

Abstract
Background
With the decreasing cost and efficiency of next generation sequencing, the technology is rapidly introduced into clinical and public health laboratory practice.
Aims
In this review, the historical background and principles of first, second and third generation sequencing are described as are the characteristics of the most commonly used sequencing instruments.
Sources
Peer reviewed literature, white papers and meeting reports.
Content & implications
Next generation sequencing is a technology that potentially could replace many traditional microbiological workflows, providing clinicians and public health specialists with more actionable information than hitherto achievable. Examples of the clinical and public health uses of the technology are provided. The challenge of comparability of different sequencing platforms is discussed. Finally, the future directions of the technology integrating it with laboratory management and public health surveillance systems, and moving it towards performing sequencing directly from the clinical specimen (metagenomics) could lead to yet another fundamental transformation of clinical diagnostics and public health surveillance.
Keywords: whole genome sequencing, next generation sequencing, short-read technology, long-read technology, surveillance, diagnostics
Introduction
DNA sequencing technologies have existed since the early 1970’s, but initially their cost, complexity, and requirement for toxic or radioactive reagents limited their use to research settings. The chain-termination methods pioneered by Sanger and colleagues (1) was more practical, and formed the basis for the first generation of automated DNA sequencers. Public health applications were first introduced in the 1990’s, such as the multilocus sequence typing scheme for Neisseria meningitidis developed by Maiden et al. (2). The first complete genome of a free-living microorganism, Haemophilus influenza, published in 1995 (3) was sequenced using the Sanger method. However, whole genome sequencing (WGS) by this technology was prohibitively expensive, cumbersome, and time-consuming. The need for high throughput sequencing technology was intensified by initiation of the Human Genome Project (HGP) in 1990 and its goal to sequence and interpret the 3.2 billion nucleotide base pairs comprising the human genome for potential medical benefits. This 3.8 billion dollar international collaboration was initially based on Sanger sequencing(4, 5).A major change occurred in the early 2000’s, when new next generation sequencing (NGS) methods using massively parallel processing brought the cost down to a fraction of the cost of Sanger sequencing, and dramatically reduced sequencing time. During the 2010’s, WGS for bacterial pathogens began migrating from research laboratories into public health practice. The trend accelerated after several high profile infectious disease events, including the cholera epidemic in Haiti following the 2010 earthquake (6), and 2011 international outbreak of E. coli O104:H4 disease associated with fenugreek sprout consumption (7, 8). Both outbreaks involved significant morbidity and mortality, which created an urgent need to understand transmission dynamics and virulence characteristics. In both situations, government laboratories and academic institutions responded rapidly with NGS technology using open sharing of data and crowd sourcing. Adoption of NGS by public health laboratories has greatly accelerated since these outbreaks, and global implementation of standardized WGS for surveillance is now well under way in public health (9, 10) (http://www.globalmicrobialidentifier.org/) and increasingly in bigger hospital laboratories.
One of the earliest applications of WGS in public health was teasing out epidemiological associations in hospital acquired infections, such as the 2010 outbreak of Acinetobacter baumannii in a British hospital (11). Within a few years, WGS began to be used more widely for elucidating and interrupting transmission pathways in hospital outbreaks, such as those caused by methicillin-resistant Staphylococcus aureus (MRSA) and carbapenem-resistant Klebsiella pneumonia (12, 13). In these examples, WGS analyses were conducted from cultured isolates. As will be described later in this chapter, WGS has the potential for rapidly providing a large amount of information from isolates, including species, strain type, antibiotic resistance, virulence, and other information for outbreak and case management. While the value of WGS for outbreak detection and investigation is clear in many situations, at current cost levels the usefulness of this approach is less clear for diagnosis and treatment of individual patients, especially considering the emergence of direct-from-specimen multi-analyte test panels. These tests have the ability to identify common pathogens in patient specimens in a highly useful timeframe (14).
The most extensive implementation of WGS in public health has been in the area of surveillance of foodborne diseases, which are both common and preventable. Globally, an estimated 1.9 billion people acquire a foodborne infection, and 715,000 die each year (15). Outbreak surveillance helps identify why people become ill, so that prevention efforts can be more precisely directed. Molecular-based real-time surveillance programs such as PulseNet have been very effective at identifying contamination problems in widely distributed food commodities that would otherwise not have been detected. Using subtyping by Pulsed Field Gel Electrophoresis (PFGE), the PulseNet surveillance in the U.S. has resulted in the prevention of millions of illnesses and saved billions of dollars in healthcare costs and lost productivity (16). Early data from surveillance of listeriosis in the U.S.A. suggests that WGS can dramatically improve upon these numbers. The number of outbreaks detected increased 36% after implementation of real-time WGS based surveillance, and likewise the number of solved outbreaks increased more than three-fold (figure 1) while the incidence of disease remained constant at approximately 0.2 cases per 100,000 (17). Similar improvements in surveillance of listeriosis were found in France after WGS implementation (18).
Figure 1.
