

Abstract
Addiction is proposed to arise from alterations in synaptic strength via mechanisms of long-term potentiation (LTP) and depression (LTD). However, the causality between these synaptic processes and addictive behaviors is difficult to demonstrate. Here we report that LTP/LTD induction altered operant alcohol self-administration, a motivated drug-seeking behavior. We first induced LTP by pairing presynaptic glutamatergic stimulation with optogenetic postsynaptic depolarization in the dorsomedial striatum, a brain region known to control goal-directed behavior. Blockade of this LTP by NMDA receptor inhibition unmasked an endocannabinoid-dependent LTD. In vivo application of the LTP-inducing protocol caused a long-lasting increase in alcohol-seeking behavior, while the LTD protocol decreased this behavior. We further identified that optogenetic LTP/LTD induction at cortical inputs onto striatal dopamine D1 receptor-expressing neurons controlled these behavioral changes. Our results demonstrate a causal link between synaptic plasticity and alcohol-seeking behavior, and that modulation of this plasticity may inspire a therapeutic strategy for addiction.
Drug addiction is a mental illness that is viewed as a transition from recreational use to compulsive drug-seeking and -taking1–3. This behavioral transition is proposed to be controlled by “drug-evoked plasticity”1–4. However, exactly how synaptic plasticity controls the adaptive changes in drug-seeking behavior remains unclear. The dorsomedial striatum (DMS), a brain region crucially involved in drug and alcohol addiction, receives glutamatergic inputs from several brain areas2, 5–7. In these neural circuits, the medial prefrontal cortex (mPFC) afferent into the DMS is essential for the control of goal-directed behaviors2, 5–7, and this connection is linked to drug or alcohol addiction2, 5, 8. For example, exposure to drugs of abuse or alcohol potentiates AMPA receptor (AMPAR)- and NMDA receptor (NMDAR)-mediated glutamatergic transmission in the DMS8–11, while pharmacological inhibition of striatal glutamatergic transmission transiently suppresses operant alcohol self-administration and cocaine relapse9, 10, 12, 13. Although these studies indicate that drugs and alcohol evoke corticostriatal plasticity, which may, in turn, contribute to drug-seeking and -taking behaviors, there has been no direct demonstration that synaptic plasticity drives addictive behaviors.
The DMS contains two types of medium spiny neurons (MSNs): D1-MSNs express dopamine D1 receptors (D1Rs) and D2-MSNs contain D2Rs14. Both neuronal types receive mPFC inputs12. While synaptic plasticity, including long-term potentiation (LTP) and depression (LTD), was observed in both D1- and D2-MSNs15, drug- or alcohol-induced plasticity was found predominantly in striatal D1-MSNs12, 16, 17. Mimicking alcohol-evoked plasticity by inducing LTP, or reversing this plasticity by inducing LTD, will provide a new understanding of how this plasticity controls alcohol-seeking behavior. LTP/LTD induction at specific neuronal circuits requires simultaneous control of both pre- and post-synaptic neurons, which can be achieved using a recently developed dual-channel optogenetic technique18, 19.
In this study, we paired optogenetic postsynaptic depolarization (oPSD) with presynaptic glutamatergic stimulation; this greatly enhanced NMDAR-mediated transmission and induced a reliable NMDAR-dependent LTP, as well as an endocannabinoid (eCB)-dependent LTD. Importantly, in vivo optogenetic delivery of the LTP protocol to the corticostriatal synapses within the DMS produced a long-lasting increase in operant self-administration of alcohol. Conversely, delivery of the LTD protocol led to a long-lasting decrease in this behavior. Furthermore, we discovered that the in vivo LTP and LTD protocols preferentially induced plasticity in D1-MSNs and that selective induction of LTP/LTD in this neuronal type produced the corresponding changes in alcohol-seeking behavior. These findings demonstrate a causal link between DMS corticostriatal synaptic plasticity and alcohol-seeking behavior and indicate that the reversal of drug-evoked synaptic plasticity may provide a novel therapeutic strategy for the treatment of alcohol use disorder.
RESULTS
oPSD facilitates induction of NMDAR-dependent LTP and eCB-LTD in the DMS
In dorsostriatal slices, LTD is the easiest form of synaptic plasticity to observe, while LTP is more difficult to detect6, 20. A D2R antagonist was thus included in the recording solution in order to prevent LTD15 and favor the induction of LTP. Field excitatory postsynaptic potentials/population spikes (fEPSP/PS) were evoked by electrical stimulation within the DMS (Fig. 1a), but these were not potentiated by electrical high-frequency stimulation (eHFS) (Fig. 1b and Supplementary Fig. 1a). This observation is consistent with previous reports6, 21. Previous studies also suggested that sufficient postsynaptic depolarization was necessary for reliable LTP induction20. Therefore, we examined whether postsynaptic depolarization by somatic current injection (iPSD) facilitated LTP induction. Using whole-cell recording, we discovered that paired eHFS and iPSD produced little LTP (Supplementary Fig. 1b). This is consistent with the notion that current injection-elicited action potentials do not back-propagate to the distal dendrites of striatal neurons20; insufficient depolarization of this region means that no LTP is generated. In contrast, optogenetics can be used to depolarize any process of postsynaptic neurons, with no limitation of their distance to the soma, and in a non-invasive manner22. We observed that optogenetic postsynaptic depolarization (oPSD) induced a higher distal dendritic calcium transient than iPSD (Supplementary Fig. 2), suggesting that oPSD produced more effective depolarization of this region.
Figure 1.
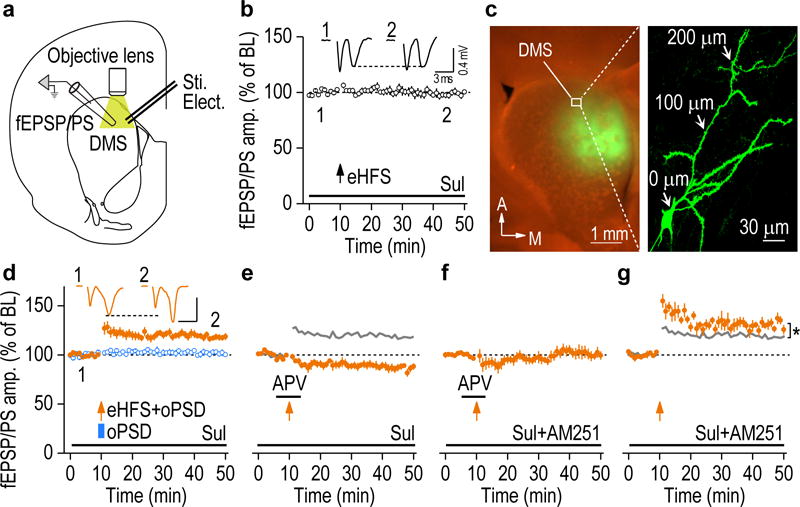
oPSD facilitated induction of NMDAR-dependent LTP and eCB-LTD in DMS slices. (a) Schematic showing the bipolar stimulating electrodes (Sti. Elect.) used to evoke fEPSP/PS and the objective lens for optogenetic depolarization. (b) Presynaptic eHFS did not potentiate fEPSP/PS (98.59 ± 2.39% of baseline [BL], t(7) = 0.59, P = 0.57; n = 8 slices, 6 rats). Inset, sample fEPSP/PS traces at time 1 and 2. Sulpiride (Sul; 20 μM) was bath-applied to prevent LTD and favor LTP induction in this and following recordings as indicated. (c) Representative fluorescent images showing C1V1-eYFP expression in the DMS (left) and in the full-length dendrites of a DMS neuron (right). The section was counter-stained with NeuroTrace (red). (d) Pairing of presynaptic eHFS and oPSD (1 sec), but not oPSD alone, induced robust LTP. eHFS+oPSD: 118.32 ± 2.96% of BL, t(9) = −6.18, P = 0.00016; n = 10 slices, 6 rats; oPSD: 101.59 ± 2.26% of BL, t(6) = −0.70, P = 0.51; n = 7 slices, 3 rats. Scale bars: 3 ms, 0.4 mV. (e) Optogenetic induction of LTP was abolished by APV (50 μM), leading to LTD (86.75 ± 3.06% of BL, t(6) = 4.33, P = 0.0049; n = 7 slices, 6 rats). The grey line is the control LTP from d for reference. (f) LTD was completely abolished by the CB1R antagonist, AM251 (3 μM) (101.32 ± 4.71% of BL, t(8) = −0.28, P = 0.79, n = 9 slices, 5 rats). (g) AM251 facilitated LTP induction (130.73 ± 2.89% of BL, t(5) = −10.62, P = 0.00013; compared with the control LTP: t(14) = −2.79, *P = 0.015; n = 6 slices, 3 rats, unpaired t test). Two-sided paired t test for b and d-g, unless otherwise stated. Data are presented as mean ± s.e.m.
Next, we assessed whether oPSD facilitates LTP induction. An adeno-associated virus (AAV) expressing a channelrhodopsin, C1V123, was infused into the DMS, resulting in C1V1 expression in the soma and distal dendrites (Fig. 1c). We found that pairing of eHFS with oPSD of DMS neurons induced a robust and reliable LTP, whereas oPSD alone did not (Fig. 1d). Furthermore, paired presynaptic stimulation and oPSD enhanced synaptic NMDAR activity and consequently Ca2+ influx through this channel (Supplementary Fig. 3). LTP was blocked by bath application of an NMDAR antagonist, APV (Fig. 1e). In addition, it was reported that dorsostriatal LTP induction also depended on D1R activation6, 15. We found that optogenetic LTP was inhibited by a D1R antagonist, SCH 23390, and was facilitated by D1R activation (Supplementary Fig. 4). Collectively, these results suggest that paired presynaptic stimulation and oPSD produced effective distal dendrite depolarization and induced robust NMDAR-dependent LTP that is strongly regulated by D1R signaling.
Surprisingly, after blockade of optogenetic LTP by APV, LTD was observed (Fig. 1e). This LTD was completely abolished by bath application of an eCB CB1 receptor (CB1R) antagonist, AM251 (Fig. 1f). This was consistent with previous reports indicating that LTD in the dorsal striatum was mediated by the CB1R24, 25. Since this eCB-LTD only emerged after LTP was blocked, we reasoned that LTP and LTD were induced simultaneously, and that LTD was masked by LTP. To assess this possibility, we bath-applied AM251 throughout the recording period; this produced a significantly greater magnitude of LTP, as compared with that recorded in the absence of AM251 (Fig. 1g). Collectively, these results suggest that both NMDAR-dependent LTP and eCB-LTD were induced simultaneously and that the LTP masked the LTD.
oPSD facilitates corticostriatal LTP in the DMS
Corticostriatal plasticity is critical for drug-seeking behaviors2, 5–7. We, therefore, examined whether oPSD facilitated LTP induction at specific corticostriatal afferents in the DMS. We expressed two channelrhodopsins simultaneously in order to selective stimulation of cortical inputs and oPSD of DMS neurons: Chronos18 was expressed in the mPFC and Chrimson18 was expressed in the DMS (Fig. 2a,b). Chronos-expressing mPFC neurons and their projections to the DMS were able to follow high-frequency (up to 50 Hz) light stimulation (Supplementary Fig. 5). We thus used light stimulation of 50 Hz for 2 sec (oHFS), paired with oPSD of DMS neurons, to induce LTP. While oHFS of corticostriatal fibers or oPSD alone caused little potentiation (Fig. 2c,d), a robust LTP was observed following paired oHFS and oPSD (Fig. 2c). This corticostriatal LTP was abolished by APV or MK801, as expected (Fig. 2e and Supplementary Fig. 6a). Surprisingly, no LTD was observed after LTP was blocked (Fig. 2e and Supplementary Fig. 6b); this contrasted with the results produced by pairing eHFS and oPSD (Fig. 1e). However, whole-cell recording detected a robust LTD in specific D1-MSNs (Supplementary Fig. 6c). This was consistent with a recent report showing that selective optogenetic stimulation of cortical inputs induced LTD only in D1-MSNs26. Collectively, these data suggest that paired oHFS and oPSD induces corticostriatal LTP, as well as LTD in D1-MSNs.
