

Abstract
The HIV-1 regulatory protein, trans-activator of transcription (Tat), interacts with opioids to potentiate neuroinflammation and neurodegeneration within the CNS. These effects may involve the C-C chemokine receptor type 5 (CCR5); however, the behavioral contribution of CCR5 on Tat/opioid interactions is not known. Using a transgenic murine model that expresses HIV-1 Tat protein in a GFAP-regulated, doxycycline-inducible manner, we assessed morphine tolerance, dependence, and reward. To assess the influence of CCR5 on these effects, mice were pretreated with oral vehicle or the CCR5 antagonist, maraviroc, prior to morphine administration. We found that HIV-1 Tat expression significantly attenuated the antinociceptive potency of acute morphine (2 – 64 mg/kg, i.p.) in non-tolerant mice. Consistent with this, Tat attenuated withdrawal symptoms among morphine-tolerant mice. Pretreatment with maraviroc blocked the effects of Tat, reinstating morphine potency in non-tolerant mice and restoring withdrawal symptomology in morphine-tolerant mice. Twenty-four hours following morphine administration, HIV-1 Tat significantly potentiated (∼3.5-fold) morphine-conditioned place preference and maraviroc further potentiated these effects (∼5.7-fold). Maraviroc exerted no measurable behavioral effects on its own. Protein array analyses revealed only minor changes to cytokine profiles when morphine was administered acutely or repeatedly; however, 24 h post morphine administration, the expression of several cytokines was greatly increased, including endogenous CCR5 chemokine ligands (CCL3, CCL4, and CCL5), as well as CCL2. Tat further elevated levels of several cytokines and maraviroc pretreatment attenuated these effects. These data demonstrate that CCR5 mediates key aspects of HIV-1 Tat-induced alterations in the antinociceptive potency and rewarding properties of opioids.
Keywords: Conditioned Place Preference, C-C motif chemokines, HIV-associated neurocognitive disorders, Maraviroc, NeuroAIDS, Neuroinflammatiory cytokines, Opiate dependence, Opiate tolerance, Precipitated withdrawal, Warm-Water Tail-Withdrawal Test
1. Introduction
There is a dynamic relationship between opioid use, human immunodeficiency virus-1 (HIV-1) acquisition, and disease progression. Worldwide, injection drug use (IDU) accounts for ∼30% of new HIV-1 infections outside of sub-Saharan Africa (WHO, 2016). Within the United States, over 3,500 new infections involved IDU in 2015 (CDC, 2016), a year in which overall drug overdose deaths rose another 11%, the majority (63%) of which involved opioids (Rudd et al., 2016). The convergence of the HIV and opioid epidemics is particularly concerning given evidence that opioid usage increases the progression of HIV-1 to acquired immune deficiency syndrome (AIDS) and promotes neurocognitive impairment in humans and non-human primates (Bokhari et al., 2011; Bell et al., 1996, 2002, 2006; Chuang et al., 2005; Donahoe et al., 1993; Kumar et al., 2004, 2006; Rivera et al., 2013). Moreover, HIV-infected individuals are at risk for the development of neuropathic pain (Malvar et al., 2015) for which prescription opioids remain a common treatment (Kremer et al., 2016; Zilliox, 2017). Pharmacological treatment for opioid abuse includes substitution therapies (e.g. methadone, buprenorphine, buprenorphine/naloxone; Moatti et al., 1988; Roux et al., 2008; Sambamoorthi et al., 2000; Woody et al., 2014), which may exert neurotoxic interactions with HIV-1 proteins (Fitting et al., 2014b). As such, the mechanisms and physiological consequences of HIV/opioid interactions need to be understood in order to improve outcomes for HIV-seropositive patients that are pharmacologically managed for pain and/or addiction.
The biological mechanisms that underlie HIV-1 and opioid interactions in the central nervous system (CNS) likely involve the HIV-1 regulatory protein, trans-activator of transcription (Tat). Tat is critical for efficient HIV replication; however, Tat is soluble and can be secreted from infected cells to exert direct and indirect neurotoxicity in vitro (reviewed in King et al., 2006; Nath et al., 2002). Tat promotes neuroinflammation via NF-κling (El-Hage et al., 2008b; Herbein et al., 2010), upregulation of proinflammatory cytokines [particularly the endogenous β chemokine ligands for the C-C “motif” chemokine receptor type 5 (CCR5): C-C chemokine ligand 3 (CCL3, also known as “macrophage inflammatory protein-1α” or MIP-1α), CCL4 (also known as “macrophage inflammatory protein-1β” or MIP-1β), and CCL5 (also known as “regulated on activation normal T-cell expressed and secreted” or RANTES); El-Hage et al., 2005; Hahn et al., 2010], and subsequent recruitment of neuroimmune cells promoting neuroinflammation. In vitro, morphine exacerbates Tat effects to activate microglia (Bokhari et al., 2009; Gupta et al., 2010; Sorrell & Hauser, 2014), increase cytokine production (Bokhari et al., 2009; El-Hage et al., 2005; Fitting et al., 2014b; Turchan-Cholewo et al., 2009), drive oxidative stress (Dalvi et al., 2016; Fitting et al., 2014a,b; Malik et al., 2011; Turchan-Cholewo et al., 2009), increase intracellular calcium (El-Hage et al., 2005; Fitting et al., 2014a,b), and promote neurotoxicity (Fitting et al., 2014a,b; Gurwell et al., 2001; Malik et al., 2011). Morphine and Tat interactions may depend on μ opioid receptors (MORs) given that neurotoxic synergy is observed in co-cultures when mixed glia express MORs, but not when they are derived from MOR−/− mice (Zou et al., 2011). These data support the notion that glial MORs are critical for the indirect neurotoxic effects of Tat.
The proinflammatory effects of HIV-1 Tat at CCR5 may directly influence opioid sensitivity. In studies of opioid-mediated antinociception in rats, activation of CCR5 or CXCR4 can rapidly (within 30 min) desensitize μ- or δ-opioid-receptors (Chen et al., 2007). Blocking actions at CCR5 in proinflammatory states may attenuate heterologous desensitization of MORs and increase therapeutic efficacy. In support, intrathecal administration of the CCR5 antagonist, maraviroc, attenuated chronic constriction injury-induced microgliosis, astrogliosis, upregulation of CCR5 protein, and mRNA expression of CCR5-ligands (CCL3, CCL4, and CCL5) in the spinal cord and dorsal root ganglion concurrent with reduced neuropathic pain (Kwiatkowski et al., 2016). Moreover, CCR5 and MORs may form functionally active heteromers. A bivalent ligand derived from a MOR agonist (oxymorphone) and a CCR5 antagonist (TAK-220) had ∼2000× greater antinociceptive potency than morphine in mice experiencing LPS-mediated inflammation (Akgün et al., 2015). Another bivalent ligand comprised of an opioid receptor antagonist (naltrexone) and maraviroc reduced the infectivity of human astrocytes when cultured with R5-tropic HIV (Arnatt et al., 2016; El-Hage et al., 2013; Yuan et al., 2013). These data suggest a dynamic relationship between MOR and CCR5 activation that may contribute to HIV pathology; however, the functional effects are poorly understood. As such, we investigated morphine tolerance, dependence, and reward in a transgenic murine model that conditionally-expresses the proinflammatory HIV-1 regulatory protein, Tat1-86. Using a transgenic mouse approach, conditional Tat expression has been demonstrated to reduce the antinociceptive potency of morphine (Fitting et al., 2012, 2016), while potentiating psychostimulant reward in acute drug withdrawal (24 h post drug administration; Paris et al., 2014a,b). We hypothesized that HIV-1 Tat expression would attenuate morphine antinociceptive potency and that the CCR5 antagonist, maraviroc, would reverse these effects. Further, we hypothesized that 24 h post morphine, Tat- and cytokine-mediated effects would be potentiated.
2. Materials and Methods
The use of mice in these studies was pre-approved by the Institutional Animal Care and Use Committee at Virginia Commonwealth University and the experiments were conducted in accordance with ethical guidelines defined by the National Institutes of Health (NIH Publication No. 85-23).
2.1. Subjects and housing
Adult male mice expressed (or did not express) an HIV-1IIIB tat1-86 transgene (N = 245) as previously described (Hauser et al., 2009; Fitting et al., 2013; Bruce-Keller et al., 2008) and were generated in the vivarium at Virginia Commonwealth University (MCV campus). Briefly, HIV-1 Tat1-86 is conditionally expressed in a CNS-targeted manner via a GFAP-driven, Tet-on promoter (activated by consumption of doxycycline-containing chow) in Tat(+) mice. Tat(−) control littermates express the Tet-on transcription factor without the tat1-86 transgene. While Tat can induce astrogliosis in transgenic mice (El-Hage et al., 2008; Hahn et al., 2015; Paris et al., 2015), this has not been observed to impair Tat production (Fitting et al., 2012; Paris et al., 2014b). Additional control experiments were carried out in adult, male, C57BL/6J mice (N = 30). All mice (∼70 days of age) were housed 4 - 5 / cage and were maintained in a temperature- and humidity-controlled room on a 12:12 h light / dark cycle (lights off at 18:00 h) with ad libitum access to food and water.
2.2. Chemicals
To induce HIV-1 Tat1-86 expression, Tat(+) transgenic mice [and Tat(−) controls] were placed on doxycycline chow (Dox Diet #2018, 6 g/kg; Harlan Laboratories, Madison, WI, USA) for 28 days, unless otherwise specified. Some mice received subcutaneous implants of placebo or morphine pellets (75 mg; National Institute on Drug Abuse, Rockville, MD, USA), the latter of which induces tolerance and dependence in the present strain of mice (Fitting et al., 2016). To precipitate morphine withdrawal, mice were administered (−)naloxone (1 mg/kg, s.c.; Sigma-Aldrich, St. Louis, MO; Fitting et al., 2016). To investigate morphine reward, mice were administered morphine sulfate (#M8777; Sigma-Aldrich) at a concentration of 10 mg/kg, i.p. (0.1 ml per 10 g body weight) which has been demonstrated to produce morphine-conditioned place preference (CPP; Zhu et al., 2015). To investigate the contribution of CCR5 to these effects, some mice were administered the CCR5-selective antagonist, maraviroc (62 mg/kg, p.o.; #376348-65-1; BOC Sciences, Shirley, NY, USA) which was dissolved in 100% DMSO, then diluted to 5% DMSO in vegetable oil. Using inter-species allometric scaling (by a factor of 12.3; Freireich et al., 1966; Reagan-Shaw et al., 2008) others have determined maraviroc (62 mg/kg, p.o.) dosing for mice from clinical formulations (Neff et al., 2010).
2.3. Surgical manipulation
Mice received subcutaneous implants of placebo or morphine (75 mg) pellets under isoflurane (2.5 %) anesthesia as previously reported (Fitting et al., 2016; Ross et al., 2008). Following surgery, mice were monitored to ensure weight gain, muscle tone, proper neurological response, and general health (Crawley and Paylor, 1997). No mice failed to recover.
2.4. Behavioral assays
Prior to all behavioral testing, mice were acclimated to the testing room for 24 h. For assessments of morphine tolerance and morphine dependence, mice received subcutaneous implants of placebo or morphine pellets. Five days later, mice were assessed for morphine tolerance in a warm-water tail-withdrawal assay. Following tail-withdrawal testing, mice underwent naloxone-precipitated withdrawal and were assessed for morphine dependence. For assessments of morphine reward, Tat-transgenic mice were assessed in a CPP paradigm with psychomotor sensitization assessed on conditioning days. Some mice were additionally assessed on a rotarod to rule out potential locomotor confounds.
2.4.1. Warm-water tail-withdrawal test
A warm-water tail-withdrawal test was conducted with a water-bath maintained at 56 ± 0.1°C as previously described (Coderre & Rollman, 1983; Fitting et al., 2016). Briefly, mice were gently wrapped in a cloth and the distal one-third of the tail was immersed in a water bath. The mice rapidly removed their tail from the bath at the first sign of discomfort and the tail-withdrawal latency was recorded. Tail-withdrawal latency was then assessed using a cumulative dosing procedure. Mice were injected with a starting dose of morphine and were tested for antinociception 20 min later. Mice that did not reach a 10 sec cut-off threshold received an additional cumulative dose of morphine and were retested. This process was repeated until the animals reached the cut-off value of 10 sec. Baseline latency ranged from 2 to 4 sec. The 10 sec maximum cutoff latency was used to prevent tissue damage. Antinociception was quantified as the percentage of maximal possible effect (%MPE): %MPE = [(Test latency – Baseline latency) / (10 – Baseline latency)] × 100 (Harris & Pierson, 1964).
2.4.2. Antagonist-precipitated withdrawal
Mice were administered an injection of the opioid receptor antagonist, naloxone (1 mg/kg, s.c.), in order to precipitate withdrawal. The primary symptom assessed was jumping from an elevated platform (32 cm high, 17 cm diameter). The proportion of mice that jumped from their individual platforms was recorded over a 10 min trial. The proportion of jumping mice is considered an index of withdrawal (Fitting et al., 2016). This test was followed by an evaluation of additional signs of withdrawal. Mice were placed in a rectangular, clear, plastic observation box (16 × 16 × 30 cm) and observed for 5 min. The concomitant number of jumps, forepaw tremors, and wet-dog shakes was recorded. The frequency of jumps, tremors, and shakes are considered additional indices of withdrawal (Fitting et al., 2016).
