

Abstract
Behavioral symptoms associated with mood disorders have been intimately linked with immunological and psychological stress. Induction of immune and stress pathways is accompanied by increased tryptophan entry into the Kynurenine (Kyn) Pathway as governed by the rate-limiting enzymes indoleamine/tryptophan 2,3-dioxygenases (DO’s: Ido1, Ido2, Tdo2). Indeed, elevated DO expression is associated with inflammation- and stress-related depression symptoms. Here we examined central (brain, astrocyte and microglia) and peripheral (lung, liver and spleen) DO expression in mice treated intraperitoneally with lipopolysaccharide (LPS) and dexamethasone (DEX) to model the response of the Kyn Pathway to inflammation and glucocorticoids. LPS-induced expression of cytokines in peripheral tissues was attenuated by DEX, confirming inflammatory and anti-inflammatory responses, respectively. Increased Kyn levels following LPS and DEX administration verified Kyn Pathway activation. Expression of multiple mRNA isoforms for each DO, which we have shown to be differentially utilized and regulated, were quantified including reference/full-length (FL) and variant (v) transcripts. LPS increased Ido1-FL in brain (~1,000-fold), a response paralleled by increased expression in both astrocytes and microglia. Central Ido1-FL was not changed by DEX; however, LPS-induced Ido1-FL was decreased by DEX in peripheral tissues. In contrast, DEX increased Ido1-v1 expression by astrocytes and microglia, but not peripheral tissues. In comparison, brain Ido2 was minimally induced by LPS or DEX. Uniquely, Ido2-v6 was LPS- and DEX-inducible in astrocytes, suggesting a unique role for astrocytes in response to inflammation and glucocorticoids. Only DEX increased central Tdo2 expression; however, peripheral Tdo2 was upregulated by either LPS or DEX. In summary, specific DO isoforms are increased by LPS and DEX, but LPS-dependent Ido1 and Ido2 induction are attenuated by DEX only in the periphery indicating that elevated DO expression and Kyn production within the brain can occur independent of the periphery. These findings demonstrate a plausible interaction between immune activation and glucocorticoids associated with depression.
Keywords: astrocyte, microglia, inflammation, stress, kynurenine
1. Introduction
The brain and immune system have a sophisticated bidirectional relationship that not only provides protection for the brain, but also affords a mechanism whereby inflammatory cytokines influence mood and behavior. Under most conditions, this interaction allows appropriate immune responses to physiological stressors. However, when inappropriate responses occur, major psychiatric sequelae may emerge (Dantzer et al., 2008). Similar to immune-related psychiatric changes, the hypothalamic pituitary adrenal (HPA) response occurring during immune activation and following psychological stress is intimately related to mental wellbeing (Capuron et al., 2006; Dantzer et al., 2008). Depression is a prime example. Major depression, the most common psychiatric disorder in the United States, presents more often as a co-morbidity to other illnesses rather than alone (Kessler et al., 2003). Major depression is frequently associated with elevated corticosteroid levels (Pace et al., 2007; Pariante, 2009, 2006) and elevated pro-inflammatory cytokines (Zorrilla et al., 2001). Defining the effect of inflammation and glucocorticoids on the brain should aid in the development of new treatments for depression and other psychiatric disorders.
Physiological challenges such as infection, cancer, heart disease or perceived psychological stress activate the immune system and increase HPA axis activity (McEwen et al., 2015). In turn, resultant pro-inflammatory cytokine and adrenocortical secretions induce the Kyn Pathway (Lawson et al., 2016). Accumulating evidence implicates increased levels of tryptophan (Trp) metabolites (kynurenines) in precipitating or mediating psychiatric symptomologies (Bradley et al., 2015; McCusker et al., 2014; Miura et al., 2008; Myint et al., 2012; O’Farrell and Harkin, 2017; Savitz, 2016; Schwieler et al., 2016). Trp availability per se does not appear to limit the Kyn Pathway in vivo as Trp concentrations remain sufficiently high in extracellular fluids (Badawy, 2015; Dantzer, 2016). Instead, Trp entry into the Kyn Pathway is governed by indoleamine/tryptophan 2,3-dioxygenases (DO’s: Ido1, Ido2 and Tdo2). It is currently accepted that Ido1 and Ido2 are upregulated by pro-inflammatory cytokines, whereas Tdo2 is controlled by glucocorticoids (Lawson et al., 2016). Although inflammatory cytokines and glucocorticoids may individually contribute to disease morbidity and mortality via DO upregulation, interacting effects of inflammatory mediators and glucocorticoids on DO expression remain poorly defined.
Increased DO expression results in Trp → Kyn metabolism and production of downstream kynurenines. An underappreciated aspect of this field is the cell-specific generation of unique kynurenines (McCusker et al., 2014). In the brain, quinolinic acid (QuinA) generated predominantly by microglia is a glutamate receptor (NMDA-R) agonist. Kynurenine acid (KynA) from neurons and astrocytes is a NMDA-R antagonist (Lawson et al., 2016). In addition to neuromodulation, kynurenines are also immunomodulatory largely by the ability of Kyn and KynA to suppress the immune response (Lawson et al., 2016). Thus, an imbalance in kynurenines is implicated in various illnesses including neurodegenerative disorders (Souza et al., 2016), autoimmune diseases (Merlo et al., 2017), cancer, (Zhai et al., 2017, 2015) schizophrenia and depression (Bradley et al., 2015; McCusker et al., 2014; Miura et al., 2008; Myint et al., 2012; O’Farrell and Harkin, 2017; Savitz, 2016; Schwieler et al., 2016).
Ido1 and Ido2 genes are separated by < 8,000 bp’s on chromosome 8 (Ball et al., 2007) while Tdo2 resides on murine chromosome 3 (Kanai et al., 2009a). It was unclear why three DO’s evolved to perform the same activity until the realization that multiple genes driven by distinct promoters endow the ability to regulate the Kyn Pathway in a cell/tissue-specific manner. Ball et al. suggested that two murine Ido2 protein bands were due to post-translational modification (Ball et al., 2007). At the same time, Metz et al. described the presence of mRNA variants for human IDO2 (Metz et al., 2007). Ball then reported Ido1 and Ido2 mRNA isoforms encoding different sized proteins (Ball et al., 2009) thereby linking the existence of mRNA isoforms to alternate protein products. The use of alternate promoters was proposed as a means to generate the full-length (FL) and truncated (variant) isoforms differing with the choice of exon used to initiate transcription. It wasn’t until 2014 that physiological attributes were linked to DO isoforms (Metz et al., 2014). Ido2 isoforms differing in the splicing of an internal exon were transcribed into FL and Δ4 proteins with ~8-fold differences in enzymatic activity. We reported that mRNA isoforms for Ido1, Ido2 and Tdo2 are expressed with both alternate choice of first exons (Class 1 and 2) and alternate splicing of internal exons (Brooks et al., 2017, 2016a, 2016b; Dostal et al., 2017); see supplementary Tables S1–S4 for isoform and qPCR assay descriptions.
The class 1 reference Ido1 (Ido1-FL) and Ido2 (Ido2-FL) transcripts are basically undetectable in naïve mouse brain and glia, albeit highly inducible (André et al., 2008; Ball et al., 2007; Brooks et al., 2016b; Browne et al., 2012; Croitoru-Lamoury et al., 2011; Dostal et al., 2017; Heisler and O’Connor, 2015; Henry et al., 2009; O’Connor et al., 2009c; Park et al., 2011; Parrott et al., 2016a; Salazar et al., 2012; Wang et al., 2010b). However, the naïve mouse brain has intrinsic Ido activity (Fu et al., 2010; Fujigaki et al., 1998; Lestage et al., 2002; Saito et al., 1992). By contrast, Ido1 and Ido2 mRNA are readily detected with assays that simultaneously quantify all isoforms, i.e. ‘Tot’ Ido expression (Brooks et al., 2017, 2016a, 2016b; Browne et al., 2012; Dostal et al., 2017; Fuertig et al., 2016; Gibney et al., 2014; Heisler and O’Connor, 2015; Liu et al., 2015; Park et al., 2011; Parrott et al., 2016a). Thus, Ido1 enzymatic activity in brain is presumably derived from Ido1 protein encoded by variant mRNA isoforms. The presence of variant Ido1 isoforms was initially illustrated with PCR assays that simultaneously quantify all Ido1 isoforms; i.e. Ido1-Tot (Fuertig et al., 2016; Gibney et al., 2014; Liu et al., 2015; Park et al., 2011). Likewise, the class 1 reference Ido2-FL expression is low in the naïve mouse brain (Ball et al., 2007; Croitoru-Lamoury et al., 2011). Ido2-Tot is more readily detected (lower Ct) (Brooks et al., 2016b) and more frequently reported (Browne et al., 2012; Fuertig et al., 2016; Heisler and O’Connor, 2015; Park et al., 2011; Parrott et al., 2016a) because Ido2-Tot assays detect both FL and variant (v) isoforms. Tdo2 isoforms (Tdo2-FL, Tdo2-v1 and Tdo2-v2) are differentially expressed and regulated (Brooks et al., 2017, 2016a, 2016b; Dostal et al., 2017), but studies reporting Tdo2-Tot are overrepresented in the literature (Fuertig et al., 2016; Gibney et al., 2014; Heisler and O’Connor, 2015; Park et al., 2011; Parrott et al., 2016a). Thus, quantifying a single DO isoform does not necessarily reflect mRNA levels or enzymatic activity. Importantly, our work highlights isoform-specific DO changes routinely missed with non-specific ‘Tot’ assays suggesting unique and unappreciated roles for the individual DO transcripts (Brooks et al., 2017, 2016a, 2016b; Dostal et al., 2017).
Data generated with preclinical animal models demonstrate that inflammation- or stress-induced depression-like motivated behaviors and cognitive deficits are DO-dependent (Dugan et al., 2016; Gibney et al., 2014; Heisler and O’Connor, 2015; O’Connor et al., 2009b, 2009c; Salazar et al., 2012). Thus, we probed the interacting effects of LPS (to model an acute inflammatory response) and DEX (to model glucocorticoid receptor activation by acute stress) as a first step towards relating specific mRNA isoforms to DO-dependent changes in physiology and behavior.
2. Materials and Methods
2.1. Animals
Male C57BL/6J mice (Bar Harbor, ME, USA), kept on a reversed 12 h light-dark cycle with ad libitum access to food and water, were individually housed for 3 weeks prior to experiments. Mice were handled for 5 consecutive days prior to treatment and were 15 weeks of age at the time of treatment. All animal procedures were approved by the Institutional Animal Care and Use Committee and performed in accordance with the Guide for the Care and Use of Laboratory Animals (National Research Council).