Figure 2.
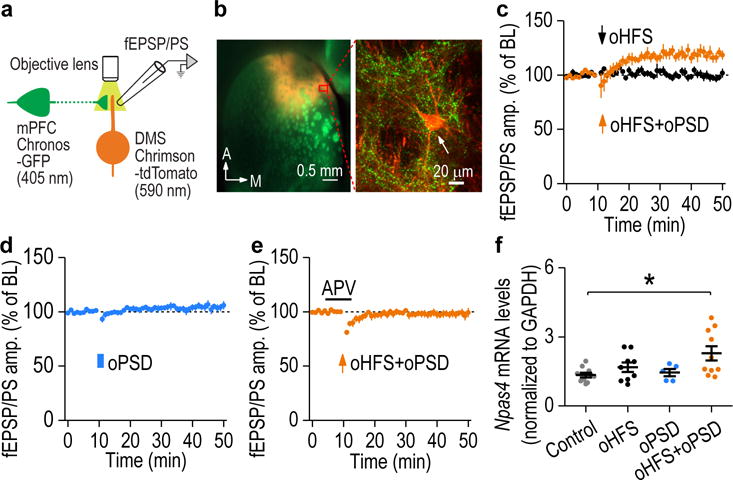
oPSD facilitated corticostriatal LTP induction in the DMS. (a) Schematic illustration of selective pre- and post-synaptic stimulation of DMS corticostriatal synapses using dual-channel optogenetics. AAV-Chronos-GFP was infused into the mPFC and AAV-Chrimson-tdTomato into the DMS of rats. Chronos and Chrimson were activated by 405- and 590-nm light, respectively. (b) Confocal fluorescent images showing Chronos-GFP-expressing mPFC fibers (green) and Chrimson-tdTomato-expressing neurons (red) in the DMS. Images shown in b is representative of 3 experiments of 3 rats. (c) Pairing oHFS with oPSD produced robust LTP in the DMS (119.06 ± 4.69% of baseline [BL], t(6) = −4.07, P = 0.0066; n = 7 slices, 4 rats). oHFS alone did not alter fEPSP/PS (99.60 ± 3.12% of BL, t(7) = 0.13, P = 0.90; n = 8 slices, 5 rats). (d) oPSD alone did not induce LTP (104.55 ± 3.15% of BL, t(6) = −1.44, P = 0.20; n = 7 slices, 3 rats). (e) Dual-channel optogenetic induction of LTP was blocked by APV (98.06 ± 3.27% of BL; t(8) = 0.59, P = 0.57; n = 9 slices, 5 rats). (f) Npas4 mRNA levels were significantly increased following paired oHFS+oPSD, but not after oHFS or oPSD only. F(3,30) = 3.86, P = 0.019; *P < 0.05; n = 10 (Control), 9 (oHFS), 5 (oPSD), and 10 (oHFS+oPSD) rats. Two-sided paired t test for c-e; one-way ANOVA followed by SNK test for f. Data are presented as mean ± s.e.m.
LTP stimulation has been demonstrated to induce expression of immediate early genes, contributing to drug addiction27, 28. We analyzed DMS slices and found that paired oHFS and oPSD, but not oHFS or oPSD alone, significantly increased mRNA levels of Npas4 (neuronal PAS domain protein 4) gene, which encodes the Npase4 protein (Fig. 2f). This immediate early gene is associated with synaptic plasticity and positive valence experience29, 30.
In vivo optogenetic induction of corticostriatal LTP in the DMS produces a long-lasting increase in operant alcohol self-administration in rats
Our ex vivo findings revealed that paired oHFS and oPSD elicited LTP in the DMS. We thus asked whether in vivo delivery of this LTP-inducing protocol (oHFS+oPSD) altered alcohol-seeking behavior. To test this possibility, rats were trained to self-administer alcohol in operant chambers. Chronos and Chrimson were expressed as described above, and optical fibers were implanted into the DMS (Fig. 3a). We found that in vivo delivery of this optogenetic protocol produced significant increases in active lever presses, alcohol deliveries, and alcohol intake at 30 min, 2 days, and 4 days (Fig. 3b and Supplementary Fig. 7a). This increased alcohol intake resulted in elevated blood alcohol concentrations (Supplementary Fig. 7b,c). In contrast, oHFS or oPSD alone did not alter alcohol-seeking behavior (Supplementary Fig. 7d,e). Together, these data suggest that in vivo delivery of this LTP-inducing protocol is sufficient to cause long-lasting enhancement of alcohol-seeking behavior. However, systemic administration of antagonists of NMDARs (MK801) or D1Rs (SCH 23390) blocked this effect of the LTP-inducing protocol on alcohol-seeking behavior (Supplementary Fig. 7f,g). Note that administration of SCH 23390 alone did not affect this behavior (Supplementary Fig. 7h). These results suggest that both NMDARs and D1Rs are required for the enhancement of alcohol-seeking behavior by in vivo LTP induction.
Figure 3.
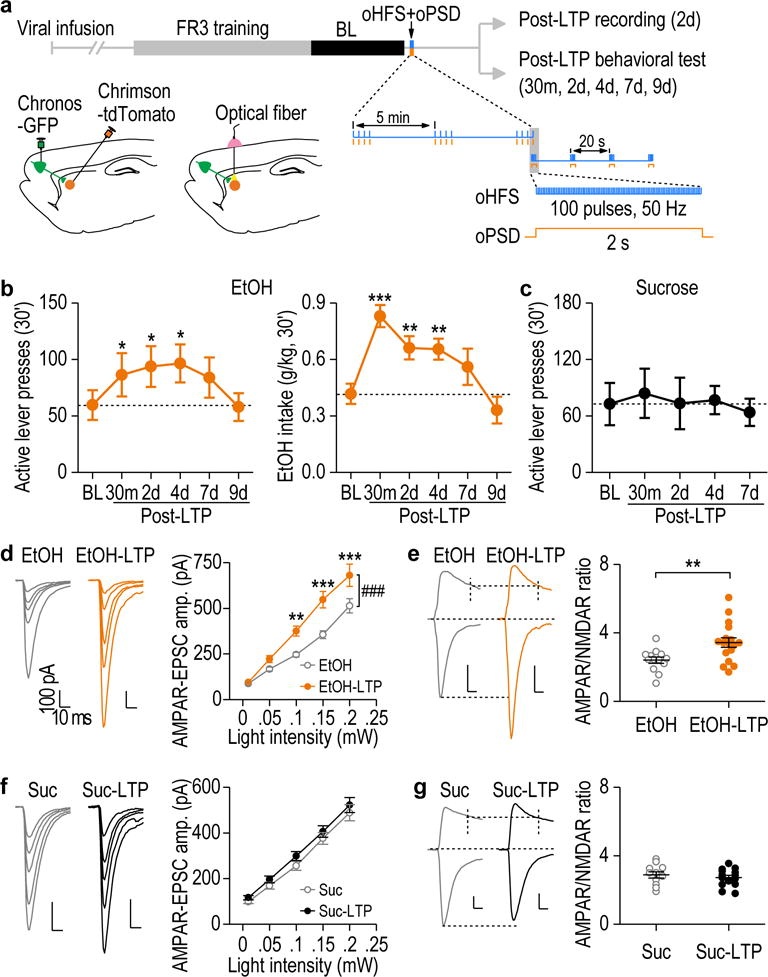
In vivo optogenetic LTP induction in the DMS produced a long-lasting increase in operant alcohol self-administration. (a) Schematic of the training procedure and in vivo LTP induction protocol. (b) In vivo LTP induction produced long-lasting increases in active lever presses (left, F(5,54) = 3.26, P = 0.012) and alcohol (EtOH) intake (right, F(5,49) = 10.91, P < 0.0001). *P < 0.05, **P < 0.01, ***P < 0.001 vs. baseline (BL); n = 14 rats. (c) In vivo LTP induction did not alter active lever presses for sucrose. F(4,18) = 1.10, P = 0.39; n = 6 rats. (d) In vivo LTP induction caused potentiation of AMPAR-EPSCs on day 2 in alcohol-administered rats. Left and middle: Sample traces of AMPAR-EPSCs. Right: Input-output curves for AMPAR-EPSCs with (EtOH-LTP) and without (EtOH) in vivo LTP induction. F(1,107) = 15.62, P = 0.0005; n = 16 neurons, 5 rats (EtOH) and 13 neurons, 3 rats (EtOH-LTP). (e) Representative traces and averaged data showing a significant increase in the AMPAR/NMDAR ratio by LTP induction. t(28) = −2.88, P = 0.0075; n = 13 neurons, 5 rats (EtOH) and 17 neurons, 3 rats (EtOH-LTP). (f) In vivo LTP induction did not change AMPAR-EPSCs from the sucrose-administered rats. F(1,99) = 1.30, P = 0.27; n = 11 neurons, 4 rats (Suc) and 16 neurons, 5 rats (Suc-LTP). (g) AMPAR/NMDAR ratio did not change in the sucrose group after in vivo LTP induction. t(23) = 0.73, P = 0.47; n = 11 neurons, 5 rats (Suc) and 14 neurons, 5 rats (Suc-LTP). Scale bars: 10 ms, 100 pA for d-g. One-way RM ANOVA for b, c; two-way RM ANOVA for d, f; Two-sided unpaired t test for e, g. Data are presented as mean ± s.e.m.
Next, we asked whether the LTP-inducing protocol specifically enhanced alcohol-seeking behavior. Another cohort of rats was trained to self-administer sucrose prior to receiving the same LTP-inducing protocol as the alcohol group. We found that the LTP protocol did not alter the active lever presses, sucrose deliveries, or sucrose intake (Fig. 3c and Supplementary Fig. 7i). We then asked why the same LTP-inducing protocol specifically promoted alcohol-seeking, but not sucrose-seeking, in rats. On day 2 post-LTP induction, both AMPAR-mediated excitatory postsynaptic currents (EPSCs) and the AMPAR/NMDAR ratio were increased in the alcohol group (Fig. 3d,e), but not in the sucrose group (Fig. 3f,g). Interestingly, prior to in vivo LTP induction, operant alcohol or sucrose self-administration had increased the AMPAR/NMDAR ratio, as compared with the water group without operant training (Supplementary Fig. 8a). The increase was slightly lower in the alcohol-treated rats than in the sucrose controls, but the difference was not statistically significant (Q = 2.86, P = 0.051, SNK test). These data suggest that while operant-training produced plasticity in alcohol and sucrose groups, in vivo delivery of the LTP-inducing protocol caused further long-lasting synaptic potentiation selectively in the alcohol group. This difference may reflect the distinct effects of alcohol and sucrose on NMDAR activity6, 31–33. To investigate this, we measured NMDAR activity in the DMS of rats that self-administered alcohol or sucrose. We found that the amplitude of NMDAR-mediated EPSCs was significantly higher in the alcohol group than in the sucrose group (Supplementary Fig. 8b,c). Furthermore, LTP was induced in DMS slices from the alcohol-treated rats, but not in those from the sucrose-drinking animals (Supplementary Fig. 8d), suggesting that alcohol-mediated facilitation of NMDAR activity34, 35 promotes subsequent ex vivo and in vivo induction of LTP. In addition, the rectification index of AMPAR-EPSCs was significantly enhanced following in vivo LTP induction (Supplementary Fig. 9), suggesting that this plasticity is mediated by an increase in calcium-permeable AMPARs.
