

Abstract
An emerging paradigm shift for disease diagnosis is to rely on molecular characterization beyond traditional clinical and symptoms-based examinations. Although genetic alterations and transcription signature were first introduced as potential biomarkers, clinical implementations of these markers are limited due to low reproducibility and accuracy. Instead, epigenetic changes are considered as an alternative approach to disease diagnosis. Complex epigenetic regulation is required for normal biological functions and it has been shown that distinctive epigenetic disruptions could contribute to disease pathogenesis. Disease-specific epigenetic changes, especially DNA methylation, have been observed, suggesting its potential as disease biomarkers for diagnosis. In addition to specificity, the feasibility of detecting disease-associated methylation marks in the biological specimens collected non-invasively, such as blood samples, has driven the clinical studies to validate disease-specific DNA methylation changes as a diagnostic biomarker. Here we highlight the advantages of DNA methylation signature for diagnosis in different diseases and discuss the statistical and technical challenges to be overcome before clinical implementation.
Keywords: DNA methylation, epigenetics, molecular diagnosis, biomarker, liquid biopsy, cancer, brain disorders
1. Introduction
Inter-individual phenotypic diversity is not sufficiently explained by the approximately 0.1 % of genetic variation between individuals (Altshuler et al., 2015). In addition, obtaining the sequence of the human genome could not account for how the cells in multicellular organisms sharing the same genetic code yet exhibit unique gene expression for their cellular functions (Waddington, 2012). Epigenetics, first introduced by C. H. Waddington in 1939, was proposed as an additional layer of gene regulation in the limited context of primary DNA sequence differences (Jenuwein and Allis, 2001; Bernstein et al., 2010; Waddington, 2012). Various epigenetic modifications including DNA methylation, histone modifications, and chromatin remodeling, can contribute to cell-type specific gene expression signature necessary for cellular function (Doi et al., 2009; Mack et al., 2016). These epigenetic modifications are heritable but reversible and dynamic, thereby not only establishing specific cellular states but also responding to changes in microenvironment, which confers cellular plasticity (Mack et al., 2016). In addition, epigenetic dysregulation could contribute to the development and progression of many diseases (Esteller, 2008; Hwang et al., 2017). For example, aberrant epigenetic programs, such as hypermethylation at CpG island promoters of non-mutated tumor suppressor genes, are recurrently identified in pediatric and adult brain tumors, leading to proliferative advantages and aggressive phenotypes during tumorigenesis (Suva et al., 2013; Mack et al., 2016). Abnormal epigenetic changes are also strongly associated with neurodegeneration by modifying disease risk, age of manifestation, and progression (Qureshi and Mehler, 2013; Hwang et al., 2017). In addition to their biological role as pathogenic factors, epigenetic marks associated with specific disease are considered as emerging biomarkers for diagnosis and predictors of treatment response and prognosis in many diseases (Paluszczak and Baer-Dubowska, 2006; Qureshi and Mehler, 2013). Intriguingly, it has been shown that the epigenetic changes can also be detected in different biologic fluids, such as blood, urine, and fecal samples (Van Neste et al., 2012; Haggarty, 2015; Diaz-Lagares et al., 2016; Jakubowski and Labrie, 2017). Among epigenetic modification assays, the DNA methylation-based assay was the first FDA approved screening test for colorectal cancer (Song et al., 2017b), suggesting that methylation analysis can be implemented in clinics as a screening test of different disease types. In this review, we will focus on the application of DNA methylation and its derivatives to diagnose various diseases and summarize the technical and analytical challenges to move their application into clinic.
2. DNA modifications in genome: cytosine modifications and beyond
The most abundant DNA modification is cytosine methylation (5-methycytosine, 5mC) that is the addition of a methyl group to the 5′ position of the cytosine pyrimidine ring. This direct chemical modification to the DNA is conserved during evolution and plays a critical role in various cellular processes. Hypermethylation at promoters, for instance, suppresses gene expression by either inhibiting the binding of transcription factors or by recruiting complex proteins known as methyl-CpG-binding domain proteins (MBDs) (Robertson, 2005; Schubeler, 2015; Yao et al., 2016). 5mC is also involved in the repression of transposable elements, contributing to genome integrity (Slotkin and Martienssen, 2007). In addition to the identification of biological functions of 5mC, the discovery of DNA methyltransferases (DNMTs) including DNMT1, DNMT3A, and DNMT3B provides the mechanism how to maintain or generate 5mC in the genome (Yoder and Bestor, 1998; Okano et al., 1999; Schubeler, 2015). In the context of disease pathogenesis, the mutations within the DNMT3B gene have been associated with the childhood onset of the neurodevelopmental disorder immunodeficiency-centromeric instability-facial anomalies (ICF), suggesting the importance of precise regulation of 5mC (Jin and Robertson, 2013). Methylation was assumed as a permanent modification once created due to the chemical stability of the methyl group and a lack of detection of demethylase and other modifications. Two independent studies, however, dramatically changed the understanding of dynamic regulation in 5mC by identifying ten-eleven translocation (TET) proteins that can oxidize 5mC to 5-hydroxymethylcytosine (5hmC) and generate further oxidative derivatives 5-formylcytosine (5fC) and 5-carboxylcytosine (5caC) (Fig. 1) (Ito et al., 2011; Plongthongkum et al., 2014; Schubeler, 2015; Yao et al., 2016). Although the overall abundance of intermediates (5hmC, 5fC and 5caC) is much less than 5mC, further investigation into their biological functions in transcriptional regulation during embryogenesis and neurodevelopment revealed the independent roles of these intermediates beyond the demethylation process (Lappalainen and Greally, 2017). Interestingly, the presence of another DNA methylation, adenine methylation (N6-methyladenine, 6mA), was recently identified in mammals even though the abundance is lower than that observed in prokaryotes (Heyn and Esteller, 2015). While host defense is the main role of 6mA in prokaryotic systems, 6mA is deemed as a suppressive mark in eukaryotes based on its significant enrichment at transposable elements (Heyn and Esteller, 2015). Recent studies support the role of 6mA in regulating neuronal gene expression (Yao et al., 2017); however, detailed functional investigation of 6mA’s role in transcriptional regulation is needed. Identification of methylation and demethylation enzymes involved in adenine methylation will be critical.
Fig. 1. Cytosine modifications in mammalian genome.
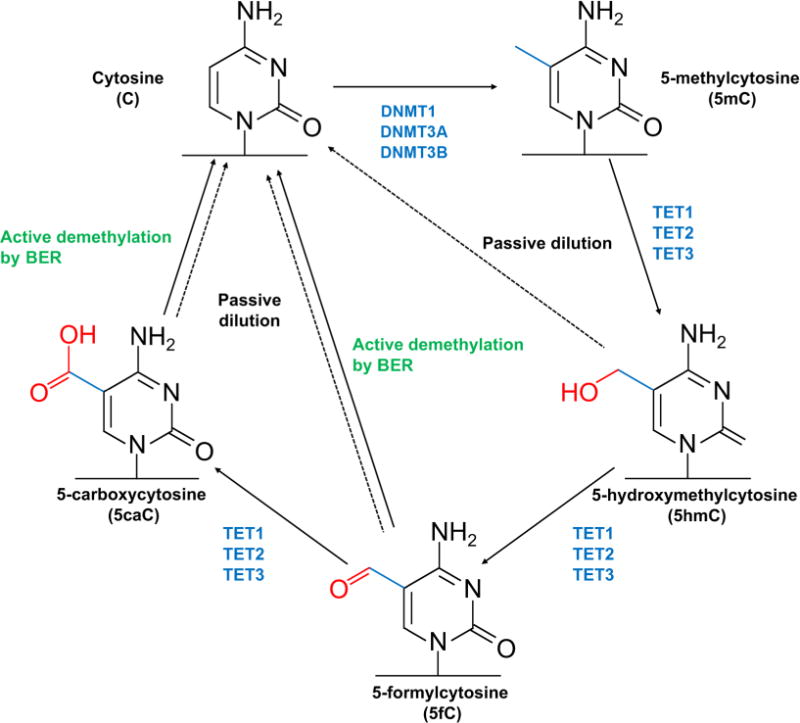
The fifth position of cytosine can be methylated by DNA methyltransferases (DNMT1, DNMT3A, and DNMT3B) to generate 5-methylcytosine (5mC). The methyl group of 5mC can be oxidized by ten-eleven translocation (TET) family enzymes (TET1, TET2, and TET3), generating 5-hydroxymethylcytosine (5hmC), 5-formlycytosine (5fC), and 5-carboxycytosine (5caC). While 5mC is maintained by the interaction between the replication machinery and DNMT1, no maintenance mechanisms exist for the oxidative derivatives; therefore, the levels of 5hmC, 5fC and 5caC are diminished over the replication (passive dilution). In addition, 5fC and 5caC can be excised by thymine DNA glycosylase (TDG), and eventually replaced with cytosine (active demethylation by base excision repair (BER)). Both passive and active demethylation mechanisms contribute to dynamics of cytosine modification.
3. Diagnostic role of DNA methylation markers in oncology
Since molecular markers are mechanistically associated with tumorigenesis, there have been multiple attempts to utilize molecular markers such as genetic, epigenetic, and transcriptomic alterations for cancer diagnosis (Esteller, 2008; Hanahan and Weinberg, 2011; Suva et al., 2013). Alterations in primary DNA sequence are primary contributors of cancer initiation, development, and progression (Hanahan and Weinberg, 2011). Although genetic instability is one of the hallmarks of cancer, a single genetic alteration cannot be reliably used as a diagnostic biomarker in that even the most frequently mutated gene, TP53, showed at best a 50% mutation rate in most tumor types (Olivier et al., 2010; Hanahan and Weinberg, 2011).
Alternatively, DNA methylation is deemed as a great biomarker for cancer diagnosis in that methylation signature is more cancer origin-specific and commonly identified within the same primary tumor types (Jenuwein and Allis, 2001; Suva et al., 2013). In addition, DNA can be isolated with high quality and sufficient yield from frozen biospecimens and formalin-fixed paraffin embedded (FFPE) tissues compared to RNA and protein, indicating that DNA methylation profiles may be a more promising diagnostic approach than RNA- or protein-based assays (Lou et al., 2014). Many studies have already proposed the advantages of DNA methylation signature in cancer diagnosis. Here, we will highlight the well-established methylation signatures for diagnosis in the most common solid tumors including colorectal, prostate, lung, and breast cancers, as well as ongoing clinical and preclinical research to evaluate the specificity and sensitivity of these markers.
3.1. SEPT9, the first FDA-approved methylation assay for colorectal cancer screening
Colorectal cancer (CRC) is the third leading cause of cancer death in the United State even though the death rate in both men and women has been dropping for several decades (Siegel et al., 2018). Early diagnosis for CRC is one of most effective strategies to increase survival rate. In this regard, the U.S. Preventive Services Task Force (USPSTF) recommends regular screening using stool-based tests (including guaiac-based fecal occult blood test (gFOBT), fecal immunochemical test (FIT) and multitargeted stool DNA test (FIT-DNA)), direct visualization tests (including colonoscopy, computed tomography (CT) colonoscopy and sigmoidoscopy), or serology tests (e.g., SEPT9 methylated DNA test), in adult aged 50 to 75 years (Ryan and Creagh, 2018). Among these screening tests, colonoscopy has the highest efficacy, but the invasive procedure can increase morbidity as well as induce psychological pain (Ryan and Creagh, 2018). Therefore, considerable effort is being made to identify novel cancer markers in biospecimens such as serum, plasma, and stool for the development of less invasive screening tests.