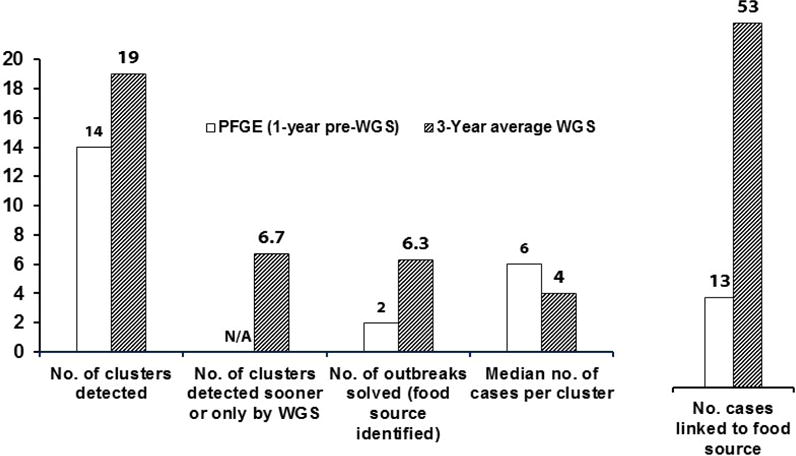
Metrics illustrating the benefits of using WGS compared to PFGE for real-time outbreak laboratory surveillance for listeriosis in the United States
NGS can be a replacing technology since the information generated from multiple traditional workflows may be combined into a single efficient WGS workflow. For example, information about species, serotype, virulence characteristics, and antibiotic resistance can be extracted from genomes in addition to phylogenetically relevant subtyping information. Information about antimicrobial resistance and virulence characteristics could be used in clinical decision making although more data is needed on the correlation between genotype and phenotype before this may be used in clinical practice. .
Sequencing Technologies
The Sanger method (“first generation” technology) was the primary sequencing technology between 1975 and 2005. Sanger sequencing produces relatively long (500–1000 bp) high quality DNA sequences, and has long been considered the gold standard for sequencing DNA. The introduction of pyrosequencing technology by 454 Life Sciences in 2005 began the “next generation sequencing” (NGS) revolution (19). This high throughput technology allowed the generation and detection of thousands to millions of short sequencing reads in a single machine run without the need for cloning. Since then, many other NGS technologies have emerged that generate both short (50 – 400 bp) and long reads (1 – 100 kb). A brief description of the main platforms and their performance is provided below. More details can be found from some excellent recent reviews (20–22). A glossary of some commonly used sequencing terminology is provided in Table 1.
Table 1.
Glossary of some commonly used sequencing terms
| Adapter | Any short piece of DNA of known sequence that one adds to the ends of their unknown DNA of interest, usually for the purpose of eventually allowing a sequencing primer to hybridise at this position |
| Amplicon sequencing | Ultra-deep sequencing of PCR products for analyzing genetic variations |
| ANI | Average nucleotide identity – An analysis method that assesses the nucleotide identity between genetic regions shared by two isolates |
| Assembly | Genome assembly is the process by which many short DNA sequence fragments, such as those generated by next generation sequencers, are reassembled into a representation of the original genomic sequence |
| Bridge amplification | A PCR technique that embeds DNA on a solid surface for sequencing. It is used by Illumina’s platforms |
| Contig | A contiguous consensus sequence derived from the assembly of many short, overlapping DNA fragments |
| cgMLST | Core genome multi-locus sequence typing - an analysis method that detects variation in genes that are present in the majority (>97%) of strains of a given species |
| Coverage (read depth) | The average number of reads that include a given nucleotide in the reconstructed sequence |
| Draft genome | Sequence of genomic DNA having lower accuracy than finished sequence; some segments are missing or in the wrong order or orientation |
| Emulsion PCR | A PCR technique that is conducted on a bead surface within tiny water bubbles floating on an oil solution. It is used by IonTorrent platforms. |
| Error rate | The per-read error rate is defined as the proportion of reads containing sequencing errors |
| Flow cell | A glass slide containing small fluidic channels, through which polymerases, nucleotides and buffers can be pumped during sequencing |
| High quality SNP | A single nucleotide polymorphism that has been verified using specific criteria such as: sequence coverage, sequence quality, and population and allelic frequency |
| Homopolymer | A DNA sequence (2 or more base pairs) consisting of the same nucleotide |
| Index (barcode) | Unique individual DNA sequences added to each sample so they can be distinguished and sorted during data analysis. Enables sequencing multiple samples per instrument run. |
| Massively parallel sequencing | High throughput DNA sequencing approaches that use the concept of miniaturized massive parallel processing to sequence 1 million to 43 billion short reads per instrument run |
| Metagenomics | The study of genetic material recovered directly from the primary samples |
| Paired-end reading | Sequencer starts reading DNA fragment at one end, finishes this direction at the specified read length, and then starts another round of reading from the opposite end of the fragment |
| Per-base sequence quality (accuracy) | The sequence quality score for each individual base position in a sequence. Typically, phred scores are used, where Q = −10log (Error Probability). A Q30, for example, means a 1 in 1000 likelihood of an incorrect base call at that position. |
| Pyrosequencing | Sequencing is performed by detecting the nucleotide incorporated using enzymatic reactions after which the substrate emits light |
| Read | A unit of continuous DNA sequence derived from target DNA |
| Reversibly-blocked terminator | A molecule added to a nucleotide to prevent addition of multiple nucleotides per sequencing cycle. Used by Illumina platforms. |
| Sanger sequencing | A low throughput sequencing method based on the selective incorporation of chain-terminating dideoxynucleotides by DNA polymerase during in vitro DNA replication |
| Semiconductor sequencing | Sequencing is performed by detection of hydrogen ions that are released during incorporation of the nucleotide. Used by IonTorrent platforms. |
| Sequencing by synthesis | Sequencing is performed by detecting the nucleotide incorporated by a DNA polymerase |
| Single-end reading | The sequencer reads a DNA fragment from only one end to the other, generating the sequence of base pairs |
| wgMLST | Whole genome multi-locus sequence typing – an analysis method that detects variation in all genes (core and accessory genes) of a given genome |
The short read technologies currently in use are collectively known as massively parallel sequencing and are often also referred to as second generation sequencing (23). They produce billions of nucleotide sequences during each run, where each genome is sequenced multiple times in small random pieces to generate very large datasets. Even though platforms have different biochemistry and arrays, the workflows include similar steps: i) DNA extraction; ii) library preparation, which usually includes shearing the DNA either mechanically or enzymatically, adding adaptors and barcodes/indexes, and amplification; iii) template preparation, either by bridge amplification or emulsion PCR; and iv) automated sequencing (Figure 2)(22).