Collectively, these results suggest that induction of corticostriatal LTP in the DMS produces a long-lasting and specific increase in operant alcohol self-administration in rats.
In vivo optogenetic delivery of an LTD-inducing protocol in the DMS produces a long-lasting decrease in alcohol-seeking behavior in rats
Having observed the link between LTP and alcohol-seeking behavior, we reasoned that reversal of alcohol-induced potentiation of corticostriatal inputs by LTD should reduce alcohol-seeking behavior. To induce LTD in vivo, we systemically administered a cocktail of MK801 and a D2R antagonist, raclopride, 30 min before delivering oHFS and oPSD (Fig. 4a). We used these two categories of antagonists because they were employed in the ex vivo LTD experiments described above (Fig. 1e and Supplementary Fig. 6c). Thirty minutes after oHFS and oPSD delivery, significant reductions in active lever presses, alcohol deliveries, and alcohol intake were observed; these reductions were maintained for at least 7 days (Fig. 4b and Supplementary Fig. 10a). Note that neither the cocktail (MK801+raclopride) plus oHFS (Supplementary Fig. 10b–d) nor the cocktail alone (Supplementary Fig. 10e–g) affected alcohol-seeking behavior. Together with our finding that oHFS+oPSD+MK801 induced no changes in alcohol consumption (Supplementary Fig. 7g), these data suggest that the in vivo LTD-inducing protocol (oHFS+oPSD+MK801+raclopride) produces a long-lasting reduction of alcohol-seeking behavior and that this induction requires D2R blockade.
Figure 4.
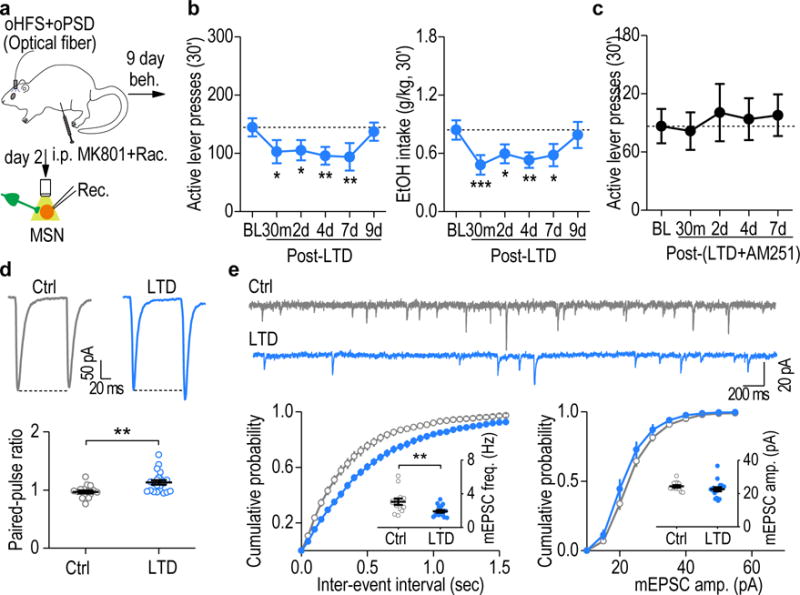
In vivo LTD induction caused a long-lasting reduction of alcohol-seeking behavior in an eCB-dependent manner. (a) Schematic of the in vivo LTD-inducing protocol (oHFS+oPSD+MK801+raclopride) and ex vivo LTD measurement. MK801 (0.1 mg/kg) and raclopride (0.01 mg/kg) were systemically administered 15 min before the optogenetic stimulation. (b) Delivery of the in vivo LTD-inducing protocol in the DMS produced a long-lasting decrease in active lever presses (left, F(5,38) = 3.89, P = 0.006) and alcohol intake (right, F(5,35) = 5.17, P = 0.0012). *P < 0.05, **P < 0.01, ***P < 0.001 vs. baseline (BL); n = 9 rats. (c) Delivery of the in vivo LTD-inducing protocol in the presence of AM251 failed to alter active lever presses. F(4,31) = 0.40, P = 0.81; n = 9 rats. (d,e) Delivery of the in vivo LTD-inducing protocol produced a long-lasting depression of glutamatergic transmission in the DMS on day 2 post-stimulation. (d) Top, sample traces showing paired-pulse ratios (100-ms inter-stimulus interval) measured in fluorescent neurons from LTD-induced rats and their controls (without light stimulation). Bottom, averaged data showing an increased paired-pulse ratio after LTD induction. t(36) = −3.31, P = 0.0021; n = 17 neurons, 3 rats (Ctrl) and 21 neurons, 5 rats (LTD). (e) Top, representative traces of mEPSCs in fluorescent neurons in LTD-induced and control rats. Bottom, cumulative distributions of inter-event intervals and amplitudes of mEPSCs. Inset, reduced frequency (left), but not amplitude (right), of mEPSCs after in vivo LTD induction. t(28) = 2.97, P = 0.006 for frequency; t(28) = 1.11, P = 0.28 for amplitude; n = 13 neurons, 3 rats (Ctrl) and 17 neurons, 3 rats (LTD). One-way RM ANOVA for b, c; Two-sided unpaired t test for d, e. Data are presented as mean ± s.e.m.
Since the LTD induction is eCB-dependent (Fig. 1e), we examined whether blockade of CB1Rs attenuated the effect of the LTD-inducing protocol on alcohol-seeking behavior. We found that systemic administration of additional AM251 completely abolished the LTD-induced reduction of active lever presses, alcohol deliveries, and alcohol intake (Fig. 4c and Supplementary Fig. 10h–i). In contrast, AM251 itself did not alter alcohol-seeking behavior (Supplementary Fig. 10j–l). Lastly, our ex vivo results further ascertained that the paired-pulse ratio was significantly increased (Fig. 4d) and that the frequency of spontaneous miniature EPSCs was decreased (Fig. 4e) 2 days after delivery of the LTD protocol, confirming a presynaptically expressed striatal eCB-LTD. These data indicate that delivery of the in vivo LTD-inducing protocol at the corticostriatal synapses within the DMS produced a long-lasting suppression of alcohol-seeking behavior.
In vivo deliveries of LTP and LTD protocols cause plasticity preferentially in DMS D1-MSNs
The DMS contains D1- and D2-MSNs, which have been reported to exert opposite effects on drug and alcohol drinking behaviors16, 36. We thus explored how LTP or LTD induction altered glutamatergic transmission in these two neuronal types.
First, to examine ex vivo LTP induction in D1- and D2-MSNs, we infused AAV-DIO-ChR2-mCherry into the DMS of Drd1a- and Drd2-Cre transgenic mice, to enable selective depolarization of D1- or D2-MSNs. Paired eHFS and oPSD induced significant LTP, which did not differ between D1- and D2-MSNs (Fig. 5a,b). This promoted us to explore how synaptic transmission changed in these two neuronal populations following in vivo LTP induction. To achieve this, we infused Chronos into the mPFC and Chrimson into the DMS of adult rats, as described above (Fig. 3a). D1-MSNs were labeled by retrograde beads infused into the substantia nigra pars reticulata (SNr) (Fig. 5c, left), whereas D2-MSNs were labeled by infusion of AAV-D2SP-eYFP (Fig. 5c, right). Two days after in vivo delivery of the LTP-inducing protocol, the AMPAR-EPSC amplitude and the AMPAR/NMDAR ratio were increased in D1-MSNs, but not in D2-MSNs (Fig. 5d,e), as compared with slices from control rats that were not exposed to light stimulation. This cell type-specific LTP induction is likely attributable to the higher GluN2B/NMDA ratio in D1-MSNs than in D2-MSNs (Fig. 5f), as alcohol-mediated enhancement of GluN2B promotes LTP induction9, 37. Collectively, these data indicate that the in vivo LTP protocol potentiated synaptic transmission selectively in D1-MSNs.
Figure 5.
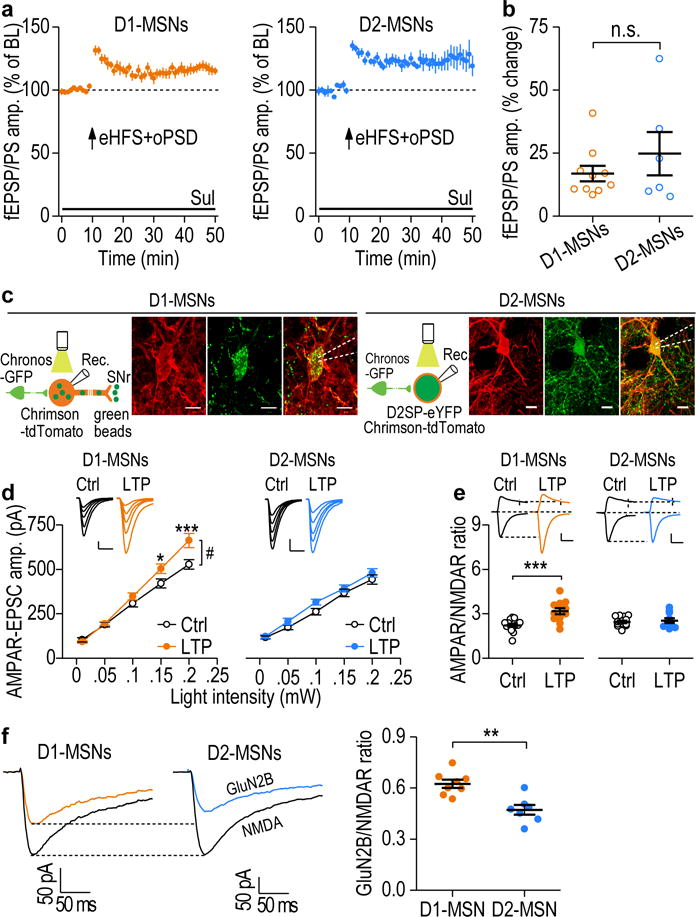
Corticostriatal LTP was preferentially induced in DMS D1-MSNs. (a) AAV-DIO-ChR2-mCherry was infused into the DMS of D1-Cre and D2-Cre mice. Pairing of eHFS and oPSD induced LTP in both D1-MSNs (116.88 ± 3.07% of baseline [BL], t(9) = −5.49, P = 0.00038; n = 10 slices, 5 mice) and D2-MSNs (124.79 ± 8.58% of BL, t(5) = −2.89, P = 0.034; n = 6 slices, 5 mice). (b) There was no significant difference (n.s.) in D1- and D2-MSN LTP. t(14) = −1.04, P = 0.32. (c) Left, experimental design and sample images of retrograde bead labeling of a D1-MSN. AAV-Chronos-GFP, AAV-Chrimson-tdTomato, and green beads were infused into the mPFC, DMS, and SNr, respectively. Right, experimental design and sample images of labeling of a D2-MSNs with D2SP-eYFP. AAV-D2SP-eYFP was infused into the DMS. Scale bar: 10 μm. (d) Left, In vivo LTP induction resulted in higher AMPAR-EPSC amplitudes in D1- (left), but not D2- (right), MSNs from LTP-induced (LTP) rats, as compared with rats that were not exposed to light stimulation (Ctrl). Both groups of rats were trained to self-administer alcohol. D1-MSNs: F(1,95) = 4.72, P = 0.04; n = 13 neurons, 4 rats (Ctrl) and 13 neurons, 5 rats (LTP). D2-MSNs: F(1,90) = 2.11, P = 0.16; n = 14 neurons, 4 rats (Ctrl) and 11 neurons, 5 rats (LTP). Scale bars: 20 ms, 100 pA. (e) Left, the AMPAR/NMDAR ratio increased in D1- (Left), but not D2- (Right), MSNs after in vivo LTP induction. D1-MSNs: t(25) = −4.43, P = 0.00016; n = 15 neurons, 5 rats (Ctrl) and 12 neurons, 5 rats (LTP). D2-MSNs: t(22) = −0.52, P = 0.61; n = 14 neurons, 5 rats (Ctrl) and 10 neurons, 5 rats (LTP). Scale bars: 20 ms, 100 pA. (f) Operant alcohol self-administration resulted in a higher GluN2B/NMDA ratio in D1-MSNs than in D2-MSNs. Left and middle, sample trace of NMDAR-EPSCs in the absence or presence of Ro 25-6981 (0.5 μM). Right, summarized data of the ratios in D1-MSNs and D2-MSNs. t(13) = 4.16, P = 0.0011; n = 8 neurons, 5 rats (D1-MSNs), n = 7 neurons, 5 rats (D2-MSNs). Two-sided paired t test for a; Two-sided unpaired t test for b, e, f; two-way RM ANOVA for d. Data are presented as mean ± s.e.m.