Aberrant DNA methylation is a key molecular event for CRC (Toyota et al., 2000; Toyooka et al., 2002; Ebert et al., 2005). The cancer-specific DNA methylation is identified not only in primary tumor tissues but also in circulating tumor DNA (ctDNA) (Lofton-Day et al., 2008), which enables the development of novel blood-based early detection tests for CRC. One of the major challenges to detect methylated targets in blood is the differentiation of target DNA from a vast excess of unmethylated or partially methylated background DNA (Lofton-Day et al., 2008). The systemic, unbiased approach using restriction enzyme-based discovery methods followed by microarray and real-time PCR testing identified three candidate genes, transmembrane protein with EGF-like and two follistatin like domains 2 (TMEFF2), nerve growth factor receptor (NGFR), and septin 9 (SEPT9), as potential biomarkers for CRC (Lofton-Day et al., 2008). The methylation levels of candidate genes were significantly higher in CRC tissue and CRC-derived DNA compared to healthy colon tissue, other tissues, and peripheral blood lymphocytes (PBLs) (Lofton-Day et al., 2008). The three methylation markers were also detected in plasam samples from CRC patients with high sensitivity: 69% for SEPT9, 65% for TMEFF2, 51% for NGFR (Lofton-Day et al., 2008). Among three markers, the SEPT9 gene was a primary focus in the following studies due to higher sensitivity than other markers. Multiple clinical studies with more than 5000 subjects validated 48.2%–95.6% sensitivity and 79.1%–99.1% specificity of the SEPT9 methylation assay (Song et al., 2017b). Compared to early-stage SEPT9 methylation assay research kits and commercialized kits (ARUP Lab LDT assay and Epi proColon 1.0), the performance of two latest commercialized assays, Epi proColon 2.0 and SensiColon, was improved and stabilized on the basis of a higher sensitivity at 71.1%–95.6% and a higher specificity at 81.5%–99% (Song et al., 2017b). The sensitivity (63.9%–80.9%) and specificity (75.5%–86.3%) of the SEPT9 methylation assay are comparable to FIT (sensitivity: 58.2%–76.5%; specificity: 94.1%–98.9%) (Johnson et al., 2014). There are still concerns on the adequacy of the end point for approval, the increase in false-positive results, and an unclear relationship between the SEPT9 assay and the improvement of disease-specific mortality (Parikh and Prasad, 2016). However, the FDA approved the SEPT9 methylation assay (Epi proColon) in 2016, since it can benefit average-risk population who refuse invasive screening test, but prefer blood-based test (Song et al., 2017b).
3.2. GSTP1, a potent diagnostic marker for early prostate cancer diagnosis
Prostate cancer (PCa) has one of the highest cancer incidence rates in men and is a leading cause of cancer death (Siegel et al., 2017). Since early diagnosis can lead to better prognosis, many screening and molecular testing have been evaluated for early detection of PCa cases (Crawford, 2009; Schroeder et al., 2009; Wolf et al., 2010). PSA (prostate-specific antigen) testing is currently available as a PCa screening test recommended for men aged 50 and older. This test measures the amount of PSA in a blood sample with PSA levels frequently elevated in the presence of PCa (Stamey et al., 1987; Catalona et al., 1994; Carter, 2000; Wolf et al., 2010). However, PSA elevation in blood is also associated with other benign (non-cancerous) conditions such as enlarged prostate (benign prostatic hyperplasia, BPH) and inflamed or infected prostate (prostatitis) (Van Neste et al., 2012). In this regard, over-diagnosis and overtreatment based on positive PSA test results can yield unnecessary and invasive diagnostic procedures to healthy people and produce a substantial socio-economic cost (Mettlin et al., 1997; Barry, 2001). Accordingly, demands on a better PCa screening test have led to the identification of more accurate and reliable molecular alternatives.
Since 1994 when the link between glutathione S-transferase Pi 1 protein (GSTP1) and PCa was discovered, hypermethylation at the CpG island promotor region of GSTP1 is frequently identified exclusively in tumor tissues, even in early stage (Lee et al., 1994; Chu et al., 2002a; Chu et al., 2002b; Maruyama et al., 2002; Bernardini et al., 2004). In addition, histological analysis using immunohistochemistry (IHC) demonstrated that the abnormal hypermethylation is significantly associated with negative expression of GSTP1 in PCa lesion compared to adjacent normal (Lee et al., 1994; Song et al., 2002), which could result from the inhibition of transcriptional factor SP1 binding (Lee et al., 1994). Due to recurrent methylation in PCa, a number of recent clinical studies have tried to evaluate the sensitivity and specificity of GSPT1 methylation assay as a novel diagnostic test for PCa. Recent meta-analysis by integrating 35 independent studies using more than 1000 patients revealed both high sensitivity (82%) and high specificity (95%) of tissue-based GSTP1 methylation assay while blood-based PSA testing exhibited high sensitivity (82.8%) but low specificity (38.6%) (Van Neste et al., 2012). However, another meta-analysis which evaluated the performance of GSTP1 methylation assay using body fluids including plasma, serum and urine, showed 89% of the pooled specificity and 52% of the pooled sensitivity (Wu et al., 2011). Low sensitivity of non-invasive GSTP1 methylation assay may be due to a technical difficulty to discriminate tumor-derived DNA fraction from excessive background DNA in blood or urine. Given that the combination of GSTP1 with other candidate genes such as adenomatous polyposis coli (APC) and retinoic acid receptor beta (RARB) in tissue-based methylation assay showed improved sensitivity (Van Neste et al., 2012), multi-target approach could resolve low sensitivity issue of non-invasive methylation assay. Despite low sensitivity, high specificity of current non-invasive GSTP1 methylation analysis can complement PSA test, contributing to the improvement of detection accuracy and management of PCa cases as well as non-cancerous symptoms in the future.
3.3. Identification of lung cancer-specific methylation signature for early diagnosis
Despite high mortality rate, lung cancer diagnosis is still challenging in that most lung cancer patients are detected at advanced stages (Liloglou et al., 2014; Siegel et al., 2017). Chest X-ray is a currently used approach to detect lung cancer (Nikolaidis et al., 2012). A solitary pulmonary nodule (SPN) is, however, detectable on reaching the reliable size of at least 9 mm after approximately 30 doubling times (Erasmus et al., 2000). Given that late diagnosis substantially links to poor survival rate, early diagnosis of lung cancer by identifying molecular signature in minimally or non-invasive specimens benefits treatment and prognosis of lung cancer (Leng et al., 2012; Nikolaidis et al., 2012).
Distinctive transcriptions in lung cancer are distinguishable from normal as well as other lung infectious disorders such as pulmonary tuberculosis, pulmonary sarcoidosis, and pneumonias (Bloom et al., 2013). Although transcriptional signatures detectable in blood are a promising biomarker for non-invasive lung cancer diagnosis, without prominent signal-to-noise ratios (SNRs) (Bloom et al., 2013), it is challenging to establish a reliable marker if the amount of tumor-derived mRNA is scarce. On the other hand, epigenetic signature, primarily DNA methylation, usually has a bimodal distribution, which facilitates analysis of sparse genetic materials in biological specimens (e.g., bronchial aspirate (BAS) or sputum), and therefore, enables early detection (Ahmed, 2010; Nikolaidis et al., 2012; Diaz-Lagares et al., 2016). In addition, progressive change in epigenetics across disease stages endows the implementation of epigenetic markers to determine the progression and stage of lung cancer (Diaz-Lagares et al., 2016).
A recent clinical study identified a novel epigenetic signature based on the four genes: branched chain amino acid transaminase 1 (BCAT1), cysteine dioxygenase type 1 (CDO1), tripartite motif containing 58 (TRIM58), and zinc finger protein 177 (ZNF177) (Diaz-Lagares et al., 2016). In this study, the hypermethylation of these four genes was recurrently detected in discovery cohorts (the Cancer Genome Atlas (TCGA) and the CURELUNG FP7 Consortium datasets) and independent cohorts, but globally absent in normal counterpart (Diaz-Lagares et al., 2016). The hypermethylation signature is also identified in minimally invasive samples such as BAS, bronchoalveolar lavage (BAL) fluid, and sputum (Diaz-Lagares et al., 2016). In addition, the expression of hypermethylated genes was downregulated, consistent with well-established mechanism of DNA methylation to silence gene expression (Herman and Baylin, 2003; Belinsky, 2004; Kwon et al., 2012; Diaz-Lagares et al., 2016). This study provides a novel epigenetic predictive model that may help to improve lung cancer diagnosis.
3.4. DNA methylation analysis for early diagnosis of breast cancer
Breast cancer is the most prevalent malignancy among women in the U.S. (Siegel et al., 2017). The advent of high-throughput assay led to intensive investigation of breast cancer at the molecular level since the early 2000s (Sorlie et al., 2001; Banerji et al., 2012; Yates et al., 2015). The molecular characterization of genomics and epigenetics for the past decades contributes to the classification of breast cancer patients into four major subtypes (including luminal A, luminal B, triple negative/basal-like and HER2 type), facilitating the establishment of better treatment for each subtype (Sorlie et al., 2001). Additionally, the development of targeted therapies such as Herceptin opened a new era toward precision medicine through molecular profiling, a promising approach to improve patients’ survival (Slamon et al., 2001). In addition to therapeutic benefits, genetic and epigenetic analyses contributed to improvement of molecular pathology based on tumor-specific gene panels (Jovanovic et al., 2010; Banerji et al., 2012; Yates et al., 2015). Mammogram-based diagnosis is a gold standard of breast cancer screening for women at age 40 (Paalman et al., 2016). However, up to 4% of false positive rate engendered an unnecessary invasive procedure to confirm breast cancer. Three key genetic mutations/amplification in estrogen receptor (ER), progesterone receptor (PR) and Erb-B2 receptor tyrosine kinase 2 (ERBB2/HER2) are initially considered as molecular candidates for diagnostic testing; however, 15%–20% of breast cancer patients represents triple-negative breast cancer (TNBC), and no genetic mutations account for TNBC (Sorlie et al., 2001; Yates et al., 2015). As such, genetic abnormalities are not ideal biomarkers for breast cancer detection through blood testing (Paalman et al., 2016).