2.4.3. Conditioned Place Preference
Behavior in the CPP test was recorded and digitally-encoded by an ANY-maze behavioral tracking system (Stoelting Co., Wood Dale, IL, USA). Morphine-CPP and locomotor sensitization were assessed simultaneously (Zhu et al., 2015). CPP was conducted as modified from previous methods (Paris et al., 2014a). The apparatus (#64101; Stoelting Co.) consisted of two black conditioning chambers (18 × 20 × 35 cm), each visually-distinguished by the presence of white circles or horizontal stripes on the chamber walls, as well as ∼30 lux difference in ambient lighting. Conditioning chambers were connected by a start box/transition chamber (10 × 20 cm). A biased conditioning design was utilized (Semenova et al., 1995). On day 1, mice were allowed to freely explore the apparatus for 10 min in order to establish an initial chamber-preference (there was no significant side preference observed across experimental groups). On days 2 - 5 mice underwent one cycle of morphine conditioning per day (receiving an i.p. saline injection paired with confinement to the preferred chamber for 30 min, followed 4 h later by an i.p. morphine, 10 mg/kg, injection paired with confinement to the less preferred chamber for 30 min). Twenty-four hours after the last morphine-conditioning cycle, mice were allowed to freely explore the apparatus in order to assess their final chamber preference. The amount of time that mice spent in the chambers or the start/transition box, as well as the distance traveled, and the frequency of rearing was recorded on each day. CPP was quantified as a difference score: CPP d-score = (time spent in the morphine-paired chamber) – (time spent in the saline-paired chamber) (Paris et al., 2014a).
2.4.4. Rotarod
Locomotor coordination was assessed on an accelerated rotarod as previously described (Paris et al., 2013). Briefly, mice were trained to balance on an immobile rotarod (3 cm in diameter and suspended 44.5 cm high; Columbus Instruments, Columbus, OH, USA) for 30 sec. Mice were then trained to navigate the task across two 30 sec fixed speed trials (10 rpm) and two 180 sec fixed speed trials (10 rpm). Lastly, mice were tested on two accelerated speed trials (180 sec max. latency at 0 - 20 rpm). The mean latency to fall from the rotarod and the maximum RPM achieved across the two accelerated trials were utilized as indices for locomotor performance. Decreased latencies to fall and lower maximal RPM on the accelerated test indicate an impaired motor phenotype.
2.5. Cytokine assay
The head of the caudate nucleus has been identified as a central reservoir for maximal HIV viral load in humans (reviewed in Nath, 2015) and we have found the striatum to host a cell population that may be selectively vulnerable to HIV-1 Tat (Schier et al., 2017). As such, cytokine analyses were conducted on caudate/putamen (dorsal striatum) in the present mouse model. Mice underwent cervical dislocation and bilateral dorsal striata were immediately dissected, flash-frozen in liquid nitrogen, and stored at -80°C until assay. At the time of assay, tissues were homogenized in IP lysis buffer (#87787; Pierce Biotechnology, Rockford, IL, USA) with a protease/phosphatase inhibitor cocktail (#04693159001; Roche, Mannheim, Germany). Protein concentrations were determined via bicinchoninic acid (BCA) assay per kit manufacturer instructions (#23224; Pierce Biotechnology).
Cytokines were assessed using a Bio-Plex Pro™ Mouse Cytokine 23-plex assay kit (#M60009RDPD; Bio-Rad Laboratories, Inc., Hercules, CA, USA) and analyzed on a Bio-Plex 200 system. Samples were diluted to a concentration of 500 μg/mL. Unknown samples and standards were incubated with fluorescent, antibody-tagged microspheres and detected via streptavidin-phycoerythrin-labeled detection antibodies. Cytokine concentrations were calculated from respective standard curves via Bio-Plex Manager 4.0 software. All samples were analyzed in duplicate. Limits of detection ranged from 2 to 20 pg/mL. Mean intra- and inter-assay coefficients of variance were 7.2 % and 5.3 %, respectively.
2.6. Statistical analyses
Median effective doses (ED50; reported with 95 % confidence intervals) were determined via non-linear regression (sigmoidal curvilinear modeling with variable slope) using a least-squares fit for each treatment group (bottom and top values constrained to 0 and 100, respectively). Dependent measures for additional behavioral analyses were assessed via ANOVA (to assess dependence) or repeated measures ANOVA (to assess psychostimulation or CPP over multiple trials) with drug condition (placebo or morphine), inhibitor condition (vehicle or maraviroc), and genotype [Tat(−) or Tat(+)] as factors. Cytokine arrays were assessed via separate three-way ANOVAs. Mice treated acutely with doxycycline (48 h) were analyzed with morphine condition (acute saline or acute morphine), inhibitor condition (vehicle or maraviroc), and genotype [Tat(−) or Tat(+)] as factors. Mice treated chronically with doxycycline for 28 days were analyzed with morphine condition (morphine-naïve, repeated morphine, or 24 h post morphine), inhibitor condition (vehicle or maraviroc), and genotype [Tat(−) or Tat(+)] as factors. Fisher's Protected Least Significant Difference post-hoc tests determined group differences following main effects. Interactions were delineated via simple main effects and main effect contrasts with alpha controlled for multiple comparisons. Analyses were considered significant when p ≤ 0.05.
3. Results
3.1. HIV-1 Tat decreased morphine potency in non-tolerant mice; tolerance or pharmacological antagonism of CCR5 attenuated Tat effects
As negative control measures before proceeding to tests involving Tat-tg mice, morphine dosing, non-specific interactions with maraviroc, and warm-water tail-withdrawal test conditions were confirmed in C57BL/6J mice. In a 52°C water bath, morphine administered at 5 mg/kg (Fig. 1A) or 10 mg/kg (Fig. 1B) produced antinociception that was present for at least 2 h and peaked at 60 min, commensurate with observations in other animal models (Altun et al., 2015; Williams et al., 2008). In a 56°C water bath, cumulative morphine-dosing produced antinociception commensurate with what we have previously observed in C57BL/6J mice (Fitting et al., 2016; Fig. 1C). Maraviroc did not significantly influence the acute time-course (Fig. 1A-B) or the antinociceptive effects of morphine in C57BL/6J mice [vehicle/morphine ED50 = 2.0 (95 % CI: 0.9 – 4.3), maraviroc/morphine ED50 = 3.0 (95% CI: 2.2 – 4.1)] (Fig. 1C).
Figure 1. Maraviroc did not significantly influence the anti-nociceptive response to morphine in C57BL/6J mice.
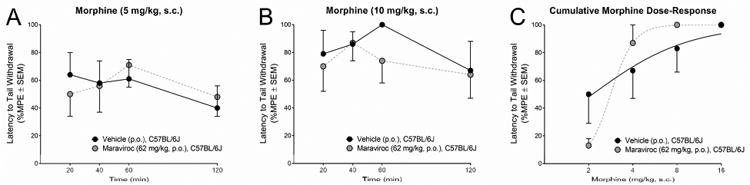
Latency to tail withdrawal (% MPE ± SEM) among C57BL/6J mice (n = 5 / group) that were administered vehicle (p.o.; black circles) or maraviroc (62 mg/kg, p.o.; blue circles) prior to (A) morphine (5 mg/kg, s.c.), (B) morphine (10 mg/kg, s.c.), or (C) a cumulative morphine dosing regimen (2 – 16 mg/kg, s.c.).
Tat(−) and Tat(+) mice were placed on doxycycline for 28 days to induce Tat expression (or not). On day 28, mice were implanted with a subcutaneous placebo pellet or morphine pellet to induce morphine tolerance. From days 28 – 32, mice were administered vehicle or maraviroc (p.o., QD). On day 33, mice received their last dose of vehicle or maraviroc and were tested 30 min later.
Tat induction, morphine tolerance, and maraviroc pretreatment significantly influenced the antinociceptive response to acute morphine (Fig. 2A-B). Among non-tolerant, placebo-pelleted mice, Tat exposure produced a modest but significant shift to the right in the antinociceptive potency of acutely-administered morphine [F(1,48) = 7.97, p < 0.05] [Fig. 2A; Tat(−)ED50 = 4.5 (95% CI: 3.6 - 5.5), Tat(+)ED50 = 6.8 (95% CI: 5.6 - 8.0)]. This effect was obviated by maraviroc pretreatment [Fig. 2B; Tat(−)ED50 = 6.0 (95% CI: 4.9 - 7.1), Tat(+)ED50 = 5.8 (95% CI: 4.9 - 6.7)]. Morphine tolerance significantly shifted the ED50 for morphine-induced antinociception to the right in all treatment groups [Fig. 2A-B; ED50 range = 17.8 - 53.8]. Notably, baseline tail-withdrawal latencies were ∼0.4 sec greater among morphine-pelleted (2.7 ± 0.1 sec) vs. placebo pelleted mice (2.3 ± 0.1 sec) [F(1,44) = 10.91, p < 0.05]. Neither Tat induction nor maraviroc pretreatment influenced baseline tail-withdrawal latencies.
Figure 2. HIV-1 Tat significantly shifted the ED50 for morphine to the right in non-tolerant mice and maraviroc pretreatment obviated this effect.
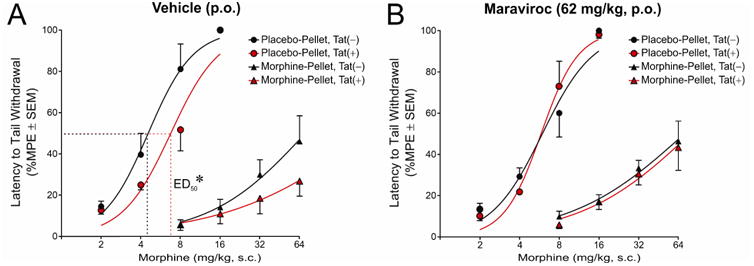
Latency to tail withdrawal (% MPE ± SEM) among Tat(−) or Tat(+) mice (n = 6 - 7 / group) that were morphine-naïve (placebo-pelleted; circles) or morphine-tolerant (morphine-pelleted; triangles). Mice [Tat(−) in black, Tat(+) in red] were administered (A) daily vehicle (p.o.) or (B) daily maraviroc (62 mg/kg, p.o.) following pelleting and were assessed for tail withdrawal reflex in response to a cumulative morphine regimen (2 - 64 mg/kg, s.c.). * indicates a significant shift in ED50, p < 0.05.
3.2. Among tolerant mice, Tat-exposure decreased primary withdrawal behavior; pharmacological antagonism of CCR5 attenuated Tat effects
Following testing for antinociception, mice were administered the opioid receptor antagonist, naloxone, and precipitated withdrawal behaviors were assessed. Tat induction, morphine tolerance, and maraviroc pretreatment significantly interacted to influence the primary measure of withdrawal, the proportion of mice that jumped from an elevated platform [F(1,44) = 6.77, p < 0.05] (Fig. 3A). Unlike other withdrawal-related behaviors assessed, elevated platform jumping was only observed among previously morphine-tolerant mice (a significant difference from their placebo-pelleted counterparts [F(1,44) = 122.27, p < 0.05]; Fig. 3A). Among morphine-tolerant mice, withdrawal- precipitated jumping was significantly attenuated by Tat exposure [Tat(+) mice significantly differed from Tat(−) controls (p = 0.03)] or maraviroc pretreatment [maraviroc-treated, Tat(−) mice significantly differed from Tat(−) controls (p = 0.005)] (Fig. 3A). However, combined Tat exposure and maraviroc pretreatment restored withdrawal-precipitated jumping [maraviroc-treated Tat(+) mice significantly differed from maraviroc-treated Tat(−) mice (p = 0.004) or vehicle-treated Tat(+) mice (p = 0.03)] (Fig. 3A).
Figure 3. HIV-1 Tat significantly attenuated withdrawal-precipitated jumping and maraviroc pretreatment reversed this effect; Tat and morphine tolerance influenced additional withdrawal symptomology.
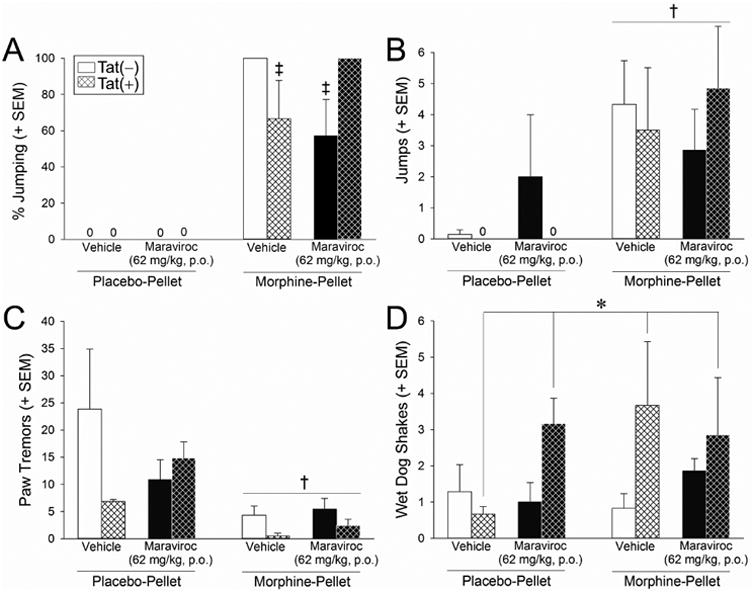
Measures of naloxone-precipitated withdrawal among Tat(−) or Tat(+) mice [n = 6 - 7 / group; Tat(−) in open bars, Tat(+) in hatched bars] that were morphine-naïve (placebo-pelleted; left bars in each panel) or morphine-tolerant (morphine-pelleted; right bars in each panel), administered daily vehicle (p.o.; white bars) or maraviroc (62 mg/kg, p.o.; black bars), and assessed for warm water tail withdrawal. (A) The proportion of mice jumping from an elevated platform over 10 min, as well as the concomitant frequency of (B) spontaneous jumping, (C) paw tremor, and (D) wet dog shakes over 5 min in an observation box, were recorded. * main effect of Tat-exposure [greater wet-dog shakes among Tat(+) mice vs. Tat(−) mice]. † main effect of prior morphine tolerance (greater frequency of spontaneous jumps and fewer simultaneous paw tremors among morphine-tolerant vs. non-tolerant mice). ‡ interaction for indicated groups to differ from morphine-tolerant Tat(−) mice or morphine-tolerant, maraviroc-treated Tat(+) mice, p < 0.05.