We next investigated whether LTD was also preferentially induced in D1-MSNs ex vivo and in vivo. To induce oPSD selectively in rat D1-MSNs, we infused a retrograde AAV encoding Cre (AAV5-Cre) into the SNr and a Cre-inducible AAV expressing Chrimson (AAV-Flex-Chrimson-tdTomato)18 into the DMS. We found that in DMS slices from alcohol-naïve rats, a protocol (eHFS+oPSD+MK801+raclopride) that was similar to that used to successfully induce LTD (Fig. 1e) caused a robust LTD in D1-MSNs; this LTD was abolished by AM251 (Fig. 6a,b). To induce oPSD specifically in D2-MSNs, we infused AAV-D2SP-ChR238 into the DMS of alcohol-naïve rats. We found that the same protocol of eHFS+oPSD+MK801+raclopride did not produce any LTD in the D2-MSNs (Fig. 6a,c), which was consistent with previous reports21, 39.
Figure 6.
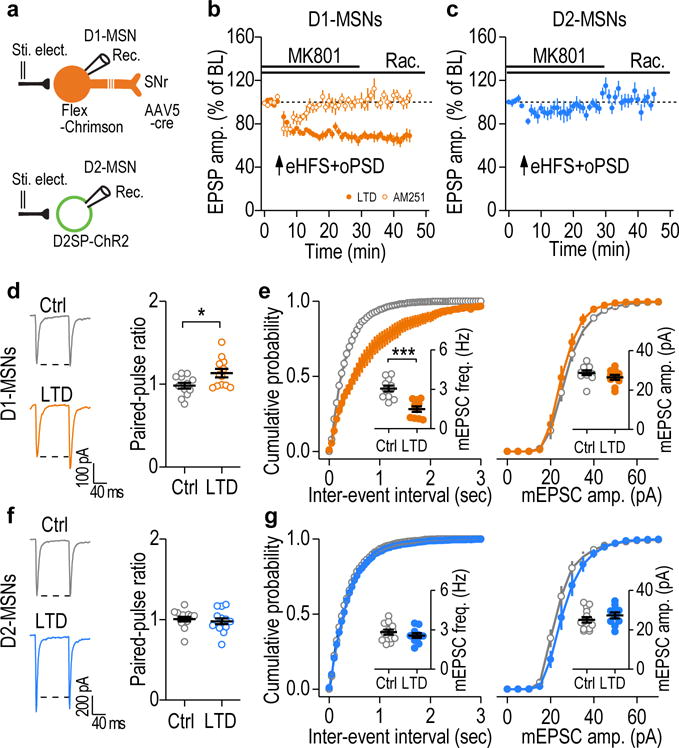
Corticostriatal LTD was preferentially induced in DMS D1-MSNs. (a) Schematic of viral infusion and whole-cell recordings of rat D1- or D2-MSNs. (b) Paired eHFS and oPSD in the presence of NMDAR and D2R antagonists caused a robust LTD in DMS D1-MSNs (66.99 ± 4.56% of baseline [BL], t(7) = 7.23, P = 0.00017; n = 8 neurons, 5 rats), which was completely abolished by AM251 (3 μM) (102.82 ± 4.28% of BL, t(6) = −0.66, P = 0.53; n = 7 neurons, 3 rats). (c) Paired eHFS and oPSD in the presence of NMDAR and D2R antagonists did not induce LTD in D2-MSNs (100.08 ± 5.15% of BL, t(7) = −0.02, P = 0.99; n = 8 neurons, 3 rats). (d) Sample traces and averaged data showing an increased paired-pulse ratio in D1-MSNs 2 days after in vivo LTD induction. t(22) = −2.45, P = 0.023; n = 12 neurons, 4 rats (Ctrl) and 12 neurons, 3 rats (LTD). (e) In vivo optogenetic LTD induction reduced the mEPSC frequency (left), but not the mEPSC amplitude (right) of DMS D1-MSNs. t(19) = 5.00, P < 0.0001 for frequency; t(19) = 1.30, P = 0.21 for amplitude; n = 11 neurons, 4 rats (Ctrl) and 10 neurons, 3 rats (LTD). (f) In vivo LTD induction did not change paired-pulse ratios in D2-MSNs. t(26) = 0.59, P = 0.56; n = 14 neurons, 4 rats (Ctrl) and 14 neurons, 3 rats (LTD). (g) In vivo LTD induction did not alter the mEPSC frequency (left, inset) or the mEPSC amplitude (right) in DMS D2-MSNs. t(22) = 0.95, P = 0.35 for frequency; t(22) = −1.11, P = 0.28 for amplitude; n = 14 neurons, 4 rats (Ctrl) and 10 neurons, 3 rats (LTD). Two-sided paired t test for b, c; Two-sided unpaired t test for d-g. Data are presented as mean ± s.e.m.
Lastly, to ascertain whether in vivo LTD induction caused glutamatergic depression in D1-MSNs, we measured corticostriatal EPSCs in DMS slices prepared two days after in vivo delivery of the LTD-inducing protocol. We found that the LTD protocol reduced the release probability, as indicated by the increased paired-pulse ratio, and reduced the mEPSC frequency in D1-, but not D2-, MSNs, as compared to neurons from control animals without LTD induction (Fig. 6d–g). These results indicate that in vivo LTD induction leads to long-lasting depression of corticostriatal inputs selectively onto DMS D1-MSNs.
Taken together, our results suggest that in vivo delivery of optogenetic LTP and LTD protocols preferentially induced plasticity in DMS D1-MSNs.
Selective LTP and LTD induction in DMS D1-MSNs produces long-lasting changes in controls alcohol-seeking behavior
Finally, we examined whether in vivo induction of corticostriatal LTP or LTD directly in DMS D1-MSNs altered alcohol-seeking behavior. For oHFS, we infused AAV-Chronos into the mPFC; for oPSD of D1-MSNs, we infused the retrograde AAV5-Cre into the SNr and AAV-Flex-Chrimson into the DMS (Fig. 7a). These infusions led to Chronos expression at the mPFC inputs and selective Chrimson expression in DMS D1-MSNs (Fig. 7b). In vivo LTP induction produced significant increases in active lever presses, alcohol deliveries, and alcohol intake at 30 min; this effect persisted for at least 2 days (Fig. 7c and Supplementary Fig. 11a). However, D1-MSN oPSD alone did not alter alcohol-seeking behavior (Fig. 7d and Supplementary Fig. 11b,c). These data suggest that in vivo corticostriatal LTP in DMS D1-MSNs caused a long-lasting potentiation of alcohol-seeking behavior.
Figure 7.
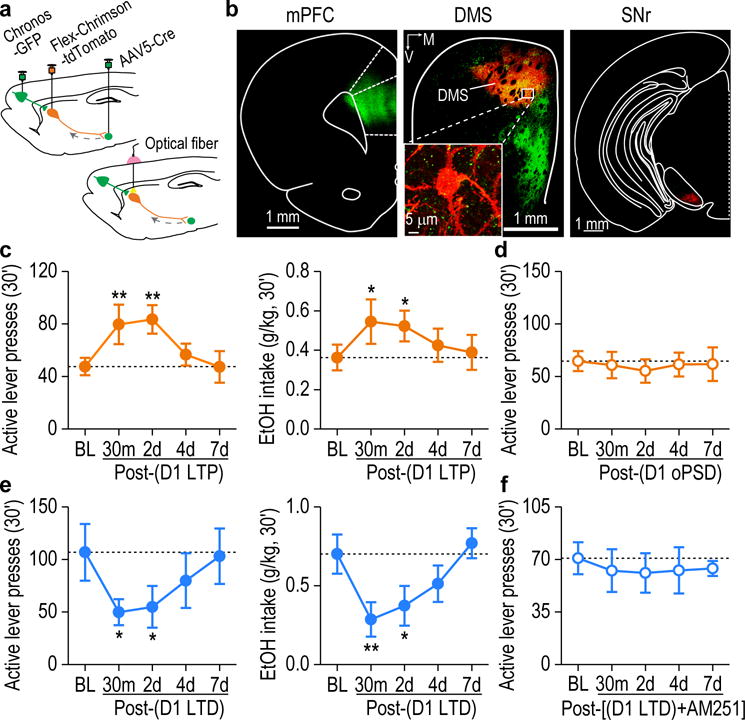
Selective in vivo LTP or LTD induction in D1-MSNs produced a long-lasting control of alcohol-seeking behavior. (a) Schematic showing viral infusion (top) and optical fiber implantation (bottom). (b) Representative fluorescent images showing that mPFC (left) and SNr infusions produced Chronos-expressing fibers (green) and Chrimson-tdTomato expression (red) in DMS D1-MSNs (middle), which project to the SNr (right). Images shown in b is representative of 3 experiments of 3 rats. (c) Paired oHFS of mPFC inputs and oPSD of D1-MSNs induced long-lasting increases in active lever presses (left, F(4,24) = 6.02, P = 0.0017) and alcohol intake (right, F(4,24) = 3.74, P = 0.017). *P < 0.05, **P < 0.01 vs. baseline (BL); n = 8 rats. (d) In vivo delivery of oPSD alone in DMS D1-MSNs did not alter active lever presses for alcohol. F(4,18) = 0.36, P = 0.83; n = 6 rats. (e) Delivery of the in vivo LTD-inducing protocol to mPFC inputs onto DMS D1-MSNs produced a long-lasting attenuation in active lever presses (left, F(4,23) = 4.07, P < 0.0001) and alcohol intake (right, F(4,15) = 6.67, P = 0.0027). *P < 0.05, **P < 0.01 vs. BL; n = 7 rats for active lever presses and n = 6 rats for alcohol intake. (f) In vivo delivery of the LTD-inducing protocol in the presence of AM251 (oHFS+oPSD+MK801+raclopride+AM251) failed to alter active lever presses for alcohol. F(4,22) = 0.12, P = 0.97, n = 7 rats. One-way RM ANOVA for c-f. Data are presented as mean ± s.e.m.