A number of recent studies investigated the potential of methylated cell-free DNA (cfDNA) as a non-invasive biomarker for breast cancer diagnosis (Cheuk et al., 2017). Methylation analysis of three genes, Ras association domain family member 1 (RASSF1A), GSTP1, and RARB, exhibited a better sensitivity for the detection of early-stage breast cancer (sensitivity: 22%–26%) than assays of two conventional serum tumor markers, carcinoembryonic antigen (CEA) and cancer antigen 15-3 (CA15-3) (sensitivity: 8%–19%) (Yamamoto et al., 2012). Although sensitivity of CEA/CA15-3 assay (sensitivity: 59%) was comparable to that of methylation analysis using the three genes (sensitivity: 55%) in the case of metastatic cancer, combined assays improved overall sensitivity (78%), implying that methylation analysis could complement conventional screening assay for breast cancer (Yamamoto et al., 2012). In addition, the promoter of SRY (sex determining region Y)-box 17 (SOX17) gene was significantly methylated in primary breast cancer tissues (86.0%) as well as cfDNA in early breast cancer cases (34.5%) and metastatic cancer cases (40.7%) (Chimonidou et al., 2013); however, methylation analysis of the SOX17 gene alone was not sufficient to detect breast cancer. Methylation assay using multi-gene panel, therefore, should be taken into account to enhance the sensitivity and specificity of breast cancer screening.
4. Molecular diagnostics with DNA methylation signature for brain disorders
Neurological and psychiatric disorders are the disease of central and peripheral nervous system, causing an increase in chronic disability and morbidity as well as mortality worldwide (Group, 2017). Despite growing population, diagnosis mainly relies on medical history, a review of symptoms and conditions, and physical examination. Some imaging techniques such as a CT scan and magnetic resonance imaging (MRI) are also used for a diagnostic test, but laboratory screening tests and genetic tests, if available, are the first-line diagnostic tests due to quick and accurate procedure. Early diagnosis is critical to delay disease progression and relieve disease-associated symptoms with proper intervention and treatment; therefore, substantial and intensive efforts are required to develop a more accurate and reliable molecular diagnostic test.
A number of genome-wide association (GWA) studies have contributed to the identification of risk loci and genetic variations associated with common neurological and psychiatric diseases including autism spectrum disorder (ASD), migraine, schizophrenia (SZ), epilepsy, Alzheimer’s disease (AD) and Parkinson’s disease (PD) (Schizophrenia Working Group of the Psychiatric Genomics, 2014; Poduri, 2015; Gormley et al., 2016; Van Cauwenberghe et al., 2016; Autism Spectrum Disorders Working Group of The Psychiatric Genomics, 2017; Chang et al., 2017). However, most of disease-associated genetic variations are too low-penetrance to be used as diagnostic markers although these large-scale studies have provided a hint to develop new drugs for the alleviation of clinical symptoms for AD (Han and Mook-Jung, 2014; Schneider et al., 2014). Since dynamic DNA methylation during both early development and adulthood confers neuronal plasticity in response to environmental stimuli (Baker-Andresen et al., 2013; Wang et al., 2016), its dysregulation is associated with pathogenesis (Grayson and Guidotti, 2013; Delgado-Morales et al., 2017). In addition, recent studies have highlighted disease-specific cell-free circulating DNA methylation patterns as the potential biomarkers (Hwang et al., 2017; Jakubowski and Labrie, 2017). We will review the latest studies that identify methylation candidates for future diagnosis of common neurological and psychiatric disorders including ASD, SZ, AD and PD.
4.1. ASD
ASD is a complex developmental disorder characterized by the disability of social communication and interaction. The improvement of clinical outcomes by early intervention has been proven by early intervention programs such as the Early Start Denver Model (ESDM) and early intervention services supported by the Individuals with Disabilities Education Act (IDEA), but diagnosis in infants is still challenging due to lack of standardized molecular screening tests. Several twin studies and family-based studies revealed high heritability of ASD (55%–80%) (Lichtenstein et al., 2010; Sandin et al., 2014), promoting GWA studies to identify genetic factors associated with ASD pathogenesis (Autism Spectrum Disorders Working Group of The Psychiatric Genomics, 2017). Among several candidate genes, the association of variants of the oxytocin receptor gene (OXTR) with ASD are validated in multiple GWA studies, and subsequent functional studies elucidated that OXTR and oxytocin (OXT; a substrate of OXTR) plays a critical role in social perceptual process and the regulation of affiliative behavior (Ylisaukko-oja et al., 2006; LoParo and Waldman, 2015). In addition to its variants, converging evidence suggested the association of promoter methylation at OXTR with ASD (Jack et al., 2012; Kumsta et al., 2013; Elagoz Yuksel et al., 2016). Another study showed hypermethylation of the OXTR promoter in fetal membrane from preterm birth, a strongly associated condition with ASD incidence, implying that OXTR methylation in fetus could be a surrogate marker of ASD (Behnia et al., 2015).
Moreover, array-based genome-wide methylation profiling using brain tissues or blood samples identified hypomethylation in several genes such as phosphatidylinositol 3-kinase catalytic subunit type 3 (PIK3C3), methylthioribose-1-phosphate isomerase (MRI1), gamma-aminobutyric acid type B receptor subunit 1 (GABBR1) and Mir124-2 in affected patients compared to healthy control, whereas other genes showed hypermethylation, including family with sequence similarity 124 member B (FAM124B) and long non-coding RNA nuclear enriched abundant transcript 1 (lnNEAT1) (Nardone et al., 2017; Wong et al., 2014). Along with differential methylation levels at specific genes, recent multivariate data analysis revealed that the global level of DNA methylation combined with the concentration of other metabolites in blood could be used as a potential classifier and predictive marker for adaptive behaviors (Howsmon et al., 2017). Although DNA methylation has not been widely studied as a diagnostic biomarker for ASD, more research can evaluate the potential of altered methylation signature frequently detected in ASD for early diagnosis in the future.
4.2. SZ
SZ is a severe mental disorder that impairs a person’s ability to think, feel and behave clearly. Current diagnosis is based on physical exam which checks whether a person has two or more of the following SZ-associated symptoms persistently: delusions, hallucinations, disorganized speech, disorganized or catatonic daze, bizarre or hyperactive behavior. Although antipsychotic medicines are prescribed to alleviate the symptoms and help normal daily life, early intervention and preventative approach before onset of disease are the main goal to manage this disease effectively. Therefore, recent many studies aim to identify blood-based molecular biomarkers for early diagnosis. Nearly 40% of molecular biomarker studies focused on protein level changes (Lai et al., 2016). In recent multi-cohort analysis, a blood-based biomarker containing an optimized set of 26 analytes showed high sensitivity (59%–91%) and specificity (65%–81%) in terms of discriminating affected patients from healthy control (Chan et al., 2015). However, another experimental target, methylation and its derivatives, drew more attention since proteins implicated in DNA modifications are frequently altered in SZ (Grayson and Guidotti, 2013). Increased level of total methylation in telencephalic GABAergic neurons is frequently detected in affected patients, which is caused by aberrant overexpression of DNA methyltransferase 1 (DNMT1) and DNMT3A (Zhubi et al., 2009). As the consequence of global hypermethylation, the mRNA expression of glutamic acid decarboxylase 1 (GAD1), an enzyme involved in GABA synthesis, is significantly reduced in brain (Akbarian and Huang, 2006). In addition, the methylation level of other genes in GAD1 regulatory network was assayed, resulting in the identification of disease-associated tissue-specific methylation enrichment at three genes, msh homeobox 1 (MSX1), cyclin D2 (CCND2), and death domain associated protein (DAXX) in hippocampus of SZ patients relative to healthy control (Ruzicka et al., 2015). Although these studies highlighted the association of abnormal methylation with SZ, further investigation to determine whether differential methylation patterns associated with SZ are replicated in blood should precede the development of DNA methylation biomarkers for early diagnosis of SZ.
4.3. AD
AD, a major form of dementia, affects estimated 48.6 million people worldwide in 2015, and is characterized by a progressive reduction in neuronal functions and the distinguishable accumulation of two different protein aggregates, amyloid plaques and neurofibrillary tangles, in brain (Sanchez-Mut and Graff, 2015). The progression, albeit there exists interpersonal variability, shows slow and irreversible patterns. Intervention in the early stage is a proven successful management of AD progression. However, in most cases, clinical diagnosis is not made in a timely manner due to a lack of solid molecular biomarkers. Deep sequencing of AD samples contributed to the identification of AD-associated genetic factors (Guerreiro et al., 2012; Vinkhuyzen et al., 2013), but there are no strong driver mutations hitherto to explain AD pathogenesis, suggesting the critical role of non-genetic factors. AD-specific alterations in DNA methylation and histone modification have been explored (De Jager et al., 2014; Sanchez-Mut and Graff, 2015). Although global hypomethylation is a general trend of AD samples, hypermethylation of two genes, sorbin and SH3 domain containing 3 (SORBS3) and Ankyrin 1 (ANK1), has been frequently reported in two AD mouse models (i.e., APPswe/dE9 and 3xTg-AD) and longitudinal studies of AD-affected samples (De Jager et al., 2014; Sanchez-Mut and Graff, 2015). On the other hand, the brain derived neurotrophic factor (BDNF) gene, showing increase in methylation in independent Asian AD cohorts, is rarely methylated in Caucasian AD population, suggesting differential methylation could be associated with genetic background (Chang et al., 2014; Carboni et al., 2015; Nagata et al., 2015). As such, one or two methylation loci are not suitable to diagnose AD in multi-genetic background population (Fransquet et al., 2018), but emerging interests in methylation profiles using a large AD cohort can lead to the identification of methylation signature associated with AD incidence in near future, which will open a new era to facilitate AD diagnosis in an early phase.
4.4. PD
PD is an adult onset neurodegenerative disease, which more than 10 million people suffer from worldwide (Jakubowski and Labrie, 2017). Better treatment outcome could be achieved by early diagnosis and intervention before severe disease progression (Pilleri and Antonini, 2015). The identification of molecular biomarkers is, hence, an ongoing interest in PD research area (Valadas et al., 2015; Yeo et al., 2015). Recent exploration of epigenetic alterations in PD samples contributed to a better understanding of PD etiology and prediction of disease development of at-risk individuals (Jakubowski and Labrie, 2017). Indeed, significant hypomethylation at the promoter of the α-synuclein (SNCA) gene in PD cases was recurrently described in three independent studies (Jowaed et al., 2010). Importantly the changes found in brain tissues could also be detected in blood samples. However, age-dependent differential methylation patterns of SNCA suggest that one biomarker is not sufficient for accurate PD diagnosis. Genome-wide DNA methylation analyses by either probe-based or sequencing-based platform have provided more than 700 candidate epigenetic loci differentially expressed in brain, blood, and saliva samples from PD cases (Chuang et al., 2017; Jakubowski and Labrie, 2017). Mitochondrial methylation change at the D-loop region, a key locus of transcription and replication of mitochondria, is considered as a plausible epigenetic mark for PD diagnosis (Jakubowski and Labrie, 2017). Relative to healthy individuals, almost all CpG and non-CpG sites of the D-loop region of mtDNA have shown a loss of DNA methylation in PD patients without the alteration of hydroxymethylation at the same region (Jakubowski and Labrie, 2017). Recurrent hypomethylation information on mtDNA can be supplemented to improve the accuracy of PD diagnosis in the case that the pattern found in brain tissues could also be detected in blood sample.
