

Abstract
Cognitive impulsivity is a heritable trait believed to represent the behavior that defines the volition to initiate alcohol drinking. We have previously shown that a neuronal Toll-like receptor 4 (TLR4) signal located in the central amygdala (CeA) and ventral tegmental area (VTA) controls the initiation of binge drinking in alcohol-preferring P rats, and TLR4 expression is upregulated by alcohol-induced corticotropin-releasing factor (CRF) at these sites. However, the function of the TLR4 signal in the nucleus accumbens shell (NAc-shell), a site implicated in the control of reward, drug-seeking behavior and impulsivity and the contribution of other signal-associated genes, are still poorly understood. Here we report that P rats have an innately activated TLR4 signal in NAc-shell neurons that co-express the α2 GABAA receptor subunit and CRF prior to alcohol exposure. This signal is not present in non-alcohol drinking NP rats. The TLR4 signal is sustained by a CRF amplification loop, which includes TLR4-mediated CRF upregulation through PKA/CREB activation and CRF-mediated TLR4 upregulation through the CRF type 1 receptor (CRFR1) and the MAPK/ERK pathway. NAc-shell Infusion of a neurotropic, non-replicating herpes simplex virus vector for TLR4-specific small interfering RNA (pHSVsiTLR4) inhibits TLR4 expression and cognitive impulsivity, implicating the CRF-amplified TLR4 signal in impulsivity regulation.
Keywords: Activated TLR4 signal, PKA/ CREB, CRF, GABAA 2, HSV siRNA vectors, impulsivity
INTRODUCTION
Cognitive impulsivity is a heritable trait generally defined as a tendency to act without thinking that correlates with drug addiction and is believed to represent the ethanol-seeking behavior, which precedes steady alcohol consumption (Beckwith and Czachowski, 2014; Oberlin and Grahame 2009). Stressor-induced elevations in the corticotropin-releasing factor (CRF) system regulate impulsivity and play a key role in the transition to escalated drug taking, including excessive ethanol drinking (Gondre-Lewis et al., 2016; Lowery-Gionta et al., 2012). However, the genes that regulate the predisposition to initiate alcohol drinking, their potential interaction at distinct brain sites, and their contribution to impulsivity, if any, are still poorly understood.
Toll-like receptors (TLRs) are largely recognized as neuroimmune signals located in neurons and glial cells (Takeda and Akira, 2015). An extensive body of literature has associated one of the TLRs, TLR4, with a lifetime of alcohol consumption and adaptation during ethanol exposure, likely involving differentially activated neuronal and glial signaling pathways. This includes the findings that systemic injection of the TLR4-specific ligand, bacterial endotoxin lipopolysaccharide (LPS) increases voluntary alcohol consumption in mice, and human alcoholics have elevated levels of plasma LPS (Alfonso-Loeches et al., 2016; Blednov et al., 2011; Breese and Knapp, 2016; Crews et al., 2017; Leclercq et al., 2012; Pandey, 2012; Pascual et al., 2011). Pharmacologic and genetic studies suggested that alcohol induces CRF signaling in the central amygdala (CeA) and it plays a significant role in the maintenance of addiction, apparently via activation of the CRF1 receptor [CRFR1] (Dedic et al., 2017; Gondre-Lewis et al., 2016; Koob et al., 2014, Lowery-Gionta et al., 2012; Phillips et al., 2015).
We have previously shown that alcohol-preferring (P) rats, which fulfill most of the criteria for an animal model of human alcohol abuse (Bell et al., 2006), have a neuronal TLR4/ monocyte chemoattractant protein 1 (MCP-1) signal located in the CeA and the ventral tegmental area (VTA) that controls the predisposition to initiate alcohol drinking and is regulated by the γ-aminobutyric acidA (GABAA) receptor α2 subunit. Significantly, however, this signal does not function in the ventral pallidum (VP), documenting the existence of dominant regulatory mechanisms at distinct brain sites (Liu et al., 2011). Furthermore, alcohol-induced CRF expression in the CeA and VTA upregulates TLR4 (June et al., 2015), establishing a potential link between stress and TLR4 expression at these brain sites. However, the role of the CRF/CRFR1 system and its interaction with the TLR4 signal in defining the predisposition of non-alcohol exposed rats to initiate alcohol drinking, particularly as it relates to impulsivity regulation, have not been investigated.
We report that in the nucleus accumbens shell (NAc-shell), a site implicated in the control of reward, drug-seeking behavior and impulsivity (Chaudhri et al., 2010; Feja et al., 2014) the levels of TLR4, CRF and α2 GABAA receptor subunit (α2), and the percentage of co-expressing neurons, are significantly higher in P, than non-alcohol preferring (NP) rats. The TLR4 signal is innately activated in P rats before exposure to alcohol, as evidenced by increased expression and nuclear localization of the phosphorylated transcription factor cAMP response element-binding protein (CREB). Activation is sustained by a CRF amplification loop that includes TLR4-induced CRF expression through protein kinase A (PKA)/CREB activation and CRF feedback regulation of TLR4 expression through CRFR1 and mitogen-activated protein kinases/ extracellular signal-regulated kinases (MAPK/ERK) activation. The TLR4 signal regulates impulsivity, as evidenced by the finding that it is markedly reduced through TLR4 inhibition by NAc-shell infusion of a neurotropic HSV vector for TLR4 siRNA (pHSVsiTLR4).
MATERIALS AND METHODS
Animals
Alcohol preferring (P) (n = 59; 3 – 4 months old; 250–550g) and non-preferring (NP) rats (n=14, 3–4 months old; 250–550g) were obtained from the Alcohol Research Center, Indiana University School of Medicine. P rats perform an operant response for access to ethanol that is not performed by the NP rats, develop both tolerance and physical dependence following excessive intake, and upon removal, show signs of withdrawal following chronic consumption (Bell et al., 2006). Animals were individually housed, maintained at an ambient temperature of 21°C and a reverse 12 h light/dark cycle and provided with food and water, ad libitum. Training and experimental sessions were conducted between 8:30 am and 5:30 pm. Treatment was approved by the IACUC of the Howard University College of Medicine and all procedures were conducted in strict adherence with the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Antibodies
The following antibodies were commercially obtained and used according to the manufacturer’s instructions: mouse anti-GAPDH [catalog (Cat.) #sc-47724; RRID: AB_627678] and mouse anti-TLR4 monoclonal antibody (Cat. #sc-293072, RRID: AB_10611320) were from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Two CRF antibodies were used, with similar results: (i) mouse anti-CRF monoclonal antibody (2B11) (Santa Cruz Biotechnology, Cat. # sc-293187, RRID: AB_2687937) that recognizes amino acids (aa) 154–196 in the human protein and (ii) rabbit polyclonal anti-CRF antibody (Bioss Antibodies, Woburn, MA, USA; Cat. # bs-0246R, RRID: AB_10885735) that recognizes amino acids 176–196 in the human protein. Other used antibodies include rabbit phospho-CREB (pCREB; Ser133; Cat. # 9198, RRID: AB_2561044), rabbit phospho-PKA (pPKA; Thr197; Cat. # 4781, RRID: AB_2300165), and rabbit phosphop44/42 MAPK (pERK1/2; Thr202/Tyr204; Cat # 4377, RRID: AB_331775) were from Cell Signaling Technology (Danvers, MA, USA). Other antibodies were goat anti-GAD67 (glutamic acid decarboxylase 1 (GAD1)) (LifeSpan BioSciences, Seattle, WA, USA, Cat. # LS-B3027-50, RRID: AB_1965223), rabbit anti-CRFR1 (Thermo Fisher Scientific, Waltham, MA, USA, Cat. # 720290, RRID: AB_2633242), rabbit anti-NF-kB p65 (Abcam, Cambridge, MA, USA, Cat. # ab32536, RRID: AB_776751), mouse beta Actin (β-Actin; Proteintech Group, Rosemont, IL, USA, Cat. # 66009-1-Ig, RRID: AB_2687938), Alexa Fluor 488 goat anti-rabbit or donkey anti-goat IgG (H+L; Cat. # A11034, RRID: AB_2576217 or Cat. # A11055, RRID: AB_142672, respectively, Thermo Fisher Scientific), and Alexa Fluor 546 goat anti-mouse or goat anti-rabbit IgG (H+L; Cat.# A11030, RRID: AB_2534089, or Cat. # A11035, RRID: AB_2534093, respectively, Thermo Fisher Scientific). Horseradish peroxidase-labeled secondary antibodies were anti-rabbit (Cat. # 7074, RRID: AB_2099233, Cell Signaling Technology) and anti-mouse IgG (Cat. # 170-6516, RRID: AB_11125547, Bio-Rad, Hercules, CA, USA). The generation and specificity of the rabbit-derived GABAA α2 antibody (W. Sieghart, Center for Brain Research, Medical University of Vienna; Vienna; Austria Cat# GABAA Receptor alpha 2, RRID: AB_2532077) was previously described; it recognizes amino acids 322–357 of the α2 protein (Liu et al., 2011). Antibody validation used the following criteria: (i) protein expression in some, but not other cell lines in the absence of non-specific reactivity, (ii) protein expression following transfection with the appropriate plasmid (positive control), (iii) loss of protein expression with siRNA knockdown (negative control), (iv) failure to inhibit protein expression with scrambled siRNA (siRNA control), and (v) above listed criteria applied to monoclonal and polyclonal antibodies that target distinct epitopes. The data are shown in Supplemental Information (SI) Figs. S1–S4 for TLR4, CRF and CRFR1 antibodies. Antibody specificity is further confirmed by data presented in the MS, notably restored protein expression upon loss of siRNA integrity (MS, Fig. 7B).
Fig. 7. pHSVsiTLR4 infusion in NAc-shell inhibits impulsivity associated with TLR4 inhibition.

(A) Mean adjusted delay scores are significantly increased (impulsivity is decreased) in P rats infused with pHSVsiTLR4 (n=8) relative to those of P rats injected with pHSVsiNC (n=13). Impulsivity is decreased on day 7 post-surgery and returns to baseline levels on day 17. (*p≤ 0.05 by ANOVA). (B) pHSVsiTLR4 infusion in the NAc-shell inhibits TLR4 expression at days 3 after injection (*p≤0.05 by ANOVA), but inhibition is no longer significant on day 16 post injection and expression is not inhibited by pHSVsiNC (p≥0.05 by ANOVA).
Cells, plasmids, transfection and reagents
SK-N-SH (human) and Neuro2a (mouse) neuroblastoma cells were from American Type Culture Collection (Manassas, VA, USA). SK-N-SH cells were grown in RPMI 1640 medium with 2 mM L-glutamine (Gibco, Gaithersburg, MD, USA), 10% fetal bovine serum (FBS; Gemini, West Sacramento, CA, USA), and 1% Penicillin/ Streptomycin (Gibco). The Neuro2a cells were grown in Dulbecco’s modified Eagle’s medium (DMEM) (Gibco) with 10% FBS and 1% Penicillin/Streptomycin. The TLR4FLAG plasmid (# 42646) is a gift from Scott Friedman, the GABRA2phGFP plasmid (# 49169) is a gift from Tija Jacob & Stephen Moss, and the pEMS1153-hCRF plasmid (#29068) is a gift from Elizabeth Simpson. The plasmids were from Addgene (Cambridge, MA, USA). They were incubated [(15 min, room temperature (RT)] with FuGENE 6 Transfection Reagent (Promega, Madison, WI, USA, Cat. # E2693) in antibiotic-free medium and added to 50–80% confluent cultures (20 µg/T-75 flask, 7 µg/T-25 flask or 2.6 µg/well of 6-well plate). The selective CRFR1 antagonist, antalarmin hydrochloride (15 nM; R & D Systems, Minneapolis, MN, USA, Cat. # 2778), the PKA-specific inhibitor H89 (10 µM; Cell Signaling Technology, Cat. # 9844), and the specific inhibitor of MAPK extracellular signaling regulated kinase (ERK) kinase (MEK), U0126 (20 µM; Promega, Cat. # V1121) were added to the cultures 24 h before cell collection. By inhibiting MEK1/2, U0126 prevents the activation of MAP kinases p42 and p44 (ERK1/2).
