mapping of cancer dependencies is now proceeding swiftly with these new small-molecule and genetic perturbation tools in hand. Earlier efforts such as the NCI-60 anticancer drug screen (Shoemaker, 2006) and focused profiling of breast cancer cell lines (Daemen et al., 2013) provided essential proof of concept. In recent years, small-molecule sensitivity datasets have been generated using much larger cancer-cell-line panels, which have the potential to better represent the diversity of patient tumors and permit the identification of context-specific dependencies. These newer datasets include pharmacological profiles of ~500 cancer cell lines tested with 24 anticancer drugs, generated as part of the Cancer Cell Line Encyclopedia project (Barretina et al., 2012); and an independent study in which 130 preclinical or clinical agents were tested across a range of 275–507 cancer cell lines (Garnett et al., 2012). Some concerns were initially raised about the reproducibility of such large-scale studies (Haibe-Kains et al., 2013). However, a recent confirmatory study (Haverty et al., 2016) as well as new integrated analysis of the two initial reports (Cancer Cell Line Encyclopedia Consortium and Genomics of Drug Sensitivity in Cancer Consortium, 2015) now point to reasonable agreement and biological consilience between the studies.
New efforts are expanding this approach even further. As one example, an effort as part of the NCI-sponsored Cancer Target Discovery and Development (CTD2) initiative has led to small-molecule sensitivity profiling of over 800 genomically characterized cancer cell lines, using a panel of 481 US Food and Drug Administration (FDA)-approved drugs, clinical candidates, and small-molecule probes (Seashore-Ludlow et al., 2015). A more recent study correlated molecular features of a larger panel of ~1,000 cancer cell lines to sensitivity to 265 anticancer drugs (Genomics of Drug Sensitivity in Cancer [GDSC] database; Iorio et al., 2016). Finally, a new cellular barcoding method called PRISM enabled the high-throughput viability assessment of 8,400 small molecules against 102 cell lines (Yu et al., 2016). Now, each rare cancer small-molecule dependency assessment can be compared against these large reference datasets (Figure 4) to prioritize dependencies that share a common cellular feature or are highly specific to the rare cancer under study, thereby deprioritizing compounds whose effects are pan-lethal. For example, these types of comparative analyses have recently been used to identify small-molecule dependencies in models of medulloblastoma (Hanaford et al., 2016) and pediatric undifferentiated sarcoma (Hong et al., 2016).
Figure 4. Large-Scale Cancer Dependency Datasets Enable the Identification of Rare Cancer Type-Relevant Vulnerabilities.
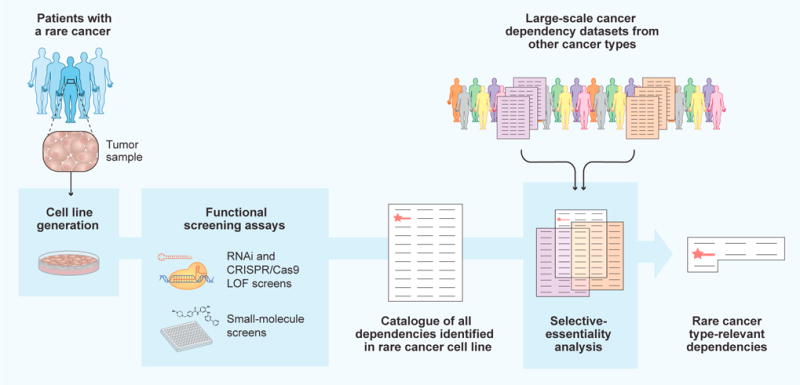
Workflow of cancer dependency discovery in rare cancers. Patient-derived cell lines are subjected to loss-of-function (LOF) screening approaches to identify a catalog of all dependencies in a given rare tumor model. To distinguish key rare cancer type-relevant dependencies (red star) from commonly essential genes or pan-lethal small molecules, large-scale cancer dependency datasets generated using other cancer types can then be used to compare and filter screening results. A list of rare cancer type-relevant dependencies, including key targets, is thus obtained.
In parallel, the application of genome-scale RNAi and CRISPR/Cas9 libraries to large genomically characterized panels of cancer cell lines provides a critical reference dataset for identifying rare tumor dependencies. Earlier work as part of Project Achilles demonstrated basic feasibility using RNAi and mapped dependencies at genome scale to 102 or 216 cell lines (Cheung et al., 2011; Cowley et al., 2014). In parallel, other efforts focused on individual tumor types, such as breast, pancreatic, and ovarian cancers (Marcotte et al., 2012, 2016). Recent proof-of-concept studies demonstrating the potential for large-scale application of CRISPR/Cas9 technology (Wang et al., 2015) have now been extended to 5 (Munoz et al., 2016), 14 (Wang et al., 2017), or 33 (Aguirre et al., 2016) cancer cell lines, leading to the discovery of PREX1 as an activator of MAPK signaling in Ras-driven acute myeloid leukemia cells, among other discoveries.
Several key principles have emerged from these studies that are relevant to rare tumors. Firstly, many clinically validated dependencies that arise from activated oncogenes (Weinstein, 2002) are readily recapitulated in these studies (Cheung et al., 2011). Thus, if a novel rare cancer model is found to be exquisitely dependent on a mutated oncogene, there may be strong rationale for therapeutic targeting of such oncogene. Second, expression-driven dependencies are prominent and include lineage-specifying transcription factors such as PAX8 and other master regulator genes (Marcotte et al., 2016; Cheung et al., 2011), suggesting potential to target the underlying tissue lineage for rare tumors in which such lineage is expendable. Also, partial copy number loss of certain genes renders cancer cells highly sensitive to further depletion (Nijhawan et al., 2012), suggesting therapeutic opportunities for rare tumors with hemizygous deletions.
Lastly, these studies point to opportunities to target previously challenging loss-of-function events in cancer, by exploiting the concept of synthetic lethality (Kaelin, 2005). One example relates to CDKN2A deletion, which is a common alteration in multiple cancer lineages. The co-deletion of the enzyme methylthioadenosine phosphorylase (MTAP), which frequently co-occurs with CDKN2A loss due to their genomic proximity, confers selective dependence on the protein arginine methyltransferase 5 (PRMT5) gene (Kryukov et al., 2016; Mavrakis et al., 2016). Other examples include paralog dependencies (also called “collateral vulnerabilities”) in which the loss of one paralogous protein is associated with a dependency on another. Examples include the ARID1B dependency in ARID1A-mutant cancers (Helming et al., 2014), the SMARCA2 vulnerability in SMARCA4-mutant cancers (Wilson et al., 2014), and the ENO2 vulnerability in glioblastomas with ENO1 deletion (Muller et al., 2012).
Now, demonstration efforts have shown basic feasibility of using these technologies to assess cancer dependencies in novel patient-derived cell lines (Hong et al., 2016; Crystal et al., 2014; Yu et al., 2014), organoids (van de Wetering et al., 2015), and xenografts (Bruna et al., 2016; Townsend et al., 2016; Gao et al., 2015; Bossi et al., 2016). Leveraging multiple technologies in unison, it should soon become increasingly feasible to assess dependencies of a given rare cancer type (even with a limited number of models), compare results to large reference datasets, and home in on therapeutically actionable dependencies that are induced by specific, shared cellular features (Figure 4).
Accelerating Research on Chordoma
One example of the convergence of the advances described in the above sections can be found in chordoma research. For this rare cancer type, an important event was the founding, in 2007, of the Chordoma Foundation (CF), which over the last decade has mobilized the chordoma research field and played key roles in several areas, described in detail below and summarized in Table 1.
Table 1.
Advances in Chordoma Research Assets
| 2007 | 2017 | Interventions | |
|---|---|---|---|
| Researcher community | Small, disconnected | >300 active researchers; robust collaboration | International Chordoma Research Workshops; networking events |
| Patient community | None | >1,800 patients | Facebook group; website; online advertising and search engine optimization; educational materials; Chordoma Community Conferences |
| Tumor tissue | Scarce, no centralized source | Centralized repository with tissue from >200 cases | Tumor donation program allows patients to donate tumor from any US hospital; CF Biobank stores and distributes tumor samples |
| Genome characterization | None | 35 cases sequenced | Chordoma Genome Project, in partnership with academic labs |
| Cell lines | 1 | 17 | Cell line prize; cell line validation pipeline; genomic characterization; publicly accessible cell-line repository |
| Patient-derived xenografts (PDX) | None | 5 | Tumor donation program coupled with rapid implantation in vivo; PDX prize; PDX validation pipeline; publicly accessible PDX repository |
| Small-molecule screening | None | >10,000 compounds screened, including all FDA-approved drugs | Collaboration with NIH Chemical Genomics Center; grants to academic investigators; collaborations with industry |
| Functional genomic screening | None | Genome-wide CRISPR/Cas9 screens; targeted shRNA screen | Grants to academic investigators |
| In vivo evaluation | None | >20 drugs evaluated | CF drug screening pipeline provides in vivo evaluation as a service to academic and industry collaborators |
| Clinical trials | 1 completed clinical trial | Pipeline of 7 new clinical trials | Clinical trials program supports trials with: funding, trial design, patient education, and outreach |
Source material: Chordoma Foundation (J. Sommer, personal communication) and www.chordomafoundation.org.