5. Challenges of developing non-invasive diagnostic epigenetic testing
Epigenetic profiling, mainly DNA methylation analysis, is a promising approach to improve current diagnostic problems in clinics (Jankowska et al., 2015). Along with the advancement of sequencing technology (Table 1), whole-genome methylation profiling allows to identify many candidate variations for precise classification of disease status in discovery phase (Paluszczak and Baer-Dubowska, 2006; Fernandez et al., 2012; Moran et al., 2016b; Hwang et al., 2017). However, in a clinical setting, not all methylation information across the genome is needed for diagnosis. How many DNA methylation sites are needed for accurate diagnosis should be determined by meta-analysis of multiple clinical studies and large prospective studies to evaluate the combined performance of methylation analysis and establish the relationship between methylation status of multi-loci and disease (Diaz-Lagares et al., 2016). In addition, scientific evidence with the limited number of samples would not be sufficient to substitute the epigenetic information for standard diagnosis methods due to population bias (Moran et al., 2016b). Epigenetic modifications could be influenced by biological age, diets, and environment. Since normal controls are usually recruited from family members or someone living in the same geographic area, the controls included in clinical studies may not represent healthy general population. In the case of rare diseases, such a situation can be more problematic. Even if the false positive rate of identified epigenetic signature in healthy groups is less than 1%, positive predictive value (PPV), the major performance parameter of a diagnostic test, would be small due to rare incidence rate of disease. External validation by integrating normal population-based studies, including human epigenome consortium and the Roadmap Epigenome Project, will fortify the specificity of epigenetic signature in discovery phase (Bernstein et al., 2010; Heyn and Esteller, 2012; Davis et al., 2018).
Table 1.
DNA methylation profiling techniques
| Method | Description |
|---|---|
| Whole-genome bisulphite sequencing (WGBS) | WGBS (aslo known as MethylC-seq) is a sequencing-based approach to profile DNA methylomes with an unbiased manner (Frommer et al., 1992). This approach utilizes the selective chemical conversion of unmethylated cytosines by treating bisulphite, allowing the detection of genome-wide differential DNA methylation regions (DMRs). WGBS is widely used to identify novel DMRs under disease status (Heyn and Esteller, 2012). |
| Methylation DNA immunoprecipitation sequencing (MeDIP-seq) | MeDIP-seq utilizes the 5mC-specific antibody to enrich genome-wide DNA methylation regions (Heyn and Esteller, 2012; Jacinto et al., 2008). Despite containing less information than WGBS, MeDIP-seq can cover large genomic regions with a substantial resolution and low-cost. In discovery phase, MeDIP-seq is a suitable alternative to detect disease-specific hypermethylated regions in a quantitative manner (Heyn and Esteller, 2012). |
| Reduced representation bisulfite sequencing (RRBS) | RRBS is a restriction enzyme based technique to reduce non-informative genomic regions. This approach utilizes a methylation-insensitive restriction enzyme (Msp I) that contains a CpG site in its recognition sequence motif, resulting in at least one DNA methylation site in each sequencing read (Heyn and Esteller, 2012). |
| Microarray-based assay | CpG-specific differential DNA methylation is frequently identified in cancer; in this case, CpG-specific array technology is a cost-effective option to identify genome-wide DMRs (Heyn and Esteller, 2012). The latest HumanMethylation 850 beadchip assay (Illumina) covers over 850,000 CpG methylation sites with high resolution, including 450,000 CpG sites of previous version and 333,265 enhancer CpG sites identified by the ENCODE and FANTOM5 projects (Moran et al., 2016a). |
| Locus-specific DNA methylation analysis | In clinic, locus-specific DNA methylation analysis is an optimal technique for disease diagnostic testing in the case that single or a small number of genes are already established as biomarkers. Target regional DNA methylation level can be detected by either sequencing-based approach such as direct bisulphite and pyrosequencing or alternatives such as methylation-specific-PCR-based and matrix-assisted laser desorption/ionization (MALDI)- mass-spectrometry-based analysis (Heyn and Esteller, 2012). |
Although biopsy is a reliable resource to determine disease status, invasive procedure not only engenders health problems but also prevents obtaining samples particularly in the case of brain disorders. Emerging evidence highlights that cfDNA identified in blood contains various tissue-originated DNA (Lehmann-Werman et al., 2016; Snyder et al., 2016). A recently introduced multi-analyte blood test, called CanerSEEK, exhibited the high performance for the early detection of 8 common cancer types using the combined assays of 16 genetic mutations in cfDNA and 8 protein biomarkers (Cohen et al., 2018). However, mutation-based screening may not be optimal for tumors having low mutation rates such as pediatric and hematological cancers. For such tumors, methylation marks can contribute to increase in sensitivity without changes in specificity. Tissue-specific methylation marks can be conserved even after detaching from the original organs, and by combining adjacent methylation marks called methylation haplotype blocks, even heterogeneous tissue samples can be differentiated by analyzing cfDNA (Guo et al., 2017) (Fig. 2). Since these unique methylation patterns are dependent on pathological progression of various disease such as diabetes, cancer, and traumatic brain, methylation profiling of cfDNA facilitates the improvement of follow-up cares beyond clinical diagnosis (Lehmann-Werman et al., 2016). In addition to methylation, 5hmC is detectable in cfDNA. Despite its low abundance, specificity of 5hmC depending on tissue origin and various cancer types elucidated its potential as a diagnostic biomarkers (Li et al., 2017; Song et al., 2017a). As such, the integration of both 5mC and 5hmC profiling can be a more reliable biomarker for diagnosis as well as follow-up of diseases.
Fig. 2. Clinical application of DNA methylation marks specific to disease status.
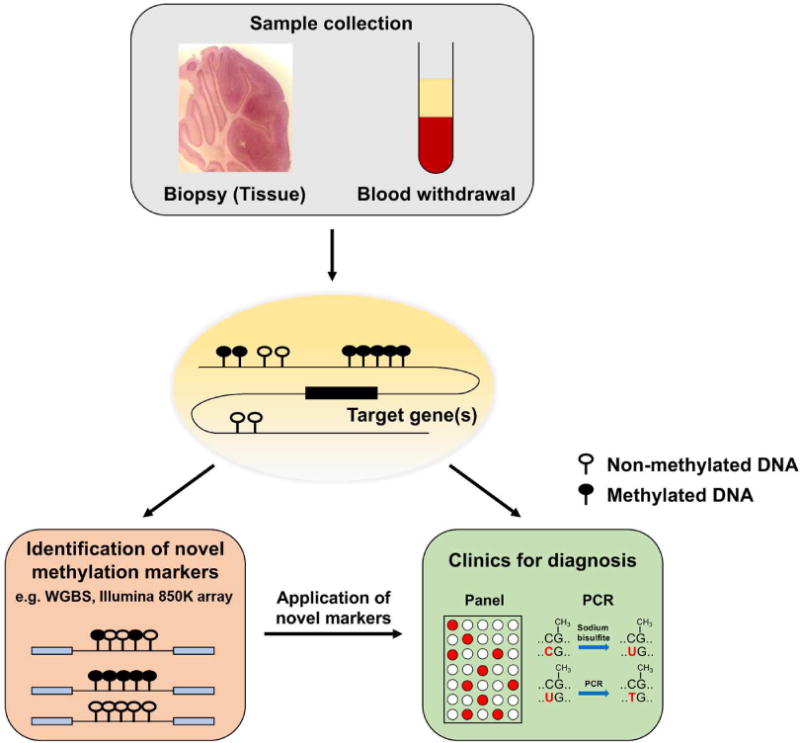
DNA methylation is specific to tissue of origin. Even cell-free DNA (cfDNA) found in the circulating system retains DNA methylation marks. However, these modifications are frequently altered during disease progression. Epigenetic alterations are one of cancer hallmarks and hypo- or hyper-methylation at particular promoter regions are biomarkers of neurodegenerative disorders. Differential DNA methylation regions (DMRs) associated with disease can be identified in an unbiased manner including whole-genome bisulfite sequencing (WGBS) and microarray-based techniques (e.g., Illumina 850K array), which can be clinically used as diagnostic markers.
6. Conclusion
Early intervention of diseases could significantly reduce medical costs in terms of hospitalization and drug. The post-sequencing era facilitates the identification of molecular classifiers of disease status and its implementation for early diagnosis. Transcriptome information produced by microarray represents the first disease-causing molecular alterations, but technical reproducibility and ambiguous interpretation are considered as limitation for clinical application. In addition, genetic mutations are not always reliable as effective sources to accurately diagnose the disease, particularly in the case that disease-associated genetic factors are not identified yet or genetic basis is complex. Epigenetics, including DNA methylation, histone code, and 3D conformation of chromatin, is an emerging candidate for disease diagnosis. DNA methylation draws more attention since the modification is stable and inherited (Heyn and Esteller, 2012; Haggarty, 2015). Its binomial patterns of disease-associated genetic regions enable to distinguish the presence of disease. In many diseases, such as cancers and neurological disorders, the feasibility of DNA methylation assay for diagnosis has been reviewed through clinical studies (Esteller, 2008; Heyn and Esteller, 2012; Nikolaidis et al., 2012; Van Neste et al., 2012; Suva et al., 2013; Haggarty, 2015; Hwang et al., 2017). For example, SEPT9 methylation analysis was the first FDA-approved blood test with a high sensitivity and specificity in for colorectal cancer screening and early detection (Song et al., 2017b). In addition, GSTP1 methylation assay is considered as an alternative diagnostic testing of prostate cancer. Compared to widely-used PSA assay, the hypermethylation of GSTP1 promoter was found to be exclusively detected in prostate cancer patients so that the methylation assay exhibited lower false positive rate consistently in numerous studies, suggesting a good biomarker of GSTP1 methylation for prostate cancer case (Van Neste et al., 2012). In other examples in lung and breast cancers, methylation status of multi-genes is informative for disease diagnosis (Atalay, 2013; Liloglou et al., 2014; Paalman et al., 2016). The presence of cancer-type specific methylation signature in blood allows the unbiased molecular diagnosis for early diagnosis, early treatment and monitoring disease progression without invasive procedure. In addition to cancer-related methylation changes, methylation signature implicated in neurological and neuropsychiatric disorders can be a promising biomarker for disease detection and accurate diagnosis (Hwang et al., 2017). A recent genome-wide methylation study highlighted that syndrome-specific DNA methylation epi-signature identified in 14 Mendelian disorders could contribute to the precise classification and diagnosis of complex neurodevelopmental syndromes (Aref-Eshghi et al., 2018). However, technical and analytic issues of methylation assays remain to be resolved before clinical application (Diaz-Lagares et al., 2016; Heyn and Esteller, 2012). More technical advance and extensive clinical studies will facilitate the successful implementation of epigenetic assay for disease diagnosis in the near future.