Immunofluorescence
Immunofluorescent staining was as previously described (Aurelian et al., 2016; June et al, 2015). Cells grown on poly-l-lysine (Sigma)-coated glass coverslips (n=5/group) were fixed with 4% paraformaldehyde (30 min; RT) and permeabilized (2 min; 4 °C) with 0.1% Triton X-100 in 0.1% sodium citrate buffer. They were exposed to primary antibody (diluted in 5% bovine serum albumin and 5% normal goat serum) overnight at 4 °C, washed in phosphate-buffered saline (PBS) with 0.1% Tween 20 and exposed to fluorochrome-labeled secondary antibodies (1 h; RT).
Free-floating (30-µm thick) frozen sections were collected as described in Supporting Information (SI). Coronal serial sections from 5 rats/group were obtained from the brain areas containing NAc, extending from +2.5 mm anterior to bregma to +1.0 mm anterior to bregma (Paxinos and Watson, 2009). For each animal, five representative sections from 1:10 series were rinsed in PBS, treated (95°C, 10 min) with Retrievagen A (BD Biosciences, San Jose, CA, USA, Cat.# 550524), cooled (20 min, RT), and blocked with 5% goat serum (90 min, RT). They were exposed to primary antibodies (overnight, 4°C) followed by the appropriate Alexa Fluor-labeled secondary antibodies (1h, RT). Z-stack images (1µm optical steps) were collected on an Olympus Fluoview FV5000 confocal microscope fitted with standard excitation and emission filters. The total number of single and double staining cells were counted in three randomly selected (×40 magnified) images of NAc-shell from each of the five studied sections. The % GAD1+, GABAA α2+ or CRF+ cells co-expressing TLR4, % GABAA α2+ cells co-expressing GAD1, and % TLR4+ or CRF+ cells co-expressing nuclear pCREB or pERK1/2 were calculated for each image. The results are expressed as mean ± SEM (Table 1).
Table 1.
Percentage of co-staining cells
| Immunoreactivity | Animal s |
% cells ± SEM |
|---|---|---|
| GAD1+ cells expressing TLR4 (SI, Fig. S7) | P rats | 93.7 ± 5.7%* |
| NP rats | 10.0 ± 1.8%* | |
|
| ||
| α2+ cells expressing TLR4 | P rats | 95.9 ± 4.9%* |
| NP rats | 9.6 ± 2.6%* | |
|
| ||
| CRF+ cells expressing TLR4 | P rats | 92.1 ± 6.7%* |
| NP rats | 14.7 ± 5.1%* | |
|
| ||
| TLR4+ cells expressing nuclear pCREB | P rats | 83.5 ± 4.8%* |
| NP rats | 6.0 ± 3.8%* | |
|
| ||
| CRF+ cells expressing nuclear pCREB | P rats | 78.5 ± 7.9%* |
| NP rats | 4.1 ± 3.9%* | |
|
| ||
| TLR4+ cells expressing nuclear pERK1/2 | P rats | 23.1 ± 7.8%* |
| NP rats | 1.1 ± 0.98%* | |
|
| ||
| CRF+ cells expressing nuclear pERK1/2 | P rats | 21.7 ± 5.3%* |
| NP rats | 0.93 ± 0.91%* | |
P≤0.001, P vs. NP
Immunoblotting
Immunoblotting was as previously described (Aurelian et al., 2016; June et al., 2015; Liu et al., 2011). Cells grown on T-75 or T-25 flasks (n=5/group) were lysed with radioimmunoprecipitation buffer (20 mm Tris-HCl (pH 7.4), 0.15 mm NaCl, 1% Nonidet P-40 (Sigma, St. Louis, MO, USA), 0.1% SDS (sodium dodecyl sulfate), 0.5% sodium deoxycholate) supplemented with protease and phosphatase inhibitor cocktails (Sigma). NAc-shell micropunches (300-µm thick) from naive P (n = 12) and NP (n=9) rats, and from P rats infused with pHSVsiTLR4 (n = 6) or pHSVsiNC (n = 4) were lysed with CelLytic MT (dialyzable mild detergent, bicine and 150 mM NaCl; Sigma Aldrich, St. Louis, MO, USA, Cat. # C3228) supplemented with protease and phosphatase inhibitor cocktails (Sigma) according to the manufacturer’s instructions. The total protein was determined by the bicinchoninic acid assay (BCA, Thermo Fisher Scientific, Waltham, MA, USA, Cat.# 23228 and Cat.# 1859078). The proteins were resolved by SDS–polyacrylamide gel electrophoresis and transferred to polyvinylidene fluoride membranes (PVDF, Bio-Rad, Cat.# 162-0177). The blots were exposed to primary antibody overnight (4°C), followed (1 h; RT) by horseradish peroxidase-labeled secondary antibodies. Detection was with the Plus-ECL kit reagents (Perkin Elmer, Waltham, MA, USA, Cat.# NEL105001EA) and quantification was by densitometric scanning with a Bio-Rad GS-700 imaging densitometer.
Small Interfering RNAs
Small interfering (si) RNAs were designed to target distinct sequences within the rat TLR4 gene (Gene bank Entry No: NC_005104.2) and α2 subunit of the rat GABAA receptor (Gene bank Entry No: NC_005113.2). A scrambled siRNA (siNC) served as control. BLAST search against EST libraries was performed to ensure that no other gene was targeted. As previously reported (Aurelian et al., 2016; Liu et al., 2011), the TLR4 siRNA is AATGCCAGGATGATGCCTC (targets nt -9 to 10) and the scrambled siRNA is GCGGCACACGTA GTAAGTT (SI, Table S1). The GABAA α2 siRNA is TAAGCTTCCATGAGGACAA (targets nt -9 to 10). The siRNAs were synthesized as 60-mer sense and antisense oligonucleotide templates (19 × 2 nt) specific to the targeted genes and 22 nt for restriction enzyme sites and hairpin structure. Synthesis was at the University of Maryland Biopolymer Core Facility and used the phosphoramidite (AB) technology. Inhibition of cognate gene expression was confirmed in RAW264.7 cells that express TLR4 and RINm5F cells that express GABAA α2. The siRNAs were transfected at a final concentration of 65 nM using the siPORT amine transfection agent (Thermo Fisher Scientific, Waltham, MA; Cat# AM4502) according to the manufacturer’s instructions, and extracts collected 72 h post-transfection were immunoblotted with TLR4 or α2 antibodies, as previously described (Aurelian et al., 2016; June et al., 2015; Liu et al., 2011; Yang et al., 2011). The CRF and scrambled (scr) siRNAs were obtained from Santa Cruz Biotechnology. The CRF siRNA (Cat. # sc-39395) is CAUGGAGAUUAUUGGGAAAtt; the scr siRNA (Cat. # sc- 37007) is UUCUCCGAACGUGUCACGUtt.
HSV-1–Based Amplicon Vectors
siRNAs were delivered with non-replicating non-toxic herpes simplex virus type 1 (HSV-1) vectors, known as amplicons. Amplicons are bacterial plasmids that contain two noncoding elements from HSV-1, an origin of DNA replication and a DNA packaging/cleavage signal, which allow replication and packaging into HSV-1 particles as a 150-kb concatamer. Numerous copies of the transgene sequences are packaged into one vector particle, thereby allowing for high expression levels. Amplicons retain the HSV naturally discriminative neurotropism. Indeed, owing to the ubiquitous receptors that it employs, HSV tends to be relatively nonselective in infection of a variety of cells in culture, including astrocytes and microglia. However, in vivo, particularly after CNS administration, it specifically targets only neurons (neurotropism). This is reflected by the ability of the virus to: (i) establish life-long infection of the peripheral ganglia sensory neurons, known as latency, and (ii) cause encephalitis, which is the most common viral encephalitis and is associated with virus-induced neuronal apoptosis. Amplicons retain the HSV natural in vivo tropism for neurons, particularly after CNS delivery (Aurelian, 2014; Berges et al., 2007; Cohen et al., 2011; de Silva and Bowers, 2011; Fiandaca et al., 2012; June et al., 2015; Liu et al., 2011; Manservigi et al., 2010; Perkins et al., 2003; Saeki, 2006; Suzuki et al., 2008; Taylor et al., 2005). The neuronal localization of the amplicons after NAc-shell infusion is shown in SI, Fig. S6.
The construction and properties of the amplicons for TLR4 siRNA (pHSVsiTLR4), GABAA α2 siRNA (pHSVsiLA2) and scrambled siRNA (pHSVsiNC) were previously described (Aurelian et al., 2016; June et al., 2015; Liu et al. 2011; Yang et al., 2011). Briefly, the pHSVsi vector used to generate the siRNA plasmids that are packaged into HSV-1 virions expresses EGFP under the direction of the HSV-1 IE4/5 immediate-early promoter. The incorporation of EGFP allows for the titration of the vector stocks and the visualization of cell transduction in culture and in the CNS. The pSUPER plasmid, which contains the RNA polymerase III-dependent H1 promoter and well-defined start of transcription and termination signals, is used to generate a second transcription unit for the synthesis of siRNA. The siRNAs were inserted into the pHSVsi vector between the BglII and HindIII sites, downstream of the RNA polymerase III-dependent H1 promoter and packaged as previously described (Aurelian et al., 2016; June et al., 2015; Liu et al. 2011; Saydam et al., 2005; Yang et al., 2011). The amplicon titers were 1 × 109, 5×108 and 2 × 108 Transducing Units (TU)/ml for pHSVsiTLR4, pHSVsiLA2 and pHSVsiNC, respectively.
Stereotaxic Procedures for amplicon or antalarmin delivery
Rats were anesthetized through isoflurane/O2 gas inhalation and positioned in a stereotaxic apparatus. The microinjection sites were in the NAc-shell and extended from +2.5 mm anterior to bregma to +1.0 mm anterior to bregma, 0.8 mm lateral to the midline in both hemispheres, and −7.2 mm into the brain from the surface of the skull (Paxinos and Watson, 2009). Because amplicons do not diffuse over long distances, we gave 3 small injections in each hemisphere spaced across the entire shell. Each site received 200 nL of PBS or amplicon (2.5 × 105 TU) delivered with a calibrated pulled glass micropipette (~20-µm tip) connected to a Picospritzer II pneumatic pressure injection apparatus (Science Products GmbH). Injections were over 30 s followed by a 1- to 2- min pause for tissue recovery before insertion of the pipette at the next site. The Institutional Animal Care and Use Committee and Biosafety Committees of Howard University approved the procedures. Transduction is effective with clusters of 20–45 transduced neurons localized in neurons at the injection sites and failing to traffic to distant brain areas (Aurelian et al., 2016; June et al., 2015; Liu et al., 2011; Yang et al., 2011).