First, a key advance was the physical and virtual unification of the scientific, medical, and patient communities, which had previously been siloed and geographically dispersed. To accomplish this, the CF established International Chordoma Research Workshops and Chordoma Community Conferences, allowing researchers, patients, and caregivers to physically gather and collaborate. Social media was used extensively to enable the virtual interaction and engagement of patients. Second, a centralized tumor biorepository, the “CF Biobank,” was established in 2012 to collect and provide access to tumor tissue, blood, and clinical data; this permitted any chordoma patient at any hospital in the US to contribute material. In addition to being banked for future use, when possible, samples are engrafted in mice to create PDX models. Third, the CF implemented a system of monetary prizes that have incentivized the development of new chordoma cell-line and PDX models, which are subsequently validated and deposited in publicly accessible repositories. Together, these efforts effectively lower the barriers to research for new and existing investigators.
Building on this new convergence of researchers and scientific resources, the Foundation initiated several projects to identify possible therapeutic targets in chordoma. In 2007, little was known of the molecular basis of chordoma. To address this gap, the Chordoma Genome Project was launched to perform whole-genome, whole-exome, and transcriptome sequencing of chordoma tumors, leading to the discovery of a common variant in the T (brachyury) gene (Pillay et al., 2012), as well as several other known cancer genes. With these genomic data now available, large-scale functional genomic and small-molecule screening efforts are underway in industry and academia to uncover dependencies linked to these underlying features (J. Sommer, Chordoma Foundation, personal communication).
Finally, to facilitate translation of novel discoveries, the CF launched additional programs. These included a centralized pipeline for conducting in vivo efficacy studies in a contract lab that continually maintains tumor-bearing animals, permitting chordoma investigators to rapidly test candidate therapies with no upfront investment in personnel or laboratory setup. In addition, a formal process for providing grant support for clinical trials was established in 2015, and a patient navigation service was created to connect patients to available trials, among other programs intended to speed clinical impact.
As a result of these efforts, foundational datasets and resources have been established, and several clinical trials have now been launched, in contrast to a single phase II trial that had been completed by 2007. Overall, the current pace of progress in chordoma research demonstrates how new structures to facilitate rare cancer target discovery and translation can have major impact.
Toward a Platform for Rare Cancer Target Discovery
As described above, important biological and translational discoveries have been made for specific rare cancer types, often by individual laboratories or institutions with disease-specific expertise (Figure 5). In recent years, this work has been advanced considerably by research consortia and disease-specific research foundations, which have coalesced communities comprising patients, scientists, and clinicians for specific rare tumor types, such as chordoma.
Figure 5. Toward a Platform for Target Discovery in Rare Cancers.
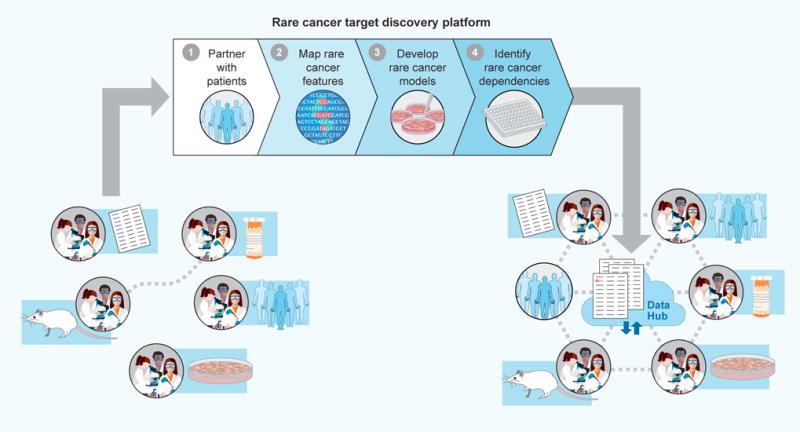
Historically, research on rare cancers has been carried out by small numbers of independent laboratories. The emergence of new technologies permits the potential implementation of new research frameworks to (1) leverage online platforms and social media to partner with patients, (2) perform genomic and cellular characterization of samples that such patients contribute, (3) create cell line, organoid, and/or PDX models from such samples, and (4) use small-molecule and CRISPR/Cas9 libraries to create “dependency maps” for each rare cancer type. Resulting target discovery findings, stored in cloud-based “data hubs,” can help further coalesce rare cancer research communities, with each laboratory contributing specific areas of expertise to the larger community.
However, despite these successes, each rare cancer community is often charged with independently establishing components that are needed (to perform genomic analysis, develop organoid models, or assemble small-molecule libraries, for example), often with extremely limited funds. As a result, there may be missed opportunities to benefit from existing expertise or to capture cost reductions that might come from scale.
Looking ahead, given the remarkable technological and social advances that have recently emerged, it is worth considering whether additional research structures might now accelerate progress. One possible framework includes technology-focused “rare cancer target discovery” platforms, acting as close partners with biology-focused rare cancer research laboratories. These platforms could support the four essential components of rare cancer target discovery described here: (1) leveraging online platforms and social media to establish meaningful partnerships with patients (providing remote opportunities to provide consent for specific research projects), (2) performing genomic and cellular characterization of samples that such patients contribute, (3) creating cell-line, organoid, and/or PDX models from such samples, and (4) using small-molecule and CRISPR/Cas9 libraries to create “dependency maps” for each rare cancer type (Figure 5). Resulting target discovery findings can help further coalesce rare cancer research communities (Figure 5).
The potential for such platforms to be transformative will rely on commitments at each step to share reagents, cell models, and data broadly. In the case of data, cloud-based rare cancer “data hubs” (Figure 5) could permit each lab to have access to aggregated rare cancer data in an environment that maintains patient privacy while flexibly and cost-effectively supporting data sharing and analysis. By sharing data and resources, each stakeholder in the rare cancer research enterprise stands to benefit: target discovery could be enabled in a more cost-effective fashion, individual laboratories and clinicians could translate key findings toward the clinic, and patients could become further empowered as important partners in the research process.
Future Directions
Here, we have presented a status update on overarching progress to target rare cancers, describing new advances in social media, genomics, ex vivo patient-derived model propagation, and cancer dependency screening with CRISPR/Cas9 reagents and small molecules. Important additional topics, not reviewed herein, include challenges in funding rare cancer research and opportunities to accelerate the drug development process, including drug development incentives and redesign of clinical trial protocols (Panageas, 2015; Casali et al., 2015).
In the case of clinical trials for rare cancers, for example, innovative approaches are emerging that may speed progress. Historically, relatively low rates of patient accrual have impeded clinical testing of therapies for rare cancers. In addition, conventional trial design based on histological subtype challenges the testing of emerging (or approved) therapies in patients whose tumors do harbor the relevant mutation, but in a distinct histology. One potential solution involves the phasing of trials to first demonstrate proof of concept using small trials that focus on homogeneous rare tumor types; indications can later be expanded to subsets of patients with common cancers (Fishman, 2013). Another approach includes the broad implementation of basket trials based on molecular criteria, such as the NCI-MATCH or NCI-COG Pediatric MATCH, as described above (NCT02465060; NCT03155620). Finally, the recent FDA approval of the immune checkpoint inhibitor pembrolizumab for solid tumors with microsatellite instability-high or mismatch repair deficient status (US Food and Drug Administration, 2017) is a key harbinger of genetic-based, tissue-/site-agnostic approvals. Removing limitations based on tissue or site in this way can mitigate the difficulties associated with using (rare) histological criteria to accrue, or treat, cancer patients.
As reviewed herein, each rare cancer type is distinct, but different rare cancer types can pose similar impediments to research progress. New preclinical research frameworks, such as those discussed here, may accelerate rare cancer research and draw research communities together to benefit patients. Looking forward, we anticipate that target discovery approaches successfully employed in rare cancer test cases may be broadly applicable to common cancer types, many of which are increasingly viewed as heterogeneous collections of “rare” diseases that require tailored treatment approaches (Figure 1A). In this way, the rapidly evolving field of rare cancer research may provide a learning system for the treatment of more common cancer types, whose increasing fragmentation according to molecular criteria may soon classify them, too, as “rare.”
Acknowledgments
The authors would like to thank Lauren Solomon, Susanna M. Hamilton, Mathias Wawer, Josh Campbell, and Zarko Boskovic for assistance with figures; Josh Sommer of the Chordoma Foundation for providing chordoma-pertinent source material and reviewing the manuscript; Gilles Frydman, David Sandak, Shubhroz Gill, Zuzana Tothova, Steven Corsello, and Paul Clemons for helpful discussions; and Joanne Kotz and Justine Levin-Allerhand for providing critical feedback on the manuscript. This work was generously supported by The Katie Moore Fund for Rare Cancer Research at the Broad Institute of Harvard and MIT, by the Chordoma Foundation, by CureSearch for Children’s Cancer, by Alex’s Lemonade Stand Foundation for Childhood Cancer, by National Cancer Institute grant P50CA101942, and by an anonymous donor.