Acknowledgments
This work was supported in part by NIH grants (NS051630, NS079625, MH102690 and NS097206 to P.J.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1000 Genomes Project Consortium. A global reference for human genetic variation. Nature. 2015;526:68–74. doi: 10.1038/nature15393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed H. Promoter methylation in prostate cancer and its application for the early detection of prostate cancer using serum and urine samples. Biomark Cancer. 2010;2010:17–33. doi: 10.4137/BIC.S3187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akbarian S, Huang HS. Molecular and cellular mechanisms of altered GAD1/GAD67 expression in schizophrenia and related disorders. Brain Res Rev. 2006;52:293–304. doi: 10.1016/j.brainresrev.2006.04.001. [DOI] [PubMed] [Google Scholar]
- Aref-Eshghi E, Rodenhiser DI, Schenkel LC, Lin H, Skinner C, Ainsworth P, Pare G, Hood RL, Bulman DE, Kernohan KD, Care4Rare Canada C. Boycott KM, Campeau PM, Schwartz C, Sadikovic B. Genomic DNA methylation signatures enable concurrent diagnosis and clinical genetic variant classification in neurodevelopmental syndromes. Am J Hum Genet. 2018;102:156–174. doi: 10.1016/j.ajhg.2017.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atalay C. Epigenetics in breast cancer. Exp Oncol. 2013;35:246–249. [PubMed] [Google Scholar]
- Autism Spectrum Disorders Working Group of The Psychiatric Genomics, C. Meta-analysis of GWAS of over 16,000 individuals with autism spectrum disorder highlights a novel locus at 10q24.32 and a significant overlap with schizophrenia. Mol Autism. 2017;8:21. doi: 10.1186/s13229-017-0137-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker-Andresen D, Ratnu VS, Bredy TW. Dynamic DNA methylation: a prime candidate for genomic metaplasticity and behavioral adaptation. Trends Neurosci. 2013;36:3–13. doi: 10.1016/j.tins.2012.09.003. [DOI] [PubMed] [Google Scholar]
- Banerji S, Cibulskis K, Rangel-Escareno C, Brown KK, Carter SL, Frederick AM, Lawrence MS, Sivachenko AY, Sougnez C, Zou L, Cortes ML, Fernandez-Lopez JC, Peng S, Ardlie KG, Auclair D, Bautista-Pina V, Duke F, Francis J, Jung J, Maffuz-Aziz A, Onofrio RC, Parkin M, Pho NH, Quintanar-Jurado V, Ramos AH, Rebollar-Vega R, Rodriguez-Cuevas S, Romero-Cordoba SL, Schumacher SE, Stransky N, Thompson KM, Uribe-Figueroa L, Baselga J, Beroukhim R, Polyak K, Sgroi DC, Richardson AL, Jimenez-Sanchez G, Lander ES, Gabriel SB, Garraway LA, Golub TR, Melendez-Zajgla J, Toker A, Getz G, Hidalgo-Miranda A, Meyerson M. Sequence analysis of mutations and translocations across breast cancer subtypes. Nature. 2012;486:405–409. doi: 10.1038/nature11154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barry MJ. Clinical practice. Prostate-specific-antigen testing for early diagnosis of prostate cancer. N Engl J Med. 2001;344:1373–1377. doi: 10.1056/NEJM200105033441806. [DOI] [PubMed] [Google Scholar]
- Behnia F, Parets SE, Kechichian T, Yin H, Dutta EH, Saade GR, Smith AK, Menon R. Fetal DNA methylation of autism spectrum disorders candidate genes: association with spontaneous preterm birth. Am J Obstet Gynecol. 2015;212:533 e531–539. doi: 10.1016/j.ajog.2015.02.011. [DOI] [PubMed] [Google Scholar]
- Belinsky SA. Gene-promoter hypermethylation as a biomarker in lung cancer. Nat Rev Cancer. 2004;4:707–717. doi: 10.1038/nrc1432. [DOI] [PubMed] [Google Scholar]
- Bernardini S, Miano R, Iori R, Finazzi-Agro E, Palmieri G, Ballerini S, Angeloni C, Orlandi A, Bellincampi L, Cortese C, Federici G. Hypermethylation of the CpG islands in the promoter region of the GSTP1 gene in prostate cancer: a useful diagnostic and prognostic marker? Clin Chim Acta. 2004;350:181–188. doi: 10.1016/j.cccn.2004.07.022. [DOI] [PubMed] [Google Scholar]
- Bernstein BE, Stamatoyannopoulos JA, Costello JF, Ren B, Milosavljevic A, Meissner A, Kellis M, Marra MA, Beaudet AL, Ecker JR, Farnham PJ, Hirst M, Lander ES, Mikkelsen TS, Thomson JA. The NIH Roadmap Epigenomics Mapping Consortium. Nat Biotechnol. 2010;28:1045–1048. doi: 10.1038/nbt1010-1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloom CI, Graham CM, Berry MP, Rozakeas F, Redford PS, Wang Y, Xu Z, Wilkinson KA, Wilkinson RJ, Kendrick Y, Devouassoux G, Ferry T, Miyara M, Bouvry D, Valeyre D, Gorochov G, Blankenship D, Saadatian M, Vanhems P, Beynon H, Vancheeswaran R, Wickremasinghe M, Chaussabel D, Banchereau J, Pascual V, Ho LP, Lipman M, O’Garra A. Transcriptional blood signatures distinguish pulmonary tuberculosis, pulmonary sarcoidosis, pneumonias and lung cancers. PLoS One. 2013;8:e70630. doi: 10.1371/journal.pone.0070630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carboni L, Lattanzio F, Candeletti S, Porcellini E, Raschi E, Licastro F, Romualdi P. Peripheral leukocyte expression of the potential biomarker proteins Bdnf, Sirt1, and Psen1 is not regulated by promoter methylation in Alzheimer’s disease patients. Neurosci Lett. 2015;605:44–48. doi: 10.1016/j.neulet.2015.08.012. [DOI] [PubMed] [Google Scholar]
- Carter HB. A PSA threshold of 4.0 ng/mL for early detection of prostate cancer: the only rational approach for men 50 years old and older. Urology. 2000;55:796–799. doi: 10.1016/s0090-4295(00)00517-3. [DOI] [PubMed] [Google Scholar]
- Catalona WJ, Richie JP, Ahmann FR, Hudson MA, Scardino PT, Flanigan RC, Dekernion JB, Ratliff TL, Kavoussi LR, Dalkin BL, Waters WB, Macfarlane MT, Southwick PC. Comparison of digital rectal examination and serum prostate specific antigen in the early detection of prostate cancer: results of a multicenter clinical trial of 6,630 men. J Urol. 1994;151:1283–1290. doi: 10.1016/s0022-5347(17)35233-3. [DOI] [PubMed] [Google Scholar]
- Chan MK, Krebs MO, Cox D, Guest PC, Yolken RH, Rahmoune H, Rothermundt M, Steiner J, Leweke FM, van Beveren NJ, Niebuhr DW, Weber NS, Cowan DN, Suarez-Pinilla P, Crespo-Facorro B, Mam-Lam-Fook C, Bourgin J, Wenstrup RJ, Kaldate RR, Cooper JD, Bahn S. Development of a blood-based molecular biomarker test for identification of schizophrenia before disease onset. Transl Psychiatry. 2015;5:e601. doi: 10.1038/tp.2015.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang D, Nalls MA, Hallgrimsdottir IB, Hunkapiller J, van der Brug M, Cai F, International Parkinson’s Disease Genomics C. Me Research T, Kerchner GA, Ayalon G, Bingol B, Sheng M, Hinds D, Behrens TW, Singleton AB, Bhangale TR, Graham RR. A meta-analysis of genome-wide association studies identifies 17 new Parkinson’s disease risk loci. Nat Genet. 2017;49:1511–1516. doi: 10.1038/ng.3955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang L, Wang Y, Ji H, Dai D, Xu X, Jiang D, Hong Q, Ye H, Zhang X, Zhou X, Liu Y, Li J, Chen Z, Li Y, Zhou D, Zhuo R, Zhang Y, Yin H, Mao C, Duan S, Wang Q. Elevation of peripheral BDNF promoter methylation links to the risk of Alzheimer’s disease. PLoS One. 2014;9:e110773. doi: 10.1371/journal.pone.0110773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheuk IW, Shin VY, Kwong A. Detection of methylated circulating DNA as noninvasive biomarkers for breast cancer diagnosis. J Breast Cancer. 2017;20:12–19. doi: 10.4048/jbc.2017.20.1.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chimonidou M, Strati A, Malamos N, Georgoulias V, Lianidou ES. SOX17 promoter methylation in circulating tumor cells and matched cell-free DNA isolated from plasma of patients with breast cancer. Clin Chem. 2013;59:270–279. doi: 10.1373/clinchem.2012.191551. [DOI] [PubMed] [Google Scholar]
- Chu D, Chuang C, Fu J, Huang S, Chia R, Sun C. Hypermethylation of the CpG islands in the promoter region flanking GSTP1 gene is a candidate plasma DNA biomarker for screening or monitoring prostate carcinoma prognosis. Am J Hum Genet. 2002a;71:232–232. [Google Scholar]
- Chu DC, Chuang CK, Fu JB, Huang HS, Tseng CP, Sun CF. The use of real-time quantitative polymerase chain reaction to detect hypermethylation of the CpG islands in the promoter region flanking the GSTP1 gene to diagnose prostate carcinoma. J Urol. 2002b;167:1854–1858. [PubMed] [Google Scholar]
- Chuang YH, Paul KC, Bronstein JM, Bordelon Y, Horvath S, Ritz B. Parkinson’s disease is associated with DNA methylation levels in human blood and saliva. Genome Med. 2017;9:76. doi: 10.1186/s13073-017-0466-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen JD, Li L, Wang Y, Thoburn C, Afsari B, Danilova L, Douville C, Javed AA, Wong F, Mattox A, Hruban RH, Wolfgang CL, Goggins MG, Dal Molin M, Wang TL, Roden R, Klein AP, Ptak J, Dobbyn L, Schaefer J, Silliman N, Popoli M, Vogelstein JT, Browne JD, Schoen RE, Brand RE, Tie J, Gibbs P, Wong HL, Mansfield AS, Jen J, Hanash SM, Falconi M, Allen PJ, Zhou S, Bettegowda C, Diaz L, Tomasetti C, Kinzler KW, Vogelstein B, Lennon AM, Papadopoulos N. Detection and localization of surgically resectable cancers with a multi-analyte blood test. Science. 2018 doi: 10.1126/science.aar3247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andriole GL, Crawford ED, Grubb RL, 3rd, Buys SS, Chia D, Church TR, Fouad MN, Gelmann EP, Kvale PA, Reding DJ, Weissfeld JL, Yokochi LA, O’Brien B, Clapp JD, Rathmell JM, Riley TL, Hayes RB, Kramer BS, Izmirlian G, Miller AB, Pinsky PF, Prorok PC, Gohagan JK, Berg CD. Mortality results from a randomized prostate-cancer screening trial. N Engl J Med. 2009;360:1310–1319. doi: 10.1056/NEJMoa0810696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis CA, Hitz BC, Sloan CA, Chan ET, Davidson JM, Gabdank I, Hilton JA, Jain K, Baymuradov UK, Narayanan AK, Onate KC, Graham K, Miyasato SR, Dreszer TR, Strattan JS, Jolanki O, Tanaka FY, Cherry JM. The encyclopedia of DNA elements (ENCODE): data portal update. Nucleic Acids Res. 2018;46:D794–D801. doi: 10.1093/nar/gkx1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Jager PL, Srivastava G, Lunnon K, Burgess J, Schalkwyk LC, Yu L, Eaton ML, Keenan BT, Ernst J, McCabe C, Tang A, Raj T, Replogle J, Brodeur W, Gabriel S, Chai HS, Younkin C, Younkin SG, Zou F, Szyf M, Epstein CB, Schneider JA, Bernstein BE, Meissner A, Ertekin-Taner N, Chibnik LB, Kellis M, Mill J, Bennett DA. Alzheimer’s disease: early alterations in brain DNA methylation at ANK1, BIN1, RHBDF2 and other loci. Nat Neurosci. 2014;17:1156–1163. doi: 10.1038/nn.3786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delgado-Morales R, Agis-Balboa RC, Esteller M, Berdasco M. Epigenetic mechanisms during ageing and neurogenesis as novel therapeutic avenues in human brain disorders. Clin Epigenetics. 2017;9:67. doi: 10.1186/s13148-017-0365-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz-Lagares A, Mendez-Gonzalez J, Hervas D, Saigi M, Pajares MJ, Garcia D, Crujerias AB, Pio R, Montuenga LM, Zulueta J, Nadal E, Rosell A, Esteller M, Sandoval J. A novel epigenetic signature for early diagnosis in lung cancer. Clin Cancer Res. 2016;22:3361–3371. doi: 10.1158/1078-0432.CCR-15-2346. [DOI] [PubMed] [Google Scholar]