Figure 2.
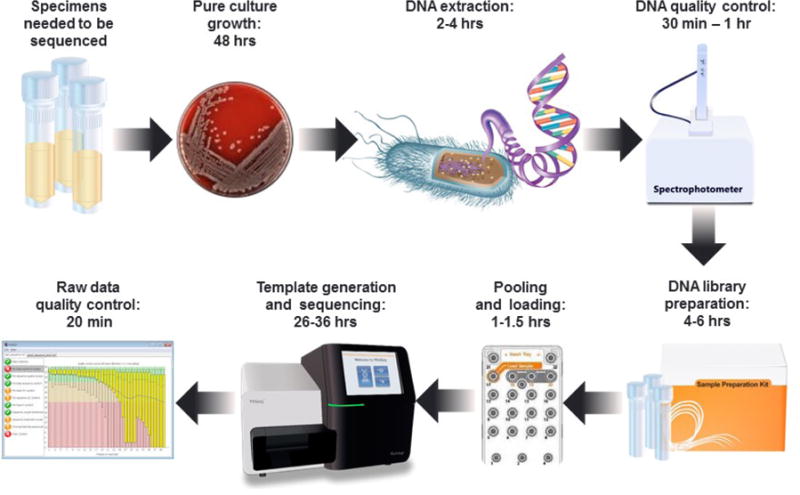
Typical WGS workflow in a clinical or public health laboratory
The short read sequencing platforms differ substantially in terms of their engineering, sequencing chemistry, output (length of reads, number of sequences), accuracy and cost (Table 2) (20). The Illumina platform, which currently occupies a vast part of the NGS market, is based on sequencing by synthesis of the complementary strand and fluorescence-based detection of reversibly-blocked terminator nucleotides (24). The platform includes multiple instruments with varying throughput and read length (Table 2).
Table 2.
Characteristics, strengths and weaknesses of commonly used sequencing platforms
| Platform \ Instrument | Throughput range (Gb)a | Read length (bp) | Strength | Weakness |
|---|---|---|---|---|
| Sanger sequencing | ||||
| ABI 3500/3730 | 0.0003 | Up to 1kb | Read accuracy and length | Cost and throughput |
| Illumina | ||||
| MiniSeq | 1.7–7.5 | 1×75 to 2×150 | Low initial investment | Run and read length |
| MiSeq | 0.3–15 | 1×36 to 2×300 | Read length, scalability | Run length |
| NextSeq | 10–120 | 1×75 to 2×150 | Throughput | Run and read length |
| HiSeq (2500) | 10–1000 | 1×50 to 2×250 | Read accuracy, throughput, low per sample cost | High initial investment, run length |
| NovaSeq 5000/6000 | 2000–6000 | 2×50 to 2×150 | Read accuracy, throughput Low per sample cost |
High initial investment, run and read length |
| IonTorrent | ||||
| PGM | 0.08–2 | Up to 400 | Read length, speed | Throughput, homopolymersc |
| S5 | 0.6–15 | Up to 400 | Read length, speed, scalability | Homopolymersc |
| Proton | 10–15 | Up to 200 | Speed, throughput | Homopolymersc |
| Pacific BioSciences | ||||
| PacBio RSII | 0.5–1b | Up to 60 kb (Average 10 kb, N50 20 kb) |
Read length, speed | High error rate and initial investment, low throughput |
| Sequel | 5–10b | Up to 60 kb (Average 10 kb, N50 20 k) |
Read length, speed | High error rate |
| Oxford Nanopore | ||||
| MInION | 0.1–1 | Up to 100 kb | Read length, portability | High error rate, run length, low throughput |
The throughput ranges are determined by available kits and run modes on a per run basis. As an example of a 15 GB throughput, thirty-five 5 MB genomes can be sequenced to a minimum coverage of 40x on the Illumina MiSeq using the v3 600 cycle chemistry.
