

Abstract
Disulfide bonds are important structural moieties of proteins; they ensure proper folding, provide stability, and proper function. With the increasing use of proteins for biotherapeutics, particularly monoclonal antibodies, which are highly disulfide bonded, it is now important to confirm the correct disulfide connectivity and to verify the presence, or absence, of disulfide bond variants in the protein therapeutics. These studies help to ensure safety and efficacy. Hence, disulfide bonds are among the critical quality attributes (CQAs) of proteins that have to be monitored closely during the biotherapeutics’ development. However, disulfide bond analysis is challenging due to the complexity of the biomolecules. Mass spectrometry has been the go-to analytical tool for the characterization of such complex biomolecules, and several methods have been reported to meet the challenging task of mapping disulfide bonds in proteins. In this review, we describe the relevant, recent mass spectrometry-based techniques and provide important considerations needed for efficient disulfide bond analysis in proteins. The review focuses on methods for proper sample preparation, fragmentation techniques for disulfide analysis, recent disulfide bond mapping methods based on the fragmentation techniques, and automated algorithms designed for rapid analysis of disulfide bonds from LC-MS/MS data. Researchers involved in method development for protein characterization can use the information herein to facilitate development of new MS-based methods for protein disulfide bond analysis. In addition, individuals doing biotherapeutics characterization, especially disulfide bond mapping in antibodies, can use this review to choose best strategies for disulfide bond assignment of their biologic products.
Keywords: Disulfide bond, mass spectrometry, liquid chromatography, software
1. INTRODUCTION
1.1 Disulfide Bond Formation and Importance in Proteins
Disulfide bonds are post-translational modifications in proteins formed between the sulfur atoms of two cysteine (Cys) residues during the biosynthesis of the proteins in the cell. The formation of the covalent bond results from oxidation of the free thiol (-SH) side chains of the Cys residues, primarily catalyzed by enzymes, including protein disulfide isomerase (PDI) and endoplasmic reticulum oxidoreductin 1 protein (Ero1p) [1,2]. A significant number of proteins contain disulfide bonds. Based on the known tertiary structures of 817 plasma proteins that contain 4594 disulfide bonds, Butera et al [3] approximated the ratio of protein-to-disulfide bond at 1:5. Hence, the approximately 2000 plasma proteins identified by Farrah et al [4] would contain about 10,000 disulfide bonds, representing an enormous number of disulfide bonds in plasma proteins, alone. Disulfide bonds are important in protein folding, and they have both structural and functional roles in the proteins.
Structurally, disulfide bonds ensure proper folding of proteins; they can lead to structural isoforms [5], and they stabilize the native high-order conformations of the proteins that are necessary to execute their biological functions [6, 7]. The concept of disulfide engineering is, therefore, an attractive choice in biotechnology as non-native disulfide bonds can be engineered into proteins to increase the protein stability. For instance, some proteins that initially lacked disulfide bonds have been shown to be more stable with engineered disulfides [8, 9], and the stability of proteins with native disulfide bonds increased with the introduction of additional disulfide bonds [10–13]. In some cases, engineered disulfide bonds increased the protein’s half-life [8, 14], reduced self-aggregation [15], and decreased immunogenicity [16]. Besides proteins, peptides with engineered disulfide bonds have also shown increased stability and half-life [17, 18]. Although the above examples demonstrate the benefits of additional disulfide bonds in proteins and peptides, not all engineered disulfide bonds produce the expected increase in protein stability [19].
Some disulfide bonds, known as allosteric disulfides, are responsible for effective biological functions of proteins, and the cleavage of such bonds would lead to a change in the protein activity [20]. The functional roles of allosteric disulfide bonds in blood and cancer cells have been extensively reviewed by Hogg et al [3, 21]. A recent report showed that reduction and alkylation of some disulfide bonds in rituximab and trastuzumab, IgG1-based drugs, increased the binding affinity of the modified drug to some Fc gamma receptor isotypes [22, 23], but also led to decreased binding to other Fc gamma receptors [23]. In some cases, mutation of Cys residues involved in disulfide bonds may have no effect on the protein’s biological activity, as is the case of an anti-CD44 IgG2 antibody where Cys to Ser mutations led to structural changes but had no impact on the binding of the protein to receptors and to complement C1 [24]. However, this may not be the case for all IgG2 antibodies.
Mapping the disulfide connectivity pattern in proteins, therefore, provides important information for research pertaining to protein stability, structure-function relationships, and any disulfide-mediated isoforms of proteins. In addition, disulfide bond characterization is of high importance during the development of biopharmaceuticals to ensure the safety and potency of biologics, which have increased dramatically in the drug market in recent years. Hence, there is an increasing demand for efficient analytical methods for accurate characterization of disulfide bonds in proteins, particularly therapeutic proteins.
1.2 Disulfide Bonds in Biotherapeutics
Protein disulfide bond characterization has become even more important in biopharmaceutical industries, due to the increasing use of recombinant proteins as biotherapeutics (biologics) for the treatment of diseases such as cancer, arthritis, asthma, and diabetes [25, 26]; and as vaccines against various diseases [27, 28]. These biologics are from a vast array of protein classes, including hormones [25, 29], monoclonal antibodies [25–26, 30], and growth factors [31, 32]. All these classes of biomolecules contain proteins that are disulfide bonded. Among these proteins, immunoglobulin gamma (IgG) antibodies, which are highly disulfide bonded (16 disulfide bonds for IgG1 and IgG4, 18 disulfide bonds for IgG2, and 25 disulfide bonds for IgG3) [33], and IgG-based therapeutics are the most prevalent in the market. As such, disulfide bonds are one of the many critical quality attributes (CAQs) of antibody-based drugs that have to be monitored throughout their development stages, as disulfide bond reduction and scrambling can occur in biotherapeutics during manufacturing [34, 35] and storage [36, 37]. Figure 1 shows the disulfide bonding patterns of the four main classes of IgGs. The presence of free Cys (reduced disulfide bonds) have been reported during manufacturing, and they can lead to formation of non-native disulfide bonds and aggregates [38, 39], and possibly cause immunogenicity and loss of biological activity of the biotherapeutics. Hence, disulfide bond characterization is necessary during biologic development to confirm the correct disulfide connectivity and to verify the presence, or absence, of disulfide bond variants in order to ensure the safety and efficacy of the drugs. In addition, regulatory agencies require comprehensive characterization of the disulfide bond pattern in biomolecules, in order to meet the quality by design (QbD) requirements for biologics [40, 41], hence the need for effective methods to map disulfide bonds in biotherapeutics.
Figure 1.
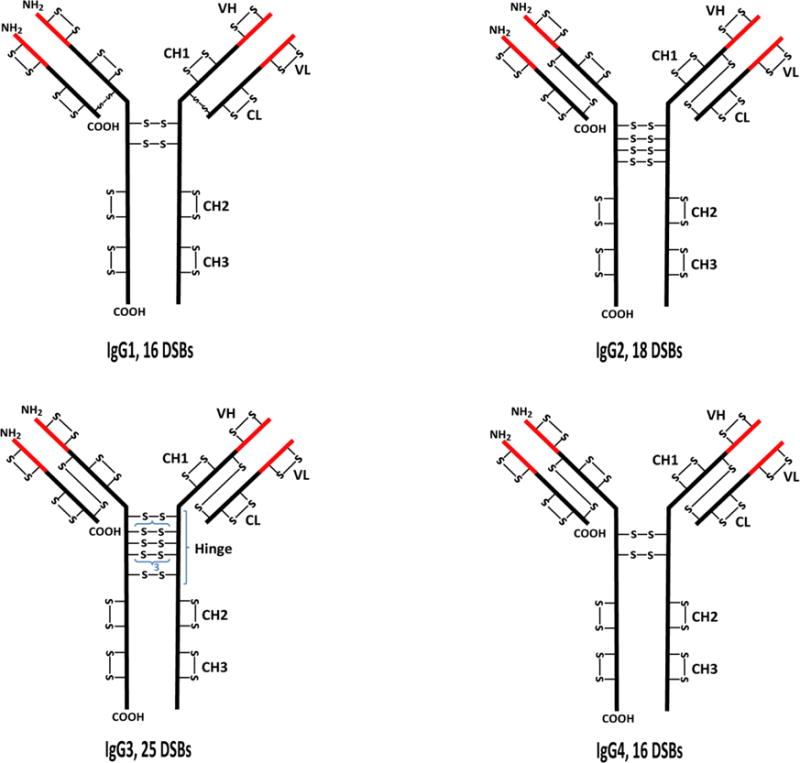
Structures showing the typical disulfide bond (DSB) patterns of the four classes of immunoglobulin gamma (IgG) antibodies. The black and red parts represent the constant (C) and variable (V) regions, respectively. H represents the heavy chain while L represents the light chain. VL and CL are domains on the light chain while VH, CH1, hinge, CH2, and CH3 are domains on the heavy chain. IgG3 has a 15-residue segment in the hinge region that is repeated 3 times; each of the segments contains 3 disulfide bonds.
1.3 Analytical Methods for Disulfide Bond Characterization
Several methods for disulfide bond analysis in proteins have been developed using a variety of analytical techniques, including NMR [42–44], X-ray crystallography [45], Edman degradation [5, 46], diagonal paper electrophoresis [47, 48], and liquid chromatography coupled to mass spectrometry (LC-MS) [49–51], which is the focus of this review. NMR and X-ray crystallography provide information about disulfide bonds at the molecular level, but they require significant amounts of highly pure samples, and they are not typically used for disulfide bond mapping due to the low throughput of solving structures by these methods. Traditional methods, such as Edman degradation and diagonal paper electrophoresis, were the prominent methods for disulfide bond mapping in the early 1960s, although Edman degradation methods (in combination with mass spectrometry) were still sparingly used in the late 2000s [5, 46]. With the advent of mass spectrometry, LC-MS and tandem mass spectrometry (LC-MS/MS) methods have become the go-to methods for disulfide bond mapping in proteins.
Bottom-up mass spectrometry is the most widely used method for disulfide bond analysis in proteins. The attractive aspects of the bottom-up approach include the availability of a variety of enzymes to digest the large biomolecules into small pieces (peptides containing intact disulfide bonds) that are easier to analyze, several soft ionization techniques, complementary fragmentation techniques, and the ability to couple mass spectrometers with LC systems for separation of the enzymatic digests prior to MS analysis. Key challenges in the use of mass spectrometry for disulfide bond analysis include proteins with high Cys content and complex disulfide connectivity, prevention of disulfide bond scrambling during sample preparation, the determination of low levels of non-native disulfide bonds, quantitation of disulfide bond-mediated protein isoforms, and choosing the appropriate method (or fragmentation technique) to assign the disulfide bonds in a protein, especially proteins containing complex disulfide patterns. Numerous methods for disulfide bond analysis have been developed, in part, to address these challenges.
In the following sections, we review the recent bottom-up mass spectrometric methods for disulfide bond analysis and provide important considerations for the steps involved. A number of reviews for disulfide bond analysis containing additional methods that were developed prior to 2007 have been reported [49–51]. Herein, we begin by taking an in-depth look at sample preparation, which is a key step in the successful mapping of native and alternative disulfide bonds in proteins. We provide several important tips to prevent the introduction of disulfide artifacts (or scrambling) during sample preparation. Finally, we discuss the fragmentation characteristics of disulfide-linked peptides upon subjection to various mass spectrometric dissociation techniques that are important for disulfide bond mapping and describe recent MS-based disulfide bond characterization methods that have been developed within the past decade.
2. SAMPLE PREPARATION FOR BOTTOM-UP MASS SPECTROMETRIC DISULFIDE BOND ANALYSIS
2.1 Overview of Sample Preparation Methods
Sample preparation is a critical step in bottom-up mass spectrometric disulfide bond analysis. In general, the analysis is usually performed using one of two ways: non-reduced (intact) analysis or reduced/intact analysis. The sample preparation workflow for the non-reduced approach, which is the most commonly used method, is shown in Figure 2. This approach requires alkylation of any free Cys residues, deglycosylation (for glycoproteins, and only if necessary; see Section 2.4 below for more details), proteolytic digestion of the protein without disulfide bond reduction, and the disulfide linked peptides are investigated to decipher the disulfide connectivity in the protein [52–54]. For the intact/reduced approach, two batches of enzymatically digested samples are prepared, one with the disulfide bonds intact (same as the previous approach) and the other with reduced disulfide bonds. The disulfide connectivity of the protein is determined by comparing the peptide map profiles (LC profiles) of the two sample batches [55, 56]. The disulfide bond analysis methods which are based on these sample preparation approaches will be discussed fully in Section 3.2 of this article.