When observed for additional, simultaneous withdrawal-precipitated behaviors the frequency of spontaneous jumping was significantly greater [F(1,44) = 10.83, p < 0.05] (Fig. 3B) and paw tremors were significantly reduced [F(1,44) = 10.79, p < 0.05] (Fig. 3C) among morphine-tolerant mice compared to placebo-pelleted controls irrespective of Tat or maraviroc exposure. There was a main effect for wet-dog shakes to be significantly greater among Tat(+) mice compared to Tat(−) mice, irrespective of morphine or maraviroc exposure [F(1,44) = 3.97, p = 0.05] (Fig. 3D).
3.3. HIV-1 Tat exposure and CCR5 antagonism attenuated the psychomotor response to morphine
Both Tat exposure and maraviroc pretreatment interacted with repeated morphine injections to influence locomotor behavior (Fig. 4AB). Among vehicle-pretreated mice, morphine administration significantly increased the distance traveled in the CPP apparatus [F(7,147) = 16.66, p < 0.05] compared to saline administration, irrespective of Tat exposure (p < 0.0001 per day; Fig. 4A). However, among maraviroc-pretreated mice, Tat-exposure and morphine administration significantly interacted [F(7,147) = 2.46, p < 0.05] such that Tat(−) mice receiving morphine demonstrated significantly greater distances traveled on days 1 (p = 0.03) and 4 (p = 0.003), compared to Tat(+) mice receiving morphine (Fig. 4B). Tat(−) mice also displayed enhanced locomotion to a significantly greater degree following morphine-administration than following saline-administration (p < 0.0001 per day); whereas, locomotion among Tat(+) mice did not differ between morphine or saline administration (Fig. 4B). Notably, Tat exposure can impair locomotion on its own; however, these effects are not usually observed in the present model until Tat has been induced for over one month (Hahn et al., 2012). To rule out non-specific effects of Tat on locomotion, some Tat(−) and Tat(+) mice from the present experiment were assessed on an accelerated rotarod after testing. No significant differences in locomotor capacity were observed following fixed speed trials (10 rpm) lasting 30 s [Tat(−): 9 ± 2, Tat(+): 12 ± 2] or 180 s [Tat(−): 56 ± 16, Tat(+): 81 ± 20], or following accelerated speed trials (0 – 20 rpm) lasting 180 s [Tat(−): 132 ± 10, Tat(+): 128 ± 14].
Figure 4. HIV-1 Tat and maraviroc significantly attenuate morphine-mediated psychomotor behavior.
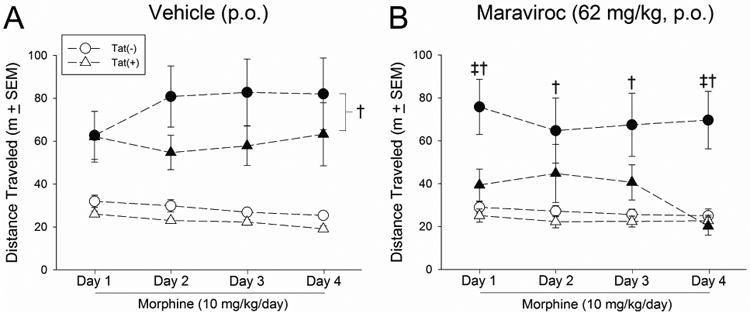
Locomotor behavior in response to saline (i.p. for 4 days; open circles) morphine (10 mg/kg, i.p. for 4 days; closed circles) was assessed during conditioning cycles among Tat(−) or Tat(+) mice that were pretreated with (A) vehicle (p.o.) or (B) maraviroc (62 mg/kg, p.o.; n = 11 - 12 / group). † main effect of morphine (greater locomotion following morphine vs. saline treatment). ‡ interaction for indicated Tat(−) groups differ from their respective Tat(+) counterparts, p < 0.05.
3.4. CCR5 antagonism potentiated Tat-related morphine reward
To assess the contribution of HIV-1 Tat on the rewarding properties of morphine, mice were assessed for morphine-CPP. Tat(−) and Tat(+) mice were exposed to two morphine conditioning cycles (saline followed by morphine) and assessed for chamber preference 24 h after the last morphine administration (Fig. 5A). Following two conditioning cycles, morphine significantly increased the time that mice spent in the previously non-preferred chamber [F(1,24) = 26.84, p < 0.05] (Fig. 5A′). However, significant differences between Tat(−) and Tat(+) mice were not observed (Fig. 5A′).
Figure 5. HIV-1 Tat potentiates morphine-conditioned place preference 24 h post morphine and maraviroc pretreatment exacerbates this effect.
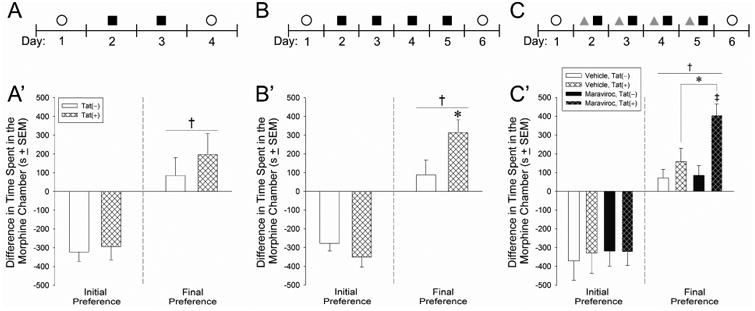
Tat(−) or Tat(+) mice (n = 11 - 14 / group) were assessed for their chamber preference (open circles) in a biased conditioned place preference (CPP) test consisting of either (A) two cycles of saline (i.p.)/morphine (10 mg/kg, i.p.) conditioning (closed squares), (B) four cycles of saline/morphine conditioning (closed squares), or (C) four cycles of vehicle (p.o.) or maraviroc pretreatment (gray triangles) followed by saline/morphine conditioning (closed squares). Initial preferences for the morphine-paired chamber (left of dashed line) are depicted followed by final CPP (right of dashed line) after (A′) two conditioning cycles, (B′) four conditioning cycles, or (C&prime) vehicle/maraviroc pretreatment prior to four conditioning cycles. Tat(−) mice are depicted in open bars, Tat(+) mice are depicted in hatched bars, vehicle (p.o.)-pretreated mice are depicted with white bars, and maraviroc (62 mg/kg, p.o.)-pretreated mice are depicted with black bars. † main effect for the final chamber preference to differ from the initial preference. * interaction for Tat(+) mice to differ from Tat(−) mice. ‡ interaction for maraviroc-pretreated Tat(+) mice to differ from all other groups, p < 0.05.
Another group of mice underwent four morphine-conditioning cycles and were assessed for final chamber preference 24 h later (Fig. 5B). Under these conditions, Tat genotype significantly interacted with the final preference trial [F(1,22) = 5.43, p < 0.05] (Fig. 5B′). Tat(−) and Tat(+) mice spent a commensurate amount of time in the least-preferred chamber prior to conditioning; but, Tat(+) mice spent a significantly greater amount of time in the morphine-paired chamber following four cycles of morphine conditioning compared to Tat(−) controls (p = 0.04; Fig. 5B′).
To assess the influence of CCR5 on Tat-potentiated CPP, Tat(−) and Tat(+) mice were pretreated with vehicle or maraviroc 30 min prior to the start of conditioning over four cycles (Fig. 5C). As previously observed, mice significantly preferred the morphine-paired chamber 24 h following conditioning, irrespective of treatment [F(1,42) = 100.80, p < 0.05] (Fig. 5C′). There was significant interaction between Tat-genotype and maraviroc condition [F(1,42) = 4.13, p < 0.05], such that Tat(+) mice spent a significantly greater amount of time in the morphine-paired chamber when pretreated with maraviroc than did any other group (p = 0.0006 – 0.02; Fig. 5C′).
3.5. Prior morphine exposure and HIV-1 Tat increase striatal cytokine content and CCR5 antagonism attenuates Tat effects
Cytokine protein expression was assessed in the dorsal striatum (caudate/putamen) of Tat(−) and Tat(+) mice. In order to parse the acute- vs. chronic influences of Tat, maraviroc, and morphine on the immune effectors examined, tissues from two separate groups of mice were assessed. The first group was acutely exposed to Tat (48 h of doxycycline to induce Tat), maraviroc (a single administration of vehicle or maraviroc, 62 mg/kg, p.o., 30 min prior to i.p. injection), and morphine (a single administration of saline or morphine, 10 mg/kg, i.p.). The second group was chronically exposed to Tat (28 d of doxycycline to induce Tat), repeated maraviroc (vehicle or maraviroc, 62 mg/kg, p.o., QD for 4 d, 30 min prior to i.p. injection), and repeated morphine (saline or morphine, 10 mg/kg, i.p., QD for 4 d). Tissues were collected either 1 h after the last treatment, or 24 h after the last treatment (the latter timeframe being commensurate with that which produced Tat-potentiated morphine-CPP).
3.5.1. Chemotactic Cytokines
Among mice that were acutely-exposed to Tat and drug manipulations (Fig. 6, left-hand panels, 48 h doxycycline exposure), acute Tat induction and acute maraviroc pretreatment significantly interacted to increase striatal CCL3, compared to vehicle- treated, Tat(−) controls [F(1,31) = 4.69, p < 0.05] (Fig. 6B, left panel, white bars). There was an additional interaction for acute morphine to significantly increase CCL5, but only among Tat(+) mice [F(1,31) = 4.59, p < 0.05] (Fig. 6G, left panel, cyan bars). Main effects were observed for acute Tat exposure to significantly increase CCL4 [F(1,31) = 15.56, p < 0.05] (Fig. 6C, left panel, white and cyan bars) and CCL11 [F(1,31) = 5.56, p < 0.05] (Fig. 6H, left panel, white and cyan bars), irrespective of maraviroc or morphine administration. Lastly, there was a main effect for morphine to significantly elevate CXCL1 [F(1,31) = 10.55, p < 0.05], irrespective of Tat or maraviroc exposure (Fig. 6D,left panel, cyan bars). Acute treatments did not otherwise influence the chemokines examined.
Figure 6. Chemokine expression in caudate/putamen is largely upregulated 24 h following repeated morphine exposure; HIV-1 Tat potentiated this effect on some analytes (outlined) which was ameliorated by maraviroc pretreatment.
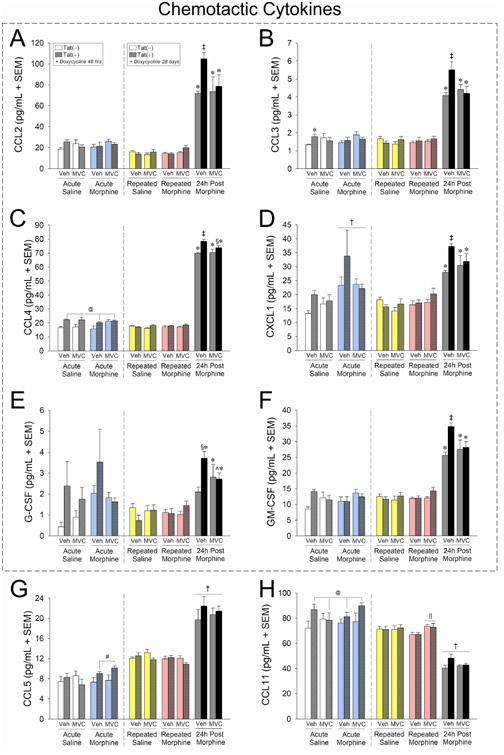
Chemotactic cytokine protein expression (pg/mL ± SEM) in dorsal striatum (caudate/putamen) of vehicle- (p.o.) or maraviroc- (62 mg/kg, p.o.) exposed mice [n = 4 – 5 / group; Tat(−) in open bars, Tat(+) in stippled bars] that had Tat expressed for 48 h or 28 days (via doxycycline). Mice were administered saline (i.p.; white bars) or morphine (10 mg/kg, i.p., QD) acutely (cyan bars), repeatedly (4 consecutive days; repeated saline in yellow bars and repeated morphine in magenta bars), or were 24 h post repeated morphine treatment (gray bars). Cytokines within the dashed box demonstrated Tat-potentiation that was ameliorated by maraviroc. @ main effect of Tat [Tat(+) mice have greater expression than Tat(−) mice, irrespective of morphine or maraviroc condition]. † main effect of morphine (indicated morphine group has greater expression than the respective saline group, irrespective of Tat or maraviroc condition). ‖ maraviroc-morphine interaction (indicated maraviroc-treated group differs from vehicle-treated control within the same morphine condition). Ģ 3-way interaction for indicated groups to differ from vehicle-treated, saline-administered, Tat(−) controls. ‡ 3-way interaction for indicated group to differ from all other groups. § 3-way interaction for indicated groups to differ from respective vehicle-treated, Tat(−) control within the 24 h post morphine group. ˆ 3-way interaction for indicated maraviroc-treated Tat(+) group be reduced compared to respective vehicle-treated Tat(+) group, p < 0.05.