Per one SMRTcell
Results in increased error rate (increased proportion of reads containing errors among all reads) which in turn results in false positive variant calling
The MiniSeq and MiSeq instruments offer low to mid sample throughput, affordable instrument pricing, user-friendly workflow with no need for automation and reasonable per sample cost ($120 per 5 MB genome) and are hence an attractive choice for diagnostic and public health laboratories. The considerably more expensive NextSeq, HiSeq and NovaSeq instruments are designed for much higher throughput reducing the per sample cost, but require additional automation for library preparation and are therefore more appropriate for sequencing core facilities. Since the best cost efficiency can only be achieved by always making full capacity sequencing runs, the choice of the appropriate sequencing platform that is in tune with laboratory’s sample throughput is critical for real-time testing. For example, a single MiSeq instrument and sequencing with the v3 chemistry would cover real-time testing in a laboratory that annually processes 4000 isolates. Of all the available NGS technologies, the Illumina data, in particular data generated on the HiSeq, generates the highest quality base calls. The IonTorrent platform from Thermo Fisher is also based on sequencing by synthesis but the detection is based on solid state pH meters measuring hydrogen ions released during DNA polymerization (semiconductor technology) (25). The PGM and S5 instruments are the IonTorrent equivalents for the Illumina MiniSeq and MiSeq; the Ion Proton is equivalent of Illumina NextSeq (Table 2). The instrument pricing is similar though the per sample cost is higher for the IonTorrent platform. Compared to the Illumina platform, the library prep process is lengthier for the IonTorrent though parts of it can be automated with the use of the IonChef sample preparation system. The IonTorrent sequencing run itself is much shorter (hours vs. days) compared to the Illumina instruments. The PGM and S5 reads are about 100 bp longer than the longest Illumina reads though only single-end reads can be generated. Because of the nature of the semiconductor sequencing, the IonTorrent platform has high error rate in base calls in long homopolymer regions. The short read platforms are commonly used for generating bacterial draft genomes for variant calling for diagnostics and infectious disease surveillance purposes. However, the lower throughput instruments are also well suited for targeted amplicon sequencing, such as detecting antimicrobial resistance determinants or 16S sequencing. Other applications include RNA sequencing to study the expression of genes and metagenomics sequencing (21).
Since the short reads from the second generation sequencing platforms tend to generate relatively fragmented genome assemblies, longer reads are desired in order to generate closed reference genomes. Longer reads are particularly useful when sequencing through complex genomic regions such as repeats and phages. Some phages can be up to 50–75 kB in length. To meet this demand, the so-called third generation sequencing platforms have been introduced (23). These technologies directly target single DNA molecules without the need for PCR amplification. The PacBio RSII platform, marketed by Pacific Biosciences, uses single molecule real-time (SMRT) sequencing technology (26). As with short read technologies, sequencing is based on synthesis utilizing nucleotides labeled with distinct fluorescent dyes but sequencing proceeds when single stranded DNA molecules are deposited into tiny wells where a single DNA polymerase molecule is immobilized. Extremely long DNA fragments of 20 kb and even longer can be obtained with run times of only few hours. However, the run throughput is low and the per sample cost high which coupled with a high instrument cost and large footprint (Table 2) render the PacBio platform more suitable for sequencing core facilities that wish to generate high quality finished genomes. The platform, as with other long-read technology, has a higher inherent error profile (11–15%) than short read technologies, necessitating a subsequent assembly algorithm that relies on error correction(27). The error rate can be reduced by increasing the number of subreads generated. However, the number of sequencing passes and the read length are a trade-off, i.e. the higher the coverage, the shorter the reads. Pacific Biosciences recently released a higher throughput and more affordable version of RSII called Sequel. The MinION device developed by the Oxford Nanopore Technologies is another long read sequencing platform that currently is not as mature as the PacBio platform. With the size of a large thumb drive, it is the smallest sequencing device available. It can plug in to a standard USB-3 port on a computer with low hardware requirement and simple configuration. MinION is the first commercial sequencing platform utilizing Nanopore technology (28). It identifies DNA bases by measuring changes in electric conductivity generated as DNA strands pass through a biological pore. The read length profile of MinION is very similar to that of PacBio but the error rate is even higher (12–38 %) though it has been improving with recent advances in chemistry (29). The current version of MinION has difficulty in sequencing GC rich regions. Unlike PacBio, the error rate cannot be improved by increasing coverage since the MinION is limited to two sequencing passes by design. Similar to PacBio, complex assembly and error correction algorithms need to be employed in order to produce high quality assemblies (29, 30). The current throughput is low and not very stable and the default run time is 48 h though data can be analyzed in real-time as the reads pass through the sequencer. The portability and the real-time sequence analysis aspect make the MinION platform an attractive option for field diagnostics applications (31). Oxford Nanopore Technologies recently announced the availability of its new higher throughput platform PromethION through an early access program. There are 48 flow cells incorporated making it equivalent to 48 MiniIONs (29).
Analytical Approaches
One of the basic assumptions in molecular epidemiology is that phylogeny approximates epidemiology, i.e. patients are more likely to be epidemiologically associated if the pathogens that made them ill are closely related phylogenetically than if they are not. Similarly, if pathogens from food or the environment are phylogenetically related to clinical isolates a causal relationship between the two is likely. The generation of reproducible, phylogenetically meaningful data through sequence analysis is central to the practical application of sequence data for outbreak detection and investigation. However, like any other subtyping or strain characterization method, this correlation between epidemiology and phylogeny is incomplete and sequencing data cannot stand on their own but should always be interpreted taking all available epidemiological and other supporting information into consideration.