2.2. Intraperitoneal (i.p.) treatments
Mice were given a single injection prior to onset of the dark cycle of either saline (Control), LPS (0.83 mg/kg, serotype 0127:B8, Sigma), DEX (10 mg/kg, 11695-4013-1, Henry Shein) or both LPS+DEX. Doses were chosen for their ability to alter cytokine expression, activate brain glucocorticoid receptors and induce depression-like behavior (O’Connor et al., 2009c; Salazar et al., 2012; Schinkel et al., 1995; Wróbel et al., 2014). Mice were euthanized by CO2 asphyxiation 5 h after treatment since LPS-induced Ido1-FL expression is optimal near this time point (André et al., 2008).
2.3. Glia enrichment (cohort 1)
Five hours after treatment and following intracardial perfusion with PBS containing 2 mM EDTA, brains were removed and gently homogenized. Whole brain homogenates were sampled prior to the enrichment of astrocytes and microglia by magnet-assisted cell separation (Miltenyi Biotec Inc.). Brain homogenate, microglia and astrocyte samples were suspended in TRIzol and stored at −80 °C until processed for gene expression. This method provides highly enriched viable cell populations (Dostal et al., 2017; Jungblut et al., 2012; Matt et al., 2016; Nikodemova and Watters, 2012). Astrocyte and microglia enrichment was further verified by comparing Glast1 and Cd11b mRNA expression. Consistent with our previous work, Glast1 expression is greater in enriched astrocytes compared to brain (6.0±0.3 fold, p<0.001) and Cd11b expression is greater in microglia compared to brain (197±12 fold, p<0.001).
2.4. Tissue collection (cohort 2)
Five hours after treatment, blood was collected prior to intracardial perfusion. Brain, liver, lung and spleen were harvested then stored at −80 °C. Brain and plasma were processed for Kyn content (Dostal et al., 2017). Peripheral tissues were processed for gene expression.
2.5. Gene expression by quantitative polymerase chain reaction (qPCR)
Methods for RNA extraction, reverse-transcription and qPCR analysis were described in detail (Dostal et al., 2017). Within each tissue or cell-type, the expression of each test gene was normalized to Gapdh using the 2−ΔΔCt method, Ct = cycle threshold (Livak and Schmittgen, 2001). Expression in the Control group was set to 1.0 and other groups expressed as fold of Control. DO gene structure, mRNA transcripts and PCR assays are found in supplementary Tables S1–S4. Custom qPCR assays were designed using the IDT PrimerQuest® Design Tool and probe-based assays were purchased from IDT (Coralville, Iowa). Because of low expression, some transcripts are ‘not detectable’ in all samples after amplification (Ct values ‘undetermined’). For analysis, a Ct value of 40 is assigned when this occurs. Frequency of this happening, raw data and n for each treatment combination are in supplementary Tables S5–S10.
2.6. HPLC
To measure Kyn, brain and plasma samples were analyzed by high pressure liquid chromatography (HPLC) with electrochemical detection using methods recently described in detail (Dostal et al., 2017). All HPLC runs resulted in standard curves with r2 ≥ 0.95. The average coefficients of variation within and between assays were ≤ 13 %.
2.7. Statistics
Data are presented as mean ± SEM representing 4–6 mice per treatment group. Following normality testing, 2-way ANOVA was performed for all data. Data that are not normally distributed were statistically analyzed after log-transformation. Significance was set at p ≤ 0.05. In the absence of a statistical interaction between treatments, significant main effects of LPS or DEX are annotated with * or ϕ, respectively. In the presence of a significant statistical interaction between LPS and DEX, data are annotated with δ to represent significant effects by post hoc analysis using the Holm-Šídák method for multiple comparisons. Graphic presentation and statistical analysis were completed using CorelDraw Graphics Suite and SigmaPlot.
3. Results
To confirm treatment effectiveness associated with this short treatment interval, changes in body weight and food intake were quantified. Both LPS and DEX reduced body weight, but only LPS reduced 5 h food intake (Table 1).
Table 1.
LPS and DEX effects on body weight and food intake.
| Control | LPS | Dex | LPS+DEX | |
|---|---|---|---|---|
|
|
|
|
|
|
| Change in body weight (g) | 0.2 ± 0.1 | −1.3±0.1 | −0.2±0.1 | −2.2±0.2*,ϕ |
| Food intake (g) | 1.3 ± 0.1 | 0.2 ± 0.2 | 1.6 ± 0.2 | 0.1 ± 0.02* |
p < 0.05 for LPS,
p < 0.05 for DEX.
3.1. DEX is anti-inflammatory in peripheral tissues, not brain, when co-administered with LPS
Fkbp5 expression was assessed to confirm glucocorticoid-receptor (GR) mediated responses. Fkbp5 is well expressed by all 4 tissues (Fig. 1A, Ct of Controls (Ctrl/−) presented above each graph). Fkbp5 expression was increased by LPS, indicating HPA axis activation. DEX increased Fkbp5 expression by all 4 tissues. LPS caused a smaller increase in Fkbp5 expression when compared to DEX. LPS+DEX did not result in greater Fkbp5 within peripheral tissues compared to DEX, suggesting saturation of the peripheral GR response by DEX alone.
Figure 1. DEX is anti-inflammatory in peripheral tissues when co-administered with LPS.

Brain, liver, lung and spleen were obtained from mice 5 h after treatment with saline (Control), LPS, DEX, or LPS+DEX to assess HPA activity by quantifying (A) Fkbp51 expression and to assess an inflammatory response by quantifying (B) Ifnγ and (C) Tnfα expression. * p ≤ 0.05 main effect of LPS. ϕ p ≤ 0.05 main effect of DEX. δ p ≤ 0.05 post hoc mean separation (brackets) in the presence of a significant LPS x DEX interaction.
Cytokine expression was quantified to verify the relative abilities of LPS and DEX to modulate an inflammatory response. Ifnγ was poorly expressed in the mouse brain and not induced by LPS or DEX. Ifnγ expression was highest in spleen (Fig. 1B, see Control Ct values). The increase in Ifnγ expression caused by LPS was abrogated by DEX in all three peripheral tissues. Tnfα expression was increased by LPS in brain, liver, lung and spleen (Fig. 1C). LPS-induced Tnfα was significantly attenuated by DEX for all three peripheral tissues, but DEX did not reduce Tnfα in brain.
3.2. Kyn levels increase in brain following LPS and in plasma following DEX
Kyn was quantified to confirm Kyn Pathway activation. Plasma Kyn was elevated 5 h after DEX administration (Fig. 2A). LPS did not increase plasma Kyn, but prevented the increase triggered by DEX. In contrast, brain Kyn was doubled by LPS (Fig. 2B). DEX decreased brain Kyn and tempered the increase produced by LPS.
Figure 2. Kyn levels increase in brain following LPS and in plasma following DEX.
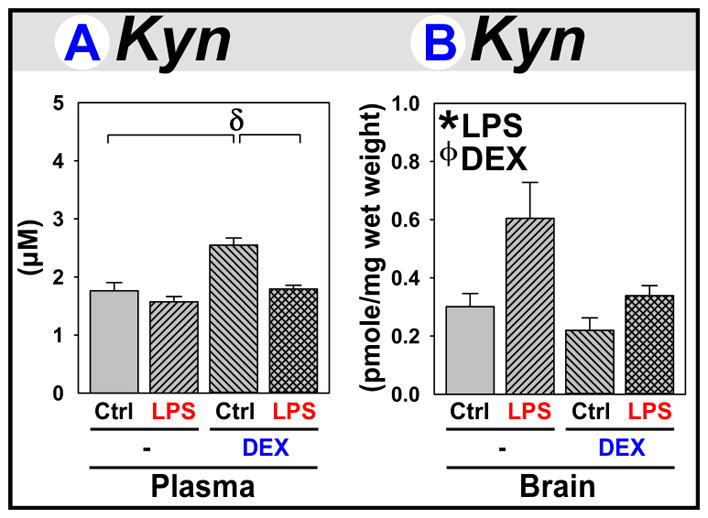
(A) Plasma and (B) brains were obtained from mice 5 h after treatment with saline (Control), LPS, DEX, or LPS+DEX to assess Kyn levels. * p ≤ 0.05 main effect of LPS. ϕ p <≤ 0.05 main effect of DEX. δ p ≤ 0.05 post hoc mean separation in the presence of a significant LPS x DEX interaction.
3.3. LPS and DEX differentially regulate Ido1 expression in brain, glia and peripheral tissues
Expression of the reference class 1 full-length Ido1 transcript (Ido1-FL), two class 2 mRNA isoforms (Ido1-v1 and Ido1-v2; class based on choice of first exon) and their combined expression (Ido1-Tot) were quantified (see supplementary materials for class designation, treatment summaries and raw data).
Ido1-FL (class 1) is poorly expressed in naïve brain, astrocytes and microglia (Fig. 3A, see Control Ct values), but strongly induced by LPS. DEX did not reduce Ido1-FL expression in these samples. In the periphery, Ido1-FL is expressed best by lungs, followed by spleens and livers from naïve mice (see Control Ct values). LPS increased Ido1-FL expression in liver, lung and spleen. Ido1-FL induction was significantly reduced by DEX in all three peripheral tissues. Thus, LPS-induced Ido1-FL is abrogated by DEX in peripheral tissues, but not in whole brain, astrocytes or microglia.
Figure 3. LPS and DEX differentially regulate Ido1 expression in brain, glia and peripheral tissues.

Brain, astrocytes, microglia, liver, lung and spleen were obtained from mice 5 h after treatment with saline (Control), LPS, DEX or LPS+DEX to assess the relative changes in the mRNA expression of (A) Ido1-FL, (B) Ido1-v1, (C) Ido1-v2 and (D) Ido1-Tot. * p ≤ 0.05 main effect of LPS. ϕ p ≤ 0.05 main effect of DEX. δ p ≤ 0.05 post hoc mean separation in the presence of a significant LPS x DEX interaction.