Antalarmin delivery was as previously described (June et al., 2015). Briefly, rats were implanted with bilateral 28-gauge guide cannulae (PlasticsOne Inc, Roanoke, VA) into the NAc shell. Antalarmin diluted in sterile PBS or PBS vehicle were infused at a rate of 0.1 µl/min using a Harvard infusion pump (0.15 µl/hemisphere). Injector tips were left in place for an additional minute to facilitate tissue accommodation of the injection volume. This was repeated over three consecutive days, for a cumulative antalarmin dose of 4 µg/animal. At the end of the injection series brains were removed and the position of the cannulae was verified by observing the surface entry point of the cannulae and the depth from the brain surface during dissection of the tissue. Extracted tissue was probed for CRF and TLR4 expression by immunoblotting.
Delay Discounting [Impulsivity]
Impulsivity, operationally defined as choosing a smaller immediate reward to the exclusion of a larger delayed reward, was quantified by the adjusted amount delay discounting (DD) assay (Aurelian et al., 2016; Gondre-Lewis et al., 2016; Oberlin and Grahame, 2009; Wilhelm and Mitchell, 2008). Operant boxes consisted of a nosepoke light, two levers, a cue light above each lever, a house light, and a 10 mL descending sipper tube for saccharin reinforcement [0.03% w/v]. Control of the operant boxes and data collection were with the MedPC IV software (MedAssociates, St. Albans, VT). Rats underwent four stages of behavioral shaping prior to testing. For stage 1, all center nose pokes are reinforced on a fixed ratio 1 (FR1) schedule with 20 s sipper access, where 1 lever press is required for sipper access. At stage 2, center nose pokes are reinforced on a FR1 schedule with 10 s sipper access, and the animal must complete 20 trials to move to the next stage. Stage 3 requires 20 trials cued with a center light illuminated for 20 s. There is a 10 s inter-trial interval. At stage 4, a nose poke and lever press is required for the 10-second sipper access, and both right and left levers are reinforced equally. There are 20 trials with a 10 s inter-trial interval in 60 min (Oberlin and Grahame, 2009).
Side bias was assessed after shaping by averaging the last 3 days’ choices on each side. The large reinforcer was assigned to the non-preferred side, to counter any initial side bias. After shaping was completed, rats were assessed at 0 s delay. This time point is used as a task to assess discrimination of reinforcer (saccharin) magnitude prior to introduction of any delay to the larger reward. Immediate reward started at 1 s of saccharin access, and was adjusted upwards and downwards by 0.1 s based on the rat’s choices. An immediate choice resulted in down-adjustment of the sipper access time by 0.1 s on the next trial, whereas a delay choice resulted in up-adjustment of the sipper access time by 0.1 s in the next trial. The total adjustments in access were restricted to a minimum of 0 s and a maximum of 2 s. Average adjusted amounts of the reward over the last 20 trials served as the measure of adjusted amount. If rats failed to complete 20 trials during shaping they were excluded from testing. All rats received 2-hour water access at the end of daily testing (Oberlin and Grahame, 2009).
P rats (n = 21) were trained in the delay discounting paradigm as previously described (Aurelian et al., 2016; Gondre-Lewis et al., 2016) and tested at 0 and 12 second delays. The P rats were randomly separated into treatment groups, and bilaterally infused with pHSVsiTLR4 (n = 13) or pHSVsiNCC (n = 8). They were tested in the delay discounting paradigm at 4, 8, 12, 16, 20 second delays for two consecutive days at each delay. On days 19–22 of Phase 2, rats in both treatment groups were tested at 12 and 16 second delays to confirm the post viral effect. The two-day data for each delay was averaged.
Statistics
Data were analyzed by appropriate ANOVAs. Significant ANOVAs were followed by Newman-Keuls post-hoc tests. Analyses were performed using the SigmaPlot 11.2 software program (Systat Software Inc., San Jose, CA).
RESULTS
TLR4 expression is higher in the NAc-shell from P than NP rats
We have previously shown that an activated TLR signal (activation defined by MCP-1 expression) is located within neurons in the CeA and VTA and it controls the predisposition to initiate alcohol drinking (binge drinking). The TLR4 signal is not located in the VP, indicating that dominant mechanisms function at distinct brain sites (June et al., 2015; Liu et al., 2011). To begin examining the potential function of the TLR4 signal in impulsivity, we therefore focused on the NAc-shell, a site implicated in impulsivity (Chaudhri et al., 2010; Feja et al., 2014). Punch biopsies were collected from P (n=7) and NP (n=4) rats that had not been previously exposed to alcohol and protein extracts were immunoblotted with antibody to TLR4, stripped and re-probed with GAPDH antibody, used as gel loading control. The results were quantitated by densitometric scanning and expressed as densitometric units normalized to GAPDH, as previously described (Aurelian et al., 2016; Gondre-Lewis et al., 2016; June et al., 2015; Liu et al., 2011; Yang et al., 2011). As shown in Fig. 1A, P rats had significantly (p≤0.05) higher levels of TLR4 than NP rats. This likely reflects the significantly higher percentage (p≤0.001) of TLR4+ GABAergic neurons in P than NP rats (93.7 ± 5.7% and 10 ± 1.8%, respectively), as evidenced by double immunofluorescent staining of NAc-shell sections with TLR4 and GAD1 antibodies (SI, Fig. S7; Table 1). The TLR4 staining was primarily located in the cell body with minimal staining seen in projections/fibers (SI, Fig. S8). Also, although TLR4 is expressed in VTA dopaminergic neurons [Tyrosine hydroxylase (TH+)] (Aurelian et al., 2016), it did not co-localize with the TH+ projections that extend from the VTA to the NAc (SI, Fig. S9).
Fig. 1. P rats have elevated levels of TLR4 and GABAA α2 and increased numbers of co-expressing cells in the NAc-shell.

(A,B) Protein extracts of micropunches collected from the NAc-shell from NP (n=4) and P (n=7) rats were immunoblotted with TLR4 (A) or α2 (B) antibodies, stripped and reprobed with antibody to GAPDH used as gel loading control. The results were quantitated by densitometric scanning and expressed as densitometric units normalized to GAPDH ± SEM. Each lane represents a distinct animal. The TLR4 and α2 levels are elevated in P compared to NP rats. (*p≤0.05 by ANOVA). (C,D) Confocal microscopy and Z-stack imaging of double immunofluorescent staining of NAc-shell sections (n=5/group) with TLR4 (red) and GABAA α2 (green) antibodies. Merged images reveal numerous α2+ neurons expressing TLR4 in the NAc-shell from P rats (C), but the numbers of TLR4+/α2+ cells are significantly lower in the NAc-shell from NP rats (D). Scale bars: 15 µm. The % co-staining cells are summarized in Table 1.
TLR4 is co-expressed with GABAA α2+ and CRF
To better understand which cells express TLR4, we followed on our previous finding that in the CeA, TLR4 signaling is downstream of the GABAA α2 subunit (Liu et al., 2011). Protein extracts of NAc-shell micropunches from P and NP rats (n=5/group) were immunoblotted with α2 antibody, and sections obtained from NAc-shell samples were stained by double immunofluorescence with α2 and TLR4 antibodies. The α2 levels were significantly (P≤0.05) higher in P than NP rats (Fig. 1B) and TLR4 colocalized with α2 in 95.9 ± 4.9% and 9.6 ± 2.6% of the cells from P and NP rats, respectively (p≤0.001) (Fig. 1C, D; Table 1). We conclude that the α2+/TLR4+ neurons at this site are GABAergic, because double immunofluorescent staining indicated that most of the α2+ cells (91.1 ± 8.3%) and TLR4+ cells (93.7 ± 5.7%) express GAD1 (SI, Fig. S7).
Interestingly, immunoblotting with monoclonal or polyclonal CRF antibodies directed against partially distinct epitopes [aa 154–196 and 176–196, respectively (M&M)] indicated that the levels of the CRF prohormone (a single ~20 kDa band) were also significantly elevated in P as compared to NP rats (Fig. 2A). Double immunofluorescent staining with TLR4 and CRF antibodies confirmed that virtually all the CRF+ cells co-expressed TLR4 (92.1 ± 6.7% and 14.7 ± 5.1% CRF+/TLR4+ cells in P and NP rats, respectively) (Fig. 2B, C; Table 1). α2 and CRF staining was both in the cell bodies and fibers (SI, Fig. S8). Collectively, the data indicate that the TLR4+ cells at this site co-express α2 and CRF, potentially indicative of molecular interaction.
Fig. 2. The levels of CRF and numbers of TLR4+/CRF+ cells are elevated in the NAc-shell from P rats.

(A) Protein extracts of the micropunches examined in Fig. 1 were immunoblotted with CRF monoclonal antibody (Santa Cruz Biotechnology, Cat. # sc-293187), stripped and re-probed with antibody to GAPDH and the results are expressed as densitometric units normalized to GAPDH ± SEM. As also shown in SI, Fig. S2A, the only detected band is ~20 kDa and its levels are significantly (*p≤0.05 by ANOVA) elevated in P as compared to NP rats. Similar results were obtained for the polyclonal antibody (Bioss Antibodies, Cat. # bs-0246R). B, C) Confocal microscopy and Z-stack imaging of double immunofluorescent staining of NAc-shell sections (n=5/group) with TLR4 (red) and CRF (Bioss Antibodies, Cat. # bs-0246R) (green) antibodies. Merged images reveal numerous CRF+ neurons expressing TLR4 in the NAc-shell from P rats (B), but the numbers of TLR4+/CRF+ cells are significantly lower in the NAc-shell from NP rats (C). Scale bars: 20 µm. The % co-staining cells are summarized in Table 1.
TLR4 regulates the CRF/CRFR1 system through PKA/CREB activation
To begin inquiring whether the TLR4/CRF co-localization is functionally relevant, mock- or TLR4-transfected neuronal (SK-N-SH) cells were examined for CRF expression by immunoblotting. SK-N-SH cells were used because they barely express TLR4, thereby enabling direct verification of the TLR4 effects through plasmid transfection. The stripped blots were re-probed with antibody to pCREB followed by antibody to activated PKA (pPKA), which is known to activate CREB through phosphorylation at Ser-133 (Ahmed et al., 2013; Sands and Palmer, 2008), and antibody to activated MAPK/ERK (pERK1/2). Cells transfected with TLR4 in the presence of the PKA-specific inhibitor H89, were studied in parallel and immunoblotting with β-Actin antibody was used as gel loading control. The results were quantitated by densitometric scanning and are expressed as mean, β-Actin-adjusted, densitometric units ± SEM. PKA/CREB activation was studied because it functions in the LPS-activated TLR4 signal and controls the transcription of genes, which contain cyclic AMP response elements (CRE) in their promoters, such as CRF (Ahmed et al., 2013; Aurelian et al., 2016; Kageyama and Suda, 2009; Wen et al., 2017). MAPK/ERK activation was studied because the PKA and MAPK/ERK pathways cross-talk (Nguyen et al., 2009).