Figure 2.
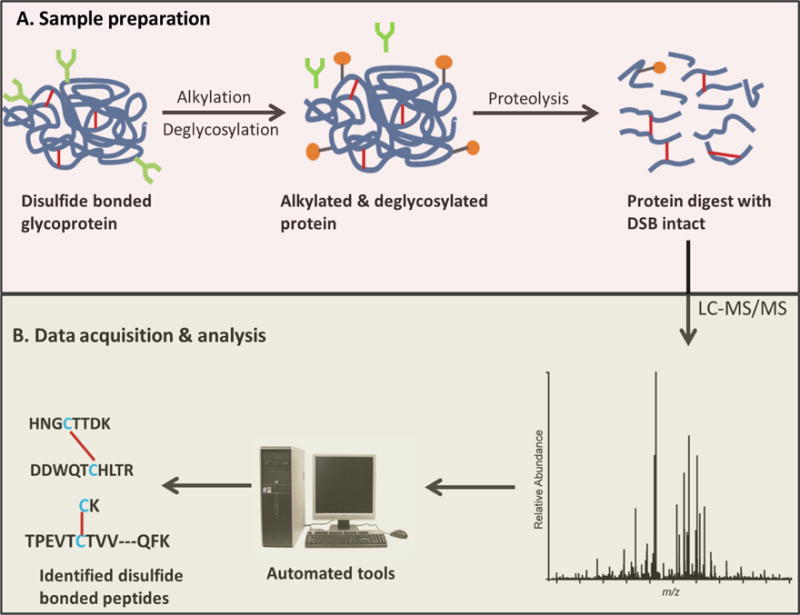
Disulfide bond analysis workflow. A. Sample preparation of non-reduced protein digest for disulfide bond analysis. Alkylation of free Cys is important to prevent disulfide shuffling (see text, Section 2.3). For glycoproteins, deglycosylation would ensure less complex data (see text, Section 2.4). B. Disulfide bond assignment from LC-MS/MS data.
2.2 Preventing Disulfide Bond Artifacts During Sample Preparation
A major requirement during sample preparation is to prevent the formation of non-native disulfide bonds (disulfide bond artifacts). To this end, a number of methods aimed at efficiently mapping disulfides in proteins without the formation of disulfide artifacts during sample preparation have been recently reported [57, 58]. Disulfide bond artifacts can be introduced during sample preparation via three main routes: (1) reaction between two free Cys residues (2) reaction between a free Cys residue and a disulfide, and (3) reaction between two Cys residues that were formerly involved in a disulfide bond[59–62]; these possible routes of disulfide bond formation are illustrated in Figure 3. These reactions are highly favored at alkaline pH [63, 64] and high temperatures [63, 65, 66], where disulfide scrambling usually occurs, especially for proteins containing free thiols. Besides pH and temperature, the thiol size, disulfide size, and the steric effects (exposure) of the thiols can also affect the formation of scrambled disulfide bonds, as shown by Kerr and coworkers [64]; but these factors cannot be controlled during sample preparation.
Figure 3.
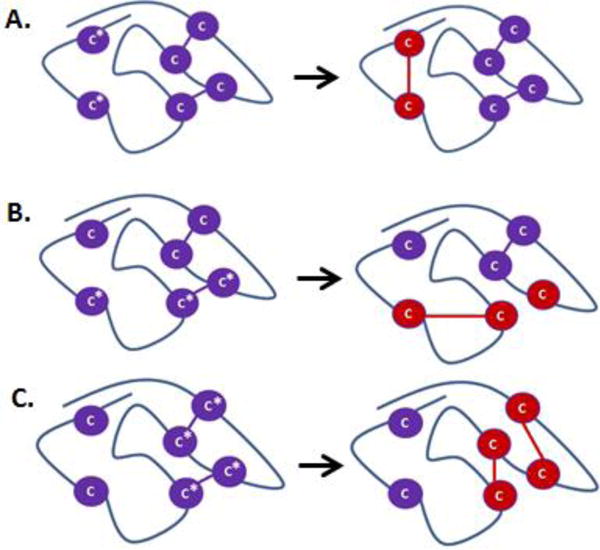
Schematic illustration of the different possible routes for formation of non-native disulfide bonds during sample preparation. Purple Cys residues are shown in native disulfide conformation; Cys residues marked with asterisks are modified into non-native conformations (red Cys residues). A. Reaction between two free Cys residues. B. Reaction between free Cys residues and Cys residues involved in a disulfide bond. C. Reaction between Cys residues that were formerly involved in disulfide bonds.
Temperature, pH, and the availability of free Cys are, therefore, the critical factors that must be controlled during sample preparation to prevent the formation of non-native disulfide bonds. Generally, samples for disulfide bond analysis are prepared at room temperature and then subjected to enzymatic digestion at 37 °C, and such low temperatures do not trigger any cysteine reaction or disulfide bond shuffling [63]. However, pH tremendously affects disulfide bond or cysteine reactivity, even at room temperature, and must be carefully controlled during sample preparation [63, 64]. Sample preparation should be done at slightly acidic pH because at alkaline pH, free thiols are deprotonated, and the resulting thiolate anions are oxidized or react with adjacent disulfide bonds (thiol/disulfide exchange) to form new, non-native disulfide bonds. The thiol groups that are more exposed to solvent will be more reactive than those that are not exposed [64].
Therefore, the first step during sample preparation for disulfide analysis is to cap any free cysteine residues to prevent formation of non-native disulfide bonds. This step is important even for proteins for which all the Cys residues are known to be in the disulfide bonded state because low levels of free Cys residues could be present. For example, although all the Cys residues in the four classes of IgG are expected to be disulfide bonded, low levels of free Cys residues have been detected in all 4 IgG classes [33], and the free Cys residues can induce disulfide artifacts if they are not alkylated or not properly alkylated. Commonly used Cys alkylating reagents include: iodoacetamide (IAM), iodoacetic acid (IAA), and N-ethyl-maleimide (NEM) [67, 68]. Rogers and coworkers showed that NEM is a more suitable alkylating agent for protein thiols than IAM and IAA because it reacts faster, requires less reagent per mole of free Cys, and is very effective at acidic pH (pH 4.3 to 7.0) [68], while the other reagents are most effective at alkaline pH (pH 8.0), where thiol/disulfide exchange and free Cys oxidation reactions can compete with alkylation and lead to low levels of non-native disulfide bonds. Recently, Lu et al [69] reported low levels of disulfide artifacts when disulfide bond mapping of RNase A was done after alkylating free thiols with IAM and IAA at pH 6.5, but no artifacts were identified when NEM was used at the same pH; indicating that IAM and IAA do not properly alkylate free thiols at slightly acidic pH conditions [69].
2.3 Digestion
Proteins can be digested either chemically or enzymatically [70]. Enzymatic digestion is the most widely used protein digestion approach, and there are a variety of enzymes to choose from, as reviewed by Switzer et al [70]. Table 1 shows different types of disulfide-linked peptides that can result from protein digestion. Selecting an appropriate enzyme for effective digestion is important because the enzyme used would determine the types of disulfide bonded peptides in the protein digest (Table 1). In selecting an enzyme, the goal is usually to choose one that produces more “simple” inter-chain disulfide-linked peptides, preferably with only one interchain disulfide bond and with bonded chains of appropriate lengths (4-15 AA residues). Such simple dipeptides can be easily mapped manually and with existing analysis software. (Software is discussed in Section 4.) The best enzyme choice for generating optimally simple disulfides can be found by conducing an, in silico digestion of the protein using different enzymes. Two important questions that can help in selecting a suitable enzyme are: (1) What are the sizes (number of disulfide bonded peptide chains and the length of each chain) of the disulfide linked peptides? and (2) How complex is the disulfide connectivity of the disulfide-linked peptides? That is, are there only interchain disulfide bonds, interchain and intrachain disulfide bonds, nested intrachain disulfide bonds? etc. If a single enzyme does not yield disulfide-bonded peptides with simple interchain disulfide bonds, a combination of enzymes can be used [71, 72]. However, it is worth noting that using multiple enzymes could lead to very short disulfide-bonded peptides, which may not be retained in reverse-phase columns, thus making disulfide assignment difficult. In such cases, a planned digestion that deliberately ensures missed cleavages in order to obtain longer peptide chains might be necessary.[72] In addition to the type of disulfide bonded peptides generated, another important consideration is the digestion efficiency of the enzyme used. Glatter et al studied several protein digestion strategies and showed that a combination of trypsin and Lys-C gives better digestion efficiency than trypsin alone [73]. Furthermore, the digestion of proteins with intact disulfide bonds might lead to low digestion efficiency, since some parts of the protein may not be well exposed to enzymes when the disulfide bonds are intact (non-reduced protein).
Table 1.
Examples of different types of disulfide bonded peptides in a vareity of proteins.
| No. | Protein | Enzyme(s) | Disulfide bonded peptides | Comment | Ref. |
|---|---|---|---|---|---|
| 1 | RNase A | Asp-N/C + trypsin |
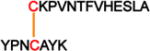
|
Two-chain DSBP with one interchain DSB | 72 |
| 2 | lgG3 | Trypsin |

|
Two-chain DSBP with two interchain DSBs (disulfide box) | 54 |
| 3 | RNase A | Trypsin |
|
Three-chain DSBP with two interchain DSBs | 72 |
| 4 | HIV, gp140 | Trypsin |

|
Four-chain DSBP with three interchain DSBs - PNGase F used for deglycosylation |
75 |
| 5 | CTT, gelatinase inhibitor | N/A |
|
Completely cyclisized DSBP | 91 |
| 6 | Chicken lysozyme | Trypsin |
|
Two-chain DSBP with both interchain and intrachain DSBs | 80 |
| 7 | rhASA | Lys-C + trypsin + Asp-N |
|
Two-chain DSBP with both interchain and intrachain DSBs | 71 |
| 8 | rhASA | Pepsin |
|
Single-chain DSBP with three intrachain DSBs (nested disulfides) | 71 |
- Aspartic acids in green ( ) represent previous asparagine residues that were converted to aspartic acids after deglycosylation with PNGase F.
- DSBP and DSB imply disulfide-bonded peptide and disulfide bond, respectively.
As mentioned earlier, the most important consideration during sample preparation for disulfide analysis is to prevent the formation of non-native disulfide bonds. Therefore, as with the case for alkylation, the pH and temperature are also critical during digestion. Proteolytic digestion is commonly done at 37°C, where no scrambling occurs. In fact, Wang et al [58] recently showed that trypsin plus Lys-C digestion can also be highly efficient at room temperature. Hence, pH is, again, a critical factor in preventing disulfide shuffling during protein digestion. Since disulfide artifacts can be introduced when samples are prepared at alkaline pH [59, 60], neutral or slightly acidic pH is preferred for protein digestions for disulfide bond mapping. Recently, Sung et al [57] investigated disulfide scrambling in lysozyme and bevacizumab (an IgG1 antibody drug) upon digestion with trypsin, trypsin plus Glu-C, and Lys-C at pH of 6 and 7; and thermolysin at pH ranging from 5 to 7. No disulfide scrambling was observed when the proteins were digested using trypsin, trypsin plus Glu-C, and Lys-C at a pH of 6. However, disulfide scrambling was observed when digestion was done at pH 7 using the same enzymes, and at pH 5 to 7 using thermolysin [57]. Similarly, low levels of disulfide scrambling have also been reported when trypsin or Lys-C plus trypsin were used for protein digestion at pH of 6.8 [71, 74, 58]. Nonetheless, disulfide scrambling at neutral or slightly acidic pH conditions may depend on a variety of conditions in addition to pH, since no scrambling was observed when disulfide mapping of bovine fetuin was performed after trypsin digestion in buffers at pH of 5.5, 6.5, and 7 [75]. In some cases, disulfide scrambling at slightly acidic pH may result from lack of effective alkylation of any free Cys residues prior to digestion. One useful strategy in determining whether a particular digestion conditions would introduce artifacts is to check the sample preparation method using a standard protein whose disulfide bonding is known, prior to analyzing an unknown protein; this strategy has been demonstrated previously [75]. It is worth noting that trypsin and Lys-C digestion at slightly acidic pH can lead to missed cleavages due to incomplete digestion, as the optimal efficiency of the enzymes are at pH 8.0 and pH range of 8.0 to 8.8, respectively [70]; therefore, a single set of digestion conditions is not necessarily optimal for every protein.