Ido1-v1 (class 2) is the only Ido1 isoform appreciably expressed in naïve mouse brains (Fig. 3B, Control Ct’s) where expression was unaltered by LPS or DEX. However, LPS increased Ido1-v1 expression in astrocytes, but not microglia. Ido1-v1 expression by astrocytes, and less so by microglia, was DEX-inducible. Elevated Ido1-v1 expression by glia without noteworthy changes in brain might seem inconsistent. However, Ido1-v1 expression in Control astrocytes and microglia is lower than brain (a brain Ct of 33.1 represents ~13-fold greater expression compared to the astrocyte Ct of 36.8 and ~42-fold greater compared to a microglia Ct of 38.5). These data indicate that most of the Ido1-v1 in brain is not derived from these LPS- and DEX-sensitive glia, but from an LPS- and DEX-insensitive cell-type(s). In peripheral tissues, LPS only increased Ido1-v1 in the lung where its induction was attenuated by DEX. LPS slightly decreased splenic Ido1-v1. Thus, DEX increased Ido1-v1 expression within astrocytes and microglia, but not in whole brain from which they were derived, indicating that detecting relevant changes in Ido1-v1 may require examination of each tissue’s component cell-types.
Ido1-v2 (class 2) expression (Fig. 3C) was essentially absent in brain, astrocytes, microglia, liver or spleen. Only in lung was Ido1-v2 strongly induced by LPS where its expression was attenuated by DEX. Thus, lung was unique in its ability to express Ido1-v2.
Ido1-Tot expression (Fig. 3D) detects all three Ido1 isoforms. Ido1-Tot in Control brains reflects expression of Ido1-v1 (Figs. 3B vs. 3D, Control Ct’s both ≈ 33). In brain, LPS increases Ido1-Tot expression ~10-fold, a marked attenuation relative to the ~1,000-fold increase in Ido1-FL. Thus, the fold-change in Ido1-FL overestimates the change in total brain Ido1. Moreover, the ability of DEX to increase Ido1 expression by astrocytes and microglia is overlooked when quantifying Ido1-Tot since only glial Ido1-v1 is affected by DEX. In liver, Ido1-Tot expression mimics Ido1-FL as only this isoform is induced by LPS and inhibited by DEX. In lung there are no differentially regulated isoforms; therefore, Ido1-Tot expression reflects the sum of all 3 isoforms. In the spleen, the Ido1 isoforms are differentially responsive to treatment; changes in Ido1-Tot reflect LPS-induced Ido1-FL as tempered by reduced Ido1-v1.
3.4. LPS and DEX regulate Ido2 transcripts in a manner distinct from Ido1
The reference transcript Ido2-FL, eight variants (Ido2-v1 thru v8) and their combined expression (Ido2-Tot) were quantified to define cell- and tissue-specific profiles. Ido2-v3 and Ido2-v7 are further differentiated based on alternate splicing of exons 5 and 4, respectively; thus, these transcripts may occur as either class 1 (Ido2-v3.1, Ido2-v7.1) or class 2 (Ido2-v3.2, Ido2-v7.2) isoforms (see Ido2 isoform description in Table S2) based on alternate choice of the first exon.
Class 1 Ido2 isoforms (Ido2-FL, Ido2-v3.1, Ido2-v4, Ido2-v7.1 and Ido2-v8) were readily detected only in liver. Hepatic expression was decreased by LPS, independently of DEX (Fig. 4A–E). Based on Control Ct values, Ido2-FL is the major hepatic isoform, Ido2-v3.1 (encoding a protein with low enzymatic activity) and Ido2-v7.1 (containing an early stop-codon, see Table S2) are detected at a lower level.
Figure 4. LPS and DEX regulate Ido2 transcripts in a manner distinct from Ido1.

Brain, astrocytes, microglia, liver, lung and spleen were obtained from mice 5 h after treatment with saline (Control), LPS, DEX or LPS+DEX to assess the relative changes in the mRNA expression of (A) Ido2-FL, (B) Ido2-v3.1, (C) Ido2-v4, (D) Ido2-v7.1, (E) Ido2-v8, (F) Ido2-v1, (G) Ido2-v2, (H) Ido2-v3.2, (I) Ido2-v5, (J) Ido2-v6 and (K) Ido2-Tot. * p ≤ 0.05 main effect of LPS. ϕ p ≤ 0.05 main effect of DEX. δ p ≤ 0.05 post hoc mean separation in the presence of a significant LPS x DEX interaction.
In contrast to the hepatic specificity of class 1 isoforms, class 2 Ido2 isoforms were detected in brain, astrocytes, microglia and all 3 peripheral tissues:
Ido2-v1 (class 2) expression is greater in brain compared to astrocytes, microglia and peripheral tissues (Fig. 4F, see Control Ct values), but Ido2-v1 expression by brain is unchanged by LPS or DEX. However, LPS increases Ido2-v1 expression in astrocytes and microglia. The LPS induction of glial Ido2-v1 is not sufficient to change whole brain expression because of lower basal expression levels of glia, indicating that a non-glial LPS/DEX-insensitive cell-types (probably neurons (Fukunaga et al., 2012)) must be the primary source of brain Ido2-v1 (mimicking Ido1-v1). In the periphery, LPS increases expression of Ido2-v1 in liver, lung and spleen. DEX abrogates the LPS effect in lung, but synergizes with LPS to further elevate Ido2-v1 in spleen. Thus, Ido2-v1 expression is LPS and DEX responsive, but in a cell-type and tissue-specific manner.
Ido2-v2 (class 2) basal expression is similar in brain, astrocytes, microglia and liver, all with greater expression than lung or spleen (Fig. 4G, see Control Ct values), indicating that glia are a major source of central Ido2-v2. Ido2-v2 expression in brain was not changed by either LPS or DEX, but slightly elevated by LPS+DEX. This induction is not evident within astrocytes or microglia. By contrast, Ido2-v2 expression is diminished in liver by LPS, unchanged in lung, but increased in spleen by LPS and DEX. Thus, Ido2-v2 expression is relatively stable in brain and glia, but regulation in the periphery is tissue-specific.
Ido2-v3.2 (class 2) is better expressed by brain when compared to astrocytes and microglia, and in the periphery its expression is greatest in the spleen then lung (Fig. 4H, Ct values). These data illustrate that neither astrocytes nor microglia are the major source of central Ido2-v3.2. Ido2-v3.2 expression is slightly increased by LPS in brain and by LPS+DEX in microglia, but not astrocytes. LPS increases Ido2-v3.2 expression in lung and spleen; DEX blocks the induction in lung. Thus, Ido2-v3.2 expression is relatively stable in brain and astrocytes, albeit inducible in microglia, lung and spleen.
Ido2-v5 (class 2) was only detected in brain, where it was increased by LPS (Fig. 4I).
Ido2-v6 (class 2) expression is greatest in the brain, but poorly expressed in astrocytes, microglia, liver, lung and spleen (Fig. 4J, Ct values). These data indicate that most of the Ido2-v6 in brain is not within these glia. Ido2-v6 expression in brain and microglia is unchanged by treatments, whereas astrocyte Ido2-v6 expression is increased by LPS and DEX. LPS also increases Ido2-v6 expression in liver, lung and spleen. DEX abrogated LPS-induced expression in liver and lung, but by itself DEX increased Ido2-v6 expression in spleen. Thus, Ido2-v6 expression is relatively stable in brain and microglia, albeit inducible in astrocytes; but LPS and DEX responsiveness in the periphery occurs in a tissue-specific manner.
Ido2-Tot expression (Fig. 4K) represents the integrated effects of LPS or DEX on Ido2 isoforms. Alone however, Ido2-Tot does not adequately define the elaborate isoform-specific regulation of Ido2 by LPS and DEX. For example, Ido2-Tot does not reflect the ability of LPS to induce Ido2-v1 and Ido2-v6 in astrocytes or Ido2-v1 in liver. Like Ido1, Ido2 expression is likely cell-type specific within each peripheral tissue. Work similar to our comparison of brain vs. astrocyte vs. microglia is needed to adequately define Ido2 regulation.
3.5. DEX upregulates Tdo2-FL while inflammation regulates Tdo2-v1 and Tdo2-v2
Tdo2 expression is widely viewed as glucocorticoid-inducible. Thus, we quantified the expression of the reference isoform Tdo2-FL, variant isoforms (Tdo2-v1, Tdo2-v2) and their combined expression (Tdo2-Tot).
Class 1 Tdo2-FL is well expressed by brain and astrocytes, less so by microglia. However, expression is highest in liver and lowest in spleen (Fig. 5A, Ct values). Tdo2-FL increases in brain and astrocytes following DEX administration, but not in microglia (p=0.054). Tdo2-FL also increases after DEX administration in all three peripheral tissues. LPS only induces Tdo2-FL expression in liver. Thus, Tdo2-FL expression is glucocorticoid-inducible across tissues and in astrocytes.
Figure 5. DEX upregulates Tdo2-FL while inflammation regulates Tdo2-v1 and Tdo2-v2.

Brain, astrocytes, microglia, liver, lung and spleen were obtained from mice 5 h after treatment with saline (Control), LPS, DEX or LPS+DEX to assess the relative changes in the mRNA expression of (A) Tdo2-FL, (B) Tdo2-v1, (C) Tdo2-v2 and (D) Tdo2-Tot. * p ≤ 0.05 main effect of LPS. ϕ p ≤ 0.05 main effect of DEX. δ p ≤ 0.05 post hoc mean separation in the presence of a significant LPS x DEX interaction.
Class 2 Tdo2-v1 and Tdo2-v2 expression are similarly regulated. Tdo2-v1/v2 are best expressed by the liver, next highest in astrocytes, then slightly lower in microglia, lung, brain and spleen (Fig. 5B–C, Ct values). Thus, astrocytes and microglia are both major sources of Tdo2-v1/v2 in brain. Tdo2-v1/v2 expression is unchanged by LPS and DEX in brain, astrocyte and microglia, but regulated in a tissue-specific manner within the periphery. Thus, Tdo2-v1/v2 expression by liver and lung is glucocorticoid-inducible (not brain or spleen), but their expression is LPS sensitive in liver and spleen (not brain or lung).
Tdo2-Tot (Fig. 5D) expression represents a composite of the three Tdo2 isoforms. In brain, Tdo2-Tot is not changed by LPS or DEX. When quantifying Tdo2-Tot, the stimulatory effect of DEX on brain Tdo2-FL is overlooked because the Tdo2-Tot assay also detects two unresponsive isoforms. With astrocytes, Tdo2-Tot expression reflects Tdo2-FL induction by astrocytes and unchanged Tdo2 expression in microglia. In liver, all Tdo2 isoforms are induced by both LPS and DEX; this is reflected by Tdo2-Tot. Similarly, in lung all 3 isoforms are DEX inducible, as is Tdo2-Tot. However, in spleen Tdo2-Tot expression is problematic. Splenic Tdo2-Tot is not significantly changed by LPS or DEX, overlooking the ability of DEX to induce Tdo2-FL and LPS to increase Tdo2-v1/v2.