We found that the levels of CRF were significantly (p≤0.01) higher in the TLR4- than mock-transfected cells, and this was accompanied by a significant (p≤0.01) increase in the levels of both pPKA and pCREB, but not pERK1/2 (Fig. 3A, B). Consistent with previous findings that CRF drives CRFR1 transcription via promoter-initiated events (Parham et al., 2004), the levels of CRFR1 were also increased in the TLR4 as compared to mock-transfected cells (Fig. 3B) but this increase was not seen in cells transfected with TLR4 in the presence of CRF siRNA (SI, Fig. S4). H89 significantly reduced the levels of both pCREB and CRF/CRFR1 induced by TLR4 transfection (Fig. 3B), supporting the interpretation that pPKA/pCREB is involved in TLR4-mediated upregulation of CRF/CRFR1. Collectively, the data indicate that the PKA/CREB pathway is a significant contributor to the ability of TLR4 to upregulate the CRF/CRFR1 system, without any apparent contribution from the MAPK pathway.
Fig. 3. CRF amplification loop sustains activated TLR4 signal; α2 contribution.

(A) Protein extracts from mock- or TLR4-transfected SK-N-SH cells (n=5 each) were immunoblotted with pCREB antibody, and the blots were sequentially stripped and immunoblotted with antibodies to pPKA, pERK1/2, TLR4 and β-Actin used as gel loading control. The results were quantitated by densitometric scanning and expressed as densitometric units normalized to β-Actin ± SEM. The levels of pCREB, pPKA and TLR4, but not pERK1/2, are significantly higher in the TLR4- than mock-transfected SK-N-SH cells (**p <0.01 by ANOVA). (B) Protein extracts from mock- or TLR4-transfected SK-N-SH cells treated or not (n=5 each) with the PKA inhibitor H89 (10 µM) were immunoblotted with antibody to pCREB and the blots were sequentially stripped and immunoblotted with antibodies to CRF (Bioss Antibodies, Cat. # bs-0246R), CRFR1 and β-Actin. The results are expressed as densitometric units normalized to β-Actin ± SEM. The levels of pCREB, CRF and CRFR1 are significantly higher in the TLR4- than mock-transfected cells, and upregulation is inhibited by H89 (*p <0.05; **p<0.01; *** p<0.001 by ANOVA). (C) Protein extracts from mock- or α2-transfected Neuro2a cells in the presence or absence of amplicons that deliver TLR4 (siTLR4) or scrambled (siNC) siRNA (n=5 each) were immunoblotted with antibody to CRF (Bioss Antibodies, Cat. # bs-0246R), the blots were sequentially stripped and immunoblotted with antibodies to TLR4 and β-Actin. Results are expressed as densitometric units normalized to β-Actin ± SEM. The levels of CRF and TLR4 are significantly higher in the α2- than mock-transfected cells and upregulation is inhibited by siTLR4, but not siNC (*p≤0.05 by ANOVA). (D) Neuro2a cells mock- or α2-transfected in the presence or absence of amplicons for TLR4 siRNA (siTLR4), α2 siRNA [siLA2 (Liu et al., 2011)] or scrambled siRNA (siNC) were stained with pCREB antibody (red) and examined for nuclear localization (activation). DAPI (blue) was used as nuclear counterstain. pCREB nuclear staining was minimal in the mock-transfected cells (CTL) and it was almost entirely cytoplasmic. α2 transfection significantly increased the % cells with nuclear pCREB staining and nuclear localization was inhibited by siTLR4 and siLA2, but not siNC (*** p≤0.001 by ANOVA).
α2 increases CRF expression through the TLR4-CREB signal
Having seen that α2 co-localizes with TLR4 and CRF in the NAc-shell, we wanted to know whether it might contribute to the ability of the TLR4 signal to increase CRF expression. The question follows on our previous finding that α2 regulates the TLR4 signal in the CeA (Liu et al., 2011), but its ability to modulate the TLR4 interaction with other genes is unknown. We used Neuro2a cells that express TLR4 in order to directly examine the contribution of α2 (introduced by transfection) to the TLR4 signal. Transfection was in the presence or absence of the amplicons that deliver siRNA specific for TLR4 (siTLR4) or scrambled siRNA control (siNC) and protein extracts collected at 48 hrs post-treatment were examined for: (i) TLR4 and CRF expression by immunoblotting and (ii) CREB activation by nuclear staining with pCREB antibody. The levels of both TLR4 and CRF were significantly higher (p≤0.05) in the α2- than mock-transfected cells, indicating that α2 contributes to CRF upregulation. siTLR4 significantly reduced the ability of α2 to upregulate CRF, but CRF was not reduced in α2-transfected cells treated with siNC, indicating that TLR4 is involved in the ability of α2 to regulate CRF expression (Fig. 3C). We conclude that the α2-activated TLR4 signal contributes to CRF upregulation, because: (i) the % cells with nuclear pCREB staining was significantly (p≤0.001) higher in α2- than mock-transfected cells (Fig. 3D; 62.2 ± 3.6% and 4.3 ± 2.1%, respectively), and (ii) pCREB nuclear translocation was significantly higher in the α2- than mock-transfected cells, an increase that was inhibited by siRNA to TLR4 or α2, but not by scrambled siRNA (Fig. 3D). However, the mechanism responsible for the α2-mediated TLR4 signal activation is still unknown, and we do not exclude the potential contribution of other signals to CRF upregulation.
CRF regulates TLR4 expression through CRFR1 and MAPK/ERK activation
To examine whether CRF can feedback on TLR4 regulation, SK-N-SH cells that express CRFR1, were transfected with a CRF plasmid in the absence or presence of the CRFR1 antagonist antalarmin, the MEK-specific inhibitor U0126, or both inhibitors, and the extracts were immunoblotted with antibodies to TLR4 followed by β-Actin. Consistent with the antibody validation data (SI, Fig. S2), the ~20 kDa CRF protein was seen in the CRF, but not mock-transfected cells (Fig. 4A) and it was associated with significantly (p≤0.01) increased levels of TLR4 expression (Fig. 4B). However, the TLR4 increase was reduced (57.2 ± 5.1%) by treatment with antalarmin or U0126 (42.8 ± 3.8%) and the levels of TLR4 in cells transfected with CRF in the presence of both inhibitors were virtually identical to those in the mock-transfected and untreated cells (100% inhibition) (Fig. 4B). The U0126 effect is through inhibition of ERK1/2 phosphorylation (activation), as evidenced by the findings that CRF transfection increased pERK1/2 expression, an increase blocked by U0126 (Fig. 4C). Significantly, the levels of pPKA were also significantly increased in the CRF-transfected cells and this increase was also inhibited by U0126 (Fig. 4C), suggesting that the MAPK/ERK pathway functions through pPKA to upregulate TLR4 in the CRF-transfected cells. Collectively, the data indicate that CRF regulates TLR4 expression both through its interaction with CRFR1 and the activation of the MAPK/ERK pathway and its effect on pPKA, defining a promiscuous, TLR4-sustaining, CRF amplification loop.
Fig. 4. CRF regulates TLR4 expression through CRFR1 and MAPK/ERK activation.

(A) Protein extracts from mock- or CRF-transfected SK-N-SH cells (n=5 each) were immunoblotted with antibodies to CRF (Bioss Antibodies, Cat. # bs-0246R) or β-Actin and the results are expressed as densitometric units normalized to β-Actin ± SEM. The levels of CRF are significantly higher in the CRF- than mock-transfected cells (****P<0.0001 CTL vs. CRF); (B) Protein extracts from mock- or CRF-transfected SK-N-SH cells untreated or treated with the CRFR1 antagonist antalarmin (15nM) or the ERK1/2 specific inhibitor U0126 (20 µM) (n=5 each) were immunoblotted with antibodies to TLR4 or β-Actin and the results are expressed as densitometric units normalized to β-Actin ± SEM. The levels of TLR4 are significantly higher in the CRF- than mock-transfected cells and this increase is reduced by treatment with antalarmin (57.2±5.1%) or U0126 (42.8±3.8%). The levels of TLR4 in CRF-transfected cells treated with both inhibitors are virtually identical to those in the mock-transfected cells (100% inhibition) (*p<0.05; **p <0.01 by ANOVA). (C) Protein extracts from mock- or CRF-transfected SK-N-SH cells treated or not (n=5 each) with U0126 (20 µM) were immunoblotted with pERK1/2 antibody, and the blots were sequentially stripped and immunoblotted with antibodies to pPKA and β-Actin. Results are expressed as densitometric units normalized to β-Actin ± SEM. The pERK1/2 and pPKA levels are significantly higher in the CRF- than mock-transfected cells, and this increase is reduced by U0126 treatment (**p<0.01 by ANOVA).
The CRF-related TLR4 signal is innately activated in the NAc-shell from P rats
To examine whether the TLR4 signal is innately activated and CRF-associated in P, but not NP, rats, NAc-shell sections from normal, non-alcohol exposed animals (n=5/group) were stained in double immunofluorescence with antibodies to TLR4 and pCREB or CRF and pCREB and examined for nuclear pCREB staining. Most of the TLR4+ cells from the P rats (83.5 ± 4.8%) had co-localized nuclear staining with pCREB antibody, as compared to only 6.0 ± 3.8% cells in the NP rats. Similar results were obtained for CRF/pCREB staining with the % of CRF+ cells with nuclear pCREB localization being 78.5 ± 7.9% and 4.1 ± 3.9% for P and NP rats, respectively (Fig. 5; Table 1). Double immunofluorescent staining with antibodies to TLR4 and NF-kB (p65) showed some cells with TLR4/p65 colocalization in the P but not NP rats, but p65 nuclear localization, which is indicative of activation, was not seen (SI, Fig. S5). Collectively, the data indicate that the TLR4 signal is innately activated in P, but not NP rats, and this is associated with activation of the CREB transcription factor and CRF expression, independent of alcohol exposure.
Fig. 5. TLR4 is innately activated in the NAc-shell from P rats.

Confocal microscopy and Z-stack imaging of double immunofluorescent staining of NAc-shell sections from P and NP rats (n=5/group) with TLR4 (red) and pCREB (green) antibodies or CRF (red) (Santa Cruz Biotechnology, Cat. # sc-293187) and pCREB (green) antibodies. Most of the TLR4+ and CRF+ cells in the NAc-shell from the P rats have co-localized nuclear staining with pCREB antibody. The numbers of TLR4+ and CRF+ cells with pCREB nuclear staining are significantly lower in the NAc-shell from NP rats (***p<0.001 by ANOVA). Scale bars: 20 µm. The % co-staining cells are summarized in Table 1.
The CRF amplification loop is present in the NAc-shell from P rats
Two series of experiments were done in order to examine whether the CRF amplification loop identified in cultured neuronal cells is also present in the NAc-shell from P, but not NP rats. First, NAc-shell micropunches collected from P rats not previously exposed to alcohol but treated (n=6) or not (n=5) with antalarmin were examined for TLR4 and CRF expression by immunoblotting, as described in M&M. Second, NAc-shell sections from non-alcohol exposed and untreated P and NP rats (n=5/ group) were stained in double immunofluorescence with antibodies to TLR4 and pERK1/2 or CRF and pERK1/2 and examined for pERK1/2 nuclear localization (activation). The levels of CRF and TLR4 were significantly (p≤0.01) lower in antalarmin-treated than untreated P rats (Fig. 6A) and pERK1/2 nuclear staining was seen in both the TLR4+ and CRF+ cells from P, but not NP rats (23.1 ± 7.8% and 1.1 ± 0.98% TLR4+/pERK+ cells and 21.7 ± 5.3% and 0.93 ± 0.91% CRF+/pERK+ cells, respectively) (Fig. 6B; Table 1). Collectively, the data indicate that the CRF amplification loop identified in the transfected neuronal cells is also present in the NAc-shell from P rats.
Fig. 6. The CRF-associated TLR4 signal is innately activated in the NAc-shell from P rats.