Among mice exposed to chronic Tat and repeated morphine, no significant differences were observed until morphine was withheld for 24 h (Fig. 6, right-hand panels, 28 d doxycycline exposure). Three-way interactions were revealed 24 h post morphine treatment for CCL2 [F(2,45) = 3.64, p < 0.05] (Fig. 6A, right panel, gray bars), CCL3 [F(2,45) = 6.14, p < 0.05] (Fig. 6B, right panel, gray bars), CCL4 [F(2,45) = 4.55, p < 0.05] (Fig. 6C, right panel, gray bars), CXCL1 [F(2,45) = 4.21, p < 0.05] (Fig. 6D, right panel, gray bars), G-CSF [F(2,45) = 5.42, p < 0.05] (Fig. 6E, right panel, gray bars), and GM-CSF [F(2,45) = 6.11, p < 0.05] (Fig. 6F, right panel, gray bars). In each case, chronic Tat significantly potentiated the increase of chemokines and maraviroc pretreatment significantly ameliorated this potentiation (see outlined chemokines in Fig. 6). Additionally, a main effect was observed for CCL5 to be increased 24 h post morphine administration, irrespective of Tat or maraviroc exposure [F(2,45) = 115.48, p < 0.05] (Fig. 6G, right panel, gray bars). Intriguingly, repeated morphine significantly reduced CCL11 [F(2,45) = 3.25, p < 0.05], an effect that was prevented by maraviroc pretreatment (Fig. 6H, right panel, magenta bars). Once morphine was withheld for 24 h, CCL11 declined in all groups (Fig. 6H, right panel, gray bars).
3.5.2. Pro-inflammatory cytokines
Acute HIV-1 Tat and maraviroc/morphine exposure (Fig. 7, left-hand panels, 48 h doxycycline exposure) influenced pro-inflammatory cytokine levels. A significant interaction was observed for acute morphine to modestly elevate IL-1α, but only in maraviroc-pretreated mice [F(1,31) = 6.11, p < 0.05] (Fig. 7G, left panel, cyan bars). Acute Tat also significantly increased IL-3 (which was attenuated by maraviroc) [F(1,31) = 3.82, p = 0.05] (Fig. 7A, left panel, white bars) and IL-9 (which was attenuated by morphine and reinstated by maraviroc) [F(1,31) = 3.96, p = 0.05] (Fig. 7C, left panel, white and cyan bars). Lastly, there was a main effect for acute Tat exposure to cause a modest, significant increase in IL-1α [F(1,31) = 7.03, p < 0.05] (Fig. 7G, left panel, white and cyan bars) and a main effect for acute maraviroc to increase IL-6 [F(1,31) = 4.07, p < 0.05] (Fig. 7B, left panel, white bars). No additional effects of acute treatments were observed on pro-inflammatory cytokines.
Figure 7. Proinflammatory cytokine expression in caudate/putamen is partly upregulated 24 h following repeated morphine exposure; HIV-1 Tat potentiated this effect on some analytes (outlined) which could be ameliorated by maraviroc pretreatment.
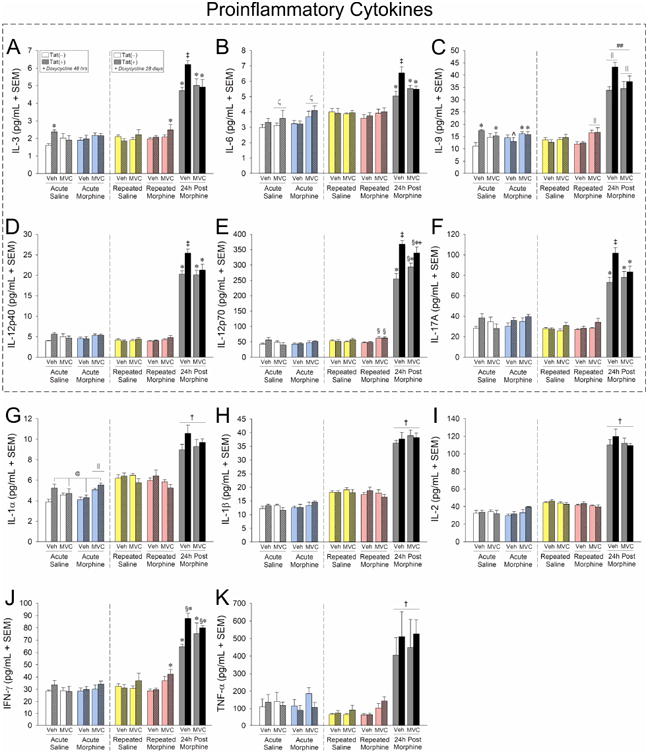
Proinflammatory cytokine protein expression (pg/mL ± SEM) in dorsal striatum (caudate/putamen) of vehicle- (p.o.) or maraviroc- (62 mg/kg, p.o.) exposed mice [n = 4 - 5 / group; Tat(−) in open bars, Tat(+) in stippled bars] that had Tat expressed for 48 h or 28 days (via doxycycline). Mice were administered saline (i.p.; white bars) or morphine (10 mg/kg, i.p., QD) acutely (cyan bars), repeatedly (4 consecutive days; repeated saline in yellow bars and repeated morphine in magenta bars), or were 24 h post repeated morphine treatment (gray bars). Cytokines within the dashed box demonstrated Tat-potentiation that was ameliorated by maraviroc. @ main effect of Tat [Tat(+) mice have greater expression than Tat(−) mice, irrespective of morphine or maraviroc condition]. † main effect of morphine (indicated morphine group has greater expression than the respective saline group, irrespective of Tat or maraviroc condition). ζ main effect of maraviroc (maraviroc-treated groups have significantly greater expression that vehicle-treated groups, irrespective of Tat or morphine condition). ## Tat-morphine interaction wherein Tat(−) and Tat(+) mice in the indicated morphine group are greater than those in other morphine groups and differ from each other (irrespective of maraviroc condition). ‖ maraviroc-morphine interaction wherein maraviroc-pretreatment differs from vehicle-pretreatment within the indicated morphine groups (irrespective of Tat condition). * 3-way interaction for indicated groups to differ from respective vehicle-treated, saline-administered, Tat(−) controls. ‡ 3-way interaction for indicated group to differ from all other groups. § 3-way interaction for indicated groups to differ from respective vehicle-treated, Tat(−) control within the 24 h post morphine group. ˆ 3-way interaction for indicated group to differ from respective vehicle-treated, acute-saline-administered, Tat(+) mice. ˆ 3-way interaction for indicated group to be greater than their maraviroc-treated, Tat(−) counterparts within the 24 h post morphine group, p < 0.05.
In mice exposed to chronic Tat and repeated maraviroc/morphine treatment, significant elevations were observed in all pro-inflammatory cytokines examined (Fig. 7, right-hand panels, 28 d doxycycline exposure). Three-way interactions were revealed 24 h post morphine treatment for IL-3 [F(2,45) = 6.73, p < 0.05] (Fig. 7A, right panel, gray bars), IL-6 [F(2,45) = 3.14, p = 0.05] (Fig. 7B, right panel, gray bars), IL-9 [F(2,45) = 5.18, p < 0.05] (Fig. 7C, right panel, gray bars), IL-12p40 [F(2,45) = 4.02, p < 0.05] (Fig. 7D, right panel, gray bars), IL-12p70 [F(2,45) = 5.43, p < 0.05] (Fig. 7E, right panel, gray bars), and IL-17A [F(2,45) = 5.65, p < 0.05] (Fig. 7F, right panel, gray bars). In each case, chronic Tat significantly potentiated the increase of proinflammatory cytokines and maraviroc pretreatment significantly ameliorated this potentiation (see outlined cytokines in Fig. 7). A 3-way interaction was also revealed for IFN-γ to be significantly increased among mice that were exposed to chronic Tat and repeated maraviroc/morphine [F(2,45) = 3.23, p < 0.05] (Fig, 7J, right panel, magenta bars). These effects were significantly exacerbated 24 h post morphine; however, this potentiation was not significantly attenuated by maraviroc-pretreatment (Fig, 7J, right panel, gray bars). Interestingly, repeated morphine also significantly increased IL-9 content in maraviroc-pretreated mice [F(2,45) = 5.63, p < 0.05] (Fig. 7C, right panel, magenta bars). Irrespective of Tat and maraviroc exposure, main effects were observed for cytokines to be increased 24 h post morphine for IL-1α [F(2,45) = 80.55, p < 0.05] (Fig. 7G, right panel, gray bars), IL-1β [F(2,45) = 288.09, p < 0.05] (Fig. 7H, right panel, gray bars), IL-2 [F(2,45) = 475.92, p < 0.05] (Fig. 7I, right panel, gray bars), and TNF-α [F(2,45) = 42.36, p < 0.05] (Fig. 7K, right panel, gray bars).
3.5.3. Anti-inflammatory cytokines
Among the anti-inflammatory cytokines assessed, only IL-10 demonstrated a significant response to acute manipulations (Fig. 8, left-hand panels, 48 h doxycycline exposure), [F(1,31) = 5.56, p < 0.05]. Acute Tat exposure significantly increased IL-10, and this effect was attenuated by maraviroc unless co-administered with acute morphine (Fig. 8A, left panel, white and cyan bars).
Figure 8. Anti-inflammatory cytokine expression in caudate/putamen is upregulated 24 h following repeated morphine exposure; HIV-1 Tat potentiated this effect on IL-10 and maraviroc pretreatment ameliorated this effect.
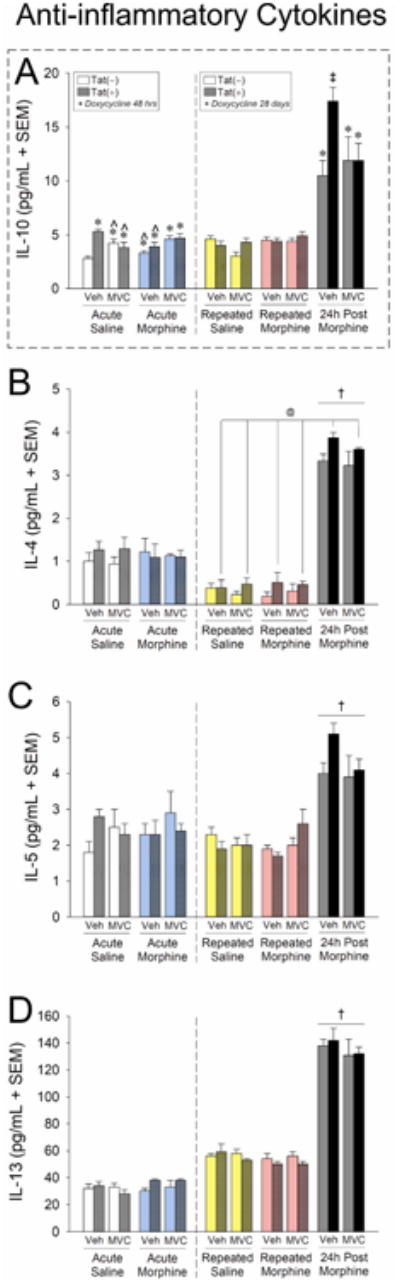
Antiinflammatory cytokine protein expression (pg/mL ± SEM) in dorsal striatum (caudate/putamen) of vehicle- (p.o.) or maraviroc- (62 mg/kg, p.o.) exposed mice [n = 4 - 5 / group; Tat(−) in open bars, Tat(+) in stippled bars] that had Tat expressed for 48 h or 28 days (via doxycycline). Mice were administered saline (i.p.; white bars) or morphine (10 mg/kg, i.p., QD) acutely (cyan bars), repeatedly (4 consecutive days; repeated saline in yellow bars and repeated morphine in magenta bars), or were 24 h post repeated morphine treatment (gray bars). The dashed box indicates IL-10 as the only anti-inflammatory cytokine assessed that demonstrated Tat-potentiation with maraviroc amelioration. @ main effect of Tat [Tat(+) mice have greater expression than Tat(−) mice, irrespective of morphine or maraviroc condition]. † main effect of morphine (indicated morphine group has greater expression than the respective saline group, irrespective of Tat or maraviroc condition). * 3-way interaction for indicated groups to differ from vehicle-treated, saline-administered, Tat(−) controls. ‡ 3-way interaction for indicated group to differ from all other groups. § 3-way interaction for indicated groups to differ from respective vehicle-treated, Tat(−) control within the 24 h post morphine group. ˆ 3-way interaction for indicated group be reduced compared to respective vehicle-treated Tat(+) group, p < 0.05.
When assessed for chronic Tat exposure and repeated maraviroc/morphine administration (Fig. 8, right-hand panels, 28 d doxycycline exposure), IL-10 was the only anti-inflammatory cytokine that demonstrated Tat-mediated potentiation 24 h post morphine that was reversible by pretreatment with maraviroc [F(2,45) = 6.46, p < 0.05] (Fig. 8A, right panel, gray bars). Other anti-inflammatory cytokines demonstrated significant main effects for enhanced protein expression 24 h post morphine, but not in a Tat- or maraviroc-sensitive manner: IL-4 [F(1,45) = 499.77, p < 0.05] (Fig. 8B, right panel, gray bars), IL-5 [F(2,45) = 3.45, p < 0.05]] (Fig. 8C, right panel, gray bars), and IL-13 [F(2,45) = 313.62, p < 0.05]] (Fig. 8D, right panel, gray bars). Additionally, a main effect for 28 d Tat exposure to significantly increase IL-4 was observed [F(1,45) = 9.13, p < 0.05], independent of maraviroc or morphine administration (Fig. 8B, right panel, yellow/magenta/gray bars).