In order to completely eliminate the possibility of disulfide scrambling, proteins can be digested using pepsin, which efficiently digests proteins at highly acidic pH (pH <2) [71, 74, 76], where the formation of non-native disulfides via free Cys reactivity is not possible. However, pepsin is less specific than trypsin or Lys-C, and it may produce very small disulfide-bonded peptides that would be difficult to separate and analyze. Nonetheless, the use of separate pepsin and trypsin (pH 6) digestions could be helpful for unambiguous assignment of disulfide bonds in cases where ambiguity in the disulfide bond pattern has been a problem [76].
2.4 Sample Preparation of Glycoproteins
Disulfide bond analysis of glycoproteins may require deglycosylation prior to mass spectrometric analysis. In order to decide whether or not a glycoprotein needs to be deglycosylated, the glycosylation sites of the protein must first be identified, and in silico digestion using an enzyme of choice (or a combination of enzymes) can be performed prior to sample preparation to determine if Cys-containing peptides that are disulfide-linked would contain occupied glycosylation sites. For some glycoproteins (e.g monoclonal antibodies) deglycosylation is not necessary because the disulfide-bonded peptides resulting from digestion using the commonly used enzymes usually do not contain a glycosylation site [65]. However, other glycoproteins may contain one or more occupied glycosylation sites near cysteine residues involved in a disulfide bond. In such cases, a deglycosylation step must be added to the sample preparation to reduce the complexity of the MS/MS data of the disulfide bonded peptides, because glycosylation cannot be simply regarded as a modification on the peptide chains [71, 75, 77]. For example, trypsin digestion of the HIV-1 Env sequence variant, C97ZA012 gp140, which contains 25 N-linked glycosylation sites and 10 disulfide bonds, resulted in seven tryptic, disulfide-linked peptides that all contain at least one occupied glycosylation site on the disulfide-linked chains [75]. One of the tryptic digests was a four-chain disulfide-linked peptide with six occupied glycosylation sites. For such complex cases,, deglycosylation is a necessary first step.
Deglycosylation is typically done enzymatically using Peptide N-glycosidase F (PNGase F) at neutral or slightly acidic pH [71, 75, 78], and the reaction can extend for several days if the protein is heavily glycosylated [75, 78]. Hence, proper Cys alkylation must be achieved to prevent disulfide shuffling during such long incubation periods. In addition, deglycosylation converts the occupied asparagine residue (N) to an aspartic acid residue (D), leading to a 0.985 Da mass change, which must be considered when calculating the molecular masses of the deglycosylated disulfide-linked peptides.
3. LIQUID CHROMATOGRAPHY-MASS SPECTROMETRIC DISULFIDE BOND ANALYSIS OF PROTEINS
3.1 Separation of Protein Digests
Disulfide bond mapping by liquid chromatography coupled to mass spectrometry requires the separation and ionization of protein digests prior to mass spectrometric analysis. The widely-used separation technique is reverse phase liquid chromatography (RPLC) by means of columns packed with either C8 or C18 stationary phases and mobile phases consisting of polar (e.g water) and non-polar (e.g acetonitrile) solvents containing modifiers such as formic acid and trifluoroacetic acid (0.01 to 0.1%, v/v). Different types of columns can be used for peptide separation, including microbore (e.g 1.0 and 2.1 mm internal diameter, i.d) [54, 79], capillary (e.g 0.5 mm i.d) [80] and nano columns (typically 0.075 mm i.d) [65]. The typical particle size is between 3 to 5 μm. Columns packed with 1.7 μm particles are becoming more common but they require much higher pressures for separation, as pressure is inversely related to the square of the particle diameter.
Peptide separation is usually followed by online electrospray ionization and tandem mass spectrometry (ESI-MS/MS) analysis. However, in some cases where the disulfide-linked peptides to be analyzed are very large and in low abundance, offline LC fractionation can be used to collect and concentrate fractions containing the disulfide-linked peptides (and carryout further digestion if necessary), prior to MS/MS analysis [5].
3.2 Fragmentation Techniques for Disulfide Bond Analysis
Collision induced dissociation (CID), where a neutral reagent gas collides with the analyte ion, and electron transfer dissociation (ETD), where an electron carrier reacts the ion of interest, are the most commonly used fragmentation techniques for mass spectrometry-based disulfide bond analysis [54, 80, 81]. Figure 4 shows CID and ETD spectra of a simple interchain disulfide-bonded peptide from IgG3 monoclonal antibody that exhibits the characteristic fragment ion peaks that result from subjecting disulfide-bonded peptides to CID (Figure 4A) and ETD (Figure 4B) fragmentation.
Figure 4.
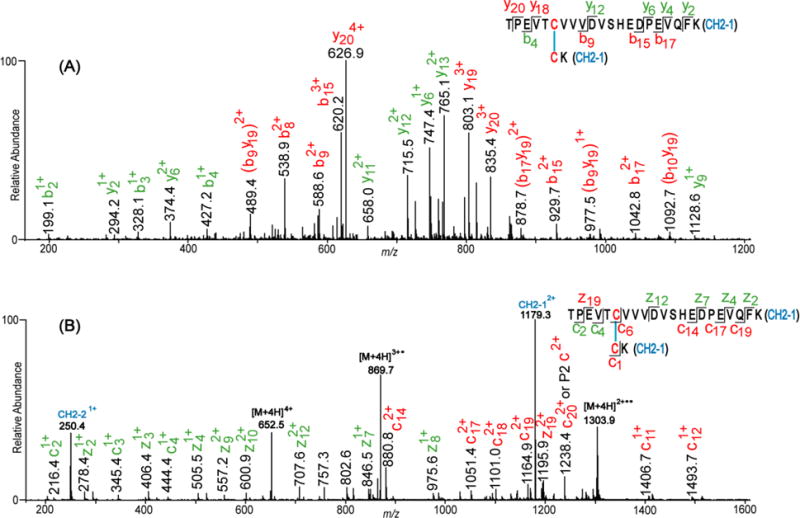
CID (A) and ETD (B) spectra of a disulfide bonded peptide from the CH2 domain of IgG3 monoclonal antibody. The spectra show the characteristic b/y (A) and c/z (B) ions resulting from the backbone cleavage of the peptides linked by the disulfide bond (DSB). Fragment ions not containing the DSB are labeled in green, and those containing the intact DSB are labeled in red. CID fragment ions resulting from two cleavage events and containing the intact DSB are in brackets. In the ETD spectrum (B), intense peaks of the Cys-containing peptides linked by the disulfide bond are observed at m/z 250.4 and 1179.3. These peaks result from the cleavage of the DSB by ETD, and they are not observed in (A).
CID typically leads to the fragmentation of the peptide backbone (amide) bonds, while leaving the disulfide bond intact; hence producing b and y ions that contain the disulfide bond (ions in red in Figure 4A), as well as ions that do not contain the disulfide bond (ions in green, Figure 4A). Although assignment of CID-generated product ions of disulfide-bonded peptides was previously based on the assumption that only one peptide bond is cleaved during CID, Clark et al [80] recently showed that cleavage of two peptide bonds (double cleavage) is common during CID fragmentation of disulfide-linked peptides. The peaks labeled in red brackets in Figure 4A are examples of this type of cleavage. Although CID does not typically cleave disulfide bonds, a few instances have been reported where thioaldehydes, persulfides, and dehydroalanine ion peaks resulting from cleavage of the C-S and S-S (disulfide) bonds are present in CID spectra [72, 82].
In contrast to the product ions generated from CID of disulfide bonded peptides, cleavage of the disulfide bond is the primary reaction pathway during ETD fragmentation. Backbone (amide) cleavage also occurs, by generation of c and z ions, although this fragmentation pathway generally occurs at a lesser extent [83, 84]. As a result, fragment peaks representing the disulfide-linked peptides (e.g peaks at m/z 250.4 and 1179.3 in Figure 4B) are usually more intense in ETD spectra than c/z ion peaks [53, 54, 83]. Peaks resulting from backbone fragmentation of the bonded peptides may or may not contain the intact disulfide bond (fragment ions labeled in red and green in Figure 4B).
Besides CID and ETD, higher-energy collisional dissociation (HCD) [85] and a dual fragmentation technique known as electron-transfer and higher-energy collision dissociation (EThcD) [86, 87] are newer fragmentation techniques available in some Orbitrap mass spectrometers, and they are gaining ground in disulfide bond analysis [69, 79, 74]. HCD has similar fragmentation pattern to beam-type CID [88], and it produces only b/y ions, while EThcD spectra exhibit product ions present in both ETD and CID spectra, and it has outperformed ETD in comparative studies [74]. In addition, ultraviolet photodissociation (UVPD) is another fragmentation technique that is being applied for disulfide bond analysis [89, 90]. Fragmentation of disulfide-linked peptides by UVPD leads to selective scission of the disulfide bond, as observed under ETD conditions [90].
3.2.1 Considerations for Selecting Fragmentation Techniques for Disulfide Bond Analysis
Choosing a suitable fragmentation technique or combination of techniques for data acquisition is vital in facilitating disulfide bond assignments from MS/MS data. The choice of a fragmentation method is typically based on the complexity of the disulfide linkages, the size of the disulfide bonded peptides, and, to a lesser extent, on the amino acid sequence of the bonded chains. Although either CID or ETD can be used to assign simple interchain disulfide bonded peptides (as shown in Figure 4), a careful choice of a fragmentation method is necessary for the assignment of complex disulfide linkages involving either intrachain or both interchain and intrachain disulfide bonds, large disulfide-bonded peptides with multiple peptide chains, as well as very small disulfide bonded peptides.
ETD is preferred for the fragmentation of peptides that are completely cyclized by a disulfide bond, as recently demonstrated by Xia and colleagues [91]. CID fragmentation of such peptides would typically not reveal any backbone sequence ions, whereas ETD leads to cleavage of the disulfide bond and the peptide backbone, thereby revealing ions that can be used to assign the disulfide bond [91]. ETD followed by a CID-MS3 step (ETD-CID-MS3), as reported by Karger et al [52], as well as a fragmentation technique involving hydroxyl radical addition followed by CID fragmentation, as reported by Durand et al [92], can also be used to map such disulfides.
For complex disulfide cases, such as nested disulfides and cysteine knots, neither ETD nor CID may be sufficient as a standalone technique to unambiguously assign the complex disulfide bond connectivity. In such cases, methods involving combined ETD and CID fragmentations would be ideal to decipher the disulfide connectivity [71, 93]. The EThcD fragmentation method could be promising in mapping these complex disulfides, but it has not yet been applied to such cases.
In terms of size, ETD is preferable for the assignment of large disulfide-bonded peptides containing three or more peptides linked by interchain disulfide bonds. Such large peptides can easily ionize in sufficiently high charge states for ETD analysis, since ETD requires highly charged ions for efficient fragmentation. Due to preferential cleavage of the disulfide bonds, ETD spectra of large multi-chain DSBPs are less complex, and they clearly reveal which peptides are directly linked to each other [75]. However, CID spectra of such large disulfide-linked peptides are complex and difficult to assign (e.g because of several double cleavages), and the spectra do not reveal which of the peptide chains are directly bonded to each other. For example, Go et al [75] assigned a three-chain disulfide-linked peptide using both CID and ETD spectra, but only ETD was used to unambiguously assign a four-chain disulfide-linked peptide. The ETD spectra of the three-chain and the four-chain disulfide-bonded peptides clearly indicated the chains that were disulfide-linked to each other [75].
On the contrary, CID is ideal for analysis of small disulfide-bonded peptides and disulfide-linked peptides with peptide chains that contain several adjacent proline residues. CID is preferable in such cases because small disulfide bonded peptides may not ionize at high charge states, which are required for ETD analysis, and peptide chains containing adjacent proline residues fragment efficiently when subjected to CID but not ETD fragmentation, due to the N-Cα ring structure of proline [94]. For example, the hinge region of IgG3 contains a small dipeptide, CPEPK linked to CPEPK, and SCDTPPPCPR disulfide-linked to SCDTPPPCPR and a recent analysis of IgG3 disulfide bonds showed that these peptide chains did not fragment well under ETD because of the small size (for the first disulfide-linked peptide) and several adjacent proline residues on the peptide chains (for both disulfide-linked peptides), but they were unambiguously assigned using CID data [54]. Hence, in addition to the size of a disulfide-linked peptide, the amino acid sequences of the Cys-containing peptides could also be a factor in selecting an efficient fragmentation technique for disulfide analysis.
Given the above explanations, it is imperative for researchers to do in silico digestion of a protein in order to study the possible types of disulfide linked peptides (see Table 1) and make a decision on which fragmentation method (or combination of methods) would be suitable for unambiguous mapping of the disulfide bonds in the protein of interest. In general, the collection of both ETD and CID data proves to be very useful, since the data sets are so complementary [53, 54].