A thorough description of bioinformatics and bioinformatic tools is the subject of another review in this series. For subtyping, two main high discriminatory phylogenetic approaches are used: high quality single nucleotide polymorphisms (hqSNP), and core genome/whole genome multilocus sequence typing (cg/wgMLST). A short introduction to the use of these approaches is given here. The hqSNP approach compares single nucleotide differences between isolates in comparison to one reference genome. This reference genome must be closely related to the isolates in the comparison to identify true phylogenetically informative SNPs for comparison. Results generated using different references and pipelines cannot directly be compared. Since the selection of the reference genome is difficult to standardize because almost all outbreaks need their own unique reference strain and there is no consensus about which of the many applications (pipelines) for hqSNP to use this method is not well suited for outbreak investigations performed at the same time in more laboratories. However, it is excellent for centralized analysis providing similar or better resolution between strains than wgMLST (9, 32, 33).
The cg/wg MLST approach is more universal. Whole genome (wg) MLST assesses the diversity of theoretically all genes in a particular genus or species (2). Core genome (cg) MLST assesses all the genes universally found in a particular genus or species (34, 35). New isolates are compared against a database of all known gene variants (an allele database) which may be used to assess the relatedness of all isolates belonging to the species it was developed for. An increasing number of such databases is being developed with some available in public domain (34–37) making the MLST approach uniquely suitable for multi laboratory outbreak investigations like the ones performed by the PulseNet networks (9).
In addition to subtyping, one consolidated WGS workflow could replace traditional characterization of pathogens, which require specialized skills and knowledge about each pathogen, expensive reagents, and complicated workflows with extensive quality control procedures. For example, current methods for full characterization of Shiga toxin-producing Escherichia coli (STEC) at the Centers for Disease Control and Prevention includes a number of phenotypic tests for species identification, ten PCR assays for virulence profiling, broth microdilution assays for antimicrobial susceptibility testing and agglutination assays with 270 pooled and individual O- and H-specific antisera for serotyping. The turn-around-time for this characterization is routinely between one and three weeks. A WGS workflow can provide more detailed information than traditional methods in a matter of days. WGS workflows have also been used to successfully identify and characterize strains in clinical settings (11, 37).
The availability of analysis tools on the web that a microbiologist without extensive bioinformatic training can use for pathogen characterization has accelerated the characterization process. Examples of such tools for in silico analysis of bacterial WGS may be found on the Center for Genomic Epidemiology (CGE) web-site (www.genomicepidemiology.org) (38–40). Among others, CGE provides virulence, resistance and serotype prediction tools (VirulenceFinder, ResFinder and SerotypeFinder) that can be used for analysis of Escherichia coli and other pathogenic bacteria (41–43). The CGE website also includes a pipeline in which multiple analyses can be performed on multiple batched sequences.
Since the 1960s DNA–DNA hybridization (DDH) has been the gold standard for species identification. In 1987, Wayne, et al. set a standard DDH similarity of 70% or greater for identification of strains to the same species (44). A WGS counterpart to DDH is Average Nucleotide Identity (ANI). This method assesses the nucleotide identity between genetic regions shared by two isolates and can unambiguously identify the species of a given isolate from its WGS. ANI values of approximately 95% correspond to 60–70% DDH values (45, 46). Twenty-eight genomic sequences from Bacillus, Burkholderia, Escherichia/Shigella, Pseudomonas, Shewanella and Streptococcus were analyzed and all showed 95% or greater ANI values when compared to isolates of the same species (45). An ANI algorithm (ANIm) can identify the species of an isolate in a matter of seconds to minutes. ANIm is run using MUMmer software which includes a rapid whole genome aligner and may use multiple reference genomes as a reference database allowing a quick comparison of the query sequence (47).
Future needs and developments
The sequencing technology, bioinformatics and the informatics infrastructure are all rapidly evolving. Therefore, a number of changes are expected to happen within the next few years that greatly will affect the implementation of sequencing in routine clinical and public health microbiology.
It is a challenge that the comparability of the sequence data generated on different platforms with different error profiles using different library preparation methods has still not been comprehensively assessed and validated. Only a handful of studies with a limited number of species, isolates and analysis methods have been performed (48–50). Systematic studies that include strain identification, characterization and subtyping by both hqSNP and cg/wgMLST are badly needed. Internationally agreed standards for sequence and analysis quality, and data interpretation for clinical, public health and regulatory action are currently missing for most applications and must be developed.
On the positive side, the cost of sequencing, data transfer and analysis will continue to decrease and the DNA purification including library preparation and the actual sequencing will become faster and more efficient. The Achilles heel of the current long read technologies, the high error rates, will continue to improve until they become as precise as the short-read technologies at which time the latter will become obsolete. The routine closure of genomes by the long read technologies will facilitate backwards compatibility with current gold standard molecular subtyping methods like multilocus variable number of tandem repeats (MLVA) and PFGE ensuring that the historic information generated by these methods is not lost.