References
- Abrams J, Conley B, Mooney M, Zwiebel J, Chen A, Welch JJ, Takebe N, Malik S, McShane L, Korn E, et al. National Cancer Institute’s Precision Medicine Initiatives for the new National Clinical Trials Network. Am Soc Clin Oncol Educ Book. 2014:71–76. doi: 10.14694/EdBook_AM.2014.34.71. [DOI] [PubMed] [Google Scholar]
- Afani L, Errihani H, Benchafai I, Lalami Y. Nasopharyngeal adenoid cystic carcinoma, a rare but highly challenging disease with unmet therapeutic needs: a case-report and review of the literature. Cancer Radiother. 2016;20:400–404. doi: 10.1016/j.canrad.2015.12.007. [DOI] [PubMed] [Google Scholar]
- Aguirre AJ, Meyers RM, Weir BA, Vazquez F, Zhang CZ, Ben-David U, Cook A, Ha G, Harrington WF, Doshi MB, et al. Genomic copy number dictates a gene-independent cell response to CRISPR/Cas9 targeting. Cancer Discov. 2016;6:914–929. doi: 10.1158/2159-8290.CD-16-0154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- American Type Culture Collection Standards Development Organization Workgroup ASN-0002. Cell line misidentification: the beginning of the end. Nat Rev Cancer. 2010;10:441–448. doi: 10.1038/nrc2852. [DOI] [PubMed] [Google Scholar]
- Arıkan M, Togral G, Hastürk AE, Aktaş E, Güngör S. Management and retrospective analysis of primary and metastatic sacral tumors and infections: evaluation with 73 cases. Eklem Hastalik Cerrahisi. 2014;25:126–132. doi: 10.5606/ehc.2014.28. [DOI] [PubMed] [Google Scholar]
- Austin CP, Brady LS, Insel TR, Collins FS. NIH molecular libraries initiative. Science. 2004;306:1138–1139. doi: 10.1126/science.1105511. [DOI] [PubMed] [Google Scholar]
- Aza-Blanc P, Cooper CL, Wagner K, Batalov S, Deveraux QL, Cooke MP. Identification of modulators of TRAIL-induced apoptosis via RNAi-based phenotypic screening. Mol Cell. 2003;12:627–637. doi: 10.1016/s1097-2765(03)00348-4. [DOI] [PubMed] [Google Scholar]
- Bandopadhayay P, Ramkissoon LA, Jain P, Bergthold G, Wala J, Zeid R, Schumacher SE, Urbanski L, O’Rourke R, Gibson WJ, et al. MYB-QKI rearrangements in angiocentric glioma drive tumorigenicity through a tripartite mechanism. Nat Genet. 2016;48:273–282. doi: 10.1038/ng.3500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, Wilson CJ, Lehár J, Kryukov GV, Sonkin D, et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012;483:603–607. doi: 10.1038/nature11003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basu A, Bodycombe NE, Cheah JH, Price EV, Liu K, Schaefer GI, Ebright RY, Stewart ML, Ito D, Wang S, et al. An interactive resource to identify cancer genetic and lineage dependencies targeted by small molecules. Cell. 2013;154:1151–1161. doi: 10.1016/j.cell.2013.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck AH, Lee CH, Witten DM, Gleason BC, Edris B, Espinosa I, Zhu S, Li R, Montgomery KD, Marinelli RJ, et al. Discovery of molecular subtypes in leiomyosarcoma through integrative molecular profiling. Oncogene. 2010;29:845–854. doi: 10.1038/onc.2009.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blay JY, Coindre JM, Ducimetière F, Ray-Coquard I. The value of research collaborations and consortia in rare cancers. Lancet Oncol. 2016;17:e62–e69. doi: 10.1016/S1470-2045(15)00388-5. [DOI] [PubMed] [Google Scholar]
- Boehm JS, Golub TR. An ecosystem of cancer cell line factories to support a cancer dependency map. Nat Rev Genet. 2015;16:373–374. doi: 10.1038/nrg3967. [DOI] [PubMed] [Google Scholar]
- Boj SF, Hwang CI, Baker LA, Chio II, Engle DD, Corbo V, Jager M, Ponz-Sarvise M, Tiriac H, Spector MS, et al. Organoid models of human and mouse ductal pancreatic cancer. Cell. 2015;160:324–338. doi: 10.1016/j.cell.2014.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bossi D, Cicalese A, Dellino GI, Luzi L, Riva L, D’Alesio C, Diaferia GR, Carugo A, Cavallaro E, Piccioni R, et al. In vivo genetic screens of patient-derived tumors revealed unexpected frailty of the transformed phenotype. Cancer Discov. 2016;6:650–663. doi: 10.1158/2159-8290.CD-15-1200. [DOI] [PubMed] [Google Scholar]
- Brait M, Izumchenko E, Kagohara LT, Long S, Wysocki PT, Faherty B, Fertig EJ, Khor TO, Bruckheimer E, Baia G, et al. Comparative mutational landscape analysis of patient-derived tumour xenografts. Br J Cancer. 2017;116:515–523. doi: 10.1038/bjc.2016.450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brastianos PK, Taylor-Weiner A, Manley PE, Jones RT, Dias-Santagata D, Thorner AR, Lawrence MS, Rodriguez FJ, Bernardo LA, Schubert L, et al. Exome sequencing identifies BRAF mutations in papillary craniopharyngiomas. Nat Genet. 2014;46:161–165. doi: 10.1038/ng.2868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruna A, Rueda OM, Greenwood W, Batra AS, Callari M, Batra RN, Pogrebniak K, Sandoval J, Cassidy JW, Tufegdzic-Vidakovic A, et al. A biobank of breast cancer explants with preserved intra-tumor heterogeneity to screen anticancer compounds. Cell. 2016;167:260–274.e22. doi: 10.1016/j.cell.2016.08.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burdett T, Hall PN, Hastings E, Hindorff LA, Junkins HA, Klemm AK, MacArthur J, Manolio TA, Morales J, Parkinson H, et al. The NHGRI-EBI Catalog of published genome-wide association studies. 2017 doi: 10.1093/nar/gkw1133. www.ebi.ac.uk/gwas. [DOI] [PMC free article] [PubMed]
- Call J, Scherzer NJ, Josephy PD, Walentas C. Evaluation of self-reported progression and correlation of imatinib dose to survival in patients with metastatic gastrointestinal stromal tumors: an open cohort study. J Gastrointest Cancer. 2010;41:60–70. doi: 10.1007/s12029-009-9111-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Call J, Walentas CD, Eickhoff JC, Scherzer N. Survival of gastrointestinal stromal tumor patients in the imatinib era: life raft group observational registry. BMC Cancer. 2012;12:90. doi: 10.1186/1471-2407-12-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callaway E. Contamination hits cell work. Nature. 2014;511:518. doi: 10.1038/511518a. [DOI] [PubMed] [Google Scholar]
- Campbell JD, Alexandrov A, Kim J, Wala J, Berger AH, Pedamallu CS, Shukla SA, Guo G, Brooks AN, Murray BA, et al. Distinct patterns of somatic genome alterations in lung adenocarcinomas and squamous cell carcinomas. Nat Genet. 2016;48:607–616. doi: 10.1038/ng.3564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Cell Line Encyclopedia Consortium; Genomics of Drug Sensitivity in Cancer Consortium. Pharmacogenomic agreement between two cancer cell line data sets. Nature. 2015;528:84–87. doi: 10.1038/nature15736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455:1061–1068. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research Network. Comprehensive molecular profiling of lung adenocarcinoma. Nature. 2014;511:543–550. doi: 10.1038/nature13385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casali PG, Bruzzi P, Bogaerts J, Blay JY, Rare Cancers Europe (RCE) Consensus Panel Rare Cancers Europe (RCE) methodological recommendations for clinical studies in rare cancers: a European consensus position paper. Ann Oncol. 2015;26:300–306. doi: 10.1093/annonc/mdu459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalfin HJ, Fabian E, Mangold L, Yeater DB, Pienta KJ, Partin AW. Role of biobanking in urology: a review. BJU Int. 2016;118:864–868. doi: 10.1111/bju.13606. [DOI] [PubMed] [Google Scholar]
- Chan-On W, Nairismägi ML, Ong CK, Lim WK, Dima S, Pairojkul C, Lim KH, McPherson JR, Cutcutache I, Heng HL, et al. Exome sequencing identifies distinct mutational patterns in liver fluke-related and non-infection-related bile duct cancers. Nat Genet. 2013;45:1474–1478. doi: 10.1038/ng.2806. [DOI] [PubMed] [Google Scholar]
- Chang ET, Adami HO. The enigmatic epidemiology of nasopharyngeal carcinoma. Cancer Epidemiol Biomarkers Prev. 2006;15:1765–1777. doi: 10.1158/1055-9965.EPI-06-0353. [DOI] [PubMed] [Google Scholar]