(A) Protein extracts of NAc-shell micropunches collected from P rats not previously exposed to alcohol but treated (n=6) or not (n=5) with antalarmin were immunoblotted with CRF antibody (Bioss Antibodies, Cat. # bs-0246R). The blots were stripped and sequentially blotted with antibodies to TLR4 and GAPDH and the results are expressed as densitometric units normalized to GAPDH ± SEM. Each lane represents a distinct animal. The levels of both CRF and TLR4 are significantly lower in the antalarmin treated than untreated rats (**p<0.01 by ANOVA). (B) Confocal microscopy and Z-stack imaging of double immunofluorescent staining of NAc-shell sections from P and NP rats (n=5/group) with CRF (red) (Santa Cruz Biotechnology, Cat. # sc-293187) and pERK1/2 (green) antibodies or TLR4 (red) and pERK1/2 (green) antibodies. TLR4+ and CRF+ cells in the NAc-shell from the P rats have co-localized nuclear staining with pERK1/2 antibody (21.7 ± 5.3% and 23.1 ± 7.8%, respectively) but co-localization is barely detectable in the NAc-shell from NP rats (0.93 ± 0.91% and 1.1 ± 0.98%, respectively) (*p<0.05 by ANOVA). Scale bars: 15 µm. The % co-staining cells are summarized in Table 1.
TLR4 in the NAc-shell regulates impulsivity
To examine whether the innately activated TLR4 signal in the NAc-shell from P rats controls impulsivity, cohorts of P rats were randomly given pHSVsiTLR4 (n = 8) or pHSVsiNC (n = 13) in the NAc-shell by bilateral stereotaxic infusion and examined by the DD assay, as described in Materials and Methods. As shown in Fig. 7A, pHSVsiTLR4 significantly increased adjusted amounts (impulsivity was decreased) above those of P rats infused with pHSVsiNC. Two Way ANOVA showed significant main effects of Treatment: F(1,178) = 28.335, p < 0.001, and Delay F(9,178) = 2.402, p = 0.014. Confirming the function of the TLR4 signal in impulsivity, the pHSVsiTLR4 effect on impulsivity reflects its inhibitory effect on TLR4 expression (Fig. 7B), which is defined by the duration of siRNA integrity/availability (Liu et al., 2011; Saydam et al., 2005). This is consistent with the antibody specificity. Indeed, both TLR4 and impulsivity were inhibited on day 3, but not day 15 post-treatment and neither TLR4 expression nor impulsivity were inhibited in animals injected with pHSVsiNC (Fig. 7B). Collectively, the data document the specificity of the TLR4 signal effect on impulsivity.
DISCUSSION
The salient feature of the data presented in this report is the finding that a TLR4 signal located in the NAc-shell α2+ GABAergic neurons is innately activated in alcohol preferring P rats. The signal is sustained through a CRF amplification loop and it regulates impulsivity. The following comments seem pertinent with respect to these findings.
The role of TLR4 in alcohol drinking was studied in different animal species/models using various methods to inhibit signaling and focusing on excessive alcohol intake. TLR4 was shown to control signaling associated with increased anxiety-related behavior during alcohol withdrawal (Crews et al., 2017) and the sedative effects of alcohol in mice (Corrigan et al., 2015; Wu et al., 2012). Although associated with side effects (locomotor activity, water intake), TLR4 inhibition in alcohol-dependent mice through intraperitoneal drug administration, also inhibited alcohol drinking (Bajo et al., 2016). Mice given intraperitoneal injections of the TLR4-specific ligand, LPS, had a prolonged increase in ethanol self-administration (Blednov et al., 2011), an effect mediated by TLR4 receptors and immune/inflammatory pathways (Wu et al., 2012), and TLR4 knockout (KO) mice evidenced reduced alcohol preference following intermittent intraperitoneal treatment (Montesinos et al., 2016). Notwithstanding, examination of KO mice has implicated TLR2, but not TLR4, in excessive alcohol drinking (Blednov et al., 2017) and a recent study concluded that TLR4 has minimal impact on the amount of consumed alcohol but it mediates the acute sedative effects, which might indicate the susceptibility to develop alcohol dependence (Harris et al., 2017).
Importantly, it is becoming increasingly evident that in response to a gene knockout the organism may upregulate one or more genes that modulate the affected function (El-Brolosy and Stainier, 2017; Teng et al., 2013). This is particularly true for genes that are related to the knockout, as is the case for TLRs, which share many properties and functions (Takeda and Akira, 2015). By contrast to the KO approach, siRNA-mediated gene knockdown examines the contribution of the targeted gene within an unaltered genetic makeup and at a specific site. The effect of the animal species and the preferential cellular tropism of the delivery vector cannot be excluded. However, knockdown lacks a compensatory response when compared to the corresponding KO mutants (El-Brolosy and Stainier, 2017). Moreover, the siRNA inhibitory effect is temporary, defined by the duration of siRNA integrity/availability (Liu et al., 2011; Saydam et al., 2005), providing a built-in control for the actual contribution of the targeted gene.
Our interest is in the genes that may contribute to the predisposition of non-alcohol exposed subjects to initiate drinking. To this end, our studies focused on alcohol-preferring (P) rats, which were developed by selective breeding rather than genetic manipulation (viz. KO or mutation), suggesting that they might best represent the unaltered genetic make-up of the non-alcohol exposed individual predisposed to initiate drinking. Relative to their alcohol-non-preferring NP counterparts, P rats display voluntary alcohol consumption (Bell et al., 2006) and have increased impulsivity, which is widely believed to represent the alcohol-seeking behavior that precedes excessive drinking (Beckwith and Czachowski, 2014; Oberlin and Grahame 2009). Using these animals and siRNA delivered with neurotropic HSV vectors (amplicons), we have previously shown that a neuronal TLR4/MCP-1 signal in the CeA and VTA regulates the predisposition to initiate alcohol drinking, and it is under the control of the GABAA α2 subunit. This TLR4 signal is likely activated in the P rats, because the chemokine MCP-1 is downstream of the LPS-activated TLR4 (Liu et al., 2013). Indeed, drinking initiation was inhibited by siRNA-mediated knockdown of TLR4, MCP1 or α2, suggesting that α2 controls an activated regulatory TLR4 signal (June et al., 2015; Liu et al., 2011). However, the α2-regulated TLR4 signal does not function in the VP, where the predisposition to initiate alcohol drinking is regulated by GABAA α1 and is unrelated to TLR4 (Liu et al., 2011; Yang et al., 2011), documenting the existence of dominant regulatory mechanisms at different brain sites, prior to alcohol exposure.
The current studies were designed to examine whether the TLR4 signal functions in the NAc-shell, a site implicated in impulsivity (Chaudhri et al., 2010; Feja et al., 2014), and define its interaction with other potentially contributing genes. We focused on the CRF/CRFR1 system, because: (i) stress is implicated in addiction and it increases CRF expression (Dedic et al., 2017; Koob et al., 2014; Phillips et al., 2015), (ii) CRFR1 is widely distributed in brain regions involved in stress and is implicated in intensification of alcohol self-administration following stress exposure (Quadros et al., 2016), (iii) alcohol-induced CRF upregulates TLR4 expression (June et al., 2015), and (iv) stress caused by maternal deprivation upregulates CRF and impulsivity (Gondre-Lewis et al., 2016). The specificity of the antibodies used to address these questions is confirmed by the validation data summarized in SI, Figs. S1–S4.
We found that TLR4 is expressed in GABAergic neurons that co-express GABAA α2 and CRF, and both the percentage of co-expressing neurons and the levels of the three co-expressed proteins are significantly higher in P than NP rats. The finding that almost all the cells in the P rats express α2, as compared to 10% in the NP rats, suggests that there may be changes in overall GABA reception or in the kinetics of the channel. A decrease in GABA feedback mechanisms in the NAc-shell would alter excitation/inhibition balance that might be favorable to habit formation of rewarding substances in the P compared to the NP rats. Conversely, although the understanding of how the different GABA subunits alter the receptor kinetics relative to function is a field in its infancy, we cannot exclude the possibility that the same level of GABA reception is happening, only with altered kinetics. Elevated levels of α2 have been associated with anxiety behaviors, which are frequently co-morbid in humans with alcoholism. Benzodiazepines used for anti-anxiety treatment bind α2 subunits, but these are by no means the only binding sites for this drug. Human genetic profiling in the Collaborative Study on Genetics of Alcoholism (COGA) identified loci associated with higher alcoholism penetrance that contain the α2 gene (Edenberg and Foroud, 2013) and taken together with our current findings, this further highlights the importance of the α2 gene and NAc subunit expression profiles.
Indicative of an interaction between TLR4 and the CRF/CRFR1 system, we also found that: (i) the levels of pPKA/pCREB and CRF/CRFR1 were elevated in TLR4-transfected neuronal cells in culture accompanied by increased pCREB nuclear translocation, and (ii) both PKA/CREB activation and CRF/CRFR1 upregulation were inhibited by the PKA inhibitor, H89. Indeed, activated PKA (pPKA, Thr197) phosphorylates CREB at Ser-133 (pCREB) and pCREB translocates to the nucleus to control the transcription of genes that contain cyclic AMP response elements (CRE) in their promoters (Ahmed et al., 2013; Sands and Palmer, 2008), such as is the case for CRF (Kageyama and Suda, 2009). Also, CRF regulates CRFR1 expression (Parham et al., 2004), a conclusion supported by our finding that CRFR1 expression is blocked by CRF siRNA (SI, Fig. S4).
Significantly, other pathways are also known to activate CREB, including MAPK/ERK, phosphatidylinositol 3-kinase (PI3K)/Akt, Ca2+/calmodulin-dependent protein kinase (CaMK) IV and protein kinase C (PKC) and pathway cross-talk is a known phenomenon (Nguyen et al., 2009). However, we found that pERK1/2 is not upregulated in the TLR4-transfected cells, and together with the finding that the H89-mediated inhibition of CREB activity in the TLR4-transfected cells is essentially absolute (90.1 ± 10.7%; Fig. 3B), the data underscore the significant role of PKA-mediated pCREB in the TLR4 part of the amplification loop. Indeed, although TLR4 is known to activate NF-kB in LPS-treated monocytes (Guha and Mackman, 2001), a conclusion supported by our finding that LPS activates NF-kB in RAW246.7 cells, NF-kB was not activated in LPS-treated neuronal (Neuro2a) cells, nor in the TLR4+ neurons in the NAc-shell, as evidenced by the failure to detect appropriate nuclear staining (SI, Fig. S5).
We conclude that CRF provides a TLR4 amplification loop, because: (i) CRF transfected neuronal cells expressed higher levels of TLR4 than mock-transfected cells, and (ii) this increase was reduced by treatment with either the CRFR1 antagonist antalarmin or the ERK1/2 inhibitor U0126, with total inhibition achieved when using both inhibitors, suggestive of an additive effect of CRFR1 and the MAPK/ERK pathway. In this context, it should be pointed out that the MAPK pathway functions through AP-1 response elements (Hollenhorst, 2012; Perkins et al., 2003) and these were implicated in CRF-mediated TLR4 upregulation (Bondeva et al., 2007). However, we found that the levels of pPKA were increased in the CRF-transfected cells, an increase inhibited by U0126, suggesting that the MAPK pathway functions through PKA in CRF-mediated TLR4 upregulation. PKA likely upregulates TLR4 through CREB and the binding of cAMP response elements in the TLR4 promoter, as previously reported (Park et al., 2016). The finding that CRF functions through MAPK, in addition to CRFR1, provides a potential explanation for the recent failure of CRFR1 antagonists to show a therapeutic effect in humans with alcohol use disorder (AUD) (Pomrenze et al., in press; Spierling and Zorrilla, 2017).