3.3 Bottom-up Methods for Disulfide Bond Analysis
A variety of bottom-up mass spectrometric methods based on the aforementioned fragmentation techniques have been reported in recent years. Figure 5 shows a schematic representation of bottom-up approaches for disulfide bond analysis. The methods generally fall under two broad categories; disulfide bond mapping from: (1) reduced and non-reduced protein digests (profile comparison), and (2) only intact (non-reduced) protein digests. The second category can be further divided into methods that require chemical or electrochemical post-column reduction of disulfide bonds, reduction of the disulfide bonds in the gas phase (ETD and EThcD fragmentations), and those that do not require any reduction (typically CID and HCD methods). In the following sections, we review the bottom-up methods that have been developed in the past decade.
Figure 5.
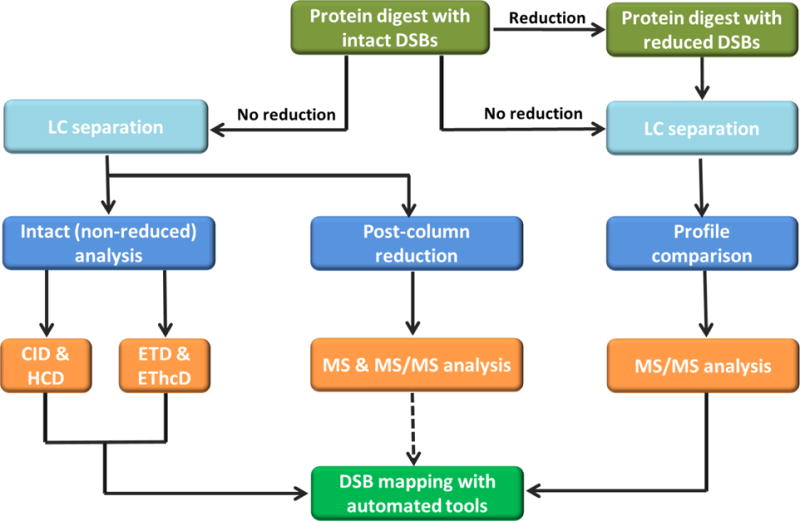
Schematic representation of bottom-up approaches for disulfide bond analysis. The methods generally fall under two broad categories: (1) disulfide bond mapping from protein digests with reduced and non-reduced disulfide bonds (profile comparison), and (2) disulfide bond mapping from only intact (non-reduced) protein digests. Non-reduced analysis can be further divided into methods that involve post-column reduction of disulfide bonds, reduction of the disulfide bonds in the gas phase (ETD and EThcD fragmentation methods), and those that do not require reduction of the disulfide bonds (CID and HCD fragmentation methods). Data analysis after post-column reduction has not been automated yet.
3.3.1 Profile Comparison: Reduced and Non-reduced Analysis
Disulfide bond mapping by profile comparison is usually done using two samples: a protein digest without reduced disulfide bonds (non-reduced sample) and a digest with reduced disulfide bonds (reduced sample). The two samples are separated in two LC runs (experiments), and peptides that are present in the ultraviolet (UV) or total ion chromatogram (TIC) profile of the non-reduced sample, but are absent in that of the reduced sample, are generally considered to be disulfide bonded, and MS/MS data can be used to assign the disulfide bonds [55, 56, 77].
Herein, we present three recent examples where complete or partial reduction of the disulfide bonds was used to assist in disulfide mapping. Researchers at Genentech used the reduced and non-reduced peptide mapping technique to assign the disulfide bonds of an unusual IgG1 antibody variant that was identified after size exclusion chromatography (SEC) [95]. Results from several experiments suggested that the mAb variant contained an extra light chain connected to the IgG1 monomer via a disulfide bond. LC-MS/MS experiments using non-reduced and reduced digests of the variant were used to confirm the disulfide linkage site of the extra light chain to the correct IgG1 monomer. In a second related example, an approach that deviates from the standard profile comparison method was reported by Klapoetke et al [96] To alleviate the need for preparing and analyzing two separate samples, these researchers conducted a partial reduction of the disulfide bonds and alkylated the resulting resulting free Cys, prior to enzymatic digestion. LC-MS/MS analysis of the mixed sample, containing partially reduced and non-reduced disulfide-linked peptides, was used to assign the disulfide bonds of the protein. The method is suitable to assign complex disulfides such as Cys knots, while simple disulfides can be mapped using a non-reduced sample [96]. A partial reduction approach has also been used by Albert et al to properly assign disulfide linkages in disulfide rich peptides. The peptides were immobilized on a stationary phase so only one disulfide linkage could be reduced at a time, and sequential alkylation steps with differential alkylating reagents were used to elucidate disulfide connectivity. [97] The challenging aspect of these approaches is to optimize the reducing agents to obtain partial reduction, as well as optimizing the alkylating agent to completely alkylate the reduced species. A shortcoming of the techniques that require disulfide reduction in the workflow is the need to prepare and analyze two samples, or to work out sample prep conditions that afford partial reduction, thereby increasing sample preparation and analysis time.
3.3.2 Intact (Non-reduced) Analysis
An alternative approach to mapping disulfide bonds is by peptide mapping of non-reduced protein digests (intact digests) without the need for reduced aliquots. The methods for disulfide bond assignment from non-reduced protein digests can be divided into three categories: methods involving post-column chemical or electrolytic reduction of the disulfide bonds, those involving gas-phase reduction of the disulfide bonds, and methods that do not involve cleavage of the disulfide bonds. In this section, we describe the methods that fall in the first two categories. For the last category, the disulfide bonds are typically assigned by peptide mapping using CID data by calculating theoretical m/z values (at different charge states) of the expected disulfide bonded peptides and searching the calculated values in the MS data; the ions are assigned by identifying the characteristic fragment ions in the MS/MS spectra [78, 80]. Several automated tools have been developed to identify disulfide bonded peptides without cleaving the disulfide bond; these tools are discussed in the automated tools section (Section 4).
3.3.2.1 Post-Column Reduction Methods
Several methods have been developed whereby non-reduced protein digests are separated by liquid chromatography followed by post-column partial reduction of disulfide bonded peptides prior to mass spectrometric characterization, typically with CID fragmentation. Post-column reduction of disulfide bonds can be done chemically by introducing a reducing agent such as Tris (2-carboxyethyl) phosphine (TCEP), by in-source reduction during ionization, or electrochemically via an increased potential on an electrochemical cell placed on the flow path from the column.
Post-column chemical reduction of disulfide-bonded proteolytic digests typically involves separation of the non-reduced protein digests by reversed phase liquid chromatography, and the eluates from the column are mixed (via a mixing-tee) with an optimized amount of a reducing agent (e.g TCEP) to partially reduce the disulfide bonds, as demonstrated by Li et al [98, 99]. Since only partial disulfide bond reduction occurs after LC separation, disulfide-linked peptides with intact disulfide bonds and their corresponding Cys-containing peptides that resulted from the disulfide bond reduction would have the same retention time and would both be detected in the same full scan mass spectrum. To identify disulfide-bonded peptides, the extracted ion chromatograms of all Cys-containing peptides are compared, and the Cys-containing peptides that have the same retention time are assigned as disulfide bonded [98, 99]. The assignments are confirmed by the presence of a disulfide-linked peptide at the same retention time with a molecular mass corresponding to the sum of the molecular masses of the two Cys-containing peptides minus 2 Da. Scrambled disulfide bonds are readily assigned in the same manner [99]. Liu and coworkers [100] used this approach to map the disulfide bonds of an IgG2 antibody, but instead of using only LC-MS data, LC-MS/MS (CID) data were used for unambiguous characterization of the LC peaks of the disulfide bonded peptides and those of their corresponding Cys-containing peptides.
Besides chemical reduction, in-source reduction during electrospray ionization can be used for partial reduction of disulfide-linked peptides, as shown by Cramer et al [79]. Post-column in-source reduction, followed by HCD analysis of the partially reduced peptides, was used to develop a method capable of assigning complex intra- and inter-chain disulfide bonds in proteins, including disulfide bonds involving closely-spaced Cys residues [79].
Disulfide bond characterization methods involving post-column electrochemical (EC) reduction of disulfide bonds prior to mass spectrometric analysis are also emerging. Cramer et al [101] recently demonstrated that disulfide bond assignment in small proteins, such as human insulin, can be done by direct infusion of the intact protein into an electrochemical cell for partial reduction of the disulfide bonds from the intact protein, followed by tandem mass spectrometry (CID fragmentation) to confirm the sequence coverage of the protein. For larger proteins (e.g human serum albumin and ribonuclease B), proteolytic digestion and LC separation of the protein digests are necessary prior to post-column EC reduction of the disulfide-bonded peptides and MS/MS fragmentation (LC-EC-MS/MS), as recently demonstrated by Switzar et al [102]. A rapid method employing on-column proteolytic digestion and electrochemical reduction was reported by Chen et al [103, 104]. In this approach, an online pepsin column is used for rapid digestion of intact proteins, and disulfide bonded peptides are partially or completely reduced using different electrochemical potentials, followed by desorption electrospray tandem mass spectrometry (DESI-MS/MS) [103]. This online digestion approach provides unprecedented digestion and reduction speeds (less than 10 mins) and very low potential for inducing disulfide artifacts due to less sample handling and pepsin digestion at low pH.
In general, although the chemical and electrochemical post column partial reduction methods do save sample preparation time (compared to the case of reduced and non-reduced samples), optimizing the reduction conditions to get sufficient signals for both the reduced and non-reduced peptides is a major challenge.
3.3.2.2 Gas phase Dissociation (Reduction) Methods
Because ETD mainly cleaves disulfide bonds (gas phase reduction) leading to intense Cys-containing peptide product ions, several disulfide bond characterization methods have been developed that take advantage of the unique ETD fragmentation pattern to analyze non-reduced protein digests for disulfide bond mapping. Karger et al [105] reported a method that combines alternating ETD/CID fragmentation (MS2) of the disulfide-bonded peptides and CID-MS3 fragmentation of the Cys-containing fragment ions that resulted from the ETD-MS2 dissociation of disulfide bonds. This method is suitable to map disulfide-bonded peptides containing complex disulfide linkages, such as nested disulfides and cysteine knots [71], as well as peptides completely cyclized by a disulfide bond [91]. However, due to the enormous data generated because of the additional CID3 step, the method may not be necessary for samples that do not contain complex disulfides. Recently, Massonnet and coworkers [93] reported a similar technique for disulfide bond assignment of peptides containing two disulfide bonds. After opening of the disulfide bonds by ETD, the generated species are separated by ion mobility prior to characterization of the separated ions by CID fragmentation [93].
Clark et al reported a much simpler ETD-based method that is suitable for samples containing simple interchain disulfide-bonded peptides, including disulfides with multiple (three to four) disulfide-linked chains [53]. The method does not require an MS3 step, and instead of searching the m/z values of all expected (known) disulfides and possible scrambled disulfides, the extracted ion chromatograms (XIC’s) of all Cys-containing peptides are generated, and the ETD spectra of the peaks in the XICs are interrogated to determine the bonding partner(s) of the Cys-containing peptides [53, 54]. Disulfide assignments are made when fragment ions of Cys-containing peptides are identified in the same ETD spectrum, and the sum of their masses minus 2 Da (mass of 2H) match the mass of the expected disulfide. The c and z ions resulting from backbone cleavage of the bonded peptides can be used to further confirm the identities of the bonded chains [54]. This XIC approach is suitable to map the disulfide bonds in proteins whose disulfide connectivity is unknown and also to quickly verify non-native or alternative disulfide bonds. Figure 6 shows XICs used to assign both correct and scrambled disulfide linkages in an IgG antibody. In addition, the XICs of Cys-containing peptides can also provide information about any free Cys in the protein without the need for differential alkylation [106, 107].
Figure 6.