3.6. Downstream Kyn Pathway enzyme regulation by LPS, DEX and LPS+DEX
Kyn production is governed by the DO’s, but Kyn is then metabolized into immune- and neuro-modulatory kynurenines associated with various pathologies. Kynurenine aminotransferase 2 (Kat2) converts Kyn into KynA, whereas kynurenine 3-monooxygenase (Kmo), kynureninase (Kynu) and 3-hydroxyanthranilate 3,4-dioxygenase (Haao) act to convert Kyn into QuinA. These downstream enzymes are expressed with tissue- and glia-specificity.
Kat2 expression is largely unchanged albeit slightly induced in the liver by DEX (Fig. 6A). Kat2 expression is ~10-fold higher (3.5 lower Ct) in astrocytes compared to microglia, reflecting the ability of astrocytes to produce KynA. Kat2 is also highly expressed in liver supporting a well-developed ability for hepatic KynA production.
Figure 6. Downstream Kyn Pathway enzyme regulation by LPS and DEX.

Brain, astrocytes, microglia, liver, lung and spleen were obtained from mice 5 h after treatment with saline (Control), LPS, DEX or LPS+DEX to assess the relative changes in the mRNA expression of (A) Kat2, (B) Kmo, (C) Kynu and (D) Haao. * p ≤ 0.05 main effect of LPS. ϕ p ≤ 0.05 main effect of DEX. δ p ≤ 0.05 post hoc mean separation in the presence of a significant LPS x DEX interaction.
Kmo, Kynu and Haao (Fig. 6B–D) expression in brain is relatively stable, albeit Haao is decreased slightly by LPS. Surprisingly, microglial expression of all three enzymes is diminished by LPS and DEX. Haao expression by astrocytes is also diminished by DEX. Microglial expression of these three enzymes is greater than that of astrocytes (see Control Ct values), reflecting microglial ability to generate QuinA. Kmo, Kynu and Haao are best expressed by the liver compared to the other 3 tissues, reflecting hepatic ability to generate QuinA and then NAD+ from Kyn. In liver, LPS increases Kmo, but both LPS and DEX decrease Kynu and Haao expression; since Kmo acts upstream of Kynu its action would be offset by lowered Kynu. In lung, Kmo is induced by LPS, but remains unchanged in spleen.
4. Discussion
Immunogenic and psychological stress and their associated changes in cytokine production and HPA axis function result in DO-dependent changes in immunophysiology, motivated behaviors and cognition (Dugan et al., 2016; Fuertig et al., 2016; Gibney et al., 2014; Heisler and O’Connor, 2015; O’Connor et al., 2009b, 2009c; Salazar et al., 2012; Talarowska and Galecki, 2016). Although inflammatory mediators affect HPA axis function and vice versa (Wang and Dunn, 1999), only recently have we started to define how inflammation and stress-related responses interact to control DO expression. The DO’s are regulated with remarkable specificity, illustrated by intricate interactions between inflammatory mediators and glucocorticoids on DO expression ex vivo using organotypic hippocampal slice cultures (Brooks et al., 2017, 2016a, 2016b). We extend those findings by describing interacting pro-inflammatory and glucocorticoid-receptor-mediated effects on Kyn Pathway activity and DO expression in vivo.
4.1. DEX is anti-inflammatory in peripheral tissues when co-administered with LPS
Fkbp5 induction was used as an index of glucocorticoid receptor (GR) activation. LPS-induced Fkbp5 reflects inflammation-induced HPA-axis activation (Browne et al., 2012; Wang and Dunn, 1999). DEX had greater effects on Fkbp5 than LPS, illustrating greater GR activation by the GR-agonist. LPS also induces a pro-inflammatory response, confirmed here by increased cytokine expression. DEX acted as an anti-inflammatory agent, blocking Ifnγ and Tnfα induction in peripheral tissues, but not brain (Fig. 1). Thus, DEX is active in the brain, but has stronger anti-inflammatory action in the periphery. DO expression will be affected directly by LPS → cytokines and by DEX → GR-activation in both the brain and periphery. DEX will alter central DO expression indirectly by limiting the peripheral, but not central, inflammatory response.
4.2. Kyn levels increase in plasma following DEX and in brain following LPS
Changes in Kyn concentrations are used to demonstrate functional alterations of Trp entry into the Kyn Pathway historically attributed to Ido1 induction. Here, DEX increased plasma Kyn (Fig. 2) confirming previous reports that GR activation is sufficient to increase circulating Kyn (Berry and Smythe, 1963; Saito et al., 1994; Young, 1981), but DEX did not change peripheral Ido1 expression. DEX did increase spleen Ido2, as well as liver, lung and spleen Tdo2 expression. The mechanism by which LPS attenuated the DEX effect on plasma Kyn is currently unclear, but our data suggest that peripheral Ido2 and/or Tdo2 are involved in GR-mediated elevations in circulating Kyn.
Since Kyn crosses the blood-brain barrier (Heisler and O’Connor, 2015), elevated plasma Kyn would expectedly increase Kyn levels in the brain; which was not the case following DEX. Instead, brain (Fig. 2, (Guo et al., 2016)) but not plasma Kyn levels are elevated 5 h after peripheral LPS. We had previously shown that neuroinflammation without peripheral inflammation increases Kyn levels in brain, not plasma (Lawson et al., 2013). These data illustrate inflammation-dependent production of Kyn within the brain initiated by LPS. This action is likely mediated by cytokines increasing Ido1 and Ido2 expression within both astrocytes and microglia (Figs. 3–4).
Both plasma and brain Kyn levels are elevated 24 h after LPS treatment (Fujigaki et al., 2001; Larkin et al., 2016; O’Connor et al., 2009c; Parrott et al., 2016a, 2016b). These findings reflect acute/subacute effects of neuroinflammation on central Kyn production along with LPS activation of the HPA axis and peripheral Kyn production. Acute psychological stress also increases both plasma and brain Kyn (Dostal et al., 2017; Kennett and Joseph, 1981; Pawlak et al., 2000). However, DEX increased only plasma Kyn (Fig. 2). One difference is stress caused neuroinflammation (Dostal et al., 2017), DEX did not (Fig. 1). Although limited to a single time point (5 h) and metabolite (Kyn), we suggest that LPS and psychological stress act via cytokines to mediate acute and subacute increases in brain Ido1/2 expression and Kyn production, whereas DEX and stress act via GR activation to mediate acute increases in peripheral Ido2/Tdo2 and Kyn production.
4.3. LPS and DEX differentially regulate Ido1 expression in brain, glia and peripheral tissues
Inflammation and stress induce depression-like behaviors. Importantly, increased brain Ido1-FL (Heisler and O’Connor, 2015; O’Connor et al., 2009c; Park et al., 2011; Salazar et al., 2012) and Ido1-Tot (Agudelo et al., 2014; Liu et al., 2015; Park et al., 2011) expression are associated with these behaviors. While stress enhances LPS-driven depression-like behavior (Couch et al., 2016), interactions between inflammation- and glucocorticoid-dependent Ido1 expression are poorly defined.
Ido1 isoforms can be generated from a single pre-mRNA containing exons 1 thru 11 with alternate splicing to retain either exon 1 (class 1) or 2 (class 2) as the first exon within the mature mRNA. Alternatively, generating pre-mRNA using distinct promotors to initiate transcription at either exon 1 or 2 would also generate isoforms. Both processes allow for generation of class 1 and 2 transcripts, but the latter allows for differential expression of the transcripts in response to varying factors as found in the current work. Clearly the naïve mouse brain expresses only class 2 Ido1-v1, whereas naïve lung and spleen express class 1 and class 2 isoforms.
Class 1 Ido1-FL is poorly expressed in naïve mouse brains, astrocytes and microglia, but central Ido1-FL is inducible by immune activation with complete Freund’s adjuvant (Kim et al., 2012), BCG (O’Connor et al., 2009a, 2009b), pI:C (Brooks et al., 2016a) and LPS (Fig. 3, (André et al., 2008; Brooks et al., 2016a; Browne et al., 2012; Godbout et al., 2008; Heisler and O’Connor, 2015; Henry et al., 2009; Martin et al., 2014; O’Connor et al., 2009c; Parrott et al., 2016a)). While several reports describe Ido1 induction by microglia (Brooks et al., 2017; Henry et al., 2009; Wang et al., 2010a), our data show astrocytes responding just as robustly to LPS as microglia. Class 2 Ido1-v1 is the major isoform expressed in naïve brain (Brooks et al., 2016b; Dostal et al., 2017), indicating that Ido1-v1 is the only isoform available to generate central Ido1 activity of unstimulated brain (Fu et al., 2010; Fujigaki et al., 1998; Lestage et al., 2002; Saito et al., 1992). Herein we demonstrate that Ido1-v1 is induced by LPS and DEX within astrocytes. Also, Ido1-v1 (not Ido1-FL) was induced by stress within astrocytes, not microglia (Dostal et al., 2017) indicating that Ido1-v1 expression by astrocytes is cytokine- and glucocorticoid-sensitive. Reports also demonstrate that inflammation (Parrott et al., 2016b) and stress (Dugan et al., 2016; Notarangelo and Schwarcz, 2017; Pawlak et al., 2000) increase brain KynA, consistent with functional changes in the brain mediated by astrocytes (Lawson et al., 2016). Current dogma suggests that inflammation increases microglial Ido1 with resultant behavioral changes (McCusker et al., 2014; Myint, 2012). Based on the current findings, astrocyte involvement in Ido1-dependent behaviors warrants re-evaluation.
We recently reported that DEX potentiated cytokine-induced Ido1-FL expression by organotypic hippocampal slice cultures (Brooks et al., 2016b). Since i.p. LPS does not appreciably access the brain, central Ido1-FL induction by inflammatory stimuli is accepted to be mediated by cytokines, especially IFNγ and TNFα (O’Connor et al., 2009a). Thus, DEX might be expected to elevate LPS-induced Ido1-FL in vivo. However, brain, astrocyte and microglial LPS-induced Ido1-FL are not further increased by DEX (Fig. 3A). This discrepancy is easily explained. Ex vivo, the cytokine concentration is set by the investigator and DEX potentiates the ability of a set cytokine level to induce Ido1-FL expression (Brooks et al., 2017, 2016a, 2016b). However, DEX decreases peripheral cytokine expression, resulting in less cytokine input to the brain. But, DEX did not attenuate central TNFα expression, thus central expression of Ido1-FL remains elevated; presumably by a synergism between DEX and central cytokines. Ido1-FL expression is increased by LPS and diminished by DEX in all three peripheral tissues, as is Ido1-v1 in lung. Within the periphery, DEX attenuates IFNγ and TNFα expression demonstrating a prototypical inflammatory/anti-inflammatory GR-mediated response. Thus, peripheral Ido1 is decreased by DEX because of the loss of peripheral cytokines.