Significantly, we found that the TLR4 signal is innately activated in the NAc-shell from P, but not NP rats, and it is apparently sustained by the CRF amplification loop detected in cultured cells, which includes both CRFR1 and MAPK activation, apparently functioning in an additive role. Indeed, most of the TLR4+ and CRF+ cells in the NAc-shell from non-alcohol treated P rats had pCREB nuclear staining, which was barely seen in the NP rats and NF-kB was not activated in the NAc-shell (SI, Fig. S5C). Antalarmin infusion into the P rats NAc-shell reduced CRF and TLR4 expression, confirming the contribution of CRFR1 to TLR4 activation. We conclude that the MAPK/ERK pathway contributes to the TLR4-CRF amplification loop also in the NAc-shell (not only in cultured cells) because the TLR4+ and CRF+ cells in the NAc-shell from P, but not NP rats, had nuclear pERK1/2 staining before exposure to alcohol. However, the activated MAPK pathway was only seen in 25% of the neurons, suggesting that CRFR1 is the major contributor to the TLR4-CRF amplification loop. Additional studies are needed to establish the additive effects of antalarmin and U0126 in the NAc-shell. Also, the activated TLR4 signal could have autocrine or paracrine effects that involve neurons and glial cells, all of which express TLR4. Indeed, increased CRF synthesis and release in the CeA regulates GABAergic transmission (Roberto et al., 2012), and it contributes to alcohol drinking and cognitive impulsivity (Silveri et al., 2013). Further studies are needed to address these questions.
Impulsivity is a multidimensional construct with a heterogeneous relationship to drug use (Badiani et al., 2011; Caswell et al., 2016; Oberlin and Grahame, 2009). A recent study using the 5-choice serial reaction time task (5-CSRTT) to measure impulsivity concluded that P rats are not intrinsically impulsive, but their strong alcohol preference reflects increased goal-directed behavior to food incentives (Pena-Oliver et al., 2015). By contrast, high alcohol-preferring mice and rats showed steeper discounting (increased impulsivity) than low preferring strains, (Beckwith and Czachowski 2014; Oberlin and Grahame, 2009; Wilhelm and Mitchell, 2008) when using delay-discounting (DD) tasks, in which impulsive individuals opt for immediate gratification that is detrimental in the long term over delayed benefits that have a more advantageous outcome in the long run (Gullo and Potenza, 2014; MacKillop et al., 2011). Moreover, alcohol-dependent individuals consistently displayed findings of impulsivity-related deficits (Jones et al., 2015; Joos et al., 2013; Lawrence et al., 2009; Petry, 2001; Rubio et al., 2008). Using the DD assay, we have previously shown that P rats have significantly increased levels of impulsivity than wild type (Aurelian et al., 2016) and NP (data not shown) rats, and it is associated with a VTA TLR4/TH signal and stress-induced CRF upregulation (Gondre-Lewis et al., 2016). Following on these findings, we show that infusion of the pHSVsiTLR4 vector into the NAc-shell from P rats blunts impulsivity, associated with TLR4 inhibition. Impulsivity was not altered by pHSVsiNC, which does not inhibit the TLR4 signal, although both amplicons have identical neurotropic properties.
The possibilities cannot be excluded that: (i) H89 may also have PKA-unrelated functions, and (ii) cis-regulatory elements and post-transcriptional and post- translational mechanisms that are still poorly understood contribute to TLR4/CRF interaction. However, the finding that TLR4 upregulates CRF through CREB activation is particularly significant, because: (i) pCREB is known to play an important role in the addiction process, (ii) the pPKA/pCREB signal is implicated in the molecular changes that underlie alcohol drinking (Asher et al., 2012; Calabrese et al., 2015; Pandey et al., 2005), (iii) CREB overexpression in the NAc has been associated with the attempt to increase reward (Larson et al., 2011), and (iv) activated CREB is known to contribute to impulsivity (Moonat et al., 2010; Robison and Nestler, 2011).
How is the TLR4 signal innately activated in the P rats and does this differ at distinct brain sites? One possible interpretation is the presence of significantly higher levels of endogenous activating ligands or adaptor proteins in P than NP rats, acquired through selective breeding and having potentially different function(s) at distinct brain sites. These could include endogenous danger-associated molecular patterns (DAMPs) released as a consequence of injury and inflammation, such as heat shock proteins, extracellular matrix molecules (hyaluronan), HMGB1, oxidized low density lipoprotein (oxLDL) and oxidized phospholipids (oxPL) or saturated fatty acids and LPS mimetic ligands of natural origin (Calabrese et al., 2015; Rocha et al., 2016; Wang et al., 2016; Zou and Crews, 2014;). We posit that α2 is involved in TLR4 signal activation, because this the case in the CeA (Liu et al., 2011), and current findings indicate that α2 transfection increases pCREB nuclear translocation and CRF expression both of which are reduced (albeit not totally eliminated) by TLR4 or α2, but not scrambled, siRNA. The data indicate that TLR4 contributes, but is not entirely responsible for CRF upregulation downstream of α2. Further studies are needed in order to better understand the mechanism of the α2-mediated TLR4-dependent upregulation of CRF and to define the other contributing pathways. However, our data are consistent with previous findings that the GABAergic response is associated with cognitive impulsivity in adolescents and young adults, and genetic variation in the GABAA α2 subunit contributes to impulsivity and a lifetime of alcohol-related problems (Dick et al., 2013; Silveri et al., 2013; Villafuerte et al., 2013).
In conclusion, we show that P rats have an innately activated TLR4 signal in the NAc-shell before exposure to alcohol, which is sustained by a CRF amplification loop and regulates impulsivity. These findings are schematically represented in Fig. 8. Together with independent results that relate impulsivity to alcohol drinking (Jones et al., 2015; Joos et al., 2013; Lawrence et al., 2009; Petry, 2001; Rubio et al., 2008), and our previous findings that: (i) P rats have significantly increased levels of impulsivity than wild type and NP rats, which is associated with a VTA TLR4/TH signal (Aurelian et al., 2016), and (ii) the TLR4 signal in the CeA and VTA predisposes to binge drinking (June et al., 2015; Liu et al., 2011), the emerging picture suggests that the TLR4 signal and its interaction with selected contributory genes at distinct brain sites, contributes to the transition to alcohol dependence. It would be interesting to better understand the ability of chemical TLR4 inhibitors to block impulsivity. Also unknown is the mechanism responsible for the innate activation of the TLR4 signal, the contribution of α2 to impulsivity regulation, and the relationship of α2 to the CRF/CRFR1 system that sustains the activated TLR4 signal are still unknown. Ongoing studies are designed to address these questions.
Fig. 8. Schematic representation of TLR4 function in impulsivity and the transition to alcohol dependence.
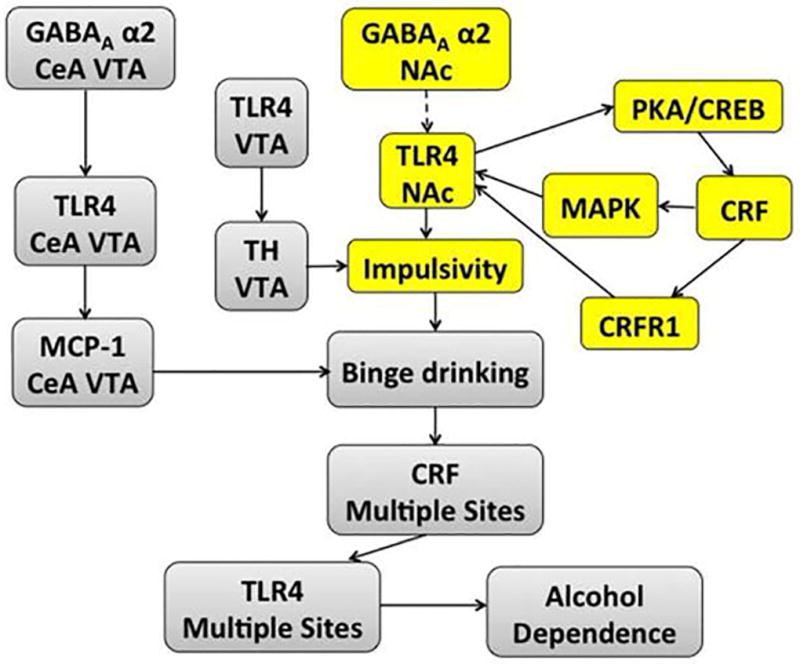
Current findings identify an innately activated TLR4 signal in the NAc-shell from P rats that is sustained by a CRF amplification loop and regulates impulsivity. The loop includes TLR4-mediated CRF upregulation through PKA/CREB activation and CRF-mediated TLR4 upregulation through CRFR1 and MAPK/ERK activation. The major contributor appears to be the CRF/CRFR1 system, because pERK1/2 is only seen in 25% of the TLR4+ and CRF+ neurons. Impulsivity is visualized as a major contributor to the initiation of alcohol drinking by previously non-alcohol exposed subjects (yellow). Dotted line represents potential involvement of α2 in the stimulation of the CRF-amplification loop in the NAc-shell. Together with previous findings that: (i) the TLR4 signal contributes to the initiation of alcohol drinking (binge drinking) at other brain sites (viz. CeA and VTA) (Liu et al., 2011), (ii) the TLR4 signal is upregulated by alcohol-induced CRF expression at these sites (June et al., 2015), and (iii) a TLR4/TH signal in the VTA is associated with increased levels of impulsivity (Aurelian et al., 2016), we posit the schematically represented emerging picture as a contributor to the transition to alcohol-dependence (grey).
Supplementary Material
Highlights.
Alcohol-preferring P rats have innately activated TLR4 signal in nucleus accumbens
TLR4 signal is located in GABAA α2+/CRF+ neurons and regulates impulsivity
CRF amplification loop sustains the activated TLR4 signal in the absence of alcohol
TLR4 upregulates CRF through PKA/CREB activation
CRF upregulates TLR4 through CRFR1 and MAPK/ERK activation
Acknowledgments
These studies were supported by Grant AA021261 (to L.A. and M.G-L) from the National Institute on Alcohol Abuse and Alcoholism.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
The authors declare no conflict of interest.
Authors contributon: LA designed the research; IB, KTW, AP, and MCG-L performed research; LA and IB contributed new reagents/analytic tools; IB, KTW and LA analyzed the data and LA and IB wrote the paper.