In contrast, we found that delivery of the in vivo LTD-inducing protocol to the mPFC input onto D1-MSNs caused sustained decreases in active lever presses, alcohol deliveries, and alcohol intake at 30 min and 2 days (Fig. 7e and Supplementary Fig. 11d). This behavioral effect was abolished by systemic administration of AM251 (Fig. 7f and Supplementary Fig. 11e,f), which confirmed that eCB signaling regulated this inhibition of alcohol-seeking behavior. These data demonstrate that eCB-LTD in D1-MSNs is required for the long-lasting decrease in alcohol-seeking behavior.
DISCUSSION
In this study, we provide evidence to suggest that alcohol intake induces glutamatergic plasticity, which can be further potentiated by in vivo LTP induction, and that this causes long-lasting enhancement of alcohol-seeking behavior (Fig. 8a). In contrast, in vivo LTD induction suppresses this plasticity and produces a long-lasting reduction of this behavior. We report that pairing HFS of corticostriatal afferents with oPSD of DMS neurons induces a robust NMDAR-dependent LTP, which masks an eCB-LTD (Fig. 8b). Furthermore, we discovered that LTP and LTD in D1-MSNs contributed to the alteration of alcohol-seeking behavior (Fig. 8b,c). These results provide a direct causal link between long-term synaptic plasticity within a given neural circuit (mPFC → DMS D1-MSNs) and alcohol-seeking behavior. Our findings also demonstrate that induction of D1-MSN LTD might be a potential therapeutic strategy for alcohol use disorder.
Figure 8.
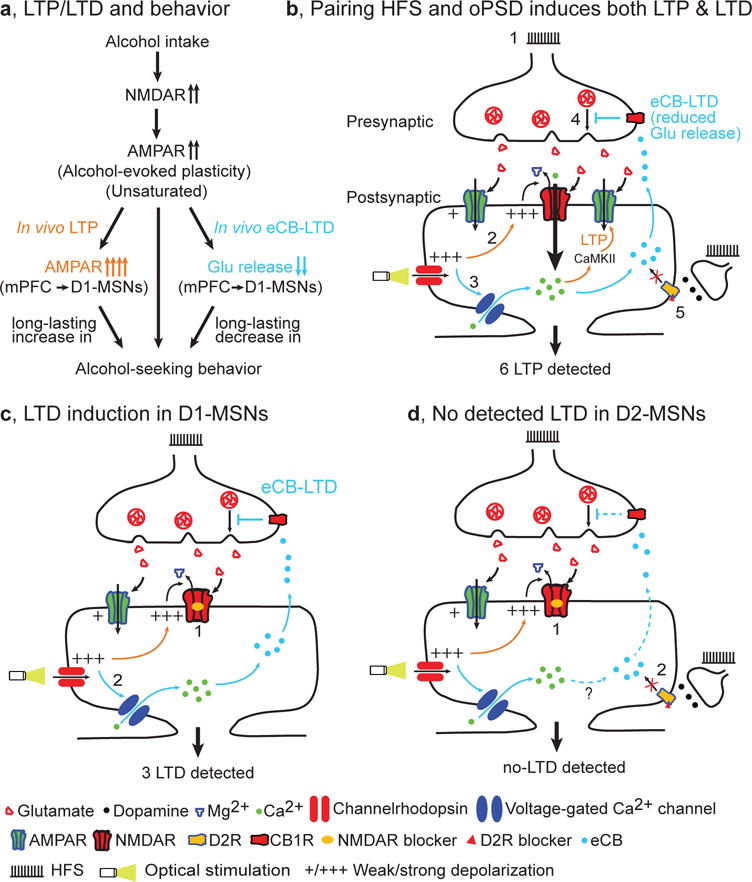
Model of bidirectional and long-lasting control of alcohol-seeking behavior by corticostriatal plasticity. (a) Alcohol intake facilitates NMDAR activity, leading to potentiation of AMPAR activity (unsaturated alcohol-evoked plasticity). This is further potentiated by in vivo LTP induction at mPFC inputs to DMS D1-MSNs, producing a long-lasting enhancement of alcohol-seeking behavior. Conversely, in vivo eCB-LTD induction at the same synapses elicited a long-lasting suppression of this behavior. (b) Paired HFS and oPSD induces both LTP and LTD, but only LTP is detected. 1, HFS causes presynaptic release of glutamate, which activates AMPARs. The resultant weak membrane depolarization is insufficient to remove the Mg2+ blockade of NMDARs and thus fails to induce LTP. 2, Optical stimulation of channelrhodopsin expressed on postsynaptic neurons causes strong membrane depolarization (oPSD). This is sufficient to remove Mg2+ blockade of NMDARs, leading to greater Ca2+ influx, activation of Ca2+/calmodulin-dependent protein kinase II (CaMKII) signaling pathways, and consequently AMPAR insertion (LTP induction). 3, oPSD also opens voltage-gated Ca2+ channels, causing Ca2+ influx and production of endocannabinoids; these are retrogradely released into the synaptic cleft, where they activate presynaptic CB1Rs. 4, CB1R activation reduces glutamate release (eCB-LTD induction). 5, D2R antagonists are used to blocking D2R-mediated eCB-LTD, which may occur following eHFS ex vivo or oHFS in vivo. 6, Since the magnitude of LTP (~31%) is greater than that of LTD (~13%), only LTP is detected. (c) LTD induction and detection in D1-MSNs. Since LTP induction is blocked by NMDAR antagonists (1) and eCB-LTD is induced (2), only eCB-LTD is detected (3). (d) No LTD is detected in D2-MSNs because LTP is blocked by NMDAR antagonists (1) and eCB-LTD is blocked by D2R antagonists (2).
oPSD facilitates LTP and LTD induction in the dorsal striatum
It has long been known that dorsostriatal LTP induction proves difficult, possibly due to insufficient depolarization of striatal neurons20, 40. In this study, oPSD was used to strongly depolarize the distal dendrites of these neurons, thus enhancing NMDAR channel opening and calcium influx, which is required for LTP induction20, 40 (Fig. 8b). Interestingly, blockade of LTP by NMDAR antagonists leads to LTD; this is consistent with a study showing that LTP blockade by memantine shifted LTP to LTD41. However, APV or MK801 was found to shift the plasticity in the current research, but not in the study by Mancini et al.41. This discrepancy may reflect the fact that eCB-LTD was induced mainly by oPSD in the presence of a D2R antagonist in the current study, and by D2R activation in the previous research. In addition, we report that dopamine D1R signaling plays a critical modulatory role in optogenetic LTP. The observations that blockade of LTP unmasks eCB-LTD and that blockade of eCB-LTD enhances the LTP magnitude suggest that paired HFS and oPSD simultaneously induces NMDAR-dependent LTP and eCB-LTD.
The current oPSD also facilitated LTP in the specific corticostriatal input when we used dual-channel optogenetics. The precise control of both pre- and post-synaptic activity allowed us to reliably induce LTP for the first time. LTP induction is known to activate the expression of immediate early genes such as Npas4, which has recently been identified as an important factor in brain plasticity30. Expression of the Npas4 gene requires Ca2+ influx and is associated with drug addiction29. Indeed, the mRNA level Npas4 gene is increased only after paired oHFS and oPSD, suggesting that oPSD predominantly facilitates LTP at corticostriatal synapses.
Optogenetic LTP induction promotes, and LTD induction suppresses, alcohol-seeking behavior
The corticostriatal circuit is believed to control goal-directed behaviors, including drug-seeking behavior2, 5–7. In vivo optogenetic induction of corticostriatal LTP enhances alcohol-seeking behavior, suggesting a link between this plasticity and the behavior. The selective effects of LTP on operant self-administration of alcohol versus sucrose may reflect the distinct activities of these two chemicals on the rats. Operant training with alcohol, but not with sucrose, enhanced NMDAR activity and facilitated subsequent in vivo and ex vivo induction of LTP. This finding is consistent with previous reports indicating that ex vivo or in vivo alcohol exposure caused long-term facilitation of NMDAR activity6, 10, 31, 32, which is required for LTP induction in the dorsal striatum6 and for operant alcohol self-administration10. Operant alcohol self-administration induced a smaller, but not significant, increase in AMPAR/NMDAR ratio than did operant sucrose training. This ratio difference might be attributable to the higher NMDAR activity in alcohol-treated rats than in sucrose controls.
How drug (e.g., cocaine)-evoked plasticity affects subsequent LTP induction is likely to depend on the degree of saturation of the plasticity. Our study reveals that operant alcohol self-administration using the FR3 schedule induced glutamatergic plasticity (increased AMPAR/NMDAR ratio). This plasticity was not saturated because the AMPAR/NMDAR ratio was further potentiated by in vivo LTP induction, and LTP was induced in slices from alcohol-drinking animals. It is known that 1‐2 day(s) withdrawal from cocaine exposure induces silent synapses that contain NMDARs but not AMPARs13, 42–44; these can mature over time (e.g., at 45 days)13, 44, 45 and potentially contribute to subsequent LTP occlusion46, 47. These studies suggest that short-term withdrawal from drug exposure induces unsaturated plasticity. Since we induced LTP 24 hours after the last alcohol exposure, it is not surprising that no occlusion was observed; this is consistent with previous reports9, 37.
Pharmacological inhibition of alcohol-evoked glutamatergic strengthening in the DMS attenuates alcohol consumption9. However, this inhibition is transient and disappears as the inhibitory compounds are metabolized. Furthermore, structural plasticity, such as an increased density of mushroom spines, has been observed following alcohol consumption48. In this study, in vivo eCB-LTD induction elicited a long-lasting decrease in alcohol-seeking behavior, indicating that this plasticity mediates more sustained behavior changes49.