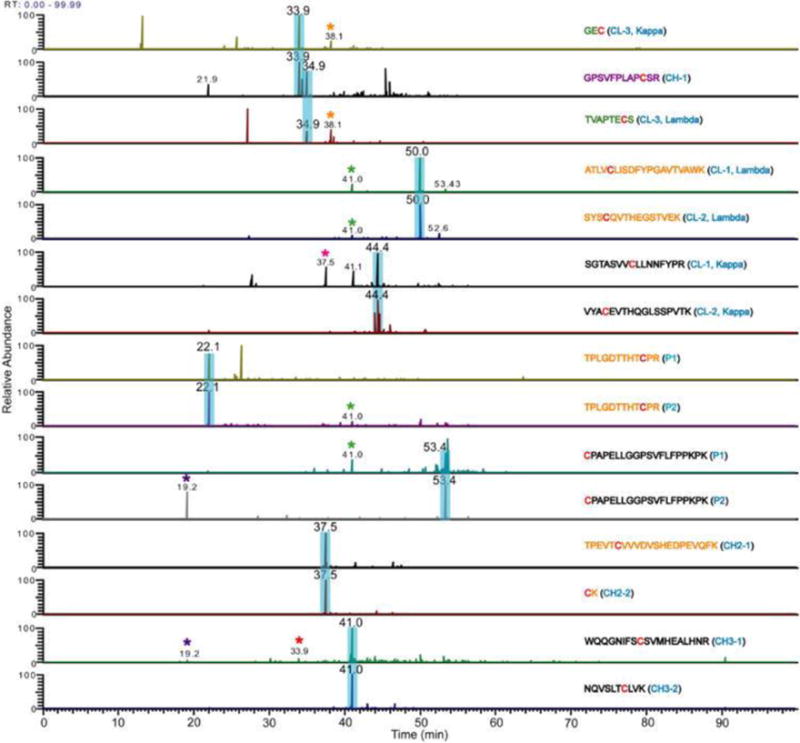
Example of Extracted Ion Chromatograms (XICs) used to identify disulfide linkages in an IgG antibody. Correct disulfide linkages are highlighted in blue. Aberrant linkages are marked with asterisks. Reproduced from Ref. 54 with permission from The Royal Society of Chemistry.
Similar to ETD fragmentation, ultraviolet photodissociation (UVPD) of simple interchain disulfide bonded peptides in the gas phase can be used to map disulfide bonds in proteins, as demonstrated by Agarwal et al [90]. After separation of non-reduced protein digests using LC, irradiation of disulfide-linked peptides, including three chain disulfide-linked peptides, with laser pulses at 266 nm in an ion-trap mass analyzer led to selective cleavage of disulfide bonds, thus revealing the disulfide bonded peptides. However, backbone cleavage of the Cys-containing peptides is not typically observed using this approach [90].
4. SOFTWARE FOR AUTOMATED DISULFIDE BOND ASSIGNMENT
Manual analysis of disulfide bonds in proteins is a challenging and time-consuming task that requires expertise in the field. As such, an increasing number of automated tools have been developed for rapid and high-throughput protein disulfide bond assignment using MS/MS data. Automated tools are urgently needed when large cohorts of samples need to be routinely analyzed, as is the case in biopharmaceutical industries. Most of the developed tools are based on the non-reduced analysis workflow, although a few tools also have the option to map disulfide bonds by comparing data from reduced and non-reduced samples.
MassMatrix, developed about a decade ago, is one of the prominent tools for automated assignment of disulfide-linked peptides from non-reduced protein digests [108]. Assignments are solely based on fragment ions from the backbone cleavage of the bonded peptides. This is a shortcoming of the tool, because only a limited number of ions are considered for disulfide bond assignment. Most importantly, fragment ions containing the disulfide bonds are not considered for disulfide bond mapping. Several tools have since been developed which address this problem. Dbond [72, 82] and MS2DB+ [109, 110] can map disulfides from MS/MS data using several characteristic fragmentation peaks of disulfide bonded peptides, including b/y or c/z ions containing intact disulfide bonds. While DBond assigns dipeptides solely from CID data, and includes ions resulting from the occasional CID cleavage of C-S and S-S bonds (resulting in thioaldehydes, persulfides and dehydroalanin ions) [72, 82], the MS2DB+ software can assign disulfides from either CID or ETD data, and the fragment ions considered for CID data include ions resulting from the loss of water or ammonia from b/y ions. For both software products, the theoretical m/z’s of all possible disulfide-bonded peptides are searched from MS1 data, and their identified precursor ions are automatically assigned by comparing theoretical and experimental MS/MS spectra of the disulfide-bonded peptides. In addition, the tools score the assigned disulfide-bonded peptides based on the number of matched fragment ions and their intensities.
A unique algorithm known as RADAR (Rapid Assignment of Disulfide Linkage via A1 ion Recognition) that assigns disulfide bonded peptides based on dimethyl labeling of the N-termini of the bonded chains was reported by Huang et al [111], and it has been applied for disulfide bond assignment in monoclonal antibodies [112]. Dimethyl-labeled peptides produce intense a1 ion signals in CID spectra, which are used to screen for disulfide bonded peptides. Because these species mostly contain two or more peptide chains, the CID spectra would have two or more a1 ion peaks, and RADAR rapidly screens for disulfide-linked peptides by first searching for such spectra that contain multiple a1 ion peaks [111]. Cys-containing peptides whose a1 ions are found in the same spectrum are considered to be disulfide bonded, and they are confirmed by matching the precursor ion mass to the molecular weight of the predicted disulfide-linked peptide, followed by assigning the fragment ions (b/y ions) resulting from fragmentation of the disulfide-bonded peptide chains [112]. Experimental workflow and sample MS2 data of RADAR are shown in Figure 7. This approach is suitable for rapid identification of disulfides, including scrambled disulfides, because prior knowledge of the disulfide connectivity is not required. Intrachain disulfides on a single peptide can be assigned in a similar manner but instead of searching for multiple a1 ions in the CID spectra, a specific (target) a1 ion corresponding to the N-terminal of the peptide can be searched for. The software is effective for disulfide mapping in complex protein mixtures (containing more than 20 disulfide bonds) and for rapid verification of disulfide scrambling [113].
Figure 7.
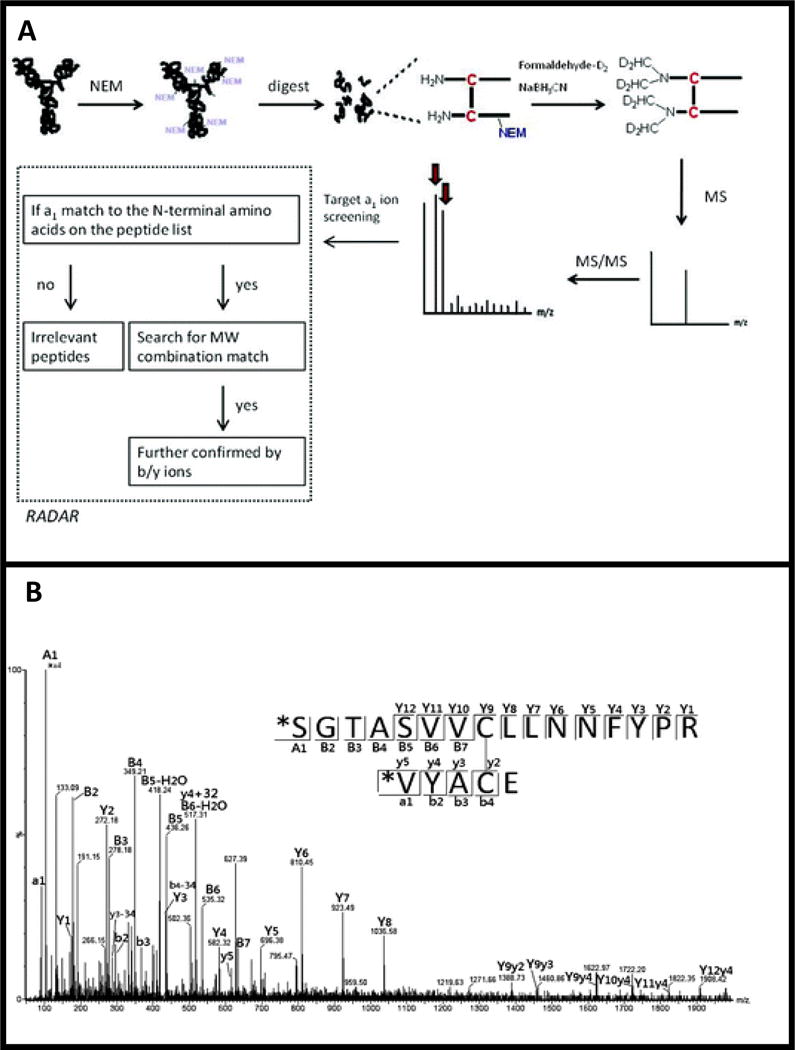
Rapid assignment of disulfide linkage via a1 ion recognition (RADAR), a1 ions are used to identify potential disulfides which are then confirmed using intact molecular weight and b/y ions. A) Experimental workflow for RADAR analysis. B) MS2 spectra used to identify disulfide bonded peptides. Adapted with permission from Ref. 112. Copyright 2012 American Chemical Society.
Tools that assign disulfide bonds from HCD and EThcD data have also been developed within the last decade. A software tool known as pLink-SS was introduced in 2015 by Lu and coworkers for automated disulfide bond assignment in proteins using HCD data [69]. The software was used to map simple and complex disulfides from a purified protein (IgG), a mixture of 10 proteins containing 74 disulfide bonds, and at the proteome level (199 DSBs in 150 proteins from E. Coli) using HCD data [69]. Another tool known as SlinkS assigns interchain and intrachain disulfide bonds by matching experimental MS/MS fragment ions from ETD and EThcD spectra to in silico fragments [74]. For assignments from ETD data, the software searches for marker ions corresponding to the masses of the disulfide-linked peptides, as well as c/z ions with and without disulfide bonds; while b/y ions are also included in the search when assigning disulfide bonds from EThcD data. The assigned disulfide-linked peptides are scored, and FDRs are calculated based on user-defined cut-off values.
Besides the aforementioned tools, several commercial software products are available for automated analysis of protein disulfide bonds connectivity using MS/MS data, particularly in protein pharmaceuticals. These include BioConfirm (Agilent Technologies), Byologic (Protein Metrics Inc), DisulfideDetect (Bruker), PepFinder, BioPharma Finder, and Pinpoint (ThermoFisher Scientific), BioPharmView (SCIEX), and BiopharmaLynx (Waters). While tools like DisulfideDetect require only non-reduced protein digest for disulfide analysis, PepFinder and BioPharma Finder can map disulfide-bonded peptides using either non-reduced protein digests or both reduced and non-reduced protein digests.
Although the automated assignment of disulfide bonded peptides has facilitated disulfide analysis of proteins, there is still room for improvement in this field. Most of the tools developed so far can readily assign simple inter-chain disulfide-linked peptides but would not readily assign disulfide-bonded peptides containing complex intertwined disulfide linkages involving both interchain and intrachain disulfide bonds, nested disulfides, and dipeptides with more than one inter-chain disulfide bond (disulfide box). Although several approaches can be utilized to manually assign the disulfide bonds of these complex cases [114], software that can readily assign such disulfide-bonded peptides remain elusive.
5. CHALLENGES IN DISULFIDE BOND ANALYSIS
Despite the existence of high resolution mass spectrometers, automated tools, and several strategies to map disulfide bonds in proteins, there are still some challenges that can be encountered during disulfide bond assignments. When non-native disulfide bonds are identified after bottom-up disulfide analysis, it is difficult to tell if they are disulfide artifacts introduced during sample preparation or if they are alternative disulfide bonds (disulfide variants) present in the sample. One way to verify if disulfide artifacts are introduced during sample preparation is to validate a method using a protein whose disulfide bond connectivity has been well characterized [78]. In addition, top-down analysis of the intact protein requires less sample handling, and it would be an important first step in pinpointing the source of non-native disulfide bonds [115]. Finally, when proteins have been identified to contain alternative disulfide bonds (disulfide-mediated isoforms), quantifying the amount of the protein that contains the native and alternative disulfide bonds remains a challenge because, many times, the disulfide-mediated isoforms would not be easily separated by standard separation techniques. State-of-the-art separation methods are a possible route forward for solving this problem, though separations of disulfide bond-mediated isoforms have yet to achieve baseline resolution [116, 117].
6. CONCLUSION
It is important to confirm the global disulfide bond structure of a particular protein and to verify whether or not any disulfide-mediated isoforms [5] or variants [95] are present, since different disulfide bond connectivity could result in different protein structures and biological functions. Several bottom-up mass spectrometry approaches have been developed for this purpose. In silico digestion is an important first step for disulfide mapping to decide what enzyme (or combination of enzymes) to use in order to get simple disulfide bonded peptides that are easy to analyze. The in silico digestion also can be used to help decide what fragmentation technique would be suitable for easy and rapid assignment of spectra of disulfide bonded peptides. The majority of disulfide mapping methods are based on CID and ETD fragmentation techniques, but with newer fragmentation techniques such as EThcD and HCD, additional disulfide mapping methods involving these techniques likely will be developed in the future. From a data analysis point of view, software tools will need to be developed for automated characterization of disulfide-bonded peptides with complex disulfide bond connectivity. Finally, quantitation of disulfide variants remains a challenge.