4. Discussion
The overall hypotheses that HIV-1 Tat expression attenuates morphine potency, CCR5 antagonism reverses these effects, and withholding morphine for 24 h exacerbates Tat's effects on cytokines were upheld. The present data revealed even greater nuance in the behavioral response to morphine regimens than was anticipated. Consistent with prior reports (Fitting et al., 2012, 2016), HIV-1 Tat expression significantly attenuated the antinociceptive potency of acute morphine injection (2 – 64 mg/kg, i.p.) and significantly attenuated aspects of withdrawal among mice made tolerant via subcutaneous implant of a morphine pellet (75 mg). The present work further extends these findings to demonstrate that pretreatment with the CCR5 antagonist, maraviroc, blocks the effects of Tat on morphine tolerance and dependence behaviors (reinstating morphine potency in non-tolerant mice and restoring aspects of withdrawal symptomology in morphine-tolerant mice). We further assessed the potential interactions of HIV-1 Tat and maraviroc on morphine reward via a CPP assay. Tat reduced the locomotor response associated with repeated, once-daily, morphine injections (10 mg/kg. i.p.) over 4 days of conditioning, but significantly potentiated (∼3.5-fold) morphine-CPP after morphine was withheld for 24 h. These results are congruent with prior findings of Tat-potentiated psychostimulant and ethanol reward (McLaughlin et al., 2014; Mediouni et al., 2015; Paris et al., 2014a,b) and a slower rate of extinction in HIV-1 transgenic rats contextually conditioned to morphine (Homji et al., 2012). Surprisingly, maraviroc further potentiated Tat's effects on morphine-mediated locomotion and CPP (∼5.7-fold), but exerted no influence on these behaviors when administered alone. Protein arrays revealed that withholding morphine for 24 h (commensurate with the timeframe of CPP assessment) markedly increased the levels of multiple categories of cytokines (proinflammatory, anti-inflammatory, and regulatory), suggesting a widespread dysregulation of immune function. Importantly, maraviroc could block or attenuate the immunological consequences of terminating morphine administration across a number of cytokines. Tat-dependent elevations in the endogenous CCR5 ligands (CCL3 and CCL4) and CCL2 (whose activation is proposed to be downstream of CCR5-dependent Tat/opioid interactions; El-Hage et al., 2008), as well as other cytokines increased by Tat, were markedly attenuated by maraviroc.
The marked increase in a large number of cytokines that was observed 24 h after withholding repeated morphine (10 mg/kg/d) was unanticipated and may be contributed to by several sources. Although immune function is generally suppressed by sustained opioid exposure (Bayer et al., 1994; Rahim et al., 2003), it can transiently rebound followed by sustained immunosuppression after opioid withdrawal (Rahim et al 2004; Eisenstein et al., 2006). Contextual cues alone (such as those used in CPP) alter immune responding once opiate conditioning is established (Hutson et al., 2017; Saurer et al., 2011), effects that involve the ventral striatum (Szczytkowski et al., 2011). Alternatively, chronic doxycycline treatment can produce alterations in the gut microbiome (Angelakis et al., 2014) which may influence immune responding and subsequent behavior. Interestingly, in this study we found that chronic doxycycline treatment upregulated some but not all chemokines and cytokines in dorsal striatum of both Tat(−) and Tat(+) mice (CCL5, IL-1α, IL-1β, IL-2, IL-6, and IL-13 were upregulated in chronic, compared to acute, doxycycline exposure). There is increasing evidence that microbial dysbiosis can influence many centrally mediated effects, including anxiety and mood (Cryan and Dinan, 2012; Foster et al., 2013). We have previously reported that HIV-1 Tat can induce bacterial translocation and enhance pro-inflammatory cytokines via TLR4 activation in the mouse colon (Guedia et al., 2016). Chronic morphine has also been reported to induce microbial dysbiosis and increase pro-inflammatory cytokines within the gut (Kang et al., 2017; Meng et al., 2013). In particular, morphine pellets have been observed to induce sepsis (Eisenstein et al., 2006; Feng et al., 2005; Hillburger et al., 1997) which may be due, in part, to intestinal stasis. Given that HIV-1 Tat has been shown to increase the permeability of the blood-brain barrier (Leibrand et al., 2017), the presence of Tat may compound the central accumulation of systemic factors including cytokines. The present findings support previous literature demonstrating MOR and CCR5 interactions on HIV-1 Tat-mediated cellular outcomes and begin to reveal the related behavioral sequelae.
The comorbidity between opioid abuse and HIV is well established. The cellular/molecular interactions between opioids and HIV co-receptors are less well understood, but may exert important effects on chemokine function. Opioids modulate CCR5 expression and signaling in vitro. Morphine or methadone upregulates CCR5 mRNA and protein expression in murine BV-2 derived microglia (Bokhari et al., 2009), human astrocytes (Mahajan et al., 2005b), human lymphoid cell lines (Miyagi et al., 2000; Suzuki et al., 2002a), or human monocyte-derived macrophages (Guo et al., 2002; Li et al., 2003) or microglia (Li et al., 2002). These effects are functional and confer susceptibility to simian immunodeficiency virus (SIV; Miyagi et al., 2000; Suzuki et al., 2002a,b) or R5-tropic HIV (Guo et al., 2002; Li et al., 2003). Importantly, CCR5 upregulation and viremia has been shown to be attenuated with opioid receptor antagonists, such as naltrexone (Guo et al., 2002), naloxone (Mahajan et al., 2005b), or the quaternary opioid antagonist, methylnaltrexone (Ho et al., 2003). These effects are, at least partly, dependent on actions at MORs given that the highly selective MOR agonist, DAMGO, upregulates CCR5 and enhances R5-HIV viremia in vitro, an effect that is blocked by the MOR-selective antagonist, CTAP (Steele et al., 2003). In addition to altered CCR5 expression, MOR and CCR5 undergo heterologous cross-desensitization (Szabo et al., 2002, 2003; Zhang et al., 2004), which may occur through convergent signaling or via direct molecular interactions involving the formation of MOR-CCR5 heteromers. Evidence for direct MOR-CCR5 dimeric/oligomeric interactions are partly based on findings that a bivalent ligand comprised naltrexone and maraviroc can reduce the infectivity of cultured human astrocytes (El-Hage et al., 2013; Yuan et al., 2013) and attenuate R5 HIV-mediated increases in CCL5, TNF-α, and IL-6 in human astrocytes (El-Hage et al., 2013). Divergent effects of the bivalent ligand were observed in cultured human microglia, which were much more dynamic in their response to morphine. Unlike astrocytes, microglia demonstrated a strong upregulation of CCR5 in response to the bivalent ligand in culture that may have accounted for the differential response between cell types (El-Hage et al., 2013). In other studies, treating a human lymphoid cell line with morphine and CCL4 (respective agonists for MOR and CCR5) increased morphine-mediated upregulation of CCR5 protein expression (Suzuki et al., 2002c). Together, our data agree with prior findings (Szabo et al., 2002, 2003; Zhang et al., 2004) supporting the notion that opioids interact with CCR5 at multiple levels, from the regulation of CCR5 expression and signaling to potential actions at MOR-CCR5 heteromers and functional effects on viremia and neuroinflammation. These present findings begin to extend these data to behavioral dysfunction in a whole-animal model.
Actions at CCR5 may represent a critical point of convergence between the viral protein, Tat, and MORs. Apart from its role in HIV entry, CCR5 signaling is essential in macrophage and microglial chemoattraction, the homing of CD8+ T cells, and the pathogenesis of CNS inflammatory diseases (Martin-Blondel et al., 2016). Foremost, Tat/opioid interactions for neurotoxicity appear to be dependent on glial MORs. While Tat demonstrates neurotoxicity in co-cultured neurons from either wildtype mice or MOR-knockout mice, these effects are potentiated by pretreatment with morphine only when neurons are co-cultured with wildtype glia (MOR-knockout glia do not potentiate toxicity; Zou et al., 2011). Second, Tat/opioid toxicity largely involves downstream neuroinflammation. In murine progenitors, Tat and opioids have interactive effects on astroglial chemokine secretion (Hahn et al., 2010), CCL5 is often the predominant central chemokine upregulated in response to Tat, an effect that can be potentiated by chronic morphine (Dutta & Roy, 2015), and inactivating CCR5 with an antibody blocks the chemoattractive effects promoted by Tat (Hahn et al., 2010). Third, CCL5 activation appears to work in concert with downstream chemokine activation, particularly CCL2. In support, morphine stimulates production of CCL5 and CCL2 in cultured peripheral blood mononuclear cells (Wetzel et al., 2000) and Tat increases CCL2 and CCL5 production in murine primary astrocyte cultures; effects that are potentiated by co-application of morphine and reversed by MOR antagonists (such as β-FNA; El-Hage et al., 2005). Striatal infusion of Tat1-86 to mice significantly increases the proportion of CCL5 immunoreactive astroglia and co-administration of morphine and Tat significantly increases astrogliosis, microgliosis, the proportion of CCL2 immunofluorescent astrocytes/macrophages/microglia, and reactive nitrosative species co-localized to these cells (El-Hage et al., 2008a). These effects on murine striatum are obviated by CCL5 knockout (El-Hage et al., 2008a) or knockout of the CCL2 receptor (El-Hage et al., 2006). Findings in primary human astrocytes are supportive with morphine downregulating CCL2 and CCL4 mRNA and upregulating their receptors in a naloxone-dependent manner (Mahajan et al., 2005b). Thus, Tat and opioids acting at MORs appear to activate CCR5 and CCR2 signaling, increasing neuroinflammation and striatal neurotoxicity. These effects may contribute to the attenuated therapeutic efficacy and increased rewarding properties of morphine that we have observed herein.
We propose that MOR-CCR5 cross-desensitization may contribute to the behavioral pathology observed (Fig. 9). When HIV-1 Tat-mediated neuroinflammation was not present [as modeled using Tat(−) control mice], cytokine concentrations were at basal levels and morphine exerted efficacious effects for tolerance, dependence, psychostimulation, and reward, presumably via actions partly mediated by MORs (Fig. 9A). The addition of maraviroc to Tat(−) mice did not influence morphine efficacy on these behavioral measures (Fig. 9B) consistent with in vitro reports wherein MOR-CCR5 bivalent ligands exert little influence when neuroinflammation is not present (Akgün et al., 2015; Portoghese et al., 2017). However, when a neuroinflammatory insult was first present [as modeled using Tat(+) mice], morphine efficacy was diminished on most measures; perhaps, due to endogenous MORs having already been cross-desensitized by activated CCR5 receptors (Fig. 9C). When maraviroc was introduced in the Tat-inflamed state, morphine efficacy was restored on most measures and even potentiated on the measure of reward. We speculate that the addition of maraviroc in this state may relieve cross-desensitization, restoring morphine signaling through its endogenous receptors (Fig. 9D). In support, a bivalent ligand (MOR agonist/CCR5 antagonist) exerted a 3100-fold increase in potency when administered to LPS-inflamed mice, relative to non-inflamed controls (Akgün et al., 2015; Portoghese et al., 2017). There was one behavioral exception to Tat's capacity to attenuate morphine efficacy and that was on the measure of reward prior to maraviroc. These divergent findings may involve the role of additional brain regions and opioid signaling beyond that involving CCR5 heterodimers (Shippenberg et al., 2009).
Figure 9. Proposed mechanism(s) of morphine/maraviroc interaction with or without HIV-1 Tat-induced neuroinflammation.
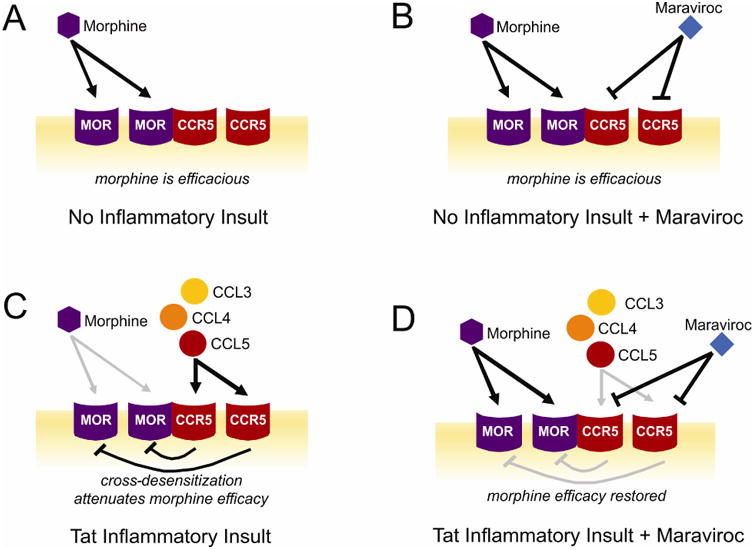
(A) In the absence of Tat-mediated neuroinflammation [as modeled using (Tat-), control mice], morphine is behaviorally efficacious, partly via actions at mu opioid receptor (MOR) oligomers or MOR-CCR5 heteromers. MOR signaling may cross-desensitize CCR5 oligomers in this state (not depicted). (B) When no prior neuroinflammatory stimulus is present, the addition of maraviroc exerts little influence on the behavioral efficacy of morphine [as modeled using (Tat-), maraviroc-treated mice]. (C) When Tat-induced neuroinflammation is present [as modeled using Tat(+) mice], there are enhanced levels of β-chemokines that bind CCR5 and may cross-desensitize MORs. When morphine is administered in this state, its behavioral efficacy is attenuated. (D) The addition of maraviroc when neuroinflammation is present may block the ability of CCR5 to cross-desensitize MORs, thereby restoring morphine efficacy [as modeled using maraviroc-treated, Tat(+) mice].