LTP and LTD in D1-MSNs affect alcohol-seeking behavior
While the present study and others15, 20 report that LTP can be induced in both D1- and D2-MSNs in slices from alcohol-naïve animals, in vivo delivery of our LTP-inducing protocol selectively causes long-lasting potentiation of corticostriatal transmission in D1-MSNs of alcohol-drinking rats. This selectivity may be attributed to the fact that alcohol consumption potentiates NMDAR activity in D1-, but not D2-, MSNs16. The current study further identified that alcohol consumption specifically potentiated GluN2B-containing NMDAR activity at the mPFC input onto D1-MSNs. Alcohol-mediated potentiation of GluN2B-NMDARs was reported to facilitate LTP induction9, 37. Our in vivo LTD-inducing protocol also caused LTD in D1-, but not D2-, MSNs because we included a D2R antagonist, which blocks LTD induction in D2-MSNs15, 21 (Fig. 8c,d). Our findings are in agreement with a recent report showing that eCB-LTD was induced at corticostriatal inputs to D1-, but not D2-, MSNs26. Given that D1-MSNs positively control alcohol consumption16, it is not surprising that induction of D1-MSN LTP produces long-lasting enhancement of operant alcohol self-administration, while LTD induction in this neuronal type reduces the same behavior. The induction of LTP/LTD by inducing oPSD selectively in D1-MSNs confirmed that synaptic plasticity in this neuronal type is sufficient to control alcohol-seeking behavior in a bi-directional manner. Therefore, blockade of striatal LTP induction and promotion of eCB-LTD in D1-MSNs may inspire a therapeutic strategy to cause a long-lasting reduction of alcohol-seeking behavior. Although optogenetic intervention cannot be immediately translated to human use, deep brain stimulation (DBS) is an FDA-approved treatment that has the potential to cause LTP46, and probably LTD. Thus, we believe that the combined use of DBS and antagonists of the NMDAR (e.g., memantine) and D2R may provide novel clinical treatments for alcohol use disorder.
In summary, we have demonstrated that optogenetic induction of bidirectional long-term synaptic plasticity at corticostriatal afferents within the DMS produced long-lasting increases or decreases in alcohol-seeking behavior. Importantly, we show that the plasticity of DMS D1-MSNs controls alcohol-seeking behavior. Our research establishes a causal link between corticostriatal synaptic potentiation and alcohol-seeking behavior and provides an evidence base for therapeutic strategies to reduce excessive alcohol consumption.
ONLINE METHODS
Reagents
AAV5-CaMKIIa-C1V1(E122T/E162T)-eYFP (3 × 1012 vg/ml), AAV8-Syn-Chronos-GFP (5.6 × 1012 vg/ml), AAV8-Syn-Chrimson-tdTomato (5.5 × 1012 vg/ml), AAV8-Syn-Flex-Chrimson-tdTomato (4.1 × 1012 vg/ml) and AAV-EF1a-DIO-ChR2-mCherry (2 × 1012 vg/ml) were purchased from the University of North Carolina Vector Core. AAV5-CAG-GCaMP6s (2.2 × 1013 vg/ml) and AAV5-CMV-Cre-eGFP (4.9 × 1012 vg/ml) were obtained from the University of Pennsylvania Vector Core. AAV8-D2SP-eYFP (2.5 × 1012 vg/ml) and AAV8-D2SP-eChR2 (H134R)-eYFP (2.5 × 1012 vg/ml) were purchased from Gene Vector and Virus Core of Stanford University School of Medicine. NBQX and APV were purchased from R&D systems. Tetrodotoxin (TTX) was obtained from Tocris. Alexa Fluor 594 was purchased from Invitrogen. MK801, sulpiride, raclopride and the other reagents were obtained from Sigma.
Animals
Male Long-Evans rats (3 months old, Harlan Laboratories) and Drd1a-Cre (D1-Cre) and Drd2-Cre (D2-Cre) transgenic mice (C57BL/6 background, 3 months old, Mutant Mouse Regional Resource Centers) were used. Both rats (2/cages) and mice (5/cage) were group-housed. All animals were kept in a temperature- and humidity-controlled environment with a light: dark cycle of 12 h (lights on at 7:00 a.m.), and food and water available ad libitum. All behavior experiments were conducted in their light cycle, and animals had no history prior to the behavior reported in this paper. All animal care and experimental procedures were approved by the Texas A&M University Institutional Animal Care and Use Committee and were conducted in agreement with the National Research Council Guide for the Care and Use of Laboratory Animals.
Stereotaxic virus infusion
The stereotaxic viral infusion was performed as described previously8. Depending on experimental design, viruses or beads were infused into the mPFC (AP: +3.2 and +2.6 mm, ML: ±0.65 mm, DV: −4.0 mm from Bregma), the DMS (AP1: +1.2 mm, ML1: ±1.9 mm, DV1: −4.7 mm; AP2: +0.36 mm, ML2: ±2.3 mm, DV2: −4.7 mm), and the SNr (AP1: −4.92 mm, ML1: ±2.3 mm, DV1: −8.3 mm; AP2: −5.5 mm, ML2: ±2.0 mm, DV2: −8.6 mm) for rats. For mice, the viruses were infused into the DMS (AP1: +1.18 mm, ML1: ±1.3 mm, DV1: −2.9 mm; AP2: +0.38 mm, ML2: ±1.55 mm, DV2: −2.9 mm from Bregma). 0.5-1 μl of the virus was infused bilaterally at a rate of 0.08 μl/min. At the end of the infusion, the injectors remained at the site for 10 min to allow for virus diffusion. Animals infused for electrophysiology were maintained in their home cages for 6–8 weeks before recordings. For animals infused with viruses for behavioral experiments, we started training them to self-administer alcohol or sucrose one week after surgery.
Slice preparation
The procedure has been described previously8, 50, 51. Briefly, coronal sections of the striatum (250 μm in thickness) were cut in an ice-cold solution containing the following (in mM): 40 NaCl, 143.5 sucrose, 4 KCl, 1.25 NaH2PO4, 26 NaHCO3, 0.5 CaCl2, 7 MgCl2, 10 glucose, 1 sodium ascorbate, and 3 sodium pyruvate, saturated with 95% O2 and 5% CO2. Slices were then incubated in a 1:1 mixture of cutting solution and external solution at 32°C for 45 min. The external solution was composed of the following (in mM): 125 NaCl, 4.5 KCl, 2 CaCl2, 1 MgCl2, 1.25 NaH2PO4, 25 NaHCO3, 15 sucrose and 15 glucose, saturated with 95% O2 and 5% CO2. Slices were maintained in external solution at room temperature until use.
Field potential recording
For LTP experiments, extracellular field recordings were conducted as previously described8. Specifically, the recording used a patch pipette filled with 1 M NaCl and was placed within the DMS. DMS slices were visualized under an epifluorescent microscope (Examiner A1, Zeiss, Germany). Bipolar stimulating electrodes were positioned 100–150 μm away from the recording electrode. Field excitatory postsynaptic potentials/population spikes (fEPSP/PS)52 were evoked by electrical stimuli through stimulating electrodes at 0.05 Hz. Picrotoxin (100 μM) was bath applied to block GABAergic transmission. The dopamine D2R antagonist, sulpiride (20 μM), was included externally for all experiments conducted in Figures 1, 5a, and Supplementary Figures 1b, 4. Optical stimuli (2 ms, 405 nm) were delivered through the objective lens to elicit fEPSP/PS. fEPSP/PS were measured using a MultiClamp 700B amplifier with Clampex 10.4 software (Molecular Devices). After a stable baseline had been established for 10 min, high-frequency stimulation (HFS) was delivered through the stimulating electrodes or objective lens to induce LTP. HFS consists of 4 trains of stimuli repeated at an interval of 20 sec. Each train contains 100 pulses at 100 Hz (electrically HFS, eHFS) or 50 Hz (optogenetic HFS, oHFS). For pairing experiments, optogenetic postsynaptic depolarization (oPSD) was induced by light stimulation (590 nm, 1 sec for eHFS or 2 sec for oHFS) of DMS neurons through the objective lens.
Whole-cell recording
In Supplementary Figures 1, 2, and 5b, we used a K+-based intracellular solution, containing (in mM): 123 potassium gluconate, 10 HEPES, 0.2 EGTA, 8 NaCl, 2 MgATP, 0.3 NaGTP (pH 7.3). All other experiments utilized the Cs-based solution, containing (in mM): 119 CsMeSO4, 8 TEA.Cl, 15 HEPES, 0.6 EGTA (10 BAPTA for Supplementary Figures 3d,e), 0.3 Na3GTP, 4 MgATP, 5 QX-314.Cl, 7 phosphocreatine, 0.05 Alexa Fluor 594 (Supplementary Figures 3a–e), and 0.1 spermine (Supplementary Figure 9). The pH was adjusted to 7.3 with CsOH. Neurons were clamped at −70 mV.
For measuring NMDAR-EPSPs in distal dendrites, Alexa Fluor 594 was infused through patch-pipettes into the recorded neurons to label their dendrites. Under the guidance of fluorescence, the stimulating electrodes were positioned close to the Alexa Fluor-labeled dendrites and were 100–150 μm away from the soma. AMPA receptor (AMPAR)-mediated EPSPs were recorded in 1.0 mM extracellular Mg2+. NBQX (10 μM) was then bath applied to block AMPAR-EPSPs. Next, simultaneous presynaptic electrical stimulation and oPSD of striatal neurons induced a response that was mediated by a C1V1-induced depolarization (Vc1v1) plus an NMDAR response (EPSPNMDA). Lastly, the EPSPNMDA component was blocked by bath application of APV (50 μM), and Vc1v1 was isolated. The optogenetic-mediated EPSPNMDA was calculated by digital subtraction of Vc1v1 from Vc1v1 + EPSPNMDA. The input-output relationships for AMPAR-mediated excitatory postsynaptic currents (EPSCs) were measured at 5 different stimulating laser powers. For measurement of the AMPAR/NMDAR ratio, the peak currents of AMPAR-mediated EPSCs were measured at a holding potential of −70 mV and the NMDAR-mediated EPSCs were estimated as the EPSCs at +40 mV at 30 ms after the peak AMPAR-EPSCs, when the contribution of the AMPAR component was minimal. The AMPA/NMDA ratio was calculated by dividing the NMDAR-EPSC by AMPAR-EPSC. To measure the GluN2B/NMDA ratio, NMDAR-EPSCs were recorded in the absence and presence of Ro 25-6981, and GluN2B-EPSCs were calculated by subtraction of these two responses. For measuring mEPSCs, we added TTX (1 μM) to the external solution to suppress action potential-driven release. The paired-pulse ratio (PPR) was calculated by dividing the second light-evoked EPSC by the first with 100-ms intervals between the two. To measure above synaptic transmission in specific D1- and D2-MSNs (Fig. 5, 6), we first patched bead-positive (D1-MSNs) or eYFP-positive neurons (D2-MSNs) within a DMS area containing strong green mPFC fibers (expressing Chronos-GFP) and red Chrimson-positive neurons. We then sequentially delivered 405-nm light to stimulate the mPFC inputs and 590-nm light to induce Chrimson-mediated oPSD. The synaptic inputs and oPSD were distinguished using 2- and 100-ms light stimulation, since the prolonged light stimulation increased the duration of oPSD, but not of synaptic transmission53. Only those neurons that received mPFC inputs and exhibited oPSD were selected for further experiments. At the end of the recording, NBQX was applied to confirm synaptic transmission induced by 405-nm light stimulation. To measure AMPAR rectification, AMPAR-EPSCs were recorded at three holding potentials of −70, 0, and +40 mV in the presence of APV (50 μM). Rectification index of the AMPAR-EPSC was calculated by plotting the EPSC magnitude at these potentials, and using the slope of the lines connecting the data between −70-0 mV and between 0–40 mV to calculate the ratio.