Acknowledgments
The authors acknowledge funding from the NIH, grant R01AI125093 and R01AI094797, to HD.
Footnotes
COMPLIANCE WITH ETHICAL STANDARDS
The authors of the paper declare that they have no conflicts of interest.
References
- 1.Woycechowsky KJ, Raines RT. Native Disulfide Bond Formation in Proteins. Curr Opin Chem Biol. 2000;4(5):533–539. doi: 10.1016/s1367-5931(00)00128-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wilkinson B, Gilbert HF. Protein Disulfide Isomerase. Biochim Biophys Acta. 2004;1699(1–2):35–44. doi: 10.1016/j.bbapap.2004.02.017. [DOI] [PubMed] [Google Scholar]
- 3.Butera D, Cook KM, Chiu J, Wong JW, Hogg PJ. Control of Blood Proteins by Functional Disulfide Bonds. Blood. 2014;123(13):2000–2007. doi: 10.1182/blood-2014-01-549816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Farrah T, Deutsch EW, Omenn GS, Campbell DS, Sun Z, Bletz JA, Mallick P, Katz JE, Malmstrom J, Ossola R, Watts JD, Lin B, Zhang H, Moritz RL, Aebersold R. A High-Confidence Human Plasma Proteome Reference Set with Estimated Concentrations in PeptideAtlas. Mol Cell Proteomics. 2011;10(9):M110.006353. doi: 10.1074/mcp.M110.006353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wypych J, Li M, Guo A, Zhang Z, Martinez T, Allen MJ, Fodor S, Kelner DN, Flynn GC, Liu YD, Bondarenko PV, Ricci MS, Dillon TM, Balland A. Structural and Functional Characterization of Disulfide Isoforms of the Human IgG2 Subclass. J Biol Chem. 2008;283(23):16194–16205. doi: 10.1074/jbc.M709987200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Alewood D, Nielsen K, Alewood PF, Craik DJ, Andrews P, Nerrie M, White S, Domagala T, Walker F, Rothacker J, Burgess AW, Nice EC. The Role of Disulfide Bonds in the Structure and Function of Murine Epidermal Growth Factor (mEGF) Growth Factors. 2005;23(2):97–110. doi: 10.1080/08977190500096061. [DOI] [PubMed] [Google Scholar]
- 7.Zhang L, Chou CP, Moo-Young M. Disulfide Bond Formation and its Impact on the Biological Activity and Stability of Recombinant Therapeutic Proteins Produced by Escherichia coli Expression System. Biotechnol Adv. 2011;29(6):923–929. doi: 10.1016/j.biotechadv.2011.07.013. [DOI] [PubMed] [Google Scholar]
- 8.Perry LJ, Wetzel R. Disulfide Bond Engineered into T4 Lysozyme: Stabilization of the Protein Toward Thermal Inactivation. Science. 1984;226(4674):555–557. doi: 10.1126/science.6387910. [DOI] [PubMed] [Google Scholar]
- 9.Mansfeld J, Vriend G, Dijkstra BW, Veltman OR, Van den Burg B, Venema G, Ulbrich-Hofmann R, Eijsink VG. Extreme Stabilization of a Thermolysin-like Protease by an Engineered Disulfide Bond. J Biol Chem. 1997;272(17):11152–11156. doi: 10.1074/jbc.272.17.11152. [DOI] [PubMed] [Google Scholar]
- 10.Kim DY, Kandalaft H, Ding W, Ryan S, van Faassen H, Hirama T, Foote SJ, MacKenzie R, Tanha J. Disulfide Linkage Engineering for Improving Biophysical Properties of Human VH Domains. Protein Eng Des Sel. 2012;25(10):581–590. doi: 10.1093/protein/gzs055. [DOI] [PubMed] [Google Scholar]
- 11.Dombkowski AA, Sultana KZ, Craig DB. Protein Disulfide Engineering. FEBS Lett. 2014;588(2):206–212. doi: 10.1016/j.febslet.2013.11.024. [DOI] [PubMed] [Google Scholar]
- 12.Seger ST, Breinholt J, Faber JH, Andersen MD, Wiberg C, Schjodt CB, Rand KD. Probing the Conformational and Functional Consequences of Disulfide Bond Engineering in Growth Hormone by Hydrogen-Deuterium Exchange Mass Spectrometry Coupled to Electron Transfer Dissociation. Anal Chem. 2015;87(12):5973–5980. doi: 10.1021/ac504782v. [DOI] [PubMed] [Google Scholar]
- 13.Go EP, Cupo A, Ringe R, Pugach P, Moore JP, Desaire H. Native Conformation and Canonical Disulfide Bond Formation are Interlinked Propertives of HIV-1 Env Glycoproteins. J Virol. 2015;90(6):2884–2894. doi: 10.1128/JVI.01953-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Le QAT, Joo JC, Yoo YJ, Kim YH. Development of Thermostable Candida antarctica Lipase B through Novel in silico Design of Disulfide Bridge. Biotechnol Bioeng. 2012;109(4):867–876. doi: 10.1002/bit.24371. [DOI] [PubMed] [Google Scholar]
- 15.Compton JR, Legler PM, Clingan BV, Olson MA, Millard CB. Introduction of a Disulfide Bond Leads to Stabilization and Crystallization of a Ricin Immunogen. Proteins. 2011;79(4):1048–1060. doi: 10.1002/prot.22933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu W, Onda M, Kim C, Xiang L, Weldon JE, Lee B, Pastan I. A Recombinant Immunotoxin Engineered for Increased Stability by Adding a Disulfide Bond has Decreased Immunogenicity. Protein Eng Des Sel. 2012;25(1):1–6. doi: 10.1093/protein/gzr053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li Y, Li X, Zheng X, Tang L, Xu W, Gong M. Disulfide Bond Prolongs the Half-Life of Therapeutic Peptide-GLP-1. Peptides. 2011;32(7):1400–1407. doi: 10.1016/j.peptides.2011.05.003. [DOI] [PubMed] [Google Scholar]
- 18.Li Y, Zheng X, Tang L, Xu W, Gong M. GLP-1 Analogs Containing Disulfide Bond Exhibited Prolonged Half-Life in vivo than GLP-1. Peptides. 2011;32(6):1303–1312. doi: 10.1016/j.peptides.2011.04.001. [DOI] [PubMed] [Google Scholar]
- 19.Siadat OR, Lougarre A, Lamouroux L, Ladurantie C, Fournier D. The Effect of Engineered Disulfide Bonds on the Stability of Drosophila Melanogaster Acetylcholinesterase. BMC Biochem. 2006;7:12. doi: 10.1186/1471-2091-7-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Azimi I, Wong JW, Hogg PJ. Control of Mature Protein Function by Allosteric Disulfide Bonds. Antioxid Redox Signal. 2011;14(1):113–126. doi: 10.1089/ars.2010.3620. [DOI] [PubMed] [Google Scholar]
- 21.Hogg PJ. Targeting Allosteric Disulphide Bonds in Cancer. Nat Rev Cancer. 2013;13(6):425–431. doi: 10.1038/nrc3519. [DOI] [PubMed] [Google Scholar]
- 22.Suzuki M, Yamanoi A, Machino Y, Kobayashi E, Fukuchi K, Tsukimoto M, Kojima S, Kohroki J, Akimoto K, Masuho Y. Cleavage of the Interchain Disulfide Bonds in Rituximab Increases its Affinity for FcγRIIIA. Biochem Biophys Res Commun. 2013;436(3):519–524. doi: 10.1016/j.bbrc.2013.05.137. [DOI] [PubMed] [Google Scholar]
- 23.Suzuki M, Yamanoi A, Machino Y, Ootsubo M, Izawa K, Kohroki J, Masuho Y. Effect of Trastuzumab Interchain Disulfide Bond Cleavage on Fcγ Receptor Binding and Antibody-Dependent Tumour Cell Phagocytosis. J Biochem. 2016;159(1):67–76. doi: 10.1093/jb/mvv074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lightle S, Aykent S, Lacher N, Mitaksov V, Wells K, Zobel J, Oliphant T. Mutations within a Human IgG2 Antibody Form Distinct and Homogeneous Disulfide Isomers but do not Affect Fc Gamma Receptor or C1q Binding. Protein sci. 2010;19(4):753–762. doi: 10.1002/pro.352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Walsh G. Biopharmaceutical Benchmarks 2014. Nat Biotechnol. 2014;32(10):992–1000. doi: 10.1038/nbt.3040. [DOI] [PubMed] [Google Scholar]
- 26.Reichert JM. Antibodies to Watch in 2016. mAbs. 2016;8(2):197–204. doi: 10.1080/19420862.2015.1125583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Michel ML, Tiollais P. Hepatitis B Vaccines: Protective Efficacy and Therapeutic Potential. Pathol Biol. 2010;58(4):288–295. doi: 10.1016/j.patbio.2010.01.006. [DOI] [PubMed] [Google Scholar]
- 28.Nascimento IP, Leite LCC. Recombinant Vaccines and the Development of New Vaccine Strategies. Braz J Med Biol Res. 2012;45(12):1102–1111. doi: 10.1590/S0100-879X2012007500142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Amoresano A, Orru S, Siciliano RA, De Luca E, Napoleoni R, Sirna A, Pucci P. Assignment of the Complete Disulphide Bridge Pattern in the Human Recombinant Follitropin β-chain. Biol Chem. 2001;382(6):961–968. doi: 10.1515/BC.2001.120. [DOI] [PubMed] [Google Scholar]
- 30.Reichert JM. Marketed Therapeutic Antibodies Compendium. mAbs. 2012;4(3):413–415. doi: 10.4161/mabs.19931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wadhwa M, Thorpe R. Haematopoietic Growth Factors and their Therapeutic Use. Thromb Haemost. 2008;99(5):863–873. [PubMed] [Google Scholar]
- 32.Calcutt NA, Jolivalt CG, Fernyhough P. Growth Factors as Therapeutics for Diabetic Neuropathy. Curr Drug Targets. 2008;9(1):47–59. doi: 10.2174/138945008783431727. [DOI] [PubMed] [Google Scholar]
- 33.Liu HC, May K. Disulfide Bond Structures of IgG Molecules: Structural Variations, Chemical Modifications and Possible Impacts to Stability and Biological Function. mAbs. 2012;4(1):17–23. doi: 10.4161/mabs.4.1.18347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Trexler-Schmidt M, Sargis S, Chiu J, Sze-Khoo S, Mun M, Kao YH, Laird MW. Identification and Prevention of Antibody Disulfide Bond Reduction During Cell Culture Manufacturing. Biotechnol Bioeng. 2010;106(3):452–461. doi: 10.1002/bit.22699. [DOI] [PubMed] [Google Scholar]
- 35.Hutterer KM, Hong RW, Lull J, Zhao X, Wang T, Pei R, Le ME, Borisov O, Piper R, Liu YD, Petty K, Apostol I, Flynn GC. Monoclonal Antibody Disulfide Reduction During Manufacturing. mAbs. 2013;5(4):608–613. doi: 10.4161/mabs.24725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yoshioka S, Aso Y, Izutsu K, Terao T. Aggregates Formed During Storage of β-Galactosidase in Solution and in the Freeze-Dried State. Pharm Res. 1993;10(5):687–691. doi: 10.1023/a:1018951530927. [DOI] [PubMed] [Google Scholar]
- 37.Chandrasekhar S, Topp EM. Thiol-Disulfide Exchange in Peptides Derived from Human Growth Hormone During Lyophilization and Storage in the Solid State. J Pharm Sci. 2015;104(4):1291–1302. doi: 10.1002/jps.24370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Andya JD, Hsu CC, Shire SJ. Mechanisms of Aggregate Formation and Carbohydrate Excipient Stabilizations of Lyophilized Humanized Monoclonal Antibody Formulations. AAPS PharmSci. 2003;5(2):E10. doi: 10.1208/ps050210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vazquez-Rey M, Lang DA. Aggregates in Monoclonal Antibody Manufacturing Processes. Biotechnol Bioeng. 2011;108(7):1494–508. doi: 10.1002/bit.23155. [DOI] [PubMed] [Google Scholar]
- 40.FDA. Q6B Specifications: Test Procedures and Acceptance Criteria for Biotechnological/Biological Products. (199) [Google Scholar]