Clearly, regulation of Ido1 expression in peripheral tissues is distinct from brain and glia (summarized in Table S1). The recognition that brain Kyn can be elevated without changes in plasma Kyn (Fig. 2) may help reconcile findings where plasma Kyn did not differ between normal and depressed subjects (Bradley et al., 2015; Hughes et al., 2012; Savitz et al., 2015). The Kyn Pathway may be elevated within the brain by the synergistic action between central cytokines and glucocorticoids on Ido1 expression, but Ido1 induction may be completely repressed in the periphery by glucocorticoids. Thus, an underappreciated interaction between low-grade inflammation and elevated glucocorticoids within the brain may drive DO-dependent clinical depression that is not reflected by circulating Kyn.
4.4. LPS and DEX regulate Ido2 transcripts in a manner distinct from Ido1
Four laboratories independently reported the existence of Ido2 (Ball et al., 2007; Metz et al., 2007; Murray, 2007; Yuasa et al., 2007). Like Ido1, Ido2 also controls Trp metabolism to Kyn (Trabanelli et al., 2014). Despite performing the same enzymatic role, Ido1 and Ido2 have non-redundant functions (Merlo et al., 2017, 2014), likely involving cell-type specific expression as reported within liver, kidney and epididymis (Ball et al., 2007). Similarly, myeloid- and plasmacytoid-dendritic cells both express Ido2, but Ido1 was detected only in myeloid-dendritic cells (Trabanelli et al., 2014). Thus, although sharing enzymatic function, Ido1 and Ido2 do not necessarily co-localize and can be differentially expressed with unique functional consequences.
Although the brain is frequently studied as a whole, a detailed evaluation of DO isoforms is needed to accurately assess mRNA changes because of regional-, cell- and isoform-specific regulation. For instance, LPS slightly increased hippocampal and frontal cortex Ido2-Tot expression (Browne et al., 2012; Park et al., 2011; Parrott et al., 2016a), although not within whole brain (Fig. 4), suggesting regional regulation. LPS and desipramine (an anti-depressant) interact to differentially modulate astrocyte and microglial Ido2 expression (Brooks et al., 2017), indicating isoform- and cell-type-specific responses. Indeed, Ido2-v1 is not increased by LPS or DEX in whole brain, but LPS induced Ido2-v1 expression in astrocytes and microglia (Fig. 4F), whereas only astrocyte Ido2-v6 expression was increased by LPS and DEX. Since expression of these isoforms in whole brain is largely unaffected by LPS or DEX, another non-responsive cell-type(s) must also express these Ido2 isoforms. Overall, changes in Ido2 expression are cell-type-specific and not necessarily evident by analysis of the resident tissue.
Of the nine Ido2 isoforms, eight were detected within the liver (Fig. 4). Here two class 2 Ido2 transcripts were increased and one was decreased by LPS, whereas LPS decreased all five class 1 Ido2 isoforms (reflected by decreased Ido2-Tot). The decrease in hepatic Ido2-Tot expression by LPS is consistent with reduced hepatic Ido2 in malaria infected mice (Ball et al., 2007). Stress also decreased hepatic expression of class 1 Ido2-FL, Ido2-v3 and Ido2-v4 (Dostal et al., 2017), but DEX alone did not regulate hepatic Ido2 (Fig. 4) suggesting that stress-induced down-regulation of hepatic Ido2 isoforms is not mediated thru glucocorticoids, but by cytokines (Dostal et al., 2017). In the lung, several Ido2 isoforms follow an expected inflammatory induction by LPS and anti-inflammatory reduction by DEX. Spleen does not follow the same pattern as several Ido2 isoforms are induced by LPS and either further induced or not changed by DEX. Likely, these changes reflect cell-type specific responses within each tissue (as seen in the brain).
The Ido2 transcripts can be generated from a single pre-mRNA (exons 1 thru 11) spliced to retain either exon 1 or 3, or by the usage of alternate promotors to initiate transcription at either exon 1 (class 1) or 3 (class 2). The latter alternative allows for differential regulation of transcripts. This is best exemplified by class 1 isoforms that were decreased by LPS only in liver. In contrast, all samples express class 2 isoforms, suggesting that only the liver efficiently uses the promoter for exon 1 (the class 1 promoter would be ~21,500 bp upstream of a promoter for exon 3 (see Table S2)). Utilizing this upstream promoter endows the ability to independently downregulate class 1 Ido2 isoforms in response to LPS, whereas class 2 isoforms are generally increased by LPS. Currently the physiological relevance of this finding is unclear. However, as visualized in Table S2, the Kyn Pathway may be elevated within the brain by both inflammation- and glucocorticoid-driven Ido2 expression, but repressed in the periphery by one or both stimuli. Thus, an underappreciated upregulation of central Ido2 may be involved in DO-dependent depression that is not evident in the periphery.
4.5. DEX upregulates Tdo2-FL while inflammation regulates Tdo2-v1 and Tdo2-v2
The first papers confirming the existence of DO isoforms reported higher Tdo2-v1 and Tdo2-v2 in mouse liver compared to brain (Kanai et al., 2009b); results we extend to include Tdo2-FL (Fig. 5, (Brooks et al., 2016b; Dostal et al., 2017)). Within the liver, Tdo2 activity is glucocorticoid-dependent (Berry and Smythe, 1963; Saito et al., 1994; Soichot et al., 2013; Young, 1981), in line with DEX elevating Tdo2 expression. Class 1 Tdo2-FL was also elevated in brain, astrocytes, liver, lung and spleen by DEX suggesting that Tdo2-FL is a glucocorticoid-sensitive Tdo2 isoform. We directly confirmed this by showing that DEX increases Tdo2-FL ex vivo (Brooks et al., 2016a, 2016b).
Tdo2-v1/v2 expression by brain and glia is unresponsive to LPS or DEX in vivo and unresponsive to the direct effects of DEX ex vivo (Brooks et al., 2016a, 2016b), at first leading us to believe that these class 2 isoforms are LPS and glucocorticoid unresponsive. However, LPS (liver, spleen) and DEX (liver, lung) increase their expression in the periphery. A putative glucocorticoid response element (GRE, Table S3) lies 5′ of the transcription start site for Tdo2-FL (Kanai et al., 2009b). Since only Tdo2-FL is directly glucocorticoid-inducible (Brooks et al., 2016a, 2016b), the promoter for Tdo2-FL probably utilizes this GRE element. However, this putative GRE element resides within exon 0 utilized as the first exon by class 2 transcripts. Corticosteroids appear unable to directly drive Tdo2-v1 and Tdo2-v2 since they initiate transcription upstream of this GRE element. Thus, inflammation- and glucocorticoid-dependent signals (not necessarily glucocorticoids per se) drive peripheral Tdo2-v1/v2 expression, whereas glucocorticoid-mediated signals likely directly drive brain and peripheral Tdo2-FL.
Tdo2 isoforms can be generated either by alternate splicing from a single pre-mRNA (exons 0a/b-1 thru 12) or by independent transcription of distinct pre-mRNA species using alternate promotors to initiate transcription at either exon 1 (class 1) or 0 (class 2); the latter allows for differential regulation of transcripts as found in the current study, but clearly all 4 tissues and glia are able to express both class 1 and 2 Tdo2 transcripts. Differential expression of Tdo2 isoforms is likely a cell-type specific phenomenon. Within the brain, Tdo2 is expressed best by neurons and astrocytes (Ohira et al., 2010; Wu et al., 2013) where Tdo2-FL induction by DEX would result in production of neuroprotective KynA. Within liver, Tdo2 is present in hepatocytes and bile ducts (Niimi et al., 1983; Streckfuss-Bomeke et al., 2014). Only within the liver and spleen does LPS induce Tdo2 expression, but it is unknown if this occurs in hepatocytes or bile ducts with different physiological consequences.
4.6. Downstream Kyn Pathway enzyme regulation
While the DO’s are rate-limiting, downstream enzymes are expressed with cellular specificity and define which downstream kynurenines are produced (supplementary Fig. S1, (Lawson et al., 2016; McCusker et al., 2014)). These kynurenines include neuroprotective and immunosuppressive metabolites (Kyn and KynA) or the neurotoxic and NAD+ precursor QuinA. Some of the actions of the kynurenines are mediated extracellularly by transmembrane receptors, requiring cellular release. Other actions are mediated by intracellular receptors. The latter case does not require release into the extracellular fluid and as such changes in the Kyn Pathway can affect the physiology of only that cell, making physiological relevance of tissue metabolites difficult to interpret. The best example, extracellular QuinA is neurotoxic via the NMDA-R (Schwarcz, 2016), but intracellular QuinA used to generate NAD+ is neuroprotective (Braidy et al., 2011). Changes in downstream enzymes could shift the relative production of these kynurenines.
Kat2 facilitates Kyn metabolism to KynA. Kat2 is preferentially expressed by astrocytes and neurons. Stress and LPS increase brain KynA without changes in Kat2 expression (Fig. 6, (Dostal et al., 2017; Dugan et al., 2016; Parrott et al., 2016b; Pawlak et al., 2000; Vecchiarelli et al., 2015)), indicating that KynA generation is dependent on Kat2 substrate availability, i.e. Kyn produced by astrocyte DO’s. Kmo, Kynu and Haao generate QuinA from Kyn. In brain, these enzymes are well-expressed by microglia. LPS and/or DEX halved Kynu and Haao expression in microglia, liver, lung and spleen, albeit doubling Kmo in liver and lung. These finding agree with other work showing minor changes in these enzymes by inflammation (Parrott et al., 2016a) and stress (Dostal et al., 2017; Vecchiarelli et al., 2015). Again, these data suggest that changes in QuinA are primarily dependent on expression of the rate-limiting DO’s.
Our data define unique expression profiles for the DO’s with minimal changes in downstream enzymes. Clearly, DO isoform expression is uniquely regulated in brain, glia and peripheral tissues. Investigators assessing chronic adaptations of DO expression should consider cell-specific regulation of the different isoforms for both preclinical and clinical studies. Whether DO isoforms map to unique cellular processes remains untested.