Calcium image
An AAV-C1V123 and an AAV-GCaMP6s54 were infused into the DMS. Whole-cell recordings were made in C1V1-expressing neurons. The GCaMP6s measures the calcium signal that is induced by current injection (iPSD), or optogenetic depolarization (oPSD) (Supplementary Figure 2). In supplementary Figures 3f–g, fluorescent Ca2+ signals were elicited by eHFS or eHFS+oPSD without whole-cell recording. The distal dendrite (~120 μm from the soma) was chosen for analysis. Ca2+ signals were acquired and analyzed with the Zen program (Zeiss) and Origin software (Origin Lab Corporation, MA), and calculated as previously described55. The fluorescence signals were quantified by measuring the mean pixel intensities of the circular regions of interest (ROI). Fluorescence intensity is expressed as ΔF/F values vs. time, where F is the baseline fluorescence and ΔF is the baseline-subtracted fluorescence.
Operant self-administration of alcohol
After one week recovery from viral infusions, Long-Evans rats were trained to self-administer a 20% alcohol solution in operant self-administration chambers as described56. Each chamber contains two levers; an active lever, in which presses result in a delivery of 0.1 ml of the alcohol solution, and an inactive lever, in which presses are recorded, but no programmed events occur. After 48-h of exposure to 20% alcohol in the home cage, and one overnight session in the chamber in which pressing the active lever delivers 0.1 ml of water in a fixed ratio 1 (FR1), operant sessions were conducted 5 days per week for two weeks in a FR1 schedule with an active lever press resulting in the delivery of 20% alcohol with sessions shortened from 3 h to 30 min. Following the first two weeks, operant sessions were run three days per week for one week, and the schedule requirement was increased to FR3. After one week of FR3 training, rats underwent surgery for optical fiber implantation. FR3 training was resumed one week after the surgery. Once a stable baseline of active lever presses was achieved, animals underwent in vivo LTP and LTD induction. Following the induction, some rats were continuously monitored with their operant behavior for 7–9 days, while other rats were euthanized at day 2 post-induction for electrophysiology recordings. To test drugs’ effect without LTP/LTD induction, we systematically administered them 30 min before the operant behavior test. Simultaneously, we also measured inactive lever presses before and after treatment (Supplementary Figure 12).
Operant self-administration of sucrose
After a one-week recovery from viral infusions, Long-Evans rats were trained to self-administer a 2% sucrose in operant chambers using the same procedure as the alcohol group described above. Optical fiber implantation was also conducted in an identical manner to the alcohol group.
Optical fiber implantation
One-week following operant training with the FR3 schedule, animals were anesthetized with isoflurane and mounted in a stereotaxic frame. An incision was made, bilateral optical fiber implants (300-nm core fiber secured to a 2.5-mm ceramic ferrule with 5-mm fiber extending past the end of the ferrule) were lowered into the DMS (AP: +0.36 mm; ML: ±2.3 mm; DV: −4.6 mm from Bregma). Implants were secured to the skull with metal screws and dental cement (Henry Schein) and covered with denture acrylic (Lang Dental). The incision was closed around the head cap and the skin vet-bonded to the head cap. Rats were monitored for one week or until they resumed normal activity.
In vivo LTP and LTD induction and operant testing
Once a stable baseline of active lever presses was attained after optical fiber implantation, an LTP/LTD-inducing protocol was delivered 30 minutes before operant testing sessions in a neutral Plexiglass chamber, with no visual cues. LTP-induction consisting of paired oHFS+oPSD used the following protocol: 100 pulses at 50 Hz of 473-nm light (2 ms) with or without constant 590-nm light for 2 sec, repeated 4 times with 20-sec intervals. The protocol was repeated three times with 5-min intervals. LTD induction employed the following protocol: animals were injected with a cocktail of MK-801 (0.1 mg/kg) and raclopride (0.01 mg/kg) 15 minutes before delivery of oHFS and oPSD. The complete LTP/LTD-inducing procedure was performed once and 30 min later animals were allowed to press levers for alcohol in a 30-min session. Operant sessions were repeated every 48 or 72 h until active lever presses returned to their levels prior to the induction.
Measurement of blood alcohol concentration (BAC)
To measure blood alcohol concentration, we used the same procedure as in Figure 3b to train two groups of rats. One week prior to LTP induction, we collected blood samples from the one side of the lateral saphenous vein57 in both groups to measure baseline BAC. Thirty minutes after LTP induction, we collected blood samples from the other side of the lateral saphenous in one group of rats. Two days after the LTP induction, we collected blood samples from the other group of rats. BAC was measured using gas chromatography as previously described58.
RNA extraction and quantitative PCR (qPCR) analysis
The rats were infused with AAV-Chronos-GFP in the mPFC and AAV-Chrimson-tdTomato in the DMS. Coronal striatal sections (250 μm) were prepared as described in Slice Preparation section above. A slice was placed in a recording chamber and perfused with the external solution at a flow rate of 3 mL/min. Optical stimuli were delivered through the objective lens to fluorescent DMS areas, which contained both GFP-expressing mPFC axons and tdTomato-expressing neurons using one of the following stimulation protocols: oHFS, oPSD, or oHFS+oPSD. All protocols were repeated three times with 5-min intervals, which is the same as the in vivo LTP-inducing protocol. Thirty minutes after completing the optogenetic stimulation, the DMS tissues from experimental and control groups were collected on ice. The RNA isolation, reverse transcription, and the qRT-PCR analyses were performed as described previously59. The mRNA level of Npas4 was normalized against the GAPDH mRNA level in the same sample and presented as fold changes over baseline using the delta-delta CT method. The following primers were used: Npas4, Forward: 5′-GAACCTCAAGGAACTGCTGC-3′, reverse: 5′- GTGCCTCCAGCAAAGAAGAC-3′; GAPDH, Forward: 5′-TGCCACTCAGAAGACTGTGG-3′, reverse: 5′-TTCAGCTCTGGGATGACCTT-3′. For each experimental condition, two slices per rat were treated, and the averaged mRNA values were used.
Histology
Rats with viral and beads infusion were perfused intracardially with 4% paraformaldehyde (PFA) in phosphate-buffered saline (PBS). The brains were taken out and put into 4% PFA/PBS solution for post-fixation overnight at 4°C, followed by dehydration in 30% sucrose solution and cryostat frozen sectioning. The brains were cut into 50-μm coronal sections. A confocal laser-scanning microscope (Fluorview-1200, Olympus) was used to image these sections with a 470-nm laser to excite eYFP and GFP and with a 593-nm laser to excite Alexa Fluor 594 and tdTomato. All images were processed using Imaris 8.3.1 (Bitplane, Zurich, Switzerland).
Data acquisition and statistics
In electrophysiology experiments, we used 184 rats and 10 D1- and D2-Cre mice, with 10 rats excluded before data collection due to virus expression in the incorrect place or expression that was too weak. In behavioral tests, we used 156 rats, among which 28 were excluded due to lack of alcohol responding in the operant setting (≤ 10 active lever presses/session), 6 were removed from data analysis due to death during surgery, and 21 were removed from the last experiments due to head cap loss. In the imaging experiments, we used 11 rats.
All data are expressed as mean ± SEM. Each experiment was replicated in 3–7 animals. The data collection was randomized. Data were obtained and analyzed by experimenters who did not know the types of treatments of the animals except Figures 3d–g, 4d, 4e, 5d, 5e, 6d–g. No data points were excluded unless specified, and the only exclusion standard was the health condition of the animal. Data from the repeated experiments for the same sub-study were pooled together for statistics. The sample size for each experiment was determined to be either at least 3 animals or 10 neurons. If animals in Figures 3d–g, 4d, 4e, 5d, 5e, 6d–g were successfully induced in vivo changes, we measured responses ex vivo in enough neurons to evaluate the effect of light stimulation. The sample size was presented as “n = x, y”, where “x” refers to the number of slices or neurons, and “y” refers to the number of animals. In electrophysiological experiments, 1–4 recordings were performed using slices from a single animal except for Figures 3d, 3e, 4d, 4e. Slice or neuron-based statistics were performed and reported for electrophysiology and animal-based statistics for behavioral data. Normal distribution was assumed and tested. Variance was estimated for most major results, and no significant difference was found between control and manipulation groups. Statistical significance was assessed in electrophysiological studies using the unpaired or paired t test, or two-way RM ANOVA followed by Student-Newman-Keuls (SNK) method. Behavioral studies were analyzed using the paired t test and one-way RM ANOVA followed by the SNK method. Two-tail tests were performed for all studies. Statistical significance was set at P < 0.05.
Life Sciences Reporting Summary
Further information on experimental design is available in the Life Sciences Reporting Summary.
Supplementary Material
Acknowledgments
We thank Drs. W. Griffith, D. DuBois, M. Han, R. Smith, Y. Zhu, Mr. J. Artz, and Ms. A Mahnke for helpful discussions and technical assistance. This research was supported by NIAAA R01AA021505 (J.W.), NIAAA U01AA025932 (J.W.), and NIDDK R01DK095013 (J.Y.J.).
Footnotes
Data availability
The data that supports the findings of this study are available from the corresponding author upon reasonable request.
Author Contributions
J.W. conceived, designed, and supervised all experiments in the study. J.W. and T.M. wrote a draft of the manuscript. J.W., T.M., and Y.C. revised the manuscript. T.M. also designed and performed electrophysiology experiments and analyzed the data. Y.C. and E.R.H. designed and performed the behavior experiments and analyzed the data. X.W. conducted behavior experiments. J.Y.J. supervised the collection and analyses of biochemical experiments. J.L. and X.G. performed and analyzed the biochemical experiments. C.H. conducted the electrophysiology experiment for virus validation and analyzed the data. Y.C. and X.Y.W. conducted confocal imaging experiments and analyzed the data. All authors discussed the results and commented on the manuscript.
Competing interests
The authors declare no competing financial interests.