- 41.Rathore AS, Winkle H. Quality by Design for Biopharmaceuticals. Nat Biotech. 2009;27(1):26–34. doi: 10.1038/nbt0109-26. [DOI] [PubMed] [Google Scholar]
- 42.Klaus W, Broger C, Gerber P, Senn H. Determination of the Disulphide Bonding Pattern in Proteins by Local and Global Analysis of Nuclear Magnetic Resonance Data. Application to Flavoridin. J Mol Biol. 1993;232(3):897–906. doi: 10.1006/jmbi.1993.1438. [DOI] [PubMed] [Google Scholar]
- 43.Mobli M, King GF. NMR Methods for Determining Disulfide Bond Connectivities. Toxicon. 2010;56(6):849–854. doi: 10.1016/j.toxicon.2010.06.018. [DOI] [PubMed] [Google Scholar]
- 44.Poppe L, Hui JO, Ligutti J, Murray JK, Schnier PD. PADLOC: A Powerful Tool to Assign Disulfide Bond Connectivities in Peptides and Proteins by NMR Spectroscopy. Anal Chem. 2012;84(1):262–266. doi: 10.1021/ac203078x. [DOI] [PubMed] [Google Scholar]
- 45.Liu D, Cowburn D. Combining Biophysical Methods to Analyze the Disulfide Bond in SH2 Domain of C-terminal Src Kinase. Biophys Rep. 2016;2(1):33–43. doi: 10.1007/s41048-016-0025-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang B, Cockrill SL. Methodology for Determining Disulfide Linkage Patterns of Closely Spaced Cysteine Residues. Anal Chem. 2009;81(17):7314–7320. doi: 10.1021/ac901161e. [DOI] [PubMed] [Google Scholar]
- 47.Perham RN. A Diagonal Paper-Electrophoretic Technique for Studying Amino Acid Sequences around the Cysteine and Cystine Residues of Proteins. Biochem J. 1967;105(3):1203–1207. doi: 10.1042/bj1051203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Milstein C, Frangion B. Disulphide Bridges of the Heavy Chain of Human Immunoglobulin G2. Biochem J. 1971;121(2):217–225. doi: 10.1042/bj1210217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gorman JJ, Wallis TP, Pitt JJ. Protein Disulfide Bond Determination by Mass Spectrometry. Mass Spectrom Rev. 2002;21(3):183–216. doi: 10.1002/mas.10025. [DOI] [PubMed] [Google Scholar]
- 50.Tsai PL, Chen SF, Huang SY. Mass Spectrometry-Based Strategies for Protein Disulfide Bond Identification. Rev Anal Chem. 2013;32(4):257–268. [Google Scholar]
- 51.Wiesner J, Resemann A, Evans C, Suckau D, Jabs W. Advanced Mass Spectrometry Workflows for Analyzing Disulfide Bonds in Biologics. Expert Rev Proteomics. 2015;12(2):115–123. doi: 10.1586/14789450.2015.1018896. [DOI] [PubMed] [Google Scholar]
- 52.Wu SL, Jiang H, Lu Q, Dai S, Hancock W, Karger BL. Mass Spectrometric Determination of Disulfide Linkages in Recombinant Therapeutic Proteins Using Online LC-MS with Electron-Transfer Dissociation. Anal Chem. 2009;81:112–122. doi: 10.1021/ac801560k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Clark DF, Go EP, Desaire H. Simple Approach to Assign Disulfide Connectivity Using Extracted Ion Chromatograms of Electron Transfer Dissociation Spectra. Anal Chem. 2013;85(2):1192–1199. doi: 10.1021/ac303124w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Lakbub JC, Clark DF, Shah IS, Zhu Z, Su X, Go EP, Tolbert TJ, Desaire H. Disulfide Bond Characterization of Endogenous IgG3 Monoclonal Antibodies Using LC-MS: an Investigation of IgG3 Disulfide-Mediated Isoforms. Anal Methods. 2016;8(31):6046–6055. doi: 10.1039/C6AY01248E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pitt JJ, Da Silva E, Gorman JJ. Determination of the Disulfide Bond Arrangement of Newcastle Disease Virus Hemagglutinin Neuraminidase. J Biol Chem. 2000;275(9):6469–6478. doi: 10.1074/jbc.275.9.6469. [DOI] [PubMed] [Google Scholar]
- 56.Zhang W, Marzilli LA, Rouse JC, Czupryn MJ. Complete Disulfide Bond Assignment of a Recombinant Immunoglublin G4 Monoclonal. Anal Biochem. 2002;311(1):1–9. doi: 10.1016/s0003-2697(02)00394-9. [DOI] [PubMed] [Google Scholar]
- 57.Sung WC, Chang CW, Huang SY, Wei TY, Huang YL, Lin YH, Chen HM, Chen SF. Evaluation of Disulfide Scrambling During the Enzymatic Digestion of Bevacizumab at Various pH Values Using Mass Spectrometry. Biochim Biophys Acta. 2016;1864(9):1188–1194. doi: 10.1016/j.bbapap.2016.05.011. [DOI] [PubMed] [Google Scholar]
- 58.Wang Y, Li H, Shameem M, Xu W. Development of a Sample Preparation Method for Monitoring Correct Disulfide Linkages of Monoclonal Antibodies by Liquid Chromatography-Mass Spectrometry. Anal Biochem. 2016;495:21–28. doi: 10.1016/j.ab.2015.11.010. [DOI] [PubMed] [Google Scholar]
- 59.Sanger F. A Disulphide Interchange Reaction. Nature. 1953;171(4362):1025–1026. doi: 10.1038/1711025a0. [DOI] [PubMed] [Google Scholar]
- 60.Ryle AP, Sanger F. Disulphide Interchange Reactions. Biochem J. 1955;60(4):535–540. [PMC free article] [PubMed] [Google Scholar]
- 61.Costantino HR, Schwendeman SP, Langer R, Klibanov AM. Deterioration of Lyophilized Pharmaceutical Proteins. Biochem (Mosc) 1998;63(3):357–363. [PubMed] [Google Scholar]
- 62.Chandrasekhar S, Moorthy BS, Xie R, Topp EM. Thiol-Disulfide Exchange in Human Growth Hormone. Pharm Res. 2016;33(6):1370–1382. doi: 10.1007/s11095-016-1879-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Monahan FJ, German JB, Kinsella JE. Effect of pH and Temperature on Protein Unfolding and Thiol/Disulfide Interchange Reactions During Heat-Induced Gelatin of Whey Proteins. J Agr Food Chem. 1995;43(1):46–52. [Google Scholar]
- 64.Kerr J, Schlosser JL, Griffin DR, Wong DY, Kasko AM. Steric Effects in Peptide and Protein Exchange with Activated Disulfides. Biomacromolecules. 2013;14(8):2822–2829. doi: 10.1021/bm400643p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wang Y, Lu Q, Wu SL, Karger BL, Hancock WS. Characterization and Comparison of Disulfide Linkages and Scrambling Patterns in Therapeutic Monoclonal Antibodies: Using LC-MS with Electron Transfer Dissociation. Anal Chem. 2011;83(8):3133–3140. doi: 10.1021/ac200128d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Creamer LK, Bienvenue A, Nilsson H, Paulsson M, van Wanroij M, Lowe EK, Anema SG, Boland MJ, Jimenez-Flores R. Heat-Induced Redistribution of Disulfide Bonds in Milk Proteins. 1. Bovine β-lactoglobulin. J Agr Food Chem. 2004;52(25):7660–7668. doi: 10.1021/jf049388y. [DOI] [PubMed] [Google Scholar]
- 67.Sechi S, Chait BT. Modification of Cysteine Residues by Alkylation. A Tool in Peptide Mapping and Protein Identification. Anal Chem. 1998;70(24):5150–5158. doi: 10.1021/ac9806005. [DOI] [PubMed] [Google Scholar]
- 68.Rogers LK, Leinweber BL, Smith CV. Detection of Reversible Protein Thiol Modifications in Tissues. Anal Biochem. 2006;358(2):171–184. doi: 10.1016/j.ab.2006.08.020. [DOI] [PubMed] [Google Scholar]
- 69.Lu S, Fan SB, Yang B, Li YX, Meng JM, Wu L, Li P, Zhang K, Zhang MJ, Fu Y, Luo J, Sun RX, He SM, Dong MQ. Mapping Native Disulfide Bonds at the Proteome Scale. Nat Methods. 2015;12(4):329–331. doi: 10.1038/nmeth.3283. [DOI] [PubMed] [Google Scholar]
- 70.Switzar L, Giera M, Niessen WMA. Protein Digestion: an Overview of the Available Techniques and Recent Development. J Proteome Res. 2013;12(3):1067–1077. doi: 10.1021/pr301201x. [DOI] [PubMed] [Google Scholar]
- 71.Ni W, Lin M, Salinas P, Savickas P, Wu SL, Karger BL. Complete Mapping of a Cystine Knot and Nested Disulfides by Recombinant Human Arylsulfatase A by Multi-Enzyme Digestion and LC-MS Analysis Using CID and ETD. J Am Soc Mass Spectrom. 2013;24(1):125–133. doi: 10.1007/s13361-012-0510-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Na S, Paek E, Choi JS, Kim D, Lee SJ, Kwon J. Characterization of Disulfide Bonds by Planned Digestion and Tandem Mass Spectrometry. Mol Biosyst. 2015;11(4):1156–1164. doi: 10.1039/c4mb00688g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Glatter T, Ludwig C, Ahrné E, Aebersold R, Heck AJR, Schmidt A. Large-Scale Quantitative Assessment of Different In-Solution Protein Digestion Protocols Reveals Superior Cleavage Efficiency of Tandem Lys-C/Trypsin Protocols over Trypsin Digestion. J Proteome Res. 2012;11(11):5145–5156. doi: 10.1021/pr300273g. [DOI] [PubMed] [Google Scholar]
- 74.Liu F, van Breukelen B, Heck AJ. Facilitation Protein Disulfide Mapping by a Combination of Pepsin Digestion, Electron Transfer Higher Energy Dissociation (EThcD), and a Dedicated Search Algorithm SlinkS. Mol Cell Proteomics. 2014;13(10):2776–2786. doi: 10.1074/mcp.O114.039057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Go EP, Hua D, Desaire H. Glycosylation and Disulfide Bond Analysis of Transiently and Stably Expressed Clade C HIV-1 gp140 Trimers in 293T Cells Identifies Disulfide Heterogeneity Present in both Proteins and Differences in O-Linked Glycosylation. J Proteome Res. 2014;13(9):4012–4027. doi: 10.1021/pr5003643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Moulaei T, Stuchlik O, Reed M, Yuan W, Pohl J, Lu W, Haugh-Krumpe L, O’Keefe BR, Wlodawer A. Topology of the Disulfide Bonds in the Antiviral Lectin Scytovirin. Protein Sci. 2010;19(9):1649–1661. doi: 10.1002/pro.445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Pike GM, Madden BJ, Melder DC, Charlesworth MC, Federspiel MJ. Simple, Automated, High Resolution Mass Spectrometry Method to Determine the Disulfide Bond and Glycosylation Patterns of a Complex Protein. J Biol Chem. 2011;286(20):17954–17967. doi: 10.1074/jbc.M111.229377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Go EP, Zhang Z, Menon S, Desaire H. Analysis of the Disulfide Bond Arrangement of the HIV Envelope Protein CON-S gp140 ΔCFI Shows Variability in the V1 and V2 Regions. J Proteome Res. 2011;10(2):578–591. doi: 10.1021/pr100764a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Cramer CN, Kelstrup CD, Olsen JV, Haselmann KF, Nielsen PK. Complete Mapping of Complex Disulfide Patterns and Closely-Spaced Cysteines by In-Source Reduction and Data-Dependent Mass Spectrometry. Anal Chem. 2017;89(11):5949–5957. doi: 10.1021/acs.analchem.7b00424. [DOI] [PubMed] [Google Scholar]