References
- Ahmed BY, Husnain O, Stafford R, Howard M, Gujar AS, Moradiya V, Patel KK, Sihotra S. Hyperphosphorylation of CREB in human dopaminergic neurons: a kinetic study of cellular distribution of total CREB and phospho-CREB following oxidative stress. Neuroreport. 2013;24:757–762. doi: 10.1097/WNR.0b013e328364d616. [DOI] [PubMed] [Google Scholar]
- Alfonso-Loeches S, Urena-Peralta J, Morillo-Bargues MJ, Gomez-Pinedo U, Guerri C. Ethanol-Induced TLR4/NLRP3 Neuroinflammatory Response in Microglial Cells Promotes Leukocyte Infiltration Across the BBB. Neurochem Res. 2016;41:193–209. doi: 10.1007/s11064-015-1760-5. [DOI] [PubMed] [Google Scholar]
- Asher O, Cunningham TD, Yao L, Gordon AS, Diamond I. Ethanol stimulates cAMP-responsive element (CRE)-mediated transcription via CRE-binding protein and cAMP-dependent protein kinase. J Pharmacol Exp Ther. 2002;301:66–70. doi: 10.1124/jpet.301.1.66. [DOI] [PubMed] [Google Scholar]
- Aurelian L. Herpes Simplex Viruses: General Features. In: Mahy BWJ, van Regenmortel MHV, editors. Encyclopedia of Virology. 3. Elsevier, ltd; 2014. pp. 383–397. [Google Scholar]
- Aurelian L, Warnock KT, Balan I, Puche A, June H. TLR4 signaling in VTA dopaminergic neurons regulates impulsivity through tyrosine hydroxylase modulation. Transl Psychiatry. 2016;6:e815. doi: 10.1038/tp.2016.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badiani A, Belin D, Epstein D, Calu D, Shaham Y. Opiate versus psychostimulant addiction: the differences do matter. Nat Rev Neurosci. 2011;12:685–700. doi: 10.1038/nrn3104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bajo M, Montgomery SE, Cates LN, Nadav T, Delucchi AM, Cheng K, Yin H, Crawford EF, Roberts AJ, Roberto M. Evaluation of TLR4 Inhibitor, T5342126, in Modulation of Ethanol-Drinking Behavior in Alcohol-Dependent Mice. Alcohol Alcohol. 2016;51:541–548. doi: 10.1093/alcalc/agw026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckwith SW, Czachowski CL. Increased delay discounting tracks with a high ethanol-seeking phenotype and subsequent ethanol seeking but not consumption. Alcohol Clin Exp Res. 2014;38:2607–2614. doi: 10.1111/acer.12523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell RL, Rodd ZA, Lumeng L, Murphy JM, McBride WJ. The alcohol-preferring P rat and animal models of excessive alcohol drinking. Addict Biol. 2006;11:270–288. doi: 10.1111/j.1369-1600.2005.00029.x. [DOI] [PubMed] [Google Scholar]
- Berges BK, Wolfe JH, Fraser NW. Transduction of brain by herpes simplex virus vectors. Mol Ther. 2007;15:20–29. doi: 10.1038/sj.mt.6300018. [DOI] [PubMed] [Google Scholar]
- Blednov YA, Benavidez JM, Geil C, Perra S, Morikawa H, Harris RA. Activation of inflammatory signaling by lipopolysaccharide produces a prolonged increase of voluntary alcohol intake in mice. Brain Behav Immun. 2011;25(Suppl 1):S92–S105. doi: 10.1016/j.bbi.2011.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blednov YA, Black M, Chernis J, Da Costa A, Mayfield J, Harris RA. Ethanol Consumption in Mice Lacking CD14, TLR2, TLR4, or MyD88. Alcohol Clin Exp Res. 2017;41:516–530. doi: 10.1111/acer.13316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bondeva T, Roger T, Wolf G. Differential regulation of Toll-like receptor 4 gene expression in renal cells by angiotensin II: dependency on AP1 and PU.1 transcriptional sites. Am J Nephrol. 2007;27:308–314. doi: 10.1159/000102551. [DOI] [PubMed] [Google Scholar]
- Breese GR, Knapp DJ. Persistent adaptation by chronic alcohol is facilitated by neuroimmune activation linked to stress and CRF. Alcohol. 2016;52:9–23. doi: 10.1016/j.alcohol.2016.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calabrese V, Cighetti R, Peri F. Molecular simplification of lipid A structure: TLR4-modulating cationic and anionic amphiphiles. Mol Immunol. 2015;63:153–161. doi: 10.1016/j.molimm.2014.05.011. [DOI] [PubMed] [Google Scholar]
- Caswell AJ, Celio MA, Morgan MJ, Duka T. Impulsivity as a Multifaceted Construct Related to Excessive Drinking Among UK Students. Alcohol Alcohol. 2016;51:77–83. doi: 10.1093/alcalc/agv070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhri N, Sahuque LL, Schairer WW, Janak PH. Separable roles of the nucleus accumbens core and shell in context- and cue-induced alcohol-seeking. Neuropsychopharmacology. 2010;35:783–791. doi: 10.1038/npp.2009.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen M, Braun E, Tsalenchuck Y, Panet A, Steiner I. Restrictions that control herpes simplex virus type 1 infection in mouse brain ex vivo. J Gen Virol. 2011;92:2383–2393. doi: 10.1099/vir.0.031013-0. [DOI] [PubMed] [Google Scholar]
- Corrigan F, Wu Y, Tuke J, Coller JK, Rice KC, Diener KR, Hayball JD, Watkins LR, Somogyi AA, Hutchinson MR. Alcohol-induced sedation and synergistic interactions between alcohol and morphine: a key mechanistic role for Toll-like receptors and MyD88-dependent signaling. Brain Behav Immun. 2015;45:245–252. doi: 10.1016/j.bbi.2014.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crews FT, Walter TJ, Coleman LG, Jr, Vetreno RP. Toll-like receptor signaling and stages of addiction. Psychopharmacology (Berl) 2017;234:1483–1498. doi: 10.1007/s00213-017-4560-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Silva S, Bowers WJ. Targeting the central nervous system with herpes simplex virus / Sleeping Beauty hybrid amplicon vectors. Curr Gene Ther. 2011;11:332–340. doi: 10.2174/156652311797415845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dedic N, Chen A, Deussing JM. The CRF family of neuropeptides and their receptors - mediators of the central stress response. Curr Mol Pharmacol. 2017 doi: 10.2174/1874467210666170302104053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dick DM, Aliev F, Latendresse S, Porjesz B, Schuckit M, Rangaswamy M, Hesselbrock V, Edenberg H, Nurnberger J, Agrawal A, Bierut L, Wang J, Bucholz K, Kuperman S, Kramer J. How phenotype and developmental stage affect the genes we find: GABRA2 and impulsivity. Twin Res Hum Genet. 2013;16:661–669. doi: 10.1017/thg.2013.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edenberg HJ, Foroud T. Genetics and alcoholism. Nat Rev Gastroenterol Hepatol. 2013;10:487–494. doi: 10.1038/nrgastro.2013.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Brolosy MA, Stainier DYR. Genetic compensation: A phenomenon in search of mechanisms. PLoS Genet. 2017;13:e1006780. doi: 10.1371/journal.pgen.1006780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feja M, Hayn L, Koch M. Nucleus accumbens core and shell inactivation differentially affects impulsive behaviours in rats. Prog Neuropsychopharmacol Biol Psychiatry. 2014;54:31–42. doi: 10.1016/j.pnpbp.2014.04.012. [DOI] [PubMed] [Google Scholar]
- Fiandaca MS, Bankiewicz KS, Federoff HJ. Gene therapy for the treatment of Parkinson's disease: the nature of the biologics expands the future indications. Pharmaceuticals (Basel) 2012;5:553–590. doi: 10.3390/ph5060553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gondre-Lewis MC, Warnock KT, Wang H, June HL, Jr, Bell KA, Rabe H, Tiruveedhula VV, Cook J, Luddens H, Aurelian L, June HL., Sr Early life stress is a risk factor for excessive alcohol drinking and impulsivity in adults and is mediated via a CRF/GABA(A) mechanism. Stress. 2016;19:235–247. doi: 10.3109/10253890.2016.1160280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guha M, Mackman N. LPS induction of gene expression in human monocytes. Cell Signal. 2001;13:85–94. doi: 10.1016/s0898-6568(00)00149-2. [DOI] [PubMed] [Google Scholar]
- Gullo MJ, Potenza MN. Impulsivity: mechanisms, moderators and implications for addictive behaviors. Addict Behav. 2014;39:1543–1546. doi: 10.1016/j.addbeh.2014.06.005. [DOI] [PubMed] [Google Scholar]
- Harris RA, Bajo M, Bell RL, Blednov YA, Varodayan FP, Truitt JM, de Guglielmo G, Lasek AW, Logrip ML, Vendruscolo LF, Roberts AJ, Roberts E, George O, Mayfield J, Billiar TR, Hackam DJ, Mayfield RD, Koob GF, Roberto M, Homanics GE. Genetic and Pharmacologic Manipulation of TLR4 Has Minimal Impact on Ethanol Consumption in Rodents. J Neurosci. 2017;37:1139–1155. doi: 10.1523/JNEUROSCI.2002-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollenhorst PC. RAS/ERK pathway transcriptional regulation through ETS/AP-1 binding sites. Small GTPases. 2012;3:154–158. doi: 10.4161/sgtp.19630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones CG, Fearnley H, Panagiotopoulos B, Kemp RI. Delay discounting, self-control, and substance use among adult drug court participants. Behav Pharmacol. 2015;26:447–459. doi: 10.1097/FBP.0000000000000149. [DOI] [PubMed] [Google Scholar]
- Joos L, Goudriaan AE, Schmaal L, De Witte NA, Van den Brink W, Sabbe BG, Dom G. The relationship between impulsivity and craving in alcohol dependent patients. Psychopharmacology (Berl) 2013;226:273–283. doi: 10.1007/s00213-012-2905-8. [DOI] [PubMed] [Google Scholar]
- June HL, Liu J, Warnock KT, Bell KA, Balan I, Bollino D, Puche A, Aurelian L. CRF-amplified neuronal TLR4/MCP-1 signaling regulates alcohol self-administration. Neuropsychopharmacology. 2015;40:1549–1559. doi: 10.1038/npp.2015.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kageyama K, Suda T. Regulatory mechanisms underlying corticotropin-releasing factor gene expression in the hypothalamus. Endocr J. 2009;56:335–344. doi: 10.1507/endocrj.k09e-075. [DOI] [PubMed] [Google Scholar]
- Koob GF, Buck CL, Cohen A, Edwards S, Park PE, Schlosburg JE, Schmeichel B, Vendruscolo LF, Wade CL, Whitfield TW, Jr, George O. Addiction as a stress surfeit disorder. Neuropharmacology. 2014;76(Pt B):370–382. doi: 10.1016/j.neuropharm.2013.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larson EB, Graham DL, Arzaga RR, Buzin N, Webb J, Green TA, Bass CE, Neve RL, Terwilliger EF, Nestler EJ, Self DW. Overexpression of CREB in the nucleus accumbens shell increases cocaine reinforcement in self-administering rats. J Neurosci. 2011;31:16447–16457. doi: 10.1523/JNEUROSCI.3070-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence AJ, Luty J, Bogdan NA, Sahakian BJ, Clark L. Impulsivity and response inhibition in alcohol dependence and problem gambling. Psychopharmacology (Berl) 2009;207:163–172. doi: 10.1007/s00213-009-1645-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leclercq S, Cani PD, Neyrinck AM, Starkel P, Jamar F, Mikolajczak M, Delzenne NM, de Timary P. Role of intestinal permeability and inflammation in the biological and behavioral control of alcohol-dependent subjects. Brain Behav Immun. 2012;26:911–918. doi: 10.1016/j.bbi.2012.04.001. [DOI] [PubMed] [Google Scholar]