Automation is critical for the routine use of the technology in clinical microbiology. Currently, the sequencing procedures are increasingly being automated in large clinical and public health laboratories. Standardized data handling and analysis is also being developed as on-line (www.genomicepidemiology.org; www.ncbi.nlm.nih.gov/pathogens/) or stand-alone applications (9). The next step in the NGS evolution will be to integrate the two, sequencing and data analysis, in one efficient workflow. The user will load the bacterial culture/specimen into the DNA preparation module of the sequencer with no manual interaction until a standardized report customized to each laboratory’s needs has been generated and stored in a Laboratory Information Management System (LIMS) database. The report will include actionable strain characterization data for the clinicians, and subtyping information for public health. This change will revolutionize infection control and public health through providing data in real-time to detect clonal spread of pathogens or plasmids in hospitals, and outbreaks in hospitals or in the outside community.
In the longer term, the improved long read technology combined with more efficient IT capacity, nucleic acid enrichment technologies, and bioinformatics will enable routine implementation of metagenomics sequencing of specimens for non-culture clinical diagnostics, true real-time infection control and public health surveillance.
Footnotes
Disclaimers:
The findings and conclusions in this presentation are those of the author and do not necessarily represent the official position of the Centers for Disease Control and Prevention
Use of trade names is for identification only and does not imply endorsement by the Centers for Disease Control and Prevention or by the U.S. Department of Health and Human Services.
Author contribution:
All authors contributed equally to the conception and writing of this review
Transparency declaration:
No conflicts of interest declared. No external funding was received to write this review.
References
- 1.Sanger F, Nicklen S, Coulson AR. DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci U S A. 1977;74(12):5463–7. doi: 10.1073/pnas.74.12.5463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Maiden MC, Bygraves JA, Feil E, Morelli G, Russell JE, Urwin R, et al. Multilocus sequence typing: a portable approach to the identification of clones within populations of pathogenic microorganisms. Proc Natl Acad Sci U S A. 1998;95(6):3140–5. doi: 10.1073/pnas.95.6.3140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fleischmann RD, Adams MD, White O, Clayton RA, Kirkness EF, Kerlavage AR, et al. Whole-genome random sequencing and assembly of Haemophilus influenzae Rd. Science. 1995;269(5223):496–512. doi: 10.1126/science.7542800. [DOI] [PubMed] [Google Scholar]
- 4.Lander ES. Initial impact of the sequencing of the human genome. Nature. 2011;470(7333):187–97. doi: 10.1038/nature09792. [DOI] [PubMed] [Google Scholar]
- 5.Tripp S, Grueber M. Economic impact of the human genome project. Battelle Memorial Institute; 2011. www.battelle.org: [Google Scholar]
- 6.Barzilay EJ, Schaad N, Magloire R, Mung KS, Boncy J, Dahourou GA, et al. Cholera surveillance during the Haiti epidemic–the first 2 years. N Engl J Med. 2013;368(7):599–609. doi: 10.1056/NEJMoa1204927. [DOI] [PubMed] [Google Scholar]
- 7.King LA, Nogareda F, Weill FX, Mariani-Kurkdjian P, Loukiadis E, Gault G, et al. Outbreak of Shiga toxin-producing Escherichia coli O104:H4 associated with organic fenugreek sprouts, France, June 2011. Clinical infectious diseases : an official publication of the Infectious Diseases Society of America. 2012;54(11):1588–94. doi: 10.1093/cid/cis255. [DOI] [PubMed] [Google Scholar]
- 8.Mellmann A, Harmsen D, Cummings CA, Zentz EB, Leopold SR, Rico A, et al. Prospective genomic characterization of the German enterohemorrhagic Escherichia coli O104:H4 outbreak by rapid next generation sequencing technology. PloS One. 2011;6(7):e22751. doi: 10.1371/journal.pone.0022751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nadon C, Van Walle I, Chinen I, Campos J, Trees E, Gilpin B, et al. PulseNet International vision for the implementation of whole genome sequencing for global foodborne disease surveillance. Eurosurveillance. 2017;22(23) doi: 10.2807/1560-7917.ES.2017.22.23.30544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Struelens M. Rapid Microbial NGS and Bioinformatics: Translation into Practice. Hamburg: Jun 9–11, 2016. ECDC Roadmap for integration of molecular and genomic typing into European level surveillance, 2016’19. URL: http://rami-ngs.org/fileadmin/rami-ngs/downloads/talks/ECDC-roadmap-for-integration-of-molecular-and-genomic-typing-into-European-level-surveillance-2016-19.pdf. [Google Scholar]
- 11.Lewis T, Loman NJ, Bingle L, Jumaa P, Weinstock GM, Mortiboy D, et al. High-throughput whole-genome sequencing to dissect the epidemiology of Acinetobacter baumannii isolates from a hospital outbreak. J Hosp Infect. 2010;75(1):37–41. doi: 10.1016/j.jhin.2010.01.012. [DOI] [PubMed] [Google Scholar]