- Cheng DT, Mitchell TN, Zehir A, Shah RH, Benayed R, Syed A, Chandramohan R, Liu ZY, Won HH, Scott SN, et al. Memorial Sloan Kettering-integrated mutation profiling of actionable cancer targets (MSK-IMPACT): a hybridization capture-based next-generation sequencing clinical assay for solid tumor molecular oncology. J Mol Diagn. 2015;17:251–264. doi: 10.1016/j.jmoldx.2014.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung HW, Cowley GS, Weir BA, Boehm JS, Rusin S, Scott JA, East A, Ali LD, Lizotte PH, Wong TC, et al. Systematic investigation of genetic vulnerabilities across cancer cell lines reveals lineage-specific dependencies in ovarian cancer. Proc Natl Acad Sci USA. 2011;108:12372–12377. doi: 10.1073/pnas.1109363108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chun HJ, Lim EL, Heravi-Moussavi A, Saberi S, Mungall KL, Bilenky M, Carles A, Tse K, Shlafman I, Zhu K, et al. Genome-wide profiles of extra-cranial malignant rhabdoid tumors reveal heterogeneity and dysregulated developmental pathways. Cancer Cell. 2016;29:394–406. doi: 10.1016/j.ccell.2016.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clevers H. Modeling development and disease with organoids. Cell. 2016;165:1586–1597. doi: 10.1016/j.cell.2016.05.082. [DOI] [PubMed] [Google Scholar]
- Cohen MH, Williams G, Johnson JR, Duan J, Gobburu J, Rahman A, Benson K, Leighton J, Kim SK, Wood R, et al. Approval summary for imatinib mesylate capsules in the treatment of chronic myelogenous leukemia. Clin Cancer Res. 2002;8:935–942. [PubMed] [Google Scholar]
- Corsello SM, Bittker JA, Liu Z, Gould J, McCarren P, Hirschman JE, Johnston SE, Vrcic A, Wong B, Khan M, et al. The Drug Repurposing Hub: a next-generation drug library and information resource. Nat Med. 2017;23:405–408. doi: 10.1038/nm.4306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowley GS, Weir BA, Vazquez F, Tamayo P, Scott JA, Rusin S, East-Seletsky A, Ali LD, Gerath WF, Pantel SE, et al. Parallel genome-scale loss of function screens in 216 cancer cell lines for the identification of context-specific genetic dependencies. Sci Data. 2014;1:140035. doi: 10.1038/sdata.2014.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crystal AS, Shaw AT, Sequist LV, Friboulet L, Niederst MJ, Lockerman EL, Frias RL, Gainor JF, Amzallag A, Greninger P, et al. Patient-derived models of acquired resistance can identify effective drug combinations for cancer. Science. 2014;346:1480–1486. doi: 10.1126/science.1254721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui Q, Feng QS, Mo HY, Sun J, Xia YF, Zhang H, Foo JN, Guo YM, Chen LZ, Li M, et al. An extended genome-wide association study identifies novel susceptibility loci for nasopharyngeal carcinoma. Hum Mol Genet. 2016;25:3626–3634. doi: 10.1093/hmg/ddw200. [DOI] [PubMed] [Google Scholar]
- Daemen A, Griffith OL, Heiser LM, Wang NJ, Enache OM, Sanborn Z, Pepin F, Durinck S, Korkola JE, Griffith M, et al. Modeling precision treatment of breast cancer. Genome Biol. 2013;14:R110. doi: 10.1186/gb-2013-14-10-r110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dagher R, Cohen M, Williams G, Rothmann M, Gobburu J, Robbie G, Rahman A, Chen G, Staten A, Griebel D, et al. Approval summary: imatinib mesylate in the treatment of metastatic and/or unresectable malignant gastrointestinal stromal tumors. Clin Cancer Res. 2002;8:3034–3038. [PubMed] [Google Scholar]
- Davis CF, Ricketts CJ, Wang M, Yang L, Cherniack AD, Shen H, Buhay C, Kang H, Kim SC, Fahey CC, et al. The somatic genomic landscape of chromophobe renal cell carcinoma. Cancer Cell. 2014;26:319–330. doi: 10.1016/j.ccr.2014.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demetri GD. Targeting c-kit mutations in solid tumors: scientific rationale and novel therapeutic options. Semin Oncol. 2001;28:19–26. [PubMed] [Google Scholar]
- Druker BJ, Talpaz M, Resta DJ, Peng B, Buchdunger E, Ford JM, Lydon NB, Kantarjian H, Capdeville R, Ohno-Jones S, et al. Efficacy and safety of a specific inhibitor of the BCR-ABL tyrosine kinase in chronic myeloid leukemia. N Engl J Med. 2001;344:1031–1037. doi: 10.1056/NEJM200104053441401. [DOI] [PubMed] [Google Scholar]
- Edris B, Espinosa I, Mühlenberg T, Mikels A, Lee CH, Steigen SE, Zhu S, Montgomery KD, Lazar AJ, Lev D, et al. ROR2 is a novel prognostic biomarker and a potential therapeutic target in leiomyosarcoma and gastrointestinal stromal tumour. J Pathol. 2012;227:223–233. doi: 10.1002/path.3986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fishbein L, Leshchiner I, Walter V, Danilova L, Robertson AG, Johnson AR, Lichtenberg TM, Murray BA, Ghayee HK, Else T, et al. Comprehensive molecular characterization of pheochromocytoma and paraganglioma. Cancer Cell. 2017;31:181–193. doi: 10.1016/j.ccell.2017.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fishman MC. Power of rare diseases: found in translation. Sci Transl Med. 2013;5:201ps11. doi: 10.1126/scitranslmed.3006800. [DOI] [PubMed] [Google Scholar]
- Foundation Medicine. Foundation Medicine announces release of molecular information from FoundationCORE to the National Cancer Institute in support of the national Cancer Moonshot and Precision Medicine Initiatives. 2016 http://investors.foundationmedicine.com/releasedetail.cfm?releaseid=977617.
- Friedman AA, Letai A, Fisher DE, Flaherty KT. Precision medicine for cancer with next-generation functional diagnostics. Nat Rev Cancer. 2015;15:747–756. doi: 10.1038/nrc4015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frydman G. Patient-driven research: rich opportunities and real risks. J Participatory Med. 2009;1:e12. [Google Scholar]
- Gao D, Vela I, Sboner A, Iaquinta PJ, Karthaus WR, Gopalan A, Dowling C, Wanjala JN, Undvall EA, Arora VK, et al. Organoid cultures derived from patients with advanced prostate cancer. Cell. 2014;159:176–187. doi: 10.1016/j.cell.2014.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao H, Korn JM, Ferretti S, Monahan JE, Wang Y, Singh M, Zhang C, Schnell C, Yang G, Zhang Y, et al. High-throughput screening using patient-derived tumor xenografts to predict clinical trial drug response. Nat Med. 2015;21:1318–1325. doi: 10.1038/nm.3954. [DOI] [PubMed] [Google Scholar]
- Garnett MJ, Edelman EJ, Heidorn SJ, Greenman CD, Dastur A, Lau KW, Greninger P, Thompson IR, Luo X, Soares J, et al. Systematic identification of genomic markers of drug sensitivity in cancer cells. Nature. 2012;483:570–575. doi: 10.1038/nature11005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garraway LA, Lander ES. Lessons from the cancer genome. Cell. 2013;153:17–37. doi: 10.1016/j.cell.2013.03.002. [DOI] [PubMed] [Google Scholar]
- Gatta G, van der Zwan JM, Casali PG, Siesling S, Dei Tos AP, Kunkler I, Otter R, Licitra L, Mallone S, Tavilla A, et al. Rare cancers are not so rare: the rare cancer burden in Europe. Eur J Cancer. 2011;47:2493–2511. doi: 10.1016/j.ejca.2011.08.008. [DOI] [PubMed] [Google Scholar]
- Gazdar AF, Gao B, Minna JD. Lung cancer cell lines: useless artifacts or invaluable tools for medical science? Lung Cancer. 2010;68:309–318. doi: 10.1016/j.lungcan.2009.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Genome Aggregation Database. gnomAD browser beta. 2017 http://gnomad.broadinstitute.org/
- Global Alliance for Genomics and Health. GENOMICS. A federated ecosystem for sharing genomic, clinical data. Science. 2016;352:1278–1280. doi: 10.1126/science.aaf6162. [DOI] [PubMed] [Google Scholar]
- Greenlee RT, Goodman MT, Lynch CF, Platz CE, Havener LA, Howe HL. The occurrence of rare cancers in U.S. adults, 1995–2004. Public Health Rep. 2010;125:28–43. doi: 10.1177/003335491012500106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haibe-Kains B, El-Hachem N, Birkbak NJ, Jin AC, Beck AH, Aerts HJ, Quackenbush J. Inconsistency in large pharmacogenomic studies. Nature. 2013;504:389–393. doi: 10.1038/nature12831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanaford AR, Archer TC, Price A, Kahlert UD, Maciaczyk J, Nikkhah G, Kim JW, Ehrenberger T, Clemons PA, Dančík V, et al. DiSCoVERing innovative therapies for rare tumors: combining genetically accurate disease models with in silico analysis to identify novel therapeutic targets. Clin Cancer Res. 2016;22:3903–3914. doi: 10.1158/1078-0432.CCR-15-3011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harbour JW, Onken MD, Roberson ED, Duan S, Cao L, Worley LA, Council ML, Matatall KA, Helms C, Bowcock AM. Frequent mutation of BAP1 in metastasizing uveal melanomas. Science. 2010;330:1410–1413. doi: 10.1126/science.1194472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris MH, DuBois SG, Glade Bender JL, Kim A, Crompton BD, Parker E, Dumont IP, Hong AL, Guo D, Church A, et al. Multi-center feasibility study of tumor molecular profiling to inform therapeutic decisions in advanced pediatric solid tumors: the individualized cancer therapy (iCat) study. JAMA Oncol. 2016 doi: 10.1001/jamaoncol.2015.5689. http://dx.doi.org/10.1001/jamaoncol.2015.5689. [DOI] [PubMed]