4.7. Conclusions
We identify Ido1, Ido2 and Tdo2 isoforms uniquely adapted to independently change in response to inflammation and glucocorticoids with elaborate cell-, tissue- and isoform-specificity in vivo. Although only the first step towards defining the biological significance of their expression, our data begin to unravel the complex interactions between inflammation and stress regarding activation of Kyn Pathway enzymes under these often comorbid conditions. Importantly, different assays are required to identify treatment-specific changes in DO expression. Our current findings provide novel evidence of a brain-specific pattern of DO induction without discernable counter-regulatory effects of glucocorticoids, but complex positive and negative interactions between inflammation and glucocorticoids within the periphery (visual summary in Tables S1–S3). These finding may define a unique mechanism pivotal to the depression susceptibility of individuals affected by chronic inflammatory disease.
Supplementary Material
Highlights.
Kynurenine is increased in brain by LPS and plasma by DEX
Ido1, Ido2 and Tdo2 expression by astrocytes and microglia are uniquely regulated
DEX attenuates cytokine induction by LPS in peripheral tissues, not brain
DEX mitigates Ido1 and Ido2 induction by LPS in peripheral tissues, not brain
Ido1, Ido2, Tdo2 are adapted to respond with cell- and tissue-specificity
Acknowledgments
This work was supported by NIH (United States) through RO1 MH101145 to RHM.
Footnotes
5. Financial disclosures
Authors declare that there are no personal or financial conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Agudelo LZ, Femenía T, Orhan F, Porsmyr-Palmertz M, Goiny M, Martinez-Redondo V, Correia JC, Izadi M, Bhat M, Schuppe-Koistinen I, Pettersson AT, Ferreira DMS, Krook A, Barres R, Zierath JR, Erhardt S, Lindskog M, Ruas JL. Skeletal muscle PGC-1α1 modulates kynurenine metabolism and mediates resilience to stress-induced depression. Cell. 2014;159:33–45. doi: 10.1016/j.cell.2014.07.051. [DOI] [PubMed] [Google Scholar]
- André C, O’Connor JC, Kelley KW, Lestage J, Dantzer R, Castanon N. Spatio-temporal differences in the profile of murine brain expression of proinflammatory cytokines and indoleamine 2,3-dioxygenase in response to peripheral lipopolysaccharide administration. J Neuroimmunol. 2008;200:90–9. doi: 10.1016/j.jneuroim.2008.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badawy AAB. Tryptophan availability for kynurenine pathway metabolism across the life span: Control mechanisms and focus on aging, exercise, diet and nutritional supplements. Neuropharmacology. 2015;112:248–263. doi: 10.1016/j.neuropharm.2015.11.015. [DOI] [PubMed] [Google Scholar]
- Ball HJ, Sanchez-Perez A, Weiser S, Austin CJD, Astelbauer F, Miu J, McQuillan JA, Stocker R, Jermiin LS, Hunt NH. Characterization of an indoleamine 2,3-dioxygenase-like protein found in humans and mice. Gene. 2007;396:203–213. doi: 10.1016/j.gene.2007.04.010. [DOI] [PubMed] [Google Scholar]
- Ball HJ, Yuasa HJ, Austin CJD, Weiser S, Hunt NH. Indoleamine 2,3-dioxygenase-2; a new enzyme in the kynurenine pathway. Int J Biochem Cell Biol. 2009;41:467–471. doi: 10.1016/j.biocel.2008.01.005. [DOI] [PubMed] [Google Scholar]
- Berry LJ, Smythe DS. Effects of bacterial endotoxins on metabolism. VI. The role of tryptophan pyrrolase in response of mice to endotoxin. J Exp Med. 1963;41:587–603. doi: 10.1084/jem.118.4.587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradley KAL, Case JAC, Khan O, Ricart T, Hanna A, Alonso CM, Gabbay V. The role of the kynurenine pathway in suicidality in adolescent major depressive disorder. Psychiatry Res. 2015;227:206–212. doi: 10.1016/j.psychres.2015.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braidy N, Guillemin GJ, Grant R. Effects of kynurenine pathway inhibition on NAD + metabolism and cell viability in human primary astrocytes and neurons. Int J Tryptophan Res. 2011;4:29–37. doi: 10.4137/IJTR.S7052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks AK, Janda TM, Lawson MA, Rytych JL, Smith RA, Ocampo-Solis C, McCusker RH. Desipramine decreases expression of human and murine indoleamine-2,3-dioxygenases. Brain Behav Immun. 2017 doi: 10.1016/j.bbi.2017.02.010. in press doi: http://dx.doi.org/10.1016/j.bbi.2017.02.010. [DOI] [PMC free article] [PubMed]
- Brooks AK, Lawson MA, Rytych JL, Yu KC, Janda TM, Steelman AJ, McCusker RH. Immunomodulatory factors galectin-9 and interferon-gamma synergize to induce expression of rate-limiting enzymes of the kynurenine pathway in the mouse hippocampus. Front Immunol. 2016a;7:1–16. doi: 10.3389/fimmu.2016.00422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks AK, Lawson MA, Smith RA, Janda TM, Kelley KW, McCusker RH. Interactions between inflammatory mediators and corticosteroids regulate transcription of genes within the Kynurenine Pathway in the mouse hippocampus. J Neuroinflammation. 2016b;13:1–16. doi: 10.1186/s12974-016-0563-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browne CA, O’Brien FE, Connor TJ, Dinan TG, Cryan JF. Differential lipopolysaccharide-induced immune alterations in the hippocampus of two mouse strains: Effects of stress. Neuroscience. 2012;225:237–248. doi: 10.1016/j.neuroscience.2012.08.031. [DOI] [PubMed] [Google Scholar]
- Capuron L, Miller A, Irwin M. Psychoneuroimmunology of depressive disorder: mechanisms and clinical implications. In: Ader R, editor. Psychoneuroimmunology. Academic Press; 2006. pp. 509–530. [DOI] [Google Scholar]
- Couch Y, Trofimov A, Markova N, Nikolenko V, Steinbusch HW, Chekhonin V, Schroeter C, Lesch KP, Anthony DC, Strekalova T. Low-dose lipopolysaccharide (LPS) inhibits aggressive and augments depressive behaviours in a chronic mild stress model in mice. J Neuroinflammation. 2016;13:108. doi: 10.1186/s12974-016-0572-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croitoru-Lamoury J, Lamoury FMJ, Caristo M, Suzuki K, Walker D, Takikawa O, Taylor R, Brew BJ. Interferon-γ regulates the proliferation and differentiation of mesenchymal stem cells via activation of indoleamine 2, 3 dioxygenase (IDO) PLoS One. 2011:6. doi: 10.1371/journal.pone.0014698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dantzer R. Role of the Kynurenine Metabolism Pathway in Inflammation-Induced Depression: Preclinical Approaches. Curr Top Behav Neurosci. 2016 doi: 10.1007/7854_2016_6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dantzer R, O’Connor JC, Freund GG, Johnson RW, Kelley KW. From inflammation to sickness and depression: when the immune system subjugates the brain. Nat Rev Neurosci. 2008;9:46–56. doi: 10.1038/nrn2297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dostal CR, Carson Sulzer M, Kelley KW, Freund GG, McCusker RH. Glial and tissue-specific regulation of Kynurenine Pathway dioxygenases by acute stress of mice. Neurobiol Stress. 2017;7:1–15. doi: 10.1016/j.ynstr.2017.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dugan AM, Parrott JM, Redus L, Hensler JG, O’Connor JC. Low-level stress induces production of neuroprotective factors in wild-type but not BDNF+/− mice: Interleukin-10 and kynurenic acid. Int J Neuropsychopharmacol. 2016;19:1–5. doi: 10.1093/ijnp/pyv089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu X, Zunich SM, O’Connor JC, Kavelaars A, Dantzer R, Kelley KW. Central administration of lipopolysaccharide induces depressive-like behavior in vivo and activates brain indoleamine 2, 3 dioxygenase in murine organotypic hippocampal slice cultures. J Neuroinflammation. 2010;7:43. doi: 10.1186/1742-2094-7-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuertig R, Azzinnari D, Bergamini G, Cathomas F, Sigrist H, Seifritz E, Vavassori S, Luippold A, Hengerer B, Ceci A, Pryce CR. Mouse chronic social stress increases blood and brain kynurenine pathway activity and fear behaviour: Both effects are reversed by inhibition of indoleamine 2,3-dioxygenase. Brain Behav Immun. 2016;54:59–72. doi: 10.1016/j.bbi.2015.12.020. [DOI] [PubMed] [Google Scholar]
- Fujigaki S, Saito K, Sekikawa K, Tone S, Takikawa O, Fujii H, Wada H, Noma A, Seishima M. Lipopolysaccharide induction of indoleamine 2,3-dioxygenase is mediated dominantly by an IFN-gamma-independent mechanism. Eur J Immunol. 2001;31:2313–8. doi: 10.1002/1521-4141(200108)31:8<2313::AID-IMMU2313>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- Fujigaki S, Saito K, Takemura M, Fujii H, Wada H, Noma A, Seishima M. Species differences in L-tryptophan-kynurenine pathway metabolism: quantification of anthranilic acid and its related enzymes. Arch Biochem Biophys. 1998;358:329–35. doi: 10.1006/abbi.1998.0861. [DOI] [PubMed] [Google Scholar]
- Fukunaga M, Yamamoto Y, Kawasoe M, Arioka Y, Murakami Y, Hoshi M, Saito K. Studies on tissue and cellular distribution of indoleamine 2,3-dioxygenase 2: the absence of IDO1 upregulates IDO2 expression in the epididymis. J Histochem Cytochem. 2012;60:854–60. doi: 10.1369/0022155412458926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibney SM, Fagan EM, Waldron A, O’Byrne J, Connor TJ, Harkin A. Inhibition of stress-induced hepatic tryptophan 2,3-dioxygenase exhibits antidepressant activity in an animal model of depressive behaviour. Int J Neuropsychopharmacol. 2014;17:917–928. doi: 10.1017/S1461145713001673. [DOI] [PubMed] [Google Scholar]