- Doi A, Park IH, Wen B, Murakami P, Aryee MJ, Irizarry R, Herb B, Ladd-Acosta C, Rho JS, Loewer S, Miller J, Schlaeger T, Daley GQ, Feinberg AP. Differential methylation of tissue- and cancer-specific CpG island shores distinguishes human induced pluripotent stem cells, embryonic stem cells and fibroblasts. Nat Genet. 2009;41:1350–U1123. doi: 10.1038/ng.471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebert MP, Mooney SH, Tonnes-Priddy L, Lograsso J, Hoffmann J, Chen J, Rocken C, Schulz HU, Malfertheiner P, Lofton-Day C. Hypermethylation of the TPEF/HPP1 gene in primary and metastatic colorectal cancers. Neoplasia. 2005;7:771–778. doi: 10.1593/neo.05235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elagoz Yuksel M, Yuceturk B, Karatas OF, Ozen M, Dogangun B. The altered promoter methylation of oxytocin receptor gene in autism. J Neurogenet. 2016;30:280–284. doi: 10.1080/01677063.2016.1202951. [DOI] [PubMed] [Google Scholar]
- Erasmus JJ, McAdams HP, Connolly JE. Solitary pulmonary nodules: Part II. Evaluation of the indeterminate nodule. Radiographics. 2000;20:59–66. doi: 10.1148/radiographics.20.1.g00ja0259. [DOI] [PubMed] [Google Scholar]
- Esteller M. Epigenetics in cancer. N Engl J Med. 2008;358:1148–1159. doi: 10.1056/NEJMra072067. [DOI] [PubMed] [Google Scholar]
- Fernandez AF, Assenov Y, Martin-Subero JI, Balint B, Siebert R, Taniguchi H, Yamamoto H, Hidalgo M, Tan AC, Galm O, Ferrer I, Sanchez-Cespedes M, Villanueva A, Carmona J, Sanchez-Mut JV, Berdasco M, Moreno V, Capella G, Monk D, Ballestar E, Ropero S, Martinez R, Sanchez-Carbayo M, Prosper F, Agirre X, Fraga MF, Grana O, Perez-Jurado L, Mora J, Puig S, Prat J, Badimon L, Puca AA, Meltzer SJ, Lengauer T, Bridgewater J, Bock C, Esteller M. A DNA methylation fingerprint of 1628 human samples. Genome Res. 2012;22:407–419. doi: 10.1101/gr.119867.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fransquet PD, Lacaze P, Saffery R, McNeil J, Woods R, Ryan J. Blood DNA methylation as a potential biomarker of dementia: A systematic review. Alzheimers Dement. 2018;14:81–103. doi: 10.1016/j.jalz.2017.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frommer M, McDonald LE, Millar DS, Collis CM, Watt F, Grigg GW, Molloy PL, Paul CL. A genomic sequencing protocol that yields a positive display of 5-methylcytosine residues in individual DNA strands. Proc Natl Acad Sci U S A. 1992;89:1827–1831. doi: 10.1073/pnas.89.5.1827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gormley P, Anttila V, Winsvold BS, Palta P, Esko T, Pers TH, Farh KH, Cuenca-Leon E, Muona M, Furlotte NA, Kurth T, Ingason A, McMahon G, Ligthart L, Terwindt GM, Kallela M, Freilinger TM, Ran C, Gordon SG, Stam AH, Steinberg S, Borck G, Koiranen M, Quaye L, Adams HH, Lehtimaki T, Sarin AP, Wedenoja J, Hinds DA, Buring JE, Schurks M, Ridker PM, Hrafnsdottir MG, Stefansson H, Ring SM, Hottenga JJ, Penninx BW, Farkkila M, Artto V, Kaunisto M, Vepsalainen S, Malik R, Heath AC, Madden PA, Martin NG, Montgomery GW, Kurki MI, Kals M, Magi R, Parn K, Hamalainen E, Huang H, Byrnes AE, Franke L, Huang J, Stergiakouli E, Lee PH, Sandor C, Webber C, Cader Z, Muller-Myhsok B, Schreiber S, Meitinger T, Eriksson JG, Salomaa V, Heikkila K, Loehrer E, Uitterlinden AG, Hofman A, van Duijn CM, Cherkas L, Pedersen LM, Stubhaug A, Nielsen CS, Mannikko M, Mihailov E, Milani L, Gobel H, Esserlind AL, Christensen AF, Hansen TF, Werge T, International Headache Genetics C. Kaprio J, Aromaa AJ, Raitakari O, Ikram MA, Spector T, Jarvelin MR, Metspalu A, Kubisch C, Strachan DP, Ferrari MD, Belin AC, Dichgans M, Wessman M, van den Maagdenberg AM, Zwart JA, Boomsma DI, Smith GD, Stefansson K, Eriksson N, Daly MJ, Neale BM, Olesen J, Chasman DI, Nyholt DR, Palotie A. Meta-analysis of 375,000 individuals identifies 38 susceptibility loci for migraine. Nat Genet. 2016;48:856–866. doi: 10.1038/ng.3598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grayson DR, Guidotti A. The dynamics of DNA methylation in schizophrenia and related psychiatric disorders. Neuropsychopharmacology. 2013;38:138–166. doi: 10.1038/npp.2012.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Group G. B. D. N. D. C. Global, regional, and national burden of neurological disorders during 1990-2015: a systematic analysis for the Global Burden of Disease Study 2015. Lancet Neurol. 2017;16:877–897. doi: 10.1016/S1474-4422(17)30299-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerreiro RJ, Lohmann E, Kinsella E, Bras JM, Luu N, Gurunlian N, Dursun B, Bilgic B, Santana I, Hanagasi H, Gurvit H, Gibbs JR, Oliveira C, Emre M, Singleton A. Exome sequencing reveals an unexpected genetic cause of disease: NOTCH3 mutation in a Turkish family with Alzheimer’s disease. Neurobiol Aging. 2012;33:1008 e1017–1023. doi: 10.1016/j.neurobiolaging.2011.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo S, Diep D, Plongthongkum N, Fung HL, Zhang K, Zhang K. Identification of methylation haplotype blocks aids in deconvolution of heterogeneous tissue samples and tumor tissue-of-origin mapping from plasma DNA. Nat Genet. 2017;49:635–642. doi: 10.1038/ng.3805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haggarty SJ. Epigenetic diagnostics for neuropsychiatric disorders: Above the genome. Neurology. 2015;84:1618–1619. doi: 10.1212/WNL.0000000000001505. [DOI] [PubMed] [Google Scholar]
- Han SH, Mook-Jung I. Diverse molecular targets for therapeutic strategies in Alzheimer’s disease. J Korean Med Sci. 2014;29:893–902. doi: 10.3346/jkms.2014.29.7.893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- Herman JG, Baylin SB. Gene silencing in cancer in association with promoter hypermethylation. N Engl J Med. 2003;349:2042–2054. doi: 10.1056/NEJMra023075. [DOI] [PubMed] [Google Scholar]
- Heyn H, Esteller M. DNA methylation profiling in the clinic: applications and challenges. Nat Rev Genet. 2012;13:679–692. doi: 10.1038/nrg3270. [DOI] [PubMed] [Google Scholar]
- Heyn H, Esteller M. An adenine code for DNA: A second life for N6-methyladenine. Cell. 2015;161:710–713. doi: 10.1016/j.cell.2015.04.021. [DOI] [PubMed] [Google Scholar]
- Howsmon DP, Kruger U, Melnyk S, James SJ, Hahn J. Classification and adaptive behavior prediction of children with autism spectrum disorder based upon multivariate data analysis of markers of oxidative stress and DNA methylation. PLoS Comput Biol. 2017;13:e1005385. doi: 10.1371/journal.pcbi.1005385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang JY, Aromolaran KA, Zukin RS. The emerging field of epigenetics in neurodegeneration and neuroprotection. Nat Rev Neurosci. 2017;18:347–361. doi: 10.1038/nrn.2017.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito S, Shen L, Dai Q, Wu SC, Collins LB, Swenberg JA, He C, Zhang Y. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science. 2011;333:1300–1303. doi: 10.1126/science.1210597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacinto FV, Ballestar E, Esteller M. Methyl-DNA immunoprecipitation (MeDIP): hunting down the DNA methylome. Biotechniques 44, 35 37 39 passim. 2008 doi: 10.2144/000112708. [DOI] [PubMed] [Google Scholar]
- Jack A, Connelly JJ, Morris JP. DNA methylation of the oxytocin receptor gene predicts neural response to ambiguous social stimuli. Front Hum Neurosci. 2012;6:280. doi: 10.3389/fnhum.2012.00280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakubowski JL, Labrie V. Epigenetic biomarkers for Parkinso’s disease: From diagnostics to therapeutics. J Parkinsons Dis. 2017;7:1–12. doi: 10.3233/JPD-160914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jankowska AM, Millward CL, Caldwell CW. The potential of DNA modifications as biomarkers and therapeutic targets in oncology. Expert Rev Mol Diagn. 2015;15:1325–1337. doi: 10.1586/14737159.2015.1084229. [DOI] [PubMed] [Google Scholar]