- Hauber AB, Gonzalez JM, Coombs J, Sirulnik A, Palacios D, Scherzer N. Patient preferences for reducing toxicities of treatments for gastrointestinal stromal tumor (GIST) Patient Prefer Adherence. 2011;5:307–314. doi: 10.2147/PPA.S20445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haverty PM, Lin E, Tan J, Yu Y, Lam B, Lianoglou S, Neve RM, Martin S, Settleman J, Yauch RL, et al. Reproducible pharmacogenomic profiling of cancer cell line panels. Nature. 2016;533:333–337. doi: 10.1038/nature17987. [DOI] [PubMed] [Google Scholar]
- Heinrich MC, Owzar K, Corless CL, Hollis D, Borden EC, Fletcher CD, Ryan CW, von Mehren M, Blanke CD, Rankin C, et al. Correlation of kinase genotype and clinical outcome in the North American Intergroup phase III trial of imatinib mesylate for treatment of advanced gastrointestinal stromal tumor: CALGB 150105 study by cancer and leukemia group B and Southwest oncology group. J Clin Oncol. 2008;26:5360–5367. doi: 10.1200/JCO.2008.17.4284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helming KC, Wang X, Wilson BG, Vazquez F, Haswell JR, Manchester HE, Kim Y, Kryukov GV, Ghandi M, Aguirre AJ, et al. ARID1B is a specific vulnerability in ARID1A-mutant cancers. Nat Med. 2014;20:251–254. doi: 10.1038/nm.3480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho AS, Kannan K, Roy DM, Morris LG, Ganly I, Katabi N, Ramaswami D, Walsh LA, Eng S, Huse JT, et al. The mutational landscape of adenoid cystic carcinoma. Nat Genet. 2013;45:791–798. doi: 10.1038/ng.2643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honeyman JN, Simon EP, Robine N, Chiaroni-Clarke R, Darcy DG, Lim II, Gleason CE, Murphy JM, Rosenberg BR, Teegan L, et al. Detection of a recurrent DNAJB1-PRKACA chimeric transcript in fibrolamellar hepatocellular carcinoma. Science. 2014;343:1010–1014. doi: 10.1126/science.1249484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong AL, Tseng YY, Cowley GS, Jonas O, Cheah JH, Kynnap BD, Doshi MB, Oh C, Meyer SC, Church AJ, et al. Integrated genetic and pharmacologic interrogation of rare cancers. Nat Commun. 2016;7:11987. doi: 10.1038/ncomms11987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houghton PJ, Morton CL, Tucker C, Payne D, Favours E, Cole C, Gorlick R, Kolb EA, Zhang W, Lock R, et al. The pediatric preclinical testing program: description of models and early testing results. Pediatr Blood Cancer. 2007;49:928–940. doi: 10.1002/pbc.21078. [DOI] [PubMed] [Google Scholar]
- Howard TP, Vazquez F, Tsherniak A, Hong AL, Rinne M, Aguirre AJ, Boehm JS, Hahn WC. Functional genomic characterization of cancer genomes. Cold Spring Harb Symp Quant Biol. 2016;81:237–246. doi: 10.1101/sqb.2016.81.031070. [DOI] [PubMed] [Google Scholar]
- Huang R, Southall N, Wang Y, Yasgar A, Shinn P, Jadhav A, Nguyen DT, Austin CP. The NCGC pharmaceutical collection: a comprehensive resource of clinically approved drugs enabling repurposing and chemical genomics. Sci Transl Med. 2011;3:80ps16. doi: 10.1126/scitranslmed.3001862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyman DM, Taylor BS, Baselga J. Implementing genome-driven oncology. Cell. 2017;168:584–599. doi: 10.1016/j.cell.2016.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ince TA, Sousa AD, Jones MA, Harrell JC, Agoston ES, Krohn M, Selfors LM, Liu W, Chen K, Yong M, et al. Characterization of twenty-five ovarian tumour cell lines that phenocopy primary tumours. Nat Commun. 2015;6:7419. doi: 10.1038/ncomms8419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- International HapMap 3 Consortium. Altshuler DM, Gibbs RA, Peltonen L, Altshuler DM, Gibbs RA, Peltonen L, Dermitzakis E, Schaffner SF, Yu F, et al. Integrating common and rare genetic variation in diverse human populations. Nature. 2010;467:52–58. doi: 10.1038/nature09298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iorio F, Knijnenburg TA, Vis DJ, Bignell GR, Menden MP, Schubert M, Aben N, Gonçalves E, Barthorpe S, Lightfoot H, et al. A landscape of pharmacogenomic interactions in cancer. Cell. 2016;166:740–754. doi: 10.1016/j.cell.2016.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ISBER. 2012 best practices for repositories collection, storage, retrieval, and distribution of biological materials for research international society for biological and environmental repositories. Biopreserv Biobank. 2012;10:79–161. doi: 10.1089/bio.2012.1022. [DOI] [PubMed] [Google Scholar]
- Izumchenko E, Meir J, Bedi A, Wysocki PT, Hoque MO, Sidransky D. Patient-derived xenografts as tools in pharmaceutical development. Clin Pharmacol Ther. 2016;99:612–621. doi: 10.1002/cpt.354. [DOI] [PubMed] [Google Scholar]
- Jeffreys AJ, Wilson V, Thein SL. Hypervariable ’minisatellite’ regions in human DNA. Nature. 1985;314:67–73. doi: 10.1038/314067a0. [DOI] [PubMed] [Google Scholar]
- Jiao Y, Pawlik TM, Anders RA, Selaru FM, Streppel MM, Lucas DJ, Niknafs N, Guthrie VB, Maitra A, Argani P, et al. Exome sequencing identifies frequent inactivating mutations in BAP1, ARID1A and PBRM1 in intrahepatic cholangiocarcinomas. Nat Genet. 2013;45:1470–1473. doi: 10.1038/ng.2813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones DT, Jäger N, Kool M, Zichner T, Hutter B, Sultan M, Cho YJ, Pugh TJ, Hovestadt V, Stütz AM, et al. Dissecting the genomic complexity underlying medulloblastoma. Nature. 2012;488:100–105. doi: 10.1038/nature11284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaelin WG., Jr The concept of synthetic lethality in the context of anticancer therapy. Nat Rev Cancer. 2005;5:689–698. doi: 10.1038/nrc1691. [DOI] [PubMed] [Google Scholar]
- Karatayli-Ozgursoy S, Bishop JA, Hillel AT, Akst LM, Best SR. Malignant salivary gland tumours of the larynx: a single institution review. Acta Otorhinolaryngol Ital. 2016;36:289–294. doi: 10.14639/0392-100X-807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz MS, Utengen A, Anderson PF, Thompson MA, Attai DJ, Johnston C, Dizon DS. Disease-specific Hashtags for online communication about cancer care. JAMA Oncol. 2016;2:392–394. doi: 10.1001/jamaoncol.2015.3960. [DOI] [PubMed] [Google Scholar]
- Kaufman B, Shapira-Frommer R, Schmutzler RK, Audeh MW, Friedlander M, Balmaña J, Mitchell G, Fried G, Stemmer SM, Hubert A, et al. Olaparib monotherapy in patients with advanced cancer and a germline BRCA1/2 mutation. J Clin Oncol. 2015;33:244–250. doi: 10.1200/JCO.2014.56.2728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knezevich SR, McFadden DE, Tao W, Lim JF, Sorensen PH. A novel ETV6-NTRK3 gene fusion in congenital fibrosarcoma. Nat Genet. 1998;18:184–187. doi: 10.1038/ng0298-184. [DOI] [PubMed] [Google Scholar]
- Kryukov GV, Wilson FH, Ruth JR, Paulk J, Tsherniak A, Marlow SE, Vazquez F, Weir BA, Fitzgerald ME, Tanaka M, et al. MTAP deletion confers enhanced dependency on the PRMT5 arginine methyltransferase in cancer cells. Science. 2016;351:1214–1218. doi: 10.1126/science.aad5214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence MS, Stojanov P, Polak P, Kryukov GV, Cibulskis K, Sivachenko A, Carter SL, Stewart C, Mermel CH, Roberts SA, et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature. 2013;499:214–218. doi: 10.1038/nature12213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee RS, Stewart C, Carter SL, Ambrogio L, Cibulskis K, Sougnez C, Lawrence MS, Auclair D, Mora J, Golub TR, et al. A remarkably simple genome underlies highly malignant pediatric rhabdoid cancers. J Clin Invest. 2012;122:2983–2988. doi: 10.1172/JCI64400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leiomyosarcoma Direct Research Foundation. Paraffin tissue block donation. 2016 http://lmsdr.org/paraffin-tissue-block-donation/