- 80.Clark DF, Go EP, Toumi ML, Desaire H. Collision Induced Dissociation Products of Disulfide-Bonded Peptides: Ions Result from the Cleavage of more than one Bond. J Am Soc Mass Spectrom. 2011;22(3):492–498. doi: 10.1007/s13361-010-0064-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wu SL, Jiang H, Hancock W, Karger BL. Identification of the Unpaired Cysteine Status and Complete Mapping of the 17 Disulfides of Recombinant Tissue Plasminogen Activator Using LC-MS with Electron Transfer Dissociation/Collision Induced Dissociation. Anal Chem. 2010;82:5296–5303. doi: 10.1021/ac100766r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Choi S, Jeong J, Na S, Lee HS, Kim HY, Lee KJ, Paek E. New Algorithm for the Identification of Intact Disulfide Linkages Based on Fragmentation Characteristics in Tandem Mass Spectra. J Proteome Res. 2010;9(1):626–635. doi: 10.1021/pr900771r. [DOI] [PubMed] [Google Scholar]
- 83.Chrisman PA, Pitteri SJ, Hogan JM, McLuckey SA. SO2−* Electron Transfer Ion/Ion Reactions with Disulfide Linked Polypeptide Ions. J Am Soc Mass Spectrom. 2005;16(7):1020–1030. doi: 10.1016/j.jasms.2005.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Gunawardena HP, Gorenstein L, Erickson DE, Xia Y, McLuckey SA. Electron Transfer Dissociation of Multiply Protonated and Fixed Charge Disulfide Linked Polypeptides. Int J Mass Spectrom. 2007;265(2–3):130–138. [Google Scholar]
- 85.Olsen JV, Macek B, Lange O, Makarov A, Horning S, Mann M. Higher-Energy C-Trap Dissociation for Peptide Modification Analysis. Nat Methods. 2007;4(9):709–712. doi: 10.1038/nmeth1060. [DOI] [PubMed] [Google Scholar]
- 86.Frese CK, Altelaar AFM, van den Toorn H, Nolting D, Griep-Raming J, Heck AJR, Mohammed S. Toward Full Peptide Sequence Coverage by Dual Fragmentation Combining Electron-Transfer and Higher-Energy Collision Dissociation Tandem Mass Spectrometry. Anal Chem. 2012;84(22):9668–9673. doi: 10.1021/ac3025366. [DOI] [PubMed] [Google Scholar]
- 87.Brunner AM, Lössl P, Liu F, Huguet R, Mullen C, Yamashita M, Zabrouskov V, Makarov A, Altelaar AFM, Heck AJR. Benchmarking Mutliple Fragmentation Methods on an Orbitrap Fusion for Top-Down Phospho-Proteoform Characterization. Anal Chem. 2015;87(8):4152–4158. doi: 10.1021/acs.analchem.5b00162. [DOI] [PubMed] [Google Scholar]
- 88.Frese CK, Altelaar AFM, Hennrich ML, Nolting D, Zeller M, Griep-Raming J, Heck AJR, Mohammed S. Improved Peptide Identification by Targeted Fragmentation Using CID, HCD and ETC on an LTQ-Orbitrap Velos. J Proteome Res. 2011;10(5):2377–2388. doi: 10.1021/pr1011729. [DOI] [PubMed] [Google Scholar]
- 89.Fung YME, Kjeldsen F, Silivra OA, Chan TWD, Zubarev RA. Facile Disulfide Bond Cleavage in Gaseous Peptide and Protein Cations by Ultraviolet Photodissociation at 157 nm. Angew Chem Int Ed. 2005;44:6399–6403. doi: 10.1002/anie.200501533. [DOI] [PubMed] [Google Scholar]
- 90.Agarwal A, Diedrich JK, Julian RR. Direct Elucidation of Disulfide Bond Partners Using Ultraviolet Photodissociation Mass Spectrometry. Anal Chem. 2011;83(17):6455–6458. doi: 10.1021/ac201650v. [DOI] [PubMed] [Google Scholar]
- 91.Cole SR, Ma X, Zhang X, Xia Y. Electron Transfer Dissociation (ETD) of Peptides Containing Intrachain Disulfide Bonds. J Am Soc Mass Spectrom. 2012;23(2):310–320. doi: 10.1007/s13361-011-0300-z. [DOI] [PubMed] [Google Scholar]
- 92.Durand KL, Tan L, Stinson CA, Love-Nkansah CB, Ma X, Xia Y. Assigning Peptide Disulfide Linkage Pattern Among Regio-Isomers via Methoxy Addition to Disulfide and Tandem Mass Spectrometry. J Am Soc Mass Spectrom. 2017;28(6):1099–1108. doi: 10.1007/s13361-017-1595-1. [DOI] [PubMed] [Google Scholar]
- 93.Massonnet P, Upert G, Smargiasso N, Gilles N, Quinton L, De Pauw E. Combined use of Ion Mobility and Collision-Induced Dissociation to Investigate the Opening of Disulfide Bridges by Electron-Transfer Dissociation in Peptides Bearing Two Disulfide Bonds. Anal Chem. 2015;87(10):5240–5246. doi: 10.1021/acs.analchem.5b00245. [DOI] [PubMed] [Google Scholar]
- 94.Mikesh LM, Ueberheide B, Chi A, Coon JJ, Syka JEP, Shabanowitz J, Hunt DF. The Utility of ETD Mass Spectrometry in Proteomic Analysis. Biochim Biophys Acta. 2006;1764(12):1811–1822. doi: 10.1016/j.bbapap.2006.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Lu C, Liu D, Liu H, Motchnik P. Characterization of Monoclonal Antibody Size Variants Containing Extra Light Chains. mAbs. 2013;5(1):102–113. doi: 10.4161/mabs.22965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Klapoetke S, Xie MH. Disulfide Bond Characterization of Human Factor Xa by Mass Spectrometry through Protein-Level Partial Reduction. J Pharmaceut Biomed. 2017;132:238–246. doi: 10.1016/j.jpba.2016.10.005. [DOI] [PubMed] [Google Scholar]
- 97.Albert A, Eksteen JI, Isaksson J, Sengee M, Hansen T, Vasskog T. General Approach to Determine Disulfide Connectivity in Cysteine-Rich Peptides by Sequential Alkylation on Solid Phase and Mass Spectrometry. Anal Chem. 2016;88:9539–9546. doi: 10.1021/acs.analchem.6b02115. [DOI] [PubMed] [Google Scholar]
- 98.Li X, Xu W, Paporello B, Richardson D, Liu H. Liquid Chromatography and Mass Spectrometry with Post-Column Partial Reduction for the Analysis of Native and Scrambled Disulfide Bonds. Anal Biochem. 2013;439(2):184–186. doi: 10.1016/j.ab.2013.04.021. [DOI] [PubMed] [Google Scholar]
- 99.Li X, Wang F, Xu W, May K, Richardson D, Liu H. Disulfide Bond Assignment of an IgG1 Monoclonal Antibody by LC-MS with Post-Column Partial Reduction. Anal Biochem. 2013;436(2):93–100. doi: 10.1016/j.ab.2013.01.020. [DOI] [PubMed] [Google Scholar]
- 100.Liu H, Lei QP, Washabaugh M. Characterization of IgG2 Disulfide Bonds with LC/MS/MS and Postcolumn Online Reduction. Anal Chem. 2016;88(10):5080–5087. doi: 10.1021/acs.analchem.5b04368. [DOI] [PubMed] [Google Scholar]
- 101.Cramer CN, Haselmann KF, Olsen JV, Nielsen PK. Disulfide Linkage Characterization of Disulfide Bond-Containing Proteins and Peptides by Reducing Electrochemistry and Mass Spectrometry. Anal Chem. 2016;88(3):1585–1592. doi: 10.1021/acs.analchem.5b03148. [DOI] [PubMed] [Google Scholar]
- 102.Switzar L, Nicolardi S, Rutten JW, Oberstein SA, Aartsma-Rus A, van der Burgt YE. In-Depth Characterization of Protein Disulfide Bonds by Online Liquid Chromatography-Electrochemistry-Mass Spectrometry. J Am Soc Mass Spectrom. 2016;27(1):50–58. doi: 10.1007/s13361-015-1258-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Zheng Q, Zhang H, Chen H. Integration of Online Digestion and Electrolytic Reduction with Mass Spectrometry for Rapid Disulfide-Containing Protein Structural Analysis. Int J Mass Spectrom. 2013;353:84–92. doi: 10.1016/j.ijms.2013.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Zhang Y, Dewald HD, Chen H. Online Mass Spectrometric Analysis of Proteins/Peptides Following Electrolytic Cleavage of Disulfide Bonds. J Proteome Res. 2011;10(3):1293–1304. doi: 10.1021/pr101053q. [DOI] [PubMed] [Google Scholar]
- 105.Jiang H, Wu SL, Karger BL, Hancock WS. Mass Spectrometric Analysis of Innovator, Counterfeit, and Follow-on Recombinant Human Growth Hormone. Biotechnol Prog. 2009;25(1):207–218. doi: 10.1002/btpr.72. [DOI] [PubMed] [Google Scholar]
- 106.Xiang T, Chumsae C, Liu H. Localization and Quantitation of Free Sulfhydryl in Recombinant Monoclonal Antibodies by Differential Labeling with 12C and 13C Iodoacetic Acid and LC-MS Analysis. Anal Chem. 2009;81(19):8101–8108. doi: 10.1021/ac901311y. [DOI] [PubMed] [Google Scholar]
- 107.Chumsae C, Gaza-Bulseco G, Liu H. Identification and Localization of Unpaired Cysteine Residues in Monoclonal Antibodies by Fluorescence Labeling and Mass Spectrometry. Anal Chem. 2009;81(15):6449–6457. doi: 10.1021/ac900815z. [DOI] [PubMed] [Google Scholar]
- 108.Xu H, Zhang L, Freitas MA. Identification and Characterization of Disulfide Bonds in Proteins and Peptides from Tandem MS Data by use of the MassMatrix and MS/MS Search Engine. J Proteome Res. 2008;7(1):138–144. doi: 10.1021/pr070363z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Murad W, Singh R, Yen TY. An Efficient Algorithmic Approach for Mass Spectrometry-Based Disulfide Connectivity Determination Using Multi-Ion Analysis. BMC Bioinformatics. 2011;12(Suppl 1):S12. doi: 10.1186/1471-2105-12-S1-S12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Murad W, Singh R. MS2DB+: A Software for Determination of Disulfide Bonds Using Multi-Ion Analysis. IEEE Trans Nanobioscience. 2013;12(2):69–71. doi: 10.1109/TNB.2012.2212029. [DOI] [PubMed] [Google Scholar]
- 111.Huang SY, Wen CH, Li DT, Hsu JL, Chen C, Shi FK, Lin YY. Assignment of Disulfide-Linked Peptides Using Automatic a1 Ion Recognition. Anal Chem. 2008;80(23):9135–9140. doi: 10.1021/ac8013725. [DOI] [PubMed] [Google Scholar]
- 112.Huang SY, Hsieh YT, Chen CH, Chen CC, Sung WC, Chou MY, Chen SF. Automatic Disulfide Bond Assignment Using a1 Ion Screening by Mass Spectrometry for Structural Characterization of Protein Pharmaceuticals. Anal Chem. 2012;84(11):4900–4906. doi: 10.1021/ac3005007. [DOI] [PubMed] [Google Scholar]
- 113.Huang SY, Chen SF, Chen CH, Huang HW, Wu WG, Sung WC. Global Disulfide Bond Profiling for Crude Snake Venom Using Dimethyl Labeling Coupled with Mass Spectrometry and RADAR Algorithm. Anal Chem. 2014;86(17):8742–8750. doi: 10.1021/ac501931t. [DOI] [PubMed] [Google Scholar]
- 114.Goyder MS, Rebeaud F, Pfeifer ME, Kalman F. Strategies in Mass Spectrometry for the Assignment of Cys-Cys Disulfide Connectivities in Proteins. Expert Rev Proteomics. 2013;10(5):489–501. doi: 10.1586/14789450.2013.837663. [DOI] [PubMed] [Google Scholar]
- 115.Bagal D, Valliere-Douglass JF, Balland A, Schnier PD. Resolving Disulfide Structural Isoforms of IgG2 Monoclonal Antibodies by Ion Mobility Mass Spectrometry. Anal Chem. 2010;82(16):6751–6755. doi: 10.1021/ac1013139. [DOI] [PubMed] [Google Scholar]
- 116.Wang Q, Lacher NA, Muralidhara BK, Schlittler MR, Aykent S, Demarest CW. Rapid and Refined Separation of Human IgG2 Disulfide Isomers Using Superficially Porous Particles. J Sep Sci. 2010;33(17–18):2671–2680. doi: 10.1002/jssc.201000230. [DOI] [PubMed] [Google Scholar]
- 117.Cao X, He Y, Smith J, Wirth MJ. Alleviating Nonlinear Behavior of Disulfide Isoforms in the Reversed-Phase Liquid Chromatography of IgG2. J Chromatogr A. 2015;1410:147–153. doi: 10.1016/j.chroma.2015.07.098. [DOI] [PMC free article] [PubMed] [Google Scholar]