- Jenuwein T, Allis CD. Translating the histone code. Science. 2001;293:1074–1080. doi: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- Jin B, Robertson KD. DNA methyltransferases, DNA damage repair, and cancer. Adv Exp Med Biol. 2013;754:3–29. doi: 10.1007/978-1-4419-9967-2_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson DA, Barclay RL, Mergener K, Weiss G, Konig T, Beck J, Potter NT. Plasma Septin9 versus fecal immunochemical testing for colorectal cancer screening: a prospective multicenter study. PLoS One. 2014;9:e98238. doi: 10.1371/journal.pone.0098238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jovanovic J, Ronneberg JA, Tost J, Kristensen V. The epigenetics of breast cancer. Mol Oncol. 2010;4:242–254. doi: 10.1016/j.molonc.2010.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jowaed A, Schmitt I, Kaut O, Wullner U. Methylation regulates alpha-synuclein expression and is decreased in Parkinson’s disease patients’ brains. J Neurosci. 2010;30:6355–6359. doi: 10.1523/JNEUROSCI.6119-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumsta R, Hummel E, Chen FS, Heinrichs M. Epigenetic regulation of the oxytocin receptor gene: implications for behavioral neuroscience. Front Neurosci. 2013;7:83. doi: 10.3389/fnins.2013.00083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon YJ, Lee SJ, Koh JS, Kim SH, Lee HW, Kang MC, Bae JB, Kim YJ, Park JH. Genome-wide analysis of DNA methylation and the gene expression change in lung cancer. J Thorac Oncol. 2012;7:20–33. doi: 10.1097/JTO.0b013e3182307f62. [DOI] [PubMed] [Google Scholar]
- Lai CY, Scarr E, Udawela M, Everall I, Chen WJ, Dean B. Biomarkers in schizophrenia: A focus on blood based diagnostics and theranostics. World J Psychiatry. 2016;6:102–117. doi: 10.5498/wjp.v6.i1.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lappalainen T, Greally JM. Associating cellular epigenetic models with human phenotypes. Nat Rev Genet. 2017;18:441–451. doi: 10.1038/nrg.2017.32. [DOI] [PubMed] [Google Scholar]
- Lee WH, Morton RA, Epstein JI, Brooks JD, Campbell PA, Bova GS, Hsieh WS, Isaacs WB, Nelson WG. Cytidine methylation of regulatory sequences near the pi-class glutathione S-transferase gene accompanies human prostatic carcinogenesis. Proc Natl Acad Sci U S A. 1994;91:11733–11737. doi: 10.1073/pnas.91.24.11733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehmann-Werman R, Neiman D, Zemmour H, Moss J, Magenheim J, Vaknin-Dembinsky A, Rubertsson S, Nellgard B, Blennow K, Zetterberg H, Spalding K, Haller MJ, Wasserfall CH, Schatz DA, Greenbaum CJ, Dorrell C, Grompe M, Zick A, Hubert A, Maoz M, Fendrich V, Bartsch DK, Golan T, Ben Sasson SA, Zamir G, Razin A, Cedar H, Shapiro AM, Glaser B, Shemer R, Dor Y. Identification of tissue-specific cell death using methylation patterns of circulating DNA. Proc Natl Acad Sci U S A. 2016;113:E1826–E1834. doi: 10.1073/pnas.1519286113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leng S, Do K, Yingling CM, Picchi MA, Wolf HJ, Kennedy TC, Feser WJ, Baron AE, Franklin WA, Brock MV, Herman JG, Baylin SB, Byers T, Stidley CA, Belinsky SA. Defining a gene promoter methylation signature in sputum for lung cancer risk assessment. Clin Cancer Res. 2012;18:3387–3395. doi: 10.1158/1078-0432.CCR-11-3049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Zhang X, Lu X, You L, Song Y, Luo Z, Zhang J, Nie J, Zheng W, Xu D, Wang Y, Dong Y, Yu S, Hong J, Shi J, Hao H, Luo F, Hua L, Wang P, Qian X, Yuan F, Wei L, Cui M, Zhang T, Liao Q, Dai M, Liu Z, Chen G, Meckel K, Adhikari S, Jia G, Bissonnette MB, Zhang X, Zhao Y, Zhang W, He C, Liu J. 5-Hydroxymethylcytosine signatures in circulating cell-free DNA as diagnostic biomarkers for human cancers. Cell Res. 2017;27:1243–1257. doi: 10.1038/cr.2017.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lichtenstein P, Carlstrom E, Rastam M, Gillberg C, Anckarsater H. The genetics of autism spectrum disorders and related neuropsychiatric disorders in childhood. Am J Psychiatry. 2010;167:1357–1363. doi: 10.1176/appi.ajp.2010.10020223. [DOI] [PubMed] [Google Scholar]
- Liloglou T, Bediaga NG, Brown BR, Field JK, Davies MP. Epigenetic biomarkers in lung cancer. Cancer Lett. 2014;342:200–212. doi: 10.1016/j.canlet.2012.04.018. [DOI] [PubMed] [Google Scholar]
- Lofton-Day C, Model F, Devos T, Tetzner R, Distler J, Schuster M, Song X, Lesche R, Liebenberg V, Ebert M, Molnar B, Grutzmann R, Pilarsky C, Sledziewski A. DNA methylation biomarkers for blood-based colorectal cancer screening. Clin Chem. 2008;54:414–423. doi: 10.1373/clinchem.2007.095992. [DOI] [PubMed] [Google Scholar]
- LoParo D, Waldman ID. The oxytocin receptor gene (OXTR) is associated with autism spectrum disorder: a meta-analysis. Mol Psychiatry. 2015;20:640–646. doi: 10.1038/mp.2014.77. [DOI] [PubMed] [Google Scholar]
- Lou JJ, Mirsadraei L, Sanchez DE, Wilson RW, Shabihkhani M, Lucey GM, Wei B, Singer EJ, Mareninov S, Yong WH. A review of room temperature storage of biospecimen tissue and nucleic acids for anatomic pathology laboratories and biorepositories. Clin Biochem. 2014;47:267–273. doi: 10.1016/j.clinbiochem.2013.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mack SC, Hubert CG, Miller TE, Taylor MD, Rich JN. An epigenetic gateway to brain tumor cell identity. Nat Neurosci. 2016;19:10–19. doi: 10.1038/nn.4190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maruyama R, Toyooka S, Toyooka KO, Virmani AK, Zochbauer-Muller S, Farinas AJ, Minna JD, McConnell J, Frenkel EP, Gazdar AF. Aberrant promoter methylation profile of prostate cancers and its relationship to clinicopathological features. Clin Cancer Res. 2002;8:514–519. [PubMed] [Google Scholar]
- Mettlin CJ, Murphy GP, Babaian RJ, Chesley A, Kane RA, Littrup PJ, Mostofi FK, Ray PS, Somers WJ, Toi A. Observations on the early detection of prostate cancer from the American Cancer Society National Prostate Cancer Detection Project. Cancer. 1997;80:1814–1817. doi: 10.1002/(sici)1097-0142(19971101)80:9<1814::aid-cncr20>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- Moran S, Arribas C, Esteller M. Validation of a DNA methylation microarray for 850,000 CpG sites of the human genome enriched in enhancer sequences. Epigenomics. 2016a;8:389–399. doi: 10.2217/epi.15.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moran S, Martinez-Cardus A, Sayols S, Musulen E, Balana C, Estival-Gonzalez A, Moutinho C, Heyn H, Diaz-Lagares A, de Moura MC, Stella GM, Comoglio PM, Ruiz-Miro M, Matias-Guiu X, Pazo-Cid R, Anton A, Lopez-Lopez R, Soler G, Longo F, Guerra I, Fernandez S, Assenov Y, Plass C, Morales R, Carles J, Bowtell D, Mileshkin L, Sia D, Tothill R, Tabernero J, Llovet JM, Esteller M. Epigenetic profiling to classify cancer of unknown primary: a multicentre, retrospective analysis. Lancet Oncol. 2016b;17:1386–1395. doi: 10.1016/S1470-2045(16)30297-2. [DOI] [PubMed] [Google Scholar]
- Nagata T, Kobayashi N, Ishii J, Shinagawa S, Nakayama R, Shibata N, Kuerban B, Ohnuma T, Kondo K, Arai H, Yamada H, Nakayama K. Association between DNA methylation of the BDNF promoter region and clinical presentation in Alzheimer’s disease. Dement Geriatr Cogn Dis Extra. 2015;5:64–73. doi: 10.1159/000375367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nardone S, Sams DS, Zito A, Reuveni E, Elliott E. Dysregulation of cortical neuron DNA methylation profile in autism spectrum disorder. Cereb Cortex. 2017;27:5739–5754. doi: 10.1093/cercor/bhx250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolaidis G, Raji OY, Markopoulou S, Gosney JR, Bryan J, Warburton C, Walshaw M, Sheard J, Field JK, Liloglou T. DNA methylation biomarkers offer improved diagnostic efficiency in lung cancer. Cancer Res. 2012;72:5692–5701. doi: 10.1158/0008-5472.CAN-12-2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okano M, Bell DW, Haber DA, Li E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell. 1999;99:247–257. doi: 10.1016/s0092-8674(00)81656-6. [DOI] [PubMed] [Google Scholar]
- Olivier M, Hollstein M, Hainaut P. TP53 mutations in human cancers: origins, consequences, and clinical use. Cold Spring Harb Perspect Biol. 2010;2:a001008. doi: 10.1101/cshperspect.a001008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paalman CH, van Leeuwen FE, Aaronson NK, de Boer AG, van de Poll-Franse L, Oldenburg HS, Schaapveld M. Employment and social benefits up to 10 years after breast cancer diagnosis: a population-based study. Br J Cancer. 2016;114:81–87. doi: 10.1038/bjc.2015.431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paluszczak J, Baer-Dubowska W. Epigenetic diagnostics of cancer–the application of DNA methylation markers. J Appl Genet. 2006;47:365–375. doi: 10.1007/BF03194647. [DOI] [PubMed] [Google Scholar]
- Parikh RB, Prasad V. Blood-based screening for colon cancer: A disruptive innovation or simply a disruption? JAMA. 2016;315:2519–2520. doi: 10.1001/jama.2016.7914. [DOI] [PubMed] [Google Scholar]
- Pilleri M, Antonini A. Therapeutic strategies to prevent and manage dyskinesias in Parkinson’s disease. Expert Opin Drug Saf. 2015;14:281–294. doi: 10.1517/14740338.2015.988137. [DOI] [PubMed] [Google Scholar]
- Plongthongkum N, Diep DH, Zhang K. Advances in the profiling of DNA modifications: cytosine methylation and beyond. Nat Rev Genet. 2014;15:647–661. doi: 10.1038/nrg3772. [DOI] [PubMed] [Google Scholar]
- Poduri A. Meta-analysis revives genome-wide association studies in epilepsy. Epilepsy Curr. 2015;15:122–123. doi: 10.5698/1535-7597-15.3.122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qureshi IA, Mehler MF. Developing epigenetic diagnostics and therapeutics for brain disorders. Trends Mol Med. 2013;19:732–741. doi: 10.1016/j.molmed.2013.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson KD. DNA methylation and human disease. Nat Rev Genet. 2005;6:597–610. doi: 10.1038/nrg1655. [DOI] [PubMed] [Google Scholar]
- Ruzicka WB, Subburaju S, Benes FM. Circuit- and diagnosis-specific DNA methylation changes at gamma-aminobutyric acid-related genes in postmortem human hippocampus in schizophrenia and bipolar disorder. JAMA Psychiatry. 2015;72:541–551. doi: 10.1001/jamapsychiatry.2015.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan EJ, Creagh EM. Emerging methods in colorectal cancer screening. Br J Surg. 2018;105:e16–e18. doi: 10.1002/bjs.10650. [DOI] [PubMed] [Google Scholar]