- Lek M, Karczewski KJ, Minikel EV, Samocha KE, Banks E, Fennell T, O’Donnell-Luria AH, Ware JS, Hill AJ, Cummings BB, et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature. 2016;536:285–291. doi: 10.1038/nature19057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis TB, Coffin CM, Bernard PS. Differentiating Ewing’s sarcoma from other round blue cell tumors using a RT-PCR translocation panel on formalin-fixed paraffin-embedded tissues. Mod Pathol. 2007;20:397–404. doi: 10.1038/modpathol.3800755. [DOI] [PubMed] [Google Scholar]
- Ley TJ, Mardis ER, Ding L, Fulton B, McLellan MD, Chen K, Dooling D, Dunford-Shore BH, McGrath S, Hickenbotham M, et al. DNA sequencing of a cytogenetically normal acute myeloid leukaemia genome. Nature. 2008;456:66–72. doi: 10.1038/nature07485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Ory V, Chapman S, Yuan H, Albanese C, Kallakury B, Timofeeva OA, Nealon C, Dakic A, Simic V, et al. ROCK inhibitor and feeder cells induce the conditional reprogramming of epithelial cells. Am J Pathol. 2012;180:599–607. doi: 10.1016/j.ajpath.2011.10.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacArthur J, Bowler E, Cerezo M, Gil L, Hall P, Hastings E, Junkins H, McMahon A, Milano A, Morales J, et al. The new NHGRI-EBI Catalog of published genome-wide association studies (GWAS Catalog) Nucleic Acids Res. 2017;45:D896–D901. doi: 10.1093/nar/gkw1133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcotte R, Brown KR, Suarez F, Sayad A, Karamboulas K, Krzyzanowski PM, Sircoulomb F, Medrano M, Fedyshyn Y, Koh JLY, et al. Essential gene profiles in breast, pancreatic, and ovarian cancer cells. Cancer Discov. 2012;2:172–189. doi: 10.1158/2159-8290.CD-11-0224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcotte R, Sayad A, Brown KR, Sanchez-Garcia F, Reimand J, Haider M, Virtanen C, Bradner JE, Bader GD, Mills GB, et al. Functional genomic landscape of human breast cancer drivers, vulnerabilities, and resistance. Cell. 2016;164:293–309. doi: 10.1016/j.cell.2015.11.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martelotto LG, De Filippo MR, Ng CK, Natrajan R, Fuhrmann L, Cyrta J, Piscuoglio S, Wen HC, Lim RS, Shen R, et al. Genomic landscape of adenoid cystic carcinoma of the breast. J Pathol. 2015;237:179–189. doi: 10.1002/path.4573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masters JR. Human cancer cell lines: fact and fantasy. Nat Rev Mol Cell Biol. 2000;1:233–236. doi: 10.1038/35043102. [DOI] [PubMed] [Google Scholar]
- Matulonis UA, Penson RT, Domchek SM, Kaufman B, Shapira-Frommer R, Audeh MW, Kaye S, Molife LR, Gelmon KA, Robertson JD, et al. Olaparib monotherapy in patients with advanced relapsed ovarian cancer and a germline BRCA1/2 mutation: a multistudy analysis of response rates and safety. Ann Oncol. 2016;27:1013–1019. doi: 10.1093/annonc/mdw133. [DOI] [PubMed] [Google Scholar]
- Mavrakis KJ, McDonald ER, 3rd, Schlabach MR, Billy E, Hoffman GR, deWeck A, Ruddy DA, Venkatesan K, Yu J, McAllister G, et al. Disordered methionine metabolism in MTAP/CDKN2A-deleted cancers leads to dependence on PRMT5. Science. 2016;351:1208–1213. doi: 10.1126/science.aad5944. [DOI] [PubMed] [Google Scholar]
- McMaster ML, Goldstein AM, Bromley CM, Ishibe N, Parry DM. Chordoma: incidence and survival patterns in the United States, 1973–1995. Cancer Causes Control. 2001;12:1–11. doi: 10.1023/a:1008947301735. [DOI] [PubMed] [Google Scholar]
- Meyerson M, Gabriel S, Getz G. Advances in understanding cancer genomes through second-generation sequencing. Nat Rev Genet. 2010;11:685–696. doi: 10.1038/nrg2841. [DOI] [PubMed] [Google Scholar]
- Mirabello L, Koster R, Moriarity BS, Spector LG, Meltzer PS, Gary J, Machiela MJ, Pankratz N, Panagiotou OA, Largaespada D, et al. A genome-wide scan identifies variants in NFIB associated with metastasis in patients with osteosarcoma. Cancer Discov. 2015;5:920–931. doi: 10.1158/2159-8290.CD-15-0125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mody RJ, Wu YM, Lonigro RJ, Cao X, Roychowdhury S, Vats P, Frank KM, Prensner JR, Asangani I, Palanisamy N, et al. Integrative clinical sequencing in the management of refractory or relapsed cancer in youth. JAMA. 2015;314:913–925. doi: 10.1001/jama.2015.10080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moffat J, Grueneberg DA, Yang X, Kim SY, Kloepfer AM, Hinkle G, Piqani B, Eisenhaure TM, Luo B, Grenier JK, et al. A lentiviral RNAi library for human and mouse genes applied to an arrayed viral high-content screen. Cell. 2006;124:1283–1298. doi: 10.1016/j.cell.2006.01.040. [DOI] [PubMed] [Google Scholar]
- Morelli MP, Calvo E, Ordoñez E, Wick MJ, Viqueira BR, Lopez-Casas PP, Bruckheimer E, Calles-Blanco A, Sidransky D, Hidalgo M, et al. Prioritizing phase I treatment options through preclinical testing on personalized tumorgraft. J Clin Oncol. 2012;30:e45–e48. doi: 10.1200/JCO.2011.36.9678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller FL, Colla S, Aquilanti E, Manzo VE, Genovese G, Lee J, Eisenson D, Narurkar R, Deng P, Nezi L, et al. Passenger deletions generate therapeutic vulnerabilities in cancer. Nature. 2012;488:337–342. doi: 10.1038/nature11331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullighan CG. Genomic characterization of childhood acute lymphoblastic leukemia. Semin Hematol. 2013;50:314–324. doi: 10.1053/j.seminhematol.2013.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullighan CG, Su X, Zhang J, Radtke I, Phillips LA, Miller CB, Ma J, Liu W, Cheng C, Schulman BA, et al. Deletion of IKZF1 and prognosis in acute lymphoblastic leukemia. N Engl J Med. 2009;360:470–480. doi: 10.1056/NEJMoa0808253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Multiple Myeloma Research Foundation. The multiple myeloma research foundation releases latest interim analysis from the CoMMpass Study to researchers and clinicians on its researcher gateway. 2015 https://www.themmrf.org/the-multiple-myeloma-research-foundation-releases-latest-interim-analysis-from-the-commpass-study-to-researchers-and-clinicians-on-its-researcher-gateway/
- Munoz DM, Cassiani PJ, Li L, Billy E, Korn JM, Jones MD, Golji J, Ruddy DA, Yu K, McAllister G, et al. CRISPR screens provide a comprehensive assessment of cancer vulnerabilities but generate false-positive hits for highly amplified genomic regions. Cancer Discov. 2016;6:900–913. doi: 10.1158/2159-8290.CD-16-0178. [DOI] [PubMed] [Google Scholar]
- National Cancer Institute. Synergizing epidemiologic research on rare cancers. 2007 https://epi.grants.cancer.gov/events/rare-cancers/
- National Cancer Institute. NCI collaborates with Multiple Myeloma Research Foundation. 2016a https://www.cancer.gov/news-events/press-releases/2016/gdc-data-expansion-mmrf.
- National Cancer Institute. SEER Cancer Statistics Review (CSR) 1975–2013. 2016b http://seer.cancer.gov/csr/1975_2013/
- NCCN Guidelines. Bone Cancer Version 2.2017. 2017a November 7, 2016 www.nccn.org.
- NCCN Guidelines. Breast Cancer Version 2.2017. 2017b April 6, 2017 www.nccn.org.
- NCCN Guidelines. Prostate Cancer Version 2.2017. 2017c February 21, 2017 www.nccn.org.
- Nijhawan D, Zack TI, Ren Y, Strickland MR, Lamothe R, Schumacher SE, Tsherniak A, Besche HC, Rosenbluh J, Shehata S. Cancer vulnerabilities unveiled by genomic loss. Cell. 2012;150:842–854. doi: 10.1016/j.cell.2012.07.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oie HK, Russell EK, Carney DN, Gazdar AF. Cell culture methods for the establishment of the NCI series of lung cancer cell lines. J Cell Biochem Suppl. 1996;24:24–31. doi: 10.1002/jcb.240630504. [DOI] [PubMed] [Google Scholar]
- Oikawa T, Wauthier E, Dinh TA, Selitsky SR, Reyna-Neyra A, Carpino G, Levine R, Cardinale V, Klimstra D, Gaudio E, et al. Model of fibrolamellar hepatocellular carcinomas reveals striking enrichment in cancer stem cells. Nat Commun. 2015;6:8070. doi: 10.1038/ncomms9070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ong CK, Subimerb C, Pairojkul C, Wongkham S, Cutcutache I, Yu W, McPherson JR, Allen GE, Ng CC, Wong BH, et al. Exome sequencing of liver fluke-associated cholangiocarcinoma. Nat Genet. 2012;44:690–693. doi: 10.1038/ng.2273. [DOI] [PubMed] [Google Scholar]
- Orphanet. Prevalence and incidence of rare diseases: bibliographic data. 2016 http://www.orpha.net/orphacom/cahiers/docs/GB/Prevalence_of_rare_diseases_by_alphabetical_list.pdf.