- 12.Harris SR, Feil EJ, Holden MT, Quail MA, Nickerson EK, Chantratita N, et al. Evolution of MRSA during hospital transmission and intercontinental spread. Science. 2010;327 doi: 10.1126/science.1182395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Snitkin ES, Zelazny AM, Thomas PJ, Stock F, Henderson DK, Palmore TN, et al. Tracking a hospital outbreak of carbapenem-resistant Klebsiella pneumoniae with whole-genome sequencing. Sci Transl Med. 2012;4(148):148ra116. doi: 10.1126/scitranslmed.3004129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Caliendo AM, Gilbert DN, Ginocchio CC, Hanson KE, May L, Quinn TC, et al. Better tests, better care: improved diagnostics for infectious diseases. Clin Infect Dis. 2013;57(Suppl 3):S139–70. doi: 10.1093/cid/cit578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.2007–2015 WFDBERG. WHO estimates of the global burden of foodborne diseases. Geneva, Switzerland: 2015. Report No.: [Google Scholar]
- 16.Scharff RL, Besser J, Sharp DJ, Jones TF, Peter GS, Hedberg CW. An economic evaluation of PulseNet: a network for foodborne disease surveillance. Am J Prev Med. 2016;50(5 Suppl 1):S66–73. doi: 10.1016/j.amepre.2015.09.018. [DOI] [PubMed] [Google Scholar]
- 17.Jackson BR, Tarr C, Strain E, Jackson KA, Conrad A, Carleton H, et al. Implementation of nationwide real-time whole-genome sequencing to enhance listeriosis outbreak detection and investigation. Clin Infect Dis. 2016;63(3):380–6. doi: 10.1093/cid/ciw242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Moura A, Tourdjman M, Leclercq A, Hamelin E, Laurent E, Fredriksen N, et al. Real-time whole-genome sequencing for surveillance of Listeria monocytogenes, France. Emerg Infect Dis. 2017;23(9):1462–70. doi: 10.3201/eid2309.170336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Margulies M, Egholm M, Altman WE, Attiya S, Bader JS, Bemben LA, et al. Genome sequencing in microfabricated high-density picolitre reactors. Nature. 2005;437(7057):376–80. doi: 10.1038/nature03959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Buermans HPJ, den Dunnen JT. Next generation sequencing technology: Advances and applications. BBA Molecular Basis of Disease. 2014;1842(10):1932–41. doi: 10.1016/j.bbadis.2014.06.015. [DOI] [PubMed] [Google Scholar]
- 21.van Dijk EL, Auger H, Jaszczyszyn Y, Thermes C. Ten years of next-generation sequencing technology. Trends Genet. 2014;30(9):418–26. doi: 10.1016/j.tig.2014.07.001. [DOI] [PubMed] [Google Scholar]
- 22.Vincent AT, Derome N, Boyle B, Culley AI, Charette SJ. Next-generation sequencing (NGS) in the microbiological world: How to make the most of your money. J Microbiol Methods. 2017;138:60–71. doi: 10.1016/j.mimet.2016.02.016. [DOI] [PubMed] [Google Scholar]
- 23.Schadt EE, Turner S, Kasarskis A. A window into third-generation sequencing. Hum Mol Gen. 2010;19(R2):R227–R40. doi: 10.1093/hmg/ddq416. [DOI] [PubMed] [Google Scholar]
- 24.Bentley DR, Balasubramanian S, Swerdlow HP, Smith GP, Milton J, Brown CG, et al. Accurate whole human Ggenome sequencing using reversible terminator chemistry. Nature. 2008;456(7218):53–9. doi: 10.1038/nature07517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Rothberg J, Myers J. Semiconductor sequencing for life. J Biomol Tech. 2011;22(Suppl):S41–S2. [Google Scholar]
- 26.Rhoads A, Au KF. PacBio Sequencing and its applications. Genomics Proteomics Bioinformatics. 2015;13(5):278–89. doi: 10.1016/j.gpb.2015.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chin C-S, Alexander DH, Marks P, Klammer AA, Drake J, Heiner C, et al. Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat Meth. 2013;10(6):563–9. doi: 10.1038/nmeth.2474. [DOI] [PubMed] [Google Scholar]
- 28.Stoddart D, Heron AJ, Mikhailova E, Maglia G, Bayley H. Single-nucleotide discrimination in immobilized DNA oligonucleotides with a biological nanopore. Proc Natl Acad Sci U S A. 2009;106(19):7702–7. doi: 10.1073/pnas.0901054106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lu H, Giordano F, Ning Z. Oxford Nanopore MinION Sequencing and Genome Assembly. Genomics proteomics bioinformatics. 2016;14(5):265–79. doi: 10.1016/j.gpb.2016.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Loman NJ, Quick J, Simpson JT. A complete bacterial genome assembled de novo using only nanopore sequencing data. Nat Meth. 2015;12(8):733–5. doi: 10.1038/nmeth.3444. [DOI] [PubMed] [Google Scholar]
- 31.Quick J, Loman NJ, Duraffour S, Simpson JT, Severi E, Cowley L, et al. Real-time, portable genome sequencing for Ebola surveillance. Nature. 2016;530(7589):228–32. doi: 10.1038/nature16996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Katz LS, Griswold T, Williams-Newkirk AJ, Wagner D, Petkau A, Sieffert C, et al. A comparative analysis of the Lyve-SET phylogenomics pipeline for genomic epidemiology of foodborne pathogens. Front Microbiol. 2017;8:375. doi: 10.3389/fmicb.2017.00375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Carleton H, Gerner-Smidt P. Whole-genome sequencing is taking over foodborne disease surveillance. Microbe. 2016;11(7):311–317. [Google Scholar]