It must be noted that the timing of morphine and Tat-dependent interactions at CCR5 is likely to be critical in determining the nature of the outcome (see Fig. 9 in Berman et al., 2006; Song et al., 2011). The relative increases in cytokines seen with acute morphine and/or Tat exposure (for example, as in CCL3, CCL4, CCL5, CXCL1, IL-1α, IL-9, and IL-10) were often smaller effects that may either be less biologically significant or may display peak activity earlier than the time examined. These effects were largely absent after sustained exposure suggesting that the CNS adapts to sustained opiate or HIV-1 Tat insults via mechanisms involving drug tolerance (Eisenstein et al., 2006) or innate immune tolerance (Cavaillon & Adib-Conquy, 2006; Biswas & Lopez-Collazo, 2009), respectively. Future investigations should additionally assess the presence of endotoxemia which may contribute to these effects (Eisenstein et al., 2006), but may be masked by downregulation of the innate response.
The present work raises several clinical considerations. Seropositive patients within the first 100 d of HIV infection demonstrate positive correlations between circulating CCL2, CCL11, GM-CSF, IL-1α, IL-6, and lateral ventricular volume as well as IL-5, IL-10, and mean diffusivity within the caudate (Ragin et al., 2015). Herein, maraviroc exerted little influence over behavioral measures on its own, but attenuated the capacity for HIV-1 Tat to dampen morphine potency. Given that up to 40% of HIV-afflicted individuals experience distal polyneuropathies, headache, and additional chronic pain states (Keswani et al., 2002; Mirsattari et al., 1999) necessitating opioid medications for relief among a sizeable percentage (∼23% of a 1400+ patient sample infected with HIV; Merlin et al., 2016), maraviroc may have potential benefits to improve opioid-based therapies. However, with chronic opioid use, the beneficial effects of maraviroc are less clear given that maraviroc was also associated with an increase in withdrawal symptomology and increased morphine reward. In addition, the neurotoxic actions of additional HIV proteins in the patient population, such as the HIV coat protein (gp120), may exert a divergent profile compared to that observed when HIV-1 Tat is expressed alone (Campbell et al., 2015; Maung et al., 2012; Mocchetti et al., 2013). The toxic effects of R5-tropic gp120 can be attenuated by morphine-mediated CCL5 upregulation, perhaps influenced by out-competition for CCR5 (Avdoshina et al., 2010). As such, future investigations may wish to target MOR-CCR5 bivalent strategies within the context of opioid dependency across a range of neuroAIDS models.
5. Conclusions
Several investigations using murine and human cell cultures have demonstrated neurotoxic interactions between the HIV-1 Tat, MORs, and CCR5. The present findings extend these data by investigating functional consequences of such interactions on morphine-mediated antinociception, tolerance, and reward in a murine model. Maraviroc blocked Tat's actions to attenuate the antinociceptive potency of acute morphine in non-tolerant mice. Intriguingly, maraviroc also potentiated the Tat-induced increase of morphine-CPP, even while it reduced the levels of many inflammatory chemokines and cytokines in the striatum including β-chemokines. Thus, while maraviroc is widely appreciated for its role in blocking HIV entry, it may be considered for additional therapeutic roles related to pain control in HIV-infected patients; albeit, caution may be warranted for individuals that are opioid-dependent or at risk for such abuse.
Highlights.
HIV Tat decreases morphine potency and CCR5 antagonism attenuates Tat effects
Tat decreases morphine withdrawal and CCR5 antagonism attenuates Tat effects
Tat exposure and CCR5 antagonism attenuate the psychomotor response to morphine
CCR5 antagonism potentiated Tat-promoted morphine conditioned place preference
24h post drug, Tat elevates central cytokines; CCR5 blockade attenuates Tat effects
Acknowledgments
This work was supported by funds from NIH: T32 DA007027 (WLD), P30 DA033934 (WLD), R01 DA036975 (WLD and HIA), R01 DA034231 (PEK and KFH), K02 DA027374 (KFH), R01 DA018633 (KFH), R01 DA033200 (KFH), and R00 DA039791 (JJP). We thank Ms. Tamara Vujanovic for her assistance with figures.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Akgün E, Javed MI, Lunzer MM, Powers MD, Sham YY, Watanabe Y, Portoghese PS. Inhibition of Inflammatory and Neuropathic Pain by Targeting a Mu Opioid Receptor/Chemokine Receptor5 Heteromer (MOR-CCR5) J Med Chem. 2015;58:8647–57. doi: 10.1021/acs.jmedchem.5b01245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altun A, Ozdemir E, Yildirim K, Gursoy S, Durmus N, Bagcivan I. The effects of endocannabinoid receptor agonist anandamide and antagonist rimonabant on opioid analgesia and tolerance in rats. Gen Physiol Biophys. 2015;34:433–40. doi: 10.4149/gpb_2015017. [DOI] [PubMed] [Google Scholar]
- Angelakis E, Million M, Kankoe S, Lagier JC, Armougom F, Giorgi R, Raoult D. Abnormal weight gain and gut microbiota modifications are side effects of long-term doxycycline and hydroxychloroquine treatment. Antimicrob Agents Chemother. 2014;58:3342–7. doi: 10.1128/AAC.02437-14. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Arnatt CK, Falls BA, Yuan Y, Raborg TJ, Masvekar RR, El-Hage N, Selley DE, Nicola AV, Knapp PE, Hauser KF, Zhang Y. Exploration of bivalent ligands targeting putative mu opioid receptor and chemokine receptor CCR5 dimerization. Bioorg Med Chem. 2016;24:5969–5987. doi: 10.1016/j.bmc.2016.09.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avdoshina V, Biggio F, Palchik G, Campbell LA, Mocchetti I. Morphine induces the release of CCL5 from astrocytes: potential neuroprotective mechanism against the HIV protein gp120. Glia. 2010;58:1630–9. doi: 10.1002/glia.21035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bayer BM, Brehio RM, Ding XZ, Hernandez MC. Enhanced susceptibility of the immune system to stress in morphine-tolerant rats. Brain Behav Immun. 1994;8:173–84. doi: 10.1006/brbi.1994.1017. [DOI] [PubMed] [Google Scholar]
- Bell JE, Arango JC, Anthony IC. Neurobiology of multiple insults: HIV-1-associated brain disorders in those who use illicit drugs. J Neuroimmune Pharmacol. 2006;1:182–91. doi: 10.1007/s11481-006-9018-2. [DOI] [PubMed] [Google Scholar]
- Bell JE, Arango JC, Robertson R, Brettle RP, Leen C, Simmonds P. HIV and drug misuse in the Edinburgh cohort. J Acquir Immune Defic Syndr. 2002;31:S35–S42. doi: 10.1097/00126334-200210012-00003. [DOI] [PubMed] [Google Scholar]
- Bell JE, Donaldson YK, Lowrie S, McKenzie CA, Elton RA, Chiswick A, Brettle RP, Ironside JW, Simmonds P. Influence of risk group and zidovudine therapy on the development of HIV encephalitis and cognitive impairment in AIDS patients. AIDS. 1996;10:493–9. doi: 10.1097/00002030-199605000-00007. [DOI] [PubMed] [Google Scholar]
- Berman JW, Carson MJ, Chang L, Cox BM, Fox HS, Gonzalez RG, Hanson GR, Hauser KF, Ho WZ, Hong JS, Major EO, Maragos WF, Masliah E, McArthur JC, Miller DB, Nath A, O'Callaghan JP, Persidsky Y, Power C, Rogers TJ, Royal W., 3rd NeuroAIDS, drug abuse, and inflammation: building collaborative research activities. J Neuroimmune Pharmacol. 2006;1:351–99. doi: 10.1007/s11481-006-9048-9. [DOI] [PubMed] [Google Scholar]
- Biswas SK, Lopez-Collazo E. Endotoxin tolerance: new mechanisms, molecules and clinical significance. Trends Immunol. 2009;30:475–87. doi: 10.1016/j.it.2009.07.009. [DOI] [PubMed] [Google Scholar]
- Bokhari SM, Hegde R, Callen S, Yao H, Adany I, Li Q, Li Z, Pinson D, Yeh HW, Cheney PD, Buch S. Morphine potentiates neuropathogenesis of SIV infection in rhesus macaques. J Neuroimmune Pharmacol. 2011;6:626–39. doi: 10.1007/s11481-011-9272-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bokhari SM, Yao H, Bethel-Brown C, Fuwang P, Williams R, Dhillon NK, Hegde R, Kumar A, Buch SJ. Morphine enhances Tat-induced activation in murine microglia. J Neurovirol. 2009;15:219–28. doi: 10.1080/13550280902913628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruce-Keller AJ, Turchan-Cholewo J, Smart EJ, Geurin T, Chauhan A, Reid R, Xu R, Nath A, Knapp PE, Hauser KF. Morphine causes rapid increases in glial activation and neuronal injury in the striatum of inducible HIV-1 Tat transgenic mice. Glia. 2008;56:1414–27. doi: 10.1002/glia.20708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell LA, Avdoshina V, Day C, Lim ST, Mocchetti I. Pharmacological induction of CCL5 in vivo prevents gp120-mediated neuronal injury. Neuropharmacology. 2015;92:98–107. doi: 10.1016/j.neuropharm.2015.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavaillon JM, Adib-Conquy M. Bench-to-bedside review: endotoxin tolerance as a model of leukocyte reprogramming in sepsis. Crit Care. 2006;10:233. doi: 10.1186/cc5055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Centers for Disease Control and Prevention. HIV Surveillance Report, 2015. [Accessed April 2017];27 http://www.cdc.gov/hiv/library/reports/hiv-surveillance.html. Published November 2016. [Google Scholar]
- Chen X, Geller EB, Rogers TJ, Adler MW. Rapid heterologous desensitization of antinociceptive activity between mu or delta opioid receptors and chemokine receptors in rats. Drug Alcohol Depend. 2007;88:36–41. doi: 10.1016/j.drugalcdep.2006.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang RY, Suzuki S, Chuang TK, Miyagi T, Chuang LF, Doi RH. Opioids and the progression of simian AIDS. Front Biosci. 2005;10:1666–1677. doi: 10.2741/1651. [DOI] [PubMed] [Google Scholar]
- Coderre TJ, Rollman GB. Naloxone hyperalgesia and stress-induced analgesia in rats. Life Sci. 1983;32:2139–2146. doi: 10.1016/0024-3205(83)90103-0. [DOI] [PubMed] [Google Scholar]
- Crawley JN, Paylor R. A proposed test battery and constellations of specific behavioral paradigms to investigate the behavioral phenotypes of transgenic and knockout mice. Horm Behav. 1997;31:197–211. doi: 10.1006/hbeh.1997.1382. [DOI] [PubMed] [Google Scholar]
- Cryan JF, Dinan TG. Mind-altering microorganisms: the impact of the gut microbiota on brain and behaviour. Nat Rev Neurosci. 2012;13:701–12. doi: 10.1038/nrn3346. [DOI] [PubMed] [Google Scholar]
- Dalvi P, Sharma H, Chinnappan M, Sanderson M, Allen J, Zeng R, Choi A, O'Brien-Ladner A, Dhillon NK. Enhanced autophagy in pulmonary endothelial cells on exposure to HIV-Tat and morphine: Role in HIV-related pulmonary arterial hypertension. Autophagy. 2016;12:2420–2438. doi: 10.1080/15548627.2016.1238551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donahoe RM, Byrd LD, McClure HM, Fultz P, Brantley M, Marsteller F, Ansari AA, Wenzel D, Aceto M. Consequences of opiate-dependency in a monkey model of AIDS. Adv Exp Med Biol. 1993;335:21–28. doi: 10.1007/978-1-4615-2980-4_4. [DOI] [PubMed] [Google Scholar]
- Dutta R, Roy S. Chronic morphine and HIV-1 Tat promote differential central nervous system trafficking of CD3+ and Ly6C+ immune cells in a murine Streptococcus pneumoniae infection model. J Neuroinflammation. 2015;12:120. doi: 10.1186/s12974-015-0341-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisenstein TK, Rahim RT, Feng P, Thingalaya NK, Meissler JJ. Effects of opioid tolerance and withdrawal on the immune system. J Neuroimmune Pharmacol. 2006;1:237–49. doi: 10.1007/s11481-006-9019-1. [DOI] [PubMed] [Google Scholar]