- Liu J, Yang AR, Kelly T, Puche A, Esoga C, June HL, Jr, Elnabawi A, Merchenthaler I, Sieghart W, June HL, Sr, Aurelian L. Binge alcohol drinking is associated with GABAA alpha2-regulated Toll-like receptor 4 (TLR4) expression in the central amygdala. Proc Natl Acad Sci U S A. 2011;108:4465–4470. doi: 10.1073/pnas.1019020108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Jiang Y, Li Y, Wang J, Fan L, Scott MJ, Xiao G, Li S, Billiar TR, Wilson MA, Fan J. TLR4 Signaling augments monocyte chemotaxis by regulating G protein-coupled receptor kinase 2 translocation. J Immunol. 2013;191:857–864. doi: 10.4049/jimmunol.1300790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowery-Gionta EG, Navarro M, Li C, Pleil KE, Rinker JA, Cox BR, Sprow GM, Kash TL, Thiele TE. Corticotropin releasing factor signaling in the central amygdala is recruited during binge-like ethanol consumption in C57BL/6J mice. J Neurosci. 2012;32:3405–3413. doi: 10.1523/JNEUROSCI.6256-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacKillop J, Amlung MT, Few LR, Ray LA, Sweet LH, Munafo MR. Delayed reward discounting and addictive behavior: a meta-analysis. Psychopharmacology (Berl) 2011;216:305–321. doi: 10.1007/s00213-011-2229-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manservigi R, Argnani R, Marconi P. HSV Recombinant Vectors for Gene Therapy. Open Virol J. 2010;4:123–156. doi: 10.2174/1874357901004010123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montesinos J, Alfonso-Loeches S, Guerri C. Impact of the Innate Immune Response in the Actions of Ethanol on the Central Nervous System. Alcohol Clin Exp Res. 2016;40:2260–2270. doi: 10.1111/acer.13208. [DOI] [PubMed] [Google Scholar]
- Moonat S, Starkman BG, Sakharkar A, Pandey SC. Neuroscience of alcoholism: molecular and cellular mechanisms. Cell Mol Life Sci. 2010;67:73–88. doi: 10.1007/s00018-009-0135-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen TV, Yao M, Pike CJ. Dihydrotestosterone activates CREB signaling in cultured hippocampal neurons. Brain Res. 2009;1298:1–12. doi: 10.1016/j.brainres.2009.08.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberlin BG, Grahame NJ. High-alcohol preferring mice are more impulsive than low-alcohol preferring mice as measured in the delay discounting task. Alcohol Clin Exp Res. 2009;33:1294–1303. doi: 10.1111/j.1530-0277.2009.00955.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey SC. TLR4-MyD88 signalling: a molecular target for alcohol actions. Br J Pharmacol. 2012;165:1316–1318. doi: 10.1111/j.1476-5381.2011.01695.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey SC, Zhang H, Roy A, Xu T. Deficits in amygdaloid cAMP-responsive element-binding protein signaling play a role in genetic predisposition to anxiety and alcoholism. J Clin Invest. 2005;115:2762–2773. doi: 10.1172/JCI24381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parham KL, Zervou S, Karteris E, Catalano RD, Old RW, Hillhouse EW. Promoter analysis of human corticotropin-releasing factor (CRF) type 1 receptor and regulation by CRF and urocortin. Endocrinology. 2004;145:3971–3983. doi: 10.1210/en.2004-0194. [DOI] [PubMed] [Google Scholar]
- Park SS, Choi H, Kim SJ, Chang C, Kim E. CREB/GSK-3β signaling pathway regulates the expression of TR4 orphan nuclear receptor gene. Mol Cell Endocrinol. 2016;423:22–29. doi: 10.1016/j.mce.2015.12.023. [DOI] [PubMed] [Google Scholar]
- Pascual M, Balino P, Alfonso-Loeches S, Aragon CM, Guerri C. Impact of TLR4 on behavioral and cognitive dysfunctions associated with alcohol-induced neuroinflammatory damage. Brain Behav Immun. 2011;25(Suppl 1):S80–91. doi: 10.1016/j.bbi.2011.02.012. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The Rat Brain in Stereotaxic Coordinates. 6. Academic Press; San Diego, CA: 2009. [Google Scholar]
- Pena-Oliver Y, Giuliano C, Economidou D, Goodlett CR, Robbins TW, Dalley JW, Everitt BJ. Correction: Alcohol-Preferring Rats Show Goal Oriented Behaviour to Food Incentives but Are Neither Sign-Trackers Nor Impulsive. PLoS One. 2015;10:e0134198. doi: 10.1371/journal.pone.0131016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perkins D, Gyure KA, Pereira EF, Aurelian L. Herpes simplex virus type 1-induced encephalitis has an apoptotic component associated with activation of c-Jun N-terminal kinase. J Neurovirol. 2003;9:101–111. doi: 10.1080/13550280390173427. [DOI] [PubMed] [Google Scholar]
- Petry NM. Delay discounting of money and alcohol in actively using alcoholics, currently abstinent alcoholics, and controls. Psychopharmacology (Berl) 2001;154:243–250. doi: 10.1007/s002130000638. [DOI] [PubMed] [Google Scholar]
- Phillips TJ, Reed C, Pastor R. Preclinical evidence implicating corticotropin-releasing factor signaling in ethanol consumption and neuroadaptation. Genes Brain Behav. 2015;14:98–135. doi: 10.1111/gbb.12189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pomrenze MB, Fetterly TL, Winder DG, Messing RO. The Corticotropin Releasing Factor Receptor 1 in Alcohol Use Disorder: Still a Valid Drug Target? Alcohol Clin Exp Res. 2017 doi: 10.1111/acer.13507. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quadros IM, Macedo GC, Domingues LP, Favoretto CA. An Update on CRF Mechanisms Underlying Alcohol Use Disorders and Dependence. Front Endocrinol (Lausanne) 2016;7:134. doi: 10.3389/fendo.2016.00134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberto M, Gilpin NW, Siggins GR. The central amygdala and alcohol: role of gamma-aminobutyric acid, glutamate, and neuropeptides. Cold Spring Harb Perspect Med. 2012;2:a012195. doi: 10.1101/cshperspect.a012195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robison AJ, Nestler EJ. Transcriptional and epigenetic mechanisms of addiction. Nat Rev Neurosci. 2011;12:623–637. doi: 10.1038/nrn3111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocha DM, Caldas AP, Oliveira LL, Bressan J, Hermsdorff HH. Saturated fatty acids trigger TLR4-mediated inflammatory response. Atherosclerosis. 2016;244:211–215. doi: 10.1016/j.atherosclerosis.2015.11.015. [DOI] [PubMed] [Google Scholar]
- Rubio G, Jimenez M, Rodriguez-Jimenez R, Martinez I, Avila C, Ferre F, Jimenez-Arriero MA, Ponce G, Palomo T. The role of behavioral impulsivity in the development of alcohol dependence: a 4-year follow-up study. Alcohol Clin Exp Res. 2008;32:1681–1687. doi: 10.1111/j.1530-0277.2008.00746.x. [DOI] [PubMed] [Google Scholar]
- Saeki Y. Stable CNS gene delivery with Sleeping Beauty armed with a high-capacity HSV virion. Mol Ther. 2006;13:457–458. doi: 10.1016/j.ymthe.2006.01.005. [DOI] [PubMed] [Google Scholar]
- Sands WA, Palmer TM. Regulating gene transcription in response to cyclic AMP elevation. Cell Signal. 2008;20:460–466. doi: 10.1016/j.cellsig.2007.10.005. [DOI] [PubMed] [Google Scholar]
- Saydam O, Glauser DL, Heid I, Turkeri G, Hilbe M, Jacobs AH, Ackermann M, Fraefel C. Herpes simplex virus 1 amplicon vector-mediated siRNA targeting epidermal growth factor receptor inhibits growth of human glioma cells in vivo. Mol Ther. 2005;12:803–812. doi: 10.1016/j.ymthe.2005.07.534. [DOI] [PubMed] [Google Scholar]
- Silveri MM, Sneider JT, Crowley DJ, Covell MJ, Acharya D, Rosso IM, Jensen JE. Frontal lobe gamma-aminobutyric acid levels during adolescence: associations with impulsivity and response inhibition. Biol Psychiatry. 2013;74:296–304. doi: 10.1016/j.biopsych.2013.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spierling SR, Zorrilla EP. Don't stress about CRF: assessing the translational failures of CRF1antagonists. Psychopharmacology (Berl) 2017;234:1467–1481. doi: 10.1007/s00213-017-4556-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki M, Chiocca EA, Saeki Y. Stable transgene expression from HSV amplicon vectors in the brain: potential involvement of immunoregulatory signals. Mol Ther. 2008;16:1727–1736. doi: 10.1038/mt.2008.175. [DOI] [PubMed] [Google Scholar]
- Takeda K, Akira S. Toll-like receptors. Curr Protoc Immunol. 2015;109 doi: 10.1002/0471142735.im1412s109. 14 12 11-10. [DOI] [PubMed] [Google Scholar]
- Taylor SW, Smith RM, Pari G, Wobeser W, Rossiter JP, Jackson AC. Herpes simplex encephalitis. Can J Neurol Sci. 2005;32:246–247. doi: 10.1017/s0317167100004054. [DOI] [PubMed] [Google Scholar]
- Teng X, Dayhoff-Brannigan M, Cheng WC, Gilbert CE, Sing CN, Diny NL, Wheelan SJ, Dunham MJ, Boeke JD, Pineda FJ, Hardwick JM. Genome-wide consequences of deleting any single gene. Mol Cell. 2013;52:485–494. doi: 10.1016/j.molcel.2013.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villafuerte S, Strumba V, Stoltenberg SF, Zucker RA, Burmeister M. Impulsiveness mediates the association between GABRA2 SNPs and lifetime alcohol problems. Genes Brain Behav. 2013;12:525–531. doi: 10.1111/gbb.12039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Su L, Morin MD, Jones BT, Whitby LR, Surakattula MM, Huang H, Shi H, Choi JH, Wang KW, Moresco EM, Berger M, Zhan X, Zhang H, Boger DL, Beutler B. TLR4/MD-2 activation by a synthetic agonist with no similarity to LPS. Proc Natl Acad Sci U S A. 2016;113:E884–893. doi: 10.1073/pnas.1525639113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen AY, Sakamoto KM, Miller LS. The role of the transcription factor CREB in immune function. J Immunol. 2010;185:6413–6419. doi: 10.4049/jimmunol.1001829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilhelm CJ, Mitchell SH. Rats bred for high alcohol drinking are more sensitive to delayed and probabilistic outcomes. Genes Brain Behav. 2008;7:705–713. doi: 10.1111/j.1601-183X.2008.00406.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Lousberg EL, Moldenhauer LM, Hayball JD, Coller JK, Rice KC, Watkins LR, Somogyi AA, Hutchinson MR. Inhibiting the TLR4-MyD88 signalling cascade by genetic or pharmacological strategies reduces acute alcohol-induced sedation and motor impairment in mice. Br J Pharmacol. 2012;165:1319–1329. doi: 10.1111/j.1476-5381.2011.01572.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang AR, Liu J, Yi HS, Warnock KT, Wang M, June HL, Jr, Puche AC, Elnabawi A, Sieghart W, Aurelian L, June HL., Sr Binge Drinking: In Search of its Molecular Target via the GABA(A) Receptor. Front Neurosci. 2011;5:123. doi: 10.3389/fnins.2011.00123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou JY, Crews FT. Release of neuronal HMGB1 by ethanol through decreased HDAC activity activates brain neuroimmune signaling. PLoS One. 2014;9:e87915. doi: 10.1371/journal.pone.0087915. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.