- Panageas KS. Clinical trial design for rare cancers: why a less conventional route may be required. Expert Rev Clin Pharmacol. 2015;8:661–663. doi: 10.1586/17512433.2015.1088382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panda PK, Wig N, Kumar S, Arava S. Invasive thymoma presenting as classic superior vena cava syndrome: a case of venous spread metastasis. BMJ Case Rep 2016. 2016 doi: 10.1136/bcr-2016-217695. http://dx.doi.org/10.1136/bcr-2016-217695. [DOI] [PMC free article] [PubMed]
- Parsons DW, Roy A, Yang Y, Wang T, Scollon S, Bergstrom K, Kerstein RA, Gutierrez S, Petersen AK, Bavle A, et al. Diagnostic yield of clinical tumor and germline whole-exome sequencing for children with solid tumors. JAMA Oncol. 2016 doi: 10.1001/jamaoncol.2015.5699. http://dx.doi.org/10.1001/jamaoncol.2015.5699. [DOI] [PMC free article] [PubMed]
- Pavia-Jimenez A, Tcheuyap VT, Brugarolas J. Establishing a human renal cell carcinoma tumorgraft platform for preclinical drug testing. Nat Protoc. 2014;9:1848–1859. doi: 10.1038/nprot.2014.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philippakis AA, Azzariti DR, Beltran S, Brookes AJ, Brownstein CA, Brudno M, Brunner HG, Buske OJ, Carey K, Doll C, et al. The Matchmaker Exchange: a platform for rare disease gene discovery. Hum Mutat. 2015;36:915–921. doi: 10.1002/humu.22858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pillay N, Plagnol V, Tarpey PS, Lobo SB, Presneau N, Szuhai K, Halai D, Berisha F, Cannon SR, Mead S, et al. A common single-nucleotide variant in T is strongly associated with chordoma. Nat Genet. 2012;44:1185–1187. doi: 10.1038/ng.2419. [DOI] [PubMed] [Google Scholar]
- Price ND, Trent J, El-Naggar AK, Cogdell D, Taylor E, Hunt KK, Pollock RE, Hood L, Shmulevich I, Zhang W. Highly accurate two-gene classifier for differentiating gastrointestinal stromal tumors and leiomyosarcomas. Proc Natl Acad Sci USA. 2007;104:3414–3419. doi: 10.1073/pnas.0611373104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pritchard CC, Mateo J, Walsh MF, De Sarkar N, Abida W, Beltran H, Garofalo A, Gulati R, Carreira S, Eeles R, et al. Inherited DNA-repair gene mutations in men with metastatic prostate cancer. N Engl J Med. 2016;375:443–453. doi: 10.1056/NEJMoa1603144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pugh TJ, Morozova O, Attiyeh EF, Asgharzadeh S, Wei JS, Auclair D, Carter SL, Cibulskis K, Hanna M, Kiezun A, et al. The genetic landscape of high-risk neuroblastoma. Nat Genet. 2013;45:279–284. doi: 10.1038/ng.2529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen SM, Bilgrau AE, Schmitz A, Falgreen S, Bergkvist KS, Tramm AM, Baech J, Jacobsen CL, Gaihede M, Kjeldsen MK, et al. Stable phenotype of B-cell subsets following cryopreservation and thawing of normal human lymphocytes stored in a tissue biobank. Cytometry B Clin Cytom. 2015;88:40–49. doi: 10.1002/cyto.b.21192. [DOI] [PubMed] [Google Scholar]
- Root DE, Hacohen N, Hahn WC, Lander ES, Sabatini DM. Genome-scale loss-of-function screening with a lentiviral RNAi library. Nat Methods. 2006;3:715–719. doi: 10.1038/nmeth924. [DOI] [PubMed] [Google Scholar]
- Ross JS, Wang K, Rand JV, Sheehan CE, Jennings TA, Al-Rohil RN, Otto GA, Curran JC, Palmer G, Downing SR, et al. Comprehensive genomic profiling of relapsed and metastatic adenoid cystic carcinomas by next-generation sequencing reveals potential new routes to targeted therapies. Am J Surg Pathol. 2014;38:235–238. doi: 10.1097/PAS.0000000000000102. [DOI] [PubMed] [Google Scholar]
- Roychowdhury S, Iyer MK, Robinson DR, Lonigro RJ, Wu YM, Cao X, Kalyana-Sundaram S, Sam L, Balbin OA, Quist MJ, et al. Personalized oncology through integrative high-throughput sequencing: a pilot study. Sci Transl Med. 2011;3:111ra121. doi: 10.1126/scitranslmed.3003161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rygaard J, Povlsen CO. Heterotransplantation of a human malignant tumour to “Nude” mice. Acta Pathol Microbiol Scand. 1969;77:758–760. doi: 10.1111/j.1699-0463.1969.tb04520.x. [DOI] [PubMed] [Google Scholar]
- Sato T, Vries RG, Snippert HJ, van de Wetering M, Barker N, Stange DE, van Es JH, Abo A, Kujala P, Peters PJ, et al. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature. 2009;459:262–265. doi: 10.1038/nature07935. [DOI] [PubMed] [Google Scholar]
- Scherer WF, Syverton JT, Gey GO. Studies on the propagation in vitro of poliomyelitis viruses. IV. Viral multiplication in a stable strain of human malignant epithelial cells (strain HeLa) derived from an epidermoid carcinoma of the cervix. J Exp Med. 1953;97:695–710. doi: 10.1084/jem.97.5.695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiber SL. Organic synthesis toward small-molecule probes and drugs. Proc Natl Acad Sci USA. 2011;108:6699–6702. doi: 10.1073/pnas.1103205108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiber SL, Kotz JD, Li M, Aubé J, Austin CP, Reed JC, Rosen H, White EL, Sklar LA, Lindsley CW, et al. Advancing biological understanding and therapeutics discovery with small-molecule probes. Cell. 2015;161:1252–1265. doi: 10.1016/j.cell.2015.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seashore-Ludlow B, Rees MG, Cheah JH, Cokol M, Price EV, Coletti ME, Jones V, Bodycombe NE, Soule CK, Gould J, et al. Harnessing connectivity in a large-scale small-molecule sensitivity dataset. Cancer Discov. 2015;5:1210–1223. doi: 10.1158/2159-8290.CD-15-0235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seltzer ED, Stolley MR, Mensah EK, Sharp LK. Social networking site usage among childhood cancer survivors–a potential tool for research recruitment? J Cancer Surviv. 2014;8:349–354. doi: 10.1007/s11764-014-0348-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharifnia T, Rusu V, Piccioni F, Bagul M, Imielinski M, Cherniack AD, Pedamallu CS, Wong B, Wilson FH, Garraway LA, et al. Genetic modifiers of EGFR dependence in non-small cell lung cancer. Proc Natl Acad Sci USA. 2014;111:18661–18666. doi: 10.1073/pnas.1412228112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoemaker RH. The NCI60 human tumour cell line anticancer drug screen. Nat Rev Cancer. 2006;6:813–823. doi: 10.1038/nrc1951. [DOI] [PubMed] [Google Scholar]
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin. 2017;67:7–30. doi: 10.3322/caac.21387. [DOI] [PubMed] [Google Scholar]
- Simonetti G, Gaviani P, Lamperti E, Innocenti A, Botturi A, Saini M, Pollo B, Silvani A. A case of medulloblastoma in adult patient affected by anaplastic oligoastrocytoma. Neurol Sci. 2016;37:1727–1730. doi: 10.1007/s10072-016-2640-8. [DOI] [PubMed] [Google Scholar]
- Somiari SB, Somiari RI. The future of biobanking: a conceptual look at how biobanks can respond to the growing human biospecimen needs of researchers. Adv Exp Med Biol. 2015;864:11–27. doi: 10.1007/978-3-319-20579-3_2. [DOI] [PubMed] [Google Scholar]
- Stein LD, Knoppers BM, Campbell P, Getz G, Korbel JO. Data analysis: create a cloud commons. Nature. 2015;523:149–151. doi: 10.1038/523149a. [DOI] [PubMed] [Google Scholar]
- Stephens PJ, Davies HR, Mitani Y, Van Loo P, Shlien A, Tarpey PS, Papaemmanuil E, Cheverton A, Bignell GR, Butler AP, et al. Whole exome sequencing of adenoid cystic carcinoma. J Clin Invest. 2013;123:2965–2968. doi: 10.1172/JCI67201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stransky N, Cerami E, Schalm S, Kim JL, Lengauer C. The landscape of kinase fusions in cancer. Nat Commun. 2014;5:4846. doi: 10.1038/ncomms5846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan SB, Dear KB, Bruzzi P, Machin D. Strategy for randomised clinical trials in rare cancers. BMJ. 2003;327:47–49. doi: 10.1136/bmj.327.7405.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanay A, Regev A. Scaling single-cell genomics from phenomenology to mechanism. Nature. 2017;541:331–338. doi: 10.1038/nature21350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Cancer Genome Atlas. NIH launches comprehensive effort to explore cancer genomics. 2005 https://cancergenome.nih.gov/newsevents/newsannouncements/news_12_13_2005.