- 34.Meric G, Yahara K, Mageiros L, Pascoe B, Maiden MC, Jolley KA, et al. A reference pan-genome approach to comparative bacterial genomics: identification of novel epidemiological markers in pathogenic Campylobacter. PloS One. 2014;9(3):e92798. doi: 10.1371/journal.pone.0092798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Moura A, Criscuolo A, Pouseele H, Maury MM, Leclercq A, Tarr C, et al. Whole genome-based population biology and epidemiological surveillance of Listeria monocytogenes. Nat Microbiol. 2016;2(3):16185. doi: 10.1038/nmicrobiol.2016.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kingry LC, Rowe LA, Respicio-Kingry LB, Beard CB, Schriefer ME, Petersen JM. Whole genome multilocus sequence typing as an epidemiologic tool for Yersinia pestis. Diagn Microbiol Infect Dis. 2016;84(4):275–80. doi: 10.1016/j.diagmicrobio.2015.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kluytmans-van den Bergh MF, Rossen JW, Bruijning-Verhagen PC, Bonten MJ, Friedrich AW, Vandenbroucke-Grauls CM, et al. Whole-genome multilocus sequence typing of extended-spectrum-beta-lactamase-producing Enterobacteriaceae. J Clin Microbiol. 2016;54(12):2919–27. doi: 10.1128/JCM.01648-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cosentino S, Voldby Larsen M, Moller Aarestrup F, Lund O. PathogenFinder–distinguishing friend from foe using bacterial whole genome sequence data. PloS One. 2013;8(10):e77302. doi: 10.1371/journal.pone.0077302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Joensen KG, Scheutz F, Lund O, Hasman H, Kaas RS, Nielsen EM, et al. Real-time whole-genome sequencing for routine typing, surveillance, and outbreak detection of verotoxigenic Escherichia coli. J Clin Microbiol. 2014;52(5):1501–10. doi: 10.1128/JCM.03617-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Thomsen MC, Ahrenfeldt J, Cisneros JL, Jurtz V, Larsen MV, Hasman H, et al. A bacterial analysis platform: an integrated system for analysing bacterial whole genome sequencing data for clinical diagnostics and surveillance. PloS One. 2016;11(6):e0157718. doi: 10.1371/journal.pone.0157718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Joensen KG, Tetzschner AM, Iguchi A, Aarestrup FM, Scheutz F. Rapid and easy in silico serotyping of Escherichia coli isolates by use of whole-genome sequencing data. J Clin Microbiol. 2015;53(8):2410–26. doi: 10.1128/JCM.00008-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kleinheinz KA, Joensen KG, Larsen MV. Applying the ResFinder and VirulenceFinder web-services for easy identification of acquired antibiotic resistance and virulence genes in bacteriophage and prophage nucleotide sequences. Bacteriophage. 2014;4(1):e27943. doi: 10.4161/bact.27943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zankari E, Hasman H, Kaas RS, Seyfarth AM, Agerso Y, Lund O, et al. Genotyping using whole-genome sequencing is a realistic alternative to surveillance based on phenotypic antimicrobial susceptibility testing. J Antimicrob Chemother. 2013;68(4):771–7. doi: 10.1093/jac/dks496. [DOI] [PubMed] [Google Scholar]
- 44.Wayne LG. International Committee on Systematic Bacteriology: announcement of the report of the ad hoc committee on reconciliation of approaches to bacterial systematics. Zentralbl Bakteriol Mikrobiol Hyg A. 1988;268(4):433–4. doi: 10.1016/s0176-6724(88)80120-2. [DOI] [PubMed] [Google Scholar]
- 45.Goris J, Konstantinidis KT, Klappenbach JA, Coenye T, Vandamme P, Tiedje JM. DNA-DNA hybridization values and their relationship to whole-genome sequence similarities. Int J Syst Evol Microbiol. 2007;57(Pt 1):81–91. doi: 10.1099/ijs.0.64483-0. [DOI] [PubMed] [Google Scholar]
- 46.Richter M, Rossello-Mora R. Shifting the genomic gold standard for the prokaryotic species definition. Proc Natl Acad Sci U S A. 2009;106(45):19126–31. doi: 10.1073/pnas.0906412106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kurtz S, Phillippy A, Delcher AL, Smoot M, Shumway M, Antonescu C, et al. Versatile and open software for comparing large genomes. Genome Biol. 2004;5(2):R12. doi: 10.1186/gb-2004-5-2-r12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Harris SR, Torok ME, Cartwright EJ, Quail MA, Peacock SJ, Parkhill J. Read and assembly metrics inconsequential for clinical utility of whole-genome sequencing in mapping outbreaks. Nature Biotechnol. 2013;31(7):592–4. doi: 10.1038/nbt.2616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Powers JG, Weigman VJ, Shu J, Pufky JM, Cox D, Hurban P. Efficient and accurate whole genome assembly and methylome profiling of E. coli. BMC Genomics. 2013;14:675. doi: 10.1186/1471-2164-14-675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Quail MA, Smith M, Coupland P, Otto TD, Harris SR, Connor TR, et al. A tale of three next generation sequencing platforms: comparison of Ion Torrent, Pacific Biosciences and Illumina MiSeq sequencers. BMC Genomics. 2012;13:341. doi: 10.1186/1471-2164-13-341. [DOI] [PMC free article] [PubMed] [Google Scholar]