- Godbout JP, Moreau M, Lestage J, Chen J, Sparkman NL, O’Connor JC, Castanon N, Kelley KW, Dantzer R, Johnson RW. Aging exacerbates depressive-like behavior in mice in response to activation of the peripheral innate immune system. Neuropsychopharmacology. 2008;33:2341–2351. doi: 10.1038/sj.npp.1301649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Y, Cai H, Chen L, Liang D, Yang R, Dang R, Jiang P. Quantitative profiling of neurotransmitter abnormalities in the hippocampus of rats treated with lipopolysaccharide: Focusing on kynurenine pathway and implications for depression. J Neuroimmunol. 2016;295–296:41–46. doi: 10.1016/j.jneuroim.2016.04.006. [DOI] [PubMed] [Google Scholar]
- Heisler JM, O’Connor JC. Indoleamine 2,3-dioxygenase-dependent neurotoxic kynurenine metabolism mediates inflammation-induced deficit in recognition memory. Brain Behav Immun. 2015;50:115–124. doi: 10.1016/j.bbi.2015.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry CJ, Huang Y, Wynne AM, Godbout JP. Peripheral lipopolysaccharide (LPS) challenge promotes microglial hyperactivity in aged mice that is associated with exaggerated induction of both pro-inflammatory IL-1β and anti-inflammatory IL-10 cytokines. Brain Behav Immun. 2009;23:309–317. doi: 10.1016/j.bbi.2008.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes MM, Carballedo A, McLoughlin DM, Amico F, Harkin A, Frodl T, Connor TJ. Tryptophan depletion in depressed patients occurs independent of kynurenine pathway activation. Brain Behav Immun. 2012;26:979–87. doi: 10.1016/j.bbi.2012.05.010. [DOI] [PubMed] [Google Scholar]
- Jungblut M, Tiveron MC, Barral S, Abrahamsen B, Knöbel S, Pennartz S, Schmitz J, Perraut M, Pfrieger FW, Stoffel W, Cremer H, Bosio A. Isolation and characterization of living primary astroglial cells using the new GLAST-specific monoclonal antibody ACSA-1. Glia. 2012;60:894–907. doi: 10.1002/glia.22322. [DOI] [PubMed] [Google Scholar]
- Kanai M, Funakoshi H, Takahashi H, Hayakawa T, Mizuno S, Matsumoto K, Nakamura T. Tryptophan 2, 3-dioxygenase is a key modulator of physiological neurogenesis and anxiety-related behavior in mice. Mol Brain. 2009a;2:8. doi: 10.1186/1756-6606-2-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanai M, Nakamura T, Funakoshi H. Identification and characterization of novel variants of the tryptophan 2,3-dioxygenase gene: differential regulation in the mouse nervous system during development. Neurosci Res. 2009b;64:111–7. doi: 10.1016/j.neures.2009.02.004. [DOI] [PubMed] [Google Scholar]
- Kennett GA, Joseph MH. The functional importance of increased brain tryptophan in the serotonergic response to restraint stress. Neuropharmacology. 1981;20:39–43. doi: 10.1016/0028-3908(81)90039-3. [DOI] [PubMed] [Google Scholar]
- Kessler RC, Berglund P, Demler O, Jin R, Koretz D, Merikangas KR, Rush AJ, Walters EE, Wang PS. The epidemiology of major depressive disorder: results from the national comorbidity survey replication (NCS-R) JAMA. 2003;289:3095–3105. doi: 10.1001/jama.289.23.3095. [DOI] [PubMed] [Google Scholar]
- Kim H, Chen L, Lim G, Sung B, Wang S, McCabe MF, Rusanescu G, Yang L, Tian Y, Mao J. Brain indoleamine 2,3-dioxygenase contributes to the comorbidity of pain and depression. J Clin Invest. 2012;122:2940–2954. doi: 10.1172/JCI61884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larkin PB, Sathyasaikumar KV, Notarangelo FM, Funakoshi H, Nakamura T, Schwarcz R, Muchowski PJ. Tryptophan 2,3-dioxygenase and indoleamine 2,3-dioxygenase 1 make separate, tissue-specific contributions to basal and inflammation-induced kynurenine pathway metabolism in mice. Biochim Biophys Acta. 2016;1860:2345–2354. doi: 10.1016/j.bbagen.2016.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawson MA, Dostal CR, Brooks AK, McCusker RH. The kynurenine (Kyn) pathway and neuroinflammation: the confused brain. In: Opp MR, editor. Primer of PsychoNeuroImmunology Research. Los Angeles, CA: 2016. pp. 95–101. [Google Scholar]
- Lawson M, Parrott JM, McCusker RH, Dantzer R, Kelley KW, O’Connor JC. Intracerebroventricular administration of lipopolysaccharide induces indoleamine-2,3-dioxygenase-dependent depression-like behaviors. J Neuroinflammation. 2013;10:87. doi: 10.1186/1742-2094-10-87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lestage J, Verrier D, Palin K, Dantzer R. The enzyme indoleamine 2, 3-dioxygenase is induced in the mouse brain in response to peripheral administration of lipopolysaccharide and superantigen. Brain Behav Immun. 2002;16:596–601. doi: 10.1016/S0889-1591(02)00014-4. [DOI] [PubMed] [Google Scholar]
- Liu Y-N, Peng Y-L, Lei-Liu Wu T-Y, Zhang Y, Lian Y-J, Yang Y-Y, Kelley KW, Jiang C-L, Wang Y-X. TNFα mediates stress-induced depression by upregulating indoleamine 2,3-dioxygenase in a mouse model of unpredictable chronic mild stress. Eur Cytokine Netw. 2015;26:15–25. doi: 10.1016/j.rasd.2014.08.015.Social. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–8. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Martin Sa, Dantzer R, Kelley KW, Woods Ja. Voluntary wheel running does not affect lipopolysaccharide-induced depressive-like behavior in young adult and aged mice. Neuroimmunomodulation. 2014;21:52–63. doi: 10.1159/000356144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matt SM, Lawson MA, Johnson RW. Aging and peripheral lipopolysaccharide can modulate epigenetic regulators and decrease IL-1b promoter DNA methylation in microglia. Neurobiol Aging. 2016;47:1–9. doi: 10.1016/j.neurobiolaging.2016.07.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCusker RH, Kavelaars A, Heijnen CJ, Dantzer R, Kelley KW. Depression, inflammation and tryptophan metabolism. In: Kusnecov A, Anisman H, editors. The Wiley-Blackwell Handbook of Psychoneuroimmunology. John Wiley & Sons, Ltd; 2014. pp. 448–468. [Google Scholar]
- McEwen BS, Bowles NP, Gray JD, Hill MN, Hunter RG, Karatsoreos IN, Nasca C. Mechanisms of stress in the brain. Nat Neurosci. 2015;18:1353–1363. doi: 10.1038/nn.4086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merlo LMF, Grabler S, DuHadaway JB, Pigott E, Manley K, Prendergast GC, Laury-Kleintop LD, Mandik-Nayak L. Therapeutic antibody targeting of indoleamine-2,3-dioxygenase (IDO2) inhibits autoimmune arthritis. Clin Immunol. 2017;179:8–16. doi: 10.1016/j.clim.2017.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merlo LMF, Pigott E, DuHadaway JB, Grabler S, Metz R, Prendergast GC, Mandik-Nayak L. IDO2 is a critical mediator of autoantibody production and inflammatory pathogenesis in a mouse model of autoimmune arthritis. J Immunol. 2014;192:2082–90. doi: 10.4049/jimmunol.1303012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metz R, DuHadaway JB, Kamasani U, Laury-Kleintop L, Muller AJ, Prendergast GC. Novel tryptophan catabolic enzyme IDO2 is the preferred biochemical target of the antitumor indoleamine 2,3-dioxygenase inhibitory compound D-1-methyl-tryptophan. Cancer Res. 2007;67:7082–7087. doi: 10.1158/0008-5472.CAN-07-1872. [DOI] [PubMed] [Google Scholar]
- Metz R, Smith C, DuHadaway JB, Chandler P, Baban B, Merlo LMF, Pigott E, Keough MP, Rust S, Mellor AL, Mandik-Nayak L, Muller AJ, Prendergast GC. IDO2 is critical for IDO1-mediated T-cell regulation and exerts a non-redundant function in inflammation. Int Immunol. 2014;26:357–367. doi: 10.1093/intimm/dxt073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miura H, Ozaki N, Sawada M, Isobe K, Ohta T, Nagatsu T. A link between stress and depression: shifts in the balance between the kynurenine and serotonin pathways of tryptophan metabolism and the etiology and pathophysiology of depression. Stress. 2008;11:198–209. doi: 10.1080/10253890701754068. [DOI] [PubMed] [Google Scholar]
- Murray MF. The human indoleamine 2,3-dioxygenase gene and related human genes. Curr Drug Metab. 2007;8:197–200. doi: 10.2174/138920007780362509. [DOI] [PubMed] [Google Scholar]
- Myint AM. Kynurenines: from the perspective of major psychiatric disorders. FEBS J. 2012;279:1375–85. doi: 10.1111/j.1742-4658.2012.08551.x. [DOI] [PubMed] [Google Scholar]
- Myint AM, Schwarz MJ, Müller N. The role of the kynurenine metabolism in major depression. J Neural Transm. 2012;119:245–251. doi: 10.1007/s00702-011-0741-3. [DOI] [PubMed] [Google Scholar]
- Niimi S, Nakamura T, Nawa K, Ichihara A. Hormonal regulation of translatable mRNA of tryptophan 2,3-dioxygenase in primary cultures of adult rat hepatocytes. J Biochem. 1983;94:1697–1706. [PubMed] [Google Scholar]
- Nikodemova M, Watters JJ. Efficient isolation of live microglia with preserved phenotypes from adult mouse brain. J Neuroinflammation. 2012;9:147. doi: 10.1186/1742-2094-9-147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Notarangelo FM, Schwarcz R. Restraint Stress during Pregnancy Rapidly Raises Kynurenic Acid Levels in Mouse Placenta and Fetal Brain. Dev Neurosci. 2017 doi: 10.1159/000455228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connor JC, André C, Wang Y, Lawson M, Szegedi SS, Lestage J, Castanon N, Kelley KW, Dantzer R. Interferon-gamma and tumor necrosis factor-alpha mediate the upregulation of indoleamine 2,3-dioxygenase and the induction of depressive-like behavior in mice in response to bacillus Calmette-Guerin. J Neurosci. 2009a;29:4200–4209. doi: 10.1523/JNEUROSCI.5032-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connor JC, Lawson MA, André C, Briley EM, Szegedi SS, Lestage J, Castanon N, Herkenham M, Dantzer R, Kelley KW. Induction of IDO by bacille Calmette-Guérin is responsible for development of murine depressive-like behavior. J Immunol. 2009b;182:3202–12. doi: 10.4049/jimmunol.0802722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connor JC, Lawson MA, André C, Moreau M, Lestage J, Castanon N, Kelley KW, Dantzer R. Lipopolysaccharide-induced depressive-like behavior is mediated by indoleamine 2,3-dioxygenase activation in mice. Mol Psychiatry. 2009c;14:511–22. doi: 10.1038/sj.mp.4002148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Farrell K, Harkin A. Stress-related regulation of the kynurenine pathway: Relevance to neuropsychiatric and degenerative disorders. Neuropharmacology. 2017;112:307–323. doi: 10.1016/j.neuropharm.2015.12.004. [DOI] [PubMed] [Google Scholar]