- The Life Raft Group. Battling the dragon. 2000 https://liferaftgroup.org/wp-content/uploads/2012/09/Dec2000newsletter.pdf.
- The New York Times. Powerful anti-cancer drug emerges from basic biology. 2001 http://www.nytimes.com/2001/05/08/science/powerful-anti-cancer-drug-emerges-from-basic-biology.html.
- The New York Times. Reseacher behind the drug Gleevec. 2009 http://www.nytimes.com/2009/11/03/science/03conv.html?pagewanted=all.
- Tiacci E, Trifonov V, Schiavoni G, Holmes A, Kern W, Martelli MP, Pucciarini A, Bigerna B, Pacini R, Wells VA, et al. BRAF mutations in hairy-cell leukemia. N Engl J Med. 2011;364:2305–2315. doi: 10.1056/NEJMoa1014209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirosh I, Izar B, Prakadan SM, Wadsworth MH, 2nd, Treacy D, Trombetta JJ, Rotem A, Rodman C, Lian C, Murphy G, et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science. 2016a;352:189–196. doi: 10.1126/science.aad0501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tirosh I, Venteicher AS, Hebert C, Escalante LE, Patel AP, Yizhak K, Fisher JM, Rodman C, Mount C, Filbin MG, et al. Single-cell RNA-seq supports a developmental hierarchy in human oligodendroglioma. Nature. 2016b;539:309–313. doi: 10.1038/nature20123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomich J, Grove Nigro K, Barr RG. Primary Angiosarcoma of the breast: a case report and review of the literature. Ultrasound Q. 2017;33:46–48. doi: 10.1097/RUQ.0000000000000274. [DOI] [PubMed] [Google Scholar]
- Townsend EC, Murakami MA, Christodoulou A, Christie AL, Köster J, DeSouza TA, Morgan EA, Kallgren SP, Liu H, Wu SC, et al. The public repository of xenografts enables discovery and randomized phase II-like trials in mice. Cancer Cell. 2016;30:183. doi: 10.1016/j.ccell.2016.06.008. [DOI] [PubMed] [Google Scholar]
- Tuveson DA, Willis NA, Jacks T, Griffin JD, Singer S, Fletcher CD, Fletcher JA, Demetri GD. STI571 inactivation of the gastrointestinal stromal tumor c-KIT oncoprotein: biological and clinical implications. Oncogene. 2001;20:5054–5058. doi: 10.1038/sj.onc.1204704. [DOI] [PubMed] [Google Scholar]
- US Food & Drug Administration. FDA approves first cancer treatment for any solid tumor with a specific genetic feature. 2017 https://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm560167.htm.
- van de Wetering M, Francies HE, Francis JM, Bounova G, Iorio F, Pronk A, van Houdt W, van Gorp J, Taylor-Weiner A, Kester L, et al. Prospective derivation of a living organoid biobank of colorectal cancer patients. Cell. 2015;161:933–945. doi: 10.1016/j.cell.2015.03.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Raamsdonk CD, Bezrookove V, Green G, Bauer J, Gaugler L, O’Brien JM, Simpson EM, Barsh GS, Bastian BC. Frequent somatic mutations of GNAQ in uveal melanoma and blue naevi. Nature. 2009;457:599–602. doi: 10.1038/nature07586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaught J, Kelly A, Hewitt R. A review of international biobanks and networks: success factors and key benchmarks. Biopreserv Biobank. 2009;7:143–150. doi: 10.1089/bio.2010.0003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagle N, Berger MF, Davis MJ, Blumenstiel B, Defelice M, Pochanard P, Ducar M, Van Hummelen P, Macconaill LE, Hahn WC, et al. High-throughput detection of actionable genomic alterations in clinical tumor samples by targeted, massively parallel sequencing. Cancer Discov. 2012;2:82–93. doi: 10.1158/2159-8290.CD-11-0184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K, Diskin SJ, Zhang H, Attiyeh EF, Winter C, Hou C, Schnepp RW, Diamond M, Bosse K, Mayes PA, et al. Integrative genomics identifies LMO1 as a neuroblastoma oncogene. Nature. 2011;469:216–220. doi: 10.1038/nature09609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Birsoy K, Hughes NW, Krupczak KM, Post Y, Wei JJ, Lander ES, Sabatini DM. Identification and characterization of essential genes in the human genome. Science. 2015;350:1096–1101. doi: 10.1126/science.aac7041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Yu H, Hughes NW, Liu B, Kendirli A, Klein K, Chen WW, Lander ES, Sabatini DM. Gene essentiality profiling reveals gene networks and synthetic lethal interactions with oncogenic Ras. Cell. 2017;168:890–903.e15. doi: 10.1016/j.cell.2017.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward E, DeSantis C, Robbins A, Kohler B, Jemal A. Childhood and adolescent cancer statistics, 2014. CA Cancer J Clin. 2014;64:83–103. doi: 10.3322/caac.21219. [DOI] [PubMed] [Google Scholar]
- Weinstein IB. Cancer. Addiction to oncogenes–the Achilles heal of cancer. Science. 2002;297:63–64. doi: 10.1126/science.1073096. [DOI] [PubMed] [Google Scholar]
- Weroha SJ, Becker MA, Enderica-Gonzalez S, Harrington SC, Oberg AL, Maurer MJ, Perkins SE, AlHilli M, Butler KA, McKinstry S, et al. Tumorgrafts as in vivo surrogates for women with ovarian cancer. Clin Cancer Res. 2014;20:1288–1297. doi: 10.1158/1078-0432.CCR-13-2611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wetterstrand KA. DNA sequencing costs: data from the NHGRI Genome Sequencing Program (GSP) 2016 www.genome.gov/sequencingcostsdata.
- Wilson BG, Helming KC, Wang X, Kim Y, Vazquez F, Jagani Z, Hahn WC, Roberts CW. Residual complexes containing SMARCA2 (BRM) underlie the oncogenic drive of SMARCA4 (BRG1) mutation. Mol Cell Biol. 2014;34:1136–1144. doi: 10.1128/MCB.01372-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worhunsky DJ, Gupta M, Gholami S, Tran TB, Ganjoo KN, van de Rijn M, Visser BC, Norton JA, Poultsides GA. Leiomyosarcoma: one disease or distinct biologic entities based on site of origin? J Surg Oncol. 2015;111:808–812. doi: 10.1002/jso.23904. [DOI] [PubMed] [Google Scholar]
- Yan H, Parsons DW, Jin G, McLendon R, Rasheed BA, Yuan W, Kos I, Batinic-Haberle I, Jones S, Riggins GJ, et al. IDH1 and IDH2 mutations in gliomas. N Engl J Med. 2009;360:765–773. doi: 10.1056/NEJMoa0808710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W, Soares J, Greninger P, Edelman EJ, Lightfoot H, Forbes S, Bindal N, Beare D, Smith JA, Thompson IR, et al. Genomics of Drug Sensitivity in Cancer (GDSC): a resource for therapeutic biomarker discovery in cancer cells. Nucleic Acids Res. 2013;41:D955–D961. doi: 10.1093/nar/gks1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu M, Bardia A, Aceto N, Bersani F, Madden MW, Donaldson MC, Desai R, Zhu H, Comaills V, Zheng Z, et al. Cancer therapy. Ex vivo culture of circulating breast tumor cells for individualized testing of drug susceptibility. Science. 2014;345:216–220. doi: 10.1126/science.1253533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu M, Selvaraj SK, Liang-Chu MM, Aghajani S, Busse M, Yuan J, Lee G, Peale F, Klijn C, Bourgon R, et al. A resource for cell line authentication, annotation and quality control. Nature. 2015;520:307–311. doi: 10.1038/nature14397. [DOI] [PubMed] [Google Scholar]
- Yu C, Mannan AM, Yvone GM, Ross KN, Zhang YL, Marton MA, Taylor BR, Crenshaw A, Gould JZ, Tamayo P, et al. High-throughput identification of genotype-specific cancer vulnerabilities in mixtures of barcoded tumor cell lines. Nat Biotechnol. 2016;34:419–423. doi: 10.1038/nbt.3460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan H, Myers S, Wang J, Zhou D, Woo JA, Kallakury B, Ju A, Bazylewicz M, Carter YM, Albanese C, et al. Use of reprogrammed cells to identify therapy for respiratory papillomatosis. N Engl J Med. 2012;367:1220–1227. doi: 10.1056/NEJMoa1203055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng S, Cherniack AD, Dewal N, Moffitt RA, Danilova L, Murray BA, Lerario AM, Else T, Knijnenburg TA, Ciriello G, et al. Comprehensive Pan-Genomic characterization of adrenocortical carcinoma. Cancer Cell. 2016;30:363. doi: 10.1016/j.ccell.2016.07.013. [DOI] [PubMed] [Google Scholar]