References
- 1.Volkow ND, Morales M. The brain on drugs: from reward to addiction. Cell. 2015;162:712–725. doi: 10.1016/j.cell.2015.07.046. [DOI] [PubMed] [Google Scholar]
- 2.Everitt BJ, Robbins TW. Drug addiction: updating actions to habits to compulsions ten years on. Annu Rev Psychol. 2016;67:23–50. doi: 10.1146/annurev-psych-122414-033457. [DOI] [PubMed] [Google Scholar]
- 3.Koob GF, Volkow ND. Neurocircuitry of addiction. Neuropsychopharmacology. 2010;35:217–238. doi: 10.1038/npp.2009.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Luscher C, Malenka RC. Drug-evoked synaptic plasticity in addiction: from molecular changes to circuit remodeling. Neuron. 2011;69:650–663. doi: 10.1016/j.neuron.2011.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yin HH, Knowlton BJ. The role of the basal ganglia in habit formation. Nat Rev Neurosci. 2006;7:464–476. doi: 10.1038/nrn1919. [DOI] [PubMed] [Google Scholar]
- 6.Lovinger DM. Neurotransmitter roles in synaptic modulation, plasticity and learning in the dorsal striatum. Neuropharmacology. 2010;58:951–961. doi: 10.1016/j.neuropharm.2010.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Balleine BW, O’Doherty JP. Human and rodent homologies in action control: corticostriatal determinants of goal-directed and habitual action. Neuropsychopharmacology. 2010;35:48–69. doi: 10.1038/npp.2009.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ma T, Barbee B, Wang X, Wang J. Alcohol induces input-specific aberrant synaptic plasticity in the rat dorsomedial striatum. Neuropharmacology. 2017;123:46–54. doi: 10.1016/j.neuropharm.2017.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang J, et al. Ethanol-mediated facilitation of AMPA receptor function in the dorsomedial striatum: implications for alcohol drinking behavior. J Neurosci. 2012;32:15124–15132. doi: 10.1523/JNEUROSCI.2783-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang J, et al. Long-lasting adaptations of the NR2B-containing NMDA receptors in the dorsomedial striatum play a crucial role in alcohol consumption and relapse. J Neurosci. 2010;30:10187–10198. doi: 10.1523/JNEUROSCI.2268-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Corbit LH, Chieng BC, Balleine BW. Effects of repeated cocaine exposure on habit learning and reversal by N-acetylcysteine. Neuropsychopharmacology. 2014;39:1893–1901. doi: 10.1038/npp.2014.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pascoli V, et al. Contrasting forms of cocaine-evoked plasticity control components of relapse. Nature. 2014;509:459–464. doi: 10.1038/nature13257. [DOI] [PubMed] [Google Scholar]
- 13.Ma YY, et al. Bidirectional modulation of incubation of cocaine craving by silent synapse-based remodeling of prefrontal cortex to accumbens projections. Neuron. 2014;83:1453–1467. doi: 10.1016/j.neuron.2014.08.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gerfen CR, Surmeier DJ. Modulation of striatal projection systems by dopamine. Annu Rev Neurosci. 2011;34:441–466. doi: 10.1146/annurev-neuro-061010-113641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Shen W, Flajolet M, Greengard P, Surmeier DJ. Dichotomous dopaminergic control of striatal synaptic plasticity. Science. 2008;321:848–851. doi: 10.1126/science.1160575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cheng Y, et al. Distinct synaptic strengthening of the striatal direct and indirect pathways drives alcohol consumption. Biol Psychiatry. 2017;81:918–929. doi: 10.1016/j.biopsych.2016.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.MacAskill AF, Cassel JM, Carter AG. Cocaine exposure reorganizes cell type- and input-specific connectivity in the nucleus accumbens. Nat Neurosci. 2014;17:1198–1207. doi: 10.1038/nn.3783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Klapoetke NC, et al. Independent optical excitation of distinct neural populations. Nature methods. 2014;11:338–346. doi: 10.1038/nmeth.2836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fenno L, Yizhar O, Deisseroth K. The development and application of optogenetics. Annu Rev Neurosci. 2011;34:389–412. doi: 10.1146/annurev-neuro-061010-113817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Surmeier DJ, Plotkin J, Shen W. Dopamine and synaptic plasticity in dorsal striatal circuits controlling action selection. Curr Opin Neurobiol. 2009;19:621–628. doi: 10.1016/j.conb.2009.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kreitzer AC, Malenka RC. Endocannabinoid-mediated rescue of striatal LTD and motor deficits in Parkinson’s disease models. Nature. 2007;445:643–647. doi: 10.1038/nature05506. [DOI] [PubMed] [Google Scholar]
- 22.Mattis J, et al. Principles for applying optogenetic tools derived from direct comparative analysis of microbial opsins. Nature methods. 2012;9:159–172. doi: 10.1038/nmeth.1808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yizhar O, et al. Neocortical excitation/inhibition balance in information processing and social dysfunction. Nature. 2011;477:171–178. doi: 10.1038/nature10360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mathur BN, Lovinger DM. Endocannabinoid-dopamine interactions in striatal synaptic plasticity. Frontiers in pharmacology. 2012;3:66. doi: 10.3389/fphar.2012.00066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gerdeman GL, Ronesi J, Lovinger DM. Postsynaptic endocannabinoid release is critical to long-term depression in the striatum. Nat Neurosci. 2002;5:446–451. doi: 10.1038/nn832. [DOI] [PubMed] [Google Scholar]
- 26.Wu YW, et al. Input- and cell-type-specific endocannabinoid-dependent LTD in the striatum. Cell reports. 2015;10:75–87. doi: 10.1016/j.celrep.2014.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Minatohara K, Akiyoshi M, Okuno H. Role of immediate-early genes in synaptic plasticity and neuronal ensembles underlying the memory trace. Front Mol Neurosci. 2015;8:78. doi: 10.3389/fnmol.2015.00078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Girault JA, Valjent E, Caboche J, Herve D. ERK2: a logical AND gate critical for drug-induced plasticity? Curr Opin Pharmacol. 2007;7:77–85. doi: 10.1016/j.coph.2006.08.012. [DOI] [PubMed] [Google Scholar]
- 29.Ye L, et al. Wiring and molecular features of prefrontal ensembles representing distinct experiences. Cell. 2016;165:1776–1788. doi: 10.1016/j.cell.2016.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sun X, Lin Y. Npas4: linking neuronal activity to memory. Trends Neurosci. 2016;39:264–275. doi: 10.1016/j.tins.2016.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang J, et al. Ethanol induces long-term facilitation of NR2B-NMDA receptor activity in the dorsal striatum: implications for alcohol drinking behavior. J Neurosci. 2007;27:3593–3602. doi: 10.1523/JNEUROSCI.4749-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kash TL, Baucum AJ, 2nd, Conrad KL, Colbran RJ, Winder DG. Alcohol exposure alters NMDAR function in the bed nucleus of the stria terminalis. Neuropsychopharmacology. 2009;34:2420–2429. doi: 10.1038/npp.2009.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Rosenmund C, Stevens CF. Definition of the readily releasable pool of vesicles at hippocampal synapses. Neuron. 1996;16:1197–1207. doi: 10.1016/s0896-6273(00)80146-4. [DOI] [PubMed] [Google Scholar]
- 34.Lovinger DM, Kash TL. Mechanisms of neuroplasticity and ethanol’s effects on plasticity in the striatum and bed nucleus of the stria terminalis. Alcohol Res. 2015;37:109–124. [PMC free article] [PubMed] [Google Scholar]
- 35.McCool BA. Ethanol modulation of synaptic plasticity. Neuropharmacology. 2011;61:1097–1108. doi: 10.1016/j.neuropharm.2010.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lobo MK, Nestler EJ. The striatal balancing act in drug addiction: distinct roles of direct and indirect pathway medium spiny neurons. Front Neuroanat. 2011;5:41. doi: 10.3389/fnana.2011.00041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wills TA, et al. GluN2B subunit deletion reveals key role in acute and chronic ethanol sensitivity of glutamate synapses in bed nucleus of the stria terminalis. Proc Natl Acad Sci U S A. 2012;109:E278–287. doi: 10.1073/pnas.1113820109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zalocusky KA, et al. Nucleus accumbens D2R cells signal prior outcomes and control risky decision-making. Nature. 2016;531:642–646. doi: 10.1038/nature17400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang Z, et al. Dopaminergic control of corticostriatal long-term synaptic depression in medium spiny neurons is mediated by cholinergic interneurons. Neuron. 2006;50:443–452. doi: 10.1016/j.neuron.2006.04.010. [DOI] [PubMed] [Google Scholar]
- 40.Calabresi P, Pisani A, Mercuri NB, Bernardi G. Long-term potentiation in the striatum is unmasked by removing the voltage-dependent magnesium block of NMDA receptor channels. Eur J Neurosci. 1992;4:929–935. doi: 10.1111/j.1460-9568.1992.tb00119.x. [DOI] [PubMed] [Google Scholar]
- 41.Mancini M, et al. Memantine alters striatal plasticity inducing a shift of synaptic responses toward long-term depression. Neuropharmacology. 2016;101:341–350. doi: 10.1016/j.neuropharm.2015.10.015. [DOI] [PubMed] [Google Scholar]
- 42.Huang YH, et al. In vivo cocaine experience generates silent synapses. Neuron. 2009;63:40–47. doi: 10.1016/j.neuron.2009.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dong Y, Nestler EJ. The neural rejuvenation hypothesis of cocaine addiction. Trends Pharmacol Sci. 2014;35:374–383. doi: 10.1016/j.tips.2014.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lee BR, et al. Maturation of silent synapses in amygdala-accumbens projection contributes to incubation of cocaine craving. Nat Neurosci. 2013;16:1644–1651. doi: 10.1038/nn.3533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ma YY, et al. Re-silencing of silent synapses unmasks anti-relapse effects of environmental enrichment. Proc Natl Acad Sci U S A. 2016;113:5089–5094. doi: 10.1073/pnas.1524739113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Creed M, Pascoli VJ, Luscher C. Addiction therapy. Refining deep brain stimulation to emulate optogenetic treatment of synaptic pathology. Science. 2015;347:659–664. doi: 10.1126/science.1260776. [DOI] [PubMed] [Google Scholar]
- 47.Creed M, Ntamati NR, Chandra R, Lobo MK, Luscher C. Convergence of reinforcing and anhedonic cocaine effects in the ventral pallidum. Neuron. 2016;92:214–226. doi: 10.1016/j.neuron.2016.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wang J, et al. Alcohol elicits functional and structural plasticity selectively in dopamine D1 receptor-expressing neurons of the dorsomedial striatum. J Neurosci. 2015;35:11634–11643. doi: 10.1523/JNEUROSCI.0003-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sidhpura N, Parsons LH. Endocannabinoid-mediated synaptic plasticity and addiction-related behavior. Neuropharmacology. 2011;61:1070–1087. doi: 10.1016/j.neuropharm.2011.05.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wei X, et al. Dopamine D1 or D2 receptor-expressing neurons in the central nervous system. Addiction biology. 2017 doi: 10.1111/adb.12512. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Huang CCY, et al. Stroke triggers nigrostriatal plasticity and increases alcohol consumption in rats. Sci Rep. 2017;7:2501. doi: 10.1038/s41598-017-02714-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yin HH, Park BS, Adermark L, Lovinger DM. Ethanol reverses the direction of long-term synaptic plasticity in the dorsomedial striatum. Eur J Neurosci. 2007;25:3226–3232. doi: 10.1111/j.1460-9568.2007.05606.x. [DOI] [PubMed] [Google Scholar]
- 53.Cruikshank SJ, Urabe H, Nurmikko AV, Connors BW. Pathway-specific feedforward circuits between thalamus and neocortex revealed by selective optical stimulation of axons. Neuron. 2010;65:230–245. doi: 10.1016/j.neuron.2009.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chen TW, et al. Ultrasensitive fluorescent proteins for imaging neuronal activity. Nature. 2013;499:295–300. doi: 10.1038/nature12354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang J, Yeckel MF, Johnston D, Zucker RS. Photolysis of postsynaptic caged Ca2+ can potentiate and depress mossy fiber synaptic responses in rat hippocampal CA3 pyramidal neurons. J Neurophysiol. 2004;91:1596–1607. doi: 10.1152/jn.01073.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.June HL, Gilpin NW. Operant self-administration models for testing the neuropharmacological basis of ethanol consumption in rats. Curr Protoc Neurosci. 2010:11–26. doi: 10.1002/0471142301.ns0912s51. Chapter 9, Unit 9 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Carnicella S, Amamoto R, Ron D. Excessive alcohol consumption is blocked by glial cell line-derived neurotrophic factor. Alcohol. 2009;43:35–43. doi: 10.1016/j.alcohol.2008.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Simms JA, Bito-Onon JJ, Chatterjee S, Bartlett SE. Long-Evans rats acquire operant self-administration of 20% ethanol without sucrose fading. Neuropsychopharmacology. 2010;35:1453–1463. doi: 10.1038/npp.2010.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zhao X, et al. Regulation of lipogenesis by cyclin-dependent kinase 8-mediated control of SREBP-1. J Clin Invest. 2012;122:2417–2427. doi: 10.1172/JCI61462. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.