- Ohira K, Hagihara H, Toyama K, Takao K, Kanai M, Funakoshi H, Nakamura T, Miyakawa T. Expression of tryptophan 2, 3-dioxygenase in mature granule cells of the adult mouse dentate gyrus. Mol Brain. 2010;3:26. doi: 10.1186/1756-6606-3-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pace TWW, Hu F, Miller AH. Cytokine-effects on glucocorticoid receptor function: Relevance to glucocorticoid resistance and the pathophysiology and treatment of major depression. Brain Behav Immun. 2007;21:9–19. doi: 10.1016/j.bbi.2006.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pariante CM. Risk factors for development of depression and psychosis: Glucocorticoid receptors and pituitary implications for treatment with antidepressant and glucocorticoids. Ann N Y Acad Sci. 2009;1179:144–152. doi: 10.1111/j.1749-6632.2009.04978.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pariante CM. The glucocorticoid receptor: part of the solution or part of the problem? J Psychopharmacol. 2006;20:79–84. doi: 10.1177/1359786806066063. [DOI] [PubMed] [Google Scholar]
- Park SE, Lawson M, Dantzer R, Kelley KW, McCusker RH. Insulin-like growth factor-I peptides act centrally to decrease depression-like behavior of mice treated intraperitoneally with lipopolysaccharide. J Neuroinflammation. 2011;8:179. doi: 10.1186/1742-2094-8-179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parrott JM, Redus L, O’Connor JC. Kynurenine metabolic balance is disrupted in the hippocampus following peripheral lipopolysaccharide challenge. J Neuroinflammation. 2016a;13:1–15. doi: 10.1186/s12974-016-0590-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parrott JM, Redus L, Santana-Coelho D, Morales J, Gao X, O’Connor JC. Neurotoxic kynurenine metabolism is increased in the dorsal hippocampus and drives distinct depressive behaviors during inflammation. Transl Psychiatry. 2016b;6:1–12. doi: 10.1038/tp.2016.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pawlak D, Takada Y, Urano T, Takada A. Serotonergic and kynurenic pathways in rats exposed to foot shock. Brain Res Bull. 2000;52:197–205. doi: 10.1016/S0361-9230(00)00252-5. [DOI] [PubMed] [Google Scholar]
- Saito K, Markey SP, Heyes MP. 6-Chloro-D,L-tryptophan, 4-chloro-3-hydroxyanthranilate and dexamethasone attenuate quinolinic acid accumulation in brain and blood following systemic immune activation. Neurosci Lett. 1994;178:211–215. doi: 10.1016/0304-3940(94)90761-7. [DOI] [PubMed] [Google Scholar]
- Saito K, Markey SP, Heyes MP. Effects of immune activation on quinolinic acid and neuroactive kynurenines in the mouse. Neuroscience. 1992;51:25–39. doi: 10.1016/0306-4522(92)90467-g. [DOI] [PubMed] [Google Scholar]
- Salazar A, Gonzalez-Rivera BL, Redus L, Parrott JM, O’Connor JC. Indoleamine 2,3-dioxygenase mediates anhedonia and anxiety-like behaviors caused by peripheral lipopolysaccharide immune challenge. Horm Behav. 2012;62:202–209. doi: 10.1016/j.yhbeh.2012.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savitz J. Role of kynurenine metabolism pathway activation in major depressive disorders. In: Dantzer R, Capuron L, editors. Current Topics in Behavioral Neurosciences. Springer International Publishing; 2016. pp. 249–267. [DOI] [PubMed] [Google Scholar]
- Savitz J, Drevets WC, Wurfel BE, Ford BN, Bellgowan PSF, Victor TA, Bodurka J, Teague TK, Dantzer R. Reduction of kynurenic acid to quinolinic acid ratio in both the depressed and remitted phases of major depressive disorder. Brain Behav Immun. 2015;46:55–59. doi: 10.1016/j.bbi.2015.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schinkel AH, Wagenaar E, Van Deemtar L, Mol CAAM, Borst P. Absence of the mdr1a P-glycoprotein in mice affects tissue distribution and pharmacokinetics of dexamethasone, digoxin, and cyclosporin A. J Clin Invest. 1995;96:1698–1705. doi: 10.1172/JCI118214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarcz R. Neuropsychopharmacology: A Tribute to Joseph T Coyle. 1. Elsevier Inc; 2016. Kynurenines and Glutamate: Multiple Links and Therapeutic Implications. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwieler L, Samuelsson M, Frye Ma, Bhat M, Schuppe-Koistinen I, Jungholm O, Johansson AG, Landén M, Sellgren CM, Erhardt S. Electroconvulsive therapy suppresses the neurotoxic branch of the kynurenine pathway in treatment-resistant depressed patients. J Neuroinflammation. 2016;13:51. doi: 10.1186/s12974-016-0517-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soichot M, Vaast A, Vignau J, Guillemin GJ, Lhermitte M, Broly F, Allorge D. Characterization of functional polymorphisms and glucocorticoid-responsive elements in the promoter of TDO2, a candidate gene for ethanol-induced behavioural disorders. Alcohol Alcohol. 2013;48:415–25. doi: 10.1093/alcalc/agt028. [DOI] [PubMed] [Google Scholar]
- Souza LC, Jesse CR, Antunes MS, Ruff JR, de Oliveira Espinosa D, Gomes NS, Donato F, Giacomeli R, Boeira SP. Indoleamine-2,3-dioxygenase mediates neurobehavioral alterations induced by an intracerebroventricular injection of amyloid-β1-42 peptide in mice. Brain Behav Immun. 2016 doi: 10.1016/j.bbi.2016.03.002. [DOI] [PubMed] [Google Scholar]
- Streckfuss-Bomeke K, Jende J, Cheng IF, Hasenfuss G, Guan K. Efficient generation of hepatic cells from multipotent adult mouse germ-line stem cells using an OP9 co-culture system. Cell Reprogr. 2014;16:65–76. doi: 10.1089/cell.2013.0057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talarowska M, Galecki P. Cognition and emotions in recurrent depressive disorders - The role of inflammation and the Kynurenine Pathway. Curr Pharm Des. 2016;22:1–8. doi: 10.2174/1381612822666151230110738. [DOI] [PubMed] [Google Scholar]
- Trabanelli S, Očadlíková D, Ciciarello M, Salvestrini V, Lecciso M, Jandus C, Metz R, Evangelisti C, Laury-Kleintop L, Romero P, Prendergast GC, Curti A, Lemoli RM. The SOCS3-independent expression of IDO2 supports the homeostatic generation of T regulatory cells by human dendritic cells. J Immunol. 2014;192:1231–40. doi: 10.4049/jimmunol.1300720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vecchiarelli HA, Gandhi CP, Hill MN. Acute psychological stress modulates the expression of enzymes involved in the Kynurenine Pathway throughout corticolimbic circuits in adult male rats. Neural Plast. 2015;2016:1–12. doi: 10.1155/2016/7215684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Dunn AJ. The role of interleukin-6 in the activation of the hypothalamo-pituitary-adrenocortical axis and brain indoleamines by endotoxin and interleukin-1β. Brain Res. 1999;815:337–348. doi: 10.1016/S0006-8993(98)01091-9. [DOI] [PubMed] [Google Scholar]
- Wang Y, Lawson MA, Kelley KW, Dantzer R. Primary murine microglia are resistant to nitric oxide inhibition of indoleamine 2,3-dioxygenase. Brain Behav Immun. 2010a;24:1249–1253. doi: 10.1016/j.bbi.2010.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Lawson M, Dantzer R, Kelley KW. LPS-induced indoleamine 2,3-dioxygenase is regulated in an interferon-gamma-independent manner by a JNK signaling pathway in primary murine microglia. Brain Behav Immun. 2010b;24:201–9. doi: 10.1016/j.bbi.2009.06.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wróbel A, Serefko A, WlaŸ P, Poleszak E. The depressogenic-like effect of acute and chronic treatment with dexamethasone and its influence on the activity of antidepressant drugs in the forced swim test in adult mice. Prog Neuro-Psychopharmacology Biol Psychiatry. 2014;54:243–248. doi: 10.1016/j.pnpbp.2014.06.008. [DOI] [PubMed] [Google Scholar]
- Wu W, Nicolazzo Ja, Wen L, Chung R, Stankovic R, Bao SS, Lim CK, Brew BJ, Cullen KM, Guillemin GJ. Expression of Tryptophan 2, 3-Dioxygenase and Production of Kynurenine Pathway Metabolites in Triple Transgenic Mice and Human Alzheimer’s Disease Brain. PLoS One. 2013:8. doi: 10.1371/journal.pone.0059749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young SN. Mechanism of decline in rat brain 5-Hydroxytryptamine after induction of liver tryptophan pyrrolase by hydrocortisone: roles of tryptophan catabolism and kynurenine synthesis. Br J Pharmacol. 1981;74:695–700. doi: 10.1111/j.1476-5381.1981.tb10480.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuasa HJ, Takubo M, Takahashi A, Hasegawa T, Noma H, Suzuki T. Evolution of vertebrate indoleamine 2,3-dioxygenases. J Mol Evol. 2007;65:705–714. doi: 10.1007/s00239-007-9049-1. [DOI] [PubMed] [Google Scholar]
- Zhai L, Dey M, Lauing KL, Gritsina G, Kaur R, Lukas RV, Nicholas MK, Rademaker AW, Dostal CR, McCusker RH, Raizer JJ, Parsa AT, Block O, Wainwright DA. The kynurenine to tryptophan ratio as a prognostic tool for glioblastoma patients enrolling in immunotherapy. J Clin Neurosci. 2015;22:1964–1968. doi: 10.1016/j.jocn.2015.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhai L, Ladomersky E, Dostal CR, Lauing K, Swoap K, Billingham LK, Gritsina G, Wu M, McCusker RH, Binder DC, Wainwright DA. Non-tumor cell IDO1 predominantly contributes to enzyme activity and response to CTLA-4/PD-L1 inhibition in mouse glioblastoma. Brain Behav Immun. 2017;62:24–29. doi: 10.1016/j.bbi.2017.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zorrilla EP, Luborsky L, McKay JR, Rosenthal R, Houldin A, Tax A, McCorkle R, Seligman DA, Schmidt K. The Relationship of Depression and Stressors to Immunological Assays: A Meta-Analytic Review. Brain Behav Immun. 2001;15:199–226. doi: 10.1006/brbi.2000.0597. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.