- Sanchez-Mut JV, Graff J. Epigenetic alterations in Alzheimer’s disease. Front Behav Neurosci. 2015;9:347. doi: 10.3389/fnbeh.2015.00347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandin S, Lichtenstein P, Kuja-Halkola R, Larsson H, Hultman CM, Reichenberg A. The familial risk of autism. JAMA. 2014;311:1770–1777. doi: 10.1001/jama.2014.4144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schizophrenia Working Group of the Psychiatric Genomics C. Biological insights from 108 schizophrenia-associated genetic loci. Nature. 2014;511:421–427. doi: 10.1038/nature13595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider LS, Mangialasche F, Andreasen N, Feldman H, Giacobini E, Jones R, Mantua V, Mecocci P, Pani L, Winblad B, Kivipelto M. Clinical trials and late-stage drug development for Alzheimer’s disease: an appraisal from 1984 to 2014. J Intern Med. 2014;275:251–283. doi: 10.1111/joim.12191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroeder FH, Hugosson J, Roobol MJ, Tammela TLJ, Ciatto S, Nelen V, Kwiatkowski M, Lujan M, Lilja H, Zappa M, Denis LJ, Recker F, Berenguer A, Maattanen L, Bangma CH, Aus G, Villers A, Rebillard X, van der Kwast T, Blijenberg BG, Moss SM, de Koning HJ, Auvinen A, Investigators E Screening and prostate-cancer mortality in a randomized European study. N Engl J Med. 2009;360:1320–1328. doi: 10.1056/NEJMoa0810084. [DOI] [PubMed] [Google Scholar]
- Schubeler D. Function and information content of DNA methylation. Nature. 2015;517:321–326. doi: 10.1038/nature14192. [DOI] [PubMed] [Google Scholar]
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin. 2017;67:7–30. doi: 10.3322/caac.21387. [DOI] [PubMed] [Google Scholar]
- Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin. 2018;68:7–30. doi: 10.3322/caac.21442. [DOI] [PubMed] [Google Scholar]
- Slamon DJ, Leyland-Jones B, Shak S, Fuchs H, Paton V, Bajamonde A, Fleming T, Eiermann W, Wolter J, Pegram M, Baselga J, Norton L. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med. 2001;344:783–792. doi: 10.1056/NEJM200103153441101. [DOI] [PubMed] [Google Scholar]
- Slotkin RK, Martienssen R. Transposable elements and the epigenetic regulation of the genome. Nat Rev Genet. 2007;8:272–285. doi: 10.1038/nrg2072. [DOI] [PubMed] [Google Scholar]
- Snyder MW, Kircher M, Hill AJ, Daza RM, Shendure J. Cell-free DNA comprises an in vivo nucleosome footprint that informs its tissues-of-origin. Cell. 2016;164:57–68. doi: 10.1016/j.cell.2015.11.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song CX, Yin S, Ma L, Wheeler A, Chen Y, Zhang Y, Liu B, Xiong J, Zhang W, Hu J, Zhou Z, Dong B, Tian Z, Jeffrey SS, Chua MS, So S, Li W, Wei Y, Diao J, Xie D, Quake SR. 5-Hydroxymethylcytosine signatures in cell-free DNA provide information about tumor types and stages. Cell Res. 2017a;27:1231–1242. doi: 10.1038/cr.2017.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song JZ, Stirzaker C, Harrison J, Melki JR, Clark SJ. Hypermethylation trigger of the glutathione-S-transferase gene (GSTP1) in prostate cancer cells. Oncogene. 2002;21:1048–1061. doi: 10.1038/sj.onc.1205153. [DOI] [PubMed] [Google Scholar]
- Song L, Jia J, Peng X, Xiao W, Li Y. The performance of the SEPT9 gene methylation assay and a comparison with other CRC screening tests: A meta-analysis. Sci Rep. 2017b;7:3032. doi: 10.1038/s41598-017-03321-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorlie T, Perou CM, Tibshirani R, Aas T, Geisler S, Johnsen H, Hastie T, Eisen MB, van de Rijn M, Jeffrey SS, Thorsen T, Quist H, Matese JC, Brown PO, Botstein D, Lonning PE, Borresen-Dale AL. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci U S A. 2001;98:10869–10874. doi: 10.1073/pnas.191367098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamey TA, Yang N, Hay AR, Mcneal JE, Freiha FS, Redwine E. Prostate-specific antigen as a serum marker for adenocarcinoma of the prostate. N Engl J Med. 1987;317:909–916. doi: 10.1056/NEJM198710083171501. [DOI] [PubMed] [Google Scholar]
- Suva ML, Riggi N, Bernstein BE. Epigenetic reprogramming in cancer. Science. 2013;339:1567–1570. doi: 10.1126/science.1230184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyooka S, Toyooka KO, Harada K, Miyajima K, Makarla P, Sathyanarayana UG, Yin J, Sato F, Shivapurkar N, Meltzer SJ, Gazdar AF. Aberrant methylation of the CDH13 (H-cadherin) promoter region in colorectal cancers and adenomas. Cancer Res. 2002;62:3382–3386. [PubMed] [Google Scholar]
- Toyota M, Shen L, Ohe-Toyota M, Hamilton SR, Sinicrope FA, Issa JP. Aberrant methylation of the Cyclooxygenase 2 CpG island in colorectal tumors. Cancer Res. 2000;60:4044–4048. [PubMed] [Google Scholar]
- Valadas JS, Vos M, Verstreken P. Therapeutic strategies in Parkinson’s disease: what we have learned from animal models. Ann N Y Acad Sci. 2015;1338:16–37. doi: 10.1111/nyas.12577. [DOI] [PubMed] [Google Scholar]
- Van Cauwenberghe C, Van Broeckhoven C, Sleegers K. The genetic landscape of Alzheimer disease: clinical implications and perspectives. Genet Med. 2016;18:421–430. doi: 10.1038/gim.2015.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Neste L, Herman JG, Otto G, Bigley JW, Epstein JI, Van Criekinge W. The epigenetic promise for prostate cancer diagnosis. Prostate. 2012;72:1248–1261. doi: 10.1002/pros.22459. [DOI] [PubMed] [Google Scholar]
- Vinkhuyzen AA, Wray NR, Yang J, Goddard ME, Visscher PM. Estimation and partition of heritability in human populations using whole-genome analysis methods. Annu Rev Genet. 2013;47:75–95. doi: 10.1146/annurev-genet-111212-133258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waddington CH. The epigenotype 1942. Int J Epidemiol. 2012;41:10–13. doi: 10.1093/ije/dyr184. [DOI] [PubMed] [Google Scholar]
- Wang Z, Tang B, He Y, Jin P. DNA methylation dynamics in neurogenesis. Epigenomics. 2016;8:401–414. doi: 10.2217/epi.15.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf AMD, Wender RC, Etzioni RB, Thompson IM, D’Amico AV, Volk RJ, Brooks DD, Dash C, Guessous I, Andrews K, DeSantis C, Smith RA. American Cancer Society guideline for the early detection of prostate cancer update 2010. CA Cancer J Clin. 2010;60:70–98. doi: 10.3322/caac.20066. [DOI] [PubMed] [Google Scholar]
- Wong CC, Meaburn EL, Ronald A, Price TS, Jeffries AR, Schalkwyk LC, Plomin R, Mill J. Methylomic analysis of monozygotic twins discordant for autism spectrum disorder and related behavioural traits. Mol Psychiatry. 2014;19:495–503. doi: 10.1038/mp.2013.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu T, Giovannucci E, Welge J, Mallick P, Tang WY, Ho SM. Measurement of GSTP1 promoter methylation in body fluids may complement PSA screening: a meta-analysis. Br J Cancer. 2011;105:65–73. doi: 10.1038/bjc.2011.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto N, Nakayama T, Kajita M, Miyake T, Iwamoto T, Kim SJ, Sakai A, Ishihara H, Tamaki Y, Noguchi S. Detection of aberrant promoter methylation of GSTP1, RASSF1A, and RARbeta2 in serum DNA of patients with breast cancer by a newly established one-step methylation-specific PCR assay. Breast Cancer Res Treat. 2012;132:165–173. doi: 10.1007/s10549-011-1575-2. [DOI] [PubMed] [Google Scholar]
- Yao B, Cheng Y, Wang Z, Li Y, Chen L, Huang L, Zhang W, Chen D, Wu H, Tang B, Jin P. DNA N6-methyladenine is dynamically regulated in the mouse brain following environmental stress. Nat Commun. 2017;8:1122. doi: 10.1038/s41467-017-01195-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao B, Christian KM, He C, Jin P, Ming GL, Song H. Epigenetic mechanisms in neurogenesis. Nat Rev Neurosci. 2016;17:537–549. doi: 10.1038/nrn.2016.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yates LR, Gerstung M, Knappskog S, Desmedt C, Gundem G, Van Loo P, Aas T, Alexandrov LB, Larsimont D, Davies H, Li YL, Ju YS, Ramakrishna M, Haugland HK, Lilleng PK, Nik-Zainal S, McLaren S, Butler A, Martin S, Glodzik D, Menzies A, Raine K, Hinton J, Jones D, Mudie LJ, Jiang B, Vincent D, Greene-Colozzi A, Adnet PY, Fatima A, Maetens M, Ignatiadis M, Stratton MR, Sotiriou C, Richardson AL, Lonning PE, Wedge DC, Campbell PJ. Subclonal diversification of primary breast cancer revealed by multiregion sequencing. Nat Med. 2015;21:751–759. doi: 10.1038/nm.3886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeo S, An KS, Hong YM, Choi YG, Rosen B, Kim SH, Lim S. Neuroprotective changes in degeneration-related gene expression in the substantia nigra following acupuncture in an MPTP mouse model of Parkinsonism: Microarray analysis. Genet Mol Biol. 2015;38:115–127. doi: 10.1590/S1415-475738120140137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ylisaukko-oja T, Alarcon M, Cantor RM, Auranen M, Vanhala R, Kempas E, von Wendt L, Jarvela I, Geschwind DH, Peltonen L. Search for autism loci by combined analysis of Autism Genetic Resource Exchange and Finnish families. Ann Neurol. 2006;59:145–155. doi: 10.1002/ana.20722. [DOI] [PubMed] [Google Scholar]
- Yoder JA, Bestor TH. A candidate mammalian DNA methyltransferase related to pmt1p of fission yeast. Hum Mol Genet. 1998;7:279–284. doi: 10.1093/hmg/7.2.279. [DOI] [PubMed] [Google Scholar]
- Zhubi A, Veldic M, Puri NV, Kadriu B, Caruncho H, Loza I, Sershen H, Lajtha A, Smith RC, Guidotti A, Davis JM, Costa E. An upregulation of DNA-methyltransferase 1 and 3a expressed in telencephalic GABAergic neurons of schizophrenia patients is also detected in peripheral blood lymphocytes. Schizophr Res. 2009;111:115–122. doi: 10.1016/j.schres.2009.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]