- El-Hage N, Bruce-Keller AJ, Knapp PE, Hauser KF. CCL5/RANTES gene deletion attenuates opioid-induced increases in glial CCL2/MCP-1 immunoreactivity and activation in HIV-1 Tat-exposed mice. J Neuroimmune Pharmacol. 2008a;3:275–85. doi: 10.1007/s11481-008-9127-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Hage N, Bruce-Keller AJ, Yakovleva T, Bazov I, Bakalkin G, Knapp PE, Hauser KF. Morphine exacerbates HIV-1 Tat-induced cytokine production in astrocytes through convergent effects on [Ca2+]i, NF-κB trafficking and transcription. PLoS One. 2008b;3:e4093. doi: 10.1371/journal.pone.0004093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Hage N, Dever SM, Podhaizer EM, Arnatt CK, Zhang Y, Hauser KF. A novel bivalent HIV-1 entry inhibitor reveals fundamental differences in CCR5-μ-opioid receptor interactions between human astroglia and microglia. AIDS. 2013;27:2181–90. doi: 10.1097/QAD.0b013e3283639804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Hage N, Gurwell JA, Singh IN, Knapp PE, Nath A, Hauser KF. Synergistic increases in intracellular Ca2+, and the release of MCP-1, RANTES, and IL-6 by astrocytes treated with opiates and HIV-1 Tat. Glia. 2005;50:91–106. doi: 10.1002/glia.20148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Hage N, Wu G, Ambati J, Bruce-Keller AJ, Knapp PE, Hauser KF. CCR2 mediates increases in glial activation caused by exposure to HIV-1 Tat and opiates. J Neuroimmunol. 2006;178:9–16. doi: 10.1016/j.jneuroim.2006.05.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng P, Meissler JJ, Jr, Adler MW, Eisenstein TK. Morphine withdrawal sensitizes mice to lipopolysaccharide: elevated TNF-alpha and nitric oxide with decreased IL-12. J Neuroimmunol. 2005;164:57–65. doi: 10.1016/j.jneuroim.2005.03.017. [DOI] [PubMed] [Google Scholar]
- Fitting S, Ignatowska-Jankowska BM, Bull C, Skoff RP, Lichtman AH, Wise LE, Fox MA, Su J, Medina AE, Krahe TE, Knapp PE, Guido W, Hauser KF. Synaptic dysfunction in the hippocampus accompanies learning and memory deficits in human immunodeficiency virus type-1 Tat transgenic mice. Biol Psychiatry. 2013;73:443–53. doi: 10.1016/j.biopsych.2012.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitting S, Knapp PE, Zou S, Marks WD, Bowers MS, Akbarali HI, Hauser KF. Interactive HIV-1 Tat and morphine-induced synaptodendritic injury is triggered through focal disruptions in Na+ influx, mitochondrial instability, and Ca²+ overload. J Neurosci. 2014a;34:12850–64. doi: 10.1523/JNEUROSCI.5351-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitting S, Scoggins KL, Xu R, Dever SM, Knapp PE, Dewey WL, Hauser KF. Morphine efficacy is altered in conditional HIV-1 Tat transgenic mice. Eur J Pharmacol. 2012;689:96–103. doi: 10.1016/j.ejphar.2012.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitting S, Stevens DL, Khan FA, Scoggins KL, Enga RM, Beardsley PM, Knapp PE, Dewey WL, Hauser KF. Morphine Tolerance and Physical Dependence Are Altered in Conditional HIV-1 Tat Transgenic Mice. J Pharmacol Exp Ther. 2016;356:96–105. doi: 10.1124/jpet.115.226407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitting S, Zou S, El-Hage N, Suzuki M, Paris JJ, Schier CJ, Rodríguez JW, Rodriguez M, Knapp PE, Hauser KF. Opiate addiction therapies and HIV-1 Tat: interactive effects on glial [Ca²+]i, oxyradical and neuroinflammatory chemokine production and correlative neurotoxicity. Curr HIV Res. 2014b;12:424–34. doi: 10.2174/1570162X1206150311161147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster JA, McVey Neufeld KA. Gut-brain axis: how the microbiome influences anxiety and depression. Trends Neurosci. 36:305–12. doi: 10.1016/j.tins.2013.01.005. 201. [DOI] [PubMed] [Google Scholar]
- Freireich EJ, Gehan EA, Rall DP, Schmidt LH, Skipper HE. Quantitative comparison of toxicity of anticancer agents in mouse, rat, hamster, dog, monkey, and man. Cancer Chemother Rep. 1966;50:219–44. [PubMed] [Google Scholar]
- Guedia J, Brun P, Bhave S, Fitting S, Kang M, Dewey WL, Hauser KF, Akbarali HI. HIV-1 Tat exacerbates lipopolysaccharide-induced cytokine release via TLR4 signaling in the enteric nervous system. Sci Rep. 2016;6:31203. doi: 10.1038/srep31203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo CJ, Li Y, Tian S, Wang X, Douglas SD, Ho WZ. Morphine enhances HIV infection of human blood mononuclear phagocytes through modulation of beta-chemokines and CCR5 receptor. J Investig Med. 2002;50:435–42. doi: 10.1136/jim-50-06-03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta S, Knight AG, Gupta S, Knapp PE, Hauser KF, Keller JN, Bruce-Keller AJ. HIV-Tat elicits microglial glutamate release: role of NAPDH oxidase and the cystine-glutamate antiporter. Neurosci Lett. 2010;485:233–6. doi: 10.1016/j.neulet.2010.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurwell JA, Nath A, Sun Q, Zhang J, Martin KM, Chen Y, Hauser KF. Synergistic neurotoxicity of opioids and human immunodeficiency virus-1 Tat protein in striatal neurons in vitro. Neuroscience. 2001;102:555–63. doi: 10.1016/s0306-4522(00)00461-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn YK, Podhaizer EM, Farris SP, Miles MF, Hauser KF, Knapp PE. Effects of chronic HIV-1 Tat exposure in the CNS: heightened vulnerability of males versus females to changes in cell numbers, synaptic integrity, and behavior. Brain Struct Funct. 2015;220:605–23. doi: 10.1007/s00429-013-0676-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn YK, Podhaizer EM, Hauser KF, Knapp PE. HIV-1 alters neural and glial progenitor cell dynamics in the central nervous system: coordinated response to opiates during maturation. Glia. 2012;60:1871–87. doi: 10.1002/glia.22403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn YK, Vo P, Fitting S, Block ML, Hauser KF, Knapp PE. β-Chemokine production by neural and glial progenitor cells is enhanced by HIV-1 Tat: effects on microglial migration. J Neurochem. 2010;114:97–109. doi: 10.1111/j.1471-4159.2010.06744.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris LS, Pierson AK. Some narcotic antagonists in the benzomorphan series. J Pharmacol Exp Ther. 1964;143:141–48. [PubMed] [Google Scholar]
- Hauser KF, Hahn YK, Adjan VV, Zou S, Buch SK, Nath A, Bruce-Keller AJ, Knapp PE. HIV-1 Tat and morphine have interactive effects on oligodendrocyte survival and morphology. Glia. 2009;57:194–206. doi: 10.1002/glia.20746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbein G, Gras G, Khan KA, Abbas W. Macrophage signaling in HIV-1 infection. Retrovirology. 2010;7:34. doi: 10.1186/1742-4690-7-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilburger ME, Adler MW, Truant AL, Meissler JJ, Jr, Satishchandran V, Rogers TJ, Eisenstein TK. Morphine induces sepsis in mice. J Infect Dis. 1997;176:183–8. doi: 10.1086/514021. [DOI] [PubMed] [Google Scholar]
- Ho WZ, Guo CJ, Yuan CS, Douglas SD, Moss J. Methylnaltrexone antagonizes opioid-mediated enhancement of HIV infection of human blood mononuclear phagocytes. J Pharmacol Exp Ther. 2003;307:1158–62. doi: 10.1124/jpet.103.056697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homji NF, Vigorito M, Chang SL. Morphine-induced conditioned place preference and associated behavioural plasticity in HIV-1 transgenic rats. Int J Clin Exp Med. 2012;5:105–23. [PMC free article] [PubMed] [Google Scholar]
- Hutson LW, Lebonville CL, Jones ME, Fuchs RA, Lysle DT. Interleukin-1 signaling in the basolateral amygdala is necessary for heroin-conditioned immunosuppression. Brain Behav Immun. 2017;62:171–79. doi: 10.1016/j.bbi.2017.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang M, Mischel RA, Bhave S, Komla E, Cho A, Huang C, Dewey WL, Akbarali HI. The effect of gut microbiome on tolerance to morphine mediated antinociception in mice. Sci Rep. 2017;7:42658. doi: 10.1038/srep42658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keswani SC, Pardo CA, Cherry CL, Hoke A, McArthur JC. HIV-associated sensory neuropathies. AIDS. 2002;16:2105–17. doi: 10.1097/00002030-200211080-00002. [DOI] [PubMed] [Google Scholar]
- King JE, Eugenin EA, Buckner CM, Berman JW. HIV tat and neurotoxicity. Microbes Infect. 2006;8:1347–57. doi: 10.1016/j.micinf.2005.11.014. [DOI] [PubMed] [Google Scholar]
- Kremer M, Salvat E, Muller A, Yalcin I, Barrot M. Antidepressants and gabapentinoids in neuropathic pain: Mechanistic insights. Neuroscience. 2016;338:183–206. doi: 10.1016/j.neuroscience.2016.06.057. [DOI] [PubMed] [Google Scholar]
- Kumar R, Torres C, Yamamura Y, Rodriguez I, Martinez M, Staprans S, Donahoe RM, Kraiselburd E, Stephens EB, Kumar A. Modulation by morphine of viral set point in rhesus macaques infected with simian immunodeficiency virus and simian-human immunodeficiency virus. J Virol. 2004;78:11425–11428. doi: 10.1128/JVI.78.20.11425-11428.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar R, Orsoni S, Norman L, Verma AS, Tirado G, Giavedoni LD, Staprans S, Miller GM, Buch SJ, Kumar A. Chronic morphine exposure causes pronounced virus replication in cerebral compartment and accelerated onset of AIDS in SIV/SHIV-infected Indian rhesus macaques. Virology. 2006;354:192–206. doi: 10.1016/j.virol.2006.06.020. [DOI] [PubMed] [Google Scholar]
- Kwiatkowski K, Piotrowska A, Rojewska E, Makuch W, Jurga A, Slusarczyk J, Trojan E, Basta-Kaim A, Mika J. Beneficial properties of maraviroc on neuropathic pain development and opioid effectiveness in rats. Prog Neuropsychopharmacol Biol Psychiatry. 2016 Jan 4;64:68–78. doi: 10.1016/j.pnpbp.2015.07.005. [DOI] [PubMed] [Google Scholar]
- Leibrand CR, Paris JJ, Ghandour MS, Knapp PE, Kim WK, Hauser KF, McRae M. HIV-1 Tat disrupts blood-brain barrier integrity and increases phagocytic perivascular macrophages and microglia in the dorsal striatum of transgenic mice. Neurosci Lett. 2017;640:136–143. doi: 10.1016/j.neulet.2016.12.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Merrill JD, Mooney K, Song L, Wang X, Guo CJ, Savani RC, Metzger DS, Douglas SD, Ho WZ. Morphine enhances HIV infection of neonatal macrophages. Pediatr Res. 2003;54:282–8. doi: 10.1203/01.PDR.0000074973.83826.4C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Wang X, Tian S, Guo CJ, Douglas SD, Ho WZ. Methadone enhances human immunodeficiency virus infection of human immune cells. J Infect Dis. 2002;185:118–22. doi: 10.1086/338011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahajan SD, Aalinkeel R, Reynolds JL, Nair BB, Fernandez SF, Schwartz SA, Nair MP. Morphine exacerbates HIV-1 viral protein gp120 induced modulation of chemokine gene expression in U373 astrocytoma cells. Curr HIV Res. 2005a;3:277–88. doi: 10.2174/1570162054368048. [DOI] [PubMed] [Google Scholar]
- Mahajan SD, Schwartz SA, Aalinkeel R, Chawda RP, Sykes DE, Nair MP. Morphine modulates chemokine gene regulation in normal human astrocytes. Clin Immunol. 2005b;115:323–32. doi: 10.1016/j.clim.2005.02.004. [DOI] [PubMed] [Google Scholar]
- Malik S, Khalique H, Buch S, Seth P. A growth factor attenuates HIV-1 Tat and morphine induced damage to human neurons: implication in HIV/AIDS-drug abuse cases. PLoS One. 2011;6:e18116. doi: 10.1371/journal.pone.0018116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malvar J, Vaida F, Sanders CF, Atkinson JH, Bohannon W, Keltner J, Robinson-Papp J, Simpson DM, Marra CM, Clifford DB, Gelman B, Fan J, Grant I, Ellis RJ CHARTER Group. Predictors of new-onset distal neuropathic pain in HIV-infected individuals in the era of combination antiretroviral therapy. Pain. 2015;156:731–9. doi: 10.1097/01.j.pain.0000461252.75089.bf. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin-Blondel G, Brassat D, Bauer J, Lassmann H, Liblau RS. CCR5 blockade for neuroinflammatory diseases--beyond control of HIV. Nat Rev Neurol. 2016;12:95–105. doi: 10.1038/nrneurol.2015.248. [DOI] [PubMed] [Google Scholar]
- Maung R, Medders KE, Sejbuk NE, Desai MK, Russo R, Kaul M. Genetic knockouts suggest a critical role for HIV co-receptors in models of HIV gp120-induced brain injury. J Neuroimmune Pharmacol. 2012;7:306–18. doi: 10.1007/s11481-011-9328-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin JP, Ganno ML, Eans SO, Mizrachi E, Paris JJ. HIV-1 Tat protein exposure potentiates ethanol reward and reinstates extinguished ethanol-conditioned place preference. Curr HIV Res. 2014;12:415–23. doi: 10.2174/1570162x1206150311160133. [DOI] [PubMed] [Google Scholar]
- Mediouni S, Jablonski J, Paris JJ, Clementz MA, Thenin-Houssier S, McLaughlin JP, Valente ST. Didehydro-cortistatin A inhibits HIV-1 Tat mediated neuroinflammation and prevents potentiation of cocaine reward in Tat transgenic mice. Curr HIV Res. 2015;13:64–79. doi: 10.2174/1570162x13666150121111548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng J, Yu H, Ma J, Wang J, Banerjee S, Charboneau R, Barke RA, Roy S. Morphine induces bacterial translocation in mice by compromising intestinal barrier function in a TLR-dependent manner. PLoS One. 2013;8:e54040. doi: 10.1371/journal.pone.0054040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merlin JS, Tamhane A, Starrels JL, Kertesz S, Saag M, Cropsey K. Factors Associated with Prescription of Opioids and Co-prescription of Sedating Medications in Individuals with HIV. AIDS Behav. 2016;20:687–98. doi: 10.1007/s10461-015-1178-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirsattari SM, Power C, Nath A. Primary headaches in HIV-infected patients. Headache. 1999;39:3–10. doi: 10.1046/j.1526-4610.1999.3901003.x. [DOI] [PubMed] [Google Scholar]
- Miyagi T, Chuang LF, Doi RH, Carlos MP, Torres JV, Chuang RY. Morphine induces gene expression of CCR5 in human CEMx174 lymphocytes. J Biol Chem. 2000;275:31305–10. doi: 10.1074/jbc.M001269200. [DOI] [PubMed] [Google Scholar]
- Moatti JP, Souville M, Escaffre N, Obadia Y. French general practitioners' attitudes toward maintenance drug abuse treatment with buprenorphine. Addiction. 1998;93:1567–75. doi: 10.1046/j.1360-0443.1998.9310156714.x. [DOI] [PubMed] [Google Scholar]
- Mocchetti I, Campbell LA, Harry GJ, Avdoshina V. When human immunodeficiency virus meets chemokines and microglia: neuroprotection or neurodegeneration? J Neuroimmune Pharmacol. 2013;8:118–31. doi: 10.1007/s11481-012-9353-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nath A. Eradication of human immunodeficiency virus from brain reservoirs. J Neurovirol. 2015;21:227–34. doi: 10.1007/s13365-014-0291-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nath A, Hauser KF, Wojna V, Booze RM, Maragos W, Prendergast M, Cass W, Turchan JT. Molecular basis for interactions of HIV and drugs of abuse. J Acquir Immune Defic Syndr. 2002;31:S62–9. doi: 10.1097/00126334-200210012-00006. [DOI] [PubMed] [Google Scholar]
- Neff CP, Ndolo T, Tandon A, Habu Y, Akkina R. Oral pre-exposure prophylaxis by anti-retrovirals raltegravir and maraviroc protects against HIV-1 vaginal transmission in a humanized mouse model. PLoS One. 2010;5:e15257. doi: 10.1371/journal.pone.0015257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paris JJ, Carey AN, Shay CF, Gomes SM, He JJ, McLaughlin JP. Effects of conditional central expression of HIV-1 tat protein to potentiate cocaine-mediated psychostimulation and reward among male mice. Neuropsychopharmacology. 2014a;39:380–8. doi: 10.1038/npp.2013.201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paris JJ, Eans SO, Mizrachi E, Reilley KJ, Ganno ML, McLaughlin JP. Central administration of angiotensin IV rapidly enhances novel object recognition among mice. Neuropharmacology. 2013;70:247–53. doi: 10.1016/j.neuropharm.2013.01.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paris JJ, Singh HD, Carey AN, McLaughlin JP. Exposure to HIV-1 Tat in brain impairs sensorimotor gating and activates microglia in limbic and extralimbic brain regions of male mice. Behav Brain Res. 2015;291:209–18. doi: 10.1016/j.bbr.2015.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paris JJ, Singh HD, Ganno ML, Jackson P, McLaughlin JP. Anxiety-like behavior of mice produced by conditional central expression of the HIV-1 regulatory protein, Tat. Psychopharmacology (Berl) 2014b;231:2349–60. doi: 10.1007/s00213-013-3385-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Portoghese PS, Akgün E, Lunzer MM. Heteromer Induction: An Approach to Unique Pharmacology? ACS Chem Neurosci. 2017;8:426–428. doi: 10.1021/acschemneuro.7b00002. [DOI] [PubMed] [Google Scholar]
- Ragin AB, Wu Y, Gao Y, Keating S, Du H, Sammet C, Kettering CS, Epstein LG. Brain alterations within the first 100 days of HIV infection. Ann Clin Transl Neurol. 2015;2:12–21. doi: 10.1002/acn3.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahim RT, Feng P, Meissler JJ, Rogers TJ, Zhang L, Adler MW, Eisenstein TK. Paradoxes of immunosuppression in mouse models of withdrawal. J Neuroimmunol. 2004;147:114–20. doi: 10.1016/j.jneuroim.2003.10.024. [DOI] [PubMed] [Google Scholar]
- Rahim RT, Meissler JJ, Zhang L, Adler MW, Rogers TJ, Eisenstein TK. Withdrawal from morphine in mice suppresses splenic macrophage function, cytokine production, and costimulatory molecules. J Neuroimmunol. 2003;144:16–27. doi: 10.1016/s0165-5728(03)00273-x. [DOI] [PubMed] [Google Scholar]
- Reagan-Shaw S, Nihal M, Ahmad N. Dose translation from animal to human studies revisited. FASEB J. 2008;22:659–61. doi: 10.1096/fj.07-9574LSF. [DOI] [PubMed] [Google Scholar]
- Rivera I, García Y, Gangwani MR, Noel RJ, Jr, Maldonado L, Kumar A, Rivera-Amill V. Identification and molecular characterization of SIV Vpr R50G mutation associated with long term survival in SIV-infected morphine dependent and control macaques. Virology. 2013;446:144–51. doi: 10.1016/j.virol.2013.07.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross GR, Gabra BH, Dewey WL, Akbarali HI. Morphine tolerance in the mouse ileum and colon. J Pharmacol Exp Ther. 2008;327:561–72. doi: 10.1124/jpet.108.143438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux P, Carrieri MP, Villes V, Dellamonica P, Poizot-Martin I, Ravaux I, Spire B MANIF2000 cohort study group. The impact of methadone or buprenorphine treatment and ongoing injection on highly active antiretroviral therapy (HAART) adherence: evidence from the MANIF2000 cohort study. Addiction. 2008;103:1828–36. doi: 10.1111/j.1360-0443.2008.02323.x. [DOI] [PubMed] [Google Scholar]
- Rudd RA, Seth P, David F, Scholl L. Increases in drug and opioid-involved overdose deaths - United States, 2010-2015. MMWR Morb Mortal Wkly Rep. 2016;65:1445–1452. doi: 10.15585/mmwr.mm655051e1. [DOI] [PubMed] [Google Scholar]
- Sambamoorthi U, Warner LA, Crystal S, Walkup J. Drug abuse, methadone treatment, and health services use among injection drug users with AIDS. Drug Alcohol Depend. 2000;60:77–89. doi: 10.1016/s0376-8716(99)00142-8. [DOI] [PubMed] [Google Scholar]
- Saurer TB, Ijames SG, Carrigan KA, Lysle DT. Neuroimmune mechanisms of opioid-mediated conditioned immunomodulation. Brain Behav Immun. 2008;22:89–97. doi: 10.1016/j.bbi.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schier CJ, Marks WD, Paris JJ, Barbour AJ, McLane VD, Maragos WF, McQuiston AR, Knapp PE, Hauser KF. Selective Vulnerability of Striatal D2 versus D1 Dopamine Receptor-Expressing Medium Spiny Neurons in HIV-1 Tat Transgenic Male Mice. J Neurosci. 2017;37:5758–5769. doi: 10.1523/JNEUROSCI.0622-17.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semenova S, Kuzmin A, Zvartau E. Strain differences in the analgesic and reinforcing action of morphine in mice. Pharmacol Biochem Behav. 1995;50:17–21. doi: 10.1016/0091-3057(94)00221-4. [DOI] [PubMed] [Google Scholar]
- Shippenberg TS, Chefer VI, Thompson AC. Delta-opioid receptor antagonists prevent sensitization to the conditioned rewarding effects of morphine. Biol Psychiatry. 2009;65:169–74. doi: 10.1016/j.biopsych.2008.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song C, Rahim RT, Davey PC, Bednar F, Bardi G, Zhang L, Zhang N, Oppenheim JJ, Rogers TJ. Protein kinase Czeta mediates micro-opioid receptor-induced cross-desensitization of chemokine receptor CCR5. J Biol Chem. 2011;286:20354–65. doi: 10.1074/jbc.M110.177303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorrell ME, Hauser KF. Ligand-gated purinergic receptors regulate HIV-1 Tat and morphine related neurotoxicity in primary mouse striatal neuron-glia co-cultures. J Neuroimmune Pharmacol. 2014;9:233–44. doi: 10.1007/s11481-013-9507-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steele AD, Henderson EE, Rogers TJ. Mu-opioid modulation of HIV-1 coreceptor expression and HIV-1 replication. Virology. 2003;309:99–107. doi: 10.1016/s0042-6822(03)00015-1. [DOI] [PubMed] [Google Scholar]
- Suzuki S, Carlos MP, Chuang LF, Torres JV, Doi RH, Chuang RY. Methadone induces CCR5 and promotes AIDS virus infection. FEBS Lett. 2002a;519:173–7. doi: 10.1016/s0014-5793(02)02746-1. [DOI] [PubMed] [Google Scholar]
- Suzuki S, Chuang AJ, Chuang LF, Doi RH, Chuang RY. Morphine promotes simian acquired immunodeficiency syndrome virus replication in monkey peripheral mononuclear cells: induction of CC chemokine receptor 5 expression for virus entry. J Infect Dis. 2002b;185:1826–9. doi: 10.1086/340816. [DOI] [PubMed] [Google Scholar]
- Suzuki S, Chuang LF, Yau P, Doi RH, Chuang RY. Interactions of opioid and chemokine receptors: oligomerization of mu, kappa, and delta with CCR5 on immune cells. Exp Cell Res. 2002c;280:192–200. doi: 10.1006/excr.2002.5638. [DOI] [PubMed] [Google Scholar]
- Szabo I, Chen XH, Xin L, Adler MW, Howard OM, Oppenheim JJ, Rogers TJ. Heterologous desensitization of opioid receptors by chemokines inhibits chemotaxis and enhances the perception of pain. Proc Natl Acad Sci U S A. 2002;99:10276–81. doi: 10.1073/pnas.102327699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo I, Wetzel MA, Zhang N, Steele AD, Kaminsky DE, Chen C, Liu-Chen LY, Bednar F, Henderson EE, Howard OM, Oppenheim JJ, Rogers TJ. Selective inactivation of CCR5 and decreased infectivity of R5 HIV-1 strains mediated by opioid-induced heterologous desensitization. J Leukoc Biol. 2003;74:1074–82. doi: 10.1189/jlb.0203067. [DOI] [PubMed] [Google Scholar]
- Szczytkowski JL, Fuchs RA, Lysle DT. Ventral tegmental area-basolateral amygdala-nucleus accumbens shell neurocircuitry controls the expression of heroin-conditioned immunomodulation. J Neuroimmunol. 2011;237:47–56. doi: 10.1016/j.jneuroim.2011.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turchan-Cholewo J, Dimayuga FO, Gupta S, Keller JN, Knapp PE, Hauser KF, Bruce-Keller AJ. Morphine and HIV-Tat increase microglial-free radical production and oxidative stress: possible role in cytokine regulation. J Neurochem. 2009;108:202–15. doi: 10.1111/j.1471-4159.2008.05756.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wetzel MA, Steele AD, Eisenstein TK, Adler MW, Henderson EE, Rogers TJ. μ-Opioid induction of monocyte chemoattractant protein-1, RANTES, and IFN-γ-inducible protein-10 expression in human peripheral blood mononuclear cells. J Immunol. 2000;165:6519–524. doi: 10.4049/jimmunol.165.11.6519. [DOI] [PubMed] [Google Scholar]
- Williams J, Haller VL, Stevens DL, Welch SP. Decreased basal endogenous opioid levels in diabetic rodents: effects on morphine and delta-9-tetrahydrocannabinoid-induced antinociception. Eur J Pharmacol. 2008;584:78–86. doi: 10.1016/j.ejphar.2007.12.035. [DOI] [PubMed] [Google Scholar]
- World Health Organization. Public health dimension of the world drug problem: Report by the Secretariat. [Accessed April 2017];2016 http://apps.who.int/gb/ebwha/pdf_files/EB138/B138_11-en.pdf?ua=1. Published January 2016.
- Woody GE, Bruce D, Korthuis PT, Chhatre S, Poole S, Hillhouse M, Jacobs P, Sorensen J, Saxon AJ, Metzger D, Ling W. HIV risk reduction with buprenorphine-naloxone or methadone: findings from a randomized trial. J Acquir Immune Defic Syndr. 2014;66:288–93. doi: 10.1097/QAI.0000000000000165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan Y, Arnatt CK, El-Hage N, Dever SM, Jacob JC, Selley DE, Hauser KF, Zhang Y. A Bivalent Ligand Targeting the Putative Mu Opioid Receptor and Chemokine Receptor CCR5 Heterodimers: Binding Affinity versus Functional Activities. Medchemcomm. 2013;4:847–851. doi: 10.1039/C3MD00080J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang N, Rogers TJ, Caterina M, Oppenheim JJ. Proinflammatory chemokines, such as C-C chemokine ligand 3, desensitize mu-opioid receptors on dorsal root ganglia neurons. J Immunol. 2004;173:594–9. doi: 10.4049/jimmunol.173.1.594. [DOI] [PubMed] [Google Scholar]
- Zhu X, Sun W, Li X, Tan S, Zhang X. Effects of spatial memory on morphine CPP and locomotor sensitization in mice. Physiol Behav. 2015;149:187–91. doi: 10.1016/j.physbeh.2015.04.045. [DOI] [PubMed] [Google Scholar]
- Zilliox LA. Neuropathic Pain. Continuum (Minneap Minn) 2017;23:512–532. doi: 10.1212/CON.0000000000000462. [DOI] [PubMed] [Google Scholar]
- Zou S, Fitting S, Hahn YK, Welch SP, El-Hage N, Hauser KF, Knapp PE. Morphine potentiates neurodegenerative effects of HIV-1 Tat through actions at μ-opioid receptor-expressing glia. Brain. 2011;134:3616–31. doi: 10.1093/brain/awr281. [DOI] [PMC free article] [PubMed] [